

Abstract
Congenital insensitivity to pain (CIP) comprises the rare heritable disorders without peripheral neuropathy that feature inability to feel pain. Fracturing and joint destruction are common complications, but lack detailed studies of mineral and skeletal homeostasis and bone histology. In 2013, discovery of a heterozygous gain-of-function mutation in SCN11A encoding voltage-gated sodium channel 1.9 (Nav1.9) established a distinctive CIP in three unrelated patients who suffered multiple painless fractures, self-inflicted mutilation, chronic diarrhea, and hyperhidrosis.
Here, we studied a mother and two children with CIP by physical examination, biochemical testing, radiological imaging including DXA, iliac crest histology, and mutation analysis.
She suffered fractures primarily of her lower extremities beginning at age two years, and had Charcot deformity of both ankles and joint hypermobility. Nerve conduction velocity and biopsy and electromyography were normal. Her children had recurrent major fractures beginning in early childhood, joint hypermobility, and chronic diarrhea. She had an excoriated external nare, and both children had hypertrophic scars from scratching. Skin collagen studies were normal. Radiographs revealed fractures and deformities. However, lumbar spine and total hip BMD Z-scores, biochemical parameters of mineral and skeletal homeostasis, and iliac crest histology of the mother (after in vivo tetracycline labeling) were normal. Genomic DNA from the children revealed a unique heterozygous missense mutation in exon 23 (c.3904C>T, p.Leu1302Phe) of SCN11A that is absent in SNP databases and alters an evolutionarily conserved amino acid.
This autosomal dominant CIP reflects the second gain-of-function mutation of SCN11A. Perhaps joint hypermobility is an unreported feature. How mutation of Nav1.9 causes fracturing remains unexplained. Lack of injury awareness is typically offered as the reason, and was supported by our unremarkable biochemical, radiological, and histological findings indicating no skeletal pathobiology. However, low-trauma fracturing in these patients suggests an uncharacterized defect in bone quality.
Keywords: Charcot arthropathy, joint hypermobility, mineral homeostasis, skeletal homeostasis
II) Introduction
Inability to feel pain includes a group of rare heritable disorders characterized by absence of pain perception.(1) The first affected individual, a “pincushion” actor, was reported by Dearborn in 1932.(2)
Up to 1983, multiple names were given to these entities, including congenital pure general analgesia,(2) congenital universal insensitiveness to pain,(3) congenital universal indifference to pain,(4) congenital indifference to pain,(5, 6) congenital insensitivity to pain,(7-9) congenital analgia,(10) congenital absence of pain,(11-14) and congenital analgesia.(15)
In 1983, with improved means to assess nerve fiber pathology, Dyck et al.(16) preferred “congenital insensitivity to pain” for this symptom if it was associated with peripheral neuropathy. These investigators distinguished it from “congenital indifference to pain” (OMIM %147430) occurring without peripheral neuropathy. Still, “insensitivity” and “indifference” to pain are used interchangeably, with the more common usage being “congenital insensitivity to pain” (CIP). Then, in 1993, Dyck(17) proposed “hereditary sensory and autonomic neuropathy” (HSAN) for the neuropathic disorders. In HSAN, mutations in 12 different genes, cause primarily sensory neuron degeneration.(18) Most HSANs are autosomal recessive (AR) disorders, but some are autosomal dominant (AD) traits.(17,19-21)
The etiology of CIP lacking peripheral neuropathy remained unknown until 2006 when Cox et al.(22) discovered three distinct homozygous nonsense mutations in SCN9A encoding the α-subunit of the voltage-gated sodium channel 1.7 (Nav1.7) in three separate consanguineous Pakistani families. These SCN9A mutations caused loss-of-function of Nav1.7, and therefore the disorder was designated “channelopathy-associated insensitivity to pain” (OMIM #243000).(20,22,23)
In 2013, Leipold et al.(24) identified in a German girl and unrelated Swedish man with CIP an identical, de novo, heterozygous, gain-of-function mutation in exon 15 of SCN11A (c.2432T>C, p.Leu811Pro) encoding Nav1.9. Both individuals with this new Nav-channelopathy suffered painless bone fractures as well as self-inflicted injuries and mutilation causing un-healing wounds. They also manifested muscle weakness, delayed motor development, failure-to-thrive, hyperhidrosis, and chronic diarrhea.(24) The following year, a Scottish girl with sporadic CIP and the same clinical phenotype was reported by Woods et al(25) to have the identical mutation (c.2432T>C, p.Leu811Pro) in SCN11A.
Despite this progress in understanding CIP, detailed assessments of mineral and skeletal homeostasis and bone pathology have not been reported. Here, we i) describe an AD Nav1.9 channelopathy (mother and two children) with CIP uniquely manifesting marked joint hypermobility, ii) identify their unique heterozygous missense (c.3904C>T, p.Leu1302Phe) defect within SCN11A, and iii) document normal mineral and skeletal homeostasis using biochemical, radiological, and histological studies.
III) Patients and Methods
Informed written consent for all research studies was obtained as sanctioned by the Human Studies Committee, Washington University School of Medicine, St. Louis, MO, USA.
A) Patients
The proposita (II-2, Figure 1) was referred at age 10 years in 1985, and was investigated as an inpatient at that time. Thirteen years later, she brought her two affected children for inpatient studies, a son (III-1) and daughter (III-2), ages 3.5 and 1.5 years, respectively; returning at ages 9.5 and 7.5 years. Subsequently, the family was lost to follow-up. Their clinical characteristics are summarized in Table 1.
Figure 1. Family Pedigree.
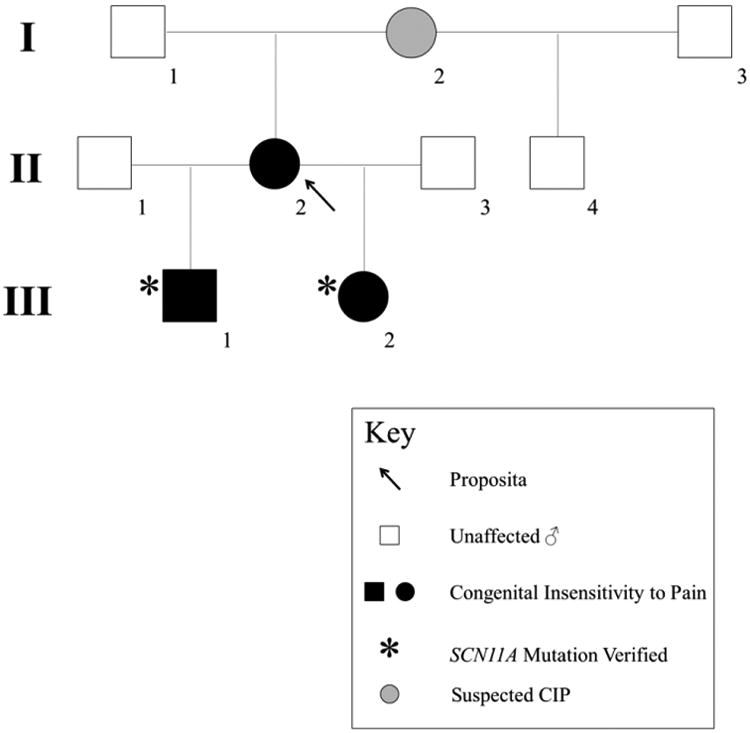
The proposita (arrow), whose DNA was unavailable for SCN11A analysis, presumably carries the same heterozygous missense mutation in SCN11A as her two affected children. Her mother (I-2) reportedly had diminished pain sensation and hypermobility but no fractures.
