

Abstract
Hypoxia is a common feature of solid tumors, particularly in glioblastoma (GBM), and known to be a poor prognosis factor in GBM patients. The growth of GBM is also associated with a marked inflammation partially characterized by an accumulation of macrophage (MΦ) of the M2 phenotype. However, the transition between M1 MΦ (antitumoral) and M2 MΦ (protumoral) phenotypes is a dynamic process. We made the assumption that oxygen (O2) availability could be a major regulator of this transition and that the intratumoral O2 gradient is of importance. We evaluated, in vivo, the impact of hypoxia on MΦ tropism and polarization in two models of human GBM, well differentiated by their degree of hypoxia. MΦ migration in the tumor was more pronounced in the more hypoxic tumor of the two GBM models. In the more hypoxic of the models, we have shown that MΦ migrated at the tumor site only when hypoxia takes place. We also demonstrated that the acquisition of the M2 phenotype was clearly an evolving phenomenon with hypoxia as the major trigger for this transition. In support of these in vivo finding, M0 but also M1 MΦ cultured in moderate or severe hypoxia displayed a phenotype close to that of M2 MΦ whose phenotype was further reinforced by severe hypoxia. These results highlight the role of hypoxia in the aggressiveness of GBM, in part, by transforming MΦ such that a protumoral activity is expressed.
Keywords: brain tumor, hypoxia, M2 macrophages, polarization, re-education
Abbreviations
- Arg1
arginase-1
- CAIX
carbonic anhydrase IX
- GBM
glioblastoma
- iNOS
inducible nitric oxide synthase
- MΦ
macrophage
- NO
nitric oxide
- O2
oxygen
- PIMO
pimonidazole
- ptO2
tissue partial pressure of oxygen
- SDF-1α
stromal cell-derived factor 1 α
- SN
supernatant
- TC
tumor core
- TS
tumor shell
- [18F]-FMISO
3-[18F]-fluoro-1-(2-nitro-1-imidazolyl)-2-propanol.
Introduction
GBM are the most frequent and aggressive primary brain tumors of the adult. Despite surgery, radiotherapy and chemotherapy, the median survival of patients is dismally low.1 GBM are highly heterogeneous tumors which can be considered as a multi-compartmental system divided into the tumor cells, the vasculature and the microenvironment. Commonly, the tumor microenvironment is a hypoxic milieu in which cell types other than tumoral cells are observed inside or close to the tumor mass such as fibroblasts, stromal cells and multiple components of the immune system.2,3
Hypoxia, formally defined as an inadequacy between O2 supply and demand, is the consequence, in tumor, of a functionally inappropriate vascularization, and/or irregular blood flow relative to the high-proliferation rate of tumor cells. Hypoxia is one main feature of GBM relative to lower grade glioma4 with a tissue partial pressure of oxygen (ptO2) demonstrated to be inferior to 10 mmHg in the tumor core (TC).5,6 Hypoxia triggers many intracellular modifications allowing adaptation to the low O2 availability as well as the potentiation of the infiltration and migration of tumor cells.7 Hypoxia is known to induce angiogenesis and to promote resistance to therapies8 and has been shown to be an independent factor for a reserved prognosis in GBM patients.9
In solid tumors, MΦ are the most abundant infiltrative inflammatory cells present in and around tumors.10 Circulating monocytes can enter tumors11 under the effect of chemokines synthetized by several cell types, including the cancerous cells themselves.12 Once entered the tissues, monocytes differentiate into MΦ under the influence of cytokines, and accumulate in specific tumor regions, such as hypoxic/necrotic areas.12 In GBM, MΦ can migrate in the tumor13,14 and have been described as the most abundant infiltrative inflammatory cells.2,15 More interestingly, it has been demonstrated that the number of MΦ is inversely correlated with the survival of GBM patients.16
Moreover, once in the tissue, MΦ are likely to undergo a rapid phenotype switching effected by local environmental signals. MΦ can be polarized into principally two main distinct phenotypes, M1 or M2, which have been respectively proposed to either restrict or promote tumor development.17,18 M1 MΦ, characterized by the expression of the inducible type of nitric oxide synthase (iNOS) are pro-inflammatory MΦ implicated in an antitumoral activity, especially by their phagocytic properties and their capacity to activate pro-inflammatory syntheses. MΦ are polarized into M1 phenotype by the presence of diverse pro-inflammatory cytokines, such as interferonγ (IFNγ), or microbial molecules, such as lipopolysaccharide (LPS).19 On the contrary, M2 MΦ, essentially characterized by a potent arginase-1 (Arg1) activity, are anti-inflammatory MΦ not only known to promote tumor development by tissue remodeling, cell proliferation and immunoregulation but also by promoting angiogenesis.20 MΦ can acquire an M2 phenotype by the presence of several anti-inflammatory molecules such as the interleukins: IL4, IL10 and/or IL13.19 The phenotype of MΦ present in tumors is not yet fully established but it seems to be a dynamic process. It has been hypothesized that M1 MΦ are found at the initiation of tumor development and that MΦ are polarized into an M2 phenotype along with the growth of the tumor.21,22 In the instance of GBM, a recent study has demonstrated that the presence of M2 MΦ increases with glioma grade and that an inverse relationship exists between patient survival and the concentration of M2 MΦ.23
Despite these results, the factors responsible for MΦ migration and polarization in GBM remain obscure. However, MΦ are likely to have an important impact on tumor development. For example, it has been recently shown that the re-education of M2 MΦ to M1 MΦ has a marked beneficial impact on glioma growth.24
Hypoxia has been proposed to be at the origin of the migration of MΦ into the TC.12 Consistent with this, the most hypoxic areas of numerous solid tumors are known to have a pronounced congregation of M2-like MΦ.25-27 Hypoxia has also been suggested to be a possible cause of MΦ polarization. Indeed, hypoxia may induce the synthesis of protumoral molecules by the tumor cells which, in turn, would promote MΦ to polarize into the M2 phenotype.28-30 Moreover, in vitro studies have also implied that hypoxia may directly act on MΦ to induce a polarization into an M2 phenotype.30,31 However, up until now, the detailed relationship between hypoxia and the M1 to M2 transition has never been formally analyzed in vivo and warrants an in-depth investigation. Using robust and pertinent models of GBM, previously shown to develop severe hypoxia32 such as that observed in patients,33 we evaluated whether GBM hypoxia, in addition to its well described effect on the infiltration of MΦ, could affect the acquisition of an M2 phenotype.
In conclusion, even if hypoxia is assumed to polarize MΦ into the M2 form, the intrinsic ability of hypoxia to drive a conversion of M1, suspected to be present at the onset of GBM development, to M2 has never been tested, to the best of our knowledge. Accordingly, we analyzed the impact of hypoxia to re-educate M1 MΦ into the M2 phenotype.
Results
Hypoxia-related MΦ migration
To determine whether hypoxia influences MΦ migration in GBM, we used two models of human GBM, U87 and U251, known to be non-hypoxic and severely hypoxic, respectively.32 CD68 immunostaining was performed to visualize the MΦ/microglial cells present in the tumor. 23 ± 5% of CD68+ cells in the tumor of the hypoxic U251 model were observed compared to only 12 ± 6% in the less hypoxic tumor implanted with the U87-MG cells (Fig. 1Aa and Ab). CD14 immunostaining was then used to differentiate MΦ (CD68+/CD14+) from microglia (CD68+/CD14−).34 21 ± 2% of CD68+ cells were also CD14+ in the U87 tumor compared to 58 ± 29% in the U251 tumor (Fig. 1Aa and Ac). To evaluate whether this migration was essentially hypoxia-dependent and not intrinsic to the tumor cells themselves, we examined the MΦ migration in U251 tumor prior to the development of hypoxia. The [18F]-FMISO µPET analysis, performed to estimate the level of hypoxia, showed no [18F]-FMISO uptake in the relatively small tumor but, in contrary, a sustained [18F]-FMISO uptake in the later stage of tumor development (Fig. 1Ba). This was also confirmed with the carbonic anhydrase IX (CAIX) (Fig. 1Bb) and the stromal cell-derived factor 1 α (SDF-1α) immunostainings (Fig. S1A), known to be up-regulated in hypoxia (Fig. S1B).35,36 Immunostainings showed that 67 ± 28% of CD68+ cells were also CD14+ in the hypoxic tumor while only 9 ± 13% were found in the pre-hypoxic tumor (Fig. 1Bc and Bd). When computing the number of MΦ (CD68+/CD14+) as a function of tumor volume, a sigmoidal relationship was observed (data not shown). These results indicate that MΦ are found in the U251 tumor at the hypoxic state. With the hypoxic U251 tumors, we also confirmed that the accumulation of MΦ occurs mainly in the hypoxic regions (Fig. S1C) which can be inferred from the SDF-1α expression marginal to the hypoxic core (Fig. S1D).
