

Abstract
Salidroside, an active ingredient isolated from Rhodiola rosea, has shown to exert protective effects against chronic hypoxia-induced pulmonary arterial hypertension (PAH). However, the underlying mechanisms were not well known. Based on our recent reports, we predicted the involvement of adenosine monophosphate-activated protein kinase (AMPK) mediated effects in salidroside regulation of PAH. Firstly, to prove the hypothesis, rats were exposed to chronic hypoxia and treated with increasing concentrations of salidroside or a selective AMPK activator-5’-aminoimidazole-4-carboxamide ribonucleoside (AICAR) for 4 weeks. After salidroside or AICAR treatment, the chronic hypoxia-induced right ventricular hypertrophy and pulmonary artery remodeling were attenuated. Then the effects of salidroside or AICAR on hypoxia-induced excess cellular proliferation and apoptosis resistance of pulmonary arterial smooth muscle cells (PASMCs), which contributed to pulmonary arterial remodeling, were investigated. Our results suggested salidroside, as well as AICAR, reversed hypoxia-induced PASMCs proliferation and apoptosis resistance while AMPK inhibitor Compound C enhanced the effects of hypoxia. To reveal the potential cellular mechanisms, activation of AMPKα1 and expression of the genes related to proliferation and apoptosis were analyzed in PASMCs after salidroside treatment under hypoxia conditions. The results demonstrated salidroside as well as AICAR might inhibit chronic hypoxia-induced PASMCs proliferation via AMPKα1-P53-P27/P21 pathway and reverse apoptosis resistance via AMPKα1-P53-Bax/Bcl-2-caspase 9-caspase 3 pathway.
Keywords: Salidroside, pulmonary arterial hypertension, AMPKα1, apoptosis, proliferation
Introduction
Pulmonary arterial hypertension (PAH) is a devastating disease characterized by increased pulmonary vascular resistance and increased pulmonary arterial pressure, ultimately leading to right ventricular failure and death [1-3]. Active vasoconstriction and vascular remodeling due to the abnormal growth, the excess cellular proliferation, and the apoptosis resistance of pulmonary arterial smooth muscle cells (PASMCs) are the main events in pathology of PAH [4,5]. Therefore, inhibition of the cell proliferation or induction of the cell apoptosis may be an efficient therapeutic strategy for PAH [6,7]. Chronic hypoxia-induced PAH is a common type of PAH, mainly secondary to the disorders of the respiratory systems and links chronic obstructive pulmonary disease (COPD) with corpulmonale. Hypoxia is also a well-established stimulus to construct animal models of PAH in which hypoxia induces the survival and proliferation of PASMCs [8].
Salidroside is a major bioactive marker substance isolated from Rhodiola rosea, which is used to relieve high altitude sickness and acute exacerbation of PAH [9]. A series of evidence has proven that salidroside has many biological properties such as anti-inflammation [10], anti-oxidation [11], anti-stress, anti-cancer, and enhancing immune effects [9,12,13]. Recently, an in vitro study has revealed the inhibitory effects of salidroside on cell proliferation in PASMCs [14]. Furthermore, our previous reports suggested that salidroside can attenuate chronic hypoxia-induced PAH via adenosine receptor 2a (A2aR) related apoptosis pathway [15]. However, knockout of A2aR could not completely block the protective effects of salidroside against PAH (data not shown), suggesting additional molecular mechanisms existing in the cellular process.
Adenosine monophosphate-activated protein kinase (AMPK), a serine/threonine protein kinase, plays a critical role in regulation of energy metabolic homeostasis by switching on catabolic pathways while switching off anabolism [16]. AMPK exists as a heterotrimeric protein with a catalytic α subunit and regulatory β and γ subunits [17]. There are two isoforms of α (α1 and α2) and β (β1 and β2) and three γ subunits (γ1, γ2, and γ3). AMPK can be activated by allosteric regulation via an increased ratio of adenosine monophosphate (AMP) to adenosine triphosphate (ATP), and its activity is maintained by the inhibition of dephosphosrylation through ADP binding and by the phsophorylation of the α subunit (T172) via upstream kinases [18-21]. AMPK is known to play a critical role in cell-regulation, the decision to enter proliferation, autophagy, or apoptosis, and other cell-fate decisions. The activation of AMPK plays an important role in cardiovascular protection [22-24] and a few studies have revealed the involvement of AMPK activation in PAH. Our previous study demonstrates elevated activation of AMPKα1 by AICAR attenuated chronic hypoxia-induced PAH via inhibiting proliferation and enhancing apoptosis in PASMCs [25]. Igata et al. reported that AICAR significantly inhibits proliferation of human aortic SMCs induced by both platelet-derived growth factor-BB (PDGF-BB) and fetal calf serum (FCS) by blocking cell cycle progression through upregulation of P53 and P21 [26]. In addition, salidroside can block progression through G0/G1 to S phase of the cell cycle which is associated with the inhibition of the P27 expression [14]. Considering that both P21 and P27 are downregulators of P53 and P53 can also modulate apoptosis by regulating expressions of Bax and Bcl-2, we predict salidroside might function by an AMPKα1-P53-P21/P27 axis to inhibit hypoxia-induced PASMCs proliferation and by an AMPKα1-p53-Bax/Bcl-2 axis to reverse hypoxia-induced apoptosis resistance, rebalancing the proliferation and apoptosis to inhibit pulmonary arterial remodeling.
In the present study, both in vivo and in vitro experiments were performed to examine the effects of salidroside on chronic hypoxia-induced PAH and roles of AMPKα1 pathways in the modulation of cell proliferation and apoptosis of PASMCs.
Materials and methods
Materials
Salidroside, AMPK agonist 5’-aminoimidazole-4-carboxamide ribonucleoside (AICAR), AMPK inhibitor Compound C, collagenase type I, 4’, 6-Diamidino-2-phenylindole dihydrochloride (DAPI) were obtained from Sigma (St Louis, MO, USA). Trizol and Sso Advanced SYBR Green Supermix were obtained from Invitrogen (Carlsbad, CA, USA) and Bio-Rad (Hercules, CA, USA), respectively. Fetal bovine serums (FBS), penicillin G, streptomycin, and Dulbecco’s Modified Eagle Medium (DMEM, high glucose) were obtained from Gibco BRL (Gaithersburg, MD, USA). Smooth Muscle Cell Growth Medium-2 (SmGM-2) was purchased from Promocell (Gaithersburg, MD, USA). The rabbit antibodies against total AMPKα1, P53, P27, P21, Bax, Casp 9, β-actin, and α-smooth muscle actin (SMA) were obtained from Abcam (Cambridge, UK). The rabbit antibodies against PCNA, α-actin, GAPDH and phosphorylated AMPKα1 were purchased from Santa Cruz (CA, USA). The rabbit antibodies against Bcl-2 and Casp 3 were purchased from Cell Signal Technology (CST, MA, USA). Cell counting kit-8 (CCK-8) was purchased from Dojindo Laboratories (Kumamoto, Japan). Alexa fluor 488-labeled goat anti-rabbit IgG (H+L) was purchased from Molecular Probes (Eugene, OR, USA).
Experimental animals and chronic hypoxia model of PAH
Adult male Sprague-Dawley rats (weight 180-220 g) were obtained from the Laboratory Animal Center of Wenzhou Medical College, Wenzhou, Zhejiang, China. Animal housing and experimental protocols were approved by Wenzhou Medical University and all the methods were carried out in accordance with the approved guidelines. The rats were randomly assigned to six groups (10 rats per group): normal control group (N), hypoxia group (H), hypoxia plus salidroside groups (2 mg/kg, 8 mg/kg, and 32 mg/kg, named by HS2, HS8, and HS32, respectively), and hypoxia plus AMPK agonist AICAR (1 mg/kg). Intraperitoneal injection was given half an hour before rats were put into hypoxia chamber. The normoxia control group was injected with saline and exposed to room air whereas the hypoxia group was exposed to 8%-11% O2 (8 hours/day) with the same injection. Salidroside or AICAR were injected at the dose according to the group destination every day. The hypoxia exposure lasts for four weeks.