Table 1. Clinical Characteristics of Nav 1.9 Channelopathy: Our 3 patients and previous reports*.
| Clinical characteristics | Our study | Leipold E, 2013 | Wood CG, 2014 | |||
|---|---|---|---|---|---|---|
| Proposita | Son | Daughter | 1 | 2 | ||
| Ethnicity | American | American | American | German | Swedish | Scottish |
| Sex | F | M | F | F | M | F |
| Age at first evaluation (years) | 10 | 3.5 | 1.5 | NR | NR | NR |
| Neurological features | ||||||
| Intelligence | Normal | Normal | Normal | Normal | Normal | Normal |
| Cranial nerves | Normal | Normal | Normal | NR | NR | NR |
| Sensation: light touch, temperature and propioception | Normal | Normal | Normal | NR | NR | NR |
| Deep tendon reflex | Normal | Normal | Normal | NR | NR | NR |
| Nerve conduction | Normal | ND | ND | Normal | ND | ND |
| Musculoskeletal features | ||||||
| Multiple painless fractures | Yes | Yes | Yes | Yes | Yes | Yes |
| Age at first fracture (years) | 2 | 2.5 | 1 | NR | NR | NR |
| Numbers of fractures | 4 | 12 | 8 | NR | NR | NR |
| Fracture sites | Tibia, fibula, talus, metatarsal | Elbow, tibia, fibula, calcaneous, cuboid, metatarsals | Ulnar, tibia, femur, calcaneous | NR | NR | NR |
| Hypermobile joints | Yes | Yes | Yes | NR | NR | NR |
| Charcot arthropathy | Yes | Yes | No | Yes | Yes | Yes |
| Muscle weakness | No | No | No | Yes | Yes | Yes |
| Neuro-cutaneous features | ||||||
| Insensitivity to pain | Yes | Yes | Yes | Yes | Yes | Yes |
| Self- inflicted injury | Yes (excoriated right alae nasi) | Yes (abraded tongue with teeth and scratched skin) | Yes (biting lip and scratched skin) | Yes | Yes | Yes |
| Skin ulcer at posterior cervical region | No | Yes (chest wall, back) | Yes | Yes | Yes | Yes |
| Slow wound healing | Yes | Yes | Yes | Yes | Yes | Yes |
| Pruritus | No | Yes | Yes | Yes | Yes | Yes |
| Intermittent body sensation of electrical pain | Yes | Yes | Yes | No | No | No |
| Excessive sweating | No | No | Yes | Yes | Yes | Yes |
| Heat/cold weather intolerance | No | Yes | Yes | Yes | Yes | Yes |
| Gastrointestinal features | ||||||
| Chronic diarrhea | Yes | Yes | Yes | Yes | Yes | Yes (during first 1-2 years of life) |
| Failure to thrive | No | No | Yes | Yes (Required parenteral nutrition) | Yes (Required parenteral nutrition) | Yes |
| Episodic abdominal pain | No | No | No | No | No | Yes |
| Perineal discomfort on passing motion or urine | No | No | No | No | Yes | Yes |
| Rectal pain from defication | No | No | No | No | Yes | Yes |
NR = No report, ND = Not done
1) Proposita
The mother (II-2) weighed 6 pounds, 3 ounces as a full-term baby born to a 14-year-old non-consanguineous mother.
Her first fracture occurred at age 2 years when her right foot swelled without a history of trauma. A radiograph showed a broken first metatarsal. Skeletal survey was reportedly otherwise normal.
At age 7 years, she fractured her left tibia and fibula after falling. The bones healed normally with casting, but soft-tissue swelling occurred frequently at her ankles. Rheumatoid arthritis was diagnosed. She was prescribed aspirin, and had some improvement in her ankle pain and swelling.
At age 8 years, she broke her right tibia, fibula, and talus during a fall from a grocery cart. She was hospitalized elsewhere, and underwent extensive evaluation. Deformities of her lower extremities and impressive hypermobility were noted. Motor power and fine motor skills were appropriate for age. Biochemical studies included “bone panels” reported as normal. Karyotype was 46, XX with a normal female variant inverted Q (type 2). Radiographs reportedly showed osteochondritis dessicans of the medial tibial plateau, and fracture at the base of the tibial spine bilaterally. Avascular changes of the talar navicular, erosions of the talus, and calcaneal fractures were found bilaterally. The diagnosis was a “collagen disorder”.
At age 10 years and referral to us, she had been without further fractures. She reported no pain. Despite numerous changes of her milk formula, chronic diarrhea without apparent malabsorption was reported during the first 2 years-of-life. Subsequently, she tolerated milk, but loose stools occurred several times daily. She also had intermittent “electric” sensations over her body once or twice daily, but sweated normally. Physical examination revealed normal weight and height, a remarkable excoriated right external nare secondary to self-mutilation, Charcot joints, and significant hypermobility (Figure 2A). Her ankles were deformed and swollen, but without tenderness. Neurological examination showed normal alertness and intelligence. Cranial nerves and corneal reflex were intact, with normal tears. Motor exam showed normal muscle bulk, strength, and tone. Gait and cerebellar testing were normal. Deep tendon reflexes (DTRs) were +2-3 and symmetrical, with down-going toes on Babinski testing. Sensory exam was normal for proprioception, light touch, and temperature. Pin prick caused some little pain. Nerve conduction velocity and electromyography studies were normal. Accordingly, we diagnosed CIP.
Figure 2. Clinical features of the family.
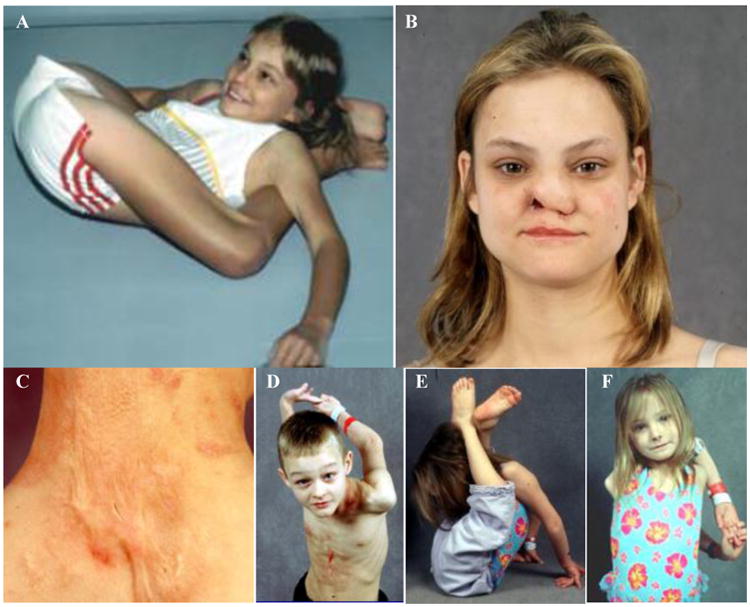
A) The proposita at age 10 years has impressive hypermobility. She could march her legs up to the base of her head, could touch her thumbs to her forearms, and demonstrated 90 degrees of internal and external rotation of her hips.
B) The proposita at age 23 years has scarring, loss of tissue, and ulceration of her right nasal ala from years of scratching.
C) Her son has a hypertrophic scar at his posterior neck from excoriations during infancy.
D) – F) The proposita's son and daughter demonstrate hypermobility.
Family history revealed that her mother (I-2) was hypermobile, bruised easily, sweated excessively, and “scratched herself until she bled”. The proposita's maternal half-brother (II-4) had a history of diminished pain sensation, but without fracturing, and did not carry the SCN11A mutation (see Results).
At age 23 and 29 years, when we studied her affected children, ulceration of her right ala nasi persisted (Figure 2B). She reported that the first time she ever felt pain was during the late stage of delivery of her first child. She continued to have the “electrical” sensations. Elsewhere, light and electron microscopy of a sural nerve biopsy was said to show: i) a normal distribution of large and small myelinated axons, ii) absence of Schwann cells and unmyelinated axons from the endoneurial connective tissue between the large myelinated axons, iii) many large myelinated axons back-to-back with an intervening parenchyma of Schwann cells and unmyelinated fibers, and iv) fewer that one unmyelinated axon for each myelinated axon (normally there are approximately 10 unmyelinated fibers for every myelinated fiber). Her joint hypermobility was now accompanied by slight genu valgum deformity.
