

Abstract
The term matrisome refers to the ensemble of proteins constituting the extracellular matrix (ECM) (core matrisome) as well as the proteins associated with the ECM. Every organ has an ECM with a unique composition that not only provides the support and anchorage for cells, but also controls fundamental cellular processes as diverse as differentiation, survival, proliferation, and polarity. The current knowledge of the matrisome of small brain vessels is reviewed with a focus on the basement membrane (BM), a specialized form of ECM located at the interface between endothelial cells, contractile cells (smooth muscle cells and pericytes), and astrocyte endfeet—a very strategic location in the communication pathway between the cerebral microcirculation and astrocytes. We discuss some of the most recent genetic data and relevant findings from experimental models of nonamyloid cerebral small vessel disease (SVD). We propose the concept that perturbations of the cerebrovascular matrisome is a convergent pathologic pathway in monogenic forms of SVD, and is likely relevant to the sporadic disease.
Keywords: Basement membrane, cerebral small vessel disease, collagen type IV, extracellular matrix, proteomics
Introduction
Non-amyloid cerebral small vessel disease (cSVD) is a heterogeneous group of disorders that refers collectively to the pathologic processes that affect the structure and function of small intracerebral vessels. The salient consequences of nonamyloid cSVD are small, deep infarctions or hemorrhages in the white and/or deep gray matter, and the development of widespread white-matter lesions. Cerebral SVD accounts for ∼25% to 30% of strokes, and ample evidence now exists that it is a leading cause of cognitive decline and disability in adults.1 Cerebral SVD is a mostly sporadic disease that appears to be driven by a complex mix of genetic and environmental factors, among which age and arterial hypertension are currently deemed the most important. However, during the past 20 years, rare Mendelian forms of cSVD, largely indistinguishable from sporadic SVD, have been characterized and the genes responsible for causing them have been identified, propelling an unprecedented leap forward in the effort to unravel the pathogenesis of these related diseases. To date, highly penetrant mutations in six distinct genes have been linked to familial cSVD, namely NOTCH3, COL4A1, COL4A2, HTRA1, TREX1, and FOXC1.2,3 These genetic findings led us to ask whether the genes associated with familial forms of cSVD represent distinct mechanisms for producing the cSVD phenotype or instead ultimately converge on a common pathologic pathway that might also be relevant to the sporadic disease.
The extracellular matrix (ECM) consists of a complex meshwork of highly crosslinked proteins that constitutes a fundamental component of the cellular microenvironment. The ECM proteins bind to cell-surface adhesion receptors, including the integrins, evoking intracellular transduction responses.4 They also have the ability to bind and regulate the distribution, activation, and presentation of a myriad of growth and secretory factors to cells.5 Hence, the ECM provides much more than support and anchorage for the cell; it also has crucial roles in the differentiation, survival, proliferation, polarity, and migration of cells.
Here, we review our current understanding of the ECM in general and in the context of cerebral blood vessels in particular, and critically examine the contribution of cerebrovascular ECM alterations in nonamyloid cSVD.
The extracellular matrix: The core matrisome and matrisome-associated proteins
What Is the Matrisome?
The name ‘matrisome’ has been coined to designate the ensemble of proteins constituting the ECM (core matrisome) plus the proteins associated with the ECM (matrisome-associated proteins).6 The ECM proteins classically contain repeats of a characteristic set of domains (e.g., EGF-like, fibronectin, laminin, and collagen triple helix repeat) that are highly conserved among different species, in both sequence and arrangement.7 Using this feature, Hynes and coworkers recently developed a bioinformatics approach that they used in combination with manual annotation to define a comprehensive repertoire of genes encoding the matrisome in murine and human genomes.6
Matrisome Categories
The core matrisome comprises 44 collagen subunits, 36 proteoglycans, and about 200 glycoproteins in humans and mice (Figure 1). Collagens, the main structural proteins of the ECM, are classified into fibrillar (collagens I to III, V, and XI) and nonfibrillar forms. Collagen subunits are characterized by a unique sequence stretch in which every third amino acid is a glycine (Gly-X-Y motif), and the structural hallmark of all collagens is their triple helix.8 Proteoglycans are glycoproteins containing attached glycosaminoglycans, which represent a significant fraction of the total mass. Proteoglycans fill the extracellular interstitial space and provide hydration by sequestering water within the tissue; they are also involved in binding many secreted factors, including growth factors, and sequestering them in the ECM.9 Glycoproteins include a myriad of proteins with various functions; among these, laminins, fibrillins, fibronectin, and vitronectin have been extensively studied in the context of normal physiology or pathology.10–13
Figure 1.
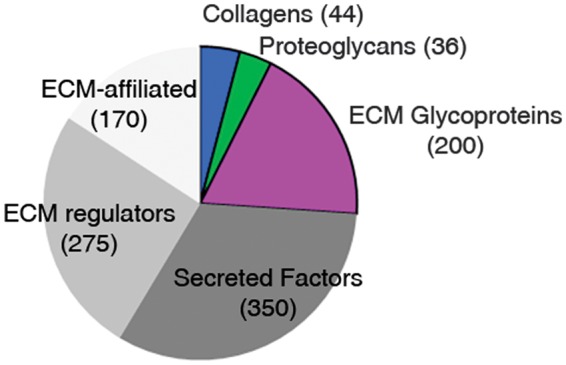
Schematic representation of the in silico matrisome. Pie chart displaying the different matrisome categories with the approximate number of genes (brackets) determined by in silico analysis of human and murine genomes. ECM, extracellular matrix.
The matrisome-associated protein category comprises almost 800 proteins divided into three categories: (1) secreted factors, including growth factors, that are bound to the ECM; (2) ECM regulators, consisting of enzymes and other proteins that modify the ECM; and (3) ECM-affiliated proteins, which either are known to be associated with ECM proteins or share some architectural similarities with ECM proteins (Figure 1).6 The ECM acts as a sink or a reservoir for almost 350 secreted factors, which bind either to glycosaminoglycans, especially heparan sulfates, or to specific domains of proteins of the core matrisome.14 Among these factors are cytokines and many of the major regulator of physiologic angiogenesis, such as vascular endothelial growth factor, platelet-derived growth factor, fibroblast growth factor, and bone morphogenic proteins, among others.15 The second category, comprising 250 to 300 proteins, includes modifiers of ECM structure and function, such as lysyl hydroxylase and lysyloxidase, which crosslink collagens, matrix metalloproteases (MMPs), HTRA serine proteases, and many other proteolytic enzymes, as well as regulators of these modifiers, including members of the tissue inhibitor of metalloproteinases (TIMP) family.16 The third category defines a heterogeneous set of about 170 ECM-affiliated proteins, considered ECM associated by some researchers, that includes annexins, complement components, lectins, and semaphorins, among others. In contrast with the core matrisome, the matrisome-associated category is less firmly established, and the list of these proteins can be expected to change with subsequent experimental analyses.17
Key Features
In vivo characterization of the matrisome from a given tissue or cell line is challenging because of the peculiar physical properties of ECM proteins. Specifically, these proteins generally have high molecular weights, are difficult to solubilize, and carry extensive post-translational modifications. Moreover, the presence of highly abundant cytoplasmic and mitochondrial proteins, which are easily solubilized, can hamper the detection of ECM proteins that may be present at comparatively low relative abundance. Recent proteomic profiling of ECMs in lung, liver, and colon revealed that each of these tissues contained about 100 to 150 ECM proteins.18
The matrisome of cerebral blood vessels
The circle of Willis gives rise to three pairs of main arteries, which divide into progressively smaller arteries that run on the surface of the brain. These pial arteries branch extensively into smaller arterioles, eventually penetrating into the brain parenchyma and ultimately terminating as an extensive capillary network. Blood is then drained through venules and veins back into dural venous sinuses at the surface.19
An Histologic View
The ECM matrix of cerebral blood vessels differs with regard to the type, location within the vessel tree, and diameter of blood vessels. Pial arteries lie within the subarachnoid spaces on top of brain tissue and are covered with the pia matter. Unlike systemic arteries, these arteries are characterized by a well-developed internal elastica lamina (which separates the endothelium from the tunica media) but no external elastica lamina, a paucity of elastic fibers in the media and a very thin adventitia (Figures 2A and 2B).20 Distinct basement membranes (BMs) cover the basal aspect of endothelial cells, facing the elastic lamina, and encircle smooth muscle cells (SMCs). The inner layer of the pia matter is anchored to the cortex via the glia limitans superficialis, which seals the entire surface of the brain. The glia limitans is composed of a meshwork of astrocyte processes covered by an outer BM that makes intimate contact with cells of the pia mater.21 As blood vessels dip into the brain, they carry with them the extensions of the pia matter, forming a cuff around the vessel. They are separated from the parenchyma by the glia limitans perivascularis, which has a structure nearly identical to that of the glia limitans superficialis (Figure 2A). The Virchow-Robin space containing pial processes corresponds to the perivascular space between the smooth muscle BM and the glial limitans. As the arteriole penetrates deeper into the parenchyma and morphs into the parenchymal arteriole, the elastica lamina becomes more and more discontinuous and the pial investment disappears; notably, the vascular and glial BMs fuse and the Virchow-Robin space disappears.22,23 At the capillary level, one-third of the circumference of the endothelial tube surface is covered by pericytes, and both cell types are wrapped by astroglial endfeet.24 Notably, there is a single, but composite, continuous BM, produced by endothelial cells, astrocytes, and possibly pericytes, that covers the basal aspect of endothelial cells and enwraps the pericytes (Figure 2C).23 The ECM structure of venules and veins is similar to that of arterioles and arteries at the same location, although the SMC coat and elastica lamina of venules and veins are discontinuous and much thinner.20,22
Figure 2.

