

Abstract
Human chaperone DnaJB6, an Hsp70 co-chaperone whose defects cause myopathies, protects cells from polyglutamine toxicity and prevents purified polyglutamine and Aβ peptides from forming amyloid. Yeast prions [URE3] and [PSI+] propagate as amyloid forms of Ure2 and Sup35 proteins, respectively. Here we find DnaJB6-protected yeast cells from polyglutamine toxicity and cured yeast of both [URE3] prions and weak variants of [PSI+] prions but not strong [PSI+] prions. Weak and strong variants of [PSI+] differ only in the structural conformation of their amyloid cores. In line with its anti-prion effects, DnaJB6 prevented purified Sup35NM from forming amyloids at 37 °C, which produce predominantly weak [PSI+] variants when used to infect yeast, but not at 4 °C, which produces mostly strong [PSI+] variants. Thus, structurally distinct amyloids composed of the same protein were differentially sensitive to the anti-amyloid activity of DnaJB6 both in vitro and in vivo. These findings have important implications for strategies using DnaJB6 as a target for therapy in amyloid disorders.
Keywords: amyloid, chaperone, heat shock protein (HSP), prion, yeast, DnaJB6
Introduction
In many mammalian diseases, including Alzheimer, Parkinson, and Huntington diseases, type 2 diabetes, and prion diseases, tissue pathology is associated with accumulation of amyloid, an insoluble, highly ordered fibrous protein aggregate. Elevating expression of protein chaperones, which help other proteins adopt and maintain their native conformations, reduces toxicity associated with amyloidogenic proteins in different model systems (1). In particular, Hsp70 and its Hsp40 (J-protein) co-chaperone partners provide broad protective effects. A major role of Hsp70 and Hsp40 is to prevent aggregation of misfolded proteins, but they also protect cells from toxicity caused by amyloid-forming proteins without preventing formation of aggregates (2, 3). These observations led to a view that toxicity is due to something other than amyloid itself, such as non-amyloid oligomeric aggregates, and that the chaperones might neutralize toxic effects by promoting incorporation of such aggregates into more benign amyloid fibers.
J-proteins are defined by a signature J-domain that mediates physical interaction with the ATPase regulatory domain of Hsp70. J-proteins have amplified through evolution more than other chaperone families. For example, Saccharomyces cerevisiae and humans each encode about a dozen Hsp70s, but yeast encodes 22 J-proteins, whereas humans have roughly 50. This difference suggests that any additional roles for Hsp70 demanded by increases in cell complexity and differentiation were met in part by an expansion in the number and diversity of J-proteins that regulate Hsp70 activity (4). Human J-proteins are categorized into DnaJA, DnaJB, and DnaJC families on the basis of structural similarities (4). The two major cytosolic yeast Hsp40 J-proteins Ydj1 and Sis1 are most closely related to the DnaJA and DnaJB families, respectively.
Elevating expression of DnaJB6b and DnaJB8, but not other DnaJ proteins, blocks aggregation and toxicity of Huntington related polyglutamine (polyQ)4 in human cells (5). DnaJB6b is a cytosolic splice variant of the C-terminally extended DnaJB6a that is localized in the nucleus and does not block toxicity. Its protective effect does not depend on J-domain function, which reflects Hsp70-independent activity and is consistent with the inability of Hsp70 to confer similar protection (5, 6). Purified DnaJB6 blocks incorporation of polyQ peptides into preformed aggregates (5–7), and sub-stoichiometric amounts of DnaJB6 prevent both the nucleation of Aβ peptides into amyloid and the incorporation of Aβ into pre-existing amyloid fibers (8). Thus, it has been proposed that DnaJB6 protects cellsfrom toxic effects of amyloid-forming proteins by binding to peptides and oligomers that have a high propensity to form amyloids and prevent their maturation into amyloid. This mechanism is distinct from those that relieve toxicity without preventing aggregation in that it neutralizes putatively toxic oligomers and blocks formation of amyloid that might continue to promote production of toxic species.
Yeast prions provide a genetic model of pathogenic amyloids useful for investigating effects of chaperones on amyloid in vivo (9). Stable propagation of prions in dividing yeast cells requires continued fragmentation of amyloid fibers into new propagons, or seeds, that are passed with cytoplasm to daughter cells (10). This fragmentation depends on the normal cellular functions of the protein disaggregation machinery that is driven by Hsp104 in cooperation with Hsp70 and its Sis1 and nucleotide exchange factor co-chaperones (11–18). Different prions depend on this machinery to different degrees, so impairing disaggregation machinery function by altering the activity of these components can affect their propagation differently (9, 19). For example, by competing with Sis1, elevating expression of Ydj1 efficiently cures cells of [URE3] prions, which are composed of Ure2 protein, but not [PSI+] prions composed of Sup35 (20, 21), and elevating Hsp104 has the opposite effects on these prions (11, 21).
The general ability of DnaJB6 to interfere with the formation of amyloid composed of different proteins suggests it could act as an anti-prion factor in yeast but in a manner mechanistically distinct from the yeast chaperones. Indeed, we find here that expressing DnaJB6b in yeast cures cells of [URE3] prions in an apparently direct manner that is clearly distinct from that of curing by Ydj1. Importantly, DnaJB6b also cured “weak” variants of [PSI+], but it was ineffective against phenotypically stronger [PSI+] variants. Thus, anti-amyloid activity of DnaJB6b in vivo was limited by the structural characteristics of different amyloids composed of the same protein. We further show that this limitation is related to its ability to block formation of Sup35NM amyloid in vitro only under conditions that produce material that is thermodynamically similar to Ure2 amyloid and that generates weak [PSI+] prions when used to infect yeast. These findings provide further insight into the remarkable properties of DnaJB6b and raise important concerns regarding use of DnaJB6 as a target for therapies in amyloid diseases.
Experimental Procedures
Yeast Strains and Growth Conditions
Yeast strains used are isogenic to 1075 (MATa, kar1-1, PDAL5::ADE2, his3Δ200, leu2Δ1, trp1Δ63, ura3-52, SUQ5, [URE3]) (22). 779-6A is the same but has ade2-1 in place of PDAL5::ADE2, is [ure-o], and has endogenous strong [PSI+] and [PIN+], both of uncertain origin. [PSI+] prion variants were cytoduced into a [rho°] version of 779-6A that had been cured of prions by passage on medium containing 3 mm guanidine-HCl to generate strains 1566, 1567, 1586, and 1587. Strains 1566 and 1567 were made using strains JW129 [PSI+]Sc4 (strong) and JW127 [PSI+]Sc37 (weak) as prion donors (23). Similarly, strains 1586 and 1587 were made using L2892 [PSI+]W (weak, here designated SL[PSI+]W) and L2885 [PSI+]S (strong, here designated SL[PSI+]S) as prion donors (24). Strains 930 and MR502, for testing complementation of SIS1 and YDJ1 in vivo functions, respectively, have been described (25).
YPAD contains 1% yeast extract, 2% peptone, 400 mg/liter (excess) adenine and 2% dextrose. To monitor prion phenotypes cells were grown on 1/2YPD (0.5% yeast extract, 2% peptone, 2% dextrose), which contains a limiting but undefined amount of adenine. To maintain plasmids, cells were grown on minimal synthetic dextrose media, which contain 2% dextrose and 0.7% yeast nitrogen base supplemented with appropriate nutrients. SRaf is 2% raffinose and 0.7% yeast nitrogen base, and SGal is 2% galactose, 2% raffinose, and 0.7% yeast nitrogen base. Limiting adenine in selective media was 10 mg/liter. Solid media are the same but contain 2% agar (Difco). Unless otherwise noted, cells were grown at 30 °C.
Plasmids
Plasmids used are listed in Table 1. pRU4 is a centromeric LEU2-marked plasmid containing the GAL1 promoter and the CYC1 terminator. It was made by replacing the GPD promoter in plasmid p415-GPD (26) with a PCR-generated GAL1 promoter fragment amplified from plasmid pGCH17 (14) using SacI/BamHI. The human J-protein cDNA clones used as the template in PCR reactions were gifts from H. Kampinga. J-protein open reading frames were cloned as BamHI/SalI fragments into either p414-GPD, p415-GPD, or p415-TEF (26) and as BamHI/XhoI into pRU4. Plasmids pRU69, pRU70, and pRU71, used for expressing C-terminal His-tagged DnaJB6b, DnaJB6b-D33N, and DnaJB6b-ΔST in Escherichia coli, respectively, were made by cloning the coding regions into pET28b as NcoI/XhoI fragments. Restriction sites and/or C-terminal c-Myc tags were added by using PCR. Point mutations were introduced by site-directed mutagenesis.
TABLE 1.