2) Son
He (III-1) was born prematurely at 33 weeks gestation following a pregnancy complicated by a motor vehicle accident. Birth weight was 4 pounds, 10 ounces, and length 19 inches. His father was healthy and without fractures.
He remained well until age 1 year when he scratched a spider bite on his posterior neck and had poor wound healing and significant scarring.
At age 2.5 years, a first fracture occurred of his right arm when he fell from a dresser. Subsequently, four surgical procedures involved the arm; one of his right elbow was complicated by a staphylococcus infection with skin ulceration that required three months to heal. He sustained five more fractures (cuboid of his right foot, right proximal tibia, right calcaneus, left cuboid, and old fractures of right metatarsals 1, 2 and 3). After a particularly active day, he had ankle pain and swelling, lasting an hour, and some stiffness. Like his mother, electrical sensations progressed through his body several times daily. He had loose and floating stools 5-6 times each day, however, evaluation was unrevealing.
At 3.5 years-of-age, when first studied by us in 1997, physical examination revealed hypermobility especially of his hips, and mild knock-knee deformity. His cranial nerves, DTRs, and muscle power were normal.
At age 9.5 years, six more fractures had occurred (total of 12 fractures), all in his lower extremities. These were typically casted for 2-3 weeks. Lumps at previous fractures lasted for about one year. He scratched causing cellulitis and staphylococcus skin infections. Numerous excoriated lesions and old scars with keloids affected his anterior chest and posterior neck (Figure 2C). Electric sensations and loose stools continued several times daily. Hot weather was poorly tolerated. A large scar of his tongue resulted from biting. Significant joint hypermobility was evident except for decreased ankle movement (Figure 2D). The right knee was enlarged and contained fluid (Figure 3A and 3B). The right ankle was also large, apparently from bony and soft tissue swellings (Figure 3C). His left ankle was large and “bony”. DTRs, motor power, and vibratory sensation were normal. Pin prick was recognized, but not painful.
Figure 3. Son at age 9 years.
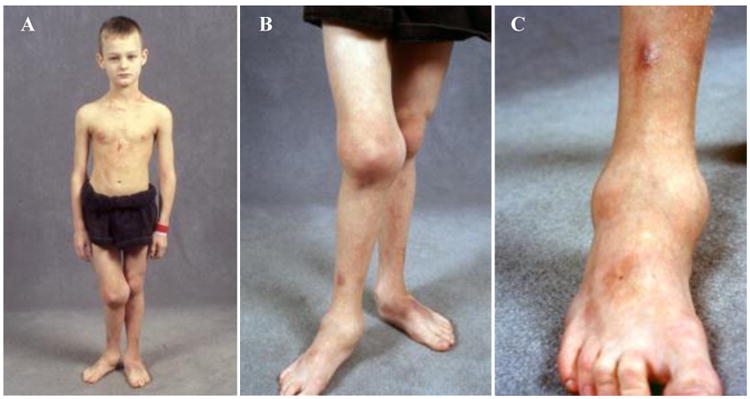
A) He has Charcot joints. Deformities affect his right leg without leg-length discrepancy.
B) His right knee appears profoundly swollen from significant bony growth and fluid.
C) His right ankle is enlarged from bony as well as a soft tissue swelling.
In 2001 in the laboratory of Dr. Peter H. Byers, University of Washington, Seattle, WA, USA), a specimen of skin showed normal type 1 and 3 collagen and pro-collagen by protein gel electrophoresis, and normal amounts of synthesized and secreted type 1 and 3 pro-collagens. Electrophoretic mobility of the pro-collagen chains and the efficiency of their secretion and conversion to collagen were similar to normal cells. These findings were against a form of osteogenesis imperfecta (OI) and Ehlers-Danlos syndrome (EDS), type 4 and type 7.
3) Daughter
She (III-2) was born at 40 weeks gestation. Birth weight was 6 pounds, 1 ounce. Soon after, gastroesophageal reflux disease and failure-to-thrive were diagnosed. Like her mother and brother, she had loose, foul-smelling stools several times daily, but extensive evaluation elsewhere was unrevealing. As a baby, she destroyed parts of her lips, fingers, and toes by chewing on them. CIP was established at ∼ age 1 year when her activities continued unchanged despite an apparent broken arm.
When first admitted to our research center at age 1.5 years, she had sustained five fractures including both calcanei, right proximal tibia, and both distal tibial metaphyses. A broken leg had been pinned. Electric sensations occurred several times daily. She was intolerant of cold, and complained of stiff ankles. Cellulitis and staphylococcal infections recurred at least yearly from scratching, but unlike her mother and brother she sweated profusely.
At age 7.5 years, her early symptoms persisted. There had been a further three fractures including both ankles and right femur, which usually healed within three weeks. Physical examination revealed a proportionately small-for-age girl. Scarring was present around her lips, hands, feet, knees, upper back and nuchal area. She had mild pectus excavatum and mild knock-knee deformity. Remarkable hypermobility was present except in her knees and ankles. Both feet could be placed behind her head (Figure 2E). With hands clasped together, she could put her arms over her head, behind her back, and down to her buttocks (Figure 2F). Her knees were wide and ankles thick with limited flexibility. She cringed, withdrew, and cried if touched by cold. Pinprick and vibration sensation was intact. Muscle power was 4/5 throughout.
B) Biochemical Studies
Biochemical studies used fasting blood and included assessments of mineral and skeletal homeostasis in our certified laboratory (see Results).
C) Radiological Studies
In 2015, all available radiological images (1985 to 2003) were reviewed and contrasted for our three patients. These included radiographs of their skulls, spines, elbows, hands, knees, ankles, and feet.
In 1997 and 2003, dual-energy X-ray absorptiometry (DXA) was performed using the QDR-4500A™ instrument (Hologic Inc., Waltham, MA, USA) at Shriners Hospital for Children; St. Louis, MO, USA. Then, in 2015, z-scores of bone mineral density (BMD) in the lumbar spine (L1-L4), hip, and wrist were re-calculated for patient age and gender using the 2005 pediatric BMD reference data of Kelly et al.(26)
D) Bone histology
When the proposita was 10-years-of-age, transiliac crest biopsy was performed using a 5 mm internal diameter Bordier trephine after she had received two 3-day “labels” of tetracycline given orally separated by approximately two weeks. The specimen was assessed using non-decalcified histology.
E) Mutation Analysis
Genomic DNA was extracted from whole blood of the two affected siblings. Each of the 26 coding exons and adjacent mRNA splice sites of SCN11A were PCR-amplified and sequenced. Forward and reverse sequencing was performed for each exon. The primer pairs designed in our laboratory and annealing temperatures are provided (Supplementary Appendix, Table 1). The sequencing electropherograms were examined visually, and also aligned using Sequencher™ software (Gene Codes Corporation, Ann Arbor, MI, USA). The missense mutation identified was validated by its absence from the single nucleotide polymorphism database (dbSNP: www.ncbi.nlm.nih.gov/SNP). Furthermore, the site of the SCN11A mutation (exon 23) was PCR-amplified and sequenced in the proposita's half-brother (II-4) who was said to have diminished pain sensation despite no history of fracture. The proposita's DNA was no longer available, although in 2003 we had performed mutation analysis of NTRK1 causing CIP with anhydrosis (also called HSAN IV), finding no mutation in the 17 coding exons or the exon/intron boundaries.(27)
IV) Results
A) Biochemical findings
Biochemical parameters of mineral and skeletal homeostasis representing all three family members were unremarkable (Table 2). The daughter's slightly high serum phosphorus level was perhaps from drinking two cans daily of PediaSure, which has a high phosphorus content
Table 2. Biochemical parameters of mineral homeostasis.