Figure 1.
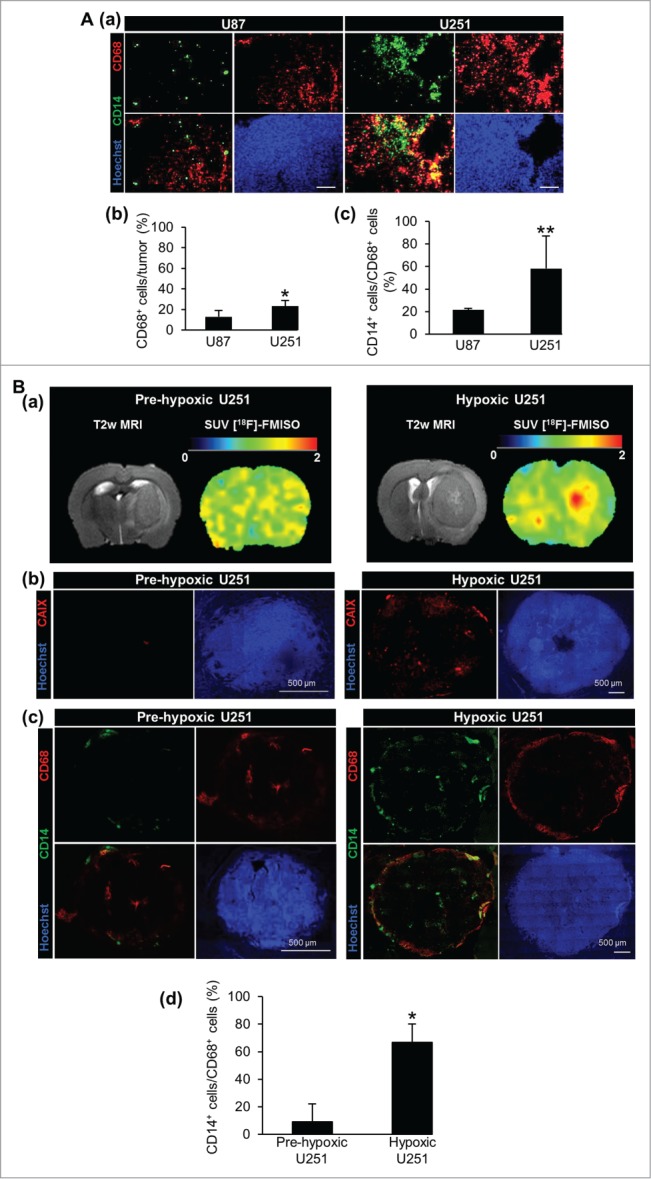
MΦ migration toward tumor models of human GBM. (A) CD68, CD14 and Hoechst 33342 immunofluorescence images (a) and their respective quantifications (b, c) in the U87 and U251 tumors. Scale bars: 100 µm. (B) T2w µMRI and [18F]-FMISO µPET images (a) and CAIX and Hoechst 33342 immunofluorescence images (b) of the U251 tumors at two different stages of tumor development. CD68, CD14 and Hoechst 33342 immunofluorescence images (c) and their respective quantifications (d) of pre-hypoxic and hypoxic U251 tumors. n = 3 animals per group and per time. Statistical significance was achieved when p < 0.05 (*) or p < 0.01 (**).
GBM oxygenation-related MΦ polarization
We then analyzed the relationships between hypoxia and MΦ polarization. Accordingly, M1 and M2 MΦ were differentiated by iNOS and Arg1 immunostaining, respectively. In the U87 tumor, iNOS staining co-localized with the CD68 signal (31±13% were CD68+/iNOS+) while Arg1 staining was not detected (2 ± 2% were CD68+/Arg1+) (Fig. 2A). In the hypoxic U251 tumor, no iNOS signal was apparent (0.6 ± 0.4% were CD68+/iNOS+) but the Arg1 signal co-localized with CD68 (55 ± 31% were CD68+/Arg1+) (Fig. 2C). MΦ polarization was also determined in the pre-hypoxic U251 tumor to ascertain that the presence of M2 MΦ is not intrinsic to the GBM cell types themselves but rather may be a function of O2 availability. No iNOS staining was visualized (1.5 ± 2% were CD68+/iNOS+) but the signal for Arg1+, though detected, was not significant (10 ± 16% were CD68+/Arg1+) (Fig. 2B).
Figure 2.

MΦ polarization in tumor models of human GBM. Representative Arg1, iNOS, CD68 and Hoechst 33342 immunofluorescence images and their respective quantifications in the U87 (A), pre-hypoxic (B) and hypoxic (C) U251 tumors. Scale bars: 100 µm. n = 3 animals per group and per time. Statistical significance was achieved when p < 0.05 (*) or p < 0.01 (**), otherwise it was not significant (NS).
Although M2 MΦ were found in the hypoxic tumor, the question that arises is whether M2 MΦ aggregation has a localization specific to the most hypoxic areas. The images of the entire volume of the hypoxic U251 tumor were taken and were divided into two regions: the TC known to be severely hypoxic and the tumor shell (TS) known to be less hypoxic and more vascularized (Fig. 3A). On this basis, iNOS and Arg1 stainings were localized and compared to that of the CD68 signal. The CD68 staining was present in the TC but also in the TS of the U251 tumor (Fig. 3B and C). In the whole tumor, iNOS signal was observed in the TS (19 ± 9% were CD68+/iNOS+) (Fig. 3B). Arg1 signal was preferentially present in the TC (63 ± 28% were CD68+/Arg1+), more particularly around pseudopalisades (Fig. 3C), and only localized in pimonidazole positive areas of this tumor (Fig. 3D). These results indicate not only that the MΦ present at the shell of this tumoral model are M0 (CD68+/iNOS−/Arg1−) but also M1 MΦ (CD68+/iNOS+/Arg1−), though the MΦ detected in the hypoxic areas present an M2 phenotype (CD68+/iNOS−/Arg1+).
Figure 3.
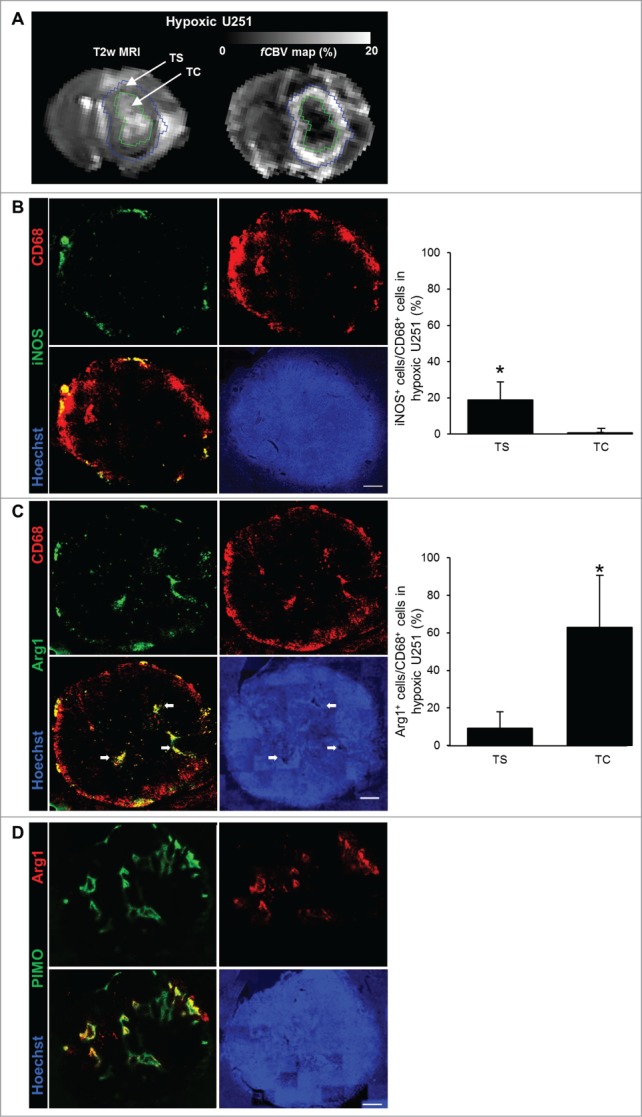
M2 MΦ distribution in the hypoxic U251 tumor. (A) Representative region of interest of the TC and TS overlayed on T2w and fCBV maps. Representative iNOS (B), Arg1 (C), CD68 and Hoechst 33342 immunofluorescence images and their quantifications in the hypoxic U251 tumor. Arrows indicate pseudopalisaded areas in the tumor. (D) Representative co-localization of Arg1+ immunofluorescence with pimonidazole staining in the hypoxic U251 tumor. Scale bars: 500 µm. n = 3 animals. Statistical significance was achieved when p < 0.05 (*).
