

Abstract
Background and Scope
Psychiatric science remains descriptive, with a categorical nosology intended to enhance inter-observer reliability. Increased awareness of the mismatch between categorical classifications and the complexity of biological systems drives the search for novel frameworks including discovery science in Big Data. In this review, we provide an overview of incipient approaches, primarily focused on classically categorical diagnoses such as schizophrenia (SZ), autism spectrum disorder (ASD) and attention-deficit/hyperactivity disorder (ADHD), but also reference convincing, if focal, advances in cancer biology, to describe the challenges of Big Data and discovery science, and outline approaches being formulated to overcome existing obstacles.
Findings
A paradigm shift from categorical diagnoses to a domain/structure-based nosology and from linear causal chains to complex causal network models of brain-behavior relationship is ongoing. This (r)evolution involves appreciating the complexity, dimensionality and heterogeneity of neuropsychiatric data collected from multiple sources (“broad” data) along with data obtained at multiple levels of analysis, ranging from genes to molecules, cells, circuits and behaviors (“deep” data). Both of these types of Big Data landscapes require the use and development of robust and powerful informatics and statistical approaches. Thus, we describe Big Data analysis pipelines and the promise and potential limitations in using Big Data approaches to study psychiatric disorders.
Conclusion
We highlight key resources available for psychopathological studies and call for the application and development of Big Data approaches to dissect the causes and mechanisms of neuropsychiatric disorders and identify corresponding biomarkers for early diagnosis.
Keywords: Neuropsychiatric disorders, psychopathology, genetics, brain image, endophenotype, Big Data, classification, inference
Introduction
Neuropsychiatric disorders, such as schizophrenia (SZ), autism spectrum disorder (ASD), attention-deficit/hyperactivity disorder (ADHD), depression and substance abuse, are among the major sources of disability world-wide. To more effectively treat neuropsychiatric disorders and identify high-risk groups for targeted interventions, understanding the causes and underlying mechanisms, whether specific or shared across disorders, is essential. Classical genetic studies have established the high heritability of neuropsychiatric disorders; for example, 75–90% of the phenotypic variance related to ADHD is ascribable to additive genetic factors and their interactions (Levy et al., 1997, Faraone et al., 2005, Hawi et al., 2015), and corresponding values are ~80% for ASD (Bailey et al., 1995, O’Roak and State, 2008) and 60–85% for SZ (Lichtenstein et al., 2009, Escudero and Johnstone, 2014). This motivated the pursuit of identifying causal genes underlying neuropsychiatric disorders. In parallel, both structural and functional brain imaging data are increasingly being collected with the goal of understanding pathophysiological processes by linking brain functions and behaviors. The general paradigm organizing these efforts is illustrated in Figure 1.
Figure 1.
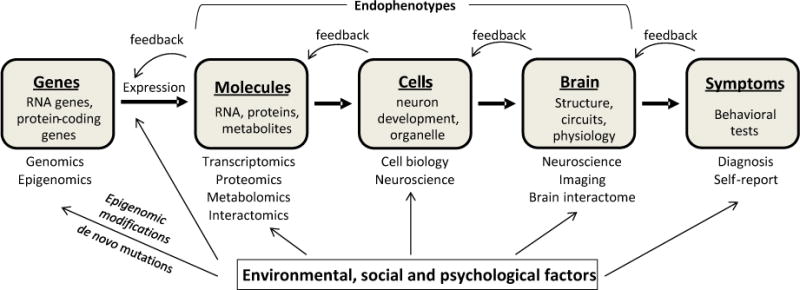
A simplified flow chart for psychiatric disorders: from genes to symptoms.
In this flow chart, results from one level (gray box) can exert feedback regulation at several levels upstream although only one level immediately upstream is shown for simplicity. Environmental impacts on each level are indicated. The studies for understanding each level and consequently the corresponding data types are listed below each level.
In this chart, variations in genome and epigenome are considered biological causes, which, by influencing gene expression at RNA and protein levels, subsequently impact molecular function and cellular metabolism. Resulting alterations in neuron structure and function are presumed to lead to the changes in neural circuits which are increasingly accessible to various imaging technologies, and which underlie distinct and often overlapping behaviors. Furthermore, developmental cues and diverse environmental factors including social and psychological influences impact these processes at multiple levels. These include de novo mutations or epigenetic modifications and regulation of gene expression or neural circuits. In this review, we briefly illustrate progress towards mechanistic understanding of neuropsychiatric disorders at multiple levels and discuss the promise and limitations of discovery science strategies applied to Big Data in meeting the challenge of identifying causal pathophysiological mechanisms.
Current status and challenges in psychopathology of mental disorders
Exponentially increasing studies in genetics and imaging in the past decade have revealed genetic variants and endophenotypes related to neuropsychiatric disorders. The extensive literature on genetics, endophenotypes and systems biology of neuropsychiatric syndromes has been elegantly reviewed (Alawieh et al., 2012, Miller and Rockstroh, 2013, Moreno-De-Luca et al., 2013, Pettersson et al., 2013, Escudero and Johnstone, 2014, Flint et al., 2014, Gaiteri et al., 2014, Glahn et al., 2014, Jeste and Geschwind, 2014, Munafo et al., 2014, Thompson et al., 2014, Tordjman et al., 2014, Hawi et al., 2015, Craddock et al., 2015, Kavanagh et al., 2015, Kiser et al., 2015). Here, our modest goal is to illustrate recent advances in studies of the pathophysiology of neuropsychiatric disorders from the perspective of Big Data approaches.