Histologic view of the extracellular matrix (ECM) along cerebral vessels in the mouse. (A) Semithin section of a pial artery and one of its branch penetrating into the brain parenchyma (toluidine blue staining). The elastic lamina (EL) (black arrows), the pia matter (red arrowheads), which enwraps the artery and covers the surface of the brain, and the glia limitans (white arrows) are shown. Brain tissue was fixed without prior transcardiac perfusion. (B) Electron micrograph of a pial artery (magnification of the boxed area in A). (From top to bottom) The pia matter (red arrowheads), a thin adventitia with collagen bundles (stars), a smooth muscle cell (SMC) with the basement membrane (BM) on its abluminal face (back arrowheads), the elastic lamina, and endothelial cells (ec) are shown. Note that BMs of the SMC luminal side and of ECs are not visible at this magnification. Brain tissue was fixed without prior transcardiac perfusion. (C) Electron micrograph of a capillary within the striatum. The single, continuous BM (black arrowheads) that covers the basal aspect of the EC and enwraps the pericyte (P) is shown. The BM is surrounded on its abluminal face by several perivascular astrocyte endfeet (pve). Brain tissue was postfixed after transcardiac perfusion of aldehydes. ec, endothelial cell; EL, elastic lamina; P, pericyte; smc, smooth muscle cell.
A Proteomic View
Two separate studies have analyzed the proteome of murine pial arteries and brain microvessels. The pial arteries used in the former study included arteries of the circle of Willis and their ramifications, surgically removed from the pia mater.25 In the latter study, brain microvessels were isolated from the mouse cortex using low speed centrifugation in a Ficoll gradient, and lysates were subsequently processed by depleting cytosolic proteins.26 Employing the commonly used gene ontology categories, 79 out of 2,188 proteins (identified by at least two distinct peptides) in the pial arteries were classified as ECM proteins, and only 27 out of 1,472 proteins (identified with a mean spectral count greater than 5) were so classified in the brain microvessels. As discussed recently by Hynes and colleagues,17 gene ontology categories are inadequate for extracting a complete list of ECM proteins from a data set. Therefore, we reanalyzed these two data sets using the matrisome repertoire defined by Hynes and colleagues.6 Consistent with the notion that every tissue contains about 100 to 150 ECM proteins, we found 150 ECM proteins in the pial artery data set, including 91 core matrisome proteins and 59 matrisome-associated proteins. Of interest, the majority of proteins (48 out of 59; 81%) annotated in this study as matrisome-associated proteins, according to Hynes and colleagues classification, had not been annotated as ECM proteins by gene ontology analysis in the original study (Figure 3A, Supplementary Table 1). A similar reanalysis of the brain microvessels data set yielded only 43 ECM proteins, including 24 core matrisome proteins and 19 matrisome-associated proteins, suggesting that the proteomic coverage in this data set is incomplete (Figure 3B, Supplementary Table 2). A comparison of matrisome proteins from the two brain vessel data sets revealed that 24 ECM proteins were common to both pial arteries and brain microvessels. Interestingly, half of the 24 shared proteins are core components of the BM (see next section). A comparison of matrisome proteins in pial arteries with those in lung and colon samples (identified by at least two peptides)17 revealed that a majority of ECM proteins in the pial artery data set (90 out of 150; 60%) have a tissue-specific expression. Conversely, only 35 out of 269 (13%) ECM proteins are found in all three tissues (Figure 3C).
Figure 3.

Characterization of the matrisome of cerebral blood vessels. (A, B). Pie charts displaying the number of matrisome proteins in murine pial arteries (A) and brain microvessels (B) determined by proteomic analysis25,26 and reclassified using the matrisome database.6 (C) Venn diagram showing the number of matrisome proteins that overlap between pial arteries, lung, and colon samples. ECM, extracellular matrix.
The Basement Membrane: A Key Specialized Form of Extracellular Matrix at the Interface Between the Vessel and Brain Parenchyma
On electron micrographs, all BMs are classically described as composed of a 50- to 100-nm-thick electron-dense layer (lamina densa) separated from the plasma membrane by an electron-lucent layer (lamina lucida). However, when tissues are processed by freeze substitution, the BM appears as a fairly uniform lamina densa that closely follows the contours of the plasma membrane, suggesting that the electron-lucent layer is a dehydratation artifact.27 All BMs are composed of a common set of interacting proteins, including at least one member of the laminin family, one or more variants of type IV collagens, heparan sulfate proteoglycans, and nidogens.28
Laminins
Laminins are large (400 to 800 kDa), cross-shaped heterotrimeric molecules consisting of α, β, and γ chains joined through a long coiled-coil domain. There are five α chains, four β chains, and three γ chains; the laminin molecules are named according to their chain composition, and a total of 16 chain combinations have been characterized biochemically.29 The laminin α chains carry the major domains that interact with cellular receptors and are considered the functionally active portion of the heterotrimer. Moreover, α chains have tissue-specific expression patterns. In cerebral blood vessels, laminin α1 is associated with the pia matter and the glia limitans, and is detected around pial and penetrating arteries but not around capillaries.30 Laminin α2 is synthetized by astrocytes and deposited at the glia limitans, although it is unclear whether it is also synthesized by SMCs.31 Laminin α4 and α5 chains are detected in the endothelial and smooth muscle BM of all cerebral blood vessels, with a discontinuous distribution at the level of venules.32 These α chains can assemble with β1 or β2 and γ1 to form Laminins-111, -121, -211, -221, 411, -421, -511, and -521. Laminin α3 is not expressed in cerebral blood vessels.10,30
Type IV collagens
Type IV collagens, the most abundant constituent of the BM, comprise up to six genetically distinct α chains designated α1(IV) to α6(IV); α chains assemble with a remarkable specificity to form only three distinct heterotrimers, [α1α1α2], [α3α4α5], and [α5α5α6], with [α1α1α2] being the predominant variant found in nearly all BMs, including those of cerebral blood vessels. The [α5α5α6] variant has been reported in association with the pia matter.33 Type IV collagens are triple-helical molecules that twist along their length and terminate in a globular noncollagenous (NC1) domain. The glycine residues within the characteristic Gly-X-Y repeats have a crucial role in helix formation and stabilization.34
Heparan sulfate proteoglycans
Heparan sulfate proteoglycans are glycoproteins with the common characteristic of covalently attached chains of heparan sulfate, a type of glycosaminoglycan. Heparan sulfate proteoglycans present in the BM include perlecan, agrin, and type XVIII collagen, which are large secreted proteins composed of multiple, functionally independent domains. They bind to many growth factors, cytokines, chemokines, enzymes, enzyme inhibitors, and ECM proteins, usually with sulfated domains within heparan sulfate chains but also with the protein core. Perlecan, agrin, and type XVIII collagen can also undergo proteolytic processing to release bioactive molecules.9 Nidogen-1 and -2 are sulfated monomeric glycoproteins that interact with a range of ECM proteins, in particular, laminins, collagen type IV, and perlecan.35
Basement membrane assembly
The BM assembles through a multistep process that is initiated by the binding of laminin to cell-surface proteins such as integrins and dystroglycan, promoting laminin polymerization and allowing recruitment of other laminin binding components into the nascent laminin scaffold. Laminin is the only component that is critical for early embryonic BM assembly.36,37 Type IV collagen polymerizes to form a second covalently stabilized network that is considered to provide structural stability to the BM. The laminin and type IV collagen networks are linked by heparan sulfate proteoglycans38 and nidogens.29 This scaffold then provides specific sites at which many other BM constituents interact to generate a fully functional BM.