Our in vivo result suggest that hypoxia is responsible for the acquisition of an M2 phenotype. However, hypoxia may drive this polarization by also acting on tumor cells. To test this latter hypothesis, we performed in vitro studies, as described below.
Effect of hypoxia-exposed GBM cells supernatants on M0 MΦ polarization
U87-MG and U251 cells were cultured in 1% O2 for 24h and the supernatant (SN) was transferred onto M0 MΦ for 24h (Fig. 4). No changes in M0 MΦ morphology were observed when the SN from U87-MG and U251 cells either in normoxia or in hypoxia was applied (Fig. 4A). In parallel, no significant effects on the expression of iNOS and Arg1 mRNA were observed in M0 MΦ cultured either with SN from U87-MG or U251 cultured in 1% or 0.2% O2 (Fig. 4B). Functionally, no significant changes in NO production were observed in M0 MΦ cultured either with SN from U87-MG or U251 cultured in 1% or 0.2% O2 for 24 h (Fig. 4C). Moreover, no changes in Arg1 activity were observed when M0 MΦ were cultured with SN from U87-MG and U251 cells cultured in 1% O2. However, M0 MΦ cultured with SN from U251 in 0.2% O2 had a significant increase in Arg1 activity (36 ± 7 µg urea/h per 106 cells), which was not observed with the U87-MG SN, compared to M0 MΦ cultured with SN from U251 in normoxia (23 ± 4 µg urea/h per 106 cells) (Fig. 4C). The cytokine profile of the U87-MG and U251 cells cultured in hypoxia failed to show any change in the expression of IFNγ and IL4 both well-known to induce the polarization of M1 and M2 MΦ, respectively37 (Fig. S2). These results indicate that severe hypoxia applied to tumor cells induced Arg1 activity in M0 MΦ, only in one GBM cell type, and independently of IFNγ and IL4 productions. However, to conclude on the hypoxic cytokine production in GBM cells, a large genomic approach would be of need to evaluate. Consequently, we postulated that hypoxia could directly influence MΦ polarization.
Figure 4.

Effect of SN of hypoxia-exposed GBM cells on MΦ polarization. (A) Phase contrast microscopy of M0 MΦ cultured with common media or with SN withdrawn from U87-MG or U251 cells cultured either in normoxia (20% O2) or in 1% or 0.2% O2 for 24 h. Scale bar: 25 µm. iNOS and Arg1 mRNA relative expressions (compared to M0 MΦ cultured with common media) (B) and NO concentration (µM per 106 cells) and Arg1 activity (µg urea/h per 106 cells) (C) in M0 MΦ cultured with SN withdrawn from U87-MG or U251 cells cultured either in normoxia (20% O2) or in 1% or 0.2% O2 for 24 h. Data were represented as the mean ± SD, n = 3 per group. Statistical significance was achieved when p < 0.05 (*), otherwise it was not significant (NS).
Hypoxia polarizes M0 toward M2 MΦ independently of GBM cells
M0 MΦ were directly cultured at different degrees of hypoxia over different time periods (Figs. 5 and S3). After 24 h in 1% or 0.2% O2, M0 MΦ presented a cell morphology similar to that of M2 MΦ (Fig. 5A). Of note is that iNOS mRNA levels increased in M0 MΦ after 24 h in 1% O2, at a level significantly less than M1 MΦ (about 70 compared to 105 times greater, respectively, when normalized to M0 MΦ in normoxia) but not significantly different from M2 MΦ. M0 MΦ cultured in 0.2% O2 for 24 h did not express iNOS mRNA as M2 MΦ (Fig. 5B). In the contrary, Arg1 mRNA was clearly expressed in M2 MΦ and M0 MΦ cultured in 1% or 0.2% O2 when normalized to M0 MΦ in normoxia (between 102 to 103 times greater) but not in M1 MΦ (Fig. 5B). Moreover, the increase in exposure time decreased the iNOS mRNA expression and increased the Arg1 mRNA expression in M0 MΦ (Fig. S3A). These results show that M0 MΦ cultured in hypoxia increase the expression of Arg1 mRNA but not iNOS mRNA. Functionally, NO was produced in M1 MΦ (230 ± 61 µM of NO per 106 cells) and was significantly different from the level of M2 MΦ and M0 MΦ cultured in 1% or 0.2% O2 (24 ± 2, 26 ± 3 and 23 ± 5 µM of NO per 106 cells, respectively). However, M0 MΦ cultured after 24h in 1% or 0.2% O2 had Arg1 activities not significantly different from M2 MΦ (33 ± 1, 62 ± 12 and 43 ± 8 µg urea/h per 106 cells, respectively) (Fig. 5C). Furthermore, the longer the time of exposure of hypoxia increased the Arg1 activity but not the NO production in M0 MΦ (Fig. S3B). These results strengthen the possibility that hypoxia directly polarizes M0 into M2 MΦ with a more efficient effect than through GBM cells.
Figure 5.

Effect of hypoxia on MΦ polarization. (A) Phase contrast microscopy of M0, M1 and M2 MΦ cultured in normoxia (20% O2) and M0 MΦ in the presence of 1% or 0.2% O2 for 24 h. Scale bar: 20 µm. iNOS and Arg1 mRNA relative expressions (compared to M0 MΦ in normoxia) (B) and NO concentration (µM per 106 cells) and Arg1 activity (µg urea/h per 106 cells) (C) in M1 and M2 MΦ in normoxia (20% O2) and M0 MΦ in 1% or 0.2% O2 for 24 h. Data were represented as the mean ±SD, n = 3 per group and per time. Statistical significance was achieved when p < 0.05 (*), otherwise it was not significant (NS).
Hypoxia reinforces the M2 phenotype and re-educates M1 MΦ toward an M2 phenotype
We have previously shown that M2 MΦ are found in hypoxic tumors and that hypoxia may drive the polarization of M0 MΦ. However, M2 MΦ could arise from the re-education of resident M1 MΦ in the tumor.