Measurements of RV hypertrophy
After hypoxia exposure, rats were anesthetized by an intraperitoneal injection of 5% chloral hydrate (400 mg/kg). Two home-made polyethylene (PE) catheters (with inside diameter as 0.9 mm and outside diameter as 1.1 mm), prefilled with heparin, were connected to the pressure transducers (PowerLab 8/30 multi-channel biological signal recording system, AD Instruments, Colorado Springs, CO, Australia), and inserted into the right ventricle and left pulmonary artery, respectively. Then the mean pulmonary arterial pressure (mPAP) and the mean carotid arterial pressure (mCAP) were recorded. Next the rats were sacrificed by bleeding to death and the hearts were dissected out, divided into right ventricle (RV), left ventricle (LV), and septum (S), washed by PBS, and weighted. The weight ratio of RV to LV plus S was calculated as an index to reflect RV hypertrophy.
Detection of pulmonary artery remodeling
The left lung of every rat was dissected longitudinally at the portapulmonis, fixed in 4% paraformaldehyde overnight, embedded in paraffin, and sectioned at 4 μm thick. After hematoxylin and eosin (HE) staining and elastic fiber staining, the structure remodeling of the pulmonary arteries were characterized by microscopic evaluation. The pulmonary arteries (external diameters of 50-200 μm) were chosen randomly and analyzed with Image-Pro Plus, Version 6.0 (Media Cybernetics, USA). The pulmonary artery medial smooth muscle cell layer (PAMT), the percentage of wall areas to total areas (WA/TA%), and the density of nuclei in the medial SMCs were calculated to evaluate pulmonary artery remodeling.
Ultrastructural examination of pulmonary arteries
The lung tissues closed to the lung hilus was sectioned into small pieces (approximately 1×1×3 mm3), fixed with 2.5% glutaraldehyde and 1% osmic acid, dehydrated with acetone, and embedded in epoxy resin 812. And then the fixed tissues were cut into ultrathin sections by ultra-microtome slices V (LKV, Stockholm, Sweden), stained with uranyl acetate and lead citrate, and examined by a Hitachi H-600 transmission electron microscopy (Hitachi, Japan) to evaluate the ultra-structural changes in pulmonary arterioles.
Isolation and cell culture of rat PASMCs
Adult male Sprague-Dawley rats were killed by intraperitoneal injection of 5% chloral hydrate followed by cervical dislocation that was approved by the guideline of Wenzhou Medical University and National Institutes of Health Standards of Animal Care. The heart and lungs were removed and pulmonary vessels were dissected in H-Hank’s buffer. After the inner and outer membranes were removed by the aids of anatomy microscope, the pulmonary arterial tissues were digested by 0.2% collagenase type I and incubated for 60-120 min at 37°C. Then the cells were cultured in SmGM-2 medium supplemented with 5% FBS, 2 ng/ml basic fibroblast growth factor, 5 μg/ml insulin, and 0.5 ng/ml epidermal growth factor maintained at 37°C with 21% O2 and 5% CO2. The smooth muscle cell was identified by microscopy and immunofluorescent and immunohistochemistry staining with anti-α-SMA antibody.
Cell proliferation analysis
The effects of salidroside on proliferation of rat PASMCs under hypoxia conditions or not were determined by a cell viability assay using a cell counting kit-8 (CCK-8, Dojindo, Japan). Rat PASMCs were seeded in 96 well plates (1×104 cells/well) and pretreated with increasing concentration of salidroside (0.5, 5, 50, 500 μmol/L), AICAR (2.0×103 μmol/L, or Compound C (40 μmol/L) before exposure to hypoxia (5% O2, 5% CO2, and 90% N2). After 24 hours of hypoxic exposure, CCK-8 (10 μl/well) was added to react with cells for 2 hours. The absorbance was examined at 450 nm by a microplate reader (ELX800, BioTek Instruments, Winooski, VT, USA). IC50 was determined according to curvilinear regression between the salidroside concentration and apoptotic index by SPSS 15.0 analysis software. Cell proliferation was also determined by counting the typan blue positive cells.
Immunohistochemistry
After blocking, the paraffin sections were incubated primary polyclonal antibodies against PCNA (1:100), AMPKα1 (1:100, phosphorylated AMPKα1 (1:50), α-SMA (1:50). After overnight incubation with primary antibodies, slides were washed with PBS and incubated with corresponding secondary HRP-linked antibodies (1:100). Lung tissues, incubated with 1% bovine serum albumin (BSA) to replace the specific primary antibody, acted as negative controls. Imaging was assessed by light microscopy and the positive staining was quantified by Image-Pro Plus 6.0 software.
Detection of apoptosis
The apoptosis were qualified using the terminal deoxyribonucleotide transferas-mediated dUTP nick end-labeling (TUNEL) assay (In Situ Cell Death Detection Kit, POD) according to the manufacture’s instructions. PASMCs cells or tissue sections were fixed with 4% paraformaldehyde in PBS (pH 7.4) for 1 hour at 25°C followed by subjected to TUNEL assay. Diaminobenzidine (DAB) was used as the chromogen and hematoxylin as the counterstain. The percentage of TUNEL-positive cells was assessed in 5 randomly selected fields in each slide.
Western blotting analysis
Lung tissues with equal weight (50 mg) were homogenized with a glass homogenizer, and lysed in pre-chilled lysis buffer by ultrasonication. PASMCs were lysed with ice-cold RIPA lysis buffer containing PMSF for 30 min. Then the lysates were centrifuged at 12000 rpm for 30 min and 15 min, respectively, at 4°C and the supernatant was collected. The protein concentrations were determined by the Bradford methods. Equal amounts of proteins (20 μg) were separated with 12% SDS-PAGE, transferred to PVDF membrane, blocked with 5% BSA, and incubated with specific primary antibodies against AMPKα1 (1:100), phosphorylated AMPKα1 (1:50), P53 (1:500), P27 (1:500), P21 (1:500), Bax (1:500), Bcl-2 (1:1000), Casp 3 (1:500), Casp 9 (1:500), β-actin (1:1000), and GAPDH (1:50), respectively. Detection of immunoreactive bands was performed using BeyoECL Plus reagents (Beyotime, China). The optical density of immunoblots was calculated with the Quantity one-4.6.2 software (Bio-Rad Laboratories, Hercules, CA, USA).
Statistical analysis
All data were expressed as mean ± standard deviation (SD). The comparison among more than 3 groups was analyzed by one-way ANOVA, followed by post hoc comparison with LSD test (equal variances assumed) or Dunnett’s T3 test (equal variances not assumed). A level of P<0.05 was considered statistically significant. SPSS version 16.0.1 was used to perform all calculations.