| Proposita | Son | Daughter | Normal range | ||||
|---|---|---|---|---|---|---|---|
| Age (years) | 10 | 23 | 3.5 | 9 | 1.5 | 7.5 | - |
| Blood | |||||||
| Calcium (mg/dL) | 9.6 | 9.2 | 9.8 | 9.6 | 10.2 | 10.6 | 8.6-10.3 |
| Ionized Calcium (mg/dL) | 4.8 | 5.2 | 5.3 | 5.1 | 5.4 | 5.4 | 4.5-5.3 |
| Phosphate (mg/dL) | 4.2 | 3.8* | 5.7 | 5.9¶ | 6.8 | 6.5¶ | 3.8-6.5 *2.7-4.7 ¶3.7-5.6 |
| Magnesium (mg/dL) | 1.8 | 1.8 | 2.3 | 2.3 | 2.4 | 2.6 | 1.6-2.2 |
| Alkaline phosphatase (U/L) | 178 | 86* | 215¶ | 173# | 351¶ | 225# | 25-100 *50-130 ¶110-320 #150-420 |
| Creatinine (mg/dL) | 0.4 | 0.6 | 0.4 | 0.5 | 0.3 | 0.4 | 0.5-1.4 |
| Albumin (g/dL) | 4.7 | 3.8 | 4.2 | 3.8 | 4.3 | 4.8 | 3.4-5.2 |
| Intact PTH (pg/mL) | - | 28 | 22 | - | 33 | - | 10-65 |
| Osteocalcin (ng/mL) | - | - | 19.6 | - | 17.8 | - | 2.8-41 |
| Urine | |||||||
| Calcium/Creatinine (mg/gm) | 192-263 | 144 | 86-103 | 98-239 | - | 217 | - |
| Phosphate/Creatinine (mg/gm) | - | - | 0.9-1.1 | 1.1-1.4 | - | 1.3 | - |
| Free deoxypyridinoline (nmoL) | - | 91.9 | - | 244 | - | 242 | - |
| Deoxypyridinoline/Creatinine | - | 6.4* | - | 19¶ | - | 32.6# | *3.0-7.4 ¶13.7-41 #10.5-45 |
B) Radiological Findings
1) Proposita
Radiographs of the proposita at age 10 years showed multiple old healed fractures of both tibias and ankles, but no osteopenia. Her ankles demonstrated marked sclerosis and significant deformities of the talus, calcaneus, and mid-tarsals consist with Charcot arthropathy (Figure 4A). At age 23 years, radiographs of her hips revealed an old avulsion fracture at the left grater trochanter, and deformity of left femoral neck. Pars articularis fractures were present at L4 and L5. Otherwise, radiographs including vertebral bodies, knees, hands, and wrists were all normal. At age 29 years, spine films showed healing of the pars articularis fractures at both L4 and L5. Bone mineral content appeared normal.
Figure 4. Radiographic studies of family members.
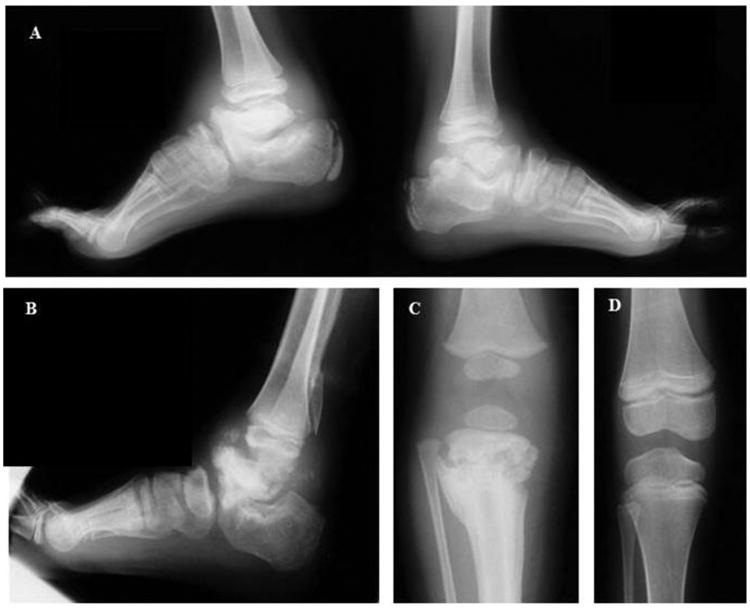
Both ankles of the proposita at age 10 years (A) and right ankle of her son at age 9.5 years (B) show marked sclerosis, flattening, fragmentation, and deformities of the talus, ankle joints, calcaneus, and mid-tarsals consist with Charcot arthropathy. There is some sclerosis and deformity of the metatarsals. The distal tibia and fibula appear normal. The daughter's knee at age 1.5 years (C) reveals a pathologic fracture with sclerosis, fragmentation, and new bone formation of the proximal tibia, but there was good healing with mild residual metaphyseal sclerosis at age 7.5 years (D)
2) Son
The son's radiographs at age 3.5 years showed no evidence of osteopenia. The right distal femur and proximal tibia were enlarged. His skull, spine, hips, hands, and wrists appeared normal. There were old fractures of both tibias and prominent right genu valgus. His right ulna and radius were subluxated inferiorly. The right calcaneus and talus were significantly sclerotic and deformed in a pattern consistent with a CIP. At age 9.5 years, his right calcaneus was almost completely destroyed (Figure 4B), and the left talus and calcaneus were deformed.
3) Daughter
At age 1.5 years, radiographs revealed minimal metaphyseal irregularity at both distal ulnas. Both tali and calcanei were significantly deformed and consistent with Charcot arthropathy. The right proximal tibia had a pathologic fracture, sclerosis, fragmentation, and new bone formation. The fibula was subluxed (Figure 4C).
At age 7.5 years, interestingly, complete healing of the right proximal tibial pathologic fracture had occurred over a 6-year period with only some minimal metaphyseal sclerosis and without joint destruction (Figure 4D). Both the talus and calcaneus showed progressive destruction. There was no evidence of osteopenia.
In summary, radiographs of all three family members showed multiple fractures primarily in their lower extremities, together with progressively worsening Charcot arthropathy in the ankle joints.
DXA BMD z-scores were normal in all three patients (Table 3).
Table 3. DXA BMD T-Scores and Z-Scores in three patients.
| BMD | T-score | Z-score | ||||
|---|---|---|---|---|---|---|
| Proposita | Proposita | Son | Daughter | |||
| Age (years) | 23 | 29 | 29 | 3.5 | 9 | 7.5 |
| Lumbar spine (L1 to L4) | - 0.4 | - 0.4 | - 0.4 | - 1.2 | - 0.1 | + 0.48 |
| Left hip | -1.1 | -1.0 | -1.0 | ND | ND | - 1.92 |
| Left forearm | + 0.2 | + 0.2 | + 0.3 | ND | ND | ND |
C) Bone histology
The proposita's iliac crest sampled at age 10 years consisted of cortical and trabecular bone with a small fragment of growth plate. The histology was normal (Figure 5).
Figure 5. Proposita's normal iliac crest biopsy.
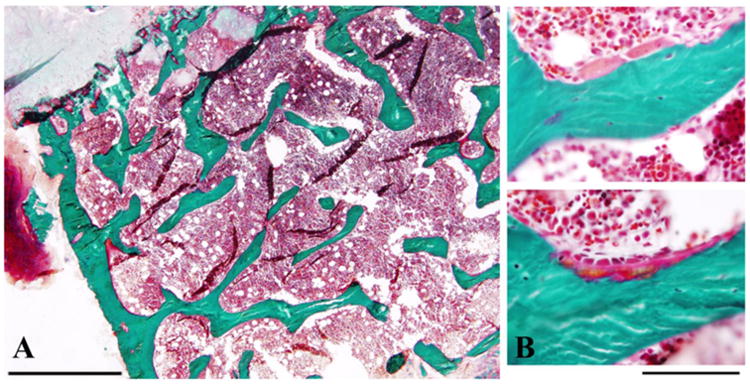
A) A low power Golder trichrome stain of iliac crest with growth plate at the top left (pale green) shows unremarkable trabecular architecture. Scale bar = 1 mm.