After having demonstrated that hypoxia is more potent to induce the M2 phenotype directly than via GBM cells, we therefore studied the question whether hypoxia could also directly re-educate M1 into M2 MΦ. MΦ were pre-polarized to either M1 or M2 phenotype for 24 h and then cultured in 1% or 0.2% O2 for different times (Fig. 6 and Fig. S4). Morphologically, no changes were observed when M2 MΦ were cultured in 1% O2 for 24 h, but M1 MΦ seemed smaller and more readily defined under phase contrast after hypoxia exposure (Fig. 6A). At the transcriptional level (Fig. 6B), no significant changes were detected in M1 MΦ cultured in 1% O2 for 24 h. However, a significant decrease in iNOS mRNA expression were observed in M1 MΦ cultured in 0.2% O2 (about 60 times greater compared to M0 in normoxia) compared to M1 MΦ in normoxia (about 104 times greater), with a level close to that of M2 MΦ in normoxia (about 30 times greater). Arg1 mRNA expression was increased in M1 MΦ cultured in 0.2% O2 (about 500 times greater), at a level close to that of M2 MΦ (about 103 times greater). Exposure of M2 MΦ to 1% or 0.2% O2 did not induce significant changes in the expression of Arg1 mRNA (Fig. 6B). Furthermore, the increase in the time the cells were exposed to hypoxia, decreased the iNOS mRNA expression in M1 MΦ and increased the Arg1 mRNA expression in M2 MΦ (Fig. S4A). Functionally, NO production in M1 MΦ significantly decreased with the decrease in O2 level (200 ± 35, 122 ± 30 and 84 ± 17 µM of NO for 106 cells in M1 MΦ in normoxia, 1% and 0.2% O2, respectively) (Fig. 6C). In the contrary, Arg1 activity increased in M1 MΦ cultured in hypoxia (38 ± 7 and 46 ± 8 µg urea/h per 106 cells in M1 MΦ in 1% and 0.2% O2, respectively) at a level not significantly different from M2 MΦ (88 ± 47 µg urea/h per 106 cells). A significantly increase in Arg1 activity was observed in M2 MΦ after 24 h of 0.2% O2 (192 ± 28 µg urea/h per 106 cells) compared to normoxia (Fig. 6C). Moreover, the Arg1 activity increased in M2 MΦ with longer the time of exposure to hypoxia (Fig. S4B). These results show that hypoxia seems to decrease iNOS mRNA expression/NO production in M1 MΦ and to reinforce M2 markers in M2 MΦ. Furthermore, M0 (Fig. S5A), M1 and M2 (Fig. S5B) MΦ cultured in 1% or 0.2% O2 and then re-oxygenated in 20% O2 for 24 h did not show a difference of iNOS and Arg1 activity compared to MΦ maintained in hypoxia. These results indicates that re-oxygenation fails to change the MΦ phenotype acquired in hypoxia.
Figure 6.

Effect of hypoxia on MΦ re-education. (A) Phase contrast microscopy of M0, M1 and M2 MΦ in normoxia (20% O2) or in 1% O2 for 24 h. Scale bar: 20 µm. iNOS and Arg1 mRNA relative expressions (compared to M0 MΦ in normoxia) (B) and NO concentration (µM per 106 cells) and Arg1 activity (µg urea/h per 106 cells) (C) in M1 and M2 MΦ in normoxia (20% O2) or in 1% or 0.2% O2 for 24 h; Data were represented as the mean ± SD, n = 3 per group and per time. Statistical significance was achieved when p < 0.05 (*), otherwise it was not significant (NS).
Discussion
Recent reports have demonstrated that hypoxia plays an important role in tumor development,6,7 treatments resistance7,8 and tumor inflammation.28,38 The inflammatory microenvironment is essential for tumoral development; particularly MΦ, markedly present in GBM.2 In this study, we have shown, in vivo, in a model of GBM that progressively evolves toward severe hypoxia, and in vitro, that GBM hypoxia is responsible for MΦ polarization, for a re-education of M1 MΦ toward an M2 phenotype and for fine-tuning the M2 phenotype.
Numerous studies have tried to correlate MΦ with hypoxia and it seems, as noted in the present investigation, that this relationship may be model or cell lines dependent.39 It should be underlined that the hypoxic signal, observed in the present study, seems to be the most important trigger for MΦ migration as well as for polarization rather than the cell type. We observed that MΦ were the most abundant in the U251 tumor that best replicates the hypoxic characteristics of human GBM.32 It is not unlikely that the two cell lines we employed may produce different cytokines and/or chemokines. Accordingly, we analyzed the evolution of MΦ migration as a function of tumoral growth with increasingly severe hypoxia. We demonstrated that this MΦ migration is oxygen-dependent. Few MΦ were observed at the early phase of U251 tumor development when hypoxia was undetectable. This oxygen-dependent phenomenon could explain the increase in MΦ numbers with increasing grade of gliomas,23,40 and is consistent with the observation that ptO2 values also fall as the glioma grade increases.5,6 Altogether, these data indicate that MΦ can accumulate in hypoxic areas of GBM and only by the presence of hypoxia.
Based on publications that have studied the relationship between the number of M2 MΦ and the grade of glioma,23,40 we have examined the possibility that hypoxia may also drive the acquisition of an M2 phenotype. Indeed, the MΦ polarization in a tumor is a relatively recent and not yet fully established concept. Here, we have shown marked differences between the two different models of glioma employed. In the early development of the U251 tumor, a time at which hypoxia had not yet developed, the proportion of M1 or M2 markers were very low. In contrast, in the late phase of the U251 tumor, MΦ present at the less hypoxic and more vascularized TS, or margin, were both M0 and M1 MΦ as described in mammary tumors,41 the only MΦ present in the highly hypoxic TC exhibited discrimatory factors for the M2 phenotype. This study demonstrate a pronounced transformation of MΦ, concomitant to the hypoxic shift, and this within the evolution of a single type of tumor.
After having shown the association between the normoxic/hypoxic transition and the acquisition of the M2 phenotype, the next question was to know whether these M2 MΦ were derived from (i) M0 MΦ, (ii) newly arrived M2 MΦ or (iii) re-educated from M1 MΦ as initially suspected.17 Although the effect of hypoxia on MΦ gene expression in tumor development has been studied,42-45 no one has described the direct effect of the O2 gradient on MΦ polarization. We cultured M0 MΦ in moderate (1% O2) and severe hypoxia (0.2% O2) to mimic the O2 gradient observed in vivo. Despite that several studies have demonstrated that MΦ polarization toward an M2 phenotype was induced by hypoxic tumor cells,27,29,30 we have shown that this hypoxia effect is independent on GBM cells. Indeed, when we performed the co-culture experiments, the production of urea was observed only with one GBM cell type and the amount of urea was no more than 36 ± 7 µg urea/h per 106 cells. These results speak about a direct effect of hypoxia on MΦ polarization. Furthermore, we observed that the more severe the hypoxia, the more importantly the M0 MΦ expressed M2 MΦ markers. These results could explain our in vivo observations in which circulating monocytes enter the tumor as M0 MΦ and then acquire M2 markers with decreased O2 levels and consequently become exclusively M2 MΦ in parallel with their accumulation in the hypoxic zones within the tumor. Factors responsible for this hypoxia-dependent MΦ polarization could be the hypoxia inducible factors, HIF-1α and HIF-2α. It has been suggested that HIF-1α was stable in M1 MΦ while HIF-2α was stabilized in M2 MΦ31 but also that HIF-1α was stable in acute hypoxia compared to HIF-2α which was stable under chronic hypoxia.46 Thus, HIF-2α seems to be involved in the establishment of an M2 phenotype under hypoxia but this needs to be demonstrated. A recent study has also indicated that M2 MΦ were promoted by the lactic acid produced by hypoxic tumor cells.30 Another hypothesis is that hypoxic M2 MΦ could also produce lactic acid which in turn reinforces the M2 phenotype. In this way, it could be interesting to focalize on this pathway and try to inhibit lactic acid production47 or intracellular transport48 in cells to inhibit or re-educate M2 MΦ in GBM.
