

Abstract
Alcoholic liver disease (ALD) remains an important health problem worldwide. The disease spectrum is featured by early steatosis, steatohepatitis (steatosis with inflammatory cells infiltration and necrosis), with some individuals ultimately progressing to fibrosis/cirrhosis. Although the disease progression is well characterized, no effective therapies are currently available for the treatment in humans. The mechanisms underlying the initiation and progression of ALD are multifactorial and complex. Emerging evidence supports that adipose tissue dysfunction contributes to the pathogenesis of ALD. In the first part of this review, we discuss the mechanisms whereby chronic alcohol exposure contributed to adipose tissue dysfunction, including cell death, inflammation and insulin resistance. It has been long known that aberrant hepatic methionine metabolism is a major metabolic abnormality induced by chronic alcohol exposure and plays an etiological role in the pathogenesis of ALD. The recent studies in our group documented the similar metabolic effect of chronic alcohol drinking on methionine in adipose tissue. In the second part of this review, we also briefly discuss the recent research progress in the field with a focus on how abnormal methionine metabolism in adipose tissue contributes to adipose tissue dysfunction and liver damage.
Keywords: Alcohol, Lipolysis, Adipose, Methylation, Methionine, Liver
Core tip: Alcoholic liver disease (ALD) remains an important health problem worldwide. The disease spectrum is featured by early steatosis, steatohepatitis (steatosis with inflammatory cells infiltration and necrosis), with some individuals ultimately progressing to fibrosis/cirrhosis. Although the disease progression is well characterized, no effective therapies are currently available for the treatment in humans. The mechanisms underlying the initiation and progression of ALD are multifactorial and complex. Emerging evidence supports that adipose tissue dysfunction contributes to the pathogenesis of ALD. In this review, we discuss the mechanisms whereby chronic alcohol exposure contributed to adipose tissue dysfunction, as well as their contribution to ALD.
ALCOHOLIC LIVER DISEASE
Sustained and excessive alcohol consumption is often accompanied with pathological changes in the liver, termed alcoholic liver disease (ALD). The spectrum of ALD encompasses steatosis, steatohepatitis (steatosis with inflammatory cells infiltration and hepatocyte necrosis), with some individuals ultimately progressing to fibrosis/cirrhosis, leading to increased risk of hepatocellular carcinoma[1-3]. The early-stage ALD, including steatosis and early steatohepatitis, is clinically reversible after termination of alcohol drinking, although the latter takes longer time for the recovery. The pathomechanism implicated in the development of ALD is complex and believed to involve multiple pathogenic factors. Although much progress has been made over last three decades of research on the mechanisms underlying the disease, ALD remains an important health problem worldwide. It ranks among the major causes of morbidity and mortality in the world, and affects millions of patients worldwide each year and there is currently no Food and Drug Administration-approved therapy available to halt or reverse this process in humans.
PATHOGENESIS OF ALD
Steatosis
Hepatic steatosis, characterized by the excessive accumulation of fat in hepatocytes, is the most common and earliest response of the liver to chronic alcohol consumption. Although “pure” steatosis is clinically considered to be a benign condition, excessive fat accumulation makes hepatocytes vulnerable to the attack of “the second hit”, such as proinflammatory cytokines and oxidative stress, leading to the progression to steatohepatitis[4,5]. The mechanisms involved in the development of alcohol-induced hepatic steatosis are multifactorial and remain to be fully elucidated. Sterol regulatory element binding proteins (SREBP)-1c, a master transcription factor controlling de novo lipogenesis, is upregulated in the liver of mice chronically exposed to ethanol-containing diet[6]. Importantly, liver-specific knockout of SREBP-1c protected mice against alcohol-induced fatty liver and liver damage[7], supporting the notion that enhanced hepatic de novo lipogenic process plays a pivotal role in alcohol-triggered fat accumulation in the liver. Moreover, chronic alcohol exposure is associated with impaired fatty acid β-oxidation, contributing to fat accumulation in hepatocytes. Suppressions of both adenosine monophosphate-activated protein kinase (AMPK) and peroxisome proliferator-activated receptor-alpha (PPAR-α), two regulatory proteins of fatty acids oxidation, are mechanistically involved in this process[8,9]. Furthermore, long-term alcohol consumption is reported to enhance uptakes of free fatty acids (FFAs) and triglyceride-rich lipoproteins by hepatocytes[10,11] and impair hepatic very-low-density lipoprotein secretion via suppressing microsomal triglyceride transfer protein activity[12], thereby contributing to fatty liver after chronic alcohol exposure.
Steatohepatitis
Steatohepatitis is characterized by fatty liver, hepatic neutrophil infiltration, and hepatocyte cell death. The stage is a prerequisite for progression to fibrosis and cirrhosis[13]. The molecular mechanism for the progression from steatosis to steatohepatitis involve complicated interactions between the direct effects of toxic ethanol metabolites on different cell types in the liver, overproduction of reactive oxygen species (ROS), and overactivated inflammatory responses[14-21]. Acetaldehyde, the major product of ethanol metabolism in the liver, plays an important role in the development of alcoholic steatohepatitis. Acetaldehyde is a reactive compound and highly toxic to hepatocytes. It binds to both proteins and DNA, leading to not only their functional changes but also activation of adaptive immune system and immune cell infiltration to the damaged liver[22,23]. Moreover, acetaldehyde also impair mitochondrial integrity and function, leading to oxidative stress and cell death[24-26]. Oxidative stress, derived from an imbalance between ROS production and cellular antioxidant capability, is believed to play a critical role in the transition from simple steatosis to steatohepatitis[27]. Many pathways have been suggested to contribute to the occurrence of oxidative stress in response to chronic ethanol exposure. In hepatocytes, the cytochrome P450 2E1 (CYP2E1) activation and mitochondria dysfunction seem to play central roles in inducing cellular oxidative stress state. CYP2E1 is highly inducible and has high catalytic activity for ethanol. During its catalytic circle, CYP2E1 generate significant amount of ROS, which can subsequently leads to cellular injury, lipid peroxidation, and mitochondrial damage[28-31]. Chronic alcohol consumption is associated with increased CYP2E1 expression and activity[32], partially resulting from increased protein stability due to decreased proteasomal degradation[33,34]. CYP2E1 activity correlates with ethanol-induced liver injury and lipid peroxidation, which was reduced by the inhibition of CYP2E1 using either chemical inhibitors or genetic knockout of CYP2E1 gene[35-38]. The detrimental effects of chronic alcohol consumption on liver mitochondria have been well documented. Long-term alcohol exposure is associated with reduced activity of key enzymes in mitochondrial respiratory chain and decreased mitochondria oxygen utilization[39,40]. Alcohol also leads to disruption between complex I and complex III of the mitochondrial electron transport chain, leading to elevated superoxide anion production[41]. Furthermore, chronic alcohol drinking is associated with damaged mitochondrial membrane integrity, possibly due to acetaldehyde accumulation, leading to defective mitochondrial GSH uptake, which sensitizes hepatocytes to TNF-α-induced cell death[42,43]. In addition to hepatocytes, accumulated evidence identified Kupffer cells (KCs) activation to be a central element in the development of steatohepatitis. Chronic alcohol exposure not only results in intestinal gram-negative bacterial overgrowth but also increases gut permeability, leading to the translocation of bacteria-derived LPS from the gut lumen to the blood[44-46]. The increased circulating LPS induces inflammatory actions in KCs in the liver via interacting with toll-like receptor (TLR)-4, resulting in production of oxidative stress and proinflammatory cytokines, including TNF-α, which plays a pivotal role in alcohol-induced hepatocyte cell death[47-50].