B) Higher magnification demonstrates normal osteoclast (top) and osteoblast (bottom) morphology. Scale bar = 100 μm.
D) Mutation analysis
A unique heterozygous mutation in exon 23 of SCN11A (c.3904C>T, p.Leu1302Phe) was discovered in both affected siblings (Figure 6A, B). This nucleotide variant was absent in the SNP database (www.ncbi.nlm.gov/SNP/) and the ExAC browser database (http://exac.broadinstitute.org/). A polymorphism in exon 12 (C1837T, rs4073113) of the son was detected. The proposita's half-brother did not carry the mutation (Figure 6C).
Figure 6. Mutation Analysis.
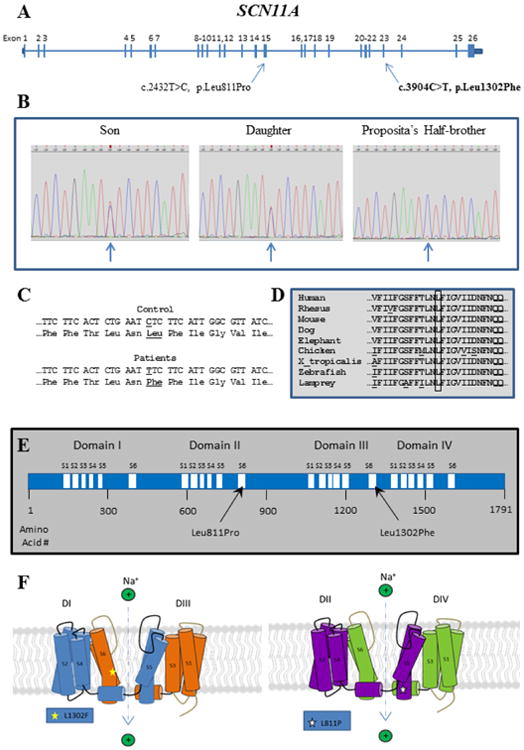
A) SCN11A gene structure showing location of mutations causing CIP.
B) Sanger sequencing electropherograms show the heterozygous SCN11A mutation in the two affected siblings. The mutation is absent in the proposita's half-brother.
C) Control and patients' DNA and amino acid sequences showing the C to T transition corresponding to CTC (Leu) to TTC (Phe) in codon 1302.
D) Evolutionary alignment of amino acid sequence in the region of exon 23 of SCN11A containing the mutation, using the alignment tool at the UCSC Genome Browser (https://genome.ucsc.edu). The boxed amino acid, leucine (L), is the affected amino acid. It is conserved throughout all species tested, indicating importance for SCN11A function.
E) SCN11A protein showing homologous Domains I–IV with transmembrane segments 1–6.
F) SCN11A protein structure: The transmembrane segments (S1-S6) are illustrated, especially the S6 segments which make up the walls of the Na+ channel, as well as the mutation (L1302F) in our CIP patients compared to the previously reported SCN11A mutation (L811P).
We found Leu1302 of SCN11A to be evolutionarily conserved (Figure 6D). A change of leucine to phenylalanine would be considered, according to PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/index.shtml), “probably damaging” with a score of 1.00 on a scale of up to 1.00. This mutation lies within transmembrane segment S6 of Domain III of SCN11A (Figure 6E), which would directly impact the sodium channel (see Discussion).
V) Discussion
Inability to feel pain leads to recurrent injuries and diminished survival.(1) Pain is perceived within the central nervous system (CNS) when a norciceptor detects and transmits a high-threshold stimulus caused by tissue injury.(28) Voltage-gated sodium (Na) channels in membranes are crucial for initiation and propagation of action potentials in excitable tissues.(29-31) To date, ten voltage-gated Na channels (Nav1.1-1.9 and Nav2.x), encoded by separate genes (SCN1ASCN5A and SCN7A-SCN11A), are expressed in mammals;(32) eight (Nav1.1-1.3, Nav1.6-1.9 and Nav2.x) in sensory neurons.(31-33) Defects in three sodium channels (Nav1.7, Nav1.8 and Nav1.9, encoded by SCN9A, SCN10A, and SCN11A, respectively) cause pain disorders in humans.(29, 34-36)
A) Physiological and Pathophysiological Role of Nav1.9 Encoded by SCN11A
Nav1.9 is highly expressed in peripheral sensory, trigeminal, and dorsal root ganglia (DRG), and in the enteric plexus.(32) It functions primarily to transmit pain signals from the periphery to the CNS. This large tetrodotoxin-resistant polypeptide comprises a 260 kDa α-subunit and an auxillary β-subunit.(33) Each α-subunit consists of six α-helical transmembrane-spanning segments (S1-S6) linked by three loops, repeated within four homologous domains (DI-DIV) (Figure 6E, F).(29,30,31,37)
In 2013, heterozygous gain-of-function mutation of SCN11A on chromosome 3p21-24 was discovered by Leipold et al(24) to cause CIP. However, only three people with CIP from SCN11A mutation have been reported -- all had sporadic disease from the identical heterozygous mutation in exon 15 (c.2432T>C, p.Leu811Pro) located at the distal end of segment 6 in domain II (S6, DII) (Figure 6E, F).(24,25,38) In the mature transmembrane Nav1.9 protein, the altered amino acid lies immediately adjacent to the channel's pore (Figure 6F). In knock-in mice, this mutant Nav1.9 caused severe cutaneous lesions at the cervical area, and reduced pain sensitivity without morphologic changes in sensory axons or small nerve fibers in the skin. Joint hypermobility was not reported in these mice.(24) This Nav1.9 Leu811Pro mutant channel in ND7/23 cells showed a prominent gain-of-function phenotype mediated by a leftward shift in the channel's activation and deactivation kinetics, and diminished the channel's inactivation thus blocking transmission of the pain signal.(24)
Interestingly, recent reports indicate that some gain-of-function mutations in SCN11A can also increase pain perception.(39,40) In 2013, Zhang and colleagues(39) reported two large Chinese families with AD episodic pain who carried separately two missense mutations in SCN11A; c.673C>T (p.Arg225Cys) and c.2423C>G (p.Ala808Gly). Both mutant Nav1.9 channels caused higher electrical activities in DRG-neurons, resulting in altered membrane potentials and the opening of the other Nav channels contributing to the pain disorder.(39) Then, in 2014, Huang and colleagues(40) sequenced SCN11A in 345 patients with familial and sporadic painful peripheral neuropathy and found 12 who carried one of eight different missense mutations [c.1142T>C (p.Ile381Thr), c.1257G>T (p.Lys419Asn), c.1744G>A (p.Ala582Thr), c.2042C>A (p.Ala681Asp), c.2524G>C (p.Ala842Pro), c.3473T>C (p.Leu1158Pro), c.4057-1G>A, c.5067C>G (p.Phe1689Leu)]. Two of these eight mutations, Ile381Thr and Leu1158Pro, were then selected for functional analysis. They conferred gain-of-function to the channel, depolarized the resting membrane potential of DRG neurons, and enhanced their spontaneous or evoked firing.(40) How gain-of-function mutations in SCN11A engender either increased or decreased pain perception is unknown.(25) Perhaps, “effect size” of the mutation determines their pathophysiologic consequences.(25)
B) CIP Families
Our study family (mother and two affected children) with a unique SCN11A mutation revealed an AD CIP. Nerve biopsy and conduction velocity and EMG investigations of the mother were normal. Since 1960, four families have been reported with AD CIP and painless fractures mainly in the lower extremities, self-mutilation, and normal sensitivity to other sensory modalities,(41-44) but they were not investigated by genetic testing, and were not reported to have joint hypermobility.