However, as proposed, M2 MΦ may originate from a re-education of M1 MΦ originally present at the initial stage of tumoral development21,22 and this re-education would be induced by cytokine stimuli.49,50,37 Here, we hypothesize that hypoxia may directly re-educate M1 MΦ, which has never been assessed in vitro to the best of our knowledge. We demonstrated that the more severe the hypoxia and the more the M1 markers were decreased, then the greater the increase in M2 markers. We also observed an augmented expression of M2 markers on M2 MΦ as an inverse function of O2 availability. These results not only indicate that hypoxia could induce the re-education from antitumoral MΦ toward protumoral MΦ but also that severe hypoxia fine-tunes the protumoral phenotype as already described in lung carcinoma.26 In this study, we have demonstrated that hypoxia directly differentiates M0 MΦ, but also re-educates M1 MΦ toward an M2 phenotype. These results differ from those of Laoui and coworkers (2014),26 who demonstrated (in a model of pulmonary carcinoma) that hypoxia is without influence on the polarization of M0 MΦ and the re-education of M1 MΦ. Although various hypotheses could explain this discrepancy, we postulate that a major possibility might be the degree of hypoxia. Indeed, in our study, we clearly show from both in vivo and in vitro experiments that the more pronounced the hypoxia, the more the M2 phenotype is present. In GBM, it has been demonstrated that, even if the median ptO2 is 7.4 mmHg, the ptO2 around pseudopalisaded areas is lower than 2.5 mmHg and even attaining near zero in the core. It is in these severe hypoxic (or near anoxic) conditions that we observe the M2 phenotype. In the study from Laoui and colleagues (2014),26 one might note that the initial ptO2 was around 6 mmHg in their tumor model, a degree of hypoxia described as being “moderate” in a general review on this subject.46 Rather than reinforce hypoxia, Laoui and coworkers induced a minor re-oxygenation (from 6 to 10 mmHg), whereas our results show that a complete re-oxygenation of MΦ failed to change the phenotype acquired in hypoxia. Concomitant to the effects of hypoxia-induced changes in the metabolism of glioma cells and also in angiogenesis, the potentiation of a protumoral inflammation should also be considered as an explanation of the results that show that hypoxia is an independent, negative prognostic factor in GBM.9,23
As a conclusion, in our study, we made the assumption that hypoxia should not be considered as a binary process but as a slowly evolving process which may directly drive the acquisition of a protumoral immunity. We used two models of GBM, highly different in terms of oxygenation and benefited from the ability to assess intratumoral hypoxia as a function of time to show that MΦ migration into the tumor as well as MΦ polarization toward a protumoral phenotype are related to the depth of hypoxia. As the GBM grows, the volume of hypoxia expands and its severity increases, hence the acquisition of the M2 phenotype (Fig. 7). However, up to now, we have been unable to characterize the effects of M2 MΦ in this model.
Figure 7.
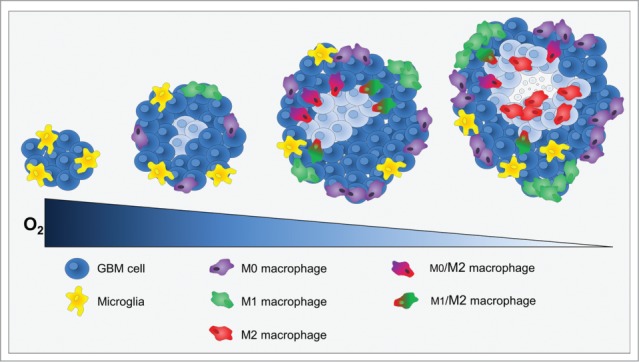
Cross-section through a theoretical GBM to indicate the position and identity of the sub-types of MΦ as a function of hypoxia development. At the onset of tumor development, only microglia was present. When hypoxia begin to take place, MΦ were attracted to the tumor site and present M0 and M1 phenotypes at the shell of the tumor. Then, M0 and M1 MΦ migrated toward hypoxic zones where they increased the M2 markers. Once arrived to hypoxic zones, MΦ were M2 cells and hypoxia fine-tunes this phenotype.
As large amounts of NO, produced by M1 MΦ,17 has been demonstrated to induce cell damage51,52 and inhibit tumor angiogenesis,53 several attempts have been made to re-educate protumoral MΦ toward antitumoral MΦ24,54-56 rather than the depletion of MΦ. Our present investigation encourages a combination of these recently developed therapies for GBM with approaches designed to alleviate intratumoral hypoxia to further potentiate the efficacy of available chemotherapeutic agents. For instance, low dose antiangiogenic therapy,57 low dose irradiation,58 or hyperoxic gases59 could all be pertinent strategies to militate against the tumoral accumulation of M2 MΦ.
Materials and Methods
GBM cell culture
Human GBM cell lines, U87-MG (ATCC) and U251 (NCI), were cultured in 1g/L glucose Dulbecco's Modified Eagle's Medium (DMEM, Sigma) supplemented with 10% fetal calf serum (Eurobio), 1 µg/mL penicillin/streptomycin (P/S, Sigma) and 2 mM glutamine (Gln, Sigma) at 37°C in wet atmosphere.
Tumor models
Tumor models consist of an orthotopic injection of human GBM cells in athymic rats (200–250 g, Charles River Laboratory). The animal investigations were performed under the current European directive (2010/63/EU). The license to investigate was given to SV (14–55) in authorized housing and laboratories (B14118001) and with the permission of the regional committee on animal ethics (CENOMEXA, 0611-02). The rats were maintained in specific pathogen-free housing and were fed γ-irradiated laboratory food and water ad libitum.
Rats were operated under anesthesia (induction in 5% and maintenance in 2% of isoflurane in 70% NO2/30% O2) and U87-MG and U251 cells were injected (5.104 cells in 3 µL in 2 mM Gln/PBS) in the right caudate-putamen.
Imaging
For all imaging experiments, animals were anesthetized as described above, and maintained in position by ear and tooth bars. Micro-magnetic resonance imaging (µMRI) was performed on a 7 Tesla horizontal magnet (Pharmascan®, Bruker, Germany). Micro-positron emission tomography (µPET) was performed on an Inveon PET-CT small animal imaging system (Siemens Healthcare) with the use of the tracer, 3-[18F]-fluoro-1-(2-nitro-1-imidazolyl)-2-propanol ([18F]-FMISO), to detect hypoxia. Imaging protocols are detailed in supplementary data.
Immunohistochemistry
At the end of the protocol, the rats were deeply anesthetized and were transcardially perfused with a 0.2 M phosphate buffer (PB)/4% paraformaldehyde (PFA, Sigma) solution 2 h after the intraperitoneal injection (80 mg/kg) of pimonidazole (Hypoxyprobe Incorporation, USA). The brain was removed and placed in 30% sucrose for 48 h and 30 µm thick freezing microtome sections were realized. Slices were blocked with PBS, 0.5% Triton, 3% BSA (Sigma) for 2 h and then incubated overnight with primary antibodies (Table S1) in PBS, 0.5% Triton, 1% BSA at 4°C. Sections were then incubated with fluorochrome-conjugated secondary antibodies (Table S1) and Hoechst 33342 (Sigma, 10 µg/mL) in PBS solution as described above.
Image analysis
Images were analyzed by in-house macros based on ImageJ software (http://imagej.nih.gov/ij/). MΦ/microglial cell density was determined as the number of positive pixels for CD68 divided by the total tumor area. MΦ density was differentiated from that of microglia by the ratio of CD14 positive pixels to CD68 positive pixels. M1 and M2 MΦ densities were characterized by the number of either iNOS or Arg1 positive pixels, respectively, within the population of CD68 positive pixels.
Bone marrow-derived MΦ culture and activation
Bone marrow was isolated from femora and tibiae of nude mice (20–25 g, Charles River Laboratory) by flushing the bones with 1 mL of Iscove's Modified Dulbecco's Media (IMDM, Sigma) containing 60% Fetal Clone II (FCII, Thermo Scientific) and 1 µg/mL P/S (Sigma). The marrow was passed through a 70 µm strainer and MΦ (M0) were selected and cultured in IMDM enriched with 15% FCII, 1 µg/mL P/S, 10 ng/mL recombinant mouse MΦ colony-stimulating factor (M-CSF, Miltenyi Biotec) and 10 ng/mL recombinant mouse Fms-related tyrosine kinase three ligand (Flt3-Ligand, Miltenyi Biotec) at 37°C in a humid atmosphere. M1 MΦ were obtained by culturing cells in 1 g/L glucose DMEM (Sigma) supplied with 15% FCII, 1 µg/mL P/S, 2 mM Gln (Sigma), 100 ng/mL LPS (Sigma) and 10 U/mL recombinant mouse IFNγ (eBioscience). M2 MΦ were obtained by culturing cells with 1g/L glucose DMEM supplemented with 15% FCII, 1% P/S, 2mM Gln (Sigma) and 50 ng/mL recombinant mouse IL4 (Miltenyi Biotec).