Familial small vessel disease caused by mutations in genes encoding matrisome proteins
Collagen Type IV-Related Small Vessel Disease
Pathogenic mutations in COL4A1 and COL4A2
As noted above, COL4A1 and COL4A2 are members of the core matrisome and major components of nearly all BMs, including those of pial and brain microvessels. Dominant mutations in COL4A1 or COL4A2 are associated with typical features of cSVD and a broad range of other disorders that most commonly involve the brain and eyes, but also affect the kidney and skeletal muscle, in both humans and mice.39 Cerebral SVD manifestations consist of microhemorrhages and macrohemorrhages that can occur at virtually every age, patchy to diffuse leukoaraiosis, dilated periventricular spaces and, less frequently, small deep infarcts. Other brain features are porencephaly and schizencephaly, which typically manifests in infants with epilepsy, motor deficits or psychomotor retardation.40 Common eye features consist of dysgenesis of the anterior segment (cornea, iris, ciliary body, and lens), including Axenfeld-Rieger anomaly, congenital cataract, and juvenile-onset glaucoma.2 Although there are no reported neuropathologic studies of patients with COL4A1 or COLA2 mutations, electron microscopy analyses of skin or kidney biopsies have shown focal defects of the BMs of vascular and epithelial cells, including areas of fragmentation, duplication, and even herniation.41 Similar defects have been described in the BM of cerebral blood vessels from Col4a1+/Δex41 mice, a well-characterized mouse model that expresses a mutant collagen alpha-1(IV) chain with a 17-amino-acid in-frame deletion.42,43 A large proportion of COL4A1 and COL4A2 mutations are missense mutations resulting in the substitution for one of the invariant glycine residues within the Gly-Xaa-Yaa repeats in the collagenous domain of α1 or α2 chains.39 Expression of COL4A1 and COL4A2 mutations is extremely variable, even between members of the same family, with part of this variability apparently related to genetic-context and environmental factors. Accumulating evidence in humans and mice suggests that allelic heterogeneity also contributes to this variable expression.43,44 Specifically, in humans, mutations clustered within 31 amino-acid residues in the N-terminus of the collagenous domain of COL4A1 are associated with a clinical subentity called HANAC (Hereditary Angiopathy with Nephropathy, Aneurysms, and Cramps)—a syndrome characterized by co-association of arterial aneurysms and eye, kidney, and skeletal manifestations.41 In mice, mutations within the triple-helical domain nearer the carboxy terminus are associated with more severe intracerebral hemorrhages than mutations nearer the amino terminus.43
Collagen type IV-related small vessel disease: a basement membrane disease?
The α-chain stoichiometry of collagen type IV predicts that heterozygous COL4A1 and COL4A2 mutations result in the production of 75% and 50% abnormal collagen IV molecules, respectively, a prediction that is consistent with the observation that COL4A2 mutations result in a disease of reduced severity compared with COL4A1 mutations.45 Several lines of evidence suggest that the pathogenicity of COL4A1 and COL4A2 mutations arises from a reduced incorporation of collagen IV in the BM. First, cells stably expressing mutant COL4A1 have reduced secretion of α1 (IV) chain as well as α2 (IV) chain, and the same holds true for cells expressing mutant COL4A2.39 Second, deposition of collagen α1 and α2 (IV) is almost absent in the Reichert’s membrane, a BM of embryonic origin, in embryos homozygous for the Col4a1Δex41 mutation.46 Also, Col4a1+/Δex41 mice have reduced extracellular COL4A1 in the BM of retinal vessels, but increased intracellular retention of COL4A1 in vascular cells.43 Third, an apparent lack of expression of mutated COL4A1 transcript has been documented in the fibroblasts of affected patients from two families carrying a frameshift and a splice site COL4A1 mutation, respectively, because of nonsense-mediated mRNA decay.47 However, the lack of an overt clinical phenotype in mice heterozygous for null alleles of Col4a1 and Col4a2 suggests that haploinsufficiency may not solely account for the disease phenotype.48 One possibility is that mutant molecules may reach the extracellular space and exert a dominant-negative effect over the various partners of collagen IV that could also compromise BM integrity and, in addition, promote abnormal cell–matrix and cell–cell interactions.
Collagen type IV interacts with all three main components of the BM, namely laminin, perlecan, and nidogen. Collagen type IV-like perlecan and nidogens are dispensable for initiation of BM assembly, but are required for maintaining BM integrity.48–50 Immunohistochemical analyses of embryos completely lacking COL4A1 and COL4A2 show reduced deposition and a patchier distribution of laminin and nidogen.48 Whether similar effects occur in patients and mice with COL4A1 or COL4A2 mutations has yet to be investigated. The role of the BM, especially that of collagen type IV, in brain vessels is essentially unknown. Nevertheless, a recent analysis of mice lacking laminin chains provides some clues. In mice with ablation of astrocytic laminin γ1, astrocytic endfoot (parenchymal) BM formation is disrupted, the blood brain barrier (BBB) is profoundly altered and multifocal hemorrhages develop in deep brain regions. Mechanistic studies show that loss of astrocytic laminin γ1 impairs SMC differentiation, with decreased expression of contractile proteins in small arteries and arterioles in deep brain regions, impaired pericyte differentiation, loss of astrocytic endfeet polarity, and a reduction in endothelial tight junction protein expression.51,52 Mice with constitutive inactivation of laminin α2 chain (dy3K/dy3K),53 which die at 4 weeks of age, exhibit breaches in the parenchymal and endothelial BM and have profound alterations in BBB development and function with limited pericyte coverage, poorly polarized astrocyte endfeet, and immature endothelial tight junctions.54 It is tempting to speculate that COL4A1 mutations might similarly affect key components of the gliovascular unit.
Collagen type IV also participates in cell-matrix interactions via cell-surface receptors; specifically, it binds to integrins containing a β1 subunit, notably α1β1 and α2β1.55 Remarkably, HANAC syndrome-associated COL4A1 mutations are clustered within 30 amino-acid residues in the N-terminus of the collagenous domain, which contains a major integrin binding site.56 Interestingly, loss of Integrin β1 in SMCs results in progressive smooth muscle degeneration and increased deposition of fibrillary collagens in the tunica media.57 Moreover, mice with postnatal inactivation of Integrin β1 in endothelial cells exhibit junctional defects in the endothelium, loss of vascular integrity and brain hemorrhages.58
Another possibility besides defects of the BM and cell-matrix interaction, suggested by both in vitro and in vivo data, is that COL4A1 or COL4A2 mutations promote intracellular retention of unfolded or misfolded mutant α1 and α2 chains and activate the unfolded protein response.43,45,59 Prolonged endoplasmic reticulum stress can eventually trigger cell death, oxidative stress, or potentially elicit an inflammatory reaction.60,61 Interestingly, Gould and colleagues recently showed in a proof of concept study that treating Col4a1+/Δex41 mice with an FDA-approved chaperone (Sodium 4-phenylbutyrate) could decrease intracellular COL4A1 accumulation and, notably, reduce intracerebral hemorrhage severity.43
CARASIL
Pathogenic mutations in HTRA1
HTRA1 is a 50-kDa secreted protein composed of a signaling peptide, an insulin growth factor binding domain, a Kazal-like protease inhibitor domain, a conserved serine protease domain, and a PDZ domain. HTRA1, classified as an ECM regulator in the matrisome-associated category, has been reported to be widely expressed, although a detailed analysis of its expression pattern in the brain has not yet been performed. Proteomic studies indicate that HTRA1 is expressed in brain vessels, with higher expression in pial arteries than in brain microvessels.25,26 Recessive mutations in HTRA1 cause CARASIL, a rare SVD initially reported in Japanese families. Clinical landmarks include cognitive deficit, motor dysfunction, and subcortical stroke leading to dementia and severe disability in the fourth decade, in association with diffuse alopecia and attacks of severe low back pain due to spondylosis deformans and disk degeneration.62 Microvascular changes in CARASIL consist of extensive loss of SMCs and deposition of fibro-hyaline material in the media, fibrous intimal proliferation, and splitting and fragmentation of the internal elastica lamina, with many vessels ultimately developing a double-barreled or split-wall appearance.63 To date, very few mutations have been reported; these include homozygous nonsense mutations as well as homozygous missense mutations in the serine protease domain, resulting in the loss of HTRA1 protein expression or protease activity.64
CARASIL: Dysregulation of transforming growth factor-β signaling?