Here, we documented in the two siblings with CIP, a novel heterozygous mutation (c.3904C>T, p.Leu1302Phe) in exon 23 of SCN11A, presumably present in their affected mother. This mutation is predicted to compromise transmembrane segment S6 in DIII of Nav1.9. The four S6 motifs form the walls of the central pore of this Na+ channel (Figure 6F).(29) In the mature transmembrane protein, the mutation would position analogously to the original Nav1.9 mutation (Leu811Pro, in S6, DII). Both defects are near the channel's pore, and in close proximity to the intracellular inactivation motif involving three hydrophobic amino acids (Ile, Phe, Met) called the IFM motif, which can block the Na+ channel. Hence, either mutation could interfere directly with the Na+ channel, or indirectly via the IFM motif, to alter Na+ transport.(29) In fact, our patients shared most clinical features of CIP reported in the three European sporadic cases (inability to feel pain, recurrent painless fractures, self-inflicted injuries, pruritus, slow wound healing, multiple scars over the body, Charcot joint, heat and cold intolerance, and chronic diarrhea),(25) however, failure-to-thrive and hyperhidrosis(25) were found only in the daughter (Table 1). Although the major consequence of the SCN11A mutation is diminished pain sensation, chronic diarrhea is common, and Nav1.9 is expressed in myenteric neurons.(45,46) Nav1.9 knockout mice have significantly increased intestinal motility, documenting that Nav1.9 functions in colonic motor activity,(47) and perhaps explaining our patients' chronic diarrhea. Hyperhidrosis in the daughter too might represent autonomic dysfunction.(24) Thus, gain-of-function mutations in SCN11A apparently engender complex effects on different neuronal subtypes, not only for pain sensation.
Uniquely, our three patients and reportedly the proposita's mother (suspected to have CIP, but DNA unavailable for testing), had marked joint hypermobility not reported previously in SCN11A mutation CIP patients or mice.(24,29) Joint hypermobility caused by ligamentous laxity is usually found in genetic disorders of connective tissue, such as EDS, OI, Marfan syndrome, or other syndromes including trisomy 21, bony dysplasias, or velocardiofacial syndrome.(48-50) Our patients did not exhibit these further clinical features of the connective tissues diseases described above, and skin fibroblast studies from the son were negative for EDS and OI. Perhaps joint hypermobility is a consequence of this SCN11A mutation (c.3904C>T, p.Leu1302Phe). All four family members with CIP had joint hypermobility, whereas the proposita's half-brother without hypermobility proved negative for the mutation. However, none of the few reports indicate SCN11A expression in bone or other connective tissues.(32,37) Our observations indicate that a potential role for SCN11A in connective tissues should be investigated. Alternatively, the joint hypermobility could be co-segregating with CIP as an independent, but uncharacterized, disorder.
C) Skeletal and Mineral Homeostasis
Skeletal complications are common in CIP and include recurrent fractures, bony non-unions and mal-unions, joint dislocations, scoliosis, leg-length discrepancy, foot deformities, auto-amputation, heterotopic ossification, avascular necrosis, osteomyelitis, and Charcot arthropathy.(43,51-54) The Charcot arthropathy, secondary to loss of sensation, is one of most prevalent complications and features chronic destruction of bones and joints alike, resulting in pathological fractures, dislocations, or subluxations.(55) The most commonly affected sites are the ankle and foot, especially the tarsometatarsal joints.(53,56,57) Infrequently, the spine is involved.(58-60) Charcot arthropathy manifests variably, depending on its clinical stage, and leads to mild to severe swelling and deformity.(56) Misdiagnosed or untreated, severe bone deformity and disability are expected in adulthood.(58,61) CIP fractures occur primarily in the femur and tibia.(51,53) In 2014, Haga and Zhang(62) reported 14 CIP patients with anhydrosis (HSAN type IV) caused by NTRK1 mutation, and identified the most common skeletal complications to be fractures (91%) and Charcot joints (29%), both involving primarily the lower limbs. Fractures were particularly frequent between ages 1-7 years.(62) Osteomyelitis, resulting from repeated minor trauma and biting of the digits, can also be found in CIP, but is usually restricted to toes and fingers, and leads to autoamputation.(53)
In our patients, recurrent painless fracturing, especially in the lower limbs, began in early childhood. These totaled 4-12 events per individual, but did not reflect their ages. The proposita and her son had genu valgus and Charcot ankles and knees. She had frequent soft tissue swelling of her ankles at age 7 years diagnosed as rheumatoid arthritis, but likely representing early Charcot arthropathy. At age 23 years, her examination showed no clinical evidence of ankle deformities. In contrast, her son's Charcot ankles and knees at age 3.5 years progressed at age 9.5 years to permanent ankle deformities. At age 7.5 years, the daughter manifested leg-length discrepancy, but without Charcot arthropathy.
Although CIP consistently features skeletal problems,(22,24,29,51-54) little is reported concerning mineral or skeletal homeostasis in this condition, and nothing is known for the NaV 1.7 and 1.9 channelopathies in particular. Four CIP reports briefly mention that “mineral homeostasis” showed normal serum calcium and phosphorus levels.(9,44,53) A study in 1971 reported reduced gastrointestinal absorption of calcium and excessive osteoid tissue on iliac crest biopsy despite normal serum calcium and phosphorus levels and normal jejunal mucosa histology.(43)
Despite the significant skeletal complications of our three patients, their fractures healed well and all had unremarkable biochemical parameters of mineral and skeletal homeostasis, no radiographic evidence of rickets or osteopenia, DXA lumbar spine and total hip BMD were unremarkable, and iliac crest biopsy of the proposita showed normal histology. Their fracturing, especially in the lower extremities, occurred early in life. Perhaps increased apprehension for harmful situations explained their diminished fracturing as they grew older. In fact, our studies spanning pediatric to adult life failed to disclose any intrinsic skeletal pathobiology. Although lack of awareness might explain the recurrent fractures, the low-trauma initial fracturing that we have sometimes encountered in CIP holds open the possibility of poor quality bone not disclosed by our skeletal investigations. Importantly, a high degree of suspicion of CIP is crucial for early diagnosis and prompt measures to prevent bone and joint destruction.
Supplementary Material
Highlights.
Nav1.9 channelopathy causes congenital insensitivity to pain with hypermobility
The heterozygous missense mutation (c.3904C>T, p.Leu1302Phe) within SCN11A is novel
Despite fracturing, mineral and skeletal homeostasis is normal
Low-trauma fracturing suggests an uncharacterized defect in bone quality
Perhaps early diagnosis can prevent bone and joint destruction
Acknowledgments
Ms. Margaret Huskey and Ms. Duan Shenghui assisted with the SCN11A sequencing. Mr. Vinieth Bijanki provided technical support, and with Mrs. Sharon McKenzie helped prepare the manuscript. We are grateful to Dr. James Coldwell for his continued interest and help concerning this family.
Funded in part by Shriners Hospitals for Children, The Clark and Mildred Cox Inherited Metabolic Bone Disease Research Fund, The Hypophosphatasia Research Fund, The Barnes-Jewish Hospital Foundation, and the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health under Award Number DK067145. The content of the manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
To be presented at the 34th Annual Meeting of the American Society for Bone and Mineral Research, October 9 - 12, 2015, Seattle, Washington, USA [J Bone Miner Res 30 (Suppl): S459, 2015].
Authors' roles: All authors helped write and approved the manuscript. VP performed the mutation analysis, reviewed the medical literature, and composed the first draft of the manuscript. SM supervised the mutation analysis and its interpretation. WHM reviewed the family's radiographs, and described the radiological nature of their skeletal disease. DVN evaluated the iliac crest histology. DW and KLC helped with the clinical studies of the patients. MPW coordinated the family investigation and manuscript preparation and completion.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Voraluck Phatarakijnirund, Email: vphatara@dom.wustl.edu.
Steven Mumm, Email: smumm@dom.wustl.edu.
William H. McAlister, Email: mcalisterw@mir.wustl.edu.
Deborah Novack, Email: Novack@wustl.edu.