Cell and hypoxic cultures
The normoxic state (20% O2) was a humidified 5% CO2/air atmosphere in an incubator and hypoxia was obtained with a humidified 5% CO2/balance N2 gas mixture in a hypoxic chamber (Invivo2 500, Ruskinn, Awel) at 37°C. For the polarization experiments, M0 MΦ were cultured either in normoxia or in hypoxia (1% or 0.2% O2) for different time periods. For the re-education experiments, M1 and M2 MΦ were activated 24 h and then cultured in hypoxia (1% or 0.2% O2) with their respective conditioning media for different time periods. GBM cell lines were cultured in the M0 MΦ growth medium, as described above, in normoxia or in 1% or 0.2% O2 for 24 h.
Reverse transcription and real-time quantitative PCR
Total RNA was isolated by the Nucleospin® RNA kit (Macherey-Nagel) following manufacter's instructions and reverse transcribed into cDNA (cDNA) with AMV® reverse transcriptase (Promega). cDNA were then quantified by real-time PCR with SYBR® Green master mix (Bio-Rad) with primers designed by Eurogentec (Table S2). Samples were run in triplicate with an amplification profile as follows: an activation stage at 95°C for 3 min following by 40 cycles at 95°C, 15sec and 60°C, 30 sec. Expression levels were determined by the ΔCt method.
Determination of nitric oxide (NO) production
NO measurement in the SN of MΦ cultures was performed by the Griess reaction.60 Briefly, the Griess reagent was prepared by mixing 2% sulphanilamide (Sigma) in 10% phosphoric acid (Sigma) and 0.2% naphthylethylene-diamine-dihydrochloride (Sigma). The reagent was added to SN and the mixture was incubated 10 min at room temperature in the dark. Each sample was assayed in duplicate, the absorbance was measured at 540 nm and NO concentration was determined with sodium nitrite as a standard.
Determination of Arg1 activity
Arg1 activity was determined by a standard colorimetric method60 in cell lysates. Briefly, cells were lysed by adding 0.1% Triton X-100 (Sigma) and 50 mM Tris-HCl (pH 7.5). 10 mM MnCl2 (Sigma) to cell samples, then heated at 56°C for 7 min to activate the enzyme. Hydrolysis of L-arginine by Arg1 was performed by incubating the mixture with 50 µmol of L-arginine (pH 9.7; Sigma) at 37°C for 2 h, and the reaction was stopped by adding an acid solution (H2SO4; H3PO4; H2O). For the determination of urea production, α-isonitrosopropiophenone (Sigma) was added and the mixture was incubated at 95°C for 30 min and then 4°C for 30 min. Each sample was assayed in duplicate, the absorbance was measured at 540 nm and urea production was determined with urea as a standard.
Statistical analyses
All data are presented as the mean ± standard deviation (SD). Statistical analyses were performed with the JMP® program (SAS institute, USA) and, unless otherwise stated, significances were calculated by nonparametric Kruskall–Wallis for multiple comparisons or Mann–Whitney U tests. Statistical significance was achieved when p < 0.05 (*) or p < 0.01 (**), otherwise they were not significant (NS).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgements
The authors wish to thank the LDM-TEP team for the [18F]-FMISO production.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Funding
This study was supported by the grants from the Conseil Régional de Basse-Normandie, the Université de Caen Basse-Normandie (UCBN), the French Ministère de l'Enseignement Supérieur et de la Recherche (MESR), the Center National de la Recherche Scientifique (CNRS), the TC2N ‘Trans Channel Neuroscience Network’ Interreg IV A 2 Mers Seas Zeeëns program, “Investing in your future” crossborder cooperation program 2007–2014 part financed by the European Union (European Regional Development Fund), the French National Agency for Research called “Investissements d'Avenir” no. ANR-11-LABEX-0018-01, the Fédération pour la Recherche sur le Cerveau (FRC), the Institut National contre le Cancer (INCa), and the Advanced Resource Center for Hadrontherapy in Europe (Archade).
References
- 1.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJB, Belanger K, Brandes A, Marosi C, Bogdahn U et al.. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005; 352:987-96; PMID:15758009; http://dx.doi.org/ 10.1056/NEJMoa043330 [DOI] [PubMed] [Google Scholar]
- 2.Charles NA, Holland EC, Gilbertson R, Glass R, Kettenmann H. The brain tumor microenvironment. Glia 2011; 59:1169-80; PMID:21446047; http://dx.doi.org/ 10.1002/glia.21136 [DOI] [PubMed] [Google Scholar]
- 3.Clavreul A, Etcheverry A, Chassevent A, Quillien V, Avril T, Jourdan M-L, Michalak S, François P, Carré J-L, Mosser J et al.. Isolation of a new cell population in the glioblastoma microenvironment. J Neurooncol 2012; 106:493-504; PMID:21928115; http://dx.doi.org/ 10.1007/s11060-011-0701-7 [DOI] [PubMed] [Google Scholar]
- 4.Hirata K, Terasaka S, Shiga T, Hattori N, Magota K, Kobayashi H, Yamaguchi S, Houkin K, Tanaka S, Kuge Y et al.. 18F-Fluoromisonidazole positron emission tomography may differentiate glioblastoma multiforme from less malignant gliomas. Eur J Nucl Med Mol Imaging 2012; 39:760-70; PMID:22307533; http://dx.doi.org/ 10.1007/s00259-011-2037-0 [DOI] [PubMed] [Google Scholar]
- 5.Rampling R, Cruickshank G, Lewis AD, Fitzsimmons SA, Workman P. Direct measurement of pO2 distribution and bioreductive enzymes in human malignant brain tumors. Int J Radiat Oncol Biol Phys 1994; 29:427-31; PMID:8005794; http://dx.doi.org/ 10.1016/0360-3016(94)90432-4 [DOI] [PubMed] [Google Scholar]
- 6.Evans SM, Judy KD, Dunphy I, Jenkins WT, Hwang W, Nelson PT, Lustig RA, Jenkins K, Magarelli DP, Hahn SM et al.. Hypoxia is important in the biology and aggression of human glial brain tumors. Clin Cancer Res 2004; 10:8177-84; PMID:15623592; http://dx.doi.org/ 10.1158/1078-0432.CCR-04-1081 [DOI] [PubMed] [Google Scholar]
- 7.Kaur B, Khwaja FW, Severson EA, Matheny SL, Brat DJ, Van Meir EG. Hypoxia and the hypoxia-inducible-factor pathway in glioma growth and angiogenesis. Neuro Oncol 2005; 7:134-53; PMID:15831232; http://dx.doi.org/ 10.1215/S1152851704001115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vaupel P, Mayer A. Hypoxia in cancer: significance and impact on clinical outcome. Cancer Metastasis Rev 2007; 26:225-39; PMID:17440684; http://dx.doi.org/ 10.1007/s10555-007-9055-1 [DOI] [PubMed] [Google Scholar]
- 9.Spence AM, Muzi M, Swanson KR, O'Sullivan F, Rockhill JK, Rajendran JG, Adamsen TCH, Link JM, Swanson PE, Yagle KJ et al.. Regional hypoxia in glioblastoma multiforme quantified with ; [18F]Fluoromisonidazole positron emission tomography before radiotherapy: correlation with time to progression and survival. Clin Cancer Res 2008; 14:2623-30; PMID:18451225; http://dx.doi.org/ 10.1158/1078-0432.CCR-07-4995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Res 2006; 66:605-12; PMID:16423985; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-4005 [DOI] [PubMed] [Google Scholar]
- 11.Franklin RA, Liao W, Sarkar A, Kim MV, Bivona MR, Liu K, Pamer EG, Li MO. The cellular and molecular origin of tumor-associated macrophages. Science 2014; 344:921-5; PMID:24812208; http://dx.doi.org/ 10.1126/science.1252510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murdoch C, Giannoudis A, Lewis CE. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood 2004; 104:2224-34; PMID:15231578; http://dx.doi.org/ 10.1182/blood-2004-03-1109 [DOI] [PubMed] [Google Scholar]