Transition from traditional dichotomous diagnoses to transdiagnostic and dimensional approaches
The development of a symptom-based nosology of mental disorders began more than a century ago and was encoded in successive editions of the Diagnostic and Statistical Manual of Mental Disorders (DSM) and the International Classification of Diseases (ICD). This necessary preliminary phase enhanced the reliability of psychiatric diagnoses, enabled familial and follow-up validation of syndromes and helped to guide existing treatments. However, the limitations of binary categorical (presence or absence) diagnoses are increasingly recognized (Simmons and Quinn, 2014). In response, the US National Institute of Mental Health (NIMH) implemented the Research Domain Criteria project (RDoC) (Cuthbert and Insel, 2013, Simmons and Quinn, 2014) with the aim of furthering the classification of mental disorders based on dimensional measurements grounded in neuroscience (http://www.nimh.nih.gov/research-priorities/rdoc/index.shtml). In the current pilot iteration of the RDoC research framework, core symptoms of mental disorders (termed “constructs”) are grouped into five major domains: negative valence system, positive valence system, cognitive systems, systems for social processes, and arousal and regulatory systems. Each domain consists of a set of related constructs, reflecting a brain system in which functions may be impaired in different psychiatric conditions (Casey et al., 2013). Different levels or units of analysis (e.g., genes, molecules, cells, circuits, physiology, behavior, and self-reports) can then be studied for each of the domains/structures. Thus, RDoC is organized as a dimensional and transdiagnostic matrix of domains/structures (rows) and units of analysis (columns) (Figure 1). However, RDoC is not intended as a replacement for DSM-like approaches, which will continue to be essential in clinical work, but as a complement to facilitate revealing the pathophysiological mechanisms underlying mental disorders.
Potential utility of endophenotypes in identifying causal genes and understanding etiological processes
Endophenotypes, also called intermediate phenotypes, were introduced in psychiatry 40 years ago by Gottesman who borrowed the term from insect genetics (Miller and Rockstroh, 2013 and references therein). In 2003, endophenotypes were redefined as “measureable components unseen by the unaided eye along the pathway between disease and distal genotype” which were presumed to be genetically less complex and closer to the levels of gene action (Gottesman and Gould, 2003). Although firm consensus on the definition of endophenotypes is lacking, in general endophenotypes should meet the following criteria: (1) association with illness, (2) heritability, (3) disease state-independence, (4) cosegregation with illness within families, (5) a higher rate in unaffected relatives than in the general population, and (6) being a reliably measured trait which exhibits disease specificity (Hasler, 2006, Chan and Gottesman, 2008, Miller and Rockstroh, 2013).
Advances in brain imaging, including structural and functional imaging, have led to the identification of several “neurophenotypes” with varying degrees of specificity and commonalities across disorders such as SZ, ASD, ADHD and intellectual disabilities (Castellanos and Tannock, 2002, Doyle et al., 2005, Viding and Blakemore, 2007, Khadka et al., 2013, Miller and Rockstroh, 2013, Port et al., 2014, Kiser et al., 2015). For example, ADHD, ASD and intellectual disabilities share similar deficits in brain functional connectivity linking regions implicated in higher order cognitive functions (e.g., the default mode network (DMN)), suggesting functional connectivity might be a plausible endophenotype (Kiser et al., 2015). Both SZ and bipolar disorder exhibited reduced resting-state functional connectivity in the DMN in medial prefrontal cortex, with abnormal recruitment in parietal cortex and frontopolar cortex/basal ganglia being unique to bipolar disorder and SZ, respectively (Ongur et al., 2010, Khadka et al., 2013, Meda et al., 2014). These findings are consistent with the observations of executive function impairments across several mental disorders including SZ, ADHD and bipolar disorder (Miller and Rockstroh, 2013, Kiser et al., 2015). Thus, endophenotype-based studies support transdiagnostic approaches.
The principal rationale for studying endophenotypes was the hope they would facilitate the identification of causal genes and the mechanistic understanding of pathophysiological processes (Miller and Rockstroh, 2013, Flint et al., 2014, Glahn et al., 2014). However, with a few exceptions (Wessa et al., 2010, Bigos et al., 2010, Jogia et al., 2011), endophenotypes have rarely been used in genetic psychopathological studies (Miller and Rockstroh, 2013). Instead, most genetic studies have continued to rely on categorical classifications. There are at least two open questions impeding the wider use of endophenotypes in genetic studies: (1) Given the potential large number of candidate endophenotypes, how can they be harnessed to increase statistical power? (2) Do the data support the hypothesis that endophenotypes will yield larger genetic effects?
