

Abstract
Background
The Tibetan cashmere goat (Capra hircus), one of the most ancient breeds in China, has historically been a critical source of meat and cashmere production for local farmers. To adapt to the high-altitude area, extremely harsh climate, and hypoxic environment that the Tibetan cashmere goat lives in, this goat has developed distinct phenotypic traits compared to lowland breeds. However, the genetic components underlying this phenotypic adaptation remain largely unknown.
Results
We obtained 118,700 autosomal SNPs through exome sequencing of 330 cashmere goats located at a wide geographic range, including the Tibetan Plateau and low-altitude regions in China. The great majority of SNPs showed low genetic differentiation among populations; however, approximately 2-3 % of the loci showed more genetic differentiation than expected under a selectively neutral model. Together with a combined analysis of high- and low-altitude breeds, we revealed 339 genes potentially under high-altitude selection. Genes associated with cardiovascular system development were significantly enriched in our study. Among these genes, the most evident one was endothelial PAS domain protein 1 (EPAS1), which has been previously reported to be involved in complex oxygen sensing and significantly associated with high-altitude adaptation of human, dog, and grey wolf. The missense mutation Q579L that we identified in EPAS1, which occurs next to the Hypoxia-Inducible Factor-1 (HIF-1) domain, was exclusively enriched in the high-altitude populations.
Conclusions
Our study provides insights concerning the population variation in six different cashmere goat populations in China. The variants in cardiovascular system-related genes may explain the observed phenotypic adaptation of the Tibetan cashmere goat.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-016-2449-0) contains supplementary material, which is available to authorized users.
Keywords: Cashmere goat, Population genetics, High altitude, Hypoxia
Background
Among the most severe environmental challenges to confront animals is the low oxygen availability of high-altitude regions such as the Tibetan Plateau, which is at an elevation above 5,000 m. The partial pressure of oxygen at 5,000 m is only approximately 50 % of the value at sea-level, and the resultant hypoxia imposes severe constraints on aerobic metabolism and leads to high-altitude illness [1–3]. Thus the mechanisms of hypoxic adaptation have become of great interest in recent years. Genome-wide scans and whole genome re-sequencing analysis for identifying selection signatures of high-altitude adaptation have been performed across a wide range of species, including human [4–6], dog [7, 8], grey wolf [9], yak [10], Tibetan antelope [11], cattle [12], pig [13], and chicken [14]. Although the convergent evolution between different species was reported in Tibetan people, dog and grey wolf with the EPAS1 (endothelial PAS domain protein 1) gene as the common selected locus [4–9], in other species, distinct loci were revealed for high-altitude adaptation, suggesting that diverse molecular mechanisms may be employed.
Cashmere goat has the broadest altitudinal range of all herbivores in mainland China [15], as it is continuously distributed from sea level (e.g., Liaoning cashmere goat) to the Tibetan Plateau (e.g., Tibetan cashmere goat). As first recorded in 3,300-2,000 BC [16], the Tibetan cashmere goat exhibits phenotypic adaptation to the hypoxia conditions of the Tibetan Plateau under long-term natural and artificial selection [17]. Pioneering work regarding the high-altitude adaptation of the Tibetan cashmere goat mainly at the anatomical, biochemical, and physiological levels began two decades ago [18–23]. These early studies showed that the Tibetan goats and the F1 progeny of Tibetan goats × imported Liaoning cashmere goats (goats that originated from Liaoning and that are imported into Tibet) had stable inheritable adaptability to live in a high-altitude environment with some adaptive traits [24]. Compared with the goats living at an altitude below 3,000 m, the Tibetan goats maintained higher adult haemoglobin (Hb) concentrations, heavier heart and lungs, and lower heart rates [18–23]. Therefore, the Tibetan cashmere goat provides an outstanding model for understanding the genetic mechanism of hypoxia adaptation and hypoxia-related disease development. However, the molecular mechanism underlying the observed phenotypic adaptation to the high-altitude environment remains largely unknown.
Our previous studies regarding the Chinese cashmere goat, based on markers from both mitochondrial DNA and microsatellites [25–27], showed a high level of genetic differentiation between the Tibetan cashmere goat and other cashmere goat breeds [27]. Hence, we speculated that the Tibetan cashmere goat carries unique adaptive alleles for the high-altitude environment [27] that have never been identified due to the limited number of polymorphic markers. The development of next-generation sequencing and the release of the first goat reference genome assembly [28] offer us a great opportunity to investigate the genetic differentiation among goat breeds in a high-throughput method. Here, we extended our previous analyses with high-throughput sequencing of exome-captured genomic DNA to detect the genetic variation of six major Chinese cashmere goat populations (Table 1). We then conducted a genome-wide screen for the selection signatures at the exome regions in two high-altitude Tibetan cashmere goat populations (Bange [BG] and Ritu [RT]) by comparing these populations with the four lowland breeds (Chaidamu [CDM], Nanjiang [NJ], Inner Mongolia [IM], and Liaoning [LN]) to identify the possible molecular basis of the high-altitude adaptation (Table 1). This study is the first comprehensive analysis of the population genetic differentiation in Chinese indigenous cashmere goats and detection of the molecular mechanisms underlying the observed phenotypic adaptation of Tibetan cashmere goats.
Table 1.
Information about samples collected from Chinese cashmere goats at different altitudes
| Breed | Locality | Sample size | Altitude (m) | P a | H b | |
|---|---|---|---|---|---|---|
| F♂ | M♀ | |||||
| Highland | ||||||
| Bange (BG) | Bange, Tibet | 27 | 33 | 4,700 | 90.9 | 23.5 |
| Ritu (RT) | Ritu, Tibet | 31 | 37 | 4,500 | 86.9 | 22.9 |
| Lowland | ||||||
| Chaidamu (CDM) | Chaidamu, Qinghai | 20 | 25 | 3,000 | 94.0 | 24.0 |
| Nanjiang (NJ) | Aksu, Xinjiang | 28 | 30 | 1,700 | 89.9 | 23.5 |
| Inner Mongolia (IM) | Erlangshan, Inner Mongolia | 14 | 25 | 1,500 | 88.8 | 23.2 |
| Liaoning (LN) | Gaizhou, Liaoning | 30 | 30 | 30 | 84.3 | 22.4 |
Note: Pa: proportion of the total SNPs that have support for both alleles in each sample; Hb: average heterozygosity = (sum of [2*p*(1-p)] for all SNP ) / (total number of SNPs), where p is the frequency of the most common allele; F♂: the sample size of female individuals; M♀: the sample size of female individuals
Results
Exome sequencing of the pooled samples
We sequenced six pools of captured exomes from goats living at different altitudes (Table 1) using a HiSeq 2000 platform, resulting in 50–84 million raw reads and 89- to 163-fold coverage for each pool (Additional file 1: Table S1). We aligned the reads to the reference genome of the domestic goat (version CHIR_1.0) and estimated the allele frequencies for all 144,046 SNPs identified by our SNP calling pipeline as described in the Methods section. In total, 118,700 autosomal SNPs were used in the downstream analysis. A strong correlation was observed between the sequence-based allele frequency and individual genotyping allele frequency estimates in each population (Pearson’s correlation test; P: P value; R: Pearson’s correlation coefficient; RBG = 0.865, PBG < 2.20E-16; RRT = 0.933, PRT < 2.20E-16; RCDM = 0.903, PCDM < 2.20E-16; RNJ = 0.928, PNJ = 2.20E-16; RIM = 0.885, P IM < 2.20E-16; RLN = 0.872, PLN < 2.20E-16; Additional file 2: Figure S1), indicating that our strategy of DNA pooling and exome capture was quite effective for estimating the allele frequency of the SNPs in the exome region at the population level.