Plasmids used in this study
| Plasmid | Description | Marker | Source |
|---|---|---|---|
| pRS314 | Empty vector | TRP1 | (57) |
| pRS315 | Empty vector | LEU2 | (57) |
| pRS426 | Empty vector, 2μ | URA3 | (58) |
| pRU4 | Empty vector, PGAL | LEU2 | This study |
| pMR287 | PGPD::YDJ1-cMyc | TRP1 | (25) |
| pMR287-D36N | PGPD::ydj1D36N-cMyc | TRP1 | (25) |
| pMR294 | PGPD::SIS1 | LEU2 | (25) |
| pMR328 | PGPD::DnaJB6b-cMyc | TRP1 | This study |
| pMR328-D33N | PGPD::DnaJB6bD33N-cMyc | TRP1 | This study |
| pMR328-ΔST | PGPD::DnaJB6bΔ154–195-cMyc | TRP1 | This study |
| pMR329 | PGPD::DnaJB6b1–154-cMyc | TRP1 | This study |
| pMR329-D33N | PGPD::DnaJB6b1–154,D33N-cMyc | TRP1 | This study |
| pMR330 | PGPD::DnaJB6b1–195-cMyc | TRP1 | This study |
| pMR330-D33N | PGPD::DnaJB6b1–195,D33N-cMyc | TRP1 | This study |
| pMR338 | PGPD::ydj11–106-cMyc | TRP1 | This study |
| pMR338-D36N | PGPD::ydj11–106,D36N-cMyc | TRP1 | This study |
| pMR345 | PTEF::YDJ1-cMyc | TRP1 | This study |
| pMR346 | PTEF::DnaJB6b-cMyc | TRP1 | This study |
| pRU13 | PGAL::DnaJB1-cMyc | LEU2 | This study |
| pRU14 | PGAL::DnaJB6b-cMyc | LEU2 | This study |
| pRU15 | PGAL::DnaJB8-cMyc | LEU2 | This study |
| pRU19 | PGAL::DnaJB7-cMyc | LEU2 | This study |
| pRU20 | PGAL::YDJ1-cMyc | LEU2 | This study |
| pYES2-HttQ103-GFP | PGAL::polyQ103-GFP, 2μ | URA3 | (30) |
| pMR18 | GST-HSP104 (E. coli) | KANR | This study |
| pET-SSA1 | SSA1 (no tag) (E. coli) | AMPR | This study |
| pPROEX-Ydj1 | His6-YDJ1 (E. coli) | AMPR | (32) |
| pKT41 | His6-URE2 (E. coli) | AMPR | (28) |
| p1339 | SUP35-NM-His6 (E. coli) | AMPR | (27) |
| pRU69 | DnaJB6b-His6 (E. coli) | AMPR | This study |
| pRU70 | dnaJB6b-D33N-His6 (E. coli) | AMPR | This study |
| pRU71 | dnaJB6b-ΔST-His6 (E. coli) | AMPR | This study |
Plasmid p1339, for expression of His-tagged Sup35NM (the prion-determining and adjacent charged domains of Sup35) in E. coli, was described previously (27). Plasmid pKT41, for expression of His-tagged Ure2 in E. coli, was described previously (28). Plasmid pYES2-HttQ103GFP, for the expression of Huntingtin polyQ103 fused to GFP under the control of the GAL promoter in yeast, was described previously (30, 31).
Plasmid pMR18, used for expression of GST-Hsp104 in E. coli, is the HSP104 open reading frame cloned into pET41c (Novagen) as a PshAI/XhoI fragment. Plasmid pET-SSA1, for expression of Ssa1p in E. coli, is the SSA1 open reading frame cloned into pET24a as an NdeI/XhoI fragment. Plasmid pPROEX-Htb-YDJ1, used for expression of HIS-tagged Ydj1p, was described previously (32).
Monitoring Prions
Cells lacking Ade2 are ade− and red when grown on limiting adenine. Ure2 represses transcription of nitrogen catabolism genes, so depletion of soluble Ure2 into [URE3] prion aggregates results in activation of such genes, such as DAL5. Our strains have ADE2 controlled by the DAL5 promoter (PDAL5), so [URE3] cells are white on limiting adenine and Ade+, whereas [ure-o] cells are red and require exogenous adenine. Strains used for monitoring [PSI+] have ade2-1, a nonsense allele that is suppressed by [PSI+] in cells with SUQ5 due to the depletion of Sup35 into prion aggregates. Thus, [PSI+] cells are white or pink and Ade+, whereas [psi−] cells are red and require adenine.
PolyQ103GFP Toxicity and Fluorescence Microscopy
Strain 779-6A was transformed with plasmids encoding polyQ103GFP and human J-proteins under the control of the GAL1 promoter or empty vectors as indicated. After overnight growth in SRaf, cultures were diluted to an A600 of 0.25 and then serially diluted and spotted onto SRaf or SGal plates. The latter induce expression of human J-proteins and/or polyQ103GFP. Plates were scanned 3 days after incubation at 30 °C. For fluorescence microscopy, transformants grown in SRaf overnight were subcultured into SGal. Cells were imaged after 4 h of incubation at 30 °C using a Nikon Eclipse Ni microscope equipped with a Q-Imaging Retiga EXi digital camera, Plan APO VC 60× oil immersion differential interference contrast optics, and short-pass GFP filter.
Curing Prions by J-protein Overexpression
Cells were cured of prions by overexpression of J-proteins by either inducible (GAL1 promoter) or constitutive (GPD or TEF1 promoter) expression. For inducible expression, transformants were first grown overnight in synthetic dextrose then diluted into SGal supplemented with the appropriate nutrients. Aliquots were removed at the indicated time points and plated onto 1/2YPD plates. Percent red (non-sectored) colonies were scored as [prion−] and plotted as a function of the number of cell divisions. For constitutive expression, both red and sectored colonies that arose on primary transformation plates were scored as curing events (see “Results”). Plates were scored and scanned after 3–5 days of incubation at 30 °C.
Western Blots
Sample preparation and Western blotting was performed as described (25). Briefly, 10 absorption units of cells were lysed in 250 μl of SDS-PAGE loading dye (0.1 m Tris-HCl, pH 6.8, 3% SDS, 1% β-mercaptoethanol, 10% glycerol, bromphenol blue) by vortexing with an equal volume of glass beads for 90 s followed by boiling for 5 min. Lysates were centrifuged for 5 min at 12,000 × g, and 5 μl of supernatant was loaded per lane onto 4–20% Criterion Tris-HCl TGX gels (Bio-Rad). c-Myc-tagged proteins were detected using rabbit anti-c-Myc (Abcam #ab9106) and goat anti-rabbit (Bio-Rad #170-6515) antibodies.
Quantifying Prion Propagons
Numbers of prion propagons per cell were determined using an established method (10). Briefly, cells were grown until colonies were just visible on YPAD plates containing 4 mm guanidine-HCl, which inhibits Hsp104 and blocks replication of prions. Entire colonies were removed, suspended in water, and spread onto synthetic dextrose plates lacking adenine. The number of resulting Ade+ colonies is an estimation of the number of propagons in the cell that gave rise to the original colony.
Protein Purification
Sup35NM was expressed, and lysates were prepared as previously described (27). Purified Sup35NM was recovered using a batch purification method in the presence of 8 m urea. It was then concentrated to 2 mm using Amicon Ultra spin concentrators and followed by buffer exchange to 6 m guanidine-HCl, 10 mm sodium phosphate pH 7.5. Aliquots were stored at −80 °C until use.
Ure2 was expressed in Rosetta2(DE3) E. coli from plasmid pKT41 as an N-terminal His6-tagged fusion (28). Cells were grown at 30 °C in 2×YT medium to an A600 of 0.4–0.6 and induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside overnight at 20 °C. Cells were lysed by incubation in 60 mm sodium phosphate, pH 7.5, 360 mm NaCl, 2.4 mm imidazole, 0.12% Triton X-100, 0.5 mg/ml lysozyme, 0.02 mg/ml each DNase I and RNase A for 20 min at room temperature. Guanidine-HCl was then added to a final concentration of 1 m from a 6 m stock. The lysate was cleared by centrifugation and filtering, then loaded onto a pre-equilibrated 5 ml Talon column (GE Healthcare). The column was washed with 50 mm sodium phosphate, pH 7.5, 300 mm NaCl, 20 mm imidazole, 1 m guanidine-HCl. Ure2 was eluted in the same buffer containing 200 mm imidazole. Peak fractions as determined by Bradford assay were pooled, concentrated, and either dialyzed to 1× PBS or diluted into reaction mixtures (see below). Fresh Ure2 protein was prepared for each experiment.
Ydj1, DnaJB6b, DnaJB6b-D33N, and DnaJB6b-ΔST were purified similarly except the guanidine-HCl was omitted. For Ydj1p, the N-terminal His6 tag was removed by digestion with AcTEV protease (Invitrogen) followed by buffer exchange to 50 mm Tris-HCl, pH 7.5, 50 mm NaCl. Talon resin (0.5 ml) was then added and incubated for 10 min to remove AcTEV protease and undigested His6-Ydj1p. Digested Ydj1p was then polished using an ANX anion exchange column (GE Healthcare).
Hsp104 was expressed in BL21(DE3)pLysS E. coli as an N-terminal GST fusion containing an enterokinase cleavage site such that upon digestion no vector-encoded sequence remained on Hsp104. Lysates were incubated with glutathione resin and washed extensively with 50 mm Tris-HCl, pH 8.0, 150 mm NaCl. The resin with bound GST-Hsp104 was then washed and suspended with EKMax buffer (50 mm Tris-HCl, pH 8.0, 1 mm CaCl2, 0.1% Tween 20). On-column cleavage was performed by the addition of EKMax (Invitrogen), and this mixture was incubated overnight at 4 °C. The eluate fractions contained digested Hsp104, and the protease was inactivated by addition of PMSF to 2 mm. This procedure was performed up to four times on the same batch of resin, as cleavage was inefficient. Fractions containing non-tagged Hsp104 were pooled and concentrated.