In response to the first question, the endophenotype ranking value (ERV) index was developed to assess the genetic utility of endophenotypes (Glahn et al., 2012). The ERV is an index based on the heritability of the illness, the heritability of the endophenotype, and their genetic correlation (see Appendix 1: Glossary). It ranges between 0 and 1, with higher ERV values representing stronger shared genetic influence on the endophenotype and the illness. In their study, ERV was assessed for a high-dimensional set of over 11,000 endophenotypes (including 37 behavioral/neurocognitive measures, 85 neuroanatomic structures and 11,337 lymphocyte-expressed genes) in 1,122 Mexican American individuals from large randomly selected extended pedigrees (Glahn et al., 2012). The top ERV-ranked endophenotypes for recurrent major depression amongst the candidate measures were the Beck Depression Inventory, bilateral ventral diencephalon volume, and RNF123 transcript level, respectively. Bivariate linkage analysis of the top-ranked endophenotypes revealed a genome-wide significant quantitative trait locus on chromosome 4p15 exhibiting pleiotropic effects on both the endophenotype (RNF123 expression) and disease risk. RNF123 encodes a ring finger protein involved in regulation of neurite outgrowth. Thus RNF123 is a novel candidate likely implicated in hippocampal neurogenesis and potentially a drug target for major depression. The successful use of the ERV metric in a genetic association study is thus encouraging (Glahn et al., 2012). Under RDoC framework, numerous endophenotypes will be derived. Adopting ERV statistics or other quantification systems in the endophenotype hunting process should help reduce the statistical burden of controlling for multiple comparisons.
With regard to the heritability of candidate endophenotypes, initial aspirations of larger effects have not borne out. Recent large-scale imaging genetic studies (Bis et al., 2012, Ikram et al., 2012, Stein et al., 2012, Taal et al., 2012) have observed small effect sizes of genetic variants on neurophenotypes of interest. For example, a novel intergenic marker 50 kilobases downstream from the KTN1 gene (rs945270) accounts for 0.52% of the variance in putamen volume (Hibar et al., 2015), which is similar to effect sizes revealed in studies based on categorical disease classification. Furthermore, full genome-wide association results explained 7–15% of phenotypic variance of putamen volume after controlling for confounding effects (Hibar et al., 2015). These results demonstrate that at least some neurophenotypes do not show markedly higher heritabilities than binary diagnoses. Instead, it appears likely that they are as polygenically controlled as psychiatric disorders (Flint and Munafo, 2007, Flint et al., 2014). This, together with the high cost of collecting multiple neurophenotypes for large samples, raises doubts about the endophenotype strategy as a shortcut to genetic discovery. However, endophenotypes grounded in neuroscience can be crucial for interpreting genetic mapping results and revealing the underlying biological mechanisms (Flint et al., 2014). As we believe that understanding physiological mechanisms should be the proximate goal in psychiatric science, this provides ample motivation for continuing to pursue informative endophenotypes.
From single major genes to polygenic control: Emerging roles of common vs. rare variants
Twin and family-based studies have provided estimates of up to 90% heritability for some psychiatric disorders (Plomin et al., 1994, Pettersson et al., 2013). The publication of the human genome sequence in 2001 and the development of new genomic technologies made genotyping practically available and thus greatly accelerated the pace of identifying genetic factors associated with the causes of mental disorders. For example, many candidate genes and genomic loci such as single nucleotide polymorphisms (SNPs) and copy number variants (CNVs) have been reported to associate with ADHD, and 10 of them (including DRD5, SLC6A3 and LPHN3) have been replicated (Arcos-Burgos et al., 2010, Ribases et al., 2011, Lionel et al., 2011, Tong et al., 2015, Hawi et al., 2015). More than 200 genes have been reported to show linkage to or association with ASD or SZ (Tordjman et al., 2014, Kavanagh et al., 2015). Of particular note, a recent large-scale cohort study revealed 108 SZ-associated genetic loci, including 83 novel loci (Schizophrenia Working Group of the Psychiatric Genomics, 2014). Although the biological meaning of these genes or genomic loci remains to be elucidated, these advances represent a large step towards attaining a mechanistic understanding of the etiologies of mental disorders.
Another area with increasing attention is the role of epigenetic factors on mental disorders. Epigenetics studies the processes that modify gene expression without changes in the DNA sequence. These epigenetic modifications include DNA methylation, histone protein modification (such as acetylation, methylation and ubiquitination) and non-coding (small or large) RNA molecules. Importantly, they respond to developmental or environmental influences. The methylome (whole genome methylation features) has been profiled in various brain tissues during the mouse life cycle and also in postmortem human brain (Davies et al., 2012, Lister et al., 2013, Montano et al., 2013, Guo et al., 2014). A study of methylation in adult mouse dentate granule neurons in vivo supported the notion that neuronal activity can modify the methylome (Guo et al., 2011). Of particular note, methylation of numerous genomic loci has been reported in peripheral tissues such as blood or postmortem brain from patients with psychiatric disorders (Aberg et al., 2014, Numata et al., 2014, Xiao et al., 2014, Ruzicka et al., 2015). This is important, since blood and skin are obtainable from patients, whereas brain tissue is only available postmortem. The recognition of dynamic epigenetic modifications in the robust control of gene expression is the basis for the hypothesis that environmental contributions to psychiatric disorders are mediated by epigenetic factors (Tordjman et al., 2014, Guintivano and Kaminsky, 2014). Indeed, findings that environmental factors such as prenatal stress exposure can alter DNA methylation and that chromatin remodeling factors are among the genes that undergo de novo mutation in patients with ASD indicate the promising future of studying epigenetic mechanisms in understanding psychiatric pathophysiology (Kiser et al., 2015).
