

Abstract
Recombinant Immunotoxins (RITs) are chimeric proteins designed to treat cancer. They are made up of an Fv or Fab that targets an antigen on a cancer cell fused to a 38 kDa portion of Pseudomonas exotoxin A (PE38). Because PE38 is a bacterial protein, it is highly immunogenic in patients with solid tumors that have normal immune systems, but much less immunogenic in patients with hematologic malignancies where the immune system is suppressed. RITs have shown efficacy in refractory hairy cell leukemia and in some children with acute lymphoblastic leukemia, but have been much less effective in solid tumors, because neutralizing antibodies develop and prevent additional treatment cycles. In this paper we will 1) review data from clinical trials describing the immunogenicity of PE38 in different patient populations, 2) review results form clinical trials using different immunosuppressive drugs and 3) describe our efforts to make new less-immunogenic RITs by identifying and removing T and B cell epitopes to hide the RIT from the immune system.
Keywords: anti-drug antibodies, neutralizing antibodies, immunotherapy, mesothelin, mesothelioma
Introduction
Protein based therapeutics have great potential to treat human diseases, but if the protein is not human, an immune response (also known as anti-therapeutic protein antibody response) can develop during treatment and neutralize the clinical effect of the agent (1). Furthermore, formation of neutralizing antibodies (Nab) against a foreign protein can be associated with serious adverse events including infusion reactions, allergic reactions, anaphylaxis, delayed hypersensitivity and autoimmunity. In addition auto-immunity to recombinant megakaryocyte growth and development factor (MGDF) and erythropoietin (EPO) (epoetin alfa), has produced antibodies to the native protein resulting in in thrombocytopenia and red cell aplasia (2, 3). Patients who develop antibodies are at higher risk of infusion-related reactions and delayed hypersensitivity, mediated by immune complexes that are deposited in tissues (1, 4).
Recombinant immunotoxins (RIT) are chimeric proteins that have shown activity in treatment of several types of cancer. They consist of a targeting portion linked to a toxin. The targeting portion is antibody derived, most commonly the Fv portion of an antibody targeting a specific antigen on tumor cells or infected cells (5). The toxin can be of plant origin or of bacterial origin like Diphtheria toxin or Pseudomonas exotoxin A (PE). RITs are now used for the treatment of several kinds of cancer and could be used for other indications (Fig. 1) (6, 7). PE is a favorable toxin for construction of RITs, because its high cell killing activity is well documented (5), its mechanism of action is well understood and it can endure many mutations without harming its activity [reviewed (8)]. PE38 kills cells by ADP-ribosylating and inactivating EF2, which is different from the mechanism by which the majority of anti-cancer agents work. For that reason, it can be combined with other chemotherapeutic agents that have different mechanisms of action and no dose reduction of either agent is required because their toxicities do not overlap (9).
Figure 1. Structural models of RITs.

The RIT SS1P consists of the disulfide-stabilized VH and VL polypeptide chains of the Fv from the antimesothelin monoclonal antibody SS1 coupled to a 38-kDa fragment of PE38. (A) SS1P. The Fv (cyan and magenta) is recombinantly connected to PE38, which is divided into domain II (gray), domain III (yellow), and part of domain Ib from native PE38. (B) SS1-LR-GGS. Deletion of domain II with GGS linker between the linker and the domain III. (C) SS1-LO10R. PE24 with six point mutations in domain III designed to eliminate binding to B cell receptor. (D) LMB-T20. PE24 with six point mutations in domain III designed to diminish T cell epitopes. All models are hypothetical arrangements based on the structures of native PE and immunoglobulin G; they do not represent actual structure determinations
While the efficacy of PE38 based RITs in tumor regression in hematological malignancies is well documented (10–12), RITs have not been as successful in the treatment of solid tumors. RITs contain a 38-kDa fragment of a bacterial toxin, which is very immunogenic in humans with normal immune systems (13–15). Over the past 15 years, much effort has been devoted to reduce the immunogenicity of these RITs. These approaches include treating patients with immunosuppressive drugs and modifying the toxin to hide it from various components of the immune system. This review will focus on the immunogenicity of PE based RITs; other RITs have been reviewed elsewhere (16).
Clinical data of PE38 based RITs
Many of the clinical trials for PE based immunotoxins used similar treatment schedules, similar ADA assays and uniform cutoffs and protocols for Nab assays. Thus it is possible to compare the results of the clinical trials in different patient populations.
The first PE-based immunotoxins that was evaluated in a clinical trial was OVB3-PE. It contained a murine antibody that targets an unknown antigen on ovarian cancer cells attached to the entire PE protein (13). OVB3-PE was administered to 23 patients and had a high level of non-specific toxicity. The immunogenicity of the RIT was evaluated by ELISA and showed that 100% of the patients that were evaluated developed antibodies against the toxin 14 days after therapy was initiated (Table 1). Human anti-mouse antibodies (HAMA) were also detected in 12/16 patients 28 days after therapy.
Table 1.
Clinical trials for PE based immunotoxins
| RIT name | Target antigen | Toxin | Indication | Immunogenicity rate after cycle 1 | Overall immunogenicity rate | Assay | Ref |
|---|---|---|---|---|---|---|---|
| Hematological malignancies | |||||||
| LMB2 | CD25 | PE38 | Leukemia, lymphoma Pediatric ALL and non–Hodgkin lymphoma |
6/35 2/23 |
10/35 3/23 |
N N |
(17) (11) |
| RFB4(dsFv)-PE38 (BL22/CAT3888) | CD22 | PE38 | Chemotherapy-resistant HCL NHL, CLL, HCL, Relapsed/refractory HCL |
1/16 ND 2/36 |
4/16 11/46 4/36 |
N N N |
(6) (26) (10) |
| Moxetumomab pasudotox(CAT-8015 or HA22) | CD22 | HCL | 1/28 | 10/28 | N and E | (19) | |
| Solid malignancies | |||||||
| LMB2 | CD25 | PE38 | Metastatic melanoma | ND | 7/8 | E | (22) |
| OVB3-PE | Ovarian antigen | PE | Refractory ovarian cancer | 12/12 | 12/12 | E | (13) |
| ERB-38 | erbB2 | PE38 | Breast cancer Cutaneous lesions of metastatic breast and colorectal cancers |
1/5 3/3 |
1/5 3/3 |
N N |
(85) (25) |
| ScFv(FRP5)-ETA | erbB2 | PE40 | Metastatic breast cancers, prostate cancers, head and neck cancer, non small cell lung cancer | 13/13 5/13 |
13/13 5/13 |
E N |
(86) |
| LMB-1 SGN-10 (BR96 sFv-PE40) | Lewis Y Lewis Y |
PE38 PE40 |
Epithelial tumors Metastatic carcinoma GBM |
33/38 20/29 ND |
33/38 ND 11/28 |
N E E |
(14) (87) (24) |
| NBI-3001 | IL4 receptor | PE38KDEL | Renal cell and non-small cell lung carcinoma | 5/11 | 11/11 | E | (88) |
| VB4-845 | EpCAM | PE40 | Carcinoma Mesothelioma, epithelial ovarian cancer, pancreatic adenocarcinoma |
27/63 30/34 18/24 |
47/61 34/34 ND |
E N N |
(23) (89) (28) |
| SS1P | Mesothelin | PE38 | Pleural mesothelioma | 19/21 | 21/21 | N | (27) |
Method to determine immunogenicity:
N = Neutralization assay
E = ELISA
ND = Not described
The results from a clinical trial evaluating the activity of LMB-1 were reported in 1996. In LMB-1, domain I is replaced by the Fv portion of an antibody to Lewis-Y. This was the first publication reporting anti-tumor activity of an immunotoxin targeting an epithelial tumor (14). In this trial, 33/39 of the patients developed neutralizing antibodies against LMB-1 three weeks after the first cycle of treatment (Table 1). The remaining 10% who did not make neutralizing antibodies after the first cycle were retreated and made antibody after the second or fourth cycle. ELISA assays indicated that eventually, 100% of the 38 patients made antibodies against the toxin moiety and 33/38 of the patients had HAMA against the Fv antibody fragment.