Ssa1 was purified as described previously for HspA1A (33). Briefly, Ssa1p was expressed without a tag in Rosetta2(DE3) E. coli and purified on DEAE- and ATP-agarose. Purified yeast chaperones were dialyzed to refolding buffer (50 mm HEPES, pH 7.5, 120 mm KCl, 13 mm MgCl2) containing 1 mm DTT and 10% glycerol, aliquoted, and stored at −80C. Purified Ydj1p was further supplemented with 10 mm ZnCl2.
In all cases proteins were purified to >95%. Purified protein concentrations were determined by Bradford assay or absorbance at 280 nm using calculated extinction coefficients (Sup35NM, 25,600 m−1cm−1; Ure2, 49,500 m−1cm−1; DnaJB6, 13,370 m−1cm−1).
Luciferase Re-activation
Purified chaperones (Hsp104, 0.75 mm; Ssa1, 1.0 mm; J-protein, 1.0 mm) were mixed in refolding buffer containing 3 mm ATP, 1 mm DTT, and 0.1 mg/ml BSA on ice to a final volume of 40 ml. Luciferase was denatured by diluting to 1 mm (from a 235 mm stock) into a refolding buffer containing 6 m urea and incubated at 30 °C for 30 min. To start the reaction, 1 ml of denatured luciferase was added and incubated at room temperature. At the indicated time points, 2-ml aliquots were removed and mixed with 50 ml of Luciferase Assay Reagent (Promega) and read in a Zylux Femtomaster Luminometer with a 5-s delay and 5-s read time.
In Vivo Complementation Assays
The ability of wild type and mutant versions of DnaJB6b to complement yeast SIS1 or YDJ1 in vivo functions was tested via plasmid shuffling as described (25).
Thioflavin-T Assays
Ure2 was purified in the presence of 1 m guanidine as described above and dialyzed overnight to 1× PBS at 4 °C. To form amyloid, purified Ure2 was diluted to 5 μm in 500 μl of PBS and shaken at 200 rpm at 37 °C. Aliquots were removed at the specified time points, mixed with thioflavin-T (Sigma, catalogue no. T-3516), and incubated at room temperature for 5–15 min without shaking. Fluorescence was measured by using an excitation of 405 nm and emission of 495–505 nm.
The manner and extent to which J-proteins affected Ure2 amyloid formation, as measured by changes in thioflavin-T fluorescence, was observed consistently with different preparations of full-length Ure2. However, we observed preparation-specific inconsistencies in lag times and overall yields. We, therefore, chose to analyze amyloid formation using the highly reproducible and quantitative SDS-resistance polyacrylamide gel mobility assay (see below).
Amyloid Preparation
Sup35-NM and Ure2 amyloid were formed as described (34). Briefly, purified Sup35-NM or freshly prepared Ure2 protein was diluted or buffer exchanged into 500 μl of prewarmed 50 mm sodium phosphate, pH 7.5, 300 mm NaCl to a final concentration of 5 mm in 2-ml round-bottom Eppendorf tubes. The addition of NaCl eliminated the propensity of DnaJB6b to precipitate.5 The tubes were then rotated end-over-end at 8 rpm for 16–24 h at the indicated temperatures.
Determination of Amyloid Thermodynamic Stability
SDS resistance and gel mobility of amyloids was performed as described previously (34). Briefly, aliquots of amyloid preparations were mixed 1:1 with SDS loading dye to give a final SDS concentration of 1.5%. Aliquots of this mixture were then incubated at the indicated temperatures for 10 min followed by brief cooling on ice. Proteins in these samples were then separated on 4–20% Tris-HCl Criterion TGX (Bio-Rad) gels and stained with Bio-Safe Coomassie G250 Stain (Bio-Rad) according to the manufacturer's instructions. Band intensity was determined using ImageJ. Data analysis and curve fitting were performed in Excel using the equation, y = (A + B)/(1 + 10(C − x)/D), where x is the temperature, y is the relative band intensity, A is the band intensity at baseline, B is the amplitude of band intensity, C is the melting temperature, and D is the width of the melting transition (34).
Results
DnaJB6b Prevents PolyQ103GFP Toxicity in Yeast
Fig. 1A shows the domain structure of human DnaJB molecular chaperones used here compared with yeast Ydj1 and Sis1. DnaJB1 is the closest human ortholog of Sis1, and it can function in place of the essential Sis1 to maintain cell viability (35). In all of these proteins the J-domains share a high degree of homology. DnaJB6b, DnaJB7, and DnaJB8 all have a serine-threonine-phenylalanine-rich region flanked by residues SSF-TST in DnaJB6, SSF-SST in DnaJB8, and SSY-TSD in DnaJB7 (5, 6) that is absent in yeast J-proteins and has roles in oligomerization and substrate recognition. For convenience we refer to this region in DnaJB6b as ST. Mutations in the GF region of DnaJB6 have been linked to myopathies (36–39). The DnaJB6 GF region functions in place of the GF region in Sis1, and when it is substituted into Sis1 these mutations influence propagation of different [PSI+] prions (40). With the exception of DnaJB1, the human J-proteins tested here share very little homology with yeast J-proteins in their C-terminal domains.
FIGURE 1.

Human DnaJB proteins protect yeast from polyQ toxicity. A, scale cartoon showing the domain structure of yeast Ydj1 and Sis1 and the human DnaJB proteins studied here. The zinc-finger and farnesylation motifs of Ydj1 are denoted by diagonal and horizontal hash marks, respectively. J is the J-domain, GF is the glycine-phenylalanine-rich domain, GM is the glycine-methionine-rich domain, ST is the serine-threonine-phenylalanine rich domain, and C is the C-terminal domain. All plasmid-expressed J-proteins contain a C-terminal c-Myc tag. B, 5-fold dilutions of wild type [PSI+] [PIN+] cells of strain 779-6A carrying plasmids encoding polyQ-GFP and the indicated J-protein, both regulated by a galactose-inducible promoter, or carrying empty vectors (ev) as indicated, were spotted onto plates containing non-inducing (Raffinose) or inducing (Galactose) medium. C, aliquots of un-induced cells used in panel B were incubated in liquid galactose cultures for 4 h and then visualized by fluorescence (GFP; extended depth focus images of Z-series using GFP filter) and differential interference contrast (DIC) microscopy. D, [PIN+] status of cells with empty vector (ev) or galactose-inducible DnaJB6b was monitored by fluorescence microscopy using plasmid-expressed Rnq1-GFP. Cells shown were grown in galactose for nine generations. The presence of fluorescent foci is diagnostic of [PIN+].
Members of the DnaJB family variably reduce polyQ aggregation and toxicity in human cells, with DnaJB6 having the strongest protective effects (5). High expression of polyQ103 Huntingtin fragments is also toxic to yeast (30, 31, 41, 42).
To test if DnaJB proteins could provide protection from this toxicity, cells were transformed with a plasmid encoding galactose-inducible polyQ103GFP and with separate plasmids expressing galactose-inducible DnaJB1, DnaJB6b, DnaJB7, DnaJB8, or empty vector. Growth of cells with the polyQ103GFP plasmid and the empty vector control was normal on non-inducing medium but was arrested on galactose plates (Fig. 1B). Using fluorescence microscopy, we observed many large polyQ103GFP aggregates per cell 4 h after inducing expression of polyQ103GFP (Fig. 1C).
As in human cells, co-expression of DnaJB proteins variably reduced polyQ103GFP toxicity. Expression of DnaJB1 did not improve growth on galactose or alter the polyQ103GFP fluorescence pattern. DnaJB6b, however, was highly effective at reducing toxicity, improving growth on galactose comparable with wild type cells that do not express polyQ103GFP. Coincidentally, DnaJB6b changed the pattern of polyQ103GFP fluorescence to one or two foci per cell. DnaJB7 and DnaJB8 also reduced toxicity noticeably, although not to the same level as DnaJB6b, with DnaJB7 having the weakest effect. Correspondingly, cells expressing DnaJB8 had a fluorescence pattern like those expressing DnaJB6 but with slightly more foci per cell, whereas fluorescence in cells expressing DnaJB7 looked more like that in cells with empty vector.
The finding that DnaJB7 modestly protected cells from polyQ103GFP was unexpected given that DnaJB7 did not prevent polyQ103GFP toxicity in human cells. The dependence of polyQ toxicity on prions in yeast (see below) is an obvious difference of the two systems. Additionally, DnaJB7 contains a putative nuclear localization signal. These observations suggest that polyQ103GFP toxicity in yeast might be mediated in part through nucleus-specific mechanisms. Perhaps DnaJB7 protects a nuclear factor from sequestration by polyQ103GFP aggregates. Alternatively, a significant portion of DnaJB7 could remain cytosolic in yeast cells. Aside from this specific difference, the relative effects these chaperones had on reducing polyQ103GFP toxicity in human cells was recapitulated in yeast.