Common vs. rare variants
Accumulating evidence from numerous genetic/genomic studies has conclusively failed to support the original hypothesis that one or a few major genes are responsible for causing psychiatric disorders. This is in contrast to medical conditions such as cystic fibrosis where the major allele of the CFTR gene accounts for 66% of cases worldwide (Bobadilla et al., 2002). For the genes associated with psychiatric disorders, the emerging trends are small effect sizes and pleiotropy (see below). The failure to find common variants with large effects led to an alternative hypothesis (Gershon et al., 2011, Bassett et al., 2010, Escudero and Johnstone, 2014, Coghill, 2014). Common variants (those with allele frequency >5%), which include most SNPs, exist in the hundreds to thousands, as revealed by genome-wide association studies (GWAS), but individual GWAS variants exhibit subtle effects and thus account for only a small contribution to the overall risk. In contrast, rare GWAS variants (allele frequency <5%), which include most CNVs and some SNPs and can be introduced through de novo mutations, can have large effects. Several examples of common and rare variants have been identified. In SZ, large-scale genotyping studies have identified a common variant in the major histocompatibility complex (MHC) as the strongest genetic risk factor (International Schizophrenia et al., 2009). A SNP in the MHC region is strongly associated with SZ, and functional evidence from a mouse model supports its implication in glutamatergic synapses and in acquired immunity (Escudero and Johnstone, 2014). By contrast, many rare variants, such as the thousands of SNPs in the DISC1 (disrupted in schizophrenia 1) gene have a minor-allele frequency less than 1% (Thomson et al., 2014). Despite their rarity, DISC1 variants can have high penetrance in brain development and function, as has been demonstrated using a mouse model (Johnstone et al., 2011). How rare variants interact with common variants in the pathophysiology of mental disorders remains to be worked out, although rare variants have been proposed to act on conditions set by common variants (Coghill, 2015).
Genetic architecture and the need for large samples to find the “missing heritability”
As the complexity of genetic interactions and polygenic control is increasingly appreciated, the concept of genetic architecture has been incorporated into psychiatric research (Munafo and Flint, 2014). Genetic architecture refers to the genotype-phenotype map, i.e., the number of genes involved, how they interact with each other and how they map to various phenotypic levels (from gene expression to metabolism/cellular structure and behavior). However, the sum of all common variants identified so far appears to account for a relatively small proportion of the variance for any major psychiatric disorder (Munafo and Flint, 2014, Munafo et al., 2014), a problem frequently called the “missing heritability.” Thus, for all complex or quantitative traits, including psychiatric disorders, the major challenge remains the identification of most, if not all, the associated common and rare variants, which is likely to require extremely large samples. Detecting a genetic variant that explains 0.05% of the phenotypic variance at a significance threshold of 0.05 with 50% statistical power can be achieved with a sample size of 50,000 (Munafo and Flint, 2011), which is typical of most consortium-based studies. To achieve adequate power (e.g., 80%), a sample size of 100,000 is required. So far, only one psychiatric study has reached this level with more than 150,000 people (Schizophrenia Working Group of the Psychiatric Genomics, 2014). However, even though this study identified 108 SZ-associated genetic loci, these are far from sufficient to account for the genetic causes of SZ (Flint and Munafo, 2014), which shows a heritability of 60–85% (Lichtenstein et al., 2009, Escudero and Johnstone, 2014).
Pleiotropic effects
The other emerging trend is awareness of pleiotropic effects for many genes associated with psychiatric disorders. Pleiotropy occurs when one gene controls or influences multiple seemingly unrelated phenotypes, likely through acting on multiple metabolic or regulatory pathways. Several large-scale SNP, CNV and exome sequencing studies have shown that many genes or genomic loci are shared among multiple disorders. For example, the examination of ASD-associated SNPs in linkage to ADHD or vice-versa has revealed many shared SNPs, consistent with their high comorbidity (Rommelse et al., 2010). In another example involving 33,332 cases (with SZ, ASD, ADHD, bipolar disorder or major depression) and 27,888 controls (Cross-Disorder Group of the Psychiatric Genomics, 2013, Cross-Disorder Group of the Psychiatric Genomics et al., 2013), substantial overlap was found across the five disorders, supporting the estimate that approximately 17% of genes involved in human complex diseases or traits have pleiotropic effects (Sivakumaran et al., 2011). Specifically, voltage-gated calcium channel signaling emerged as a common mechanism across all five disorders (Cross-Disorder Group of the Psychiatric Genomics, 2013). Given the centrality of calcium signaling for synaptic function (Catterall and Few, 2008) and the implication of one particular variant of the L-type voltage-gated calcium channel, CACNA1C, the endophenotype approach may be optimal for dissecting the causal cascade from calcium channel/signaling genes to neural and brain endophenotypes and ultimately to symptoms (Thimm et al., 2011, Miller and Rockstroh, 2013, Kiser et al., 2015). Based on the proposed role of common and rare variants in disease effect sizes, it is conceivable that most common variants (such as genes related to calcium signaling or glutamatergic neurotransmission) will have some degrees of pleiotropic influences in disease comorbidity, with rare variants exhibiting more specific effects.