Although it has been shown that HTRA1 is involved in various pathologic conditions in addition to CARASIL, its physiologic role remains unclear. HTRA1 possesses serine protease activity, and a variety of potential substrates have been identified.65 Members of the transforming growth factor-β (TGF-β) signaling pathway have received particular attention, but experimental studies have generated conflicting results. Early studies reported that HTRA1 inhibited TGF-β family members, showing that the inhibitory effects on TGF-β signaling required the proteolytic activity of HTRA1.66 Subsequent studies suggested that HTRA1 antagonizes TGF-β signaling by cleaving pro TGF-β1 intracellularly in the reticulum endoplasmic,67 or by cleaving type II and type III TGF-β receptors extracellularly.68 These findings, combined with immunohistochemical analyses of postmortem brain samples from two CARASIL patients, led to the hypothesis that CARASIL is associated with upregulation of TGF-β signaling in cerebral vessels.67 However, these earlier studies were not without limitations, especially their use of cells that vastly overexpressed HTRA1. Recently, Beaufort and colleagues revisited this issue using embryonic fibroblasts from HTRA1-null mice and in vitro assays using physiologic quantities of HTRA1. Remarkably, they came to very different conclusions. Specifically, they provided evidence that HTRA1 has a facilitating role in TGF-β pathway activation, and that LTBP1 (latent TGF-β binding protein 1), an ECM protein with a key role in TGF-β bioactivation,69 is a physiologic target of HTRA1.70 Further studies are warranted to confirm these results in brain vessels, especially since the levels of active TGF-β, which is itself a matrisome protein, are tightly controlled by a number of matrisome proteins and the composition of the matrisome is unique to each tissue.
There is ample evidence that TGF-β signaling has a key role in vessel development and maintenance, acting in both endothelial and SMCs. Moreover, both up- and downregulation of this pathway have been implicated in several vascular pathologies.71 For example, hereditary hemorrhagic telangiectasia, or Osler–Rendu–Weber syndrome, is caused by loss-of-function mutations in members of the TGF-β pathway. However, enhanced TGF-β signaling has emerged as the common final pathway in the pathogenesis of familial forms of thoracic aortic aneurysm.72 Whether dysregulated TGF-β signaling contributes to the cerebrovascular manifestations of CARASIL has yet to be investigated.
Perturbations of the matrisome as a possible proximate cause in familial small vessel disease
CADASIL
Early perturbations of the cerebrovascular matrisome
CADASIL is an archetypal SVD, considered the most frequent hereditary cSVD.73 The disease is caused by dominant mutations in NOTCH3, a heterodimeric receptor predominantly expressed in vascular SMCs that is a critical regulator of developmental formation of small arteries.74 The pathology of cerebral vessels is roughly similar to that of sporadic forms of SVD except that CADASIL is characterized by the presence of pathognomonic deposits of granular osmiophilic material within the BM of SMCs and pericytes that can protrude into the interstitial tissue around these cells.73 The majority of CADASIL mutations are missense mutations that lead to the gain or loss of a cysteine residue in the NOTCH3 extracellular domain (Notch3ECD).75,76 Regardless of whether they concomitantly impair NOTCH3 receptor activity, these mutations alter Notch3ECD itself in a way that promotes its multimerization and aberrant accumulation in granular osmiophilic material.77 Importantly, vascular Notch3ECD and granular osmiophilic material deposits have emerged as the earliest pathologic features in patients and mice with CADASIL.78 Recently, we showed that excess levels or multimerization of mutant Notch3ECD facilitates interactions with key components of the cerebrovascular ECM and promotes their accumulation and sequestration in Notch3ECD-containing deposits. Among these components are the metalloproteinase inhibitor TIMP3, a matrisome-associated regulator that modifies the ECM, and vitronectin, a glycoprotein of the core matrisome. Both TIMP3 and vitronectin have been shown to abnormally accumulate in the brain vessels not only of patients but also of preclinical models, at an early stage of the disease. Moreover, we found evidence for increased TIMP3 activity in brain microvessels from both TgNotch3R169C mice, a well-established preclinical model of CADASIL, and patients with CADASIL.77 Other ECM proteins, such as clusterin, endostatin (a proteolytic fragment derived from collagen XVIII), and LTBP1, have been shown to accumulate in Notch3ECD-containing deposits in patients with CADASIL.79,80 However, these latter proteins appear to accumulate at a later stage of disease progression (AJ, unpublished).77
CADASIL: Cerebrovascular dysfunction arising from a change in the matrisome?
These matrisome changes may affect the BBB; however, it is yet unclear whether BBB disruption has a role in the pathogenesis of CADASIL. Histopathologic studies on human postmortem tissues showed focal breaches of the BBB at the sites of microbleeds or infarcts, but no evidence of generalized BBB leakage at this end stage of the disease.81 Moreover, immunohistochemical and transmission electron microscopy analyses, as well as systemic injections of fluorescent tracers failed to reveal alterations of the BBB integrity in TgNotch3R169C mice (JR and AJ, unpublished data).82,83
However, CADASIL patients exhibit alterations in cerebral hemodynamics and cerebrovascular reactivity.73 Studies of CADASIL mouse models suggest that cerebrovascular dysfunction is an early disease mechanism in the pathophysiology of CADASIL.78 Specifically, cerebral blood flow autoregulation and functional hyperemia are impaired and myogenic responses of pial and parenchymal arteries are reduced in TgNotch3R169C mice.82,84 A recent study from Dabertrand and colleagues suggests that increased TIMP3 activity might be a proximate cause of cerebrovascular deficits in this preclinical model of CADASIL. TIMP3 is the only TIMP that becomes tightly bound to the ECM after secretion and is thus in a position to exert tissue-specific effects.85,86 Notably, TIMP3 is the only TIMP that can inhibit TACE (tumor necrosis factor-α-converting enzyme), which mediates ectodomain shedding of transmembrane receptors and ligands, including tumor necrosis factor-α and ligands of the epidermal growth factor receptor family (EGFR).87 Specifically, Dabertrand et al.84 showed that reduced myogenic responses of pial and parenchymal arteries in the TgNotch3R169C mice were caused by functional upregulation of Kv1 channels at the plasma membrane of arterial SMC, and further showed that these defects could be rescued by applying the EGFR agonist HB-EGF. Given that activation of EGFR with the agonist HB-EGF has been reported to suppress KV1 channel activity in cerebral arteries by enhancing KV1 channel endocytosis,88 we speculate that increased TIMP3 activity may lead to decreased myogenic tone in brain arteries by reducing TACE/HB-EGF/EGFR–mediated KV channel endocytosis and increasing Kv channel activity.
The TIMP3 dysregulation may also contribute to small vessel pathology through its well-known MMP inhibitory activity, and abnormally elevated TIMP3 activity may lead to vessel fibrosis. Also, several reports have shown that high levels of TIMP3 are proapoptotic in a variety of cell types, including vascular SMCs, and the importance of TIMP3 has been shown in inflammatory pathologies and autoimmune diseases.86,89 Additional studies are warranted to test these hypotheses and investigate whether and how other perturbations of the matrisome, such as the abnormal accumulation of vitronectin, contribute to CADASIL pathophysiology.
FOXC1-Associated Small Vessel Disease
Pathogenic mutations in a transcription factor expressed in blood vessels
Autosomal-dominant mutations or gene-dosage changes, including deletions or duplications, in FOXC1 result in a variety of malformations in the anterior ocular segment that ultimately lead to glaucoma and malformations of the cerebellar posterior fossa.90,91 Recently, French et al.3 reported that patients with FOXC1-attributable Axenfeld-Rieger syndrome have a higher incidence of stroke and highly penetrant magnetic resonance imaging features of cSVD, including white-matter hyperintensities, dilated perivascular spaces, and lacunes.3 FOXC1, a member of the forkhead family of transcription factors, has a broad expression pattern and regulates, in a dose-dependent manner, an array of fundamental processes, including the early development of the heart, blood vessels and somites, as well as ocular and genitourinary systems.92,93 FOXC1 is expressed in the pericytes and endothelial cells of blood vessels in the brain, and in the periocular mesenchyme of the eye. In the mouse, conditional deletion of Foxc1 in pericytes produces multifocal brain microhemorrhages toward the end of gestation, preceded by focal breakdown of BBB integrity and hyperplasia of both endothelial cells and pericytes, suggestive of increased angiogenic activity.94 Furthermore, conditional deletion of Foxc1 in neural crest cells, which give rise to most of the periocular mesenchyme, phenocopies the eye anomalies associated with the human disease Axenfeld-Rieger syndrome, notably including corneal angiogenesis; by contrast, the normal cornea is avascular.95
FOXC1 mutations and the matrisome?