PolyQ toxicity in yeast is influenced when [PSI+] prions, [PIN+] prions (formed of the protein Rnq1), or both are present (30, 41). The ability of J-proteins to reduce the toxicity also can be affected by these prions. For example, increasing Sis1 reduces toxicity in [PIN+] cells but not in cells with both [PIN+] and [PSI+] (41). Our strains propagate both [PSI+] (a strong variant, see below) and [PIN+], and we found that neither is affected by expression of DnaJB6b (Fig. 1D and below). Therefore, DnaJB6b does not affect polyQ toxicity by curing cells of either of these prions. Our data, however, do not rule out the possibility that DnaJB6b might affect the resident prions in ways that reduce toxicity without reducing stable propagation of the prions. Further investigation will be needed to define precisely how DnaJB6b reduces the toxicity.
DnaJB6b Cures Cells of [URE3] Differently than Ydj1
Our strains that lack the [URE3] prion, designated [ure-o], do not express Ade2, so they require exogenous adenine and form red colonies on limiting adenine. Cells that propagate [URE3] prions express Ade2, so they grow without adenine and are white on limiting adenine (see “Experimental Procedures”; Fig. 2B).
FIGURE 2.

Human DnaJB proteins cure yeast of [URE3] differently than Ydj1. A, strain 1075 grown in non-inducing raffinose medium lacking adenine was transferred to galactose medium containing adenine to induce expression of the indicated human J-proteins or Ydj1 as indicated, and cells were plated on 1/2YPD over the course of nine generations. [URE3] curing is plotted as percentage of cells retaining [URE3] as a function of generations (ev, empty vector control). B, representative sections of 1/2YPD plates of cells at the nine generation time point of the experiment shown in panel A. White colonies propagate [URE3], and red [ure-o] colonies do not. C, Western blot probed with c-Myc-specific antibodies showing relative expression levels of indicated c-Myc-tagged J-proteins after nine generations in medium without (−) or with (+) galactose. The slowest migrating band in each lane is the full-length protein. The same area of the membrane stained by Amido Black is shown as a loading and transfer control. D, representative 2-cm2 sections of primary transformation plates of strain 1075 transformed by plasmids encoding GPD-driven expression of the indicated J-proteins (ev+ and ev− are [URE3] and [ure-o] cells, respectively, with empty vector). D36N (Ydj1) and D33N (DnaJB6b) indicate the Asp to Asn substitution in the conserved HPD motif that disrupts Hsp70 interaction. E, Western blot probed with c-Myc-specific antibodies showing relative expression level of Ydj1 or DnaJB6b from cells used in panel D. F, representative sections of transformation plates as in panel D except cells carry plasmids encoding proteins regulated by the TEF promoter. G, representative sections of transformation plates as in panel D except cells carry plasmids encoding proteins with indicated truncations (Δ) or mutations. The arrow points to the plate with colonies that arose from a streak of cells from the colonies shown in the inset. Red colonies are [ure-o] cells from the sectors in the original colonies. For F89I and F93L, colonies are on 1/2YPD plates from the six generation time point of an experiment similar to that shown in panel B.
We tested whether DnaJB family proteins could inhibit propagation of [URE3] by expressing them from a galactose-inducible promoter in [URE3] cells (strain 1075) and monitoring loss of [URE3] as a function of generations, i.e. cell doublings. Soon after inducing Ydj1, [ure-o] cells began to arise, and prion loss continued roughly linearly over the course of nine generations (Fig. 2A, black line).
Among the DnaJB family of proteins, DnaJB6b and DnaJB8 cured [URE3] most effectively (Fig. 2A, red and green lines, respectively), and DnaJB1 had a weak effect (Fig. 2A, blue line), but DnaJB7 did not cure [URE3] (Fig. 2A, yellow line). These relative effects on [URE3] propagation again resemble the way these chaperones affect polyQ103GFP aggregation in human cells. Differences in levels of expressed J-proteins were not great (Fig. 2C) nor did they correlate with these effects (e.g. DnaJB8 cured better than either DnaJB1 or DnaJB7 but was less abundant than both), so differences in curing were more likely due to differences in protein activity than abundance.
We focused on DnaJB6b because it was most effective at inhibiting polyQ103GFP toxicity and prion propagation. Notably, although cells expressing Ydj1 or DnaJB6b had a similar proportion of cured cells after nine generations, the difference in kinetics of loss of [URE3] suggests that DnaJB6b and Ydj1 cure by distinct mechanisms.
As a more efficient way to monitor impaired propagation of [URE3], we transformed strain 1075 with plasmids encoding DnaJB6b or Ydj1 controlled by the strong constitutive GPD promoter. Although this approach does not allow direct observation of curing kinetics, each transformant colony represents an individual population where overexpression began in the cell founding the colony. Thus, we can monitor hundreds of trials simultaneously. Red sectors in a colony represent curing events. Most colonies transformed with GPD-Ydj1 were entirely red or sectored, but the degree of sectoring among them was heterogeneous (Fig. 2D), which likely reflects wide variation in the number of propagons per cell (see Table 2). Colonies with fewer sectors could be from founding cells that contained a high number of propagons. Two additional controls are consistent with earlier data suggesting that Ydj1 curing depends on interaction with Hsp70 (15, 25, 43). Ydj1 containing the D36N point mutation that disrupts interaction with any Hsp70 fails to cure [URE3], and co-expressing Sis1, which allows Sis1 to compete more effectively with Ydj1 for interaction with Hsp70, greatly reduced the extent of prion loss (Fig. 2D).
TABLE 2.
Prion seeds per cell
For each determination the number of [PSI+] cells in 8–18 colonies grown on guanidine-containing medium were counted.
| Strain/Prion | Seeds per cell |
|---|---|
| 779–6A [PSI+] | 506 ± 195 |
| 1566 [PSI+]Sc4 | 512 ± 156 |
| 1587 SL[PSI+]S | 417 ± 198 |
| 1567 [PSI+]Sc37 | 39 ± 18 |
| 1586 SL[PSI+]W | 6 ± 4 |
| 1075 [URE3] | 76 ± 57 |
In contrast to Ydj1 transformants, cells transformed with GPD-driven DnaJB6b formed uniformly red colonies, which indicates that most cells had lost [URE3] by the time visible colonies formed regardless of the number of propagons in the founding cell. Also unlike Ydj1, neither the D33N mutation (homologous to D36N in Ydj1) nor co-expression of Sis1 prevented curing by DnaJB6b, suggesting this curing did not require interaction of DnaJB6b with Hsp70. The abundance of the Ydj1 and DnaJB6b proteins was similar (Fig. 2E), and similar results were obtained when expression was driven by the weaker TEF1 promoter (Fig. 2F), indicating that the difference in curing was not due to a difference in amount of protein. These findings align with work in mammalian cell models of Huntington disease showing that DnaJB6b mutants unable to interact with Hsp70 can still block polyQ aggregation and toxicity (5, 6).
We also made and expressed mutated versions of DnaJB6b to test if other regions of DnaJB6b were important for curing cells of [URE3] (Fig. 2G). We found the ST region was required for curing of [URE3]. Additionally, deleting the C-terminal domain resulted in inefficient curing that was only noticeable as red sectors on the edges of primary transformants that became obvious upon subculturing. Thus, the CTD was important but not essential for the curing. These results differ from those with Ydj1, as truncated versions of Ydj1 that retain an intact J-domain still cure [URE3] efficiently (43) and are again consistent with the importance of both the ST and C-terminal regions for preventing polyQ aggregation and toxicity in human cells (5).
We also assessed the importance of specific residues in the GF region for the curing. The F89I and F93L mutations in the GF region of DnaJb6b are associated with certain muscular dystrophies, although it is unclear how they disrupt DnaJB6b activity (37). When Sis1 has its GF domain replaced by that of a DnaJB6b GF domain with either of these mutations, it is altered in its ability to promote propagation of certain yeast prions (40). As Sis1 is a component of the disaggregation machinery that is required for fragmentation of prion fibers and different prions rely on this activity to different degrees, this difference in ability to support different prions could be due to reduced ability of Sis1 to cooperate in the fragmentation process. We found that neither F89I nor F93L disrupted the ability of DnaJB6b to cure cells of [URE3] (Fig. 2G). These results provide evidence that the curing does not depend on a related function of DnaJB6b and are consistent with the conclusion that the inhibitory effects of DnaJB6b on [URE3] are direct rather than through inhibition of fragmentation.
D33N Disrupts Cooperation of DnaJB6b with Hsp70; DnaJB6bΔST Retains Function
Mutations in the universally conserved HPD motif of J-proteins disrupt interaction between J-proteins and Hsp70. To confirm that the D33N mutation disrupted this interaction, we assessed the ability of DnaJB6bD33N to interact with Hsp70 in vivo. Cells lacking Ydj1 grow very slowly at 30 °C and do not grow at 37 °C. Heterologous J-proteins and even isolated J-domains can restore growth (44), suggesting that J-domain regulation of Hsp70 is a more important function of Ydj1 in vivo than substrate binding. Among all of our DnaJB6b mutants tested, only the full-length protein with the D33N substitution failed to restore growth when expressed in the ydj1Δ cells (Fig. 3A). Thus, DnaJB6b-D33N does not cooperate with Hsp70 in vivo. DnaJB6bΔST restored normal growth, showing that it functionally interacts with Hsp70.