Fibrosis
Liver fibrogenesis is a wound-healing response to chronic liver injury. It is featured by excessive extracellular deposition of collagen and other extracellular matrix proteins, mainly derived from activated hepatic stellate cells (HSCs)[51-53]. The major stimuli for HSCs activation during chronic alcohol consumption include acetaldehyde[54,55], the main ethanol metabolite, and proinflammatory cytokines produced by KCs in response to gut-derived products via LPS-TLR4 interactions[56,57].
ADIPOSE TISSUE REGULATES WHOLE BODY LIPID HOMEOSTASIS
Adipose tissue plays a central role in regulating whole body lipid and energy homeostasis. The modern concept viewed adipose tissue as a complex, essential, and highly active metabolic and endocrine organ, not only as a reservoir for energy storage[58,59]. Adipose tissue communicates with other tissues and organs, including the liver, to integrate total body lipid homeostasis via both controlling circulating FFAs levels and synthesizing and releasing a host of secreted molecules, collectively designated as adipokines, including leptin, adiponectin, resistin, to name a few[58,60]. Adipose tissue stores excess energy in the form of triglycerides (TGs) and releases it in the form of FFAs, a process called lipolysis, to meet other tissues or organs’ energy requirements[61]. Under physiological conditions, lipid storage and release are both coordinated and tightly regulated so that lipid fuels are stored during postprandial periods and released during fasting states. When the regulation of TG storage and FFAs release by adipose tissue is perturbed, particularly when release of FA becomes dissociated from energy requirements in extra-adipose tissues, plasma FA levels are elevated and excessive storage of TGs in these tissues, such as the liver, ensues, leading to hepatic steatosis (fatty liver). The critical role of adipose tissue in regulating hepatic lipid homeostasis can be best manifested by the facts that both long-term fasting and lipodystrophy (adipose tissue deficiency) results in fatty liver[62-64]. Insulin plays a dominant role in suppressing adipose tissue lipolysis during postprandial periods[65,66]. Therefore, adipose tissue insulin resistance is associated with elevated circulating FFAs levels due to uncontrolled lipolysis.
ADIPOSE TISSUE DYSFUNCTION IN ALD
Although it has been well-established that chronic alcohol consumption exerts a detrimental effect on hepatic fat synthesis and disposal, leading to the development of hepatic steatosis, emerging evidence supports that adipose tissue dysfunction also plays an important role in the pathogenesis of ALD. In the clinic setting, it has been reported that visceral fat accumulation is positively related to the onset of alcoholic liver damage and body mass index represents an independent risk factor for fibrosis in alcoholic patients[67-70]. Moreover, adipose tissue inflammation is correlated with the severity of pathological features in the liver of patients with ALD[69]. Experimentally, long-term alcohol consumption is associated with adipose tissue oxidative stress, insulin resistance, inflammation, adipocyte cell death, and adiponectin decline[71-75]. Chronic alcohol feeding results in hyper-lipolysis (degradation of TGs) in adipose tissue, leading to elevated circulation FFAs concentrations and a significant loss of white adipose tissue[10]. A recent study demonstrated that moderate obesity and alcohol synergistically induced steatohepatitis[76], further supporting the critical role of adipose tissue (dys)/function in the development of ALD. Importantly, both rosiglitazone (a PPAR-γ agonist mainly targeting adipocytes)[77] and recombinant adiponectin (an adipokine exclusively secreted by adipocytes)[78] improved ALD, suggesting that improving adipose tissue function represents a potential therapeutic approach for ALD.
EFFECTS OF CHRONIC ALCOHOL CONSUMPTION ON ADIPOSE TISSUE FUNCTION
Oxidative stress, cell death, and inflammation
Although the liver is the major organ for ethanol metabolism, accumulated evidence supports that chronic alcohol feeding is also associated with increased oxidative stress, cell death, and inflammatory response in adipose tissue. Upregulation of CYP2E1 seems to play an important role in these events. Chronic ethanol feeding of rats causes increased expression and enzymatic activity of CYP2E1 in adipocytes/adipose tissue[72,79], leading to oxidative stress induction, which is evidenced by elevated 4-HNE production and protein carbonyls. When fully-differentiated 3T3-L1 adipocytes with CYP2E1 overexpression were exposed to ethanol-containing medium, increased oxidative stress was observed[72]. In contrast, overexpression of antisense CYP2E1 in adipocytes prevented ethanol-triggered oxidative stress[72]. Adipocyte cell death plays a pivotal role in triggering adipose tissue inflammatory responses[80,81]. Chronic alcohol consumption results in CYP2E1/Bid cascade-dependent adipocyte cell death in adipose tissue of rats, which contributes to adipose tissue inflammation in response to chronic alcohol exposure in that both Cyp2e1 and Bid knockout mice are protected from adipose tissue inflammation[72]. Interestingly, C1q, a component of the classical pathway of complement, seems to represent a critical link between cell death and inflammation in the setting of chronic alcohol consumption[72].
Adipokines
Alcohol consumption is known to disrupt adipokine release from adipose tissue[82,83]. Adiponectin and leptin are key adipokines that modulate hepatic lipid homeostasis. Reduced circulating leptin and adiponectin levels are observed in chronic alcohol exposed rodents, which contribute to the development of ALD[72,84-86]. Via binding with adiponectin receptor on hepatocytes, adiponectin activates AMPK pathway to stimulate FA oxidation, leading to reduced fat accumulation in the liver[87,88]. Indeed, either exogenous adiponectin administration or endogenously stimulating adiponectin production attenuate alcohol induced fatty liver in mice[78,84,89,90]. Mechanisms underlying alcohol-triggered adiponectin decline are multifactorial, including oxidative stress[79,91], hyperhomocysteinemia (HHcy)[84], and heme-oxygenase-1 dependent pathway[92]. Other than adiponectin, leptin is another adipokine reported to be affected by chronic alcohol consumption and contribute to ALD. Animal studies showed that chronic alcohol consumption decreases circulating leptin levels[80,93,94], which is associated with adipose tissue mass reduction. Importantly, exogenous leptin administration restored plasma leptin reduction triggered by chronic alcohol feeding and alleviated hepatic steatosis[95]. It is noteworthy here that clinical investigations support that moderate alcohol consumption is associated with increased adiponectin levels[95,96].
Lipolysis
Uncontrolled adipose tissue lipolysis, mainly due to insulin resistance, plays an etiological role in the development of obesity-related non-alcoholic fatty liver disease[97-100]. Insulin signaling in adipocytes plays a central role in controlling FFAs release by adipose tissue via lipolysis[65,66]. Peripheral insulin resistance derived from obesity compromises the suppressive effect of insulin on lipolysis, leading to increased exposure of the liver to circulating FFAs with subsequent development of fatty liver. The effect of chronic alcohol consumption on adipose tissue insulin sensitivity remains to be fully characterized; however, existing evidence supports that chronic alcohol consumption is associated with adipose tissue insulin resistance. In rats, chronic ethanol feeding results in impaired insulin-stimulated glucose transport in adipocytes, which is associated with a disruption of insulin-mediated Cbl/TC10 signaling and actin polymerization[97]. Moreover, chronic ethanol feeding compromises the suppression of the anti-lipolytic effects of insulin in adipocytes in both rats and mice, leading to enhanced triglyceride degradation in adipose tissue[10,73]. Interestingly, in comparison to subcutaneous fat, visceral white adipose tissue seems to be more susceptible to alcohol-induced lipolysis enhancement, which may involve increased acetaldehyde production[100]. In contrast to adipose tissue insulin resistance in obese animals which is associate with an increase in body weight and adipocyte size, chronic alcohol consumption leads to reduced adipose tissue mass and adipocyte size[10], indicating that distinct mechanism(s) are involved in the initiation of adipose tissue insulin resistance. Adipose triglyceride lipase and hormone sensitive lipase (HSL) are two critical enzymes catalyzing fatty acids release from the adipose tissue. Both enzymes are found to be activated in mice chronically fed with alcohol. Interestingly, chronic alcohol feeding has no effect on plasma catecholamine and insulin levels, while PTEN and SOC3, two negative regulators of insulin signaling pathway, are up-regulated, leading to insulin resistance[10].