At first glance, there is no overt relationship between FOXC1 mutations and the matrisome. Nevertheless, studies in neural crest-deleted Foxc1−/− mice provide one possible mechanistic link between matrisome perturbations and the vascular phenotype induced by FOXC1 mutations. Specifically, Seo et al.95 found that expression of the MMPs, MMP3, MMP9, and MMP19—all ECM regulators of the matrisome-associated category—is increased in the cornea of neural crest-deleted Foxc1−/− mice. Notably, these data suggest that the increased expression of MMPs, which are known to cleave and release ECM-sequestered vascular endothelial growth factor and enhance vascular endothelial growth factor bioavailability,96 is likely a contributing factor to the enhanced angiogenesis in the cornea of mutant mice. A second clue comes from experiments in zebrafish, where knockdown of Foxc1 produces similar eye abnormalities and brain hemorrhages.97 Interestingly, loss of foxC1 leads to focal disruptions of vascular BM integrity, with impaired expression of laminin 1-1-1. These findings have led us to speculate that FOXC1-related cSVD manifestations actually arise from vascular BM defects through deregulation of the availability of growth factors.
Matrisome changes in sporadic small vessel disease: Consequent pathologic findings or proximate cause?
Pathologic Findings
Cerebrovascular changes associated with age and vascular risk factors are commonly referred to as hyalinosis, fibrohyalinosis, or fibrinoid necrosis, although the meanings of each of these terms are somewhat different.98 A closer look at histopathologic data reveals that vessel changes consist of major alterations of the ECM associated with a more or less severe degeneration of SMCs from the tunica media, with or without exudation of plasma proteins within the vessel wall. Changes of ECM may range from thickening of the BMs, splitting and duplication of the elastic lamina and, notably, marked deposition of normally occurring types of collagens and possibly other components of the ECM in the intima, media, and adventitia.99,100 That said, it is difficult to draw conclusions about the primary site and mechanism of these ECM changes since they are postmortem observations at the end stage of the disease. In particular, it is conceivable that all or part of the increased ECM deposition is a compensatory response to SMC degeneration that serves as a repair mechanism.
Matrisome Changes: Proximate Cause?
The microvascular changes in CARASIL or CADASIL are generally considered to be qualitatively similar to those observed in sporadic SVD; however, the reverse is also true, meaning that specific perturbations of the matrisome can produce such vascular lesions. Moreover, experimental studies examining the effects of aging and arterial hypertension—the two most important risk factors for cSVD—have highlighted vascular ECM changes at the early stage of the disease in mouse or rat models. During aging, cerebral arterioles undergo SMC atrophy and exhibit an increase in the ratio of nondistensible (collagen and BM) ECM to distensible (smooth muscle and elastin) components.101 Hypertension commonly produces narrowing of the arterial lumen (inward remodeling), especially at the level of small distal arteries, with or without medial thickening (hypertrophy). Inward remodeling has been extensively documented in both peripheral and cerebral arteries, and angiotensin II has been shown to have a key role in this effect.102,103 Inward remodeling is thought to result from both changes in SMC contractility and in ECM composition, with a potential contribution of MMP activation.104 Reorganization of the ECM, with an increase in deposition of collagens and fibronectin in the tunica media, an increase in collagen-to-elastin ratio, and disorganization and fragmentation of elastic fibers, has been clearly documented in the peripheral arteries of models with chronic hypertension.105 However, evidence of ECM changes at the level of brain arteries remains scarce,106,107 although it should be noted that this aspect has been so far poorly investigated.
The dominant view is that arterial remodeling during chronic hypertension represents both an adaptive process that reduces stress on the vessel wall and protects the downstream vascular bed from increased arterial pressure, and a deleterious process that ultimately results in arterial stenosis and stiffness, which may contribute to reductions in resting cerebral blood flow, impaired vasodilation, and increased susceptibility to ischemic injury.108 Increased deposition of ECM in cerebral vessels, whether proximate cause or consequent pathologic finding, is likely to contribute to the long-term deleterious effects of hypertension.
Concluding remarks
Cerebral SVD is a heterogeneous group of disorders that are likely multifactorial in nature with different ultimate causes acting through specific pathways. Nevertheless, with recent advances in identifying major genetic causes and pathophysiologic mechanisms of familial cSVD has come the emerging view that perturbation of the cerebrovascular matrisome could be a convergent pathway that drives the functional and structural alterations of small brain vessels and disease manifestations. Specifically, in collagen type IV-related SVD and CARASIL, genetic alterations of a matrisome protein are clearly the primary event. In CADASIL and FOXC1-related SVD, the data suggest that specific perturbations of the matrisome are the proximate cause. Importantly, studies in animals and humans support the possibility that this may also apply to age- and hypertension-related SVD. Despite these provocative connections, a better characterization of matrisome alterations using more sensitive tools and further experimental studies are needed to strengthen the link between matrisome alterations and cSVD.
Though unexplained, it is not unusual for mutations in a gene with a vascular expression broader than the brain, for example NOTCH3, to lead to a brain-restricted phenotype. Also, within the brain, it is essentially unknown why lesions of cSVD predominantly affect small penetrating arteries and parenchymal arterioles, particularly in the deep cerebral white matter. The vessel-specific expression pattern of the matrisome may hold the key to these unanswered questions. Every organ has an ECM with a unique composition, and it is very likely that brain and peripheral arteries have distinct matrisomes. On the basis of a preliminary characterization of the matrisome of pial arteries and brain microvessels, it is tempting to speculate that penetrating arteries, intraparenchymal arterioles, and brain capillaries also harbor unique repertoires of matrisome proteins. Moreover, unlike pial arteries, penetrating and parenchymal arterioles are bottlenecks of flow in the cerebral angioarchitecture.109 Therefore, it is conceivable that lesions could affect brain vessels with a specific spatial pattern if expression of a specific set of matrisome proteins is required for the pathogenic process. Accordingly, identification of those matrisome proteins with a vessel-specific expression pattern might provide insight into differential manifestations of vascular pathologies.
A significant proportion of matrisome proteins is presumably expressed within the vascular BM, especially at the capillary level. The BM is located at the interface between endothelial cells, contractile cells (SMCs and pericytes), and astrocyte endfeet—a very strategic location within the gliovascular unit. Therefore, changes in the composition of the BM are in a position to exert profound effects on all components of the gliovascular unit as well as on the brain parenchyma itself that may account for the broad range of manifestations in cSVD.
In conclusion, the ECM of cerebral blood vessels is a key component of the gliovascular unit that warrants greater research attention in the future, in both physiologic and pathologic settings.
Supplementary Material
Acknowledgement
The authors gratefully acknowledge David Hill-Eubanks for editorial assistance.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported in part by a grant from the Fondation Leducq (Transatlantic Network of Excellence on the Pathogenesis of Small Vessel Disease of the Brain) to AJ and MTN, National Institutes of Health (P01-HL-095488, R01-HL-044455, R01-HL-098243) and Totman Medical Research Trust to MTN.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
AJ and MTN wrote the manuscript and generated the figures. IH and JR provided assistance in preparing the manuscript and IH performed the proteomic analysis.