FIGURE 3.

The D33N mutation disrupts cooperation of DnaJB6b with Hsp70, but ΔST does not; only DnaJB1 can replace Sis1. A, wild type and mutant versions of DnaJB6b regulated by the GPD promoter were expressed from plasmids in ydj1Δ cells. Cultures were diluted to an A600 of 1.0 and serially diluted, and replicate dilutions of cells were spotted onto plates and incubated for 3 days at the indicated temperatures (ev, empty vector). B, urea-denatured luciferase was diluted into refolding buffer containing the indicated J-proteins as described under “Experimental Procedures.” Curves are the averages of three experiments; errors bars represent S.D. C, transformants (six each) expressing indicated J-proteins from TRP1 plasmids in sis1Δ cells carrying SIS1 on a URA3 plasmid were grown on medium containing uracil and lacking tryptophan (−Trp) and then replica-plated onto medium containing FOA, which kills cells expressing Ura3. Aside from the Sis1 control (bottom row), only cells expressing DnaJB1 were able to lose the URA3 plasmid and support growth in the absence of Sis1 on FOA.
We further tested the ability of these mutants to cooperate with Hsp70 in an Hsp70-dependent protein refolding assay in vitro that requires J-protein cooperation (see Fig. 3B). Compared with Ydj1, reactions containing DnaJB6b had robust refolding activity, whereas those containing DnaJB6bD33N had none. Reactions containing DnaJB6bΔST had partial activity, which was expected as this region contributes to DnaJB6b function (5). Together, these results confirm that interaction of DnaJB6b with Hsp70 is not needed for the prion curing and that deleting the ST region does not destroy DnaJB6b function in vivo or in vitro.
We also tested the ability of the human J-proteins to functionally replace Sis1, which is essential for viability. DnaJB1 was the only DnaJB family member that supported growth in the absence of Sis1 (Fig. 3C), which reflects significant divergence of function of these non-complementing paralogs.
DnaJB6b Blocks Growth of Ure2 Amyloid
On the basis of its demonstrated ability to prevent amyloid formation in vitro (6, 7), we considered that DnaJB6b cures cells of [URE3] by blocking growth of Ure2 amyloid. Purified Ure2 forms amyloid spontaneously after a short lag period when nucleation of amyloid fibers occurs (45) (Fig. 4A, black line). Others showed that adding equimolar or greater Ydj1 at the start of this reaction extends the lag and reduces the yield of Ure2 amyloid significantly (46).
FIGURE 4.
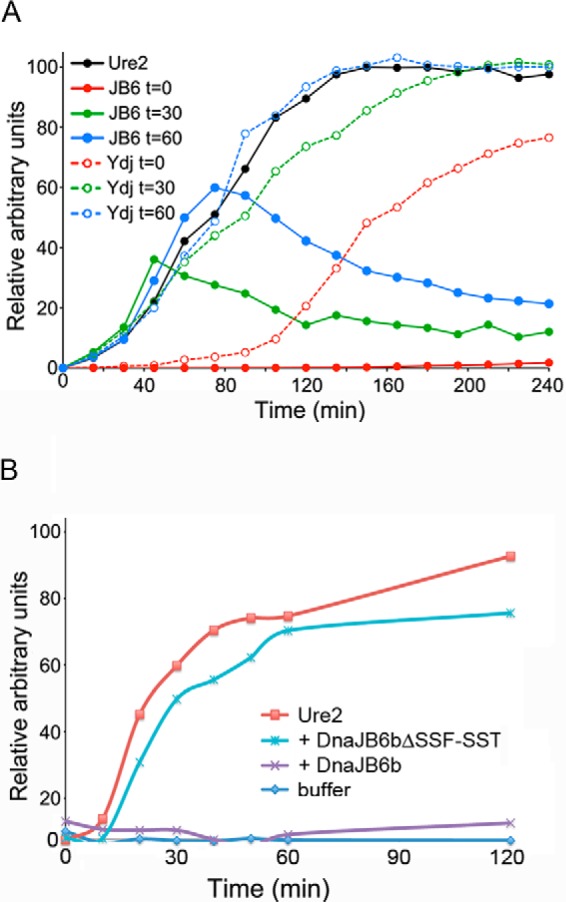
DnaJB6b blocks formation of Ure2 amyloid in vitro. A, amyloid formation of purified Ure2 at 23 °C was monitored by change in thioflavin-T fluorescence. Ure2 alone (black line) or with a 2-fold molar excess of DnaJB6b (solid lines) or Ydj1 (dashed lines) added at the start (t = 0) (red lines) or 30 (green lines) or 60 (blue lines) min after initiating the reaction. B, amyloid formation of a separate preparation of purified Ure2 alone (red line) or incubated with sub-stoichiometric amounts (1:5) of DnaJB6b (purple line) or DnaJB6bΔST (blue line) was monitored by thioflavin-T fluorescence. Panels A and B show representative individual experiments where background was subtracted, and data were normalized to the maximum fluorescence of Ure2 incubated alone, which was set at 100%.
We reproduced those results here using a 2-fold molar excess of Ydj1 (Fig. 4A, red dashed line). Also in line with earlier data, the addition of Ydj1 30 or 60 min after the initiation of the reaction, when amyloid fibers are rapidly growing, had little effect on amyloid yield or rate of growth (Fig. 4A, green and blue dashed lines). These data suggest Ydj1 can delay initiation of amyloid formation but does not have a strong effect on subsequent growth.
In clear contrast, when a similar 2-fold excess of DnaJB6b was added at the beginning of the reaction (Fig. 4A, red solid line), no Ure2 amyloid was formed for the 4-h duration of the experiment. Strikingly, when DnaJB6b was added 30 or 60 min after initiating the reaction, growth of the amyloid was arrested (Fig. 4A, green and blue solid lines, respectively). In some experiments we observed a noticeable decrease in the fluorescence signal in the reactions containing DnaJB6b that we cannot explain. We do not think it was caused by disassembly of the amyloid because it was not observed consistently, and DnaJB6b did not affect the amount, SDS resistance, or thermostability of arrested Ure2 amyloid prepared in other experiments (see below and “Experimental Procedures”). Additionally, we do not see degradation of Ure2 in other experiments that contain DnaJB6b for longer time courses (see below), so we do not think the decrease is attributable to degradation. These results suggest that DnaJB6b can cure yeast of [URE3] by blocking growth of the pre-existing Ure2 prion fibers in [URE3] cells.
Earlier work with Aβ amyloid suggests that substoichiometric levels of DnaJB6b block amyloid formation by binding pre-amyloid oligomers and that this activity depends on the ST region (8). Although the 2:1 molar ratio of DnaJB6b to Ure2 used above more likely represents conditions in cells overexpressing DnaJB6b, we repeated some experiments using a 1:5 molar ratio of DnaJB6b to Ure2. This substoichiometric amount of DnaJB6b also inhibited Ure2 amyloid formation for the 2-h duration of this experiment. Moreover, it depended on the ST region (Fig. 4B). Together our findings extend the diversity of proteins whose amyloid formation is blocked by DnaJB6b and highlight DnaJB6b as a general amyloid-inhibiting factor.
DnaJB6b Cures Weak but Not Strong Variants of [PSI+]
To continue investigating the diversity of anti-amyloid activity of DnaJB6b, we tested if it could cure cells of [PSI+], a prion composed of Sup35. Several “variants” of [PSI+] have been characterized and are classified broadly as having strong or weak Ade+ phenotypes. Strong variants confer whiter color and faster growth without adenine, whereas weak variants produce cells with a detectable pink color that often coincides with mitotic instability, especially after transient heat shock (47).
[PSI+] prion variants differ by physical characteristics of their amyloid structures. There is an inverse relationship between thermodynamic stability of prion fibers and strength of phenotype. Fibers with higher thermostability result in a weaker phenotype because they are not fragmented as readily, so they produce fewer prion seeds that provide fewer ends to recruit and deplete soluble Sup35 (48).
In addition to the endogenous strong [PSI+] variant in our wild type strain, we used previously described strong and weak variants that arose de novo (SL[PSI+]S and SL[PSI+]W) or after transfection using amyloid assembled in vitro ([PSI+]Sc4 and [PSI+]Sc37; see “Experimental Procedures”). The structural basis for variation of [PSI+] phenotypes is determined by variations in the Sup35 amyloid core (23, 27, 49).
We expressed DnaJB6b in cells carrying these variants by using strong constitutive expression as above for [URE3] and found that DnaJB6b effectively cured both weak variants of [PSI+] but did not cure the strong variants (Fig. 5). The presence of infrequent red [psi−] colonies in the empty vector controls of weak [PSI+] variants is likely due to inherent mitotic instability of these prions, which would be expected to increase after the heat shock of the transformation procedure. Thus, different prions composed of the same Sup35 protein were differentially sensitive to the anti-amyloid activity of DnaJB6b. Ydj1 did not cure cells of any of these [PSI+] prion variants, which uncovers another distinction in the ways Ydj1 and DnaJB6b antagonize prions.
FIGURE 5.