Potential of using sequencing technologies in disease allele identification
As the cost of sequencing has dropped, it has become feasible to sequence the entire genome or the exome (the entirety of protein coding segments) (Fromer et al., 2014, Purcell et al., 2014) which will expand our limited collection of SNP-only data in GWAS to genetic variants like nucleotide insertions or deletions. In addition, sequencing data directly reveals allelic differences in protein-coding genes or RNA-coding genes, in contrast to SNPs which in most of cases represent non-coding genomic loci. Thus sequencing-based genotyping methods are expected to greatly facilitate the identification of all common and most rare variants involved in disease. Second, profiling the emerging epigenome (such as the methylome and the histone code) in psychiatric patients (Kofink et al., 2013, Kiser et al., 2015) may not only help reveal the epigenetic contributions to psychiatric etiology but also provide a potential means to dissect the effects of gene by environment interactions (G × E). It is well recognized that G × E plays an important role in phenotypic expression, but the actual contribution of G × E in psychiatric diseases is difficult to measure due to multiple concerns including multiple statistical testing problems and statistical power limitations (Munafo et al., 2014). As discussed above, epigenetic modification has been regarded as a major mechanism to explain environmental impacts on gene expression. Now that hundreds of genes have been shown to be associated with psychiatric diseases, candidate gene-based epigenetic influences can be studied to directly address the contributions of G × E in those diseases. On the other hand, whether incorporating environmental factors into GWAS can increase the power of identifying the genetic causes of neuropsychiatric disorders is still debated (Munafo et al., 2014). Because of reduced genotyping cost and the relatively high cost of incorporating environmental factors in study design, there seems to be a valid argument that integrating G × E in GWAS will not effectively help identify the common variants associated with or responsible for psychiatric diseases (Munafo et al., 2014). However, it should also be noted that rapid advances in epigenome profiling may provide a promising cost-effective high throughput technology to assess genome-wide G × E effects and ultimately reveal epigenetic control mechanisms in neuropsychiatric disorders.
Shift from causal chains to causal network models of psychopathology
As reflected in Figure 1, the simple version of the general paradigm regarding causal-effect relationships of gene-brain-behaviors assumes a linear causal model: genetic/epigenetic alleles → molecular and cellular phenotypes (including gene expression, metabolism and neuron development and function) → neural circuit → behavior → symptoms. While this model has the advantage of simplicity, the consensus is that Mendelian genes with large effects do not exist for major psychiatric disorders (Miller and Rockstroh, 2013). Four lines of evidence support this conclusion. First, although common and rare variants may interact with each other in the emergence of psychopathology, environmental factors likely impact multiple levels of this chain model. For example, prenatal exposure to various stresses may lead to de novo mutations in genes associated with mental disorders or modify gene expression through non-coding RNA molecules (Serretti and Fabbri, 2013). The postnatal environment may also impact brain plasticity through epigenetic or other biological mechanisms. Therefore, multi-level regulation by environmental contributors indicates that a more complex network is almost certainly involved in etiologies of psychiatric disorders. Second, there is no direct evidence that each of the cascades in the chain model is the cause of the next cascade. It has even been proposed that genetic, environmental and all other factors such as psychological and other biological phenomena can intervene at all points or levels throughout etiology (Miller and Rockstroh, 2013, Cannon and Keller, 2006). Thus, a model relying on a single chain of causal events cannot explain this complex interrelationship. Third, the widespread occurrence of feedback regulation, i.e., the outcome of one level from an input can positively or negatively regulate the input itself or upstream events, leads to tight regulatory loops. One can envision that intermediate responses at molecular/cellular/circuit levels or brain disorder symptoms themselves might feedback-regulate gene expression involved in the control of pathophysiology. This kind of feedback regulation would also increase the complexity of the etiology of psychiatric disorders. Taken together, it has become increasingly accepted that psychiatric disorders likely involve complex regulatory networks instead of serial causal chain models.