Results
Effects of salidroside on chronic hypoxia-induced PAH and pulmonary artery remodeling in rats
To investigate the effects of salidroside on chronic hypoxia-induced PAH, rats were exposed to chronic hypoxia and treated with increasing concentrations of salidroside (2, 8, and 32 mg/kg) or AMPK agonist AICAR (1 mg/kg) for 4 weeks. mPAP, mCAP, and the weight ratio of RV to LV plus S were calculated. As shown in Figure 1, mPAP and RV/(LV+S) were increased by hypoxia (H group), demonstrating PAH and RV hypertrophy were successfully induced by chronic hypoxia. After salidroside treatment, the increase was inhibited and the treatment of 8 mg/kg exerts the strongest inhibitory effects. Similarly, AICAR also inhibited the increase of mPAP and RV/LV+S. The results suggested salidroside could improve hypoxia-induced PAH might via an AMPK axis. In contrast, there was no significant difference in mCAP among all the groups.
Figure 1.

Salidroside inhibited chronic hypoxia-induced PAH in rats. A. Rats were exposed to normoxia (N), hypoxia (H), hypoxia with increasing concentrations of salidroside (2 mg/kg, 8 mg/kg, and 32 mg/kg, named as HS2, HS8, and HS32, respectively), or hypoxia with AICAR (1 mg/kg) for 4 weeks. Mean pulmonary arterial pressure (mPAP) and mean carotid arterial pressure (mCAP) were examined. Next, the hearts were dissected out, divided into right ventricle (RV), left ventricle (LV), and septum (S), washed by PBS, and weighted. The weight ratio of RV to LV plus S was calculated as an index to reflect RV hypertrophy. B. Representative pictures of PAP waves in the N, H, HS2, HS8, and HS32 groups. #p<0.05, ##p<0.01 vs. N group; *p<0.05, **p<0.01 vs. the H group; n=8.
Then the effects of salidroside on the pulmonary vascular remodeling, a main characteristic of PAH, was examined. After hemotoxylin and eosin (HE) and elastic fiber staining on pulmonary arteries (external diameters of 50-200 μm), the percentage of wall areas to total areas (WA/TA%), the pulmonary artery medial smooth muscle cell layer (PAMT), and the density of nuclei in the medial SMCs were calculated. The prelonged hypoxia exposure resulted in significant increase in WA/TA%, PAMT%, and the density of nuclei in the medial SMCs in H group compared with N group (Figure 2). However, these increases were inhibited by treating with salidroside or AICAR, revealing the pulmonary arterial remodeling induced by chronic hypoxia was attenuated by salidroside and AICAR. In addition, an elevated inflammatory response could be observed in H group after HE staining, and was attenuated by salidroside and AICAR.
Figure 2.

Salidroside improves hypoxia induced pulmonary artery remodeling. (A) Rats were exposed to normoxia (N), hypoxia (H), hypoxia with increasing concentrations of salidroside (2 mg/kg, 8 mg/kg, and 32 mg/kg, named as HS2, HS8, and HS32, respectively), or hypoxia with AICAR (1 mg/kg) for 4 weeks. After hematoxylin and eosin (HE) staining and elastic fiber staining, percentage of wall areas to total areas (WA/TA%), pulmonary artery medial smooth muscle cell layer (PAMT), and the density of nuclei in the medial SMCs were determined. (B, C) Representative photomicrograph of pulmonary artery remodeling analyzed by HE staining (B) (×100), and elastic fiber staining (C) (×400). #p<0.05, ##p<0.01 vs. the N group; p<0.05, **p<0.01 vs. the H group; n=8.
Furthermore, an examination with electron microscope was carried out to verify the inhibitory effects of salidroside on hypoxia-induced pulmonary arterial remodeling at ultrastructural level. In pulmonary arterial smooth muscle cells under normoxia condition, cell membrane and nucleus membrane were smooth, mitochondrial crista was integrated and without vacuolus, and the cells were alignment (Figure 3, N). After hypoxia exposure, the shapes of PAMSCs were not consistent, mitochondrial crista was interrupted and full of vacuoles, and the cells were disorganized (Figure 3, H1-3). The collagenous fibers were excess proliferated and the internal and external elastic lamina were twisted. There were no immigrated muscular vessels smooth muscle cells in pulmonary artery. Compared with hypoxia group (H), in hypoxia plus salidroside group, PASMCs were well organized and the abnormal growth of collagenous fiber was reduced obviously (Figure 3, HS). And the interrupt of mitochondrial crista and the amount of vacuoles were decreased. The results suggested salidroside attenuated hypoxia induced pulmonary arterial remodeling.
Figure 3.

The effect of salidroside on hypoxia induced pulmonary arterial remodeling at ultrastructure level. Rats were exposed to normoxia (N), hypoxia (H), hypoxia with salidroside (32 mg/kg, named as HS) for 4 weeks. The ultrathin sections of lung tissues in N, H, and HS groups were observed under a Hitachi H-600 transmission electron microscopy. Three fields in H group were observed, and named as H-1, H-2, and H-3, respectively. Purple arrow: pulmonary arterial smooth muscle cell (PASMC); Red arrow: collagenous fiber; Yellow arrow: organelle; Blue arrow: muscular vessels smooth muscle cell.
Effects of salidroside on cell proliferation and apoptosis in PASMCs in vivo
The pulmonary artery remodeling under hypoxia condition is mainly caused by the elevated cell proliferation and impaired cell apoptosis of PASMCs, which is the major component of the vascular media and the main effectors of the physiological response(s) during pulmonary vascular remodeling [2,27]. The immunohistochemical staining with anti-PCNA antibody was used to evaluate PASMCs proliferation in rat. Hypoxia induced an obvious increase of PCNA expression in PASMCs in rats, indicating an excess proliferation after hypoxia exposure (Figure 4A and 4B). Consistent with the phenotype changes, salidroside or AICAR treatment attenuated the excess proliferation, demonstrating by a decreased expression of PCNA in hypoxia plus salidroside groups and hypoxia plus AICAR groups compared with the N group (Figure 4A and 4B). Next, a TUNEL assay was performed to elucidate the effects of salidroside of hypoxia induced apoptosis resistance of PASMCs in vivo. As shown in Figure 4C and 4D, an impaired apoptosis was observed in H group, and salidroside reversed the apoptosis resistance in a dose-dependent manner. AICAR treatment also exhibited a promoting effect on PASMCs apoptosis under hypoxia exposure.
Figure 4.
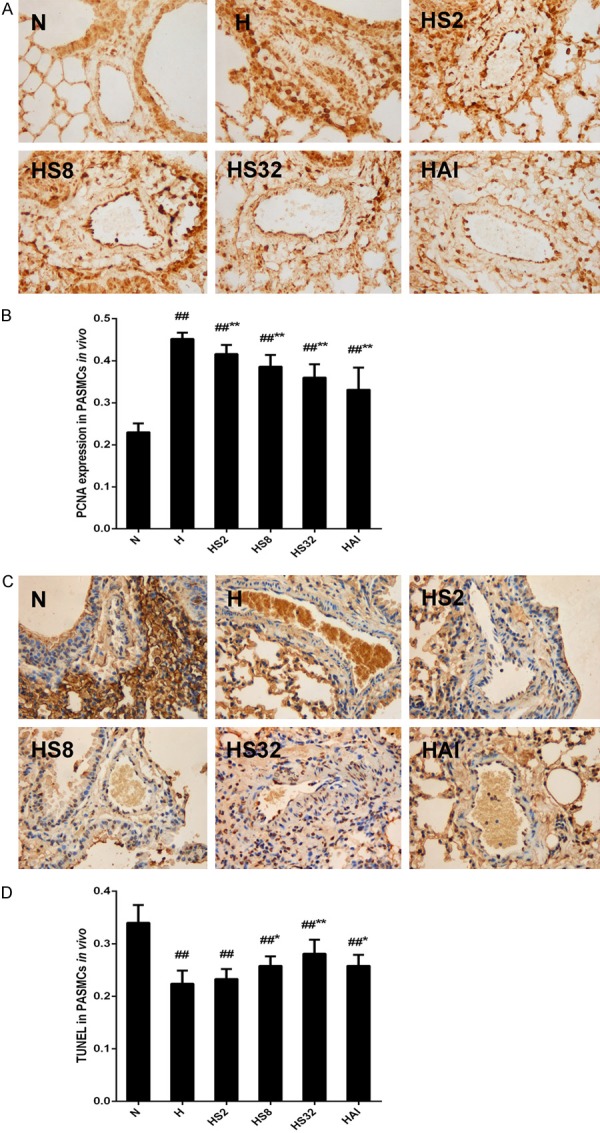
Effects of salidroside and hypoxia on PCNA expression and cell apoptosis in PASMCs in rats. Rats were exposed to normoxia (N), hypoxia (H), hypoxia with increasing concentrations of salidroside (2 mg/kg, 8 mg/kg, and 32 mg/kg, named as HS2, HS8, HS32, respectively), or hypoxia with AICAR (1 mg/kg) for 4 weeks. Lung tissue sections in each group were examined by immunochemical analysis with anti-PCNA antibody (A, B) or TUNEL assay (C, D) (×100). The PCNA expression and the apoptotic cells were quantified with Image-Pro Plus, Version 6.0. #p<0.05, ##p<0.01 vs. the N group; *p<0.05, **p<0.01 vs. the H group; n=6.