DnaJB6b cures weak but not strong variants of [PSI+]. Different pairs of isogenic yeast strains harboring the indicated [PSI+] variant (see “Experimental Procedures”) were transformed with an empty vector (top row) or a plasmid expressing GPD-driven DnaJB6b (bottom row). Representative 2-cm2 sections of primary transformation plates incubated for 3 days at 30 °C are shown.
DnaJB6b Selectively Inhibits Growth of Sup35 Amyloid Fibers with Higher Thermal Stability
One possibility to explain why DnaJB6b did not cure strong [PSI+] variants is that the higher number of propagons per cell that is characteristic of strong variants of [PSI+] (see Table 2) is above a threshold that can be eliminated by DnaJB6b. Another is that DnaJB6b could block formation of some Sup35 amyloid structures but not others.
To test the latter hypothesis, we first prepared Sup35NM amyloid overnight at 4 °C (NM-4) or 37 °C (NM-37) with and without DnaJB6b. Others showed that NM-4 amyloid fibers are less thermodynamically stable and produce strong variants of [PSI+] when used to infect yeast, whereas NM-37 fibers are more stable and produce weak prions (48). To compare the thermodynamic stabilities of our resulting NM-4 and NM-37 reaction products, we assessed their temperature-dependent SDS resistance using polyacrylamide gel mobility as described previously (34).
We found the thermostabilities of NM-4 and NM-37 fibers formed in our hands (Fig. 6A, left, and B, solid lines; Tm = 54 ± 2 °C and 78 ± 7 °C, respectively) were very similar to those reported earlier (56 °C and 77 °C; see Ref. 34). The addition of a 2-fold molar excess of DnaJB6b did not significantly affect the formation or thermal stability of amyloid when reactions were incubated at 4 °C (Fig. 6A, top right and B, gray dashed line; Tm =53 ± 2 °C), consistent with the inability of DnaJB6b to cure strong [PSI+] prions in vivo.
FIGURE 6.

DnaJB6b blocks formation of Sup35NM amyloid at 37 °C but not 4 °C. A, representative portions of Coomassie-stained gels showing the SDS resistance and mobility of Sup35NM made in the absence or presence of DnaJB6b at 4 °C or 37 °C as indicated. B, band intensities from gels as in panel A are plotted as a function of temperature. Gray lines, 4 °C; black lines, 37 °C; solid lines, without DnaJB6b; dashed lines, with DnaJB6b. Data were fitted to a sigmoidal equation. Curves are the average fits of three independent experiments, and error bars indicate S.D. C and D are plots of gel mobility as in panel B with increasing amounts of DnaJB6b, as indicated, at 4 °C and 37 °C, respectively.
Remarkably, however, DnaJB6b prevented formation of SDS-resistant material in identical reaction mixtures incubated at 37 °C (Fig. 6A, bottom right). We then assessed the effects of adding increasing amounts of DnaJB6b to the reactions. Even though a 5-fold molar excess of DnaJB6b did not affect formation of Sup35NM amyloid at 4 °C (Fig. 6C), an equimolar ratio of DnaJB6b abolished formation of amyloid at 37 °C (Fig. 6D).
Taken together, our results show that DnaJB6b selectively inhibited formation of Sup35NM amyloid only under conditions that produce fibers with the higher thermal stability that is characteristic of weak prions. Although we have not ruled out the possibility that the lower seed number of weak [PSI+] variants contributes to their sensitivity to curing by DnaJB6b, all our data align with the conclusion that the selective curing of the weak variants is due to differences in how DnaJB6b recognizes or acts on structurally distinct Sup35 amyloids of the different variants.
Stability of Ure2 Amyloid Is Similar to NM-37 and Is Not Affected by Assembly Reaction Temperature
Because DnaJB6b cured cells of [URE3] and weak [PSI+] variants, but not strong [PSI+] variants, we anticipated that the thermostability of Ure2 amyloid would be more similar to NM-37 than to NM-4. We evaluated the effect of DnaJB6b on Ure2 amyloid formation as in Fig. 6 but used Ure2 in place of NM. Specifically, we incubated purified Ure2 with or without DnaJB6b overnight (18 h) at 23 °C and then assessed the thermostability of the resulting products.
Ure2 amyloid that formed in the absence of DnaJB6 consistently produced a sigmoidal relationship between relative band intensity and temperature, with a calculated Tm of 79 ± 4 °C (Fig. 7, A and B, solid line). The products of this reaction exhibited a melting curve very similar to what was observed for NM-37.
FIGURE 7.

Thermodynamic stability of Ure2 amyloid resembles that of Sup35NM-37 and is not affected by reaction temperature. A, representative portion of a Coomassie-stained gel showing the SDS resistance and mobility of purified Ure2 incubated overnight alone (left) or with DnaJB6b (right) at 23 °C. Products of the reactions were incubated for 10 min at the indicated temperatures (Temp) immediately before loading on the gel. B, band intensities from gels as run in panel A are plotted as a function of temperature. Solid line, Ure2 alone, dashed line, Ure2 with DnaJB6b. Data were fitted to a sigmoidal equation. Curves are the average fits of three independent experiments, and error bars indicate S.D. C, a plot as in panel B was derived from data as in panel A except that overnight reactions were incubated at 4 °C (Ure2–4) without (solid lines) or with (dashed lines) DnaJB6b. D, as in panel C, except that overnight reactions were incubated at 37 °C (Ure2-37) without (solid lines) or with (dashed lines) DnaJB6b.
Products of similar reactions that contained a 2-fold excess of DnaJB6b gave rise to a melting curve profile that was roughly flat and linear (Fig. 7B, dashed line), indicative of the lack of SDS resistant material. These results are consistent with the experiments using thioflavin-T that show DnaJB6b prevented Ure2 from forming amyloid.
Although the relationship between the temperature at which Sup35NM amyloid forms and the thermodynamic stability of resulting fibers is well established, similar investigation into the effect of reaction temperature on Ure2 amyloid stability is lacking. To assess whether reaction temperature influences the stability of Ure2 amyloid, we prepared Ure2 amyloid at 4 °C and 37 °C and compared it to amyloid made at 23 °C using the SDS-resistance gel mobility assay. Similar preparations containing DnaJB6b were also made.
The thermodynamic stability of Ure2 amyloid prepared at each of the temperatures was similar (Fig. 7, solid lines; Ure2–4 Tm = 83 ± 10 °C; Ure2-37 Tm = 70 ± 12 °C). Additionally, DnaJB6b blocked formation of Ure2 amyloid at all temperatures (Fig. 7, dashed lines). These findings suggest that using different temperatures did not help Ure2 adopt amyloid conformations that escape the anti-amyloid activity of DnaJB6b and show that DnaJB6b retains its ability to inhibit amyloid formation at 4 °C. The latter result shows that the inability of DnaJB6b to inhibit formation of Sup35NM at 4 °C is not due simply to a loss of this activity at the lower temperature.
Discussion
We show that overexpression of the human J-protein DnaJB6b cured yeast of [URE3] prions very effectively. We also show that DnaJB6b blocked spontaneous formation of Ure2 amyloid fibers and the growth of pre-existing fibers in vitro. The ability of DnaJB6b to block growth of Ure2 amyloid suggests it can cure cells of [URE3] by blocking growth of the Ure2 prion fibers that are already present in [URE3] cells. These results are consistent with the way DnaJB6 inhibits formation of amyloid by polyQ and Aβ peptides in vitro, and this conclusion is in line with the way overexpression of DnaJB6b is proposed to block polyQ aggregation in mammalian cells (6, 7). The clear differences in the kinetics of curing by DnaJB6b and Ydj1, the involvement of other chaperones in the curing by Ydj1, and the inability of mutations that disrupt cooperation of DnaJB6b with chaperones to prevent curing suggest that Ydj1 cures cells of [URE3] by a different mechanism.
Many of the anti-prion effects of altering abundance or activity of chaperones and co-chaperones can be explained by disrupted function of the Hsp104-driven fragmentation machinery, a system unique to yeast prions. Here, however, our data are more consistent with DnaJB6b acting directly on prion fibers to prevent their continued growth. Our findings that DnaJB6b-D33N cures [URE3] and that increasing Sis1 expression does not interfere with curing by wild type DnaJB6b are consistent with DnaJB6b acting independently of this machinery. The failure of the F89I and F93L mutations to disrupt curing, even though similar mutations in Sis1 disrupt its ability to promote propagation of prions (40), is also consistent with DnaJb6b acting directly. If DnaJB6b does not significantly inhibit the fragmentation of prion fibers, then the non-growing fibers in cells expressing DnaJB6b might be increasingly fragmented until they no longer function efficiently as prion seeds and perhaps are then handled by protein refolding or degradation pathways. Such a scenario could be consistent with the observed lag before cells induced to express DnaJB6b begin to lose [URE3] followed by the accelerating rate of [URE] loss once curing begins.
We have not ruled out, however, that the variable effectiveness of DnaJB6b on different prion variants is due to different consequences of an inhibition of prion fiber fragmentation. Although our data and that of others (5–7) show the predominant effect of DnaJB6b on amyloid formation is to prevent initiation and assembly of amyloid fibers, it might also be able to inhibit fragmentation of fibers in yeast cells. Yeast prion variants that produce fewer seeds per cell and are more strongly dependent on fragmentation for their propagation could be more sensitive to such inhibition and be cured more effectively by DnaJB6b. More experiments will be needed to test such a mechanism.
