

Abstract
In analogy with many other proteins, Na+/Ca2+ exchangers (NCX) adapt an inverted twofold symmetry of repeated structural elements, while exhibiting a functional asymmetry by stabilizing an outward-facing conformation. Here, structure-based mutant analyses of the Methanococcus jannaschii Na+/Ca2+ exchanger (NCX_Mj) were performed in conjunction with HDX-MS (hydrogen/deuterium exchange mass spectrometry) to identify the structure-dynamic determinants of functional asymmetry. HDX-MS identified hallmark differences in backbone dynamics at ion-coordinating residues of apo-NCX_Mj, whereas Na+or Ca2+ binding to the respective sites induced relatively small, but specific, changes in backbone dynamics. Mutant analysis identified ion-coordinating residues affecting the catalytic capacity (kcat/Km), but not the stability of the outward-facing conformation. In contrast, distinct “noncatalytic” residues (adjacent to the ion-coordinating residues) control the stability of the outward-facing conformation, but not the catalytic capacity. The helix-breaking signature sequences (GTSLPE) on the α1 and α2 repeats (at the ion-binding core) differ in their folding/unfolding dynamics, while providing asymmetric contributions to transport activities. The present data strongly support the idea that asymmetric preorganization of the ligand-free ion-pocket predefines catalytic reorganization of ion-bound residues, where secondary interactions with adjacent residues couple the alternating access. These findings provide a structure-dynamic basis for ion-coupled alternating access in NCX and similar proteins.
Inverted twofold symmetry of repeated structural elements, which evolved via gene duplication and fusion events, represents a common motif among multi-helical proteins including enzymes, receptors, pumps, transporters, and channels. This structural set-up is particularly frequent in transporters1,2. This can be explained by the requirement to have isoenergetic alternating access, where the thermodynamic barrier can be lowered by “adjusting” the equilibrium constant close to unity2,3,4. This hypothetical set-up is especially appealing for symmetric small ligands, such as ions, although many transporters stabilize either the inward- or outward-facing state to fulfill physiological demands3,4,5,6,7,8. However, the underlying structure-dynamic mechanisms remain unclear.
Na+/Ca2+ exchanger (NCX) proteins extrude Ca2+ from the cell5,6 to control Ca2+ homeostasis7,8. NCX proteins catalyze the exchange (3Na+:1Ca2+)9, whereas the Na+ and Ca2+ ions are transported in separate steps10, consistent with the alternating access mechanism11,12. NCX orthologs vary as much as 104-fold in the turnover rates of the transport cycle13,14,15,16 to match dynamic swings in cytosolic Ca2+ levels7,8. Despite these kinetic differences, NCXs share a common ability to stabilize the outward-facing (extracellular) access13,17. This is required to shape the physiological values of Km at opposite sides of the membrane7,8,13,17. Although kinetics16,17 and structural studies18 revealed that under steady-state conditions the outward-facing conformation is stabilized in the presence of Na+ or Ca2+ ions, it has remained unclear whether the ligand-free NCX_Mj is already asymmetric in the absence of ions or ligand binding shifts the equilibrium in favor of the outward-facing conformation.
NCX proteins contain ten transmembrane helices that form two hubs (TM1-TM5 and TM6-TM10) with inverted twofold symmetry1,2,5,6,7,8. The crystal structure of ion-bound NCX_Mj depicts the outward-facing (extracellular) conformation (Fig. 1A) with four ion-binding sites, (Sext, Smid, Sint, and SCa), where twelve residues are involved in ion coordination18 (Fig. 1B). According to the structural data, the Sint and Sext sites have high selectivity for Na+, whereas Smid and SCa lack selectivity for ion binding. The ion pocket of NCX_Mj encompasses highly conserved α1 and α2 repeats with inverted topology, where twelve ion-coordinating residues (four in TM2 and TM7, and two in TM3 and TM8) and two helix-breaking signature sequences (GTSLPE) form the ion-pocket18,19,20,21 (Fig. 2A). According to the original interpretation of the crystallographic data, Sext, Smid, and Sint are occupied by 3Na+ ions, where E54, E213, D240, and N81 coordinate one Na+ ion at Smid, and 1Ca2+ occupies SCa18. According to this interpretation, D240 and N81 solely belong to Smid (Fig. 1B,D) and thus, are involved in Na+ binding/transport activities. Molecular dynamics simulations and ion-flux analyses offer an alternative interpretation, suggesting that 3Na+ ions occupy Sext, Sint, and SCa, whereas Ca2+ occupies SCa22 (Fig. 1C,D). According to this point of view, Smid does not bind either Na+ or Ca2+ ions and one water molecule is bound to protonated D240.
Figure 1. Structure and ion-exchange reactions of NCX_Mj.

(A) Crystal structure of NCX_Mj (PDB 3V5U) in cartoon representation. Helices 1–5 (TM1-5) are orange and helices 6–10 (TM6-10) are pink. Purple and green spheres represent Na+ and Ca2+ ions, respectively. (B) Ion coordination as suggested by the crystal structure of NCX_Mj18. (C) 3Na+ ion coordination as suggested by molecular dynamics simulations and ion-flux assays22. (D) Ca2+ binding site. (E) Schematic representation of the ion-flux assay for Na+/45Ca2+ exchange or Ca2+/45Ca2+ exchange and the ping-pong mechanism describing the exchange reactions10,16,17. The red, green, and purple arrows represent Ca2+ -entry, Ca2+ -exit, and Na+ -exit steps of the transport cycle, respectively. The dotted arrows represent the reactions of the transport cycle, which negligibly contribute to the observed ion-exchange reactions under the given experimental conditions. The K′d and K”d values represent the dissociation constants for Ca2+ binding to NCX_Mj at the cytosolic and extracellular sides, respectively. The rate contributions to the observed kcat and Km values of the exchange reactions are expressed in the equations described in Table S1.
Figure 2. HDX-MS analysis of NCX_Mj.

(A) Sequences of the α-repeats. Ion-coordinating residues are in red. Helix-breaking residues are in blue. (B) The heat map at 1,200 sec is overlaid on the crystal structure of NCX_Mj (PDB 3V5U) for the apo, Na+ -bound, and Ca2+ -bound forms. The color key indicates the HDX level. The numberings indicate regions with deuterium information (not taking into account the N-terminus amino acids of peptides without amide hydrogens). (C) Deuterium uptake plots for regions 48–52 and 231–237 and for peptides encompassing ion-coordinating residues. Data are presented as the mean ± SD (n = 3). Plots of regions 48–52 and 231–237 were calculated from the difference of deuteration between two overlapping peptides (47–59 & 53–59 and 215–230 & 215–237, respectively). (D) Mass spectra of peptide 215–230 in the presence of Ca2+. Note the characteristic EX1 kinetics bimodal isotope distribution, and the intensity of the one at high masses increasing with time relative to the one at a low masses.
The present work was undertaken to evaluate the mechanistic role of inverted pair residues within the ion pocket of NCX_Mj by exploring especially suited kinetic analyses (Fig. 1E, S1,S2, and Table S1) of overexpressed NCX_Mj mutants possessing a right-side-out orientation13,22. In parallel, HDX MS (hydrogen/deuterium exchange mass spectrometry) experiments were performed on purified NCX_Mj proteins to monitor the backbone dynamics within the ion-binding pocket. In general, HDX-MS measures the exchange of backbone amide hydrogen with deuterium in solvent, where the measured HDX is related to the solvent’s accessibility and the backbone folding/unfolding dynamics (flexible regions take up more deuterium than do the rigid domains)23,24,25. Since HDX-MS can detect small and slow conformational changes in the presence or absence of ligand26,27, this technique is well suited for monitoring the folding/refolding dynamics of interest in NCX_Mj.