Supplementary Material
Supplementary material for this paper can be found at http://jcbfm.sagepub.com/content/by/supplemental-data
References
- 1.Pantoni L. Cerebral small vessel disease: from pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol 2010; 9: 689–701. [DOI] [PubMed] [Google Scholar]
- 2.Joutel A. Pathogenic aspects of hereditray small vessel disease of the brain. In: Pantoni L, Gorelick PB. (eds). Cerebral Small Vessel Disease, Cambridge University Press, 2014, pp. 64–81. [Google Scholar]
- 3.French CR, Seshadri S, Destefano AL, Fornage M, Arnold CR, Gage PJ, et al. Mutation of FOXC1 and PITX2 induces cerebral small-vessel disease. J Clin Invest 2014; 124: 4877–4881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell 2002; 110: 673–687. [DOI] [PubMed] [Google Scholar]
- 5.Hynes RO. The extracellular matrix: not just pretty fibrils. Science 2009; 326: 1216–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Naba A, Clauser KR, Hoersch S, Liu H, Carr SA, Hynes RO. The matrisome: in silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol Cell Proteomics 2012; 11: M111.014647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jung J, Ryu T, Hwang Y, Lee E, Lee D. Prediction of extracellular matrix proteins based on distinctive sequence and domain characteristics. J Comput Biol 2010; 17: 97–105. [DOI] [PubMed] [Google Scholar]
- 8.Ricard-Blum S. The collagen family. Cold Spring Harb Perspect Biol 2011; 3: a004978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sarrazin S, Lamanna WC, Esko JD. Heparan sulfate proteoglycans. Cold Spring Harb Perspect Biol 2011; 3: pii:a004952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hallmann R, Horn N, Selg M, Wendler O, Pausch F, Sorokin LM. Expression and function of laminins in the embryonic and mature vasculature. Physiol Rev 2005; 85: 979–1000. [DOI] [PubMed] [Google Scholar]
- 11.Ramirez F, Dietz HC. Fibrillin-rich microfibrils: Structural determinants of morphogenetic and homeostatic events. J Cell Physiol 2007; 213: 326–330. [DOI] [PubMed] [Google Scholar]
- 12.Preissner KT, Reuning U. Vitronectin in vascular context: facets of a multitalented matricellular protein. Semin Thromb Hemost 2011; 37: 408–424. [DOI] [PubMed] [Google Scholar]
- 13.Schwarzbauer JE, DeSimone DW. Fibronectins, their fibrillogenesis, and in vivo functions. Cold Spring Harb Perspect Biol 2011; 3: pii:a005041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hynes RO, Naba A. Overview of the matrisome—an inventory of extracellular matrix constituents and functions. Cold Spring Harb Perspect Biol 2012; 4: a004903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell 2011; 146: 873–887. [DOI] [PubMed] [Google Scholar]
- 16.Bonnans C, Chou J, Werb Z. Remodelling the extracellular matrix in development and disease. Nat Rev Mol Cell Biol 2014; 15: 786–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Naba A, Hoersch S, Hynes RO. Towards definition of an ECM parts list: an advance on GO categories. Matrix Biol 2012; 31: 371–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.The Matrisome Project http://matrisomeproject.mit.edu/.
- 19.Cipolla MJ. The cerebral circulation. In: Granger DN, Granger J. (eds). Integrated Systems Physiology: From Molecule to Function, San Rafael, CA: Morgan & Claypool Life Sciences, 2010, pp. 1–59. [Google Scholar]
- 20.Frederickson RG, Low FN. Blood vessels and tissue space associated with the brain of the rat. Am J Anat 1969; 125: 123–145. [DOI] [PubMed] [Google Scholar]
- 21.Peters A, Palay SL, de Webster HF. The meninges. The Fine Structure of the Nervous System: Neurons and Their Supporting Cells, Oxford University Press, 1991, pp. 395–406. [Google Scholar]
- 22.Krisch B, Leonhardt H, Oksche A. Compartments and perivascular arrangement of the meninges covering the cerebral cortex of the rat. Cell Tissue Res 1984; 238: 459–474. [DOI] [PubMed] [Google Scholar]
- 23.Zhang ET, Inman CB, Weller RO. Interrelationships of the pia mater and the perivascular (Virchow-Robin) spaces in the human cerebrum. J Anat 1990; 170: 111–123. [PMC free article] [PubMed] [Google Scholar]
- 24.Mathiisen TM, Lehre KP, Danbolt NC, Ottersen OP. The perivascular astroglial sheath provides a complete covering of the brain microvessels: An electron microscopic 3D reconstruction. Glia 2010; 58: 1094–1103. [DOI] [PubMed] [Google Scholar]
- 25.Badhwar A, Stanimirovic DB, Hamel E, Haqqani AS. The proteome of mouse cerebral arteries. J Cereb Blood Flow Metab 2014; 34: 1033–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chun HB, Scott M, Niessen S, Hoover H, Baird A, Yates J, et al. The proteome of mouse brain microvessel membranes and basal lamina. J Cereb Blood Flow Metab 2011; 31: 2267–2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chan FL, Inoue S, Leblond CP. The basement membranes of cryofixed or aldehyde-fixed, freeze-substituted tissues are composed of a lamina densa and do not contain a lamina lucida. Cell Tissue Res 1993; 273: 41–52. [DOI] [PubMed] [Google Scholar]
- 28.Yurchenco PD. Basement membranes: cell scaffoldings and signaling platforms. Cold Spring Harb Perspect Biol 2011; 3: pii:a004911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hohenester E, Yurchenco PD. Laminins in basement membrane assembly. Cell Adh Migr 2013; 7: 56–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sixt M, Engelhardt B, Pausch F, Hallmann R, Wendler O, Sorokin LM. Endothelial cell laminin isoforms, laminins 8 and 10, play decisive roles in T cell recruitment across the blood-brain barrier in experimental autoimmune encephalomyelitis. J Cell Biol 2001; 153: 933–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yousif LF, Di Russo J, Sorokin L. Laminin isoforms in endothelial and perivascular basement membranes. Cell Adh Migr 2013; 7: 101–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu C, Ivars F, Anderson P, Hallmann R, Vestweber D, Nilsson P, et al. Endothelial basement membrane laminin alpha5 selectively inhibits T lymphocyte extravasation into the brain. Nat Med 2009; 15: 519–527. [DOI] [PubMed] [Google Scholar]
- 33.Urabe N, Naito I, Saito K, Yonezawa T, Sado Y, Yoshioka H, et al. Basement membrane type IV collagen molecules in the choroid plexus, pia mater and capillaries in the mouse brain. Arch Histol Cytol 2002; 65: 133–143. [DOI] [PubMed] [Google Scholar]
- 34.Khoshnoodi J, Pedchenko V, Hudson BG. Mammalian collagen IV. Microsc Res Tech 2008; 71: 357–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ho MSP, Böse K, Mokkapati S, Nischt R, Smyth N. Nidogens-Extracellular matrix linker molecules. Microsc Res Tech 2008; 71: 387–395. [DOI] [PubMed] [Google Scholar]
- 36.Smyth N, Vatansever HS, Murray P, Meyer M, Frie C, Paulsson M, et al. Absence of basement membranes after targeting the LAMC1 gene results in embryonic lethality due to failure of endoderm differentiation. J Cell Biol 1999; 144: 151–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miner JH, Li C, Mudd JL, Go G, Sutherland AE. Compositional and structural requirements for laminin and basement membranes during mouse embryo implantation and gastrulation. Development 2004; 131: 2247–2256. [DOI] [PubMed] [Google Scholar]
- 38.Behrens DT, Villone D, Koch M, Brunner G, Sorokin L, Robenek H, et al. The epidermal basement membrane is a composite of separate laminin- or collagen IV-containing networks connected by aggregated perlecan, but not by nidogens. J Biol Chem 2012; 287: 18700–18709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuo DS, Labelle-Dumais C, Gould DB. COL4A1 and COL4A2 mutations and disease: insights into pathogenic mechanisms and potential therapeutic targets. Hum Mol Genet 2012; 21: R97–R110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yoneda Y, Haginoya K, Kato M, Osaka H, Yokochi K, Arai H, et al. Phenotypic spectrum of COL4A1 mutations: porencephaly to schizencephaly. Ann Neurol 2013; 73: 48–57. [DOI] [PubMed] [Google Scholar]
- 41.Plaisier E, Gribouval O, Alamowitch S, Mougenot B, Prost C, Verpont MC, et al. COL4A1 mutations and hereditary angiopathy, nephropathy, aneurysms, and muscle cramps. N Engl J Med 2007; 357: 2687–2695. [DOI] [PubMed] [Google Scholar]