The finding that DnaJB6b cured only weak variants of [PSI+] was initially surprising because DnaJB6b had a general anti-amyloid activity for other amyloid-forming proteins, and, in particular, because all variants of [PSI+] prions are composed of Sup35. On the whole, our data are consistent with the simple explanation that the specificity in [PSI+] variant curing is related to the physical characteristics of the different amyloids. DnaJB6b blocked formation of Sup35NM amyloid at 37 °C, conditions that normally produce fibers that create DnaJB6b-curable weak [PSI+] variants when used to infect yeast. However, it did not block formation of Sup35NM amyloid at 4 °C, which produces strong [PSI+] variants that are not cured by DnaJB6b. Notably, NM-4 amyloid, which was insensitive to inhibition by DnaJB6b, is also more highly infectious than NM-37 amyloid (50).
These observations are in line with the idea that differences in phenotypic variation of Sup35 prions are encoded in the particular folds of their amyloid cores (23, 27). Our data imply that action of DnaJB6b is limited to only some of these structurally distinct prions even though they are composed of the same protein.
We also demonstrate that, unlike Sup35NM, the thermal stability of Ure2 amyloid is not affected by the temperature at which the fibers are prepared, which suggests that the structures of Ure2 amyloid formed at the different temperatures are similar. Accordingly, we found that DnaJB6b blocked formation of Ure2 amyloid at all tested temperatures. Compared with Sup35, the lower amino acid diversity in the Ure2 amyloid-forming domain or lack of an adjacent charged domain might provide possible explanations for the reduced structural variation. Thus, although phenotypic variants of [URE3] exist (51, 52), which could reflect differences in how they interact with the disaggregation machinery, Ure2 might simply be unable to adopt conformations that escape DnaJB6b because of restrictions on secondary structures exerted by its primary sequence.
Recent studies have noted genetic similarities between [URE3] and weak [PSI+] variants. For example, mutations in Sis1, which plays a crucial role in prion fragmentation, disrupt mitotic stability of [URE3] and weak [PSI+] variants but do not affect strong [PSI+] variants (25, 53–55). Additionally, elevating the Hsp70 Ssa1 cures cells of [URE3] and weak [PSI+] but not strong [PSI+] (24, 56). We show here that NM-37 amyloid is thermodynamically similar to Ure2 amyloid prepared at any temperature, with a Tm of 70–80 °C, whereas NM-4 has a Tm of ∼55 °C. These results provide evidence for a structural basis of the genetic similarities and support a view that sturdier amyloids depend more stringently on the molecular chaperone machinery for their fragmentation.
The ability of DnaJB6 to protect human cells from polyQ toxicity and to prevent polyQ and Aβ peptides from forming amyloid makes it an attractive therapeutic target for amyloidoses, such as Huntington disease (8). The differential sensitivity of structurally distinct amyloids of the same protein to inhibition by DnaJB6b should raise concerns when considering the potential of DnaJB6b or other similar proteins for such uses. This caveat becomes especially concerning when one considers that for Sup35NM, amyloids resistant to DnaJB6b are more highly infectious than other forms (50) and are more effectively selected for when DnaJB6b expression is elevated.
Author Contributions
M. R. conceived the ideas for experimental approaches, conducted most experiments, analyzed results, and wrote the paper with D. C. M. R. S. initiated the project, analyzed the data, conducted kinetic curing experiments, performed amyloid assays, and contributed insightful comments about the paper. B.-L. R. conducted seed count the experiments. D. C. M. conceived ideas for the project, analyzed the data, and constructed the strains.
Acknowledgments
We thank our National Institutes of Health and NIDDK colleagues for helpful discussions, R. Wickner and L. Greene for the gift of plasmids, Sue Liebman and Jonathan Weissman for yeast strains, and Harm Kampinga for cDNA clones of human chaperones.
This work was supported by the Intramural Program of the National Institutes of Health, NIDDK. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
M. Reidy, R. Sharma, B.-L. Roberts, and D. C. Masison, unpublished observation.
- polyQ
- polyglutamine.
References
- 1. Nagai Y., Fujikake N., Popiel H. A., and Wada K. (2010) Induction of molecular chaperones as a therapeutic strategy for the polyglutamine diseases. Curr. Pharm. Biotechnol. 11, 188–197 [DOI] [PubMed] [Google Scholar]
- 2. Warrick J. M., Chan H. Y., Gray-Board G. L., Chai Y., Paulson H. L., and Bonini N. M. (1999) Suppression of polyglutamine-mediated neurodegeneration in Drosophila by the molecular chaperone HSP70. Nat. Genet. 23, 425–428 [DOI] [PubMed] [Google Scholar]
- 3. Douglas P. M., Treusch S., Ren H. Y., Halfmann R., Duennwald M. L., Lindquist S., and Cyr D. M. (2008) Chaperone-dependent amyloid assembly protects cells from prion toxicity. Proc. Natl. Acad. Sci. U.S.A. 105, 7206–7211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kampinga H. H., and Craig E. A. (2010) The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat. Rev. Mol. Cell Biol. 11, 579–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hageman J., Rujano M. A., van Waarde M. A., Kakkar V., Dirks R. P., Govorukhina N., Oosterveld-Hut H. M., Lubsen N. H., and Kampinga H. H. (2010) A DNAJB chaperone subfamily with HDAC-dependent activities suppresses toxic protein aggregation. Mol. Cell 37, 355–369 [DOI] [PubMed] [Google Scholar]
- 6. Gillis J., Schipper-Krom S., Juenemann K., Gruber A., Coolen S., van den Nieuwendijk R., van Veen H., Overkleeft H., Goedhart J., Kampinga H. H., and Reits E. A. (2013) The DNAJB6 and DNAJB8 protein chaperones prevent intracellular aggregation of polyglutamine peptides. J. Biol. Chem. 288, 17225–17237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Månsson C., Kakkar V., Monsellier E., Sourigues Y., Härmark J., Kampinga H. H., Melki R., and Emanuelsson C. (2014) DNAJB6 is a peptide-binding chaperone which can suppress amyloid fibrillation of polyglutamine peptides at substoichiometric molar ratios. Cell Stress Chaperones 19, 227–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Månsson C., Arosio P., Hussein R., Kampinga H. H., Hashem R. M., Boelens W. C., Dobson C. M., Knowles T. P., Linse S., and Emanuelsson C. (2014) Interaction of the molecular chaperone DNAJB6 with growing amyloid-β 42 (Aβ42) aggregates leads to sub-stoichiometric inhibition of amyloid formation. J. Biol. Chem. 289, 31066–31076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Reidy M., and Masison D. C. (2014) Yeast prions help identify and define chaperone interaction networks. Curr. Pharm. Biotechnol. 15, 1008–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cox B., Ness F., and Tuite M. (2003) Analysis of the generation and segregation of propagons: entities that propagate the [PSI+] prion in yeast. Genetics 165, 23–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chernoff Y. O., Lindquist S. L., Ono B., Inge-Vechtomov S. G., and Liebman S. W. (1995) Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+]. Science 268, 880–884 [DOI] [PubMed] [Google Scholar]
- 12. Paushkin S. V., Kushnirov V. V., Smirnov V. N., and Ter-Avanesyan M. D. (1996) Propagation of the yeast prion-like [psi+] determinant is mediated by oligomerization of the SUP35-encoded polypeptide chain release factor. EMBO J. 15, 3127–3134 [PMC free article] [PubMed] [Google Scholar]
- 13. Glover J. R., and Lindquist S. (1998) Hsp104, Hsp70, and Hsp40: a novel chaperone system that rescues previously aggregated proteins. Cell 94, 73–82 [DOI] [PubMed] [Google Scholar]
- 14. Hung G. C., and Masison D. C. (2006) N-terminal domain of yeast Hsp104 chaperone is dispensable for thermotolerance and prion propagation but necessary for curing prions by Hsp104 overexpression. Genetics 173, 611–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Higurashi T., Hines J. K., Sahi C., Aron R., and Craig E. A. (2008) Specificity of the J-protein Sis1 in the propagation of 3 yeast prions. Proc. Natl. Acad. Sci. U.S.A. 105, 16596–16601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tipton K. A., Verges K. J., and Weissman J. S. (2008) In vivo monitoring of the prion replication cycle reveals a critical role for Sis1 in delivering substrates to Hsp104. Mol. Cell 32, 584–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tessarz P., Mogk A., and Bukau B. (2008) Substrate threading through the central pore of the Hsp104 chaperone as a common mechanism for protein disaggregation and prion propagation. Mol. Microbiol. 68, 87–97 [DOI] [PubMed] [Google Scholar]
- 18. Reidy M., Miot M., and Masison D. C. (2012) Prokaryotic chaperones support yeast prions and thermotolerance and define disaggregation machinery interactions. Genetics 192, 185–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sporn Z. A., and Hines J. K. (2015) Hsp40 function in yeast prion propagation: amyloid diversity necessitates chaperone functional complexity. Prion 9, 80–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wickner R. B. (1994) Evidence for a prion analog in S. cerevisiae: the [URE3] non-Mendelian genetic element as an altered URE2 protein. Science 264, 566–569 [DOI] [PubMed] [Google Scholar]