Unlike LMB-1, which was chemically fused to the toxin, LMB2 is a recombinant immunotoxin in which an Fv is directly fused to PE38. Results of trials with LMB-2 that targets CD25 were first reported in 2000 (17). LMB-2 was used to treat leukemia and lymphoma patients. Clinical evaluation showed several complete and partial responses; however, 10/35 of the patients developed Nabs, which prevented further treatment. Six of the patients developed Nabs after the first cycle of treatment (Table 1). Three of the patients that had Nabs also demonstrated immunogenicity related side effects including one anaphylactic reaction (1/35) and other allergic reactions (grade 2–3) (2/35) (17). These adverse events provided a justification to avoid further dosing once Nabs developed. However this was the only documented evidence of immunogenicity related side effects. Such reactions were not associated with any other RIT trial, despite antibody formation.
Moxetumomab pasudotox (also known as CAT-8015 or HA22) is a PE38-based immunotoxin that is currently being evaluated in phase III trials. It targets CD22 and has produced complete remissions in many patients with refractory hematological malignancies (11, 18). Part of its high efficacy, compared to patients with solid tumors, is attributed to a low rate of immunogenicity; only 1/28 hairy cell leukemia (HCL) patients made Nabs after the first treatment cycle and a total of 10/28 developed Nabs during the entire trial (19) (Table 1). This low rate of immunogenicity is attributed to the immune status of the patients. Patients with HCL have usually been treated with Cladribine, which kills many normal immune cells in the bone marrow. In addition the leukemia cells infiltrate the marrow causing more immunosuppression. Figure 2A shows that Nab formation is delayed in patients with HCL after treatment with BL22 or Moxetumomab, and is even more suppressed in patients with acute lymphoblastic leukemia (ALL) and chronic lymphocytic leukemia (CLL), who have had many cycles of immunosuppressive chemotherapy.
Figure 2. Neutralizing antibody formation for PE38 RITs.

Summary of immunogenicity rate during treatment cycles from nine clinical trials. Immunogenicity rate was evaluated using functional Nab assay with a cut point of 75% neutralization in all trials (17, 19, 22, 27, 28, 38) and unpublished data. (A) HCL (n=146), ALL (n=49) and CLL patients (n=50) treated with Moxetumomab paseudotox or BL22. (B) Patients treated with SS1P by bolus (QOD x 3) (n=21) or continues infusion (n=24) over the course of 10 days. (C) Immunogenicity of LMB-2 in hematological (n=35) and melanoma patients (n=7). (D) Immunogenicity in patients treated with SS1P as a monotherapy (n=21) or in SS1P combined with Pentostatin and Cyclophosphamide (n=11). All patients (with the exaptation of continues infusion group) were treated in a similar schedule of 3x QOD bolus per cycle, and 21-day intervals between cycles.
In clinical trials in patients with solid tumors who have normal immune function, antibodies usually appear quite rapidly. It has been suggested that continuous treatment with an immunogenic protein may induce tolerance to the therapeutic protein (20). However the mode of administration of SS1P did not affect its immunogenicity. Patients treated with continuous infusion of SS1P for 10 days had a moderate decrease of 13% in the rate of Nabs after the first cycle compared with bolus infusions (QOD X 3) (Fig. 2B). Similarly, clinical trials with other RITs (containing plant based toxin) did not find any significant difference in immunogenicity between continuous and bolus treatment (21).
Interestingly, while the hematological patients treated with LMB-2 had a relatively low rate of immunogenicity onset, LMB-2 has also been given to patients with melanoma with normal immune function to try and kill suppressor T cells. The data in Figure 2C shows that LMB2 is very immunogenic in the melanoma patients with 92% of patients making antibodies after the first treatment cycle (22) and much less immunogenic in the immune-suppressed leukemia patients with 17% immunogenicity after the first cycle. This high rate of immunogenicity in solid tumors occurs even though LMB-2 is able to kill many CD25 positive cells and those cells are required for the immune response. The higher rate of immunogenicity observed in patients with solid tumors exemplifies the multi factorial basis of the immunogenicity response.
Effect of site of injection on immunogenicity
RITs are almost always administered intravenously (I.V.) to reach all tumor cells in the body. However, in clinical trials where the immunotoxin was injected directly into the tumor site, decreased immunogenicity was observed. VB4-845 that targets EpCA was administered with a catheter into the bladder, and had a significantly diminished rate of immunogenicity (27/63 after the first cycle) (23) (Table 1). Also, NBI-3001 (IL4-PE38) that was administered into brain tumors by slow infusion over several days had a low rate of immunogenicity (overall 11/28 patients made ADA throughout the study) (24). On the other hand, ScFv (FRP5)-ETA targeting erbB2 was injected directly to cutaneous lesions of metastatic breast and colorectal tumors and caused immunogenicity in 3/3 patients that were examined. Interestingly, despite the presence of Nabs during the second treatment, the therapeutic effect of the RIT at the local site was still observed (25). These results indicate that when the tumor is compartmentalized as in the case of bladder and brain, there is decreased immunogenicity, while injecting cutaneous tumors that have many immune cells in their surroundings, will result in increased immunogenicity.
Side effects related to immunogenicity
Out of the hundreds of patients treated with PE38 based RITs only a single anaphylactic reaction was reported and occurred immediately after the first infusion of the immunotoxin (17). Several of the trials reported grade 1, 2 or 3 allergic reactions and were associated with skin rashes that were easily managed by steroids (17, 26–28).
In a few of the trials, the patients were treated with one additional cycle after Nabs developed. This was usually due to logistical reasons because neutralizing antibody data was not available before scheduled retreatment. Even though many of these patients were treated while already having Nab, no major infusion related toxicities were observed during the second cycle. As reported above, anaphylactic and allergic reactions were more common immediately after the first treatment and less common after the second. This suggests that the allergic response was a result of pre-existing immunity from previous environmental exposure.
Mesothelin
Mesothelin is a GPI linked cell surface protein that was discovered and its gene cloned in our laboratory in a search for a new therapeutic target on ovarian cancers (29, 30). Mesothelin is only expressed on normal mesothelial cells, but is highly expressed on many malignancies including ovarian cancers, mesothelioma and cancers of the pancreas, stomach, lung and bile duct (9, 31–33). This makes it an excellent target for antibody-based therapies. SS1P is a RIT that targets mesothelin and is the focus of many of the deimmunization efforts that are described below.
Formation of the immunogenicity response
The central event in the formation of both humoral and cell mediated immune responses is activation of lymphocytes. Antibody responses can be defined as T cell dependent responses and T cell independent responses. The latter is most commonly a response to a unique antigenic repetitive sequence. This kind of antibody responses is weaker, faster and mostly composed of IgM antibodies (as opposed to IgG antibodies in T cell dependent responses); also there is little immunological memory (34). Due to the nature of the immunogenicity response observed in patients that were treated with RITs, which includes the finding of highly specific neutralizing IgG, a long lasting memory response and also the longer duration required to mount the response, we conclude that the immune response against PE38 based RITs is T cell dependent.
Figure 3 shows a cartoon summarizing some of the cells (APCs, T cells, B cells, and antibody secreting plasma cells) involved in the immune response to the PE38 region of RITs like SS1P. The primary T cell dependent immune response starts with engulfment of the RIT by an APC in the periphery. The APC then moves to peripheral immune center (lymph nodes). Inside the APC, the RIT is processed in the endosomal compartments to peptides and some of these peptides bind to HLA II molecules. In the lymph nodes, naïve CD4+ T cells are exposed to the presentation of PE38 derived peptides that are presented by HLA class II and the few naïve T cells that have T cell receptors that can recognized these peptides will proliferate and differentiate to effector TH2 cells (Fig. 3, step 1).