ABERRANT METHIONINE METABOLISM AND ALCOHOL-INDUCED ADIPOSE TISSUE DYSFUNCTION
Methionine metabolism abnormality in ALD: Aberrant hepatic methionine metabolism is a major metabolic abnormality induced by chronic alcohol exposure and plays an etiological role in the pathogenesis of ALD[101-104]. As illustrated in Figure 1, intracellular methionine metabolism involves two major pathways, transmethylation reaction and transsulfuration reaction. The first step in methionine metabolism is the formation of S-adenosylmethionine (SAM) in a reaction catalyzed by methionine adenosyltransferase. Under physiological conditions, most of the SAM generated per day is used in transmethylation reactions in which methyl groups are added to a vast number of molecules, including DNA, RNA, phospholipids, histones, and other proteins, via specific methyltransferases. In this process, SAM is converted to S-adenosylhomocysteine (SAH), followed by homocysteine (Hcy) and cysteine, a precursor for glutathione biosynthesis, via transsulfuration pathway. SAH is a potent competitive inhibitor of most methyltransferases studied and decreased SAM:SAH ratio has been widely employed as an indicator of suppressed transmethylation reactions[105,106]. While chronic alcohol exposure leads to hepatic SAM deficiency, both SAH and Hcy are increased in the liver in response to alcohol[101,104,107]. HHcy is associated with ER stress induction, leading to hepatocyte dysfunction[108], while increased intracellular SAH level enhances the sensitivity of hepatocytes to TNF-α-induced hepatotoxicity[104], both contributing to the pathogenesis of ALD.
Figure 1.
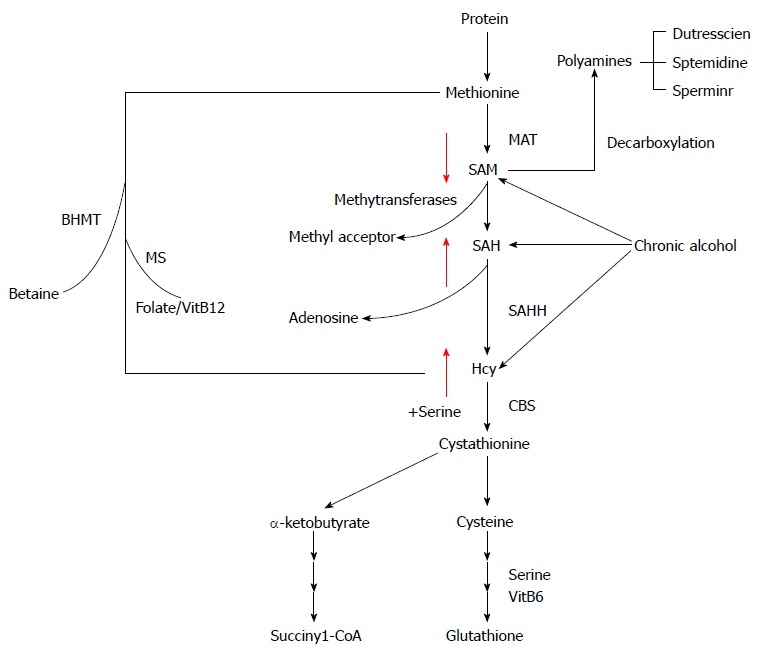
Intracellular methionine metabolism. Chronic alcohol consumption causes SAM deficiency, but enhancement of homocysteine and SAH. MAT: Methionine adenosyl-transferase; SAM: S-adenosylmethionine; SAH: S-adenosylhomocysteine; Hcy: Homocysteine; CBS: Cystathionine beta synthase; SAHH: S-adensylmonocysteine hydrolase; MS: Methionine synthase; BHMT: Betaine-homocysteine methyltransferase.
Aberrant adipose methionine metabolism and hyper-lipolytic response in adipose tissue: Studies using both genetic and dietary animal models demonstrated that HHcy is associated with adipose tissue dysfunction[71,108-112], suggesting that methionine metabolism regulates adipose tissue function. We are the first to report that, similar to its effect on the liver, chronic alcohol feeding induces methionine metabolism abnormality in adipose tissue, which is characterized by SAM deficiency, and accumulation of Hcy and SAH[71,113], leading to significant decrease of the SAM/SAH ratio, a strong indicator of inhibitory transmethylation reactions (hypomethylation). HSL is considered a rate-limiting lipase for adipose tissue lipolysis[114]. Upon lipolytic hormone stimulation, such as with catecholamine, cAMP/PKA-mediated-phosphorylations in certain serine residues activate HSL[115]. In contrast, protein phosphatase 2A -catalyzed dephosphorylation at Ser660 leads to HSL inactivation[116,117]. PP2A is a heterotrimeric protein phosphatase. The catalytic and scaffold subunits of PP2A are ubiquitously expressed and have remarkable sequence conservation within eukaryotes. Interestingly, accumulating evidence reveals that PP2A activation is under control of intracellular methylation status[118,119]. Carboxyl methylation of the PP2A catalytic subunit, catalyzed by PP2A-specific methyltransferaseleucine carboxyl methyltransferase-1, plays a critical role in regulating holoenzyme assembly[120]. These previous studies provide rational for us to posit that altered intracellular methylation status in adipocytes may affect adipose tissue lipolytic response. In a very recent study, we provided evidence supporting that intracellular hypomethylation status in adipocytes in the setting of chronic alcohol feeding contributes to adipose tissue hyper-lipolytic response in ALD via suppressing PP2A activity, leading to HSL overactivation[114] (Figure 2). Our data support that rectification of methionine metabolism through dietary supplementation of betaine protects against alcohol-induced liver damage, at least partially via improving adipose tissue function. Taken together, the recent research in our group suggest that aberrant methionine metabolism in adipocytes contributes to alcohol-elicited adipose tissue dysfunction and liver damage.
Figure 2.
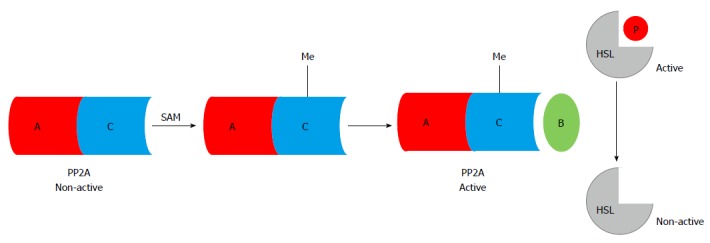
S-adenosylmethionine-dependent methylation reactions are required for protein phosphatase 2A activation, which dephosphorylates (inhibits) hormone sensitive lipase. Chronic alcohol consumption induces intracellular hypomethylation status in adipocytes, which suppresses PP2A activity, leading to uncontrolled HSL activation. SAM: S-adenosylmethionine; Me: Methyl group; PP2A: Protein phosphatase 2A; HSL: Hormone sensitive lipase.