Deborah Wenkert, Email: wenkert@i1.net.
Karen L. Clements, Email: kclements@shrinenet.org.
Michael P. Whyte, Email: mwhyte@shrinenet.org.
References
- 1.Nagasako EM, Oaklander AL, Dworkin RH. Congenital insensitivity to pain: an update. Pain. 2003;101:213–9. doi: 10.1016/S0304-3959(02)00482-7. [DOI] [PubMed] [Google Scholar]
- 2.Dearborn G. A case of congenital general pure analgesia. J Nerv Ment Dis. 1932;75:612–5. [Google Scholar]
- 3.Ford FR, Wilkins L. Congenital universal insensitiveness to pain. Bull Johns Hopkins Hosp. 1938;62:448–66. [Google Scholar]
- 4.Boyd DA, Nie LW. Congenital universal indifference to pain. Arch Neurol Psychiatry. 1949;61:402–12. doi: 10.1001/archneurpsyc.1949.02310100066005. [DOI] [PubMed] [Google Scholar]
- 5.Arbuse DI, Cantor MB, Barenberg PA. Congenital indifference to pain. J Pediatr. 1949;35(2):221–6. doi: 10.1016/s0022-3476(49)80236-8. [DOI] [PubMed] [Google Scholar]
- 6.Ingwersen OS. Congenital indifference to pain. Report of a case. J Bone Joint Surg Br. 1967;49(4):704–9. [PubMed] [Google Scholar]
- 7.McMurray GA. Experimental study of a case of insensitivity to pain. Arch Neurol Psych. 1950;64:650–67. doi: 10.1001/archneurpsyc.1950.02310290046005. [DOI] [PubMed] [Google Scholar]
- 8.Sternbach RA. Congenital insensitivity to pain: A Critique. Psychol Bull. 1963;60(3):252–64. [Google Scholar]
- 9.Thrush DC. Congenital insensitivity to pain. A clinical, genetic and neurophysiological study of four children from the same family. Brain. 1973;96(2):369–86. doi: 10.1093/brain/96.2.369. [DOI] [PubMed] [Google Scholar]
- 10.Hellstrom B. Congenital analgia. A case report. Acta Paediatr. 1962;135:83–7. [PubMed] [Google Scholar]
- 11.Winkelmann RK, Lambert EH, Hayles AB. Congenital absence of pain. Arch Dermatol. 1962;85:325–39. doi: 10.1001/archderm.1962.01590030023004. [DOI] [PubMed] [Google Scholar]
- 12.Morris D. Congenital Absence of Pain. Post grad Med J. 1962;38(445):640–1. doi: 10.1136/pgmj.38.445.640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fath MA, Hassanein MR, James JI. Congenital absence of pain. A family study. J Bone Joint Surg Br. 1983;65(2):186–8. doi: 10.1302/0301-620X.65B2.6186667. [DOI] [PubMed] [Google Scholar]
- 14.Manfredi M, Bini G, Cruccu G, Accornero N, Berardelli A, Medolago L. Congenital absence of pain. Arch Neurol. 1981;38(8):507–11. doi: 10.1001/archneur.1981.00510080069010. [DOI] [PubMed] [Google Scholar]
- 15.Gutman D, Benderli A, Laufer D, Levi J. Congenital analgesia. Report of a case. Oral Surg Oral Med Oral Pathol. 1975;39(6):867–9. doi: 10.1016/0030-4220(75)90106-1. [DOI] [PubMed] [Google Scholar]
- 16.Dyck PJ, Mellinger JF, Reagan TJ, Horowitz SJ, McDonald JW, Litchy WJ, et al. Not ‘indifference to pain’ but varieties of hereditary sensory and autonomic neuropathy. Brain. 1983;106:373–90. doi: 10.1093/brain/106.2.373. [DOI] [PubMed] [Google Scholar]
- 17.Dyck PJ. Neuronal atrophy and degeneration predominantly affecting peripheral sensory and autonomic neurons. In: Dyck PJ, Thomas PK, Griffin JW, Low PA, Poduslo JF, editors. Peripheral neuropathy. 3rd. W.B Saunders; Philadelphia: 1993. pp. 1065–1093. [Google Scholar]
- 18.Davidson GL, Murphy SM, Polke JM, Laura M, Salih MAM, Muntoni F, et al. Frequency of mutations in the genes associated with hereditary sensory and autonomic neuropathy in a UK cohort. J Neurol. 2012;259:1673–85. doi: 10.1007/s00415-011-6397-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sandroni P, Martin DP, Bruce BK, Rome JD. Congenital idiopathic inability to perceive pain: A new syndrome of insensitivity to pain and itch preserves small fibers. Pain. 2006;122:210–5. doi: 10.1016/j.pain.2006.01.033. [DOI] [PubMed] [Google Scholar]
- 20.Verhoeven K, Timmerman V, Mauko B, Thomas R, Pieber TR, Jonghe PD, et al. Recent advances in hereditary sensory and autonomic neuropathies. Curr Opin Neurol. 2006;19:474–80. doi: 10.1097/01.wco.0000245370.82317.f6. [DOI] [PubMed] [Google Scholar]
- 21.McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University; Baltimore, MD: Sep 30, 2014. Online Mendelian Inheritance in Man, OMIM®. World Wide Web URL: http://omim.org/ [Google Scholar]
- 22.Cox JJ, Reimann F, Nicholas AK, Thornton G, Roberts E, Springell K, et al. An SCN9A channelopathy causes congenital inability to experience pain. Nature. 2006;444:894–8. doi: 10.1038/nature05413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Waxman SG. Nav1.7, its mutations, and the syndromes that they cause. Neurology. 2007;69:505–7. doi: 10.1212/01.wnl.0000268068.02343.37. [DOI] [PubMed] [Google Scholar]
- 24.Leipold E, Liebmann L, Korenke GC, Heinrich T, Giesselmann S, Baets J, et al. A de novo gain-of-function mutation in SCN11A causes loss of pain perception. Nat Genet. 2013;45:1399–404. doi: 10.1038/ng.2767. [DOI] [PubMed] [Google Scholar]
- 25.Woods CG, Babiker MO, Horrocks I, Tolmie J, Kurth I. The phenotype of congenital insensitivity to pain due to the NaV1.9 variant p.L811P. Eur J Hum Genet. 2015;23(5):561–3. doi: 10.1038/ejhg.2014.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kelly TL, Specker BL, Binkley T, et al. Pediatric BMD reference database for U.S. white children. Bone. 2005;36(Suppl 1):30. [Google Scholar]
- 27.Wenkert D, McAlister WH, Coldwell JG, Mumm S, Whyte MP. Dominantly-inherited congenital insensitivity to pain manifesting with recurrent fractures: a new entity. Journal of Bone and Mineral Research. 2003;18(Suppl 2):S388. [Google Scholar]
- 28.Meyer RA, Ringkam M, Campbell JN, Raja SN. Peripheral mechanisms of cutaneous nociceptor. In: McMahon SB, Koltzenburg M, editors. Wall and Melzack's Textbook of pain. 5th. Philadelphia, PA: Elsevier/Churchill Livingston; 2006. pp. 3–34. [Google Scholar]
- 29.Bennett DL, Woods CG. Painful and painless channelopathies. Lancet Neurol. 2014;13:587–99. doi: 10.1016/S1474-4422(14)70024-9. [DOI] [PubMed] [Google Scholar]
- 30.Dib-Hajj SD, Tyrrell L, Waxman SG. Structure of the sodium channel gene SCN11A: Evidence for intron-to-exon conversion model and implications for gene evolution. Mol Neurobiol. 2002;26:235–50. doi: 10.1385/MN:26:2-3:235. [DOI] [PubMed] [Google Scholar]
- 31.Dib-Haji SD, Cummins TR, Black JA, Waxman SG. Sodium channels in normal and pathological pain. Annu Rev Neurosci. 2010;33:325–47. doi: 10.1146/annurev-neuro-060909-153234. [DOI] [PubMed] [Google Scholar]