Genetic diversity analysis
The proportion of polymorphic loci and the average heterozygosity ranged from 84.3 % to 94.0 % and from 22.4 % to 24.0 %, respectively, with the lowest heterozygosity value in LN and the highest heterozygosity in CDM (Table 1), indicating the lowest diversity in LN and the highest in CDM. A phylogenetic tree based on all SNPs showed a clear separation among all populations (Fig. 1a). The topology structure of the tree was consistent with that of our previous result based on the mitochondrial DNA and microsatellite markers from the same cashmere breeds [25, 27]. The two Tibetan cashmere populations, BG and RT, formed one branch and differed greatly from other cashmere breeds. In the main branch of the lowland breeds, LN and NJ populations were grouped together, while CDM and IM were genetically distinct. Among all the branches of the tree, CDM had the shortest branch and LN had the longest one, which corresponded to the highest diversity in CDM and the lowest diversity in LN, consistent with the heterozygosity analysis (Table 1).
Fig. 1.
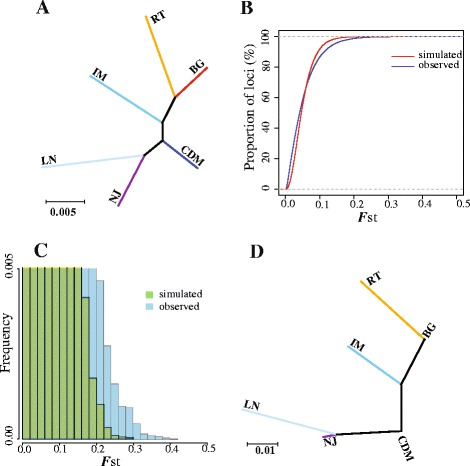
Phylogenetic analysis and Fst simulation test. a Phylogenetic tree analysis based on all markers. b Cumulative distribution of observed and simulated (assuming neutrality) Fst values. c Histogram of Fst values in the simulated and observed datasets (note the truncated y-axis). d Phylogenetic tree based on those global SNPs showing significant genetic differentiation
Because both natural selection and artificial selection have shaped the goat genomes and because genetic adaptations can occur due to many different variables, we first focused on the genetic differentiation in a global scenario to obtain a more comprehensive understanding of the selection patterns among all six populations. We estimated the global Fst for each identified SNP, and this estimation was termed the global Fst analysis. To evaluate the population diversity we calculated the average genetic differentiation coefficient (Fst), the average overall genetic diversity (Ht) and the average diversity within a population (Hs), which were 0.0531, 0.2116 and 0.2007, respectively. This calculation indicated that only 5.31 % of the total genetic diversity was partitioned among breeds and that most of the variability (94.69 %) was within populations (Hs = 0.2007).
Global Fst analysis reveals regions under selection across six populations
To elucidate whether the observed genetic differentiation is primarily driven by drift or selection, a simulation test was conducted. The simulation was based on six subpopulations, each with an effective population size of 23,231 individuals who had been separated for 3,000 generations, and on the average simulated Fst, which was identical to that observed in our global Fst dataset (mean Fst = 0.0531). Generally, the simulated data were consistent with the observed data (Fig. 1b). More than half (58.56 % and 55.15 % for the observed and simulated data, respectively) of the loci had Fst values less than 0.05; however, a significant number of loci (P = 1.10E-16) had extreme Fst values in the observed data compared with the simulation (Fig. 1c). Only 66 loci in the observed data had Fst values greater than 0.2953, whereas none was found in the simulated data. Furthermore, 2.44 % of the loci in the observed data (n = 1,415) had Fst values greater than 0.1723, whereas the corresponding value in the simulated data were 0.5 % (n = 290). Thus, approximately 2-3 % of the loci in our exome re-sequencing data showed greater genetic differentiation than expected under a selectively neutral model. Therefore, a threshold of the top 2.5 % was used to search for the loci under selection, and 1,449 SNPs, which overlapped with 880 genes, were found in the global Fst analysis.
Gene Ontology (GO) analysis of the 880 genes revealed 33 significantly enriched GO categories. Among these categories, the most significantly enriched GO categories were cell adhesion (GO:0007155, P_FDR = 6.90E-06), regulation of molecular function (GO:0065009, P_FDR = 1.79E-06) and multicellular organismal development (GO:0007275, P_FDR = 4.69E-04) (Additional file 3: Table S2). We also found some interesting GO categories, such as response to external stimulus (GO:0009605, P_FDR = 2.20E-02), response to cell communication involved in cardiac conduction (GO:0086065, P_FDR =3.04E-2), blood vessel development (GO:0001568, P_FDR = 3.04E-02), and circulatory system process (P_FDR = 4.49E-02), which may be related to adaptation the local environment adaptation.
Genetic differentiation between the high- and low-altitude goat breeds
Given that the BG and RT populations have adapted to a high-altitude environment for thousands of years, we sought to identify the candidate loci adaptive to high altitude. The phylogenetic tree that was built from the top 2.5 % outlier SNPs of the global Fst analysis showed a similar structure but with even clearer separation of the six goat populations (Fig. 1d) than the tree based on all SNPs (Fig. 1a). The new tree showed that the Tibetan cashmere goat populations (BG and RT) became even more divergent from other lowland breeds (Fig. 1d); however, the results were insufficient for determining whether the genetic differentiation resulted from high-altitude adaptation. Therefore, we evaluated the levels of population differentiation of BG and RT from other breeds to identify the loci showing evidence of adaption specific to the Tibetan goat using the statistic di [8] and χ2 test (see Methods section for details). By combining the di (di > 13.5) and χ2 test (FDR-adjusted p < 0.01) as the high-lowland analysis, we obtained 1,134 outlier SNPs that were genetically differentiated between highland and lowland goat populations (Additional file 4: Table S3; Fig. 2).
Fig. 2.
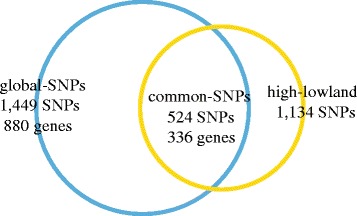
Overlap of SNPs from the global SNP dataset and high-lowland dataset
By comparing the candidate loci potentially under selection across all breeds to those genetically differentiated loci between highland and lowland populations, we identified 524 common SNPs that overlapped with 339 genes (termed as the common SNP dataset; Fig. 2; Additional file 5: Table S4). GO enrichment analysis with the 339 common genes showed a significant overrepresentation of the categories related to regulation of actin filament-based process (GO:0032970, P_FDR = 0.0266), cell adhesion (GO:0007155, P_FDR = 0.0365), regulation of molecular function (GO:0065009, P_FDR = 0.0465), regulation of multicellular organismal process (GO:0051239, P_FDR = 0.0465) and cardiac muscle cell action potential involved in contraction (GO:0086002, P_FDR = 0.0476; Table 2). Of the 339 common genes, 31 candidate genes were previously associated with high-altitude adaptation [9] and 66 were involved in cardiovascular system according to literature mining (Additional file 5: Table S4). Further protein-protein interaction network analysis showed that many hypoxia-related genes or cardiovascular system-related genes were observed in the protein interaction network (P = 8.83E-3) (Additional file 6: Figure S2). Based on the protein-protein interaction network, we identified that the ‘hub’ genes were hypoxia and cardiovascular system-related genes, including sirtuin type 1 (SIRT1), intercellular cell adhesion molecule-1 (ICAM1), endothelin receptor type A (EDNRA), Yamaguchi sarcoma viral-related oncogene homolog 1 (YES1), and plakoglobin (JUP) (Additional file 6: Figure S2).
Table 2.
Functional gene categories enriched for the common SNP dataset GO ID
| Term description | Number of genes | P value | FDR | |
|---|---|---|---|---|
| GO:0032970 | regulation of actin filament-based process | 15 | 2.11E-06 | 0.0266 |
| GO:0007155 | cell adhesion | 24 | 7.20E-06 | 0.0365 |
| GO:0065009 | regulation of molecular function | 54 | 1.23E-05 | 0.0465 |
| GO:0051239 | regulation of multicellular organismal process | 50 | 1.43E-05 | 0.0465 |
| GO:0086002 | cardiac muscle cell action potential involved in contraction | 5 | 2.27E-05 | 0.0476 |
GO enrichment analysis was also conducted with the remaining 925 specific SNPs (601 genes) from the global SNP dataset only. Interestingly, categories that were associated with the cellular component organization and biological adhesion were still significantly enriched, but the categories related to cardiovascular system were not (Additional file 7: Table S5). This finding indicated that genes in the categories of cellular component organization and biological adhesion were under selection among all goat breeds, whereas genes related to cardiovascular system were under selection in the high-altitude goat breeds.