Effects of salidroside on activation of AMPKα1 in pulmonary artery in hypoxia-PAH rats under hypoxia conditions
The in vivo studies suggested salidroside and AICAR, an AMPK agonist, exhibited the similar protective effects against chronic hypoxia-induced PAH, pulmonary artery remodeling, and the excess proliferation and apoptosis-resistance of PAMSCs. Combined with the results in our previous study that activation of AMPKα1 by AICAR exerted protective effects against PAH [25], we speculated that salidroside might attenuate hypoxia-induced PAH via activating AMPKα1. Firstly, immunohistochemistry analysis was performed to investigate the expression of total AMPKα1 and phosphorylated AMPKα1 in pulmonary arteries (Figure 5). Chronic hypoxia leaded to a remarked increase in total AMPKα1 and phosphorylated AMKPα1. Salidroside elevated a higher expression of total and phosphorylated AMPKα1 in hypoxia plus salidroside groups than that in H group and functioned in a dose-dependent manner. AICAR also elevated a significant increase of total and phosphorylated AMPKα1 expressions, similar to the high dose of salidroside (32 mg/kg, HS32 group). Secondly, the expressions of total and phosphorylated AMPKα1 in lung tissues of rats were analyzed by western blotting. Consistent with the immunohistochemical staining results in pulmonary arteries, hypoxia increased significantly both the total and phosphorylated AMPKα1 expressions, whereas salidroside and AICAR treatments enhanced the increases further (Figure 5D).
Figure 5.
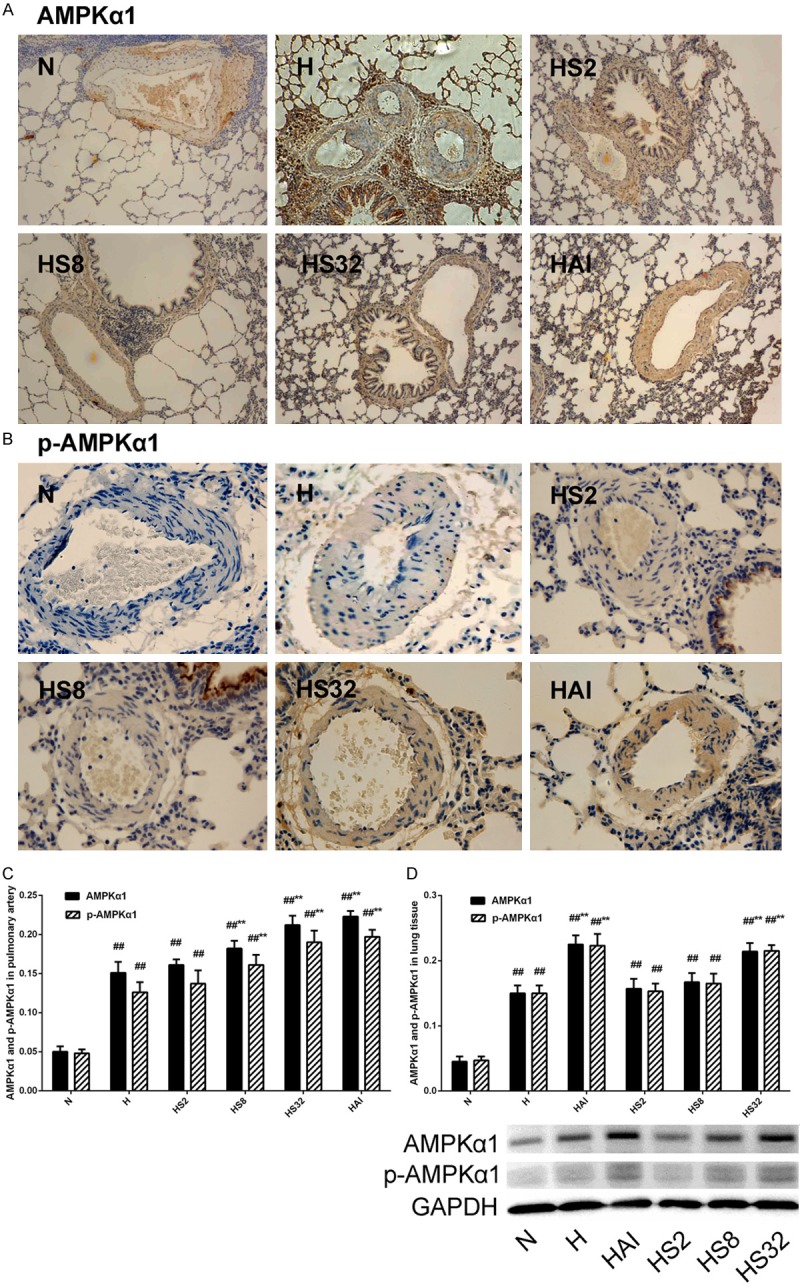
Effects of hypoxia and salidroside on AMPKα1 expression and phosphorylation in PASMCs in rats. Rats were treated with normoxia (N), hypoxia (H), hypoxia with increasing concentrations of salidroside (HS2, HS8, and HS32), or hypoxia with AMPK agonist AICAR (HAI). AMPKα1 expression (×100) and phosphorylation were examined by immunohistochemical staining (×400) or western blotting #p<0.05, ##p<0.01 vs. the N group; **p<0.01 vs. the H group; n=6.