Increasing efforts have been directed towards the construction/discovery of regulatory networks for psychiatric disorders. This involves predominantly systems or network analysis. Compared to the reductionist approach, which assumes that human behavior can be explained by breaking it down into smaller components or parts, the holistic approach emphasizes the whole rather than the constituent parts. In other words, the holistic approach is based on the notion that any given level of explanation for human behavior cannot be reduced to the one below. Therefore, the holistic view seeks to provide a systems level explanation for complex behavior. Studies have suggested that not only psychopathological factors (from cause to outcome) need to be viewed as systems but also the interrelationship between smaller components within a part or unit. For example, human brain connectomes or transcriptomes have been extensively studied using this framework. The holistic view derived from systems analysis can provide a systems level explanation for complex behavior by deciphering the normal functioning of the whole system in the brain and predicting systems’ responses to perturbations caused by drugs and interventions (Alawieh et al., 2012). Furthermore, studies have suggested that not only psychopathological factors (from cause to outcome) but also the interrelationship between smaller components within a part or unit need to be viewed as a system. There are numerous examples of systems analysis for each level, such as gene coexpression, protein-protein interaction networks and brain connectomes, each leading to new insights into mechanisms of psychiatric dysfunction (Alawieh et al., 2012, Gaiteri et al., 2014, Kitchen et al., 2014, Deco and Kringelbach, 2014). Interestingly, a recent study integrating transcriptomes to genomes in patients with SZ revealed that 50 genes undergoing damaging de novo mutations in prefrontal cortex during fetal development form a specific network (Gaiteri et al., 2014). Because those genes are known to function in neuronal migration, synaptic transmission and signaling, this integrated systems analysis suggests that by mapping neurodevelopmental processes in time and space in the brain, pathophysiological mechanisms of neuropsychiatric diseases can be revealed (e.g., disruptions of fetal prefrontal cortical neurogenesis as a psychopathological mechanism for SZ in this study). In another recent study integrating phenotypes and genotypes, a functional gene network involving 159 ASD-associated de novo single nucleotide variants and CNVs was constructed (Chang et al., 2015). This network contains four major network clusters including synapse function, ion channels, neuronal signaling and chromatin modification, thus providing a link from genotype to phenotype in ASD psychopathology.
In summary, numerous studies at various levels from genes to behaviors and to symptoms are leading to insights into the causes and underlying mechanisms of major psychiatric disorders. However, to construct disease stage- and patient-relevant pathophysiological networks that could best explain the etiology of those psychiatric disorders and provide the diagnosis and treatment strategies for researchers and clinicians, more data need to be generated and existing and new data need to be analyzed using an integrated approach (Medland et al., 2014).
Big Data approaches in neuropsychiatric studies
Why do we need Big Data approaches?
While research into neuropsychiatric diseases has traditionally consisted of mostly small-scale studies conducted in individual labs, the complexity of brain-behavior relationships requires large-scale consortium-based science (National Research Council, 2013). This entails a conceptual switch from tightly-designed hypothesis-driven studies to discovery-based, hypothesis-generating research (Van Horn and Gazzaniga, 2002). We note that although discovery-based approaches are initially explicitly hypothesis-free, if high-throughput hypothesis generation and prioritization are targeted, they can elevate hypothesis testing to a new level and powerfully complement traditional hypothesis-driven studies (Geschwind and Konopka, 2009).
Discovery-based approaches are frequently termed Big Data approaches, because they involve analyses of large-scale data sets. Big Data include large-scale homogeneously designed studies as well as “long-tailed” highly variant datasets (Ferguson et al., 2014). In neuropsychiatric studies, Big Data can be further divided into “broad” and “deep” data. “Broad” data are collected from multiple sources (labs/institutions/consortia) using different standards and thus are complex and heterogeneous when combined; “deep” data are collected at multiple levels ranging from genes to molecules and cells to circuits and ultimately behaviors and symptoms (Fig. 1). “Broad” data allow one to make population level inference, while “deep” data are needed for personalized medicine. We note that obtaining reasonable statistical power (e.g., 80%) to detect a common variant accounting for 0.05% of phenotypic variance will likely require sample sizes of 100,000 (Munafo and Flint, 2011). Clearly, if broad and deep data are further combined and stored in large-scale databases, it is pragmatically impossible to use traditional statistical approaches to extract and analyze these complex, heterogeneous and high-dimensional large-scale Big Data to reveal the most useful and robust insights into the causes and mechanisms of neuropsychiatric disorders. Thus, hypothesis-generating and discovery-based Big Data approaches offer an indispensable high-throughput analytical tool for data mining and pattern recognition. It is hoped that integrative analyses of data from many different levels will result in a new pathophysiology-based disease classification approach which will enable personalized interventions (Insel, 2014).
Currently available Big Data for studies of neuropsychiatric disorders
An ever-increasing number of openly shared genomic, neuroscience, brain imaging and psychiatric databases (Table 1) can be used for studying neuropsychiatric disorders. As the fine-grained nature of those datasets offers ample opportunities for scientists and clinicians to make inferences which might not be revealed without Big Data approaches, we briefly illustrate some of these databases in this section.
Table 1.