The present HDX-MS and kinetic studies revealed that the asymmetric preorganization of inverted pair residues in the ligand-free ion pocket predefines the catalytic reorganization of key ion-coordinating residues, where distinct “noncatalytic” residues stabilize the outward-facing conformation.
Results
In order to envisage the backbone dynamics within the ion-binding pocket and assign the binding sites occupied by each ion, HDX-MS analysis23,24,25,26,27 was performed on apo and ion-bound NCX_Mj. This approach allows one to map not only the backbone dynamics in the absence of Na+ or Ca2+ —it also permits quantitative evaluation of local changes in backbone flexibility upon ion binding at respective sites. Highly purified preparations of NCX_Mj protein were used in all these experiments. MS identification of peptic peptides resulted in 26.7% coverage (Fig. S3), where three peptides cover the 41–59 area. Furthermore, two of them overlap, which led us to consider that three regions of rather small size (5–6 amides), namely, 41–46, 48–52, and 54–59 almost completely cover TM2. Eight peptides enabled us to cover the 195–264 area (from TM7 to the beginning of TM9). The peptides contain ten out of twelve ion-coordinating residues (except S77 and N81) and both α-repeats, including the GTSLPE signature sequence (Fig. 2A, S3, S4).
HDX-MS reveals unusual HDX kinetics within the ion-binding pocket of NCX_Mj
The deuteration levels after 1,200 sec of exchange with and without ions are colored on the structure of NCX_Mj (Fig. 2B), and the corresponding deuterium uptake plots of each region or peptide at 15, 120, and 1,200 sec are shown in Fig. 2C. Interestingly, most peptides exhibited mixed EX1 and EX2 exchange kinetics of deuterium uptake. The EX1 kinetics exhibit hallmark bimodal isotope distribution, with the intensity of the one at high masses increasing with time relative to the one at low masses. EX1 kinetics implies cooperative unfolding events at specific segments of the ion-binding pocket and is quite unusual for conformational transitions that occur under physiologically related conditions (pH, ionic strength, temperature, among others)28. This indicates a local refolding rate slower than the labeling rate. For example, the presence of Ca2+ induced typical EX1 kinetics of HDX for peptide 215–230 (the end of TM7 and the beginning of TM8) (Fig. 2D). In contrast, the classical EX2 kinetics shows one binomial envelope moving towards higher masses with deuteration time28. A minor contribution of EX2 kinetics can also be detected because there is a slight shift towards higher masses in addition to the relative increase in the intensity of the high mass envelope (Fig. 2D).
HDX-MS analysis of apo NCX_Mj protein
For Na+-free samples, Na+ was replaced by choline and for Ca2+-free samples, EDTA was added. Interestingly, in the absence of Na+ and Ca2+ ions, there are marked differences in the local backbone dynamics at key ion-coordinating residues in apo-NCX_Mj (Fig. 2B,C). For example, the 41–59 region, belonging to TM2 encompassing the α1-repeat, displayed distinct deuterium incorporations along the backbone of apo NCX. Overlapping peptides 47–59 and 53–59 enabled us to calculate a low deuterium uptake in region 48–52 (not more than 26% D), in contrast with a much higher deuterium uptake observed in region 54–59 (Fig. 2B,C). This dramatic change in deuterium uptake within TM2 may be induced by the tilting effect of P53, a phenomenon that has already been observed in transmembrane helices29. In sharp contrast with the 41–59 region, a pronounced deuterium uptake was measured in the 205–214 area of apo-NCX_Mj (Fig. 2B,C), which covers TM7 encompassing the α2-repeat. The significance of these findings is that in apo-NCX_Mj the backbone of the ion-coordinating residues, located on TM2 (S51 and E54), is much more rigid than the backbone of the ion-coordinating residues on TM7 (T209, E213, and D240).
HDX-MS analysis of NCX_Mj protein in the presence of Na+ or Ca2+
The ion-bound species of NCX_Mj were studied using saturating concentrations for Na+ (100 mM) or Ca2+ (2 mM). In general, the exchange rate is higher for the apo form than for either the Na+ or Ca2+ bound forms (Fig. 2B,C). This is also true for peptide 215–230, which does not contain residues directly participating in ion ligation and, as mentioned, exhibits EX1 kinetics in the presence of Ca2+, indicating the local destabilization effect of Ca2+. This may be related to the ion-exchange mechanism. At 1,200 sec, the exchange of the relevant peptides (Fig. 2B,C) is similar for the Na+ and Ca2+ -bound forms. Interestingly, in the presence of Na+ or Ca2+, deuterium was not at all incorporated into the 48–52 region (no HDX in its five amides). This behavior may be related to the presence of Na+ ligands A47, T50, and S51 in Sint and the presence of Ca2+ ligands T50, S51, and E54 in SCa, thus, stabilizing the backbone conformation in this region upon ion binding. Assuming the occupation of Smid by Na+ 18 (Fig. 1B), one may expect that Na+ binding at Smid will result in a major decrease in deuterium uptake for peptide 238–245 encompassing D240 (Fig. 2B,C); however, either Na+ or Ca2+ equally protects this region, as compared with the apo form. In fact, at 15 and 120 sec, Ca2+ protects this peptide more than Na+ does.
The two GTSLPE signature sequences on TM2 and TM7 (residues 49–54 and 208–213), although being “symmetry-related”, exhibit marked differences in HDX kinetics (Fig. 2B,C). The α1-repeat, at residues 49–54, exhibits relatively low deuterium uptake in both the apo and bound forms and is highly protected by the presence of either Na+ or Ca2+. In contrast, the α2-repeat at residues 208–213 (peptide 205–214) is much more dynamic in both the apo and bound forms and ligand binding results in less protection. This may represent the functional asymmetry of NCX_Mj13, implemented in an asymmetric arrangement of “catalytic residues” involved in transition state stabilization.
Asymmetric contributions of inverted pair residues to ion-transport activities
For structure-based mutational analyses of ion-transport mechanisms, the initial rates of Na+i or Ca2+i-dependent 45Ca2+ -uptake were measured in right-side-oriented isolated vesicles derived from the plasma membranes of E. coli cells containing overexpressed NCX_Mj mutants13,22. In this experimental set-up the extracellular side matches the extravesicular orientation, where the kinetic parameters of the Na+/Ca2+ and Ca2+/Ca2+ exchange reactions at the ‘extracellular’ (K″m and V″max) and ‘cytosolic’ (K′m and V′max) sides can be measured by varying ion concentrations at the intravesicular and extravesicular compartments (Fig. 1E and Table S1). Single-point mutations to alanine were performed for ion-coordinating pair residues, namely, S77/S236, S51/S210, T50/T209, D240/N81, and E54/E213 (Fig. 3A). Unfortunately, the alanine-substitution approach cannot be applied for the A47/A206 pair18. The N73/N232 pair was also subjected to alanine substitutions. Although N73 and N232 do not directly coordinate the ions, they are located at inverted entries to the ion pocket (Fig. 3A) and form hydrogen bonding with T209 and T50, respectively.