CONCLUSION
Despite its high prevalence, ALD has received limited attention during past decades and no major breakthrough in terms of its clinical management. Emerging evidence shows that adipose tissue plays an important role in both initiation and progression of liver damage induced by chronic alcohol consumption. However, the exact underlying cellular/molecular mechanisms involved in adipose tissue dysfunction in ALD remain to be fully elucidated. Future efforts in identifying the common factors that promote dysfunctions in both adipose tissue and the liver in response to chronic alcohol consumption will pave the way for the discovery of new therapeutic approach.
Footnotes
Supported by National Institutes of Health NIAAA, No. R01 AA017442.
Conflict-of-interest statement: The authors declare no conflict of interest.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: May 28, 2015
First decision: August 25, 2015
Article in press: November 25, 2015
P- Reviewer: Abenavoli L, Gonzalez-Reimers E, Mattner J, Takahashi T, Wang JS, Xu CF S- Editor: Ji FF L- Editor: A E- Editor: Jiao XK
References
- 1.Adachi M, Brenner DA. Clinical syndromes of alcoholic liver disease. Dig Dis. 2005;23:255–263. doi: 10.1159/000090173. [DOI] [PubMed] [Google Scholar]
- 2.Tilg H, Day CP. Management strategies in alcoholic liver disease. Nat Clin Pract Gastroenterol Hepatol. 2007;4:24–34. doi: 10.1038/ncpgasthep0683. [DOI] [PubMed] [Google Scholar]
- 3.Grant BF, Dufour MC, Harford TC. Epidemiology of alcoholic liver disease. Semin Liver Dis. 1988;8:12–25. doi: 10.1055/s-2008-1040525. [DOI] [PubMed] [Google Scholar]
- 4.Stewart S, Jones D, Day CP. Alcoholic liver disease: new insights into mechanisms and preventative strategies. Trends Mol Med. 2001;7:408–413. doi: 10.1016/s1471-4914(01)02096-2. [DOI] [PubMed] [Google Scholar]
- 5.Reddy JK, Rao MS. Lipid metabolism and liver inflammation. II. Fatty liver disease and fatty acid oxidation. Am J Physiol Gastrointest Liver Physiol. 2006;290:G852–G858. doi: 10.1152/ajpgi.00521.2005. [DOI] [PubMed] [Google Scholar]
- 6.You M, Fischer M, Deeg MA, Crabb DW. Ethanol induces fatty acid synthesis pathways by activation of sterol regulatory element-binding protein (SREBP) J Biol Chem. 2002;277:29342–29347. doi: 10.1074/jbc.M202411200. [DOI] [PubMed] [Google Scholar]
- 7.Ji C, Chan C, Kaplowitz N. Predominant role of sterol response element binding proteins (SREBP) lipogenic pathways in hepatic steatosis in the murine intragastric ethanol feeding model. J Hepatol. 2006;45:717–724. doi: 10.1016/j.jhep.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 8.You M, Matsumoto M, Pacold CM, Cho WK, Crabb DW. The role of AMP-activated protein kinase in the action of ethanol in the liver. Gastroenterology. 2004;127:1798–1808. doi: 10.1053/j.gastro.2004.09.049. [DOI] [PubMed] [Google Scholar]
- 9.Fischer M, You M, Matsumoto M, Crabb DW. Peroxisome proliferator-activated receptor alpha (PPARalpha) agonist treatment reverses PPARalpha dysfunction and abnormalities in hepatic lipid metabolism in ethanol-fed mice. J Biol Chem. 2003;278:27997–28004. doi: 10.1074/jbc.M302140200. [DOI] [PubMed] [Google Scholar]
- 10.Zhong W, Zhao Y, Tang Y, Wei X, Shi X, Sun W, Sun X, Yin X, Sun X, Kim S, et al. Chronic alcohol exposure stimulates adipose tissue lipolysis in mice: role of reverse triglyceride transport in the pathogenesis of alcoholic steatosis. Am J Pathol. 2012;180:998–1007. doi: 10.1016/j.ajpath.2011.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Z, Dou X, Li S, Zhang X, Sun X, Zhou Z, Song Z. Nuclear factor (erythroid-derived 2)-like 2 activation-induced hepatic very-low-density lipoprotein receptor overexpression in response to oxidative stress contributes to alcoholic liver disease in mice. Hepatology. 2014;59:1381–1392. doi: 10.1002/hep.26912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sugimoto T, Yamashita S, Ishigami M, Sakai N, Hirano K, Tahara M, Matsumoto K, Nakamura T, Matsuzawa Y. Decreased microsomal triglyceride transfer protein activity contributes to initiation of alcoholic liver steatosis in rats. J Hepatol. 2002;36:157–162. doi: 10.1016/s0168-8278(01)00263-x. [DOI] [PubMed] [Google Scholar]
- 13.Bataller R, Rombouts K, Altamirano J, Marra F. Fibrosis in alcoholic and nonalcoholic steatohepatitis. Best Pract Res Clin Gastroenterol. 2011;25:231–244. doi: 10.1016/j.bpg.2011.02.010. [DOI] [PubMed] [Google Scholar]
- 14.Lieber CS. Alcoholic fatty liver: its pathogenesis and mechanism of progression to inflammation and fibrosis. Alcohol. 2004;34:9–19. doi: 10.1016/j.alcohol.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 15.Cederbaum AI, Lu Y, Wu D. Role of oxidative stress in alcohol-induced liver injury. Arch Toxicol. 2009;83:519–548. doi: 10.1007/s00204-009-0432-0. [DOI] [PubMed] [Google Scholar]
- 16.Leung TM, Nieto N. CYP2E1 and oxidant stress in alcoholic and non-alcoholic fatty liver disease. J Hepatol. 2013;58:395–398. doi: 10.1016/j.jhep.2012.08.018. [DOI] [PubMed] [Google Scholar]
- 17.Nassir F, Ibdah JA. Role of mitochondria in alcoholic liver disease. World J Gastroenterol. 2014;20:2136–2142. doi: 10.3748/wjg.v20.i9.2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fernández-Checa JC, Kaplowitz N, García-Ruiz C, Colell A. Mitochondrial glutathione: importance and transport. Semin Liver Dis. 1998;18:389–401. doi: 10.1055/s-2007-1007172. [DOI] [PubMed] [Google Scholar]
- 19.Mandrekar P, Ambade A, Lim A, Szabo G, Catalano D. An essential role for monocyte chemoattractant protein-1 in alcoholic liver injury: regulation of proinflammatory cytokines and hepatic steatosis in mice. Hepatology. 2011;54:2185–2197. doi: 10.1002/hep.24599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wheeler MD, Kono H, Yin M, Nakagami M, Uesugi T, Arteel GE, Gäbele E, Rusyn I, Yamashina S, Froh M, et al. The role of Kupffer cell oxidant production in early ethanol-induced liver disease. Free Radic Biol Med. 2001;31:1544–1549. doi: 10.1016/s0891-5849(01)00748-1. [DOI] [PubMed] [Google Scholar]
- 21.Uesugi T, Froh M, Arteel GE, Bradford BU, Thurman RG. Toll-like receptor 4 is involved in the mechanism of early alcohol-induced liver injury in mice. Hepatology. 2001;34:101–108. doi: 10.1053/jhep.2001.25350. [DOI] [PubMed] [Google Scholar]