- 42.Gould DB, Phalan FC, van Mil SE, Sundberg JP, Vahedi K, Massin P, et al. Role of COL4A1 in small-vessel disease and hemorrhagic stroke. N Engl J Med 2006; 354: 1489–1496. [DOI] [PubMed] [Google Scholar]
- 43.Jeanne M, Jorgensen J, Gould DB. Molecular and genetic analysis of collagen type IV mutant mouse models of spontaneous intracerebral hemorrhage identify mechanisms for stroke prevention. Circulation 2015; 131: 1555–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kuo DS, Labelle-Dumais C, Mao M, Jeanne M, Kauffman WB, Allen J, et al. Allelic heterogeneity contributes to variability in ocular dysgenesis, myopathy and brain malformations caused by Col4a1 and Col4a2 mutations. Hum Mol Genet 2014; 23: 1709–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Murray LS, Lu Y, Taggart A, Van Regemorter N, Vilain C, Abramowicz M, et al. Chemical chaperone treatment reduces intracellular accumulation of mutant collagen IV and ameliorates the cellular phenotype of a COL4A2 mutation that causes haemorrhagic stroke. Hum Mol Genet 2014; 23: 283–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gould DB, Phalan FC, Breedveld GJ, van Mil SE, Smith RS, Schimenti JC, et al. Mutations in Col4a1 cause perinatal cerebral hemorrhage and porencephaly. Science 2005; 308: 1167–1171. [DOI] [PubMed] [Google Scholar]
- 47.Lemmens R, Maugeri A, Niessen HWM, Goris A, Tousseyn T, Demaerel P, et al. Novel COL4A1 mutations cause cerebral small vessel disease by haploinsufficiency. Hum Mol Genet 2013; 22: 391–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pöschl E, Schlötzer-Schrehardt U, Brachvogel B, Saito K, Ninomiya Y, Mayer U. Collagen IV is essential for basement membrane stability but dispensable for initiation of its assembly during early development. Development 2004; 131: 1619–1628. [DOI] [PubMed] [Google Scholar]
- 49.Costell M, Gustafsson E, Aszódi A, Mörgelin M, Bloch W, Hunziker E, et al. Perlecan maintains the integrity of cartilage and some basement membranes. J Cell Biol 1999; 147: 1109–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bader BL, Smyth N, Nedbal S, Miosge N, Baranowsky A, Mokkapati S, et al. Compound genetic ablation of nidogen 1 and 2 causes basement membrane defects and perinatal lethality in mice. Mol Cell Biol 2005; 25: 6846–6856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen Z-L, Yao Y, Norris EH, Kruyer A, Jno-Charles O, Akhmerov A, et al. Ablation of astrocytic laminin impairs vascular smooth muscle cell function and leads to hemorrhagic stroke. J Cell Biol 2013; 202: 381–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yao Y, Chen Z-L, Norris EH, Strickland S. Astrocytic laminin regulates pericyte differentiation and maintains blood brain barrier integrity. Nat Commun 2014; 5: 3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Miyagoe Y, Hanaoka K, Nonaka I, Hayasaka M, Nabeshima Y, Arahata K, et al. Laminin alpha2 chain-null mutant mice by targeted disruption of the Lama2 gene: a new model of merosin (laminin 2)-deficient congenital muscular dystrophy. FEBS Lett 1997; 415: 33–39. [DOI] [PubMed] [Google Scholar]
- 54.Menezes MJ, McClenahan FK, Leiton CV, Aranmolate A, Shan X, Colognato H. The extracellular matrix protein laminin α2 regulates the maturation and function of the blood-brain barrier. J Neurosci 2014; 34: 15260–15280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Leitinger B, Hohenester E. Mammalian collagen receptors. Matrix Biol 2007; 26: 146–155. [DOI] [PubMed] [Google Scholar]
- 56.Parkin JD, San Antonio JD, Pedchenko V, Hudson B, Jensen ST, Savige J. Mapping structural landmarks, ligand binding sites, and missense mutations to the collagen IV heterotrimers predicts major functional domains, novel interactions, and variation in phenotypes in inherited diseases affecting basement membranes. Hum Mutat 2011; 32: 127–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Turlo KA, Scapa J, Bagher P, Jones AW, Feil R, Korthuis RJ, et al. β1-integrin is essential for vasoregulation and smooth muscle survival in vivo. Arterioscler Thromb Vasc Biol 2013; 33: 2325–2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yamamoto H, Ehling M, Kato K, Kanai K, van Lessen M, Frye M, et al. Integrin β1 controls VE-cadherin localization and blood vessel stability. Nat Commun 2015; 6: 6429. [DOI] [PubMed] [Google Scholar]
- 59.Gould DB, Marchant JK, Savinova OV, Smith RS, John SWM. Col4a1 mutation causes endoplasmic reticulum stress and genetically modifiable ocular dysgenesis. Hum Mol Genet 2007; 16: 798–807. [DOI] [PubMed] [Google Scholar]
- 60.Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol 2011; 13: 184–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chaudhari N, Talwar P, Parimisetty A, Lefebvre d’Hellencourt C, Ravanan P. A molecular web: endoplasmic reticulum stress, inflammation, and oxidative stress. Front Cell Neurosci 2014; 8: 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hara K, Shiga A, Fukutake T, Nozaki H, Miyashita A, Yokoseki A, et al. Association of HTRA1 mutations and familial ischemic cerebral small-vessel disease. N Engl J Med 2009; 360: 1729–1739. [DOI] [PubMed] [Google Scholar]
- 63.Oide T, Nakayama H, Yanagawa S, Ito N, Ikeda S-I, Arima K. Extensive loss of arterial medial smooth muscle cells and mural extracellular matrix in cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL). Neuropathology 2008; 28: 132–142. [DOI] [PubMed] [Google Scholar]
- 64.Nozaki H, Nishizawa M, Onodera O. Features of cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy. Stroke 2014; 45: 3447–3453. [DOI] [PubMed] [Google Scholar]
- 65.An E, Sen S, Park SK, Gordish-Dressman H, Hathout Y. Identification of novel substrates for the serine protease HTRA1 in the human RPE secretome. Invest Ophthalmol Vis Sci 2010; 51: 3379–3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Oka C, Tsujimoto R, Kajikawa M, Koshiba-Takeuchi K, Ina J, Yano M, et al. HtrA1 serine protease inhibits signaling mediated by Tgfbeta family proteins. Dev 2004; 131: 1041–1053. [DOI] [PubMed] [Google Scholar]
- 67.Shiga A, Nozaki H, Yokoseki A, Nihonmatsu M, Kawata H, Kato T, et al. Cerebral small-vessel disease protein HTRA1 controls the amount of TGF-beta1 via cleavage of proTGF-beta1. Hum Mol Genet 2011; 20: 1800–1810. [DOI] [PubMed] [Google Scholar]
- 68.Graham JR, Chamberland A, Lin Q, Li XJ, Dai D, Zeng W, et al. Serine protease HTRA1 antagonizes transforming growth factor-β signaling by cleaving its receptors and loss of HTRA1 in vivo enhances bone formation. PLoS ONE 2013; 8: e74094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ge G, Greenspan DS. BMP1 controls TGFbeta1 activation via cleavage of latent TGFbeta-binding protein. J Cell Biol 2006; 175: 111–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Beaufort N, Scharrer E, Kremmer E, Lux V, Ehrmann M, Huber R, et al. Cerebral small vessel disease-related protease HtrA1 processes latent TGF-β binding protein 1 and facilitates TGF-β signaling. Proc Natl Acad Sci USA 2014; 111: 16496–16501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jakobsson L, van Meeteren LA. Transforming growth factor β family members in regulation of vascular function: in the light of vascular conditional knockouts. Exp Cell Res 2013; 319: 1264–1270. [DOI] [PubMed] [Google Scholar]
- 72.Gillis E, Van Laer L, Loeys BL. Genetics of thoracic aortic aneurysm: at the crossroad of transforming growth factor-β signaling and vascular smooth muscle cell contractility. Circ Res 2013; 113: 327–340. [DOI] [PubMed] [Google Scholar]
- 73.Chabriat H, Joutel A, Dichgans M, Tournier-Lasserve E, Bousser MG. Cadasil. Lancet Neurol 2009; 8: 643–653. [DOI] [PubMed] [Google Scholar]
- 74.Fouillade C, Monet-Lepretre M, Baron-Menguy C, Joutel A. Notch signalling in smooth muscle cells during development and disease. Cardiovasc Res 2012; 95: 138–146. [DOI] [PubMed] [Google Scholar]
- 75.Joutel A, Vahedi K, Corpechot C, Troesch A, Chabriat H, Vayssiere C, et al. Strong clustering and stereotyped nature of Notch3 mutations in CADASIL patients. Lancet 1997; 350: 1511–1515. [DOI] [PubMed] [Google Scholar]