Figure 3. Mutational effects of pair residues on the intrinsic equilibrium (Kint) of bidirectional Ca2+ movements (Ca2+/Ca2+ exchange).

(A) Topological positions of pair residues within the ion pocket of NCX_Mj. Matching pair residues are marked with the same color. (B) The Km and Vmax values of the Na+/Ca2+ and Ca2+/Ca2+ exchange reactions were measured by varying the concentrations of 45Ca2+ (20–2000 μM) in the assay medium at fixed (saturating) concentrations of intravesicular Na+ (160 mM) or Ca2+ (250 μM), as described in Methods (see also Figure S1). The bars represent the mean ± SE. (C) The Km values of the Ca2+/Ca2+ exchange reaction were measured at the ‘extracellular’ (K″m) and ‘cytosolic’ (K′m) sides by varying the Ca2+ concentrations in one compartment and keeping the fixed (saturating) concentrations of Ca2+ in the trans compartment. (D) The Kint values were calculated as the K′m/K″m ratio and plotted vs the kcat values for the same mutant. The kcat values were calculated as kcat = Vmax/[E]t. The expression levels of [E]t were normalized by using the GFP assay (see Methods).
In general, the tested mutations have rather minor effects on the apparent affinity of Ca2+ transport, exhibiting less than twofold changes in the K″m values of the Na+/Ca2+ and Ca2+/Ca2+ exchange activities, whereas they have diverse effects on the V″max values (Fig. 3B). The RA index (representing the ratio of V″max values for a given pair of pair residues) reveals the asymmetric effects of alanine substitutions on the ion exchange activities, with RA values ranging from 2 to 32 (Table 1). Although the tested pair residues display RA ≫ 1 values (reflecting their asymmetric contributions), the S77/S236 pair has an extraordinarily high RA value for both the Na+/Ca2+ and Ca2+/Ca2+ exchanges, which is at least 10 times higher than the RA value imposed by any other pair residues (Table 1).
Table 1. Relative effects of alanine mutations on the ion-exchange capacity in a given pair of inverted residues.
| Mutated pair residues | RA values (V″max1/V″max2) Na/Ca exchange | RA values (V″max1/V″max2) Ca/Ca exchange |
|---|---|---|
| S236A/S77A | 31.3 ± 3.6 | 32.8 ± 2.3 |
| N81A/D240A | 3.5 ± 0.4 | 4.5 ± 0.3 |
| T50A/T209A | 2.5 ± 0.3 | 3.0 ± 0.3 |
| S210A/S51A | 2.2 ± 0.3 | 1.9 ± 0.3 |
| E213A/E54A | 1.8 ± 0.4 | 1.6 ± 0.3 |
| N73A/N232A | 2.1 ± 0.3 | 1.7 ± 0.3 |
The V″max1 and V″max2 ratios of the Na+/Ca2+ and Ca2+/Ca2+ exchange reactions were measured in mutants of matching pair residues as described in Figs 3 and 4 (see also Methods). The V″max1/V″max2 ratios are presented as the index of asymmetry (RA) for a given pair of residues. The numbers represent the mean ± SE.
Mutational effects of ion pocket residues on the intrinsic equilibrium of bidirectional Ca2+ movements
Previous studies have shown that the K′m/K″m ratio of the NCX-mediated Ca2+/Ca2+ exchange reaction accounts for the intrinsic equilibrium (Kint) of bidirectional Ca2+ movements16,17. The Kint ≃ 0.15 observed for NCX_Mj represents the intrinsic asymmetry of bidirectional ion movements (see Table S1 and Fig. S2), suggesting that the rate of Ca2+ movement from the cytosolic to the extracellular space is 5–7 times faster than Ca2+ movement in the opposite direction16. According to this analytical approach, the plotting of Kint vs. kcat values provides valuable information about mutational effects on rate-equilibrium relationships underlying the bidirectional Ca2+ movements. Thus, the K′m and K″m values of Ca2+/Ca2+ exchange were measured for each mutant (Fig. 3C) and the resulting Kint values were plotted against the kcat values for the same mutant (Fig. 3D). A striking finding is that alanine substitutions of ion-coordinating residues have very little (if any) effect on the intrinsic asymmetry of bidirectional Ca2+ movements (Kint = 0.1–0.35), whereas the same mutations have rather dramatic effects on the turnover rates (kcat = 0.01–0.8 s−1) of Ca2+/Ca2+ exchange (Fig. 3D). In sharp contrast, the alanine-substitutions of N73A or N232A increase the Kint value from 0.15 ± 0.05 to 2.8 ± 0.2, whereas they have relatively minor effects on the kcat value (Fig. 3D). These data support the notion that the mutation of N73 or N232 stabilizes the inward-facing access conformation without appreciably affecting the catalytic capacity (kcat/Km). Collectively, the present data indicate that N73 and N232 control Kint by affecting the ground/occluded states of Ca2+ with very little (if any) effects on the transition state.
Ion-coordinating residues stabilizing the transition state of Ca2+ transport
Besides A47 and A206 (which coordinate Na+ at Sint and Sext, respectively, through their backbone carbonyls), all ion-coordinating side chains were replaced by alanine to evaluate the mutational effects on the kcat/Km values of the Na+/Ca2+ or the Ca2+/Ca2+ exchange reactions (Figs 3 and 4). The K″m and kcat values of 45Ca2+ -uptake were measured by varying [45Ca2+]o at saturating [Na+]i or [Ca2+]i (Fig. 1E, S1 and Table S1). E54A and E213A exhibit a 25–50-fold decrease in their kcat/Km values (Figs 3B and 4A,B), whereas the polar (uncharged) residues, D240 (protonated, Smid), S51 (Sint), T209 (Sext), and S77 (Sext) exhibit 10–25% of the WT kcat/Km values for both exchange reactions (Figs 3B and 4A,B). The other polar residues (S236, S210, T50, and N81) have minor (if any) effects on either the kcat or Km values (Figs 3B and 4A,B). Interestingly, N81A retains the WT kcat and Km values for both ion-exchange reactions (Figs 3B and 4A).
Figure 4. Identification of distinct residues stabilizing the Ca2+ transition state.