The non-synonymous SNPs on EPAS1 is associated with the high-altitude adaptation in a larger goat population
After filtering out genes with 120 LOC symbols, as many as 235 of 404 common SNPs were located at the protein-coding regions: 101, at introns; three, at upstream regions; 11, at untranslated regions (UTRs); and 54, at the intergenic regions. Among these 235 coding SNPs, 86 non-synonymous SNPs were found in 73 genes, with one to four SNPs per gene. Among these 73 candidate genes, four genes, EPAS1, EDNRA, SIRT1 and ryanodine receptor 1 (RYR1), are hypoxia-related genes according to a study by Zhang [9] (Additional file 5: Table S4). As many as 14 genes, including EPAS1, EDNRA, SIRT1, RYR1, desmoglein 2 (DSG2), cardiomyopathy associated 5 (CMYA5), receptor-type tyrosine-protein phosphatase eta (PTPRJ), FUT1, heart development protein with EGF-like domains 1 (HEG1), tyrosine phosphatase receptor-type Z polypeptide 1 (PTPRZ1), PAS domain-containing serine/threonine kinase (PASK), sialic acid-binding Ig-like lectin 1 sialoadhesin (SIGLEC1), Niemann-Pick disease type C1 gene-like 1 (NPC1L1) and nestin (NES), were previously reported to be associated with the cardiovascular system through literature mining (Additional file 5: Table S4). Non-synonymous amino acid replacements were assigned to categories according to changes in different physicochemical properties [29]. Of these genes, eight contain at least one relevant amino acid change that altered their physicochemical properties. These genes were EPAS1, PTPRJ, EDNRA, FUT1, NES, NPC1L1, PASK, and PTPRZ1 (Additional file 5: Table S4; Additional file 6: Figure S2).
The EPAS1 gene was the first gene that drew our attention because it was involved in complex oxygen sensing and was significantly associated with high-altitude adaption in dog and human [7, 8, 30]. This gene’s non-synonymous SNP (chr11: 28306765), which was identified as a potential target for selection at high altitude, causes a Gln 579-Leu (Q579L) mutation in the translated EPAS1 protein, based on GenBank annotation (Fig. 3a, Additional file 5: Table S4). The major allele of this SNP in high-altitude breeds is “T”, whereas the major allele in lowland breeds is the same as the reference allele “A”. Interestingly, the increasing frequency of the variant allele “T” with the elevated altitude was confirmed in the extended goat populations, with rare frequency in lowland breeds (Changjiangsnajiaozhou [CWG]: 0 %; LN; 7.5 %; Guangfeng [GF]; 11.11 %), moderate frequency in midland breeds (IM: 26.67 %; NJ: 35 %; CDM: 15.3 %), but high frequency in highland breeds (BG: 73.14 %; RT: 55.36 %; Langkazi [LKZ]: 59.09 %; Fig. 3b). In addition, this mutation occurred next to a well-defined protein domain (Hypoxia-Inducible Factor-1 domain, Fig. 3a). To further evaluate the functional significance of this variant, we aligned the mutant EPAS1 protein with its ortholog proteins in seventeen different vertebrates (Fig. 3a). The comparison revealed that Q579L mutated an evolutionary conserved amino acid, which is invariant among all the terrestrial animals that we examined. Statistical analysis of the blood test results in for the BG population revealed a statistically significant association between the genotype and the mean corpuscular Hb concentration (MCHC) (ANOVA F-test: P = 0.0417; Fig. 3c). All these results implied that EPAS1 plays a critical role in high-altitude adaptation in the Chinese indigenous cashmere goat.
Fig. 3.
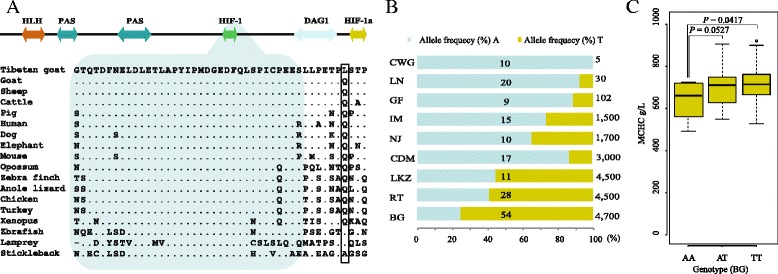
EPAS1 mutation in the coding regions. a EPAS1 protein sequence analysis. The protein coordinate is based on NCBI RefSeq XP_005686651.1. The upper panel shows the Pfam domains of the protein. The double arrows represent domains of goat EPAS1. The orthologous protein sequences from 17 vertebrates are aligned with the mutant residues shown in the box. Sheep, ENSOARP00000006140; cattle, ENSBTAP00000004836; pig, ENSSSCP00000009011; human, ENSP00000406137; dog, ENSCAFP00000003819; elephant, ENSLAFP00000010336; mouse, ENSMUSP00000024954; opossum, ENSMODP00000001136; zebra finch, ENSTGUP00000004086; anole lizard, ENSACAP00000004025; turkey, ENSGACP00000015093; xenopus, ENSXETP00000031612; zebrafish, ENSDARP00000074832; lamprey, ENSPMAP00000000148; stickleback, ENSGACP00000015093. (HLH) helix-loop-helix domain; (PAS) Per-Arnt-Sim; (HIF) hypoxia-inducible factor; (CTAD) C-terminal transactivation domain; (DAG1) Dystroglycan (Dystrophin-associated glycoprotein 1), the blue shade represent the HIF-1 domain. b Percentages of the reference allele and variant allele in larger population samples that were genotyped with Sanger sequencing technology. The number on the bar represents the sample size. The altitude information is shown at the right side of the bars. c Association analysis between EPAS1 genotypes (mutant allele, T; reference allele, A) and the MCHC in BG population. The ANOVA F-test was performed, and we found a significant association between the genotypes and MCHC
The second candidate gene is PTPRJ (also called CD148 or DEP1), which is a member of the protein tyrosine phosphatase (PTP) family and which is present in all hematopoietic lineages [31]. Mutations in this gene can lead to defects in vascular development [32]. Six outlier SNPs were located in the PTPRJ gene; one in the 3’-UTR and five in the coding region. Of these five SNPs, four were non-synonymous, and three SNPs changed the physicochemical properties of the amino acid residues. However, multi-species alignment of the non-synonymous substitutions encoded in the PTPRJ genes showed that none of these four non-synonymous SNPs were present at evolutionary conserved sites. Surprisingly, the same type of SNPs occurred in the NPC1L1, PTPRZ1, NES, and PASK gene. For the EDNRA and FUT1, the non-synonymous substitutions were present at extremely evolutionary conserved sites; however, the variants in EDNRA and FUT1 did not present increased allele frequencies in different goat populations as the altitude increased. Therefore, no further validation was conducted on these genes (Additional file 8: Figure S3).
Discussion
Genetic diversity among the Chinese cashmere goat populations
Using more than 100 K SNPs extracted from more than 300 goats of major cashmere breeds in China, an obvious separation across these breeds was observed. Phylogenetic analysis separated the Tibetan cashmere goat populations (BG and RT) from other groups. This result is consistent with previous reports from our own studies and other studies based on microsatellite and mitochondrial data [33, 34]. The other four Chinese cashmere goat breeds were grouped into three sub-clusters, with LN and NJ populations in one cluster and CDM and IM populations in two distinct branches (Table 1). These results are consistent with the breeding history of both CDM and NJ cashmere goats because they both originated from a cross between the local goat breeds (maternal) and LN (paternal). The genetic exchanges between LN and NJ, LN and CDM cashmere goat breeds for improving cashmere quality and quantity probably have led to a close relationship among these cashmere goat breeds.
We estimated the global Fst value for each identified SNP; the average Fst value was 0.0531, indicating that a small portion of the genetic diversity in the total population was attributed to differences among the populations. In addition, all six breeds had a high proportion of polymorphic loci and a similar average heterozygosity value, suggesting that a high level of variation has been maintained within these Chinese cashmere goat breeds. Our overall global Fst value was slightly lower than those of previous studies of Chinese cashmere goats (0.063 [27]) and of other Chinese indigenous goats (0.056 [33], 0.054 [34]). The relatively lower Fst value in our analysis can be explained by the fact that we used exome SNPs, which have a lower rate of substitution than intergenic SNPs.