Currently available databases for studying neuropsychiatric disorders. The databases are categorized into three types: Psychiatric disorders-focused, human brain-oriented and human genetics-related. The availability of the data at various levels is indicated by the + sign. These levels include genetic (various genotyping and epigenetic profiles), molecular (transcriptome, proteome and metabolome), brain imaging, and behavior/symptoms.
| Databases | Genetic data | Molecular data | Brain Imaging | Behavior or symptom |
|---|---|---|---|---|
| Psychiatric disorder-oriented | ||||
| Psychiatric Genome Consortium (PGC) | + | + | ||
| The Enhancing NeuroImaging Genetics through Meta-Analysis (ENIGMA) Consortium | + | + | + | |
| International Schizophrenia Consortium (ISC) | + | + | ||
| Autism Consortium | + | |||
| The Autism Sequencing Consortium | + | + | ||
| National Database for Autism Research (NDAR) | + | + | + | |
| Simons Simplex Collection | + | + | ||
| Autism Genetics Resource Exchange | + | + | ||
| ADHD-200 Consortium | + | + | ||
| Autism Brain Imaging Data Exchange | + | + | ||
| Human brain-focused | ||||
| Scalable Brain Atlas | + | |||
| The Human Brain Atlas (MSU) | + | |||
| 1000 Functional Connectomes Project (FCP) | + | |||
| Nathan Kline Institute-Rockland Sample | + | + | + | |
| Human Connectome Project | + | + | + | |
| Allen Brain Atlas | + | |||
| Human Brain Transcriptome (HBT) | + | |||
| Human genetics-related | ||||
| Human Genome Project | + | |||
| Gene Ontology (GO) | + | |||
| Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathways | + | |||
| The ENCyclopedia Of DNA Elements (ENCODE) Project | + | |||
| Human Gene Expression (HuGe) Index | + | |||
| Human Interactome Project | + | |||
Psychiatric disorder-oriented imaging genetic databases
The most comprehensive genetic database for psychiatric disorders is the Psychiatric Genome Consortium (PGC; http://www.med.unc.edu/pgc). Since its launch in early 2007, the PGC has stored genetic data for more than 170,000 subjects and has become the largest psychiatric consortium in history. Within the umbrella of the consortium, individual working groups (such as the Schizophrenia Working Group) and Cross-Disorder Group perform mega-analyses of genetic association with various psychiatric disorders (Schizophrenia Working Group of the Psychiatric Genomics, 2014, Cross-Disorder Group of the Psychiatric Genomics, 2013, Cross-Disorder Group of the Psychiatric Genomics et al., 2013). In brain imaging, the Enhancing NeuroImaging Genetics through Meta-Analysis (ENIGMA) Consortium (http://enigma.ini.usc.edu/) collects data from institutions worldwide. So far, imaging data from more than 12,826 subjects have been analyzed. The first ENIGMA project led to the identification of common variants associated with hippocampal volume or intracranial volume (Thompson et al., 2014). This imaging genetics consortium has also established several working groups, covering GWAS, diseases (such as ASD, ADHD, SZ) and methods development. It also carries out joint work with consortia such as PGC and the International League Against Epilepsy. Databases specifically focused on disorders include the Autism Consortium (http://autismconsortium.org/), Autism Sequencing Consortium (https://www.autismspeaks.org/site-wide/autism-sequencing-consortium), National Database for Autism Research (NDAR; https://ndar.nih.gov/), Autism Genetics Resource Exchange (AGRE; http://agre.autismspeaks.org/), Simons Simplex Collection (http://sfari.org/resources/simons-simplex-collection), International Schizophrenia Consortium (ISC; http://pngu.mgh.harvard.edu/isc/), and Autism Brain Imaging Data Exchange (ABIDE; http://fcon_1000.projects.nitrc.org/indi/abide/), and ADHD-200 Consortium (http://fcon_1000.projects.nitrc.org/indi/adhd200/).
Neuroscience-related databases
To provide easily accessible brain atlases, two brain-focused neuroimaging databases have been developed: Scalable Brain Atlas (providing brain atlases, imaging data and topologies for human, rat and mouse; http://scalablebrainatlas.incf.org/main/index.php), and Human Brain Atlas (displaying a combination of MRI images and stained sections for cell bodies or for nerve fibers in human brains; https://www.msu.edu/~brains/brains/human/index.html). To enhance the pace of discovery science for human brain function, the 1000 Functional Connectomes Project (FCP) (http://fcon_1000.projects.nitrc.org/) was launched in 2009 to generate and collect resting state functional and structural magnetic resonance imaging (fMRI) data from more than 1,000 individuals. The Nathan Kline Institute-Rockland Sample (http://fcon_1000.projects.nitrc.org/indi/pro/nki.html) is an on-going project aiming to collect detailed phenotypic and brain imaging data from 1,000 individuals from the Rockland County, NY community across most of the lifespan. An attractive feature of this project is that DNA samples are also provided, making it possible to relate genotypes to brain imaging and other phenotypes. The Human Connectome Project (http://www.humanconnectome.org/) is a signature NIH effort to map the structural and functional connectomes of the human brain in up to 1200 young adults consisting of monozygotic or dizygotic twins and their siblings. Deidentified data are periodically made available to the scientific community either through download, Amazon Web Services (https://db.humanconnectome.org/) or by ordering physical hard drives (Connectome-in-a-box) at cost. A version of the Human Connectome Project focused on typical development is expected to be launched in the near future.