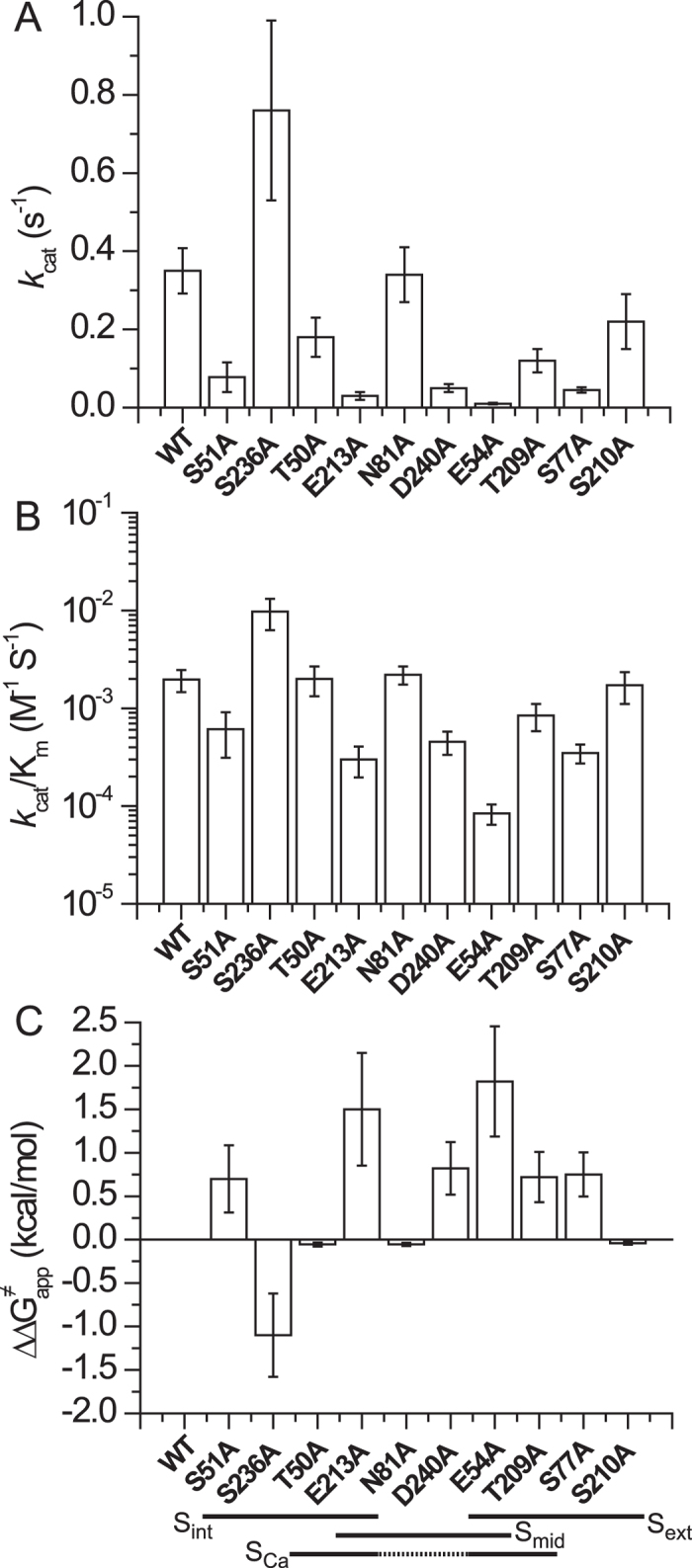
(A) The Km and Vmax values of the Ca2+/Ca2+ exchange reaction were measured by varying the extravesicular Ca2+ concentrations (20–2000 μM) at saturating intravesicular Ca2+ (250 μM) (see Methods and Fig. S1). The kcat values were calculated as described in the legend of Fig. 3D (see also Methods). The bars represent the mean ± SE. (B) The kcat/Km values were derived from experimentally observed kcat and Km values, as outlined in panel A. The bars represent the mean ± SE. (C) Experimentally derived kcat and Km values of the Ca2+/Ca2+ exchange were used for calculating the ∆∆G≠app values according to the equation ∆∆G≠app = −2.303RT•log[(kcat/Km)mut/(kcat/Km)wt. The bars represent the mean ± SE.
According to the original interpretation of the crystallographic data, E54, E213, D240, and N81 coordinate one Na+ ion at the Smid site, where D240 and N81 are mono-dentate residues that exclusively belong to the Smid site18 (Fig. 5A). However, this interpretation has been recently challenged22 by molecular dynamics simulations and pH dependency analysis of exchange reactions in different mutants, although the N81 mutant was not tested in this study. To further assess the potential role of N81 ion binding/transport activities, the pH dependency of Ca2+/Ca2+ and Na+/Ca2+ exchanges were analyzed in N81A. The pH dependency curves of N81A for both ion-exchange reactions are very similar to those of WT NCX_Mj (Fig. 5B), suggesting that the side chain of N81 does not affect the ionization of neighboring catalytic residues, namely, E54, E213, and D240 (Fig. 4C). These results provide independent evidence for inability of the Smid site to bind either Na+ nor Ca2+ in the ground state, in agreement with previous studies22.
Figure 5. pH dependence of the ion-exchange reactions for the N81A mutant.
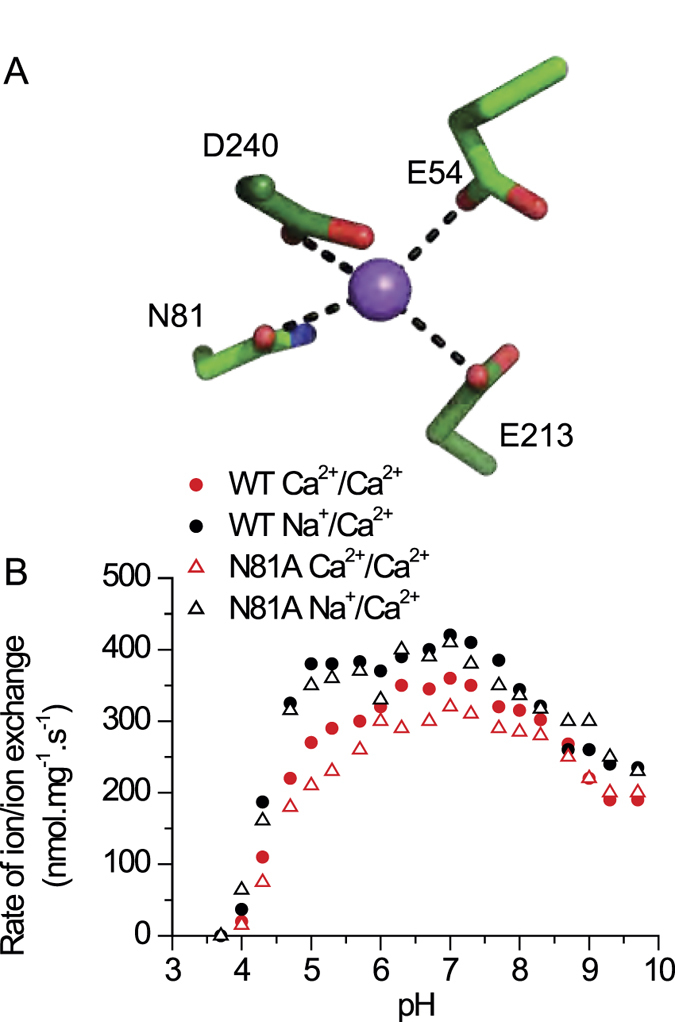
(A) Na+ coordination at Smid, as originally suggested by the X-ray data of NCX_Mj. (B) The initial rates (t = 5 sec) of the Na+/Ca2+ and Ca2+/Ca2+ exchange reactions were measured as in the experiments shown in Figs 3 and 4, in an assay medium with 500 μM 45CaCl2. The intravesicular Na+ and Ca2+ values were 160 mM and 250 μM. The pH of the assay medium was controlled with a 20 mM MES/MOPS/Tris buffer.