Population differentiation pattern of cashmere goat populations
The topological structure of the phylogenetic tree based on the top 2.5 % SNPs of the global Fst analysis (Fig. 1d) slightly differed from that based on all SNPs (Fig. 1a), showing a relatively high level of genetic differentiation among all breeds (Fig. 1d). GO enrichment analysis showed that genes with remarkable genetic differentiation among breeds were enriched in many different categories (localization, cell adhesion and system development, and so on), thus indicating that the population differentiation pattern is complex and that the factor affecting the differentiation pattern is not unique. Both natural and artificial selection have contributed to the genetic differentiation in these cashmere goat breeds.
Since being subject to both artificial and natural selection for a long time, especially strong and directional selection recently, the evolutionary direction of the cashmere goat has been in line with human needs. China has a 2,500-year history of cashmere production: an estimated 10,000 tons of cashmere is produced per year [28]. Indeed, a few genes associated with cashmere traits displayed significant differentiation among the different goat breeds. For example, the global Fst analysis indicated that the Desmoglein 3 (DSG3) gene had the highest Fst value (0.41) among the goat breeds; the gene plays an important role in maintaining the normal structure and function of hair [35]. GO categories such as skin development (P = 2.72E-2) may be relative to the differential cashmere production in these breeds (Additional file 3: Table S2).
In addition, many genes with high Fst values in the global Fst analysis play a role in the cardiovascular system. The GO categories previously reported to be associated with high-altitude adaptation in pig [36], such as blood vessel development, vasculature development, and cardiac conduction, were significantly enriched (P < 1E-4) in our analysis. The GO categories previously reported to be associated with high-altitude adaptation in grey wolf [9], such as response to external stimulus, were also significantly enriched in our study (P < 2.26E-02). Further analysis was performed to determine whether the above loci presenting significant genetic differentiation, such as cardiovascular system-related genes, are associated with high-altitude adaptation.
Cardiovascular system-related genes play an important role in the high-altitude adaptation
Adaptation to a hypoxic environment manifests as a large phenotypic shift observed in the Tibetan cashmere goat compared with the goats living at low altitudes. Previous studies have showen that the Tibetan cashmere goat has a higher adult Hb concentration, a heavier heart, lung and trachea, lower heart rate, and a high rate of arrhythmia [18–20]. These adaptive phenotypic traits enable these animals to survive in the Tibetan Plateau. In the present study, we found molecular evidences in line with the phenotypic adaptations of the cardiovascular system.
First, genes related to cardiovascular system development were genetically differentiated in the global Fst analysis (Additional files 3 and 7: Table S2 and Table S5, Table 2). Subsequently, we identified 66 (19.64 %) genes in the common SNP dataset that were previously identified as cardiovascular system development-related genes (Additional file 5: Table S4). High-altitude stresses include hypoxia, decreasing temperature, low humidity, and increasing ultraviolet radiation [37]; cold temperature and low oxygen are the two most remarkable factors that make survival at high-altitudes challenging [38]. Moving materials (especially oxygen) and regulating body temperature are the two most important functions of the cardiovascular system [39]. Thus, environment stresses of different altitudes are key factors that simulate the differentiation of goat population, and cardiovascular system-related genes may play an important role in adaptation to high-altitude environments in goats.
High-altitude adaptation may be due to multiple genes that act in concert with one another. Many cardiovascular system-related genes in the protein-protein interaction networks analysis are hub genes (Additional file 6: Figure S2) that are thought to play crucial roles in networks [40]. For example, SIRT1 encodes a member of the sirtuin protein family, which can deacetylate and activate HIF2A in cultured cells subjected to hypoxia [41, 42]. ICAM1 can be induced by hypoxia in aortic endothelial cells and cardiac myocytes [43–46]. EDNRA and YES1 were shown to be involved in high-altitude adaptation [5, 8, 47]; EDNRA encodes the receptor of endothelin-1, a peptide that plays a key role in potent and long-lasting vasoconstriction [48], whereas YES1 encodes a protein with tyrosine kinase activity that belongs to the src family [49]. YES is realted to HSP27, which can be specifically upregulated through HIF1 signaling under the hypoxic conditions [50]. JUP encodes a major cytoplasmic protein that is the only known common component of the intracellular plaques of adhesions, predominantly desmosomes and adherens junctions [51]. Mutations in this gene have been associated with arrhythmogenic right ventricular cardiomyopathy [52, 53]. However, none of these hub genes except for EDNRA contained non-synonymous SNPs.
Validation of EPAS1 in a larger population
High-altitude human populations (Tibetans, Andeans, and Ethiopians) on different continents show distinct patterns of adaptation to high-altitude hypoxia [54], and different species on the same continent also show different patterns of adaptation to high-altitude hypoxia [55]. Tibetan cashmere goats display several key adaptive features for coping with the harsh environment at a high altitude. One of these features is that Tibetan cashmere goats display a higher than expected Hb concentration relative to their lowland counterparts and to acclimatized lowland goats. In contrast to Tibetan human and Tibetan pig populations, which show decreased Hb levels at high altitude, this feature is a crucial adaptation mechanism for goats that have adapted to a high-altitude environment [18]. The elevation of Hb at high altitude has also been observed in Tibetan yak and sheep [55].
The EPAS1 gene is the most prominent candidate gene identified in this study. EPAS1, a member of the transcription factor family characterized by a basic helix-loop-helix (bHLH) and by a (Per-AhR-Arnt-Sim) PAS domain, plays a prominent role in process of adaptation to hypoxia and in the promotion of survival during hypoxic stress. EPAS1 represents a “hot spot” of high-altitude adaptation research since several previously published studies provided evidence of an association between a few SNPs near or in EPAS1 and the high-altitude adaption [8, 9, 12, 30, 56]. The EPAS1 polymorphisms in the native Tibetan people were found to be associated with their lower Hb concentrations [30]. Several non-synonymous mutations on EPAS1 have been identified in the Tibetan dog, including a key amino acid mutation thought to be associated with the lower blood flow resistance [7].
However, EPAS1 is pleiotropic; therefore, other responses to hypoxia may be similarly affected. A few individuals living at low altitudes have been reported to have gain of function mutations in the HIF domains of EPAS1; these individuals exhibit excessive erythrocytosis and elevated Hb levels [57].
In our study, we also discovered a novel non-synonymous mutation, Q579L, on EPAS1 in high-altitude goat populations; multi-species alignment indicated that this mutation is near to a well-defined domain (HIF) and is present at an evolutionary conserved site (Fig. 3a). Mutations in this domain of human EPAS1 are associated with erythrocytosis and elevated Hb levels [58, 59]. Goat EPAS1 Q579 corresponds to human EPAS1 Q557. A study in humans demonstrated that a novel mutation in human EPAS1 (Q557*) affects EPAS1 prolyl hydroxylation and VHL (von Hippel-Lindau tumor suppressor) protein binding, resulting in reduced EPAS1 ubiquitination but intact transcriptional activity and activation activity [60]. Further screen of this allele in additional Tibetan and lowland goat populations further confirmed a large divergence between low- and high-altitude goat populations (Fig. 3b). Furthermore, a statistically significant association between the genotypes and MCHC was found (Fig. 3c), implying the biological relevance of the mutant allele in the elevated MCHC levels of the Tibetan cashmere goat.
Taken together, these results indicate that EPAS1 may play a key role in the adaptation of Tibetan cashmere goats to a high-altitude environment at the molecular level; however, the adaptation patterns may not as the same in human and dog.
Conclusions
Our study provides insights concerning the high-altitude adaptation of the important goat breeds in China, which is crucial for the sustainable utilization of cashmere goat. A high level of variation has been maintained within the Chinese cashmere goat breeds. Genes involved in the cardiovascular system development and the missense mutation of EPAS1 may play a crucial role in the high-altitude adaptation of Tibetan cashmere goats.