- 22.Svegliati-Baroni G, Baraona E, Rosman AS, Lieber CS. Collagen-acetaldehyde adducts in alcoholic and nonalcoholic liver diseases. Hepatology. 1994;20:111–118. doi: 10.1016/0270-9139(94)90142-2. [DOI] [PubMed] [Google Scholar]
- 23.You M, Crabb DW. Recent advances in alcoholic liver disease II. Minireview: molecular mechanisms of alcoholic fatty liver. Am J Physiol Gastrointest Liver Physiol. 2004;287:G1–G6. doi: 10.1152/ajpgi.00056.2004. [DOI] [PubMed] [Google Scholar]
- 24.Zhou Z, Sun X, Kang YJ. Ethanol-induced apoptosis in mouse liver: Fas- and cytochrome c-mediated caspase-3 activation pathway. Am J Pathol. 2001;159:329–338. doi: 10.1016/S0002-9440(10)61699-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bradham CA, Plümpe J, Manns MP, Brenner DA, Trautwein C. Mechanisms of hepatic toxicity. I. TNF-induced liver injury. Am J Physiol. 1998;275:G387–G392. doi: 10.1152/ajpgi.1998.275.3.G387. [DOI] [PubMed] [Google Scholar]
- 26.Román J, Colell A, Blasco C, Caballeria J, Parés A, Rodés J, Fernández-Checa JC. Differential role of ethanol and acetaldehyde in the induction of oxidative stress in HEP G2 cells: effect on transcription factors AP-1 and NF-kappaB. Hepatology. 1999;30:1473–1480. doi: 10.1002/hep.510300623. [DOI] [PubMed] [Google Scholar]
- 27.Meagher EA, Barry OP, Burke A, Lucey MR, Lawson JA, Rokach J, FitzGerald GA. Alcohol-induced generation of lipid peroxidation products in humans. J Clin Invest. 1999;104:805–813. doi: 10.1172/JCI5584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Porter TD, Coon MJ. Cytochrome P-450. Multiplicity of isoforms, substrates, and catalytic and regulatory mechanisms. J Biol Chem. 1991;266:13469–13472. [PubMed] [Google Scholar]
- 29.Rendic S, Di Carlo FJ. Human cytochrome P450 enzymes: a status report summarizing their reactions, substrates, inducers, and inhibitors. Drug Metab Rev. 1997;29:413–580. doi: 10.3109/03602539709037591. [DOI] [PubMed] [Google Scholar]
- 30.Guengerich FP. Uncommon P450-catalyzed reactions. Curr Drug Metab. 2001;2:93–115. doi: 10.2174/1389200013338694. [DOI] [PubMed] [Google Scholar]
- 31.Lewis DF, Pratt JM. The P450 catalytic cycle and oxygenation mechanism. Drug Metab Rev. 1998;30:739–786. doi: 10.3109/03602539808996329. [DOI] [PubMed] [Google Scholar]
- 32.Lieber CS. Cytochrome P-4502E1: its physiological and pathological role. Physiol Rev. 1997;77:517–544. doi: 10.1152/physrev.1997.77.2.517. [DOI] [PubMed] [Google Scholar]
- 33.Gonzalez FJ. The 2006 Bernard B. Brodie Award Lecture. Cyp2e1. Drug Metab Dispos. 2007;35:1–8. doi: 10.1124/dmd.106.012492. [DOI] [PubMed] [Google Scholar]
- 34.Roberts BJ, Song BJ, Soh Y, Park SS, Shoaf SE. Ethanol induces CYP2E1 by protein stabilization. Role of ubiquitin conjugation in the rapid degradation of CYP2E1. J Biol Chem. 1995;270:29632–29635. doi: 10.1074/jbc.270.50.29632. [DOI] [PubMed] [Google Scholar]
- 35.Bai J, Cederbaum AI. Adenovirus-mediated expression of CYP2E1 produces liver toxicity in mice. Toxicol Sci. 2006;91:365–371. doi: 10.1093/toxsci/kfj165. [DOI] [PubMed] [Google Scholar]
- 36.Bardag-Gorce F, Yuan QX, Li J, French BA, Fang C, Ingelman-Sundberg M, French SW. The effect of ethanol-induced cytochrome p4502E1 on the inhibition of proteasome activity by alcohol. Biochem Biophys Res Commun. 2000;279:23–29. doi: 10.1006/bbrc.2000.3889. [DOI] [PubMed] [Google Scholar]
- 37.Bell LN, Temm CJ, Saxena R, Vuppalanchi R, Schauer P, Rabinovitz M, Krasinskas A, Chalasani N, Mattar SG. Bariatric surgery-induced weight loss reduces hepatic lipid peroxidation levels and affects hepatic cytochrome P-450 protein content. Ann Surg. 2010;251:1041–1048. doi: 10.1097/SLA.0b013e3181dbb572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Butura A, Nilsson K, Morgan K, Morgan TR, French SW, Johansson I, Schuppe-Koistinen I, Ingelman-Sundberg M. The impact of CYP2E1 on the development of alcoholic liver disease as studied in a transgenic mouse model. J Hepatol. 2009;50:572–583. doi: 10.1016/j.jhep.2008.10.020. [DOI] [PubMed] [Google Scholar]
- 39.Bailey SM, Cunningham CC. Contribution of mitochondria to oxidative stress associated with alcoholic liver disease. Free Radic Biol Med. 2002;32:11–16. doi: 10.1016/s0891-5849(01)00769-9. [DOI] [PubMed] [Google Scholar]
- 40.Bailey SM, Pietsch EC, Cunningham CC. Ethanol stimulates the production of reactive oxygen species at mitochondrial complexes I and III. Free Radic Biol Med. 1999;27:891–900. doi: 10.1016/s0891-5849(99)00138-0. [DOI] [PubMed] [Google Scholar]
- 41.Hoek JB, Cahill A, Pastorino JG. Alcohol and mitochondria: a dysfunctional relationship. Gastroenterology. 2002;122:2049–2063. doi: 10.1053/gast.2002.33613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lluis JM, Colell A, García-Ruiz C, Kaplowitz N, Fernández-Checa JC. Acetaldehyde impairs mitochondrial glutathione transport in HepG2 cells through endoplasmic reticulum stress. Gastroenterology. 2003;124:708–724. doi: 10.1053/gast.2003.50089. [DOI] [PubMed] [Google Scholar]
- 43.Colell A, García-Ruiz C, Miranda M, Ardite E, Marí M, Morales A, Corrales F, Kaplowitz N, Fernández-Checa JC. Selective glutathione depletion of mitochondria by ethanol sensitizes hepatocytes to tumor necrosis factor. Gastroenterology. 1998;115:1541–1551. doi: 10.1016/s0016-5085(98)70034-4. [DOI] [PubMed] [Google Scholar]
- 44.Enomoto N, Schemmer P, Ikejima K, Takei Y, Sato N, Brenner DA, Thurman RG. Long-term alcohol exposure changes sensitivity of rat Kupffer cells to lipopolysaccharide. Alcohol Clin Exp Res. 2001;25:1360–1367. [PubMed] [Google Scholar]
- 45.Enomoto N, Ikejima K, Yamashina S, Hirose M, Shimizu H, Kitamura T, Takei Y, Sato And N, Thurman RG. Kupffer cell sensitization by alcohol involves increased permeability to gut-derived endotoxin. Alcohol Clin Exp Res. 2001;25:51S–54S. doi: 10.1097/00000374-200106001-00012. [DOI] [PubMed] [Google Scholar]
- 46.Tsukamoto H, Takei Y, McClain CJ, Joshi-Barve S, Hill D, Schmidt J, Deaciuc I, Barve S, Colell A, Garcia-Ruiz C, et al. How is the liver primed or sensitized for alcoholic liver disease? Alcohol Clin Exp Res. 2001;25:171S–181S. doi: 10.1097/00000374-200105051-00029. [DOI] [PubMed] [Google Scholar]