To quantify the relative contributions of the ion-coordinating residues to transport catalysis, the observed ∆∆G≠app values of the Ca2+/Ca2+ exchange reactions are presented for relevant mutants (∆∆G≠app = −2.303RT•log[(kcat/Km)mut/(kcat/Km)wt]) (Fig. 4C). Notably, several ion-coordinating residues (S236, S210, T50, and N81) make negligible (if any) contributions to catalysis (∆∆G≠app < 0.3 kcal/mol), whereas E54 and E213 are major residues that stabilize the transition-state, each contributing ∆∆G≠app = 1.5–1.8 kcal/mol (Fig. 4C). Four ion-coordinating residues (D240, S51, S77, and T209) make moderate contributions to ion-transport activity (∆∆G≠app = 0.7–0.8 kcal/mol), suggesting that these polar residues also participate in stabilizing the transition state. Thus, the side chains of two negatively charged residues (E54 and E213) and of four uncharged (polar) residues (protonated D240, S51, S77, and T209) stabilize the Ca2+ transition state (Fig. 4C).
Proline/glycine residues of GTSLPE limit ion transport capacity
The glycine and proline residues in the signature sequences 49-GTSLPE-54 (TM2) and 208-GTSLPE-213 (TM7) are not directly involved in Na+ or Ca2+ coordination, although these residues are highly conserved among NCXs and other membrane proteins18,30. This is especially interesting in light of the fact that G49, G208, P53, and P212 are located near the key functional residues E54 and E213. In general, mutations of glycine and proline residues have no appreciable effect on the Km values of either ion-exchange reaction (Fig. 6A). Although G208C largely retains the WT properties, G49C exhibits ~10-fold decrease in the kcat/Km values (Fig. 6B,C). Thus, G49C and G208C have different effects on ion transport activities, although these two glycine residues represent symmetrically related positions in the inverted α1/α2 repeats of NCX_Mj. This finding is in agreement with the different levels of HDX (Fig. 2B,C) and ligand protection at the signature GTSLPE repeats on TM2 and TM7 (Fig. 6E). Interestingly, P53C and P212C retain only ~3% and ~12% of the WT kcat/Km values, respectively (Fig. 6B,C), thereby suggesting that both proline residues may restrict the backbone flexibility within the ion-binding pocket, consequently affecting the ion transport activities (Fig. 6D). Although G208C does not affect the ion transport activities, the other three helix-breaking residues (G49, P53, and P212) are actively involved in ion transport activities with ∆∆G≠app = 0.5–1.8 kcal/mol (Fig. 6D,E). This suggests that the structure-encoded energy of backbone folding/unfolding at critical residues controls the transition state’s stabilization.
Figure 6. Mutational effects of GTSLPE proline and glycine on ion-exchange activities.

(A) The Km values of the Na+/Ca2+ and Ca2+/Ca2+ exchange reactions were measured by varying the concentrations of 45Ca2+ (20–2000 μM) in the assay medium and using saturating concentrations of intravesicular Na+ (160 mM) or Ca2+ (250 μM), as described in Methods (see also Fig. S1). The bars represent the mean ± SE. (B) The kcat values were calculated as kcat = Vmax/[E]t, where [E]t was normalized by using the GFP assay. The bars represent the mean ± SE. (C) The kcat/Km values were derived from experimentally observed kcat and Km values as outlined in panels (A) and (B). The bars represent the mean ± SE. (D) Experimentally derived values of kcat and Km of the Ca2+/Ca2+ exchange reaction were used for calculating the ∆∆G≠app values according to the equation ∆∆G≠app = −2.303RT•log[(kcat/Km)mut/(kcat/Km)wt. The bars represent the mean ± SE. (E) The difference between the HDX profile of the apo and Na+-bound forms of NCX_Mj at 1200 sec are overlaid on the crystal structure of NCX_Mj (PDB 3V5U). The color key indicates the difference in the percentage of deuterium incorporation between the Na+-bound and apo forms. The blue labeling corresponds to regions with less deuterium incorporation in the Na+-bound form compared with the apo form. The results are nearly identical for the Ca2+- and Na+-bound species.
Discussion
The present work was undertaken to analyze the structure-dynamic determinants of ion-transport catalysis and the intrinsic asymmetry of bidirectional movements in the NCX_Mj protein as related to the inverted twofold symmetry of ion-coordinating pair residues. For this purpose, structure-based kinetic analyses of mutants were performed by testing the effect of alanine mutations on the catalytic capacity (kcat /Km) and on the intrinsic equilibrium of bidirectional Ca2+ movements (Kint = K′m/K″m) by assaying the Ca2+/Ca2+ exchange reaction in E. coli-derived cell-membrane vesicles containing overexpressed NCX_Mj proteins with a right-side-out orientation. In another set of experiments the backbone dynamics of purified NCX_Mj were investigated in the presence or absence of Na+ or Ca2+ ions using HDX-MS, with the goal of tracking the protein backbone dynamics in the ligand-free and ion-bound pocket of NCX_Mj.
In general, HDX-MS is the technique of choice for investigating the structure-dynamic features of local backbone dynamics in apo- and ligand-bound proteins, although the outcome of valuable information depends very much on the sequence coverage in the mass spectrometry experiments. The observed sequence coverage (26.7%) of the NCX_Mj protein is quite low (Fig. S3) compared with that obtained with soluble proteins, mainly due to the detergent that often protects the protein from the protease under conditions suitable for HDX (quick digestions at low pHs and low temperatures). However, the regions of interest (TM2, TM7, and TM8), containing ten out of twelve ion-coordinating residues (except S77 and N81) on the α1 and α2 repeats, are well covered in the HDX-MS experiments. Notably, TM4, TM5, TM6, TM9, and TM10 neither contain any ion-binding residues nor are expected to undergo any significant conformational changes during the alternative access. Thus, the missing information from these sites is less relevant within the scope of the present work. It is somewhat regrettable that the present HDX-MS studies did not cover the TM1/TM6 segments, since these helices may represent the moving gating bundle associated with alternative access. However, this drawback seems to be tolerable as well, since the scope of the present work does not focus on the ion-coupled “sliding” of the gating bundle. Although the present studies provide a clue for identifying the structure-dynamic determinants controlling the ion-coupled sliding of the gating bundle (see below), more dedicated research is required for elucidating the relevant mechanism.
The HDX-MS experiments described here demonstrate that the “symmetric” repeating structures exhibit inherently asymmetrical dynamics, observed in the absence of Na+ or Ca2+ (Fig. 2). These variances in the pattern of backbone dynamics include the strictly conserved α-repeats within the ion-binding pocket, where the local backbone dynamics are much more constrained at T50 and S51 (both of these residues belong to the 48–52 region showing low HDX) (Fig. 2B,C). This implies a unique pattern of dynamic preorganization of catalytic residues in the absence of a bound ligand. Moreover, three helix-breaking residues (G49, P53, and P212), located nearby E54 and E213, actively contribute to the helix folding/refolding dynamics at critical residues, thereby limiting their ion-transport activities (Fig. 6). Notably, G208 is located on a very flexible segment of TM7B (Fig. 2), which may explain why the mutation of this residue does not limit ion transport activities in NCX_Mj. These findings, in conjunction with recent findings27, raise the possibility that the ion-binding pocket of mammalian NCX1 is more flexible than that of NCX_Mj. The reason for this could lie in the structure-dynamic and functional differences in the helix-breaking signature sequences as well as the length of helices involved in ion coordination.
Next, we examined how ligand binding affects the structure dynamics. The data revealed a fairly moderate effect of ion binding on the HDX profiles (Fig. 2). However, these changes are highly specific to previously resolved binding sites. Notably, the strength and location of Na+ and Ca2+-dependent effects on deuterium uptake are comparable, thereby suggesting that both ions rigidify local backbone dynamics similarly (Fig. 2). The emergence of EX1 kinetics upon ligand binding (Fig. 2D) is intriguing. It is tempting to speculate that upon ligand binding, alternative access proceeds through multiple local unfolding/refolding steps requiring low activation energies rather than through a few large, concerted conformational changes, requiring high activation energies.