Methods
Sample collection and preparation
Genomic DNA was isolated from ear tissue of 330 goats collected from six locations (LN, 30 m; IM, 1500 m; NJ, 1700 m; CDM, 3000 m; RT, 4500 m, BG, 4700 m, Table 1). The DNA isolated from each individual per sample location was quantified using a Nanodrop 2000 (Thermo Fisher Scientific, DE) and pooled in equimolar concentrations.
The peripheral blood samples from 60 BG cashmere goats (Tibetan goats) were collected and tested individually at Tibet Autonomous Region People’s Hospital within 24 hours using a Sysmex XE-2100 Autoanalyzer (Sysmex Corporation, Kobe, Japan). Hb and MCHC values were measured by blood tests. The 60 BG cashmere goats whose Hb levels were measured were the same goats used in the exome re-sequencing.
The validation in the larger population included the following samples: Tibetan goat (BG, 54; RT, 28; LKZ, 11), CDM cashmere goat (17), NJ cashmere goat (10), IM cashmere goat (17), GF goat (9), LN cashmere goat (20), and CWG goat (10). All animal experiments in this study were fully approved by the Animal Care and Use Committee of the Ministry of Agriculture of the People’s Republic of China.
Exome sequencing and reads mapping
For each DNA pool, a targeted exome library with an insert size of 150–200 bp was constructed by an exome capture strategy, using a GenCap custom exome enrichment kit (MyGenostics, Beijing, China). An Illumina HiSeq 2000 platform was used to generate paired-end 100-bp raw reads for each enriched library according to the manufacturer’s protocol. Quality filtering of the raw reads was performed using NGSQC Tookit v2.3.3 [61]. For each sample, we discarded reads that did not have an overall quality score of 20 using the PHRED33 scale for at least 70 % of the bases or for any base with quality less than 20. Only paired reads were used for further analysis. The final depth of coverage of the six populations was between 83-155X.
SNP detection
After removing low quality reads, we used BWA (v0.7.10) [62] software with the default setting to separately map the paired-end reads that passed quality filtration separately from each of the six populations (Table 1) to the goat reference genome assembly CHIR_1.0 [28]. Duplicated reads were removed and mapping statistics were calculated using the SAMtools package (v0.1.19) [62]. Then, the SNPs were called using the SAMtools pipeline on a per-population basis [63]. We filtered the resulting SNPs using the following steps: 1) positions with read coverage lager than 200 were filtered to avoid calling SNPs in parts of the exome with excess coverage, likely the duplicated regions; 2) SNP positions with a read depth of at least 60 in the union of all reads from all populations were kept for further analysis; and 3) the positions that had less than 10 supporting reads for the variant allele were removed. The major SNPs from the BG population were selected as reference SNPs. ANNOVAR (v2014-07-22) was used to annotate the variants after filtering [64]. We validated the sequence-based allele frequency estimates by individual genotyping of the same goats for pooled sequencing. A total of 53 SNPs were selected for the validation and all were successfully genotyped in the six goat populations (Additional file 9: Table S6).
Genetic differentiation analysis across populations using global Fst values
We estimated the allele frequency of each SNP site for each population by analyzing read count data for the sequencing pool. For each SNP site that had a minimum of 30 reads from each of the six sampled breeds, global Fst values across six populations were calculated using the method from Nei (1987, p.191) [65]. To describe the expected sampling distribution of Fst values under a perfect neutral model, a simulation was conducted using the POWSIM software [66]. This program mimics a large base population segregating for a specified number of independent, selectively neutral loci with defined allele frequencies under the Wright-Fisher model, and then split the base population into s subpopulations of equal effective size (Ne) through random sampling of 2Ne genes. After drift for t generations for each subpopulation, the expected degree of divergence is Fst = 1 - (1- 1/2Ne)t [65].
To reduce the sampling bias of Fst, 57,948 SNPs with the minimum coverage ≥ 30 for each of the six sampled populations were used to perform the simulation. Over all these SNPs, the average frequency of the most common allele was ~0.8619, and the average Fst was 0.0531.
The objective evidence from archaeological excavations and historical records demonstrated that Tibetan cashmere goat was first recorded at Chamdo County in Tibet and has resided in the plateau area since 4,000-5,000 years ago [16, 67, 68]. In addition, China has a 2,500-year history of cashmere production [28, 69]. However, the effective population sizes of the base sampled populations are unknown, and as discussed in a study by Sangeet [70], exact values of Ne have little effect on Fst distribution [70]. Moreover, for the bezoar, the ancestor of goat, females become sexually mature at 2–3 years [71], while for the Chinese cashmere goats, females become sexually mature at 150–180 days [68, 72]. Therefore, we set the generation time to one year and the t to 3000 generations for the Chinese cashmere goat. According to the above-mentioned information and formula, we tested various combinations of Ne and t to reach Fst ≈ 0.0531 and simulated a relative large base population where a single biallelic locus segregated at a frequency of 0.8619. The base population was split into six subpopulations of effective size Ne = 23,231, and then these subpopulations were allowed to drift apart for t = 3,000 generations to arrive at an expected Fst = 0.0531 for neutral loci, with 1 allele sampled from each subpopulation. If we had used Ne = 23,231 and t = 3,000, the simulation would take a long time (several weeks); therefore, we changed the Ne and t values to 2,749 and 300, respectively. These values can produce almost an identical Fst distribution. After Fst was calculated and this process was repeated 57,948 times, we compared the distribution of simulated Fst values with the observed values. The largest simulated Fst value was Fst =0.2953, and the SNPs exhibiting Fst > 0.2953 were considered under directional selection.
Genetic differentiation between highland and lowland populations
To define the candidate positions that have undergone directional selection during high-altitude adaption, 57,948 SNPs with minimum coverage ≥ 30 for each of the six populations were used in the high-lowland Fst analysis. Six goat populations were divided into two groups based on the altitude; the highland group (BG + RT) consisted of goat populations originating from Tibet, and the lowland group (CDM + NJ + IM + LN) consisted of goat populations originating from other locations. To retrieve candidate SNPs under selection in highland breeds, the di values, which combined the Fst value for each breed were calculated for each SNP as described in Akey et al. (2010). A general statistic di [73] was based on all eight comparisons between the highland and lowland groups. χ2 test [70] was used to compare the allelic frequency between highland and lowland breeds. We performed a χ2 test, as a second empirical outlier analysis, combining the highland population group (BG + RT) as the case population and treating the other lowland population group (CDM + NJ + IM + LN) as a control group. In total, 1,449 SNPs that were within the top 2.5 % of the empirical distribution were identified in each analysis. Due to a smaller effective population size than the autosomes, X chromosomal and unplaced regions were not included in the analysis. The complete workflow of the data analysis is presented in Additional file 10; Figure S4.
Phylogeny analysis
Based on the genetic distance matrix that was generated from the population allele frequency data of SNPs in the six populations, the neighbor-joining phylogenetic trees was constructed using POPTREE software [74]. The phylogenetic trees were displayed by TreeView [75].
Candidate SNPs genotyping in a large population and EPAS1 mutation analysis
The primers for polymerase chain reaction (PCR) were designed for genotyping of the individual goat to measure the accuracy of the site frequency survey using a pooled strategy. After PCR amplification, Sanger sequencing technology was employed to verify the target SNPs in ten different populations.
The protein domains of EPAS1 were predicted by the Pfam web service [76]. The full-length amino acid sequences of EPAS1 in 17 representative vertebrates species were retrieved from Ensembl revision 78, and only one-to-one orthologs were included (Fig. 3). A non-synonymous mutation of interest at EPAS1 (chr11: 28306765) was genotyped on a larger population using traditional Sanger sequencing technology. All PCR primers used in this study are listed in Additional file 9: Table S6.
Enrichment analysis
GO categories and function association networks were defined with the STRING v9.1 program using standard parameters [77], and removal of the redundant GO terms from this list was performed with REVIGO using standard parameters [78]. In all tests, the whole set of autosomal genes was appointed as the background and FDR adjusted P values, which indicate the significance of the overlap between various gene sets, were calculated. Only the terms with a FDR-adjusted P value < 0.05 were considered statistically significant and were listed. Genes with LOC symbols were not used in the enrichment analysis.