- 13.Badie B, Schartner JM. Flow cytometric characterization of tumor-associated macrophages in experimental gliomas. Neurosurgery 2000; 46:957-62; PMID:10764271; http://dx.doi.org/ 10.1097/00006123-200004000-00035 [DOI] [PubMed] [Google Scholar]
- 14.Strik HM, Stoll M, Meyermann R. Immune cell infiltration of intrinsic and metastatic intracranial tumours. Anticancer Res 2004; 24:37-42; PMID:15015573 [PubMed] [Google Scholar]
- 15.Hussain SF, Yang D, Suki D, Aldape K, Grimm E, Heimberger AB. The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses. Neuro Oncol 2006; 8:261-79; PMID:16775224; http://dx.doi.org/ 10.1215/15228517-2006-008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu-Emerson C, Snuderl M, Kirkpatrick ND, Goveia J, Davidson C, Huang Y, Riedemann L, Taylor J, Ivy P, Duda DG et al.. Increase in tumor-associated macrophages after antiangiogenic therapy is associated with poor survival among patients with recurrent glioblastoma. Neuro Oncol 2013; 15:1079-87; PMID:23828240; http://dx.doi.org/ 10.1093/neuonc/not082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol 2002; 23:549-55; PMID:12401408; http://dx.doi.org/ 10.1016/S1471-4906(02)02302-5 [DOI] [PubMed] [Google Scholar]
- 18.Chang CI, Liao JC, Kuo L. Macrophage arginase promotes tumor cell growth and suppresses nitric oxide-mediated tumor cytotoxicity. Cancer Res 2001; 61:1100-6; PMID:11221839 [PubMed] [Google Scholar]
- 19.Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest 2012; 122:787-95; PMID:22378047; http://dx.doi.org/ 10.1172/JCI59643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Biswas SK, Chittezhath M, Shalova IN, Lim J-Y. Macrophage polarization and plasticity in health and disease. Immunol Res 2012; 53:11-24; PMID:22418728; http://dx.doi.org/ 10.1007/s12026-012-8291-9 [DOI] [PubMed] [Google Scholar]
- 21.Biswas SK, Sica A, Lewis CE. Plasticity of macrophage function during tumor progression: regulation by distinct molecular mechanisms. J Immunol 2008; 180:2011-7; PMID:18250403; http://dx.doi.org/ 10.4049/jimmunol.180.4.2011 [DOI] [PubMed] [Google Scholar]
- 22.Huang Y, Snuderl M, Jain RK. Polarization of tumor-associated macrophages: a novel strategy for vascular normalization and antitumor immunity. Cancer Cell 2011; 19:1-2; PMID:21251607; http://dx.doi.org/ 10.1016/j.ccr.2011.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prosniak M, Harshyne LA, Andrews DW, Kenyon LC, Bedelbaeva K, Apanasovich TV, Heber-Katz E, Curtis MT, Cotzia P, Hooper DC. Glioma grade is associated with the accumulation and activity of cells bearing M2 monocyte markers. Clin Cancer Res 2013; 19:3776-86; PMID:23741072; http://dx.doi.org/ 10.1158/1078-0432.CCR-12-1940 [DOI] [PubMed] [Google Scholar]
- 24.Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, Olson OC, Quick ML, Huse JT, Teijeiro V et al.. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med 2013; 19:1264-72; PMID:24056773; http://dx.doi.org/ 10.1038/nm.3337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lewis C, Murdoch C. Macrophage responses to hypoxia: implications for tumor progression and anti-cancer therapies. Am J Pathol 2005; 167:627-35; PMID:16127144; http://dx.doi.org/ 10.1016/S0002-9440(10)62038-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Laoui D, Van Overmeire E, Di Conza G, Aldeni C, Keirsse J, Morias Y, Movahedi K, Houbracken I, Schouppe E, Elkrim Y et al.. Tumor hypoxia does not drive differentiation of tumor-associated macrophages but rather fine-tunes the M2-like macrophage population. Cancer Res 2014; 74:24-30; PMID:24220244; http://dx.doi.org/ 10.1158/0008-5472.CAN-13-1196 [DOI] [PubMed] [Google Scholar]
- 27.Zhang J, Cao J, Ma S, Dong R, Meng W, Ying M, Weng Q, Chen Z, Ma J, Fang Q et al.. Tumor hypoxia enhances non-small cell lung cancer metastasis by selectively promoting macrophage M2 polarization through the activation of ERK signaling. Oncotarget 2014;5:9664-77; PMID:25313135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tafani M, Di Vito M, Frati A, Pellegrini L, De Santis E, Sette G, Eramo A, Sale P, Mari E, Santoro A et al.. Pro-inflammatory gene expression in solid glioblastoma microenvironment and in hypoxic stem cells from human glioblastoma. J Neuroinflammation 2011; 8:32; PMID:21489226; http://dx.doi.org/ 10.1186/1742-2094-8-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tripathi C, Tewari BN, Kanchan RK, Baghel KS, Nautiyal N, Shrivastava R, Kaur H, Bhatt MLB, Bhadauria S. Macrophages are recruited to hypoxic tumor areas and acquire a pro-angiogenic M2-polarized phenotype via hypoxic cancer cell derived cytokines Oncostatin M and Eotaxin. Oncotarget 2014; 5:5350-68; PMID:25051364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Colegio OR, Chu N-Q, Szabo AL, Chu T, Rhebergen AM, Jairam V, Cyrus N, Brokowski CE, Eisenbarth SC, Phillips GM et al.. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014; 513:559-63; PMID:25043024; http://dx.doi.org/ 10.1038/nature13490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Takeda N, O'Dea EL, Doedens A, Kim J, Weidemann A, Stockmann C, Asagiri M, Simon MC, Hoffmann A, Johnson RS. Differential activation and antagonistic function of HIF-α isoforms in macrophages are essential for NO homeostasis. Genes Dev 2010; 24:491-501; PMID:20194441; http://dx.doi.org/ 10.1101/gad.1881410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Corroyer-Dulmont A, Pérès EA, Petit E, Durand L, Marteau L, Toutain J, Divoux D, Roussel S, MacKenzie ET, Barré L et al.. Noninvasive assessment of hypoxia with 3-; [18F]-fluoro-1-(2-nitro-1-imidazolyl)-2-propanol (; [18F]-FMISO): a PET study in two experimental models of human glioma. Biol Chem 2013; 394:529-39; PMID:23399636; http://dx.doi.org/ 10.1515/hsz-2012-0318 [DOI] [PubMed] [Google Scholar]
- 33.Hou H, Krishnamurthy Nemani V, Du G, Montano R, Song R, Gimi B, Swartz HM, Eastman A, Khan N. Monitoring oxygen levels in orthotopic human glioma xenograft following carbogen inhalation and chemotherapy by implantable resonator-based oximetry. Int J Cancer 2014; 136:1688-96; PMID:25111969; http://dx.doi.org/ 10.1002/ijc.29132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guillemin GJ, Brew BJ. Microglia, macrophages, perivascular macrophages, and pericytes: a review of function and identification. J Leukoc Biol 2004; 75:388-97; PMID:14612429; http://dx.doi.org/ 10.1189/jlb.0303114 [DOI] [PubMed] [Google Scholar]
- 35.Rempel SA, Dudas S, Ge S, Gutiérrez JA. Identification and localization of the cytokine SDF1 and its receptor, CXC chemokine receptor 4, to regions of necrosis and angiogenesis in human glioblastoma. Clin Cancer Res 2000; 6:102-11; PMID:10656438 [PubMed] [Google Scholar]