- 21. Moriyama H., Edskes H. K., and Wickner R. B. (2000) [URE3] prion propagation in Saccharomyces cerevisiae: requirement for chaperone Hsp104 and curing by overexpressed chaperone Ydj1p. Mol. Cell. Biol. 20, 8916–8922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sharma D., and Masison D. C. (2008) Functionally redundant isoforms of a yeast Hsp70 chaperone subfamily have different antiprion effects. Genetics 179, 1301–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tanaka M., Chien P., Naber N., Cooke R., and Weissman J. S. (2004) Conformational variations in an infectious protein determine prion strain differences. Nature 428, 323–328 [DOI] [PubMed] [Google Scholar]
- 24. Mathur V., Hong J. Y., and Liebman S. W. (2009) Ssa1 overexpression and [PIN(+)] variants cure [PSI(+)] by dilution of aggregates. J. Mol. Biol. 390, 155–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Reidy M., Sharma R., Shastry S., Roberts B. L., Albino-Flores I., Wickner S., and Masison D. C. (2014) Hsp40s specify functions of Hsp104 and Hsp90 protein chaperone machines. PLoS Genet. 10, e1004720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Funk M., Niedenthal R., Mumberg D., Brinkmann K., Rönicke V., and Henkel T. (2002) Vector systems for heterologous expression of proteins in Saccharomyces cerevisiae. Methods Enzymol. 350, 248–257 [DOI] [PubMed] [Google Scholar]
- 27. Gorkovskiy A., Thurber K. R., Tycko R., and Wickner R. B. (2014) Locating folds of the in-register parallel β-sheet of the Sup35p prion domain infectious amyloid. Proc. Natl. Acad. Sci. U.S.A. 111, E4615–E4622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Umland T. C., Taylor K. L., Rhee S., Wickner R. B., and Davies D. R. (2001) The crystal structure of the nitrogen regulation fragment of the yeast prion protein Ure2p. Proc. Natl. Acad. Sci. U.S.A. 98, 1459–1464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Deleted in proof
- 30. Meriin A. B., Zhang X., He X., Newnam G. P., Chernoff Y. O., and Sherman M. Y. (2002) Huntington toxicity in yeast model depends on polyglutamine aggregation mediated by a prion-like protein Rnq1. J. Cell Biol. 157, 997–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhao X., Park Y. N., Todor H., Moomau C., Masison D., Eisenberg E., and Greene L. E. (2012) Sequestration of Sup35 by aggregates of huntingtin fragments causes toxicity of [PSI+] yeast. J. Biol. Chem. 287, 23346–23355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sharma D., and Masison D. C. (2011) Single methyl group determines prion propagation and protein degradation activities of yeast heat shock protein (Hsp)-70 chaperones Ssa1p and Ssa2p. Proc. Natl. Acad. Sci. U.S.A. 108, 13665–13670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Genest O., Reidy M., Street T. O., Hoskins J. R., Camberg J. L., Agard D. A., Masison D. C., and Wickner S. (2013) Uncovering a region of heat shock protein 90 important for client binding in E. coli and chaperone function in yeast. Mol. Cell 49, 464–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tanaka M., and Weissman J. S. (2006) An efficient protein transformation protocol for introducing prions into yeast. Methods Enzymol. 412, 185–200 [DOI] [PubMed] [Google Scholar]
- 35. Lopez N., Aron R., and Craig E. A. (2003) Specificity of class II Hsp40 Sis1 in maintenance of yeast prion [RNQ+]. Mol. Biol. Cell 14, 1172–1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Harms M. B., Sommerville R. B., Allred P., Bell S., Ma D., Cooper P., Lopate G., Pestronk A., Weihl C. C., and Baloh R. H. (2012) Exome sequencing reveals DNAJB6 mutations in dominantly inherited myopathy. Ann. Neurol. 71, 407–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sarparanta J., Jonson P. H., Golzio C., Sandell S., Luque H., Screen M., McDonald K., Stajich J. M., Mahjneh I., Vihola A., Raheem O., Penttilä S., Lehtinen S., Huovinen S., Palmio J., Tasca G., Ricci E., Hackman P., Hauser M., Katsanis N., and Udd B. (2012) Mutations affecting the cytoplasmic functions of the co-chaperone DNAJB6 cause limb-girdle muscular dystrophy. Nat. Genet. 44, 450–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nigro V., and Savarese M. (2014) Genetic basis of limb-girdle muscular dystrophies: the 2014 update. Acta Myol. 33, 1–12 [PMC free article] [PubMed] [Google Scholar]
- 39. Ruggieri A., Brancati F., Zanotti S., Maggi L., Pasanisi M. B., Saredi S., Terracciano C., Antozzi C., D Apice M. R., Sangiuolo F., Novelli G., Marshall C. R., Scherer S. W., Morandi L., Federici L., Massa R., Mora M., and Minassian B. A. (2015) Complete loss of the DNAJB6 G/F domain and novel missense mutations cause distal-onset DNAJB6 myopathy. Acta Neuropathol. Commun. 3, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Stein K. C., Bengoechea R., Harms M. B., Weihl C. C., and True H. L. (2014) Myopathy-causing mutations in an HSP40 chaperone disrupt processing of specific client conformers. J. Biol. Chem. 289, 21120–21130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gokhale K. C., Newnam G. P., Sherman M. Y., and Chernoff Y. O. (2005) Modulation of prion-dependent polyglutamine aggregation and toxicity by chaperone proteins in the yeast model. J. Biol. Chem. 280, 22809–22818 [DOI] [PubMed] [Google Scholar]
- 42. Gong H., Romanova N. V., Allen K. D., Chandramowlishwaran P., Gokhale K., Newnam G. P., Mieczkowski P., Sherman M. Y., and Chernoff Y. O. (2012) Polyglutamine toxicity is controlled by prion composition and gene dosage in yeast. PLoS Genet. 8, e1002634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sharma D., Stanley R. F., and Masison D. C. (2009) Curing of yeast [URE3] prion by the Hsp40 cochaperone Ydj1p is mediated by Hsp70. Genetics 181, 129–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sahi C., and Craig E. A. (2007) Network of general and specialty J protein chaperones of the yeast cytosol. Proc. Natl. Acad. Sci. U.S.A. 104, 7163–7168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Taylor K. L., Cheng N., Williams R. W., Steven A. C., and Wickner R. B. (1999) Prion domain initiation of amyloid formation in vitro from native Ure2p. Science 283, 1339–1343 [DOI] [PubMed] [Google Scholar]
- 46. Lian H. Y., Zhang H., Zhang Z. R., Loovers H. M., Jones G. W., Rowling P. J., Itzhaki L. S., Zhou J. M., and Perrett S. (2007) Hsp40 interacts directly with the native state of the yeast prion protein Ure2 and inhibits formation of amyloid-like fibrils. J. Biol. Chem. 282, 11931–11940 [DOI] [PubMed] [Google Scholar]
- 47. Newnam G. P., Birchmore J. L., and Chernoff Y. O. (2011) Destabilization and recovery of a yeast prion after mild heat shock. J. Mol. Biol. 408, 432–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tanaka M., Collins S. R., Toyama B. H., and Weissman J. S. (2006) The physical basis of how prion conformations determine strain phenotypes. Nature 442, 585–589 [DOI] [PubMed] [Google Scholar]
- 49. Wong S. H., and King C. Y. (2015) Amino acid proximities in two Sup35 prion strains revealed by chemical cross-Linking. J. Biol. Chem. 290, 25062–25071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ohhashi Y., Ito K., Toyama B. H., Weissman J. S., and Tanaka M. (2010) Differences in prion strain conformations result from non-native interactions in a nucleus. Nat. Chem. Biol. 6, 225–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Schlumpberger M., Prusiner S. B., and Herskowitz I. (2001) Induction of distinct [URE3] yeast prion strains. Mol. Cell. Biol. 21, 7035–7046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Brachmann A., Baxa U., and Wickner R. B. (2005) Prion generation in vitro: amyloid of Ure2p is infectious. EMBO J. 24, 3082–3092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Harris J. M., Nguyen P. P., Patel M. J., Sporn Z. A., and Hines J. K. (2014) Functional diversification of hsp40: distinct j-protein functional requirements for two prions allow for chaperone-dependent prion selection. PLoS Genet. 10, e1004510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kirkland P. A., Reidy M., and Masison D. C. (2011) Functions of yeast Hsp40 chaperone Sis1p dispensable for prion propagation but important for prion curing and protection from prion toxicity. Genetics 188, 565–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Stein K. C., and True H. L. (2014) Structural variants of yeast prions show conformer-specific requirements for chaperone activity. Mol. Microbiol. 93, 1156–1171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Schwimmer C., and Masison D. C. (2002) Antagonistic interactions between yeast [PSI+] and [URE3] prions and curing of [URE3] by Hsp70 protein chaperone Ssa1p but not by Ssa2p. Mol. Cell. Biol. 22, 3590–3598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sikorski R. S., and Hieter P. (1989) A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122, 19–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Christianson T. W., Sikorski R. S., Dante M., Shero J. H., and Hieter P. (1992) Multifunctional yeast high copy number shuttle vectors. Gene 110, 119–122 [DOI] [PubMed] [Google Scholar]