Effects of salidroside on proliferation and apoptosis of rat PASMCs under hypoxia conditions in vitro
The in vivo studies illustrated salidroside exert protective effects against chronic hypoxia induced PAH via inhibiting cell proliferation and increasing apoptosis of PASMCs, and activating AMPKα1. Then in vitro studies were carried out to verify the effects of salidroside and the mechanisms. PASMCs were firstly isolated from rat and identified (Figure S1). To determine the effects of salidroside on proliferation, rat PASMCs were treated with an increasing concentrations of salidroside (0.5, 5, 50, 500 μmol/L), AMPK agonist AICAR (2.0×103 μmol/L), or AMPK inhibitor Compound C (40 μmol/L) under hypoxia conditions for 24 hours. The cell viability was analyzed by CCK-8 kit. Hypoxia caused a significant increase of cell proliferation and the increase was inhibited by salidroside in a dose-dependent manner and by AICAR while Compound C induced a more excess proliferation than that in hypoxia (H) group (Figure 6A). Meanwhile, cell growth was also evaluated by typan blue staining and the results were consistent with CCK-8 results (Figure 6B). Next, the effects of salidroside and hypoxia on cell apoptosis in rat PASMCs were analyzed with a TUNEL assay (Figure 6C and 6D). After hypoxia exposure, only 1.83% cells were TUNEL positive, about 35% to that in N group. There were 16%~18% cells were positive after salidroside or AICAR treatment under hypoxia conditions which was more than 3-fold to that in N group. The treatment of Compound C combined with hypoxia inhibited the apoptosis, even below than that in H group. Finally, the expressions of total and phosphorylated AMPKα1 were examined by western blotting in rat PAMSCs in vitro, which were treated with salidroside, AICAR, or Compound C under hypoxia conditions. A marked increase of total and phosphorylated AMPKα1 was observed in H group compared with N group (Figure 6E). Salidroside and AICAR upregulated higher expressions compared with H group while Compound C inhibited hypoxia induced total and phosphorylated AMPKα1 expressions.
Figure 6.
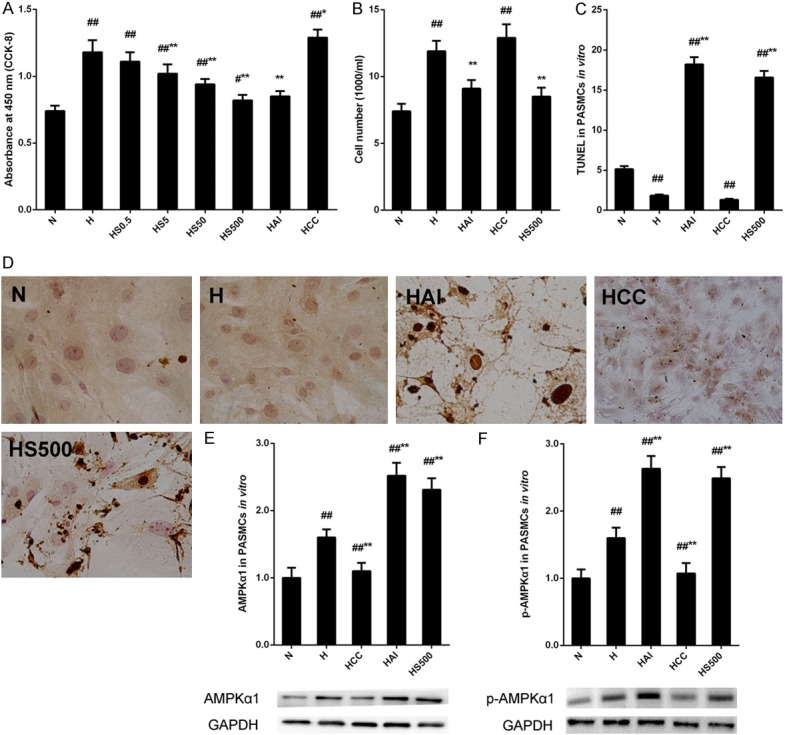
The effects of salidroside and hypoxia on proliferation and apoptosis in PASMCs in vitro. (A) Rat PASMCs cells were exposed to hypoxia and treated with increasing concentrations of salidroside (0-500 μmol/L), AICAR (2.0×10-3 μmol/L) or Compound C (2.0 μmol/L) and proliferation was analyzed by a CCK-8 assay. (B-D) PASMCs cells were treated with salidroside (500 μmol/L), AICAR or Compound C under hypoxia conditions. Typan blue staining (B) and TUNEL assay (C, D ×100) were used to analyze cell growth and cell apoptosis of PASMCs. (E, F) AMPKα1 expression (E) and phosphorylation (F) were examined by western blotting and quantified (C). ##p<0.01 vs. the N group; **p<0.01 vs. the H group; n=6.
Effects of salidroside on the genes related to proliferation and apoptosis in PASMCs under hypoxia conditions in vitro
The in vivo and in vitro studies revealed the critical role of AMPKα1 in the effects of salidroside on cell proliferation and apoptosis in PAH, however, the detailed mechanisms were unclear. The previous studies indicated that activation of AMPKα1 could inhibit cell proliferation via P53-independent pathway [14,26]. Then the expressions of P53 and its downstream effectors, P27 and P21, were detected by western blotting after that the PASMCs were treated with salidroside, AICAR, Compound C under hypoxia conditions. All the protein expressions were downregulated by hypoxia alone or hypoxia with compound C that were reversed by AICAR or salidroside, revealing an AMPKα1-P53-P27/P21 axis in regulating of cell proliferation (Figure 7A-C). The cell apoptosis might also be triggered by a P53-dependent manner, thus apoptosis related proteins were examined. After treatments, the pro-apoptotic factor, Bax, was decreased by hypoxia or hypoxia combined with compound C and increased by AICAR or salidroside under hypoxia conditions, whereas the anti-apoptotic factor, Bcl-2, exert a opposite trend (Figure 7D-F). The activity and cleavage of caspase 9 and 3 were further analyzed. Their activity and cleavage were reduced by hypoxia alone or hypoxia combined with compound C and elevated by salidroside or AICAR after hypoxia treatment (Figure 7G-J). These results suggested that salidroside might reverse hypoxia-induced apoptosis resistance via an AMPKα1-P53-Bax/Bcl-2-caspase 9-caspase 3 pathway.
Figure 7.
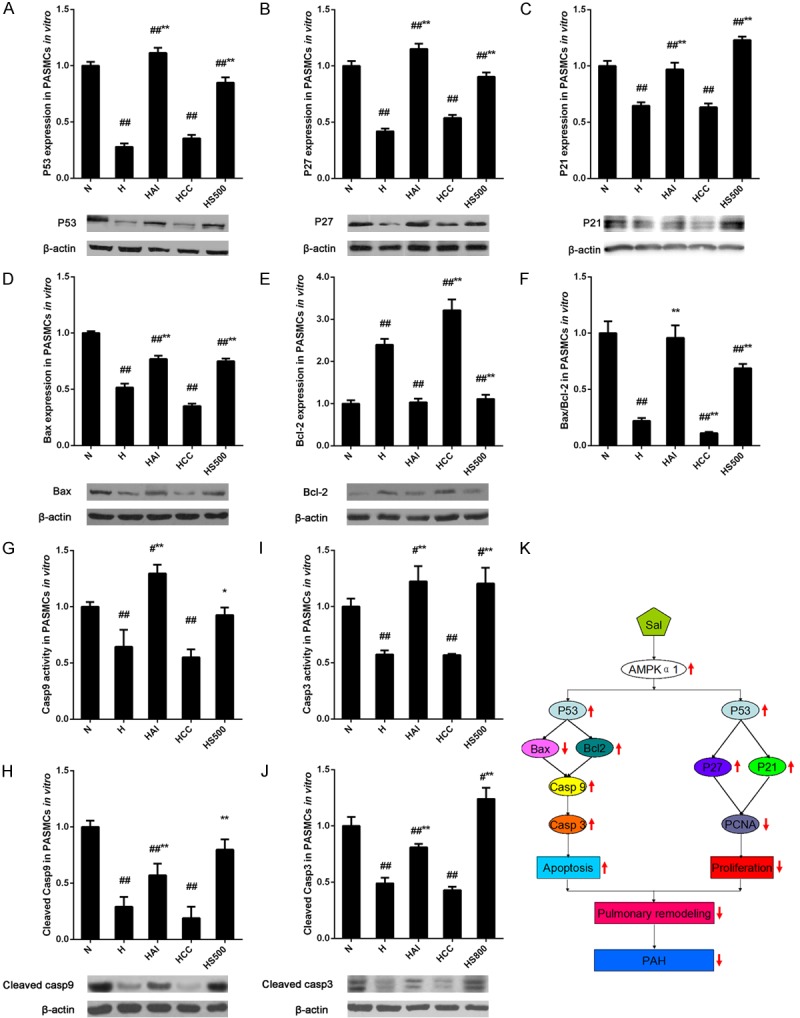
Effects of hypoxia and salidroside on the cell proliferation and apoptosis related genes in PASMCs. (A-E) Rat PASMCs were treated with normoxia (N), hypoxia (H), hypoxia with salidroside (500 μmol/l, HS500), hypoxia with AMPK agonist AICAR (HAI), or hypoxia with AMPK inhibitor Compound C (HCC) and determined by western blotting with antibodies against P53 (A), P27 (B), P21 (C), Bax (D), and Bcl-2 (E). (F) Protein expression ratio of Bax to Bcl-2. (G-J) The activity and cleavage of caspase 9 and 3 in the cells treated as above were detected. (K) A schematic modelillustrating the proposed mechanisms of the protective role of salidroside against hypoxia-induced PAH through AMPKα1-P53-dependent pathways. ##p<0.01 vs. the N group; **p<0.01 vs. the H group; n=6.