Figure 3. Antibody mediated response to protein immunogen.

(Step 1) The RIT is engulfed by an APC and inside the APC, the RIT is processed in endosomal compartments and some high affinity peptides bind to HLA II molecules. The peptides- HLA class II complex is transported to the membrane of the APC and is presented. A specific T cell receptor recognizes the complex and interacts with the APC via co stimulatory signals resulting in T cell activation and differentiation. (Step 2) At the same time, B cells that have a B cell receptor that can recognize conformational epitopes on PE38 encounters PE38. Crosslinking of the B cell receptor initiates an activation cascade of the B cell and homing to the lymph node. The activated B cell also processes whole antigen and presents PE38 derived peptides. Once a cognate B and TH cell meet co-activation will occur. TH2 cells start secreting cytokines that help the B cell to activate. (Step 3) Activated B cell start affinity maturation and (Step 4) class switching and evolve to a memory or a secreting plasma cell.
At the same time, B cells that have a B cell receptor that can recognize conformational epitopes on the surface of PE38 encounter PE38 in the periphery or lymph node. Crosslinking of the B cell receptor activates the B cell and also causes it to home to the lymph node. The activated B cells, like APCs, process the RIT and present PE38 derived peptides to T cell. The B cell moves to the T cell-zone in the lymph node, which improves the chances of finding a cognate TH2 cell. Once a cognate B and TH cell meet, co-activation will occur (Fig. 3, step 2). TH2 cells start secreting cytokines that help activate the B cell and, promote affinity maturation and class switching (Fig. 3, step 3). As a consequence, the B cell evolves to a memory cell or an antibody secreting plasma cell (Fig. 3, step 4). Hence, maturation of an antibody secreting B cell is almost always dependent on the presence of an activated TH cell (35). The activation of B and T cell lymphocytes is complex and involves numerous additional steps that are reviewed elsewhere (36). A secondary (“recall” response) is a faster and stronger immune response than primary activation and is composed of memory B and T cells. This response does not require the first step of APC presentation (Fig. 3, step 1).
Efforts to reduce immunogenicity of RITs
The most striking observation in the clinical trials described above is that 2–5 cycles of treatment is required to obtain major clinical response including complete remissions (37). HCL patients that were treated with Moxetumomab pasudotox and made Nabs demonstrated a correlation between how early the patients made antibodies and the outcome of the treatment. In other words, many patients that made antibodies during the first, second or third cycle of treatment only obtained partial responses while patients that did not make Nabs until later stages and could receive more cycles, had a higher rate of complete responses and better outcomes.
Furthermore, when SS1P was combined with Pentostatin and Cyclophosphamide to kill T and B cells and suppress anti-drug antibodies, more treatment cycles could be given to most of the patients and major tumor responses were observed in several patients with advanced refractory mesothelioma (38). These findings indicate that producing less immunogenic immunotoxins would be of great clinical value.
Combination of RITs with immune suppression therapy
LMB-1 is a RIT that includes a PE38 toxin that is conjugated to a murine antibody that targets Lewis Y antigen. In 2004 we hypothesized that elimination of B cells by rituximab, would diminish the development of human antibodies to LMB-1. Five patients were treated with rituximab followed by LMB-1. The development of human antibodies against LMB-1 was detected using a serum neutralization and ELISA assay. We were surprised to find that while the circulating CD20/CD19+ B cells were reduced by 99.9%, all of the patients (5/5) developed neutralizing antibodies to the immunotoxin by day 21 of drug administration (39).
Based on the Rituximab data, it was clear that elimination of peripheral B cells is not sufficient and addition of an agent that will eliminate T cell could be of beneficial. To that end, we evaluated treatment of 10 refractory mesothelioma patients with combination therapy with Pentostatin and Cyclophosphamide. We found that this combination delayed the formation of neutralizing antibodies to SS1P. Only 2/10 patients made Nabs after the first cycle, and 6/10 after the second cycle (Fig. 2D). One patient did not make Nabs throughout the entire treatment that included 6 cycles. This patient had a remarkable response to the treatment with 74% tumor shrinkage that was maintained for more than 15 months (38). The toxicity observed in the trial described above was moderately severe and expected from known side effects of Pentostatin and Cyclophosphamide.
Similarly, LMB-2 has been evaluated in combination with Fludarabine and Cyclophosphamide (FC) in patients with adult T-cell leukemia (ATL). FC was chosen, because it is widely used in transplant patients to prevent GVHD and it also helped in preventing tumor regrowth after response to LMB-2. This combination was found to be very effective in prevention of Nabs formation; none of the patients had neutralizing antibodies after the first, second or third cycle. One patient had Nabs after the fourth cycle, two patients after the fifth cycle and one more after the seventh cycle. The total immunogenicity rate in the trial was 29% (4/17) (40). Similar to the Pentostatin and Cyclophosphamide trial, the dose-limiting toxicity was attributed to the moderately severe side effects of the chemotherapy. For these reasons alternative immune modulating combinations with RITs are being explored.
In studies on mice or dogs, Siegall et al. showed that combination of CTLA4-IG that is used for immune suppression of a PE40 containing RIT could lead block antibody formation against the RIT in mice and dogs and also prevented hypersensitivity reactions in dogs. This combination enabled additional immunotoxin cycles, which resulted in enhanced antitumor activity against colon carcinoma in syngeneic rat models (41). Similarly, Onda et al. showed monotherapy of mice with tofacitinib suppressed antibody responses to an immunotoxin. Mechanistic investigation revealed that tofacitinib treatment led to lower numbers of CD127+ pro-B cells, fewer germinal center B cells and impaired formation of germinal centers (42). Because normal Ig levels were still present during tofacitinib treatment, we conclude this agent specifically reduced ADAs.
Recent experiments in mice have focused on elimination of pre-existing antibodies and plasma cells, which would allow treatment of patients that have pre-existing antibodies from environmental exposure to Pseudomonas exotoxin and also allow repeated dosing. Bortezomib is a proteasome inhibitor that targets both short- and long-lived plasma cells as a result of their high rates of Ig production (43). We found that the combination of SS1P with Bortezomib reduced ADA formation by 50% compared to SS1P with no immune suppression. Addition of Pentostatin and Cyclophosphamide to Bortezomib reduced the ADA by 88% (44).
Modification in PE38 to reduce immunogenicity
Several strategies have been pursued to modify the structure of RITs to overcome their immunogenicity. Some studies have also addressed the immunogenicity of the murine antibody, while most have concentrated on reducing the immunogenicity of the toxin itself.
Framework exchange and humanization of antibody fragment
Framework exchange and humanization focused on the antibody portion of a RIT B3(Fv)-PE38 targeted against Lewis-Y (45). The variable domains of the heavy (VH) and light (VL) chains of the murine B3 Fv were aligned with their best human homolog to identify framework residues that differ. Six residues were humanized and a new RIT was produced and tested for immunogenicity. Binding assays of monkey’s serum that contained anti-RIT antibodies to the humanized immunotoxin showed that the humanized RIT lost some of its epitopes (45). Currently the new generation of RITs contains a humanized Fab or Fv (31, 46). However, the majority of the ADA that have been found against RITs react with the bacterial toxin moiety (47) and humanization of the bacterial toxin is an important and challenging task.
PEGylation
Covalent attachment of Polyethylene glycol (PEG) to therapeutic proteins has been found to be useful in “masking” the immunogenic conformational epitopes in the protein from the host’s immune system. It also increases its hydrodynamic size, which prolongs its circulation time by reducing renal clearance (48). PEGylation of RIT was performed in the 1990s (49–51); however PEGylation has not been successful.