Alanine substitutions have identified three different groups of residues having diverse effects on the catalytic capacity (kcat/Km) and intrinsic equilibrium (Kint) of bidirectional Ca2+-movements (Figs 3 and 4). In the first group, the alanine mutations of four ion-coordinating residues (S236, S210, T50, and N81) have no appreciable effects either on the kcat/Km or Kint values, thereby suggesting that these residues neither control the ion-transport catalysis nor the intrinsic equilibrium of bidirectional ion movements. In the second group of residues (S51, E54, S77, T209, E213, and D240) the alanine substitutions affect the kcat/Km but not the Kint value (Figs 3 and 4B). Thus, these residues control ion-transport catalysis without affecting the intrinsic equilibrium of bidirectional ion movements. In the third group of residues (N73 and N232), the alanine mutations have no appreciable effects on the kcat/Km values, but result in up to 20-fold changes in Kint, thereby revealing inward-facing conformation stabilization (Kint = 2–3) in these mutants (Fig. 3D).
Although N73 and N232 are not directly involved in ion coordination, according to the crystal structure, N73 and N232 can form a hydrogen bond with T209 and T50, respectively. Since the T209 and T50 side chains contribute to the Na+ sites, whereas their backbone carbonyls form the Ca2+ site (Figs 1C,D and 7A), the N73A and N232A mutations may affect the stability of the Ca2+ site and thus, may affect the asymmetry of bidirectional ion movements (Fig. 7A). The mechanistic significance of this rationale is that N73 and N232 may control one or more occluded states of ion-bound species, while favorably stabilizing the outward-facing conformation during alternating access. Therefore, N73 and N232 may play a critical role in coupling the conformational rearrangement of cytosolic and extracellular gates upon Ca2+ or Na+ binding.
Figure 7. Proposed mechanism underlying the catalytic preorganization for Ca2+ -bound species in the NCX_Mj antiporter.

(A) Inverted pair residues (shown in matching colors) contributing to the ion-transport activities, bidirectional ion movements, and local backbone dynamics at key functional residues are represented as sticks. (B) On the basis of the available experimental data, it is proposed that the backbone carbonyls of T209 and T50 and side chains of E54 and E213 bind Ca2+ in the ground state, where N73 and N232 form a hydrogen bond with T209 and T50, respectively (Fig. 1D). According to the available data, the side chains of E54, E213, D240, S51, T209, and S77 stabilize Ca2+ in the transition state, where ∆Gb = RTlnKm; ∆G≠app = −RTln(kcat/Km); ∆G≠cat = −RTlnkcat.
In general, the observed ∆∆G≠app values (0.5–1.8 kcal/mol) for “catalytic residues” (Fig. 4D), based on the kcat/Km measurements, are too small to result in large conformational changes in the local backbone dynamics of the ion-binding pocket. This is in good agreement with the observed small changes in deuterium uptake within the ion-binding pocket upon Ca2+ or Na+ binding, observed in the HDX-MS experiments (Figs 2, 3, and S4). Notably, E54 and E213 make the most prominent contributions to transition-state stabilization (∆∆G≠app = 1.5–1.8 kcal/mol), in conjunction with four polar residues, namely, S51, S77, T209, and D240 (Fig. 4D). These findings suggest that a polar-electrostatic mechanism governs catalytic reorganization31,32, involving the asymmetric contribution of inverted pair residues to the stabilization of the transition state (Fig. 4A,D and Fig. 7). Notably, the sum of the observed ∆∆G≠app values of “catalytic” ion-coordinating residues adds up to ~5.8 kcal/mol (Figs 4C and 6D), which approaches the experimentally derived value of ∆G≠app = 4.9 kcal for WT NCX_Mj (Fig. 7). Careful computational studies are required for evaluating the physical meaning of the observed ∆∆G≠app values in relation to ∆G≠app and ∆G≠cat. Although the exact mechanism underlying ion-coupled alternative access remains to be resolved, the present findings are consistent with the notion that asymmetric preorganization of key residues in apo-NCX_Mj predefines the differential contributions of matching pair residues toward ion occlusion and the reoganization of catalytic residues (Fig. 7B). This information may provide a structure-dynamic basis for elucidating the exact mechanisms underlying ion-coupled alternating access in NCX and similar proteins, since the present findings provide a clue for determining the future experimental design and impose physical restrictions for computational studies.
Collectively, our data support the notion that the α1-repeat (at TM2) serves as a rigid anchor around which the catalytic residues E54 (SCa), E213 (SCa), protonated D240 (Smid), S51 (Sint), S77 (Sext), and T209 (Sext) stabilize the transition state (Figs 4C, 7, and S4). Therefore, the six catalytic residues belonging to all four ion-binding sites take part in the transition state (Fig. 7B), whereas in the ground state only the SCa site is involved in Ca2+ binding (Fig. 1C,D). The helix-breaking signature sequences (GTSLPE) on α1 and α2 also contribute to transition-state stabilization, presumably because they lopsidedly restrict the backbone dynamics within the ion-binding pocket and thereby, pre-establish the dynamic asymmetry in the ligand-free protein (Figs 1 and 6). Collectively, Ca2+ ligation by “rigid residues” (E54 and S51) may largely predefine the positioning of catalytic residues in the transition state (thereby governing the asymmetric nature of catalytic reorganization), whereas the other catalytic residues (E213, D240, T209, and S77) may possess a higher degree of freedom for Ca2+ interaction in the transition state (Figs 2 and 7). Based on stereochemical considerations, it is tempting to speculate that two water molecules may contribute to the stabilization of the Ca2+ transition state through S77 and/or D240 (Fig. 7).
In conclusion, we found hallmark differences in the local backbone dynamics at key ion-coordinating residues within the ligand-free ion-pocket of apo-NCX_Mj, whereas Na+ or Ca2+ binding to the respective sites induced relatively small, but specific changes in the backbone dynamics. In addition, the catalytic capacity and the intrinsic equilibrium of bidirectional ion movements are controlled by asymmetric contributions of inverted pair residues, thereby revealing a specific preorganization of ion-coordinating and helix-breaking residues within the ion-binding pocket. Thus, inverted structural elements in NCX_Mj are asymmetrically preorganized at the single-residue level, thus providing a structure-dynamic basis for asymmetric contributions to the catalytic reorganization. The currently revealed asymmetric nature of ligand-free preorganization and the catalytic reorganization may provide a clue for assessing the mechanism underlying ion-coupled alternating access. Taking into account the structural similarities of NCX_Mj to other proteins belonging to the superfamily of Ca2+/CA antiporters7,8,18,19,20,22, the present findings may have general significance for attaining a better understanding of the mechanisms underlying alternating access and transport catalysis in secondary transport systems.
Methods
Construct preparation
DNA encoding the WT NCX_Mj was amplified by PCR from a Methanocaldococcus jannaschii cDNA library (DSMZ) and ligated between the NcoI and BamHI restriction sites of a pET-28a plasmid13,22. Mutations were introduced by QuickChange mutagenesis (Stratagene) and were confirmed by sequencing.