Discussion
In the previous study, we have revealed the protective effects of salidroside against chronic hypoxia-induced PAH with a mice model and unveiled the important role of A2aR related mitochondria-dependent apoptotic pathway. Unfortunately, other molecular mechanisms were not investigated. The present research confirmed the effects of salidroside with a chronic hypoxia-induced PAH model of rat and provided insight on physiological significance of AMPK activation, by which salidroside attenuated hypoxia-induced PAH. Furthermore, to explore the underlying mechanisms of AMPK activation, we examined the effects of AMPK activation on the cell proliferation and apoptosis related genes and the results suggested that salidroside rebalanced the cell proliferation and apoptosis to improve chronic hypoxia-induced PAH via AMPKα1-P53 dependent pathways.
PAH is debilitating disease, leads ultimately to right ventricular failure and death, and characterized by increased pulmonary vascular resistance and increased pulmonary arterial pressure [1-3]. Pulmonary arterial remodeling is the main event in pathogenesis of PAH, which is mostly due to the abnormal growth, excess proliferation, and apoptosis resistance in PASMCs [4,28,29]. Therefore, inhibition of the cell proliferation or induction of the cell apoptosis may be an efficient therapeutic strategy for PAH. Salidroside, a pharmacologically active ingredient purified from Rhodiola which is used to relieve high altitude sickness and acute exacerbation of PAH, has been revealed to possess multiple pharmacological activities including antiproliferative effects in PASMCs [9,14]. Chen et al. reported that salidroside significantly inhibits the proliferation and DNA synthesis of PASMCs induced by platelet-derived growth factor (PDGF)-BB in a dose- and time-dependent manner without cell cytotoxicity [14]. Both in our recent and present studies, we uncovered that salidroside could inhibit chronic hypoxia induced PAH. And the results of immunohistochemical analysis with anti-PCNA antibody and TUNEL assay demonstrated that salidroside inhibited cell proliferation and reversed apoptosis resistance in PASMCs under hypoxia conditions in rats. The in vitro results of CCK-8 assay, typan blue staining, and TUNEL assay also clarified the conclusions.
AMPK is a critical regulator of energy metabolic homeostasis at the cellular whole organism levels that has been highly conserved during evolution [16] and play a critical role in preventing cardiovascular disease including heart disease [30], atherogenesis [31], neointima formation [32,33], and hypertension [34,35]. AMPK activation has been shown to reduce myocardial ischemic injury [36,37], inhibit myocardial hypertrophy [17,38,39], and impede the transition from cardiac hypertrophy to heart failure [30]. Furthermore, AMPK has been revealed to inhibit cardiac hypertrophy by promoting autophagy [40]. Activation of AMPK by metformin also inhibit development of PAH by suppressing vascular remodeling in animal model [41] and inhibited PASMCs proliferation in vitro [42]. Our previous study also showed AMPK activated by AICAR can attenuate hypoxia induced PAH and pulmonary arterial remodeling via inhibiting cell proliferation and enhance apoptosis of PASMCs in rats [25]. Based on the reports, we predicted that AMPK activation might be involved in salidroside regulation of chronic hypoxia induced PAH. In vivo experiments in our study showed that the inhibitory effects of salidroside on hypoxia induced PAH, pulmonary arterial remodeling, and PASMCs proliferation and apoptosis resistance was similar with AICAR. Consistent with the in vivo results, salidroside as well as AICAR inhibited proliferation and reversed apoptosis resistance in PASMCs in vitro while Compound C, an AMPK inhibitor, exhibited an opposite effects. These results illustrated that salidroside exert protective effects against chronic hypoxia induced PAH might via AMPK activation. Both catalytic α subunit of AMPK are expressed in VSMCs while AMPKα1 is the predominant isoform contributing to total AMPKα and its activation in VSMCs [32]. We determined effects of salidroside on AMPKα1 expression and phosphorylation under hypoxia condition. The results indicated hypoxia upregulated AMPKα1 expression and phosphorylation, revealing a compensatory upregualtion of AMPKα1 activation responding to hypoxia. And salidroside as well as AICAR elevated further upregulation of AMPKα1 expression and phosphorylation after hypoxia exposure while Compound C inhibited it.
AMPK activation and its function have been addressed in smooth muscle cells (SMCs). Igata et al. reported that the AMPK activator AICAR significantly inhibits proliferation of human aortic SMCs induced by both platelet-derived growth factor-BB (PDGF-BB) and fetal calf serum (FCS) by blocking cell cycle progression through upregulation of P53 and P21 [26]. Recently, a potent anti-tumor agent, β-lapachone was shown to inhibit FCS- or PDGF-induced proliferation of VSMCs and to reduce neointimal formation in balloon-injured rat carotid arteries through the LKB1-AMPK-P53-P21 pathway [43]. And also in vitro studies suggested that salidroside can block progression through G0/G1 to S phase of the cell cycle which is associated with the elevation of the P27 expression [14]. In the present study, we have found that salidroside induced AMPKα1 activation might inhibit PASMCs proliferation through a P53-P21/P27-PCNA pathway and enhance cell apoptosis via a P53-Bax/Bcl-2-caspase 9-caspase 3 pathway to attenuate hypoxia induced pulmonary arterial remodeling and consequently improve PAH (Figure 7K). In addition, AMPK activation can trigger PI3K/Akt signaling to inhibit cell proliferation [44] and blocking the PASMCs proliferation by salidroside is associated with the suppression of the AKT signaling pathway [14]. Then the AMPK-PI3K/Akt axis might also play a critical role in salidroside inhibition of PASMCs proliferation thatwill be clarified in our future work.
In summary, salidroside exhibited protective effects against chronic hypoxia induced PAH and pulmonary arterial remodeling partially by AMPK activation. The underlying mechanism might be that AMPKα1 activation induced by salidroside rebalancing PASMCs proliferation and apoptosis via AMPKα1-P53-P21/P27-PCNA and AMPKα1-P53-Bax/Bcl-2-caspase 9-caspase 3 pathways under hypoxia conditions. The evidence provided a significant insight of the protective effects of salidroside against PAH and the underlying mechanisms may become a theoretical basis for the use of salidroside in the treatment of clinical hypoxic pulmonary hypertension.