Identification of B cell epitopes
B cell epitope are usually located at a small number of distinct sites on the surface of a protein. Roscoe et al. identified B cell epitopes in primates and human patients by exposing overlapping synthetic peptides of the RIT to serum from monkeys or patients that were treated with RIT (52, 53). However, this approach could not locate discontinuous conformational B cell epitopes, which are the major neutralizing epitopes. Therefore, Onda et al. set out to identify the conformational murine B cell epitopes in a PE38-containing RIT (54). They immunized mice and isolated monoclonal antibodies that reacted with conformational epitopes on PE38, and used those antibodies to determine the number of epitopes on PE38. They found that PE38 contains seven major conformational epitopes located in specific positions on the protein and not distributed over the entire surface of PE38. The finding that the epitopes are clustered enabled them to determine the precise location of most of the epitopes by mutating amino acids, which have large side chains to alanine or glycine and showing that specific antibody binding to the selected epitope was abolished or greatly reduced. They used this information to construct an immunotoxin that contained mutations that greatly reduced immunogenicity when examined in 3 strains of mice. Furthermore the mutated RIT (8M) retained excellent cytotoxic and anti-tumor activity. These experiments constitute proof that removing B cell epitopes can greatly diminish immunogenicity. However because there are differences between mouse and human immune systems, human B cell epitopes needed to be identified (54, 55).
Deletion of domain II of PE38
Concurrent with research identifying and eliminating B cell epitopes in PE38, experiments were also carried out to investigate the susceptibility of RITs to lysosomal protease digestion. We found that domain II of PE38 was very protease sensitive and that almost all of domain II could be removed without loss of activity as long as the furin cleavage site (amino acids 274–284) was retained. This strategy had the additional benefit of removing immunogenic B and T cell epitopes along with protease cleavage sites (56). Protease resistance reduces the protein processing in the endosome and late endosome, and reduces the number of peptides that can be presented on MHC II molecules to activate T cells. The resulting mutant (designated LR for lysosome protease resistance) (Fig. 1B) was tested in three strains of mice and showed a greatly decreased antibody response (57).
Identification and modification of human B cell epitopes
To identify human B cell epitopes and thereby reduce the immunogenicity of RITs in humans, Liu et al isolated and analyzed anti-immunotoxin antibodies from patients treated with immunotoxins SS1P and HA22. They used M13 phage display to express Fvs isolated from B cells of patients with anti-PE38 antibodies (58), Biotinylated-RIT was used as the antigen for selection of phage expressing Fvs that bound to PE38. They identified the residues that make up the B cell-epitopes by measuring their binding to variant RITs with point mutations that removed large bulky amino acids like arginine. Then they constructed a variant RIT with a deletion of domain II and seven point mutations in domain III that modified human B cell epitopes (HA22-LO10) (Fig. 1b). This RIT had significantly reduced reactivity with human antisera (Fig. 4) and yet retained good cytotoxic and antitumor activity (59). To increase the size and decrease immunogenicity of HA22-LR-LO10, we replaced the mouse Fv with a humanized Fab, which decreased the immunogenicity as well as increased the half-life in circulation by increasing the size of the RIT to 72kDa (46).
Figure 4. Antigenicity of SS1P and SS1-LO10 to human antisera.
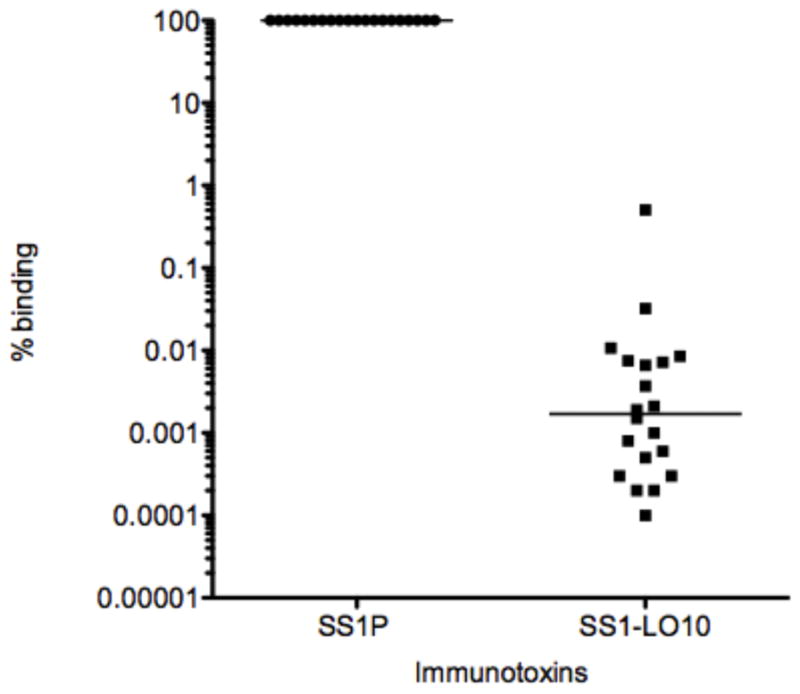
Ratio of binding of SS1-LO10 to binding of SS1P from 20 patients treated with SS1P. p<0.0001, Wilcoxon signed rank test.
RG7787
In a recent collaboration with Roche (Basel, Switzerland, http://www.roche.com), we used this approach to develop a new immunotoxin against mesothelin named RG7787. This deimmunized RIT has a humanized Fab version of the SS1 antibody attached to the B cell epitope deficient toxin, similar to HA22-LR-LO10 described above (60). To enhance the decreased activity of SS1-LO10, they introduced R456A to replace R458A, because R458A was the main mutation responsible for the low activity. Because both amino acids R456 and R458 play a role in the immunogenicity of epitope H3, replacement of R458A with R456A in RG7787 still maintained low antigenicity, however the cytotoxic activity of RG7787 was significantly improved (9). RG7787 has shown excellent anti-tumor activity in animal models (9, 31) Clinical testing for RG7787 started late in 2014 and immunogenicity results should be available in early 2016.
Identification of T cell epitopes
In 2009, elimination of T cell epitopes began to be a well-accepted strategy to deimmunize protein therapeutics (61–67). Yeung et al. showed that elimination of a T cell epitopes in the protein IFNβ eliminated the ADA response in BALB/c mice (68). Most previous studies in this field used silico algorithms to identify T cell epitopes or at least narrow the epitope candidates; however, there was little experimental evidence that this approach was useful (69). Therefore, we decided to use an experimental approach to identify T cell epitopes, based on the work from the laboratory of A. Sette (69). For our studies we used PBMCs from 50 naïve donors that represent the HLA distribution of the typical patient population in the western world. The PBMCs were cultured with intact immunotoxin to allow its uptake and processing into peptides and stimulation of T cells. After in vitro expansion, T cell epitopes were identified by re-stimulation of the specifically expanded cells with 15-mer peptides spanning the sequence of PE38 (70). T cell activation was detected using IL-2 ELISpot (71). We used IL-2 because it supports T-cell activation, differentiation, and memory and is a less specialized cytokine than IL-4 or IFN-γ (72). We identified eight epitopes in PE38 (73). One epitope in domain II was immuno-dominant and extremely promiscuous. It was present in 21/50 donors (71). The presence of all the epitopes was confirmed using samples from 16 cancer patients previously treated with PE38 containing RITs and who had mounted an immune response to the protein. In addition no new epitopes were detected after immunization, although the number of epitopes detected in any single patient was higher than the number detected in naïve donors (73). These results strongly indicate that PE38 has eight T cell epitopes and other regions of the protein are immunologically silent. Interestingly, T cell epitope mapping of PBMCs from immunized HCL patients showed some epitopes are missing (73), perhaps due to the chemotherapy these patients had received. Further work will address the difference in response of HCL patients.