- 36.Zagzag D, Esencay M, Mendez O, Yee H, Smirnova I, Huang Y, Chiriboga L, Lukyanov E, Liu M, Newcomb EW. Hypoxia- and vascular endothelial growth factor-induced stromal cell-derived factor-1α/CXCR4 expression in glioblastomas: one plausible explanation of scherer's structures. Am J Pathol 2008; 173:545-60; PMID:18599607; http://dx.doi.org/ 10.2353/ajpath.2008.071197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pelegrin P, Surprenant A. Dynamics of macrophage polarization reveal new mechanism to inhibit IL-1beta release through pyrophosphates. EMBO J 2009; 28:2114-27; PMID:19536133; http://dx.doi.org/ 10.1038/emboj.2009.163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murat A, Migliavacca E, Hussain SF, Heimberger AB, Desbaillets I, Hamou M-F, Rüegg C, Stupp R, Delorenzi M, Hegi ME. Modulation of angiogenic and inflammatory response in glioblastoma by hypoxia. PLoS One 2009; 4:e5947; PMID:19536297; http://dx.doi.org/ 10.1371/journal.pone.0005947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chiang C-S, Fu SY, Wang S-C, Yu C-F, Chen F-H, Lin C-M, Hong J-H. Irradiation promotes an M2 macrophage phenotype in tumor hypoxia. Front Oncol 2012; 2:89; PMID:22888475; http://dx.doi.org/ 10.3389/fonc.2012.00089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Komohara Y, Ohnishi K, Kuratsu J, Takeya M. Possible involvement of the M2 anti-inflammatory macrophage phenotype in growth of human gliomas. J Pathol 2008; 216:15-24; PMID:18553315; http://dx.doi.org/ 10.1002/path.2370 [DOI] [PubMed] [Google Scholar]
- 41.Movahedi K, Laoui D, Gysemans C, Baeten M, Stangé G, Van den Bossche J, Mack M, Pipeleers D, In't Veld P, De Baetselier P et al.. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res 2010; 70:5728-39; PMID:20570887; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-4672 [DOI] [PubMed] [Google Scholar]
- 42.Leek RD, Talks KL, Pezzella F, Turley H, Campo L, Brown NS, Bicknell R, Taylor M, Gatter KC, Harris AL. Relation of hypoxia-inducible factor-2α (HIF-2α) expression in tumor-infiltrative macrophages to tumor angiogenesis and the oxidative thymidine phosphorylase pathway in human breast cancer. Cancer Res 2002; 62:1326-9; PMID:11888900 [PubMed] [Google Scholar]
- 43.Burke B, Giannoudis A, Corke KP, Gill D, Wells M, Ziegler-Heitbrock L, Lewis CE. Hypoxia-induced gene expression in human macrophages: implications for ischemic tissues and hypoxia-regulated gene therapy. Am J Pathol 2003; 163:1233-43; PMID:14507633; http://dx.doi.org/ 10.1016/S0002-9440(10)63483-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fang H-Y, Hughes R, Murdoch C, Coffelt SB, Biswas SK, Harris AL, Johnson RS, Imityaz HZ, Simon MC, Fredlund E et al.. Hypoxia-inducible factors 1 and 2 are important transcriptional effectors in primary macrophages experiencing hypoxia. Blood 2009; 114:844-59; PMID:19454749; http://dx.doi.org/ 10.1182/blood-2008-12-195941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Doedens AL, Stockmann C, Rubinstein MP, Liao D, Zhang N, DeNardo DG, Coussens LM, Karin M, Goldrath AW, Johnson RS. Macrophage expression of HIF-1α suppresses T cell function and promotes tumor progression. Cancer Res 2010; 70:7465-75; PMID:20841473; http://dx.doi.org/ 10.1158/0008-5472.CAN-10-1439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koh MY, Powis G. Passing the baton: the HIF switch. Trends Biochem Sci 2012; 37:364-72; PMID:22818162; http://dx.doi.org/ 10.1016/j.tibs.2012.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sonveaux P, Végran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, De Saedeleer CJ, Kennedy KM, Diepart C, Jordan BF et al.. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest 2008; 118:3930-42; PMID:19033663; http://dx.doi.org/ 10.1172/JCI36843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miranda-Gonçalves V, Honavar M, Pinheiro C, Martinho O, Pires MM, Pinheiro C, Cordeiro M, Bebiano G, Costa P, Palmeirim I et al.. Monocarboxylate transporters (MCTs) in gliomas: expression and exploitation as therapeutic targets. Neuro Oncol 2013; 15:172-88; PMID:23258846; http://dx.doi.org/ 10.1093/neuonc/nos298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stout RD, Jiang C, Matta B, Tietzel I, Watkins SK, Suttles J. Macrophages sequentially change their functional phenotype in response to changes in microenvironmental influences. J Immunol 2005; 175:342-9; PMID:15972667; http://dx.doi.org/ 10.4049/jimmunol.175.1.342 [DOI] [PubMed] [Google Scholar]
- 50.Dehne N, Tausendschön M, Essler S, Geis T, Schmid T, Brüne B. IL-4 reduces the proangiogenic capacity of macrophages by down-regulating HIF-1α translation. J Leukoc Biol 2014; 95:129-37; PMID:24006507; http://dx.doi.org/ 10.1189/jlb.0113045 [DOI] [PubMed] [Google Scholar]
- 51.Goodman JE, Hofseth LJ, Hussain SP, Harris CC. Nitric oxide and p53 in cancer-prone chronic inflammation and oxyradical overload disease. Environ Mol Mutagen 2004; 44:3-9; PMID:15199542; http://dx.doi.org/ 10.1002/em.20024 [DOI] [PubMed] [Google Scholar]
- 52.Weigert A, Brüne B. Nitric oxide, apoptosis and macrophage polarization during tumor progression. Nitric Oxide 2008; 19:95-102; PMID:18486631; http://dx.doi.org/ 10.1016/j.niox.2008.04.021 [DOI] [PubMed] [Google Scholar]
- 53.Singh S, Gupta AK. Nitric oxide: role in tumour biology and iNOS/NO-based anticancer therapies. Cancer Chemother Pharmacol 2011; 67:1211-24; PMID:21544630; http://dx.doi.org/ 10.1007/s00280-011-1654-4 [DOI] [PubMed] [Google Scholar]
- 54.Hagemann T, Lawrence T, McNeish I, Charles K, Kulbe H, Thompson RG, Robinson SC, Balkwill FR. “Re-educating” tumor-associated macrophages by targeting NF-kappaB. J Exp Med 2008; 205:1261-8; PMID:18490490; http://dx.doi.org/ 10.1084/jem.20080108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Buhtoiarov IN, Sondel PM, Wigginton JM, Buhtoiarova TN, Yanke EM, Mahvi DA, Rakhmilevich AL. Anti-tumour synergy of cytotoxic chemotherapy and anti-CD40 plus CpG-ODN immunotherapy through repolarization of tumour-associated macrophages. Immunology 2011; 132:226-39; PMID:21039467; http://dx.doi.org/ 10.1111/j.1365-2567.2010.03357.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Garris C, Pittet MJ. Therapeutically reeducating macrophages to treat GBM. Nat Med 2013; 19:1207-8; PMID:24100977; http://dx.doi.org/ 10.1038/nm.3355 [DOI] [PubMed] [Google Scholar]
- 57.Huang Y, Yuan J, Righi E, Kamoun WS, Ancukiewicz M, Nezivar J, Santosuosso M, Martin JD, Martin MR, Vianello F et al.. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc Natl Acad Sci U S A 2012; 109:17561-6; PMID:23045683; http://dx.doi.org/ 10.1073/pnas.1215397109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Klug F, Prakash H, Huber PE, Seibel T, Bender N, Halama N, Pfirschke C, Voss RH, Timke C, Umansky L et al.. Low-dose irradiation programs macrophage differentiation to an iNOS+/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell 2013; 24:589-602; PMID:24209604; http://dx.doi.org/ 10.1016/j.ccr.2013.09.014 [DOI] [PubMed] [Google Scholar]
- 59.Stuhr LEB, Raa A, Oyan AM, Kalland KH, Sakariassen PO, Petersen K, Bjerkvig R, Reed RK. Hyperoxia retards growth and induces apoptosis, changes in vascular density and gene expression in transplanted gliomas in nude rats. J Neurooncol 2007; 85:191-202; PMID:17557137; http://dx.doi.org/ 10.1007/s11060-007-9407-2 [DOI] [PubMed] [Google Scholar]
- 60.Reiner NE, editor. Macrophages and Dendritic Cells: Methods and Protocols. New York, NY: Humana Press; 2009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.