- 76.Dichgans M, Ludwig H, Muller-Hocker J, Messerschmidt A, Gasser T. Small in-frame deletions and missense mutations in CADASIL: 3D models predict misfolding of Notch3 EGF-like repeat domains. Eur J Hum Genet 2000; 8: 280–285. [DOI] [PubMed] [Google Scholar]
- 77.Monet-Leprêtre M, Haddad I, Baron-Menguy C, Fouillot-Panchal M, Riani M, Domenga-Denier V, et al. Abnormal recruitment of extracellular matrix proteins by excess Notch3 ECD: a new pathomechanism in CADASIL. Brain 2013; 136: 1830–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Joutel A. Pathogenesis of CADASIL: transgenic and knock-out mice to probe function and dysfunction of the mutated gene, Notch3, in the cerebrovasculature. Bioessays 2011; 33: 73–80. [DOI] [PubMed] [Google Scholar]
- 79.Arboleda-Velasquez JF, Manent J, Lee JH, Tikka S, Ospina C, Vanderburg CR, et al. Hypomorphic Notch 3 alleles link Notch signaling to ischemic cerebral small-vessel disease. Proc Natl Acad Sci USA 2011; 108: E128–E135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kast J, Hanecker P, Beaufort N, Giese A, Joutel A, Dichgans M, et al. Sequestration of latent TGF-β binding protein 1 into CADASIL-related Notch3-ECD deposits. Acta Neuropathol Commun 2014; 2: 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tikka S, Baumann M, Siitonen M, Pasanen P, Pöyhönen M, Myllykangas L, et al. CADASIL and CARASIL. Brain Pathol 2014; 24: 525–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Joutel A, Monet-Lepretre M, Gosele C, Baron-Menguy C, Hammes A, Schmidt S, et al. Cerebrovascular dysfunction and microcirculation rarefaction precede white matter lesions in a mouse genetic model of cerebral ischemic small vessel disease. J Clin Invest 2010; 120: 433–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cognat E, Cleophax S, Domenga-Denier V, Joutel A. Early white matter changes in CADASIL: evidence of segmental intramyelinic oedema in a pre-clinical mouse model. Acta Neuropathol Commun 2014; 2: 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dabertrand F, Krøigaard C, Bonev AD, Cognat E, Dalsgaard T, Domenga-Denier V, et al. Potassium channelopathy-like defect underlies early-stage cerebrovascular dysfunction in a genetic model of small vessel disease. Proc Natl Acad Sci USA 2015; 112: E796–E805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Brew K, Nagase H. The tissue inhibitors of metalloproteinases (TIMPs): an ancient family with structural and functional diversity. Biochim Biophys Acta 2010; 1803: 55–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Khokha R, Murthy A, Weiss A. Metalloproteinases and their natural inhibitors in inflammation and immunity. Nat Rev Immunol 2013; 13: 649–665. [DOI] [PubMed] [Google Scholar]
- 87.Nagase H, Visse R, Murphy G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc Res 2006; 69: 562–573. [DOI] [PubMed] [Google Scholar]
- 88.Koide M, Penar PL, Tranmer BI, Wellman GC. Heparin-binding EGF-like growth factor mediates oxyhemoglobin-induced suppression of voltage-dependent potassium channels in rabbit cerebral artery myocytes. Am J Physiol Heart Circ Physiol 2007; 293: H1750–H1759. [DOI] [PubMed] [Google Scholar]
- 89.Baker AH, Zaltsman AB, George SJ, Newby AC. Divergent effects of tissue inhibitor of metalloproteinase-1, -2, or -3 overexpression on rat vascular smooth muscle cell invasion, proliferation, and death in vitro. TIMP-3 promotes apoptosis. J Clin Invest 1998; 101: 1478–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nishimura DY, Searby CC, Alward WL, Walton D, Craig JE, Mackey DA, et al. A spectrum of FOXC1 mutations suggests gene dosage as a mechanism for developmental defects of the anterior chamber of the eye. Am J Hum Genet 2001; 68: 364–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Aldinger KA, Lehmann OJ, Hudgins L, Chizhikov VV, Bassuk AG, Ades LC, et al. FOXC1 is required for normal cerebellar development and is a major contributor to chromosome 6p25.3 Dandy-Walker malformation. Nat Genet 2009; 41: 1037–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kume T, Deng KY, Winfrey V, Gould DB, Walter MA, Hogan BL. The forkhead/winged helix gene Mf1 is disrupted in the pleiotropic mouse mutation congenital hydrocephalus. Cell 1998; 93: 985–996. [DOI] [PubMed] [Google Scholar]
- 93.Winnier GE, Kume T, Deng K, Rogers R, Bundy J, Raines C, et al. Roles for the winged helix transcription factors MF1 and MFH1 in cardiovascular development revealed by nonallelic noncomplementation of null alleles. Dev Biol 1999; 213: 418–431. [DOI] [PubMed] [Google Scholar]
- 94.Siegenthaler JA, Choe Y, Patterson KP, Hsieh I, Li D, Jaminet S-C, et al. Foxc1 is required by pericytes during fetal brain angiogenesis. Biol Open 2013; 2: 647–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Seo S, Singh HP, Lacal PM, Sasman A, Fatima A, Liu T, et al. Forkhead box transcription factor FoxC1 preserves corneal transparency by regulating vascular growth. Proc Natl Acad Sci USA 2012; 109: 2015–2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ferrara N. Binding to the extracellular matrix and proteolytic processing: two key mechanisms regulating vascular endothelial growth factor action. Mol Biol Cell 2010; 21: 687–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Skarie JM, Link BA. FoxC1 is essential for vascular basement membrane integrity and hyaloid vessel morphogenesis. Invest Ophthalmol Vis Sci 2009; 50: 5026–5034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ogata J, Yamanishi H, Ishibashi-Ueda H. Pathology of cerebral small vessel disease. In: Pantoni L, Gorelick PB. (eds). Cerebral Small Vessel Disease, Cambridge University Press, 2014, pp. 4–15. [Google Scholar]
- 99.Farkas E, de Vos RAI, Donka G, Jansen Steur EN, Mihály A, Luiten PGM. Age-related microvascular degeneration in the human cerebral periventricular white matter. Acta Neuropathol (Berl) 2006; 111: 150–157. [DOI] [PubMed] [Google Scholar]
- 100.Zhang WW, Olsson Y. The angiopathy of subcortical arteriosclerotic encephalopathy (Binswanger’s disease): immunohistochemical studies using markers for components of extracellular matrix, smooth muscle actin and endothelial cells. Acta Neuropathol (Berl) 1997; 93: 219–224. [DOI] [PubMed] [Google Scholar]
- 101.Hajdu MA, Heistad DD, Siems JE, Baumbach GL. Effects of aging on mechanics and composition of cerebral arterioles in rats. Circ Res 1990; 66: 1747–1754. [DOI] [PubMed] [Google Scholar]
- 102.Intengan HD, Schiffrin EL. Structure and mechanical properties of resistance arteries in hypertension: role of adhesion molecules and extracellular matrix determinants. Hypertension 2000; 36: 312–318. [DOI] [PubMed] [Google Scholar]
- 103.Pires PW, Dams Ramos CM, Matin N, Dorrance AM. The effects of hypertension on the cerebral circulation. Am J Physiol Heart Circ Physiol 2013; 304: H1598–H1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Martinez-Lemus LA, Zhao G, Galiñanes EL, Boone M. Inward remodeling of resistance arteries requires reactive oxygen species-dependent activation of matrix metalloproteinases. Am J Physiol Heart Circ Physiol 2011; 300: H2005–H2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lemarié CA, Tharaux P-L, Lehoux S. Extracellular matrix alterations in hypertensive vascular remodeling. J Mol Cell Cardiol 2010; 48: 433–439. [DOI] [PubMed] [Google Scholar]
- 106.Izzard AS, Graham D, Burnham MP, Heerkens EH, Dominiczak AF, Heagerty AM. Myogenic and structural properties of cerebral arteries from the stroke-prone spontaneously hypertensive rat. Am J Physiol Heart Circ Physiol 2003; 285: H1489–H1494. [DOI] [PubMed] [Google Scholar]
- 107.Baumbach GL, Walmsley JG, Hart MN. Composition and mechanics of cerebral arterioles in hypertensive rats. Am J Pathol 1988; 133: 464–471. [PMC free article] [PubMed] [Google Scholar]
- 108.Iadecola C, Davisson RL. Hypertension and cerebrovascular dysfunction. Cell Metab 2008; 7: 476–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Nishimura N, Schaffer CB, Friedman B, Lyden PD, Kleinfeld D. Penetrating arterioles are a bottleneck in the perfusion of neocortex. Proc Natl Acad Sci USA 2007; 104: 365–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.