To eliminate the epitopes in domain II, we exploited the deletion in SS1P-LR-GGS and introduced point mutations in domain III to eliminate the epitopes in domain III. The point mutations were determined by alanine scanning mutagenesis. Once an alanine variant that eliminated T cell activation was identified, we constructed the mutant RIT and tested its cytotoxic activity. We also stimulated PBMC with the mutant RIT and re stimulated with the mutant peptides to be sure that the point mutation did not introduce a new T cell epitope. Two of the epitopes (epitope 2 and epitope 6) were difficult to solve by alanine scanning, because the mutant constructs lost cytotoxic activity or did not eliminate the epitopes. To solve epitope 6, we collaborated with the Baker lab that uses Rosetta computational protein design methods combined with an HLA binding algorithm to identify point mutations that disrupt binding to HLA II molecules but still maintain cytotoxic activity (74). Epitope 2 was not easily resolved. We found that a combination of two point mutations in amino acids R494 and R505 were able to diminish the T cell responses significantly. Unfortunately, the cytotoxic activity was reduced two-fold by one of these mutations.
T cell deimmunized RITs
The point mutations described above were combined into two new immunotoxins that have their T cell epitopes removed or suppressed. LMB-T18 targets CD22 (73) and LMB-T20, targets mesothelin (75). Each contains the same mutated toxin which is shown in Figure 1D. T cell epitope mapping of LMB-T20 showed that peptides that did not induce responses after stimulation with SS1P also did not generate responses after stimulation with LMB-T20 (Fig. 5) indicating that cryptic or new epitopes did not emerge as a result of altered antigen processing in LMB-T20. Compared to the parental immunotoxin SS1P, LMB-T20 has a 81%% decrease in immunogenicity (p>0.0001 in 2-way ANOVA) (as assessed by T cell activation analysis) and is, in most cases, much more cytotoxic than SS1P on cancer cell lines expressing mesothelin as well as mesothelioma cells recently isolated from patients (75). LMB-T20 also promoted complete regressions in tumor-bearing mice. We believe LMB-T20 is an excellent candidate for further clinical development as a RIT with low immunogenicity, because the removal of T cell epitopes is now thought to be more effective than the removal of B cell epitopes.
Figure 5. T cell activation of SS1P and LMB-T20.

Stimulation of PBMC from 10 donors with LMB-T20 and SS1P show a decrease in T cell activation. PBMC from 10 naïve donors were stimulated with either SS1P or LMB-T20. After 14 days of in vitro expansion cells that were re-stimulated with either 111 peptides spanning the sequence of PE38 or 76 peptides spanning the sequence of LMB-T20. T cell activation was detected using IL-2 ELISpot. All positive peptides were assayed twice for each PBMC sample and each assay was run in triplicate. Response strength is shown in the Spot Forming cell/million cells (SFC/1E6) ladder on the right. Black stars represent peptides that were deleted in LMB-T20 and red stars represent peptides that are different from wild type. This figure to be published in Mazor et al., Mol. Cancer Res., In press (75).
Elimination of T cell epitopes in BALB/c mice
To determine the effect of T cell epitope removal in an animal with an intact immune system, we mapped the T cell epitopes of PE38 in immune competent BALB/c mice and found that these mice recognize two epitopes in PE38. One corresponds to the human immunodominant T cell epitope and the other to a subdominant epitope; both are eliminated in LMB-T20. We immunized mice with LMB-T20 and did not observe T cell activation or the development of anti-drug antibodies as detected by both ELISA and drug neutralizing assays (76). This result confirms the validity of our efforts to lower immunogenicity by T cell epitope removal.
Comparison of experimental and in silico predicted epitopes in PE38
HLA binding algorithms are frequently used to predict T cell epitopes. We have computed the binding of PE38 peptides using HLA binding prediction algorithms and compared the predicted and experimentally identified T-cell epitopes. We defined an epitope as one to four continues positive peptides. A total of 9 epitopes were identified based on the new criteria. We found that the prediction for individual donors did not correlate well with the experimental data. Furthermore, prediction of T-cell epitopes in an HLA heterogenic population revealed that the two strongest epitopes were predicted at multiple cutoffs, however, the third epitope was not found at any cutoff and overall 4/9 epitopes were missed at several cutoffs (77). We conclude that MHC class-II binding predictions are not yet sufficient to accurately predict the T-cell epitopes in PE38 and should be supplemented by experimental work. We are currently working to understand why the algorithm missed epitope 3 and several other epitopes.
B and T shared epitopes
In the process of identifying point mutations that eliminate T cell epitopes, we found that two of these were the same mutations that had deleted B cell epitopes; these are R427A and R505A (Fig. 1C and 1D). This means that not only the T and B cell epitopes are in common but also that a common single point mutation can eliminate both epitopes. R505A and R427A have very high ASAs (150Å and 142Å, respectively) indicating those arginine amino acids are located on the surface of the molecule. Since B cell epitopes are known to contain bulky hydrophilic amino acid like arginine (78–80) it is not surprising that mutations that diminish T cell epitopes, also diminish B cell epitopes. Reports have shown that important epitopes may be shared by B and T cells (81–83) and it has been suggested that there is a functional link between B and T cells (84).
Conclusions and future directions
The process of deimmunizing PE38 by the introduction of multiple mutations to interfere with either B or T cell epitopes involves a difficult balancing act between the reduction of immunogenicity and the integrity and potency of toxin activity. In some cases, like the deletion of domain II, the activity was increased in most cell lines. In other cases, some of the mutations caused a reduction in cytotoxic activity and could not be incorporated in the final deimmunized RIT. For that reason, both B cell and T cell deimmunized RITs represent a compromise between the best mutation for immunogenicity and the best mutation for potency. This means that the immunogenicity is not completely resolved. To overcome this compromise, it would be useful to design a new RIT that has both its B cell and T cell epitopes diminished. In this way, most of the steps during the immune response (Fig. 3) would be interrupted. We believe that our new immunotoxins with B or T cell epitopes removed will be effective in cancer treatment.
Acknowledgments
We thank Dr. B. K. Sathyanarayana for editing Figure 1 and to Dr. Alan Hoofring for editing Figure 3. We thank Dr. Robert J. Kreitman for providing unpublished immunogenicity data that is shown in Figure 2A.