Data access
All raw sequencing data from this study have been deposited in the Sequence Read Archive (SRA) database at the NCBI under accession number: SRX1009748, SRX1013181, SRX1013324, SRX1013922, SRX1013947, and SRX1013952.
Acknowledgements
The project was supported by the National Natural Science Foundation of China (No: 31272403) and the Agricultural Science and Technology Innovation Program of China (ASTIP-IAS01). LJ was supported by the Elite Youth Program in Chinese Academy of Agricultural Sciences.
Abbreviations
- EPAS1
Endothelial PAS domain protein 1
- GO
Gene ontology
- SIRT1
Sirtuin type 1
- ICAM1
Intercellular cell adhesion molecule-1
- EDNRA
Endothelin receptor type A
- YES1
Yamaguchi sarcoma viral related oncogene homolog 1
- JUP
Plakoglobin
- UTR
Untranslated region
- RYR1
Ryanodine receptor 1
- DSG2
Desmoglein 2
- CMYA5
Cardiomyopathy associated 5
- PTPRJ
Receptor-type tyrosine-protein phosphatase eta
- HEG1
Heart development protein with EGF-like domains 1
- PTPRZ1
Tyrosine phosphatase receptor-type Z Polypeptide 1
- PASK
PAS domain containing serine/threonine kinase
- SIGLEC1
Sialic acid binding Ig-like lectin 1 sialoadhesin
- NPC1L1
Niemann-Pick disease type C1 gene-like 1
- NES
Nestin
- MCHC
Mean corpuscular haemoglobin concentration
- DSG3
Desmoglein 3
- VHL
Von Hippel-Lindau tumor suppressor
Additional files
Whole exome sequencing results for six goat populations. (XLSX 11 kb)
Positive correlation between site frequencies using a pooling strategy and Sanger sequencing technology for Tibetan Bange cashmere goat (BG, a), Tibetan Ritu cashmere goat (RT, b), Chaidamu cashmere goat (CDM, c), Nanjiang cashmere goat (NJ, d), Inner Mongolia cashmere goat (IM, e), and Liaoning cashmere goat (LN, f). (PDF 197 kb)
Functional gene categories enriched for the global SNP dataset. (XLSX 12 kb)
Outlier SNPs genetically differentiated between highland and lowland goat populations. (XLS 214 kb)
SNPs in common SNP dataset. Notes: The amino acids physicochemical properties involved in the non-synonymous mutations were characterized according to the definition provided in the study by Moilanen JS and Majamaa K [29]. Green denotes genes related to high-altitude adaptation that were listed in the study by Zhang [9]; grey denotes genes related to hypoxia and the cardiovascular system in GO categories; yellow denotes genes related to the cardiovascular system according to the literature. (XLSX 87 kb)
Protein-protein interaction network with the common SNP dataset. Notes: Proteins without connections were removed for clarity. Red circle, proteins related to hypoxia and the cardiovascular system from the GO categories; yellow circle, proteins related to the cardiovascular system from the literature; green dot, proteins related to hypoxia adaptation listed in a study by Zhang [9]. “line”: interaction with medium confidence; “bold line”: interaction with high confidence. (PDF 405 kb)
Functional gene categories enriched for the global SNP specific dataset. (XLSX 10 kb)
The multi-species alignment of non-synonymous substitutions encoded in the NPC1L1, NES, PTPRJ, PTPRZ1, FUT1, EDNRA, and PASK genes. (PDF 332 kb)
Primer and SNP information for verification using Sanger technology. (XLSX 13 kb)
Workflow of data analysis. (PDF 352 kb)
Footnotes
Competing interests
The authors declare that there are no competing interests.
Authors’ contributions
SS performed most of the bioinformatics analysis. SS, NY, QJZ, YBP and XHH carried out the sample collection and DNA extraction. SS, MY, XXL and KZD participated in the functional validation. WJG, NY, YHM and LJ designed and coordinated the studies. SS and LJ analysed the data and wrote the paper. All authors have read and approved the final manuscript.
Contributor Information
Shen Song, Email: songs_193@163.com.
Na Yao, Email: yaona820226@126.com.
Min Yang, Email: yangmindk@163.com.
Xuexue Liu, Email: lxx_caas@163.com.
Kunzhe Dong, Email: dkzhe1987@163.com.
Qianjun Zhao, Email: zhaoqianjun@caas.cn.
Yabin Pu, Email: pyb@iascaas.net.cn.
Xiaohong He, Email: hexiaohong@caas.cn.
Weijun Guan, Email: wjguan86@iascaas.net.cn.
Ning Yang, Email: nyang@cau.edu.cn.
Yuehui Ma, Email: yuehui.ma@263.net.
Lin Jiang, Email: jianglin@caas.cn.
References
- 1.Quirouette R, Arch B. Air pressure and the building envelope. Ottawa: Canada Mortgage and Housing Corporation; 2004. [Google Scholar]
- 2.Storz JF, Sabatino SJ, Hoffmann FG, Gering EJ, Moriyama H, Ferrand N, et al. The molecular basis of high-altitude adaptation in deer mice. PLoS Genet. 2007;3:e45. doi: 10.1371/journal.pgen.0030045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Taylor AT. High-altitude illnesses: physiology, risk factors, prevention, and treatment. Rambam Maimonides Med J. 2011;2:e0022. doi: 10.5041/RMMJ.10022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lorenzo FR, Huff C, Myllymaki M, Olenchock B, Swierczek S, Tashi T, et al. A genetic mechanism for Tibetan high-altitude adaptation. Nat Genet. 2014;46:951–6. doi: 10.1038/ng.3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Simonson TS, Yang Y, Huff CD, Yun H, Qin G, Witherspoon DJ, et al. Genetic evidence for high-altitude adaptation in Tibet. Science. 2010;329:72–5. doi: 10.1126/science.1189406. [DOI] [PubMed] [Google Scholar]
- 6.Peng Y, Yang Z, Zhang H, Cui C, Qi X, Luo X, et al. Genetic variations in Tibetan populations and high-altitude adaptation at the Himalayas. Mol Biol Evol. 2011;28:1075–81. doi: 10.1093/molbev/msq290. [DOI] [PubMed] [Google Scholar]
- 7.Gou X, Wang Z, Li N, Qiu F, Xu Z, Yan D, et al. Whole-genome sequencing of six dog breeds from continuous altitudes reveals adaptation to high-altitude hypoxia. Genome Res. 2014;24:1308–15. doi: 10.1101/gr.171876.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang GD, Fan RX, Zhai W, Liu F, Wang L, Zhong L, et al. Genetic convergence in the adaptation of dogs and humans to the high-altitude environment of the tibetan plateau. Genome Biol Evol. 2014;6:2122–8. doi: 10.1093/gbe/evu162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang W, Fan Z, Han E, Hou R, Zhang L, Galaverni M, et al. Hypoxia adaptations in the grey wolf (Canis lupus chanco) from Qinghai-Tibet Plateau. PLoS Genet. 2014;10:e1004466. doi: 10.1371/journal.pgen.1004466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qiu Q, Zhang G, Ma T, Qian W, Wang J, Ye Z, et al. The yak genome and adaptation to life at high altitude. Nat Genet. 2012;44:946–9. doi: 10.1038/ng.2343. [DOI] [PubMed] [Google Scholar]
- 11.Ge RL, Cai Q, Shen YY, San A, Ma L, Zhang Y, et al. Draft genome sequence of the Tibetan antelope. Nat Commun. 2013;4:1858. doi: 10.1038/ncomms2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Newman JH, Holt TN, Cogan JD, Womack B, Phillips JA, 3rd, Li C, et al. Increased prevalence of EPAS1 variant in cattle with high-altitude pulmonary hypertension. Nat Commun. 2015;6:6863. doi: 10.1038/ncomms7863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li M, Jin L, Ma J, Tian S, Li R, Li X. Detecting mitochondrial signatures of selection in wild Tibetan pigs and domesticated pigs. Mitochondrial DNA. 2014;27:747–52. doi: 10.3109/19401736.2014.913169. [DOI] [PubMed] [Google Scholar]
- 14.Wang MS, Li Y, Peng MS, Zhong L, Wang ZJ, Li QY, et al. Genomic Analyses Reveal Potential Independent Adaptation to High Altitude in Tibetan Chickens. Mol Biol Evol. 2015;32:1880–9. doi: 10.1093/molbev/msv071. [DOI] [PubMed] [Google Scholar]
- 15.FAOSTAT. Final 2012 Data and Preliminary 2013 Data for live animals. In., vol. http://faostat.fao.org/site/573/DesktopDefault.aspx?PageID=573#ancor; 2014.