E. coli-derived vesicles containing overexpressed NCX_Mj mutants
Expression vectors were transformed into E. coli BL21 (DE3) pLysS competent cells. Cells were grown, harvested, homogenized, and lysed by French-Press13,22. Cell lysates were loaded onto a three-step sucrose gradient and membrane vesicles were stored in 5 mM Mops-Tris pH 7.4, 250 mM sucrose at −80 °C until use13,17. The expression levels of NCX_Mj were evaluated using the GFP-assay. The orientation of NCX_Mj in E. coli-derived cell-membrane vesicles was tested using an antibody against the 6xHis-tag13.
GFP-assay for evaluating the expression levels of NCX_Mj
The ratio of NCX-Mj expression to the total vesicle protein was approximated by calculating the concentration of GFP by its absorbance at 488 nm (e.c. = 56000) and dividing it by the value determined by the Lowry assay. The GFP absorbance signal at 488 nm was isolated from vesicle noise using a correction factor determined by measuring non-expressing vesicles13.
Protein purification
NCX_Mj was overexpressed and purified as outlined before13,18,33. Briefly, membranes were isolated from the cell lysate by ultracentrifugation and after membrane protein extraction with 20 mM DDM (n-Dodecyl β-D-maltoside), the supernatant was loaded onto a Talon Co2+ affinity column. Protein was then desalted to eliminate imidazole with a buffer containing 2 mM DDM and digested overnight with TEV protease. Following a second passage through the Co2+ column (with a buffer containing 2 mM DDM) to eliminate the His-tag and TEV protease, the protein was purified on Superdex-200 pre-equilibrated with 0.5 mM DDM buffer. Concentrated (1–2.5 mg/ml) preparations of purified proteins (>95% purity, judged by SDS-PAGE) were stored at −80 °C in buffer containing 0.5 mM DDM.
45Ca2+-uptake assay in E. coli-derived vesicles
The initial rates of the Na+/Ca2+ and Ca2+/Ca2+ exchange reactions were assayed by measuring 45Ca2+-uptake in the E. coli-derived vesicles containing the overexpressed WT or mutants of NCX_Mj, as previously described13,17,22. Briefly, Na+ (160 mM) or Ca2+ (250 μM)-loaded vesicles were rapidly diluted 25–50-fold at 35 °C in an assay medium containing 20 mM MOPS/Tris, pH 6.5, 100 mM KCl, and 5–2000 μM 45CaCl2. The 45Ca2+-uptake was quenched by cold buffer containing 10 mM EGTA and filtrated on GF/C filters, as outlined before13,17,22. The Km and Vmax values were measured in at least three independent experiments (data are presented as mean ± SEM) with GraFit 7.1 (Erithacus Software, Ltd.)10,13,17. The kcat was calculated as kcat = Vmax/[E]t, where [E]t was determined by the GFP assay (see above).
Peptide analysis by tandem mass spectrometry (MS/MS) after NCX_Mj proteolysis
LC-MS/MS analyses were performed under H/D exchange compatible conditions and data analysis was performed with MStools34. The system consisted of injection and switching valves mounted with a desalting cartridge (peptide Opti-Trap Micro from Optimize Technologies) and an analytical column (Jupiter C18, 0.5 × 50 mm, 5 μm, 300 Å, Phenomenex). NCX_Mj was digested using a porcine pepsin (Sigma) solution at 0.23 mg/mL in 100 mM glycine-HCl, pH 2.5 in an ice-water bath for 2 min. Desalting (3 min) was driven by a Shimadzu LC20-AD pump isocratically (0.4% formic acid in water at 100 μL/min). Gradient was separated using the HPLC system (Agilent Technologies 1200) at a flow rate of 15 μL/min. Gradient elution (from 10% B to 40% B in 25 min), followed by 1 min gradient to 95% B, was used for separation. The solvents used were A–0.4% formic acid, 2% acetonitrile in water, and solvent B–0.4% formic acid in 95% acetonitrile. The outlet of the analytical column was directly connected to an electrospray ionization (ESI) source of an Apex-ULTRA Qe Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometer (Bruker Daltonics) equipped with a 9.4 T superconducting magnet. ESI-FT-ICR MS was calibrated externally using arginine clusters, resulting in a mass accuracy below 2 ppm. For LC-MS/MS the instrument was operated in data-dependent mode by selecting the six most intense ions in each MS scan for an MS/MS analysis using collisionally induced dissociation in a quadrupole. The data were searched by MASCOT (MatrixScience) against single-protein databases containing the NCX sequence and against a database containing pepsin (to exclude possible autolysis products).
Hydrogen/Deuterium exchange Mass Spectrometry (HDX-MS)
HDX-MS experiments were fully automated using a PAL autosampler (CTC Analytics). This controlled the start of the exchange and quench reactions, the proteolysis temperature (4 °C), the injection of the deuterated peptides, as well as management of the injection and washing valves; it also triggered the acquisition of the mass spectrometer and HPLC pumps. A Peltier-cooled box (4 °C) contained two Rheodyne automated valves, a desalting cartridge (peptide Opti-Trap Micro from Optimize Technologies) and a HPLC column (Jupiter 4μm Proteo, Phenomenex). HDX MS reactions were carried out using different forms of WT NCX_Mj (apo, with Na+ or Ca2+) at a concentration of 40 μM. Deuteration was initiated by a 5-fold dilution of the protein samples (10 μL) with the same buffer in D2O (40 μL). The proteins were deuterated for 15 sec, or for 2 or 20 min at 4 °C. Back-exchange quench and digestion were performed using 50 μL of porcine pepsin (Sigma) solution at 0.23 mg/mL in 100 mM glycine-HCl, pH 2.5 at 4 °C for 2 min. The peptides were desalted for 6 min using a HPLC pump (Agilent Technologies) with 0.03% TFA in water, at a flow rate of 100 μL/min. The peptides were separated using another HPLC pump (Agilent Technologies) at 50 μL/min for 6 min with 15–50% gradient B (Buffer A: TFA 0.03% in water; Buffer B: acetonitrile 95%, TFA 0.03% in water), followed by 9 min at 50% B and 1 min at 100% B. The peptide masses were measured using an electrospray-TOF mass spectrometer (Agilent 6210) in the 300–1300 m/z range. Each deuteration experiment was conducted in triplicate. The Mass Hunter (Agilent Technologies) software was used for data acquisition. The HD Examiner software (Sierra Analytics) was used for HDX-MS data processing. MSTools was used for visualization and presentation of the HDX-MS data34.
Additional Information
How to cite this article: Giladi, M. et al. Asymmetric Preorganization of Inverted Pair Residues in the Sodium-Calcium Exchanger. Sci. Rep. 6, 20753; doi: 10.1038/srep20753 (2016).
Supplementary Material
Acknowledgments
This work was partially funded by the USA-Israel Binational Foundation Research Grant #2009-334, and the Israel Science Foundation Grant #825/14 to DK. The support of the Fields Estate foundation to DK is highly appreciated.
Footnotes
Author Contributions M.G., L.A., E.F., P.M. and D.K. designed the experiments. L.A., M.G., P.M., L.D., and R.H. conducted the experiments. M.G., L.A., R.H., E.F., P.M. and D.K. analyzed and interpreted the experimental data. D.K. wrote the manuscript.