This research was supported in part by the Intramural Research Program of NIH, National Cancer Institute, Center for Cancer Research and a Cooperative Research and Development Agreement (#2791) with Hoffman-LaRoche Inc./F. Hoffman-LaRoche Ltd
References
- 1.Brennan FR, et al. Safety and immunotoxicity assessment of immunomodulatory monoclonal antibodies. mAbs. 2010;2:233–255. doi: 10.4161/mabs.2.3.11782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li J, et al. Thrombocytopenia caused by the development of antibodies to thrombopoietin. Blood. 2001;98:3241–3248. doi: 10.1182/blood.v98.12.3241. [DOI] [PubMed] [Google Scholar]
- 3.Casadevall N, et al. Pure red-cell aplasia and antierythropoietin antibodies in patients treated with recombinant erythropoietin. N Engl J Med. 2002;346:469–475. doi: 10.1056/NEJMoa011931. [DOI] [PubMed] [Google Scholar]
- 4.Jahn EM, Schneider CK. How to systematically evaluate immunogenicity of therapeutic proteins - regulatory considerations. N Biotechnol. 2009;25:280–286. doi: 10.1016/j.nbt.2009.03.012. [DOI] [PubMed] [Google Scholar]
- 5.Wilkins DK, Mayer A. Development of antibodies for cancer therapy. Expert Opin Biol Ther. 2006;6:787–796. doi: 10.1517/14712598.6.8.787. [DOI] [PubMed] [Google Scholar]
- 6.Kreitman RJ, et al. Efficacy of the anti-CD22 recombinant immunotoxin BL22 in chemotherapy-resistant hairy-cell leukemia. N Engl J Med. 2001;345:241–247. doi: 10.1056/NEJM200107263450402. [DOI] [PubMed] [Google Scholar]
- 7.Kennedy PE, et al. Anti-HIV-1 immunotoxin 3B3(Fv)-PE38: enhanced potency against clinical isolates in human PBMCs and macrophages, and negligible hepatotoxicity in macaques. J Leukoc Biol. 2006;80:1175–1182. doi: 10.1189/jlb.0306139. [DOI] [PubMed] [Google Scholar]
- 8.Weldon JE, Pastan I. A guide to taming a toxin--recombinant immunotoxins constructed from Pseudomonas exotoxin A for the treatment of cancer. FEBS J. 2011;278:4683–4700. doi: 10.1111/j.1742-4658.2011.08182.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hollevoet K, Mason-Osann E, Liu XF, Imhof-Jung S, Niederfellner G, Pastan I. In vitro and in vivo activity of the low-immunogenic antimesothelin immunotoxin RG7787 in pancreatic cancer. Mol Cancer Ther. 2014;13:2040–2049. doi: 10.1158/1535-7163.MCT-14-0089-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kreitman RJ, et al. Phase II trial of recombinant immunotoxin RFB4(dsFv)-PE38 (BL22) in patients with hairy cell leukemia. J Clin Oncol. 2009;27:2983–2990. doi: 10.1200/JCO.2008.20.2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wayne AS, et al. Anti-CD22 immunotoxin RFB4(dsFv)-PE38 (BL22) for CD22-positive hematologic malignancies of childhood: preclinical studies and phase I clinical trial. Clin Cancer Res. 2010;16:1894–1903. doi: 10.1158/1078-0432.CCR-09-2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kreitman RJ. Recombinant immunotoxins for the treatment of chemoresistant hematologic malignancies. Curr Pharm Des. 2009;15:2652–2664. doi: 10.2174/138161209788923949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pai LH, et al. Clinical evaluation of intraperitoneal Pseudomonas exotoxin immunoconjugate OVB3-PE in patients with ovarian cancer. J Clin Oncol. 1991;9:2095–2103. doi: 10.1200/JCO.1991.9.12.2095. [DOI] [PubMed] [Google Scholar]
- 14.Pai LH, Wittes R, Setser A, Willingham MC, Pastan I. Treatment of advanced solid tumors with immunotoxin LMB-1: an antibody linked to Pseudomonas exotoxin. Nat Med. 1996;2:350–353. doi: 10.1038/nm0396-350. [DOI] [PubMed] [Google Scholar]
- 15.Hassan R, Bera T, Pastan I. Mesothelin: a new target for immunotherapy. Clin Cancer Res. 2004;10:3937–3942. doi: 10.1158/1078-0432.CCR-03-0801. [DOI] [PubMed] [Google Scholar]
- 16.Becker N, Benhar I. Antibody-based immunotoxins for the treatment of cancer. Antibodies. 2012;1:39–69. [Google Scholar]
- 17.Kreitman RJ, et al. Phase I trial of recombinant immunotoxin anti-Tac(Fv)-PE38 (LMB-2) in patients with hematologic malignancies. J Clin Oncol. 2000;18:1622–1636. doi: 10.1200/JCO.2000.18.8.1622. [DOI] [PubMed] [Google Scholar]
- 18.Kreitman RJ, Pastan I. Antibody fusion proteins: anti-CD22 recombinant immunotoxin moxetumomab pasudotox. Clin Cancer Res. 2011;17:6398–6405. doi: 10.1158/1078-0432.CCR-11-0487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kreitman RJ, et al. Phase I trial of anti-CD22 recombinant immunotoxin moxetumomab pasudotox (CAT-8015 or HA22) in patients with hairy cell leukemia. J Clin Oncol. 2012;30:1822–1828. doi: 10.1200/JCO.2011.38.1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tamura K, Kanazawa T, Suzuki M, Shioya A, Morikawa A. Successful induction of immune tolerance by continuous infusion of recombinant factor VIII in a haemophilia A patient with high-inhibitor titres. Haemophilia. 2006;12:100–102. doi: 10.1111/j.1365-2516.2006.01181.x. [DOI] [PubMed] [Google Scholar]
- 21.Sausville EA, et al. Continuous infusion of the anti-CD22 immunotoxin IgG-RFB4-SMPT-dgA in patients with B-cell lymphoma: a phase I study. Blood. 1995;85:3457–3465. [PubMed] [Google Scholar]
- 22.Powell DJ, Jr, et al. Administration of a CD25-directed immunotoxin, LMB-2, to patients with metastatic melanoma induces a selective partial reduction in regulatory T cells in vivo. J Immunol. 2007;179:4919–4928. doi: 10.4049/jimmunol.179.7.4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kowalski M, et al. A Phase I study of an intravesically administered immunotoxin targeting EpCAM for the treatment of nonmuscle-invasive bladder cancer in BCGrefractory and BCG-intolerant patients. Drug Des Devel Ther. 2010;4:313–320. doi: 10.2147/DDDT.S14071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weber F, et al. Safety, tolerability, and tumor response of IL4-Pseudomonas exotoxin (NBI-3001) in patients with recurrent malignant glioma. J Neurooncol. 2003;64:125–137. doi: 10.1007/BF02700027. [DOI] [PubMed] [Google Scholar]
- 25.Azemar M, et al. Regression of cutaneous tumor lesions in patients intratumorally injected with a recombinant single-chain antibody-toxin targeted to ErbB2/HER2. Breast Cancer Res Treat. 2003;82:155–164. doi: 10.1023/b:brea.0000004371.48757.19. [DOI] [PubMed] [Google Scholar]
- 26.Kreitman RJ, et al. Phase I trial of recombinant immunotoxin RFB4(dsFv)-PE38 (BL22) in patients with B-cell malignancies. J Clin Oncol. 2005;23:6719–6729. doi: 10.1200/JCO.2005.11.437. [DOI] [PubMed] [Google Scholar]
- 27.Hassan R, et al. Phase 1 study of the antimesothelin immunotoxin SS1P in combination with pemetrexed and cisplatin for front-line therapy of pleural mesothelioma and correlation of tumor response with serum mesothelin, megakaryocyte potentiating factor, and cancer antigen 125. Cancer. 2014;120:3311–3319. doi: 10.1002/cncr.28875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kreitman RJ, Hassan R, Fitzgerald DJ, Pastan I. Phase I trial of continuous infusion anti-mesothelin recombinant immunotoxin SS1P. Clin Cancer Res. 2009;15:5274–5279. doi: 10.1158/1078-0432.CCR-09-0062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chang K, Pastan I. Molecular cloning of mesothelin, a differentiation antigen present on mesothelium, mesotheliomas, and ovarian cancers. Proc Natl Acad Sci USA. 1996;93:136–140. doi: 10.1073/pnas.93.1.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chowdhury PS, Chang K, Pastan I. Isolation of anti-mesothelin antibodies from a phage display library. Mol Immunol. 1997;34:9–20. doi: 10.1016/s0161-5890(97)00011-4. [DOI] [PubMed] [Google Scholar]