- 16.K-c C, Xu P, Allan S, Lu L. The formation of Chinese civilization: an archaeological perspective. New Haven CT: Yale University Press; 2005. [Google Scholar]
- 17.Wang Y, Wang J, Zi XD, Huatai CR, Ouyang X, Liu LS. Genetic diversity of Tibetan goats of Plateau type using microsatellite markers. Archiv Fur Tierzucht-Archives of Animal Breeding. 2011;54:188–97. [Google Scholar]
- 18.Renzheng JJHMG, Ciren B. Comparison on several hematologic value of goat in Tibet Plateau at different altitude. Southwest China J Agr Sci. 1992;1:014. [Google Scholar]
- 19.Yangxi O, Jie W, Yong W, Yong Z. Seasonal Variety of Blood physiological and Biochemical Targets of Tibetan Goat. J. Southwest Univ. National. (Nat. Sci. Ed.) 1992;3:007. [Google Scholar]
- 20.Huang W. Some physiological parameters of domestic animals at various altitudes in Xizang. Beijing, China: Proceedings of symposium on Qinghai-Xizang (Tibet) Plateau; 1980. pp. 182–83. [Google Scholar]
- 21.Li MH, Li K, Zhao SH. Diversity of Chinese indigenous goat breeds: a conservation perspective - A review. Asian-Australasian J Anim Sci. 2004;17:726–32. doi: 10.5713/ajas.2004.726. [DOI] [Google Scholar]
- 22.Zhang CJ, Wang Y, Lu FS. Study on electrocardiogram of Qinghai native goat. Progress Vet Med. 2004;4:021. [Google Scholar]
- 23.Zhong WJ, Xi YO, Yong W. Comparative studies on the ecoligical characteristies of Plateau-Type and Mountain-Valley-Type Tibetan goats. Journal of Southwest Nationalities College (Natural Science Edition). 1994;3:293-98.
- 24.Wen S, Wang Q, Wang D, Jiang Z, Hu Y. Liaoning cashmere goat adapted to the high altitude environment and cross with the Tibetan cashmere goat. Prataculture & Animal Husbandry. 1995;4:42-48.
- 25.Wu YP, Guan WJ, Zhao QJ, He XH, Pu YB, Huo JH, et al. A fine map for maternal lineage analysis by mitochondrial hypervariable region in 12 Chinese goat breeds. Anim Sci J. 2009;80:372–80. doi: 10.1111/j.1740-0929.2009.00659.x. [DOI] [PubMed] [Google Scholar]
- 26.Zhao Y, Zhao R, Zhao Z, Xu H, Zhao E, Zhang J. Genetic diversity and molecular phylogeography of Chinese domestic goats by large-scale mitochondrial DNA analysis. Mol Biol Rep. 2014;41:3695–704. doi: 10.1007/s11033-014-3234-2. [DOI] [PubMed] [Google Scholar]
- 27.Di R, Vahidi SM, Ma YH, He XH, Zhao QJ, Han JL, et al. Microsatellite analysis revealed genetic diversity and population structure among Chinese cashmere goats. Anim Genet. 2011;42:428–31. doi: 10.1111/j.1365-2052.2010.02072.x. [DOI] [PubMed] [Google Scholar]
- 28.Dong Y, Xie M, Jiang Y, Xiao N, Du X, Zhang W, et al. Sequencing and automated whole-genome optical mapping of the genome of a domestic goat (Capra hircus) Nat Biotechnol. 2013;31:135–41. doi: 10.1038/nbt.2478. [DOI] [PubMed] [Google Scholar]
- 29.Moilanen JS, Majamaa K. Phylogenetic network and physicochemical properties of nonsynonymous mutations in the protein-coding genes of human mitochondrial DNA. Mol Biol Evol. 2003;20:1195–210. doi: 10.1093/molbev/msg121. [DOI] [PubMed] [Google Scholar]
- 30.Yi X, Liang Y, Huerta-Sanchez E, Jin X, Cuo ZX, Pool JE, et al. Sequencing of 50 human exomes reveals adaptation to high altitude. Science. 2010;329:75–8. doi: 10.1126/science.1190371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de la Fuente-Garcia MA, Nicolas JM, Freed JH, Palou E, Thomas AP, Vilella R, et al. CD148 is a membrane protein tyrosine phosphatase present in all hematopoietic lineages and is involved in signal transduction on lymphocytes. Blood. 1998;91:2800–09. [PubMed] [Google Scholar]
- 32.Takahashi T, Takahashi K, St John PL, Fleming PA, Tomemori T, Watanabe T, et al. A mutant receptor tyrosine phosphatase, CD148, causes defects in vascular development. Mol Cell Biol. 2003;23:1817–31. doi: 10.1128/MCB.23.5.1817-1831.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ling YH, Zhang XD, Yao N, Ding JP, Chen HQ, Zhang ZJ, et al. Genetic differentiation of chinese indigenous meat goats ascertained using microsatellite information. Asian-Australas J Anim Sci. 2012;25:177–82. doi: 10.5713/ajas.2011.11308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xiang-Long L, Valentini A. Genetic diversity of Chinese indigenous goat breeds based on microsatellite markers. J Anim Breed Genet. 2004;121:350–55. doi: 10.1111/j.1439-0388.2004.00465.x. [DOI] [Google Scholar]
- 35.Koch PJ, Mahoney MG, Cotsarelis G, Rothenberger K, Lavker RM, Stanley JR. Desmoglein 3 anchors telogen hair in the follicle. J Cell Sci. 1998;111(Pt 17):2529–37. doi: 10.1242/jcs.111.17.2529. [DOI] [PubMed] [Google Scholar]
- 36.Dong K, Yao N, Pu Y, He X, Zhao Q, Luan Y, et al. Genomic scan reveals loci under altitude adaptation in Tibetan and Dahe pigs. PLoS One. 2014;9:e110520. doi: 10.1371/journal.pone.0110520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gallagher SA, Hackett PH. High-altitude illness. Emerg Med Clin North Am. 2004;22:329–55. doi: 10.1016/j.emc.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 38.Hainsworth R, Drinkhill MJ, Rivera-Chira M. The autonomic nervous system at high altitude. Clin Auton Res. 2007;17:13–9. doi: 10.1007/s10286-006-0395-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pittman RN. In: Regulation of Tissue Oxygenation. Morgan & Claypool Life Science: San Rafael (CA); 2011. [PubMed]
- 40.Bork P, Jensen LJ, von Mering C, Ramani AK, Lee I, Marcotte EM. Protein interaction networks from yeast to human. Curr Opin Struct Biol. 2004;14:292–9. doi: 10.1016/j.sbi.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 41.Leiser SF, Kaeberlein M. A role for SIRT1 in the hypoxic response. Mol Cell. 2010;38:779–80. doi: 10.1016/j.molcel.2010.06.015. [DOI] [PubMed] [Google Scholar]
- 42.Lim JH, Lee YM, Chun YS, Chen J, Kim JE, Park JW. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol Cell. 2010;38:864–78. doi: 10.1016/j.molcel.2010.05.023. [DOI] [PubMed] [Google Scholar]
- 43.Zund G, Uezono S, Stahl GL, Dzus AL, McGowan FX, Hickey PR, et al. Hypoxia enhances induction of endothelial ICAM-1: role for metabolic acidosis and proteasomes. Am J Physiol. 1997;273:C1571–80. doi: 10.1152/ajpcell.1997.273.5.C1571. [DOI] [PubMed] [Google Scholar]
- 44.Van Seventer GA, Shimizu Y, Horgan KJ, Shaw S. The LFA-1 ligand ICAM-1 provides an important costimulatory signal for T cell receptor-mediated activation of resting T cells. J Immunol. 1990;144:4579–86. [PubMed] [Google Scholar]
- 45.Hayasaki Y, Nakajima M, Kitano Y, Iwasaki T, Shimamura T, Iwaki K. ICAM-1 expression on cardiac myocytes and aortic endothelial cells via their specific endothelin receptor subtype. Biochem Biophys Res Commun. 1996;229:817–24. doi: 10.1006/bbrc.1996.1886. [DOI] [PubMed] [Google Scholar]