Acknowledgements
This study was supported by the National Natural Science Foundation of ChinaGrants (No. 81473406), the Natural Science Foundation of Zhejiang Province Grants (LY13H010003), and Science and Technology Project of Wenzhou (No. Y20100190), Project of Zhejiang Province Health Department (No. 2012ZDA035).
Disclosure of conflict of interest
None.
Authors’ contribution
The experiments were conceived and designed by MYC, LXW, and XYH, and mainly performed by MYC, HC, and CY. The reagents/materials/analysis tools were prepared and provided by PLW, YYF, XMX, and RF. The Figures were prepared by MYC, HC, CLX, and YFC. The data were analyzed by MYC, HC, and XYH. The manuscript was written by MYC, LXW, and XYH. All authors reviewed the manuscript.
Supporting Information
References
- 1.Jeffery TK, Wanstall JC. Pulmonary vascular remodeling: a target for therapeutic intervention in pulmonary hypertension. Pharmacol Ther. 2001;92:1–20. doi: 10.1016/s0163-7258(01)00157-7. [DOI] [PubMed] [Google Scholar]
- 2.Humbert M, Morrell NW, Archer SL, Stenmark KR, MacLean MR, Lang IM, Christman BW, Weir EK, Eickelberg O, Voelkel NF, Rabinovitch M. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43:13S–24S. doi: 10.1016/j.jacc.2004.02.029. [DOI] [PubMed] [Google Scholar]
- 3.Chin KM, Rubin LJ. Pulmonary arterial hypertension. J Am Coll Cardiol. 2008;51:1527–1538. doi: 10.1016/j.jacc.2008.01.024. [DOI] [PubMed] [Google Scholar]
- 4.Tuder RM, Abman SH, Braun T, Capron F, Stevens T, Thistlethwaite PA, Haworth SG. Development and pathology of pulmonary hypertension. J Am Coll Cardiol. 2009;54:S3–9. doi: 10.1016/j.jacc.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 5.Walsh-Sukys MC, Tyson JE, Wright LL, Bauer CR, Korones SB, Stevenson DK, Verter J, Stoll BJ, Lemons JA, Papile LA, Shankaran S, Donovan EF, Oh W, Ehrenkranz RA, Fanaroff AA. Persistent pulmonary hypertension of the newborn in the era before nitric oxide: practice variation and outcomes. Pediatrics. 2000;105:14–20. doi: 10.1542/peds.105.1.14. [DOI] [PubMed] [Google Scholar]
- 6.McMurtry MS, Archer SL, Altieri DC, Bonnet S, Haromy A, Harry G, Bonnet S, Puttagunta L, Michelakis ED. Gene therapy targeting survivin selectively induces pulmonary vascular apoptosis and reverses pulmonary arterial hypertension. J Clin Invest. 2005;115:1479–1491. doi: 10.1172/JCI23203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Courboulin A, Barrier M, Perreault T, Bonnet P, Tremblay VL, Paulin R, Tremblay E, Lambert C, Jacob MH, Bonnet SN, Provencher S, Bonnet S. Plumbagin reverses proliferation and resistance to apoptosis in experimental PAH. Eur Respir J. 2012;40:618–629. doi: 10.1183/09031936.00084211. [DOI] [PubMed] [Google Scholar]
- 8.Stenmark KR, Fagan KA, Frid MG. Hypoxiainduced pulmonary vascular remodeling: cellular and molecular mechanisms. Circ Res. 2006;99:675–691. doi: 10.1161/01.RES.0000243584.45145.3f. [DOI] [PubMed] [Google Scholar]
- 9.Panossian A, Wagner H. Stimulating effect of adaptogens: an overview with particular reference to their efficacy following single dose administration. Phytother Res. 2005;19:819–838. doi: 10.1002/ptr.1751. [DOI] [PubMed] [Google Scholar]
- 10.Guan S, Feng H, Song B, Guo W, Xiong Y, Huang G, Zhong W, Huo M, Chen N, Lu J, Deng X. Salidroside attenuates LPS-induced pro-inflammatory cytokine responses and improves survival in murine endotoxemia. Int Immunopharmacol. 2011;11:2194–2199. doi: 10.1016/j.intimp.2011.09.018. [DOI] [PubMed] [Google Scholar]
- 11.Qu ZQ, Zhou Y, Zeng YS, Lin YK, Li Y, Zhong ZQ, Chan WY. Protective effects of a Rhodiola crenulata extract and salidroside on hippocampal neurogenesis against streptozotocininduced neural injury in the rat. PLoS One. 2012;7:e29641. doi: 10.1371/journal.pone.0029641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Darbinyan V, Kteyan A, Panossian A, Gabrielian E, Wikman G, Wagner H. Rhodiola rosea in stress induced fatigue--a double blind crossover study of a standardized extract SHR-5 with a repeated low-dose regimen on the mental performance of healthy physicians during night duty. Phytomedicine. 2000;7:365–371. doi: 10.1016/S0944-7113(00)80055-0. [DOI] [PubMed] [Google Scholar]
- 13.Ming DS, Hillhouse BJ, Guns ES, Eberding A, Xie S, Vimalanathan S, Towers GH. Bioactive compounds from Rhodiola rosea (Crassulaceae) Phytother Res. 2005;19:740–743. doi: 10.1002/ptr.1597. [DOI] [PubMed] [Google Scholar]
- 14.Chen C, Tang Y, Deng W, Huang C, Wu T. Salidroside blocks the proliferation of pulmonary artery smooth muscle cells induced by plateletderived growth factor‑BB. Mol Med Rep. 2014;10:917–922. doi: 10.3892/mmr.2014.2238. [DOI] [PubMed] [Google Scholar]
- 15.Huang X, Zou L, Yu X, Chen M, Guo R, Cai H, Yao D, Xu X, Chen Y, Ding C, Cai X, Wang L. Salidroside attenuates chronic hypoxia-induced pulmonary hypertension via adenosine A2a receptor related mitochondria-dependent apoptosis pathway. J Mol Cell Cardiol. 2015;82:153–166. doi: 10.1016/j.yjmcc.2015.03.005. [DOI] [PubMed] [Google Scholar]
- 16.Lage R, Dieguez C, Vidal-Puig A, Lopez M. AMPK: a metabolic gauge regulating wholebody energy homeostasis. Trends Mol Med. 2008;14:539–549. doi: 10.1016/j.molmed.2008.09.007. [DOI] [PubMed] [Google Scholar]
- 17.Tian R, Musi N, D’Agostino J, Hirshman MF, Goodyear LJ. Increased adenosine monophosphate-activated protein kinase activity in rat hearts with pressure-overload hypertrophy. Circulation. 2001;104:1664–1669. doi: 10.1161/hc4001.097183. [DOI] [PubMed] [Google Scholar]
- 18.Chen L, Jiao ZH, Zheng LS, Zhang YY, Xie ST, Wang ZX, Wu JW. Structural insight into the autoinhibition mechanism of AMP-activated protein kinase. Nature. 2009;459:1146–1149. doi: 10.1038/nature08075. [DOI] [PubMed] [Google Scholar]
- 19.Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Makela TP, Alessi DR, Hardie DG. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J Biol. 2003;2:28. doi: 10.1186/1475-4924-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D, Schlattner U, Wallimann T, Carlson M, Carling D. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol. 2003;13:2004–2008. doi: 10.1016/j.cub.2003.10.031. [DOI] [PubMed] [Google Scholar]