- 32.Levinson SR, Luo S, Henry MA. The role of sodium channels in chronic pain. Muscle Nerve. 2012;46(2):155–65. doi: 10.1002/mus.23314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chahine M, Ziane R, Vijayaragavan K, Okamura Y. Regulation of Nav channels in sensory neurons. Trends Pharmacol Sci. 2005;26:496–50. doi: 10.1016/j.tips.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 34.Houlden H. Extending the clinical spectrum of pain channelopathies. Brain. 2012;135:313–6. doi: 10.1093/brain/aws007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Faber CG, Lauria G, Merkies ISJ, Cheng X, Han C, Ahn HS, et al. Gain-of-function Nav1.8 mutations in painful neuropathy. Proc Natl Acad Sci USA. 2012;109(47):19444–9. doi: 10.1073/pnas.1216080109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang J, Yang Y, Zhao P, Gerrits MM, Hoeijmakers JG, Bekelaar K, et al. Small-fiber neuropathy Nav1.8 mutation shifts activation to hyperpolarized potentials and increases excitability of dorsal root ganglion neurons. J Neurosci. 2013;33(35):14087–97. doi: 10.1523/JNEUROSCI.2710-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brunklaus A, Ellis R, Reavey E, Semsarian C, Zuberi SM. Genotype phenotype associations across the voltage-gated sodium channel family. J Med Genet. 2014;51:650–8. doi: 10.1136/jmedgenet-2014-102608. [DOI] [PubMed] [Google Scholar]
- 38.Cox JJ, Wood JN. No pain, more gain. Nat Genet. 2013;45:1271–2. doi: 10.1038/ng.2810. [DOI] [PubMed] [Google Scholar]
- 39.Zhang XY, Wen J, Yang W, Wang C, Gao L, Zheng LH, et al. Gain-of-function mutations in SCN11A cause familial episodic pain. Am J Hum Genet. 2013;93:957–66. doi: 10.1016/j.ajhg.2013.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang J, Han C, Estacion M, Vasylyev D, Hoeijmakers JG, Gerrits MM, et al. Gain-of-function mutations in sodium channel Na(v)1.9 in painful neuropathy. Brain. 2014;137:1627–42. doi: 10.1093/brain/awu079. [DOI] [PubMed] [Google Scholar]
- 41.Ervin FR, Sternbach RA. Hereditary insensitivity to pain. Trans Am Neurol Assoc. 1960;85:70–4. [PubMed] [Google Scholar]
- 42.Comings DE, Amromin GD. Autosomal dominant insensitivity to pain with hyperplastic myelinopathy and autosomal dominant indifference to pain. Neurology. 1974;24:838–48. doi: 10.1212/wnl.24.9.838. [DOI] [PubMed] [Google Scholar]
- 43.Haaxma R, Korver MF, Willemse J. Congenital indifference to pain associated with a defect in calcium metabolism. Acta Neurol Scand. 1971;47(2):194–208. doi: 10.1111/j.1600-0404.1971.tb07476.x. [DOI] [PubMed] [Google Scholar]
- 44.Landrieu P, Said G, Allaire C. Dominantly transmitted congenital indifference to pain. Ann Neurol. 1990;27(5):574–8. doi: 10.1002/ana.410270520. [DOI] [PubMed] [Google Scholar]
- 45.Dib-Hajj SD, Tyrrell L, Black JA, Waxman SG. NaN, a novel voltage-gated Na channel, is expressed preferentially in peripheral sensory neurons and down-regulated after axotomy. Proc Natl Acad Sci USA. 1995;95:8963–8. doi: 10.1073/pnas.95.15.8963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rugiero F, Mistry M, Sage D, Black JA, Waxman SG, Crest M, et al. Selective expression of a persistent tetrodotoxin-resistant Na+ current and NaV1.9 subunit in myenteric sensory neurons. J Neurosci. 2003;23:2715–25. doi: 10.1523/JNEUROSCI.23-07-02715.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Copel C, Clerc N, Osorio N, Delmas P, Mazet B. The Nav1.9 channel regulates colonic motility in mice. Front Neurosci. 2013;7:58. doi: 10.3389/fnins.2013.00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hakim A, Grahame R. Joint hypermobility. Best Pract Res Clin Rheumatol. 2003;17:989–1004. doi: 10.1016/j.berh.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 49.Grahame R, Hakim AJ. Hypermobility. Curr Opin Rheumatol. 2008;20:106–10. doi: 10.1097/BOR.0b013e3282f31790. [DOI] [PubMed] [Google Scholar]
- 50.Clinch J, Deere K, Sayers A, Palmer S, Riddoch C, Tobias JH, et al. Epidemiology of generalized joint laxity (hypermobility) in fourteen-year-old children from the UK: a population-based evaluation. Arthritis Rheum. 2011;63:2819–27. doi: 10.1002/art.30435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bar-On E, Weigl D, Parvari R, Katz K, Weitz R, Steinberg T. Congenital insensitivity to pain. Orthopaedic manifestations. J Bone Joint Surg Br. 2002;84(2):252–7. doi: 10.1302/0301-620x.84b2.11939. [DOI] [PubMed] [Google Scholar]
- 52.Dimon JH, 3rd, Funk FJ, Jr, Wells RE. Congenital indifference to pain with associated orthopedic abnormalities. South Med J. 1965;58:524–9. doi: 10.1097/00007611-196504000-00029. [DOI] [PubMed] [Google Scholar]
- 53.Guidera KJ, Multhopp H, Ganey T, Ogden JA. Orthopaedic manifestations in congenitally insensate patients. J Pediatr Orthop. 1990;10(4):514–21. [PubMed] [Google Scholar]
- 54.Greider TD. Orthopedic aspects of congenital insensitivity to pain. Clin Orthop Relat Res. 1983;172:177–85. [PubMed] [Google Scholar]
- 55.Sinacore DR, Withrington NC. Recognition and management of acute neuropathic (Charcot) arthropathies of the foot and ankle. J Orthop Sports Phys Ther. 1999;29(12):736–46. doi: 10.2519/jospt.1999.29.12.736. [DOI] [PubMed] [Google Scholar]
- 56.Sommer TC, Lee TH. Charcot foot: the diagnostic dilemma. Am Fam Physician. 2001;64:1591–8. [PubMed] [Google Scholar]
- 57.Jones EA, Manaster BJ, May DA, Disler DG. Neuropathic osteoarthropathy: diagnostic dilemmas and differential diagnosis. Radiographics. 2000;20:S279–9. doi: 10.1148/radiographics.20.suppl_1.g00oc22s279. [DOI] [PubMed] [Google Scholar]
- 58.Cassidy RC, Shaffer WO. Charcot arthropathy because of congenital insensitivity to pain in an adult. Spine J. 2008;8:691–5. doi: 10.1016/j.spinee.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 59.Erdil M, Bilsel K, Imren Y, Ceylan HH, Tuncay I. Total hip arthroplasty in a patient with congenital insensitivity to pain: a case report. J Med Case Rep. 2012;6:190. doi: 10.1186/1752-1947-6-190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hirsch E, Moye D, Dimon JH., 3rd Congenital indifference to pain: long-term follow-up of two cases. South Med J. 1995;88(8):851–7. doi: 10.1097/00007611-199508000-00014. [DOI] [PubMed] [Google Scholar]
- 61.Abdulla M, Khaled SS, Khaled YS, Kapoor H. Congenital insensitivity to pain in a child attending a paediatric fracture clinic. J Pediatr Orthop B. 2014;23:406–10. doi: 10.1097/BPB.0000000000000051. [DOI] [PubMed] [Google Scholar]
- 62.Zhang Y, Haga N. Skeletal complications in congenital insensitivity to pain with anhidrosis: a case series of 14 patients and review of articles published in Japanese. J Orthop Sci. 2014;19:827–31. doi: 10.1007/s00776-014-0595-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.