- 46.Ziegelstein RC, He C, Hu Q. Hypoxia/reoxygenation stimulates Ca2 + −dependent ICAM-1 mRNA expression in human aortic endothelial cells. Biochem Biophys Res Commun. 2004;322:68–73. doi: 10.1016/j.bbrc.2004.07.080. [DOI] [PubMed] [Google Scholar]
- 47.Yang L, Wang Y, Zhang Z, He S. Comprehensive transcriptome analysis reveals accelerated genic evolution in a Tibet fish, Gymnodiptychus pachycheilus. Genome Biol Evol. 2015;7:251–61. doi: 10.1093/gbe/evu279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Itoh S, Sasaki T, Asai A, Kuchino Y. Prevention of delayed vasospasm by an endothelin ETA receptor antagonist, BQ-123: change of ETA receptor mRNA expression in a canine subarachnoid hemorrhage model. J Neurosurg. 1994;81:759–64. doi: 10.3171/jns.1994.81.5.0759. [DOI] [PubMed] [Google Scholar]
- 49.Lynch G, Kohler S, Leser J, Beil M, Garcia-Marin LJ, Lutz MP. The tyrosine kinase yes regulates actin structure and secretion during pancreatic acinar cell damage in rats. Pflugers Arch. 2004;447:445–51. doi: 10.1007/s00424-003-1188-7. [DOI] [PubMed] [Google Scholar]
- 50.Whitlock NA, Agarwal N, Ma JX, Crosson CE. Hsp27 upregulation by HIF-1 signaling offers protection against retinal ischemia in rats. Invest Ophthalmol Vis Sci. 2005;46:1092–8. doi: 10.1167/iovs.04-0043. [DOI] [PubMed] [Google Scholar]
- 51.Whittock NV, Eady RA, McGrath JA. Genomic organization and amplification of the human plakoglobin gene (JUP) Exp Dermatol. 2000;9:323–6. doi: 10.1034/j.1600-0625.2000.009005323.x. [DOI] [PubMed] [Google Scholar]
- 52.McKoy G, Protonotarios N, Crosby A, Tsatsopoulou A, Anastasakis A, Coonar A, et al. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease) Lancet. 2000;355:2119–24. doi: 10.1016/S0140-6736(00)02379-5. [DOI] [PubMed] [Google Scholar]
- 53.Protonotarios NI, Tsatsopoulou AA, Gatzoulis KA. Arrhythmogenic right ventricular cardiomyopathy caused by a deletion in plakoglobin (Naxos disease) Card Electrophysiol Rev. 2002;6:72–80. doi: 10.1023/A:1017943323473. [DOI] [PubMed] [Google Scholar]
- 54.Beall CM. Andean, Tibetan, and Ethiopian patterns of adaptation to high-altitude hypoxia. Integr Comp Biol. 2006;46:18–24. doi: 10.1093/icb/icj004. [DOI] [PubMed] [Google Scholar]
- 55.Ai H, Yang B, Li J, Xie X, Chen H, Ren J. Population history and genomic signatures for high-altitude adaptation in Tibetan pigs. BMC Genomics. 2014;15:834. doi: 10.1186/1471-2164-15-834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xu XH, Huang XW, Qun L, Li YN, Wang Y, Liu C, et al. Two functional loci in the promoter of EPAS1 gene involved in high-altitude adaptation of Tibetans. Sci Rep. 2014;4:7465. doi: 10.1038/srep07465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Percy MJ, Beer PA, Campbell G, Dekker AW, Green AR, Oscier D, et al. Novel exon 12 mutations in the HIF2A gene associated with erythrocytosis. Blood. 2008;111:5400–2. doi: 10.1182/blood-2008-02-137703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Percy MJ. Familial erythrocytosis arising from a gain-of-function mutation in the HIF2A gene of the oxygen sensing pathway. Ulster Med J. 2008;77:86–8. [PMC free article] [PubMed] [Google Scholar]
- 59.Percy MJ, Furlow PW, Lucas GS, Li X, Lappin TR, McMullin MF, et al. A gain-of-function mutation in the HIF2A gene in familial erythrocytosis. N Engl J Med. 2008;358:162–8. doi: 10.1056/NEJMoa073123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Taieb D, Barlier A, Yang C, Pertuit M, Tchoghandjian A, Rochette C, et al. Somatic gain-of-function HIF2A mutations in sporadic central nervous system hemangioblastomas. J Neurooncol. 2015. [DOI] [PubMed]
- 61.Patel RK, Jain M. NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLoS One. 2012;7:e30619. doi: 10.1371/journal.pone.0030619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27:2987–93. doi: 10.1093/bioinformatics/btr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nei M. Molecular evolutionary genetics. Columbia university press. New York, NY, USA: 1987.
- 66.Ryman N, Palm S. POWSIM: a computer program for assessing statistical power when testing for genetic differentiation. Mol Ecol Notes. 2006;6:600–02. doi: 10.1111/j.1471-8286.2006.01378.x. [DOI] [PubMed] [Google Scholar]
- 67.Fan B, Chen SL, Kijas JH, Liu B, Yu M, Zhao SH, et al. Phylogenetic relationships among Chinese indigenous goat breeds inferred from mitochondrial control region sequence. Small Ruminant Res. 2007;73:262–66. doi: 10.1016/j.smallrumres.2006.12.007. [DOI] [Google Scholar]
- 68.Tu Y, Jiang Y, Han Z, Feng W. The sheep and goat breeds in China. Shanghai: Shanghai Science and Technology Press; 1989. p. 51. [Google Scholar]
- 69.White C. The Cashmere Shawl; An Eastern Fiction Volume 1: London: Henry Colburn. 1840. [Google Scholar]
- 70.Lamichhaney S, Martinez Barrio A, Rafati N, Sundstrom G, Rubin CJ, Gilbert ER, et al. Population-scale sequencing reveals genetic differentiation due to local adaptation in Atlantic herring. Proc Natl Acad Sci U S A. 2012;109:19345–50. doi: 10.1073/pnas.1216128109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nomura K, Yonezawa T, Mano S, Kawakami S, Shedlock AM, Hasegawa M, et al. Domestication process of the goat revealed by an analysis of the nearly complete mitochondrial protein-encoding genes. PLoS One. 2013;8:e67775. doi: 10.1371/journal.pone.0067775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shi Y, Wang S, Bai S, Huang L, Hou Y. Postnatal ovarian development and its relationship with steroid hormone receptors in JiNing Grey goats. Anim Reprod Sci. 2015;154:39–47. doi: 10.1016/j.anireprosci.2015.01.001. [DOI] [PubMed] [Google Scholar]
- 73.Akey JM, Ruhe AL, Akey DT, Wong AK, Connelly CF, Madeoy J, et al. Tracking footprints of artificial selection in the dog genome. Proc Natl Acad Sci U S A. 2010;107:1160–5. doi: 10.1073/pnas.0909918107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Takezaki N, Nei M, Tamura K. POPTREE2: Software for constructing population trees from allele frequency data and computing other population statistics with Windows interface. Mol Biol Evol. 2010;27:747–52. doi: 10.1093/molbev/msp312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Page RD. Visualizing phylogenetic trees using TreeView. Curr Protoc Bioinformatics. 2002;Unit 6:2. doi: 10.1002/0471250953.bi0602s01. [DOI] [PubMed] [Google Scholar]
- 76.Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, et al. The Pfam protein families database. Nucleic Acids Res. 2012;40:D290–301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, et al. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41:D808–15. doi: 10.1093/nar/gks1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Supek F, Bosnjak M, Skunca N, Smuc T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One. 2011;6:e21800. doi: 10.1371/journal.pone.0021800. [DOI] [PMC free article] [PubMed] [Google Scholar]