- 47.Miller AM, Horiguchi N, Jeong WI, Radaeva S, Gao B. Molecular mechanisms of alcoholic liver disease: innate immunity and cytokines. Alcohol Clin Exp Res. 2011;35:787–793. doi: 10.1111/j.1530-0277.2010.01399.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Szabo G, Mandrekar P, Petrasek J, Catalano D. The unfolding web of innate immune dysregulation in alcoholic liver injury. Alcohol Clin Exp Res. 2011;35:782–786. doi: 10.1111/j.1530-0277.2010.01398.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Szabo G. Gut-liver axis in alcoholic liver disease. Gastroenterology. 2015;148:30–36. doi: 10.1053/j.gastro.2014.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mandrekar P, Szabo G. Signalling pathways in alcohol-induced liver inflammation. J Hepatol. 2009;50:1258–1266. doi: 10.1016/j.jhep.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fujii H, Kawada N. Fibrogenesis in alcoholic liver disease. World J Gastroenterol. 2014;20:8048–8054. doi: 10.3748/wjg.v20.i25.8048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Suh YG, Jeong WI. Hepatic stellate cells and innate immunity in alcoholic liver disease. World J Gastroenterol. 2011;17:2543–2551. doi: 10.3748/wjg.v17.i20.2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mormone E, George J, Nieto N. Molecular pathogenesis of hepatic fibrosis and current therapeutic approaches. Chem Biol Interact. 2011;193:225–231. doi: 10.1016/j.cbi.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reyes-Gordillo K, Shah R, Arellanes-Robledo J, Hernández-Nazara Z, Rincón-Sánchez AR, Inagaki Y, Rojkind M, Lakshman MR. Mechanisms of action of acetaldehyde in the up-regulation of the human α2(I) collagen gene in hepatic stellate cells: key roles of Ski, SMAD3, SMAD4, and SMAD7. Am J Pathol. 2014;184:1458–1467. doi: 10.1016/j.ajpath.2014.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Szuster-Ciesielska A, Plewka K, Daniluk J, Kandefer-Szerszeń M. Zinc supplementation attenuates ethanol- and acetaldehyde-induced liver stellate cell activation by inhibiting reactive oxygen species (ROS) production and by influencing intracellular signaling. Biochem Pharmacol. 2009;78:301–314. doi: 10.1016/j.bcp.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 56.Paik YH, Schwabe RF, Bataller R, Russo MP, Jobin C, Brenner DA. Toll-like receptor 4 mediates inflammatory signaling by bacterial lipopolysaccharide in human hepatic stellate cells. Hepatology. 2003;37:1043–1055. doi: 10.1053/jhep.2003.50182. [DOI] [PubMed] [Google Scholar]
- 57.Gobejishvili L, Barve S, Breitkopf-Heinlein K, Li Y, Zhang J, Avila DV, Dooley S, McClain CJ. Rolipram attenuates bile duct ligation-induced liver injury in rats: a potential pathogenic role of PDE4. J Pharmacol Exp Ther. 2013;347:80–90. doi: 10.1124/jpet.113.204933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ahima RS, Flier JS. Adipose tissue as an endocrine organ. Trends Endocrinol Metab. 2000;11:327–332. doi: 10.1016/s1043-2760(00)00301-5. [DOI] [PubMed] [Google Scholar]
- 59.Frühbeck G, Gómez-Ambrosi J, Muruzábal FJ, Burrell MA. The adipocyte: a model for integration of endocrine and metabolic signaling in energy metabolism regulation. Am J Physiol Endocrinol Metab. 2001;280:E827–E847. doi: 10.1152/ajpendo.2001.280.6.E827. [DOI] [PubMed] [Google Scholar]
- 60.Piya MK, McTernan PG, Kumar S. Adipokine inflammation and insulin resistance: the role of glucose, lipids and endotoxin. J Endocrinol. 2013;216:T1–T15. doi: 10.1530/JOE-12-0498. [DOI] [PubMed] [Google Scholar]
- 61.Suganami T, Tanaka M, Ogawa Y. Adipose tissue inflammation and ectopic lipid accumulation. Endocr J. 2012;59:849–857. doi: 10.1507/endocrj.ej12-0271. [DOI] [PubMed] [Google Scholar]
- 62.Gan SK, Watts GF. Is adipose tissue lipolysis always an adaptive response to starvation?: implications for non-alcoholic fatty liver disease. Clin Sci (Lond) 2008;114:543–545. doi: 10.1042/CS20070461. [DOI] [PubMed] [Google Scholar]
- 63.Safar Zadeh E, Lungu AO, Cochran EK, Brown RJ, Ghany MG, Heller T, Kleiner DE, Gorden P. The liver diseases of lipodystrophy: the long-term effect of leptin treatment. J Hepatol. 2013;59:131–137. doi: 10.1016/j.jhep.2013.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Reue K, Phan J. Metabolic consequences of lipodystrophy in mouse models. Curr Opin Clin Nutr Metab Care. 2006;9:436–441. doi: 10.1097/01.mco.0000232904.82038.db. [DOI] [PubMed] [Google Scholar]
- 65.Chakrabarti P, Kandror KV. Adipose triglyceride lipase: a new target in the regulation of lipolysis by insulin. Curr Diabetes Rev. 2011;7:270–277. doi: 10.2174/157339911796397866. [DOI] [PubMed] [Google Scholar]
- 66.Frühbeck G, Méndez-Giménez L, Fernández-Formoso JA, Fernández S, Rodríguez A. Regulation of adipocyte lipolysis. Nutr Res Rev. 2014;27:63–93. doi: 10.1017/S095442241400002X. [DOI] [PubMed] [Google Scholar]
- 67.Hart CL, Morrison DS, Batty GD, Mitchell RJ, Davey Smith G. Effect of body mass index and alcohol consumption on liver disease: analysis of data from two prospective cohort studies. BMJ. 2010;340:c1240. doi: 10.1136/bmj.c1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tsai J, Ford ES, Zhao G, Li C, Greenlund KJ, Croft JB. Co-occurrence of obesity and patterns of alcohol use associated with elevated serum hepatic enzymes in US adults. J Behav Med. 2012;35:200–210. doi: 10.1007/s10865-011-9353-5. [DOI] [PubMed] [Google Scholar]
- 69.Loomba R, Bettencourt R, Barrett-Connor E. Synergistic association between alcohol intake and body mass index with serum alanine and aspartate aminotransferase levels in older adults: the Rancho Bernardo Study. Aliment Pharmacol Ther. 2009;30:1137–1149. doi: 10.1111/j.1365-2036.2009.04141.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shen Z, Li Y, Yu C, Shen Y, Xu L, Xu C, Xu G. A cohort study of the effect of alcohol consumption and obesity on serum liver enzyme levels. Eur J Gastroenterol Hepatol. 2010;22:820–825. doi: 10.1097/MEG.0b013e3283328b86. [DOI] [PubMed] [Google Scholar]
- 71.Sebastian BM, Roychowdhury S, Tang H, Hillian AD, Feldstein AE, Stahl GL, Takahashi K, Nagy LE. Identification of a cytochrome P4502E1/Bid/C1q-dependent axis mediating inflammation in adipose tissue after chronic ethanol feeding to mice. J Biol Chem. 2011;286:35989–35997. doi: 10.1074/jbc.M111.254201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Song Z, Zhou Z, Deaciuc I, Chen T, McClain CJ. Inhibition of adiponectin production by homocysteine: a potential mechanism for alcoholic liver disease. Hepatology. 2008;47:867–879. doi: 10.1002/hep.22074. [DOI] [PubMed] [Google Scholar]