- 31.Alewine C, Xiang L, Yamori T, Niederfellner G, Bosslet K, Pastan I. Efficacy of RG7787, a next-generation mesothelin-targeted immunotoxin, against triple-negative breast and gastric cancers. Mol Cancer Ther. 2014;13:2653–2661. doi: 10.1158/1535-7163.MCT-14-0132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hassan R, Kreitman RJ, Pastan I, Willingham MC. Localization of mesothelin in epithelial ovarian cancer. Appl Immunohistochem Mol Morphol. 2005;13:243–247. doi: 10.1097/01.pai.00000141545.36485.d6. [DOI] [PubMed] [Google Scholar]
- 33.Tang Z, Qian M, Ho M. The role of mesothelin in tumor progression and targeted therapy. Anticancer Agents Med Chem. 2013;13:276–280. doi: 10.2174/1871520611313020014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alberts B, Jonson A, Lewis J, Raff M, Roberts K, Walter P. Molecular biology of the cell. 4. New York: Garland Science; 2002. [Google Scholar]
- 35.Owen JA, Punt J, Stranford SA, Jones PP, Kuby J. Kuby Immunology. 2013. [Google Scholar]
- 36.Parker DC. T cell-dependent B cell activation. Ann Rev Immunol. 1993;11:331–360. doi: 10.1146/annurev.iy.11.040193.001555. [DOI] [PubMed] [Google Scholar]
- 37.Kreitman RJ. Immunoconjugates and new molecular targets in hairy cell leukemia. Hematology. 2012;2012:660–666. doi: 10.1182/asheducation-2012.1.660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hassan R, et al. Major cancer regressions in mesothelioma after treatment with an anti-mesothelin immunotoxin and immune suppression. Sci Transl Med. 2013;5:208ra147. doi: 10.1126/scitranslmed.3006941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hassan R, Williams-Gould J, Watson T, Pai-Scherf L, Pastan I. Pretreatment with rituximab does not inhibit the human immune response against the immunogenic protein LMB-1. Clin Cancer Res. 2004;10:16–18. doi: 10.1158/1078-0432.ccr-1160-3. [DOI] [PubMed] [Google Scholar]
- 40.Kreitman RJ, et al. Complete remissions of adult T-cell leukemia with anti-CD25 recombinant immunotoxin LMB-2 and chemotherapy to block immunogenicity. Clin Cancer Res. 2015 doi: 10.1158/1078-0432.CCR-15-1412. pii:clincanres.1412.2015, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Siegall CB, et al. Prevention of immunotoxin-induced immunogenicity by coadministration with CTLA4Ig enhances antitumor efficacy. J Immunol. 1997;159:5168–5173. [PubMed] [Google Scholar]
- 42.Onda M, et al. Tofacitinib suppresses antibody responses to protein therapeutics in murine hosts. J Immunol. 2014;193:48–55. doi: 10.4049/jimmunol.1400063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meister S, et al. Extensive immunoglobulin production sensitizes myeloma cells for proteasome inhibition. Cancer Res. 2007;67:1783–1792. doi: 10.1158/0008-5472.CAN-06-2258. [DOI] [PubMed] [Google Scholar]
- 44.Manning ML, Mason-Osann E, Onda M, Pastan I. Bortezomib reduces pre-existing antibodies to recombinant immunotoxins in mice. J Immunol. 2015;194:1695–1701. doi: 10.4049/jimmunol.1402324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Benhar I, Padlan EA, Jung SH, Lee B, Pastan I. Rapid humanization of the Fv of monoclonal antibody B3 by using framework exchange of the recombinant immunotoxin B3(Fv)-PE38. Proc Natl Acad Sci USA. 1994;91:12051–12055. doi: 10.1073/pnas.91.25.12051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bera TK, Onda M, Kreitman RJ, Pastan I. An improved recombinant Fab-immunotoxin targeting CD22 expressing malignancies. Leuk Res. 2014;38:1224–1229. doi: 10.1016/j.leukres.2014.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nagata S, Pastan I. Removal of B cell epitopes as a practical approach for reducing the immunogenicity of foreign protein-based therapeutics. Adv Drug Deliv Rev. 2009;61:977–985. doi: 10.1016/j.addr.2009.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Inada Y, et al. Biomedical and biotechnological applications of PEG- and PM-modified proteins. Trends Biotechnol. 1995;13:86–91. doi: 10.1016/S0167-7799(00)88912-X. [DOI] [PubMed] [Google Scholar]
- 49.Kuan CT, Wang QC, Pastan I. Pseudomonas exotoxin A mutants. Replacement of surface exposed residues in domain II with cysteine residues that can be modified with polyethylene glycol in a site-specific manner. J Biol Chem. 1994;269:7610–7616. [PubMed] [Google Scholar]
- 50.Benhar I, Wang QC, FitzGerald D, Pastan I. Pseudomonas exotoxin A mutants. Replacement of surface-exposed residues in domain III with cysteine residues that can be modified with polyethylene glycol in a site-specific manner. J Biol Chem. 1994;269:13398–13404. [PubMed] [Google Scholar]
- 51.Tsutsumi Y, Onda M, Nagata S, Lee B, Kreitman RJ, Pastan I. Site-specific chemical modification with polyethylene glycol of recombinant immunotoxin anti-Tac(Fv)-PE38 (LMB-2) improves antitumor activity and reduces animal toxicity and immunogenicity. Proc Natl Acad Sci USA. 2000;97:8548–8553. doi: 10.1073/pnas.140210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Roscoe DM, Pai LH, Pastan I. Identification of epitopes on a mutant form of Pseudomonas exotoxin using serum from humans treated with Pseudomonas exotoxin containing immunotoxins. Eur J Immunol. 1997;27:1459–1468. doi: 10.1002/eji.1830270624. [DOI] [PubMed] [Google Scholar]
- 53.Roscoe DM, Jung SH, Benhar I, Pai L, Lee BK, Pastan I. Primate antibody response to immunotoxin: serological and computer-aided analysis of epitopes on a truncated form of Pseudomonas exotoxin. Infect Immun. 1994;62:5055–5065. doi: 10.1128/iai.62.11.5055-5065.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Onda M, et al. Characterization of the B cell epitopes associated with a truncated form of Pseudomonas exotoxin (PE38) used to make immunotoxins for the treatment of cancer patients. J Immunol. 2006;177:8822–8834. doi: 10.4049/jimmunol.177.12.8822. [DOI] [PubMed] [Google Scholar]
- 55.Onda M, Beers R, Xiang L, Nagata S, Wang QC, Pastan I. An immunotoxin with greatly reduced immunogenicity by identification and removal of B cell epitopes. Proc Natl Acad Sci USA. 2008;105:11311–11316. doi: 10.1073/pnas.0804851105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Weldon JE, et al. A protease-resistant immunotoxin against CD22 with greatly increased activity against CLL and diminished animal toxicity. Blood. 2009;113:3792–3800. doi: 10.1182/blood-2008-08-173195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hansen JK, Weldon JE, Xiang L, Beers R, Onda M, Pastan I. A recombinant immunotoxin targeting CD22 with low immunogenicity, low nonspecific toxicity, and high antitumor activity in mice. J Immunother. 2010;33:297–304. doi: 10.1097/CJI.0b013e3181cd1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Siegel DL. Translational applications of antibody phage display. Immunol Res. 2008;42:118–131. doi: 10.1007/s12026-008-8044-y. [DOI] [PubMed] [Google Scholar]
- 59.Liu W, et al. Recombinant immunotoxin engineered for low immunogenicity and antigenicity by identifying and silencing human B-cell epitopes. Proc Natl Acad Sci USA. 2012;109:11782–11787. doi: 10.1073/pnas.1209292109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Alewine C, Hassan R, Pastan I. Advances in anticancer immunotoxin therapy. Oncologist. 2015;20:176–185. doi: 10.1634/theoncologist.2014-0358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Harding FA, et al. A beta-lactamase with reduced immunogenicity for the targeted delivery of chemotherapeutics using antibody-directed enzyme prodrug therapy. Mol Cancer Ther. 2005;4:1791–1800. doi: 10.1158/1535-7163.MCT-05-0189. [DOI] [PubMed] [Google Scholar]