- 21.Xiao B, Sanders MJ, Underwood E, Heath R, Mayer FV, Carmena D, Jing C, Walker PA, Eccleston JF, Haire LF, Saiu P, Howell SA, Aasland R, Martin SR, Carling D, Gamblin SJ. Structure of mammalian AMPK and its regulation by ADP. Nature. 2011;472:230–233. doi: 10.1038/nature09932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ewart MA, Kennedy S. AMPK and vasculoprotection. Pharmacol Ther. 2011;131:242–253. doi: 10.1016/j.pharmthera.2010.11.002. [DOI] [PubMed] [Google Scholar]
- 23.Wang Y, Gao E, Tao L, Lau WB, Yuan Y, Goldstein BJ, Lopez BL, Christopher TA, Tian R, Koch W, Ma XL. AMP-activated protein kinase deficiency enhances myocardial ischemia/reperfusion injury but has minimal effect on the antioxidant/antinitrative protection of adiponectin. Circulation. 2009;119:835–844. doi: 10.1161/CIRCULATIONAHA.108.815043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim AS, Miller EJ, Young LH. AMP-activated protein kinase: a core signalling pathway in the heart. Acta Physiol (Oxf) 2009;196:37–53. doi: 10.1111/j.1748-1716.2009.01978.x. [DOI] [PubMed] [Google Scholar]
- 25.Huang X, Fan R, Lu Y, Yu C, Xu X, Zhang X, Liu P, Yan S, Chen C, Wang L. Regulatory effect of AMP-activated protein kinase on pulmonary hypertension induced by chronic hypoxia in rats: in vivo and in vitro studies. Mol Biol Rep. 2014;41:4031–4041. doi: 10.1007/s11033-014-3272-9. [DOI] [PubMed] [Google Scholar]
- 26.Momcilovic M, Hong SP, Carlson M. Mammalian TAK1 activates Snf1 protein kinase in yeast and phosphorylates AMPactivated protein kinase in vitro. J Biol Chem. 2006;281:25336–25343. doi: 10.1074/jbc.M604399200. [DOI] [PubMed] [Google Scholar]
- 27.Mandegar M, Fung YC, Huang W, Remillard CV, Rubin LJ, Yuan JX. Cellular and molecular mechanisms of pulmonary vascular remodeling: role in the development of pulmonary hypertension. Microvasc Res. 2004;68:75–103. doi: 10.1016/j.mvr.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 28.Pietra GG, Capron F, Stewart S, Leone O, Humbert M, Robbins IM, Reid LM, Tuder RM. Pathologic assessment of vasculopathies in pulmonary hypertension. J Am Coll Cardiol. 2004;43:25S–32S. doi: 10.1016/j.jacc.2004.02.033. [DOI] [PubMed] [Google Scholar]
- 29.Yuan JX, Rubin LJ. Pathogenesis of pulmonary arterial hypertension: the need for multiple hits. Circulation. 2005;111:534–538. doi: 10.1161/01.CIR.0000156326.48823.55. [DOI] [PubMed] [Google Scholar]
- 30.Beauloye C, Bertrand L, Horman S, Hue L. AMPK activation, a preventive therapeutic target in the transition from cardiac injury to heart failure. Cardiovasc Res. 2011;90:224–233. doi: 10.1093/cvr/cvr034. [DOI] [PubMed] [Google Scholar]
- 31.Dong Y, Zhang M, Wang S, Liang B, Zhao Z, Liu C, Wu M, Choi HC, Lyons TJ, Zou MH. Activation of AMP-activated protein kinase inhibits oxidized LDL-triggered endoplasmic reticulum stress in vivo. Diabetes. 2010;59:1386–1396. doi: 10.2337/db09-1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Song P, Wang S, He C, Wang S, Liang B, Viollet B, Zou MH. AMPKalpha2 deletion exacerbates neointima formation by upregulating Skp2 in vascular smooth muscle cells. Circ Res. 2011;109:1230–1239. doi: 10.1161/CIRCRESAHA.111.250423. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 33.Nagata D, Takeda R, Sata M, Satonaka H, Suzuki E, Nagano T, Hirata Y. AMP-activated protein kinase inhibits angiotensin IIstimulated vascular smooth muscle cell proliferation. Circulation. 2004;110:444–451. doi: 10.1161/01.CIR.0000136025.96811.76. [DOI] [PubMed] [Google Scholar]
- 34.Wang S, Liang B, Viollet B, Zou MH. Inhibition of the AMP-activated protein kinasealpha2 accentuates agonist-induced vascular smooth muscle contraction and high blood pressure in mice. Hypertension. 2011;57:1010–1017. doi: 10.1161/HYPERTENSIONAHA.110.168906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Buhl ES, Jessen N, Pold R, Ledet T, Flyvbjerg A, Pedersen SB, Pedersen O, Schmitz O, Lund S. Long-term AICAR administration reduces metabolic disturbances and lowers blood pressure in rats displaying features of the insulin resistance syndrome. Diabetes. 2002;51:2199–2206. doi: 10.2337/diabetes.51.7.2199. [DOI] [PubMed] [Google Scholar]
- 36.Russell RR 3rd, Li J, Coven DL, Pypaert M, Zechner C, Palmeri M, Giordano FJ, Mu J, Birnbaum MJ, Young LH. AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J Clin Invest. 2004;114:495–503. doi: 10.1172/JCI19297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Young LH. AMP-activated protein kinase conducts the ischemic stress response orchestra. Circulation. 2008;117:832–840. doi: 10.1161/CIRCULATIONAHA.107.713115. [DOI] [PubMed] [Google Scholar]
- 38.Chan AY, Dolinsky VW, Soltys CL, Viollet B, Baksh S, Light PE, Dyck JR. Resveratrol inhibits cardiac hypertrophy via AMP-activated protein kinase and Akt. J Biol Chem. 2008;283:24194–24201. doi: 10.1074/jbc.M802869200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li HL, Yin R, Chen D, Liu D, Wang D, Yang Q, Dong YG. Long-term activation of adenosine monophosphate-activated protein kinase attenuates pressure-overload-induced cardiac hypertrophy. J Cell Biochem. 2007;100:1086–1099. doi: 10.1002/jcb.21197. [DOI] [PubMed] [Google Scholar]
- 40.Li Y, Chen C, Yao F, Su Q, Liu D, Xue R, Dai G, Fang R, Zeng J, Chen Y, Huang H, Ma Y, Li W, Zhang L, Liu C, Dong Y. AMPK inhibits cardiac hypertrophy by promoting autophagy via mTORC1. Arch Biochem Biophys. 2014;558:79–86. doi: 10.1016/j.abb.2014.06.023. [DOI] [PubMed] [Google Scholar]
- 41.Agard C, Rolli-Derkinderen M, Dumas-de-La-Roque E, Rio M, Sagan C, Savineau JP, Loirand G, Pacaud P. Protective role of the antidiabetic drug metformin against chronic experimental pulmonary hypertension. Br J Pharmacol. 2009;158:1285–1294. doi: 10.1111/j.1476-5381.2009.00445.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu Y, Liu L, Zhang Y, Wang G, Han D, Ke R, Li S, Feng W, Li M. Activation of AMPK inhibits pulmonary arterial smooth muscle cells proliferation. Exp Lung Res. 2014;40:251–258. doi: 10.3109/01902148.2014.913092. [DOI] [PubMed] [Google Scholar]
- 43.Zhang M, Dong Y, Xu J, Xie Z, Wu Y, Song P, Guzman M, Wu J, Zou MH. Thromboxane receptor activates the AMP-activated protein kinase in vascular smooth muscle cells via hydrogen peroxide. Circ Res. 2008;102:328–337. doi: 10.1161/CIRCRESAHA.107.163253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Memmott RM, Dennis PA. Akt-dependent and -independent mechanisms of mTOR regulation in cancer. Cell Signal. 2009;21:656–664. doi: 10.1016/j.cellsig.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.