- 73.Kang L, Chen X, Sebastian BM, Pratt BT, Bederman IR, Alexander JC, Previs SF, Nagy LE. Chronic ethanol and triglyceride turnover in white adipose tissue in rats: inhibition of the anti-lipolytic action of insulin after chronic ethanol contributes to increased triglyceride degradation. J Biol Chem. 2007;282:28465–28473. doi: 10.1074/jbc.M705503200. [DOI] [PubMed] [Google Scholar]
- 74.Chen X, Sebastian BM, Nagy LE. Chronic ethanol feeding to rats decreases adiponectin secretion by subcutaneous adipocytes. Am J Physiol Endocrinol Metab. 2007;292:E621–E628. doi: 10.1152/ajpendo.00387.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tang H, Sebastian BM, Axhemi A, Chen X, Hillian AD, Jacobsen DW, Nagy LE. Ethanol-induced oxidative stress via the CYP2E1 pathway disrupts adiponectin secretion from adipocytes. Alcohol Clin Exp Res. 2012;36:214–222. doi: 10.1111/j.1530-0277.2011.01607.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xu J, Lai KK, Verlinsky A, Lugea A, French SW, Cooper MP, Ji C, Tsukamoto H. Synergistic steatohepatitis by moderate obesity and alcohol in mice despite increased adiponectin and p-AMPK. J Hepatol. 2011;55:673–682. doi: 10.1016/j.jhep.2010.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sun X, Tang Y, Tan X, Li Q, Zhong W, Sun X, Jia W, McClain CJ, Zhou Z. Activation of peroxisome proliferator-activated receptor-γ by rosiglitazone improves lipid homeostasis at the adipose tissue-liver axis in ethanol-fed mice. Am J Physiol Gastrointest Liver Physiol. 2012;302:G548–G557. doi: 10.1152/ajpgi.00342.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.West M. Dead adipocytes and metabolic dysfunction: recent progress. Curr Opin Endocrinol Diabetes Obes. 2009;16:178–182. doi: 10.1097/med.0b013e3283292327. [DOI] [PubMed] [Google Scholar]
- 79.Lafontan M. Adipose tissue and adipocyte dysregulation. Diabetes Metab. 2014;40:16–28. doi: 10.1016/j.diabet.2013.08.002. [DOI] [PubMed] [Google Scholar]
- 80.Rogers CQ, Ajmo JM, You M. Adiponectin and alcoholic fatty liver disease. IUBMB Life. 2008;60:790–797. doi: 10.1002/iub.124. [DOI] [PubMed] [Google Scholar]
- 81.Nicolás JM, Fernández-Solà J, Fatjó F, Casamitjana R, Bataller R, Sacanella E, Tobías E, Badía E, Estruch R. Increased circulating leptin levels in chronic alcoholism. Alcohol Clin Exp Res. 2001;25:83–88. [PubMed] [Google Scholar]
- 82.You M, Considine RV, Leone TC, Kelly DP, Crabb DW. Role of adiponectin in the protective action of dietary saturated fat against alcoholic fatty liver in mice. Hepatology. 2005;42:568–577. doi: 10.1002/hep.20821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Otaka M, Konishi N, Odashima M, Jin M, Wada I, Matsuhashi T, Ohba R, Watanabe S. Effect of alcohol consumption on leptin level in serum, adipose tissue, and gastric mucosa. Dig Dis Sci. 2007;52:3066–3069. doi: 10.1007/s10620-006-9635-x. [DOI] [PubMed] [Google Scholar]
- 84.Tan X, Sun X, Li Q, Zhao Y, Zhong W, Sun X, Jia W, McClain CJ, Zhou Z. Leptin deficiency contributes to the pathogenesis of alcoholic fatty liver disease in mice. Am J Pathol. 2012;181:1279–1286. doi: 10.1016/j.ajpath.2012.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.You M, Rogers CQ. Adiponectin: a key adipokine in alcoholic fatty liver. Exp Biol Med (Maywood) 2009;234:850–859. doi: 10.3181/0902-MR-61. [DOI] [PubMed] [Google Scholar]
- 86.Marra F, Bertolani C. Adipokines in liver diseases. Hepatology. 2009;50:957–969. doi: 10.1002/hep.23046. [DOI] [PubMed] [Google Scholar]
- 87.Shen Z, Liang X, Rogers CQ, Rideout D, You M. Involvement of adiponectin-SIRT1-AMPK signaling in the protective action of rosiglitazone against alcoholic fatty liver in mice. Am J Physiol Gastrointest Liver Physiol. 2010;298:G364–G374. doi: 10.1152/ajpgi.00456.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ajmo JM, Liang X, Rogers CQ, Pennock B, You M. Resveratrol alleviates alcoholic fatty liver in mice. Am J Physiol Gastrointest Liver Physiol. 2008;295:G833–G842. doi: 10.1152/ajpgi.90358.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhang X, Wang Z, Li J, Gu D, Li S, Shen C, Song Z. Increased 4-hydroxynonenal formation contributes to obesity-related lipolytic activation in adipocytes. PLoS One. 2013;8:e70663. doi: 10.1371/journal.pone.0070663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mandal P, Pritchard MT, Nagy LE. Anti-inflammatory pathways and alcoholic liver disease: role of an adiponectin/interleukin-10/heme oxygenase-1 pathway. World J Gastroenterol. 2010;16:1330–1336. doi: 10.3748/wjg.v16.i11.1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Santolaria F, Pérez-Cejas A, Alemán MR, González-Reimers E, Milena A, de la Vega MJ, Martínez-Riera A, Gómez-Rodríguez MA. Low serum leptin levels and malnutrition in chronic alcohol misusers hospitalized by somatic complications. Alcohol Alcohol. 2004;38:60–66. doi: 10.1093/alcalc/agg015. [DOI] [PubMed] [Google Scholar]
- 92.Calissendorff J, Brismar K, Röjdmark S. Is decreased leptin secretion after alcohol ingestion catecholamine-mediated? Alcohol Alcohol. 2004;39:281–286. doi: 10.1093/alcalc/agh054. [DOI] [PubMed] [Google Scholar]
- 93.Joosten MM, Witkamp RF, Hendriks HF. Alterations in total and high-molecular-weight adiponectin after 3 weeks of moderate alcohol consumption in premenopausal women. Metabolism. 2011;60:1058–1063. doi: 10.1016/j.metabol.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 94.Joosten MM, Beulens JW, Kersten S, Hendriks HF. Moderate alcohol consumption increases insulin sensitivity and ADIPOQ expression in postmenopausal women: a randomised, crossover trial. Diabetologia. 2008;51:1375–1381. doi: 10.1007/s00125-008-1031-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jou J, Choi SS, Diehl AM. Mechanisms of disease progression in nonalcoholic fatty liver disease. Semin Liver Dis. 2008;28:370–379. doi: 10.1055/s-0028-1091981. [DOI] [PubMed] [Google Scholar]
- 96.Jacome-Sosa MM, Parks EJ. Fatty acid sources and their fluxes as they contribute to plasma triglyceride concentrations and fatty liver in humans. Curr Opin Lipidol. 2014;25:213–220. doi: 10.1097/MOL.0000000000000080. [DOI] [PubMed] [Google Scholar]