- 62.Cantor JR, Yoo TH, Dixit A, Iverson BL, Forsthuber TG, Georgiou G. Therapeutic enzyme deimmunization by combinatorial T-cell epitope removal using neutral drift. Proc Natl Acad Sci USA. 2011;108:1272–1277. doi: 10.1073/pnas.1014739108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Moise L, Song C, Martin WD, Tassone R, De Groot AS, Scott DW. Effect of HLA DR epitope de-immunization of Factor VIII in vitro and in vivo. Clin Immunol. 2012;142:320–331. doi: 10.1016/j.clim.2011.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cizeau J, Grenkow DM, Brown JG, Entwistle J, MacDonald GC. Engineering and biological characterization of VB6-845, an anti-EpCAM immunotoxin containing a T-cell epitope-depleted variant of the plant toxin bouganin. J Immunother. 2009;32:574–584. doi: 10.1097/CJI.0b013e3181a6981c. [DOI] [PubMed] [Google Scholar]
- 65.Salvat RS, Choi Y, Bishop A, Bailey-Kellogg C, Griswold KE. Protein deimmunization via structure-based design enables efficient epitope deletion at high mutational loads. Biotechnol Bioeng. 2015;112:1306–1318. doi: 10.1002/bit.25554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.De Groot AS, Martin W. Reducing risk, improving outcomes: bioengineering less immunogenic protein therapeutics. Clin Immunol. 2009;131:189–201. doi: 10.1016/j.clim.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 67.Tangri S, et al. Rationally engineered therapeutic proteins with reduced immunogenicity. J Immunol. 2005;174:3187–3196. doi: 10.4049/jimmunol.174.6.3187. [DOI] [PubMed] [Google Scholar]
- 68.Yeung VP, Chang J, Miller J, Barnett C, Stickler M, Harding FA. Elimination of an immunodominant CD4+ T cell epitope in human IFN-beta does not result in an in vivo response directed at the subdominant epitope. J Immunol. 2004;172:6658–6665. doi: 10.4049/jimmunol.172.11.6658. [DOI] [PubMed] [Google Scholar]
- 69.Koren E, et al. Clinical validation of the “in silico” prediction of immunogenicity of a human recombinant therapeutic protein. Clin Immunol. 2007;124:26–32. doi: 10.1016/j.clim.2007.03.544. [DOI] [PubMed] [Google Scholar]
- 70.Oseroff C, et al. Molecular determinants of T cell epitope recognition to the common Timothy grass allergen. J Immunol. 2010;185:943–955. doi: 10.4049/jimmunol.1000405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mazor R, et al. Identification and elimination of an immunodominant T-cell epitope in recombinant immunotoxins based on Pseudomonas exotoxin A. Proc Natl Acad Sci USA. 2012;109:E3597–3603. doi: 10.1073/pnas.1218138109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tassignon J, et al. Monitoring of cellular responses after vaccination against tetanus toxoid: comparison of the measurement of IFN-gamma production by ELISA, ELISPOT, flow cytometry and real-time PCR. J Immunol Methods. 2005;305:188–198. doi: 10.1016/j.jim.2005.07.014. [DOI] [PubMed] [Google Scholar]
- 73.Mazor R, et al. Recombinant immunotoxin for cancer treatment with low immunogenicity by identification and silencing of human T-cell epitopes. Proc Natl Acad Sci USA. 2014;111:8571–8576. doi: 10.1073/pnas.1405153111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.King C, et al. Removing T-cell epitopes with computational protein design. Proc Natl Acad Sci USA. 2014;111:8577–8582. doi: 10.1073/pnas.1321126111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mazor R, et al. Recombinant immunotoxin with T cell epitope mutations that greatly reduce immunogenicity for treatment of mesothelin expressing tumors. Mol Cancer Ther. 2015 doi: 10.1158/1535-7163.MCT-15-0532. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mazor R, Crown D, Addissie S, Jang Y, Kaplan G, Pastan I. Elimination of murine and human T cell epitopes in recombinant immunotoxin eliminates neutralizing and anti-drug antibodies in vivo. Cell Mol Immunol. 2015 doi: 10.1038/cmi.2015.91. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mazor R, Tai CH, Lee B, Pastan I. Poor correlation between T-cell activation assays and LA-DR binding prediction algorithms in an immunogenic fragment of Pseudomonas exotoxin A. J Immunol Methods. 2015 doi: 10.1016/j_jim.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Onda M, et al. Recombinant immunotoxin against B-cell malignancies with no immunogenicity in mice by removal of B-cell epitopes. Proc Natl Acad Sci USA. 2011;108:5742–5747. doi: 10.1073/pnas.1102746108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ansari HR, Raghava GP. Identification of conformational B-cell Epitopes in an antigen from its primary sequence. Immunome Res. 2010;6:6. doi: 10.1186/1745-7580-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bogan AA, Thorn KS. Anatomy of hot spots in protein interfaces. J Mol Biol. 1998;280:1–9. doi: 10.1006/jmbi.1998.1843. [DOI] [PubMed] [Google Scholar]
- 81.Kaliyaperumal A, Michaels MA, Datta SK. Naturally processed chromatin peptides reveal a major autoepitope that primes pathogenic T and B cells of lupus. J Immunol. 2002;168:2530–2537. doi: 10.4049/jimmunol.168.5.2530. [DOI] [PubMed] [Google Scholar]
- 82.Ratto-Kim S, et al. Identification of Immunodominant CD4-Restricted Epitopes Co-Located with Antibody Binding Sites in Individuals Vaccinated with ALVAC-HIV and AIDSVAX B/E. PLoS One. 2015;10:e0115582. doi: 10.1371/journal.pone.0115582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Barnett BC, Burt DS, Graham CM, Warren AP, Skehel JJ, Thomas DB. I-Ad restricted T cell recognition of influenza hemagglutinin. Synthetic peptides identify multiple epitopes corresponding to antibody-binding regions of the HA1 subunit. J Immunol. 1989;143:2663–2669. [PubMed] [Google Scholar]
- 84.Steede NK, Rust BJ, Hossain MM, Freytag LC, Robinson JE, Landry SJ. Shaping T cell - B cell collaboration in the response to human immunodeficiency virus type 1 envelope glycoprotein gp120 by peptide priming. PLoS One. 2013;8:e65748. doi: 10.1371/journal.pone.0065748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pai-Scherf LH, et al. Hepatotoxicity in cancer patients receiving erb-38, a recombinant immunotoxin that targets the erbB2 receptor. Clin Cancer Res. 1999;5:2311–2315. [PubMed] [Google Scholar]
- 86.von Minckwitz G, et al. Phase I clinical study of the recombinant antibody toxin scFv(FRP5)-ETA specific for the ErbB2/HER2 receptor in patients with advanced solid malignomas. Breast Cancer Res. 2005;7:R617–626. doi: 10.1186/bcr1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Posey JA, et al. A phase I trial of the single-chain immunotoxin SGN-10 (BR96 sFv-PE40) in patients with advanced solid tumors. Clin Cancer Res. 2002;8:3092–3099. [PubMed] [Google Scholar]
- 88.Garland L, et al. Phase I trial of intravenous IL-4 pseudomonas exotoxin protein (NBI-3001) in patients with advanced solid tumors that express the IL-4 receptor. J Immunother. 2005;28:376–381. doi: 10.1097/01.cji.0000162782.86008.ml. [DOI] [PubMed] [Google Scholar]
- 89.Hassan R, et al. Phase I study of SS1P, a recombinant anti-mesothelin immunotoxin given as a bolus I.V. infusion to patients with mesothelin-expressing mesothelioma, ovarian, and pancreatic cancers. Clin Cancer Res. 2007;13:5144–5149. doi: 10.1158/1078-0432.CCR-07-0869. [DOI] [PubMed] [Google Scholar]