References
- Forrest L. R. Structural biology. (Pseudo-)symmetrical transport. Science 339, 399–401 (2013). [DOI] [PubMed] [Google Scholar]
- Forrest L. R. Structural Symmetry in Membrane Proteins. Annu. Rev. Biophys. 44, 311–337 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller R., Ziegler C. & Schneider D. When two turn into one: evolution of membrane transporters from half modules. Biol. Chem. 395, 1379–1388 (2014). [DOI] [PubMed] [Google Scholar]
- Boudker O. & Verdon G. Structural perspectives on secondary active transporters. Trends Pharmacol. Sci. 31, 418–426 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philipson K. D. & Nicoll D. A. Sodium-calcium exchange: a molecular perspective. Ann. Rev. Physiol. 62, 111–133 (2000). [DOI] [PubMed] [Google Scholar]
- Blaustein M. P. & Lederer W. J. Sodium/calcium exchange: its physiological implications. Physiol. Rev. 79, 763–854 (1999). [DOI] [PubMed] [Google Scholar]
- Khananshvili D. The SLC8 gene family of sodium-calcium exchangers (NCX)-structure, function, and regulation in health and disease. Mol. Aspects Med. 34, 220–235 (2013). [DOI] [PubMed] [Google Scholar]
- Khananshvili D. Sodium-calcium exchangers (NCX): molecular hallmarks underlying the tissue-specific and systemic functions. Pflugers Arch. EJP. 466, 43–60 (2014). [DOI] [PubMed] [Google Scholar]
- Reeves J. P. & Hale C. C. The stoichiometry of the cardiac sodium-calcium exchange system. J. Biol. Chem. 259, 7733–7739 (1984). [PubMed] [Google Scholar]
- Khananshvili D. Distinction between the two basic mechanisms of cation transport in the cardiac Na+ -Ca2+ exchange system. Biochemistry 29, 2437–2442 (1990). [DOI] [PubMed] [Google Scholar]
- Jardetzky O. Simple allosteric model for membrane pumps. Nature 211, 969–970 (1966). [DOI] [PubMed] [Google Scholar]
- Forrest L. R., Kramer R. & Ziegler C. The structural basis of secondary active transport mechanisms. Biochim. Biophys. Acta 1807, 167–188 (2011). [DOI] [PubMed] [Google Scholar]
- Baazov D., Wang X. & Khananshvili D. Time-resolved monitoring of electrogenic Na+ -Ca2+ exchange in the isolated cardiac sarcolemma vesicles by using a rapid-response fluorescent probe. Biochemistry 38, 1435–1445 (1999). [DOI] [PubMed] [Google Scholar]
- Niggli E. & Lederer W. J. Molecular operations of the sodium-calcium exchanger revealed by conformation currents. Nature 349, 621–624 (1991). [DOI] [PubMed] [Google Scholar]
- Hilgemann D. W., Nicoll D. A. & Philipson K. D. Charge movement during Na+ translocation by native and cloned cardiac Na+/Ca2+ exchanger. Nature 352, 715–718 (1991). [DOI] [PubMed] [Google Scholar]
- Almagor L. et al. Functional asymmetry of bidirectional Ca2+ -movements in an archaeal sodium-calcium exchanger (NCX_Mj). Cell calcium 56, 276–284 (2014). [DOI] [PubMed] [Google Scholar]
- Khananshvili D., Weil-Maslansky E. & Baazov D. Kinetics and mechanism: modulation of ion transport in the cardiac sarcolemma sodium-calcium exchanger by protons, monovalent, ions, and temperature. Ann. N. Y. Acad. Sci. 779, 217–235 (1996). [DOI] [PubMed] [Google Scholar]
- Liao J. et al. Structural insight into the ion-exchange mechanism of the sodium/calcium exchanger. Science 335, 686–690 (2012). [DOI] [PubMed] [Google Scholar]
- Waight A. B. et al. Structural basis for alternating access of a eukaryotic calcium/proton exchanger. Nature 499, 107–110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishizawa T. et al. Structural basis for the counter-transport mechanism of a H+/Ca2+ exchanger. Science 341, 168–172 (2013). [DOI] [PubMed] [Google Scholar]
- Wu M. et al. Crystal structure of Ca2+/H+ antiporter protein YfkE reveals the mechanisms of Ca2+ efflux and its pH regulation. Proc. Natl. Acad. Sci. USA. 110, 11367–11372 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinelli F. et al. Sodium recognition by the Na+/Ca2+ exchanger in the outward-facing conformation. Proc. Natl. Acad. Sci. USA. 111, E5354–5362 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giladi M., Lee S. Y., Hiller R., Chung K. Y. & Khananshvili D. Structure-dynamic determinants governing a mode of regulatory response and propagation of allosteric signal in splice variants of Na+/Ca2+ exchange (NCX) proteins. Biochem. J. 465, 489–501 (2015). [DOI] [PubMed] [Google Scholar]
- Konermann L., Pan J. & Liu Y. H. Hydrogen exchange mass spectrometry for studying protein structure and dynamics. Chem. Soc. Rev. 40, 1224–1234 (2011). [DOI] [PubMed] [Google Scholar]
- Mehmood S., Domene C., Forest E. & Jault J. M. Dynamics of a bacterial multidrug ABC transporter in the inward- and outward-facing conformations. Proc. Natl. Acad. Sci. USA. 109, 10832–10836 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey M. et al. Conformational dynamics of the bovine mitochondrial ADP/ATP carrier isoform 1 revealed by hydrogen/deuterium exchange coupled to mass spectrometry. J. Biol. Chem. 285, 34981–34990 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalmers M. J., Busby S. A., Pascal B. D., West G. M. & Griffin P. R. Differential hydrogen/deuterium exchange mass spectrometry analysis of protein-ligand interactions. Expert Rev. Proteomics 8, 43–59 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weis D. D., Wales T. E., Engen J. R., Hotchko M. & Ten Eyck L. F. Identification and characterization of EX1 kinetics in H/D exchange mass spectrometry by peak width analysis. J. Am. Soc. Mass Spectrom. 17, 1498–1509 (2006). [DOI] [PubMed] [Google Scholar]
- Demmers J. A., Haverkamp J., Heck A. J., Koeppe R. E. 2nd & Killian J. A. Electrospray ionization mass spectrometry as a tool to analyze hydrogen/deuterium exchange kinetics of transmembrane peptides in lipid bilayers. Proc. Natl. Acad. Sci. USA. 97, 3189–3194 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- John S. A., Liao J., Jiang Y. & Ottolia M. The cardiac Na+ -Ca2+ exchanger has two cytoplasmic ion permeation pathways. Proc. Natl. Acad. Sci. USA. 110, 7500–7505 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warshel A. et al. Electrostatic basis for enzyme catalysis. Chem. Rev. 106, 3210–3235 (2006). [DOI] [PubMed] [Google Scholar]
- Kamerlin S. C. & Warshel A. At the dawn of the 21st century: Is dynamics the missing link for understanding enzyme catalysis? Proteins 78, 1339–1375 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giladi M., Hiller R., Hirsch J. A. & Khananshvili D. Population shift underlies Ca2+ -induced regulatory transitions in the sodium-calcium exchanger (NCX). J. Biol. Chem. 288, 23141–23149 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavan D. & Man P. MSTools-Web based application for visualization and presentation of HDX-MS data. Int. J. Mass Spectrom. 302, 53–58 (2011). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.