- 97.Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115:1343–1351. doi: 10.1172/JCI23621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Combs TP, Marliss EB. Adiponectin signaling in the liver. Rev Endocr Metab Disord. 2014;15:137–147. doi: 10.1007/s11154-013-9280-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sebastian BM, Nagy LE. Decreased insulin-dependent glucose transport by chronic ethanol feeding is associated with dysregulation of the Cbl/TC10 pathway in rat adipocytes. Am J Physiol Endocrinol Metab. 2005;289:E1077–E1084. doi: 10.1152/ajpendo.00296.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhang W, Zhong W, Sun X, Sun Q, Tan X, Li Q, Sun X, Zhou Z. Visceral white adipose tissue is susceptible to alcohol-induced lipodystrophy in rats: role of acetaldehyde. Alcohol Clin Exp Res. 2015;39:416–423. doi: 10.1111/acer.12646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Barak AJ, Beckenhauer HC, Kharbanda KK, Tuma DJ. Chronic ethanol consumption increases homocysteine accumulation in hepatocytes. Alcohol. 2001;25:77–81. doi: 10.1016/s0741-8329(01)00168-9. [DOI] [PubMed] [Google Scholar]
- 102.Mato JM, Cámara J, Fernández de Paz J, Caballería L, Coll S, Caballero A, García-Buey L, Beltrán J, Benita V, Caballería J, et al. S-adenosylmethionine in alcoholic liver cirrhosis: a randomized, placebo-controlled, double-blind, multicenter clinical trial. J Hepatol. 1999;30:1081–1089. doi: 10.1016/s0168-8278(99)80263-3. [DOI] [PubMed] [Google Scholar]
- 103.Lu SC, Huang ZZ, Yang H, Mato JM, Avila MA, Tsukamoto H. Changes in methionine adenosyltransferase and S-adenosylmethionine homeostasis in alcoholic rat liver. Am J Physiol Gastrointest Liver Physiol. 2000;279:G178–G185. doi: 10.1152/ajpgi.2000.279.1.G178. [DOI] [PubMed] [Google Scholar]
- 104.Song Z, Zhou Z, Uriarte S, Wang L, Kang YJ, Chen T, Barve S, McClain CJ. S-adenosylhomocysteine sensitizes to TNF-alpha hepatotoxicity in mice and liver cells: a possible etiological factor in alcoholic liver disease. Hepatology. 2004;40:989–997. doi: 10.1002/hep.20412. [DOI] [PubMed] [Google Scholar]
- 105.Mato JM, Alvarez L, Ortiz P, Pajares MA. S-adenosylmethionine synthesis: molecular mechanisms and clinical implications. Pharmacol Ther. 1997;73:265–280. doi: 10.1016/s0163-7258(96)00197-0. [DOI] [PubMed] [Google Scholar]
- 106.Chiang PK, Gordon RK, Tal J, Zeng GC, Doctor BP, Pardhasaradhi K, McCann PP. S-Adenosylmethionine and methylation. FASEB J. 1996;10:471–480. [PubMed] [Google Scholar]
- 107.Ji C, Kaplowitz N. Betaine decreases hyperhomocysteinemia, endoplasmic reticulum stress, and liver injury in alcohol-fed mice. Gastroenterology. 2003;124:1488–1499. doi: 10.1016/s0016-5085(03)00276-2. [DOI] [PubMed] [Google Scholar]
- 108.Gupta S, Kruger WD. Cystathionine beta-synthase deficiency causes fat loss in mice. PLoS One. 2011;6:e27598. doi: 10.1371/journal.pone.0027598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mikael LG, Wang XL, Wu Q, Jiang H, Maclean KN, Rozen R. Hyperhomocysteinemia is associated with hypertriglyceridemia in mice with methylenetetrahydrofolate reductase deficiency. Mol Genet Metab. 2009;98:187–194. doi: 10.1016/j.ymgme.2009.05.011. [DOI] [PubMed] [Google Scholar]
- 110.Li Y, Zhang H, Jiang C, Xu M, Pang Y, Feng J, Xiang X, Kong W, Xu G, Li Y, et al. Hyperhomocysteinemia promotes insulin resistance by inducing endoplasmic reticulum stress in adipose tissue. J Biol Chem. 2013;288:9583–9592. doi: 10.1074/jbc.M112.431627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Li Y, Jiang C, Xu G, Wang N, Zhu Y, Tang C, Wang X. Homocysteine upregulates resistin production from adipocytes in vivo and in vitro. Diabetes. 2008;57:817–827. doi: 10.2337/db07-0617. [DOI] [PubMed] [Google Scholar]
- 112.Wang Z, Dou X, Yao T, Song Z. Homocysteine inhibits adipogenesis in 3T3-L1 preadipocytes. Exp Biol Med (Maywood) 2011;236:1379–1388. doi: 10.1258/ebm.2011.011234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Dou X, Xia Y, Chen J, Qian Y, Li S, Zhang X, Song Z. Rectification of impaired adipose tissue methylation status and lipolytic response contributes to hepatoprotective effect of betaine in a mouse model of alcoholic liver disease. Br J Pharmacol. 2014;171:4073–4086. doi: 10.1111/bph.12765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Holm C, Kirchgessner TG, Svenson KL, Fredrikson G, Nilsson S, Miller CG, Shively JE, Heinzmann C, Sparkes RS, Mohandas T. Hormone-sensitive lipase: sequence, expression, and chromosomal localization to 19 cent-q13.3. Science. 1988;241:1503–1506. doi: 10.1126/science.3420405. [DOI] [PubMed] [Google Scholar]
- 115.Anthonsen MW, Rönnstrand L, Wernstedt C, Degerman E, Holm C. Identification of novel phosphorylation sites in hormone-sensitive lipase that are phosphorylated in response to isoproterenol and govern activation properties in vitro. J Biol Chem. 1998;273:215–221. doi: 10.1074/jbc.273.1.215. [DOI] [PubMed] [Google Scholar]
- 116.Wood SL, Emmison N, Borthwick AC, Yeaman SJ. The protein phosphatases responsible for dephosphorylation of hormone-sensitive lipase in isolated rat adipocytes. Biochem J. 1993;295(Pt 2):531–535. doi: 10.1042/bj2950531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kinney BP, Qiao L, Levaugh JM, Shao J. B56alpha/protein phosphatase 2A inhibits adipose lipolysis in high-fat diet-induced obese mice. Endocrinology. 2010;151:3624–3632. doi: 10.1210/en.2010-0245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sontag JM, Nunbhakdi-Craig V, Sontag E. Leucine carboxyl methyltransferase 1 (LCMT1)-dependent methylation regulates the association of protein phosphatase 2A and Tau protein with plasma membrane microdomains in neuroblastoma cells. J Biol Chem. 2013;288:27396–27405. doi: 10.1074/jbc.M113.490102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Jackson JB, Pallas DC. Circumventing cellular control of PP2A by methylation promotes transformation in an Akt-dependent manner. Neoplasia. 2012;14:585–599. doi: 10.1593/neo.12768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Stanevich V, Jiang L, Satyshur KA, Li Y, Jeffrey PD, Li Z, Menden P, Semmelhack MF, Xing Y. The structural basis for tight control of PP2A methylation and function by LCMT-1. Mol Cell. 2011;41:331–342. doi: 10.1016/j.molcel.2010.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]