To help map human brain activity-related genes, two databases have been developed: Allen Brain Atlas (an interactive, genome-wide image database of gene expression in mouse and human brain; http://www.brain-map.org/), and Human Brain Transcriptome (HBT, providing transcriptome data and associated metadata for 16 regions of the developing and adult human brain; http://hbatlas.org/). Thus, linking candidate genes (identified from GWAS and other genotyping studies) to specific brain regions can now be done relatively easily, which will help reveal the regulatory mechanisms of genes implicated in psychiatric pathophysiology.
Human genome and interactome databases
Besides those psychiatric disorder-oriented and neuroscience-related databases, several human genetics or epigenetics related databases have been developed. These databases deal with whole genomes and transcriptomes and thus are useful for understanding the functions of psychiatric disorder-related genes and the networks involving those gene products. For example, the Human Genome Project (HGP) stores the sequence information for all human genes, Gene Ontology (GO) assigns functional categories for candidate genes such as biological process, molecular function and subcellular location, and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways provide a list of pathways (biochemical and/or signaling) for human genes. The Human Gene Expression (HuGe) Index (http://zlab.bu.edu/HugeIndex/index.htm) provides expression patterns for human genes in normal organs, tissues and cells. The ENCODE (ENCyclopedia Of DNA Elements) Project (http://www.genome.gov/encode/) aims to identify all regulatory elements in the human genome sequence. The Human Interactome Project (http://interactome.dfci.harvard.edu/H_sapiens/index.php) has stored high-quality binary protein-protein interactions validated by experimental evidence with a long-term goal of constructing a reference map of human protein-protein interactome networks.
In summary, the above diverse databases are providing resources at genetic, molecular, and brain imaging levels which will be useful for revealing the mechanisms of genetic contributions to psychopathology. Clearly, it is essential to integrate these complex datasets into psychiatric genetic and molecular studies using Big Data analysis pipelines as described below.
Big Data analysis pipelines
Although Big Data approaches are discovery-oriented and focused on generating novel testable hypotheses, their success depends on the insights these approaches can provide after integrated analyses of various types of data. This highlights the critical importance of integrating vast and complex genomic, brain imaging, and/or behavioral data to reveal gene-brain-behavior linkages underlying psychiatric dysfunction (McIntyre et al., 2014), and presents exciting challenges for the development of data integration and mining tools (Hussong et al., 2013). In this section, we focus on Big Data analysis pipelines with a special emphasis on large-scale genomic and/or neuroimaging data. Figure 2 provides a general flow chart of Big Data approaches. We address each step of analysis pipelines with a focus on how statistical thinking can help tackle some challenging problems in Big Data analyses.
Figure 2.
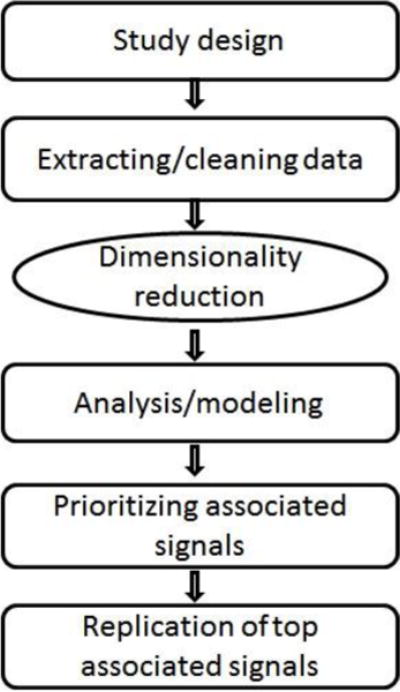
Big Data analysis pipeline.
Study design
Big Data approaches require careful planning of data selection, quality control, statistical modeling approaches, and replication/validation strategies. The biggest challenge in large-scale data analysis is to ask good questions and formulate an intelligent experimental design for the available data (Engert, 2014). This requires a collaborative effort from an interdisciplinary team of researchers with specialized knowledge in domain science, statistics, and computer science.
Extracting/cleaning data
This is an essential data processing step that involves putting raw data into an appropriate format for statistical modeling. One of the main challenges in large-scale data analysis is to remove systematic biases due to experimental variations. For example, in genomic data preprocessing, technical factors such as batch effects and dye effects should be removed. Other factors such as possible contamination of DNA samples should also be investigated (Hunt et al., 2013). In neuroimaging data preprocessing, this involves removing biases introduced in imaging data acquisition and controlling for experimental variations if the images are aggregated from multiple studies. Although the preprocessing step is extremely time consuming, it is highly recommended that statisticians, who perform statistical modeling of the data, participate in data preprocessing or at least know intimately how the data were preprocessed. Downstream analysis results are highly influenced by decisions made in upstream preprocessing steps. We also note that traditional methods for checking potential data errors, mislabeling, and detecting outlier are well developed and suited for small-scale data settings (Dasu and Johnson, 2013), but extension of those methods to massive data settings is still under development.
Dimensionality reduction
High dimensionality in Big Data entails phenomena such as spurious correlations and noise accumulation (Fan et al., 2014). Spurious correlation refers to the tendency for high magnitude correlations to occur frequently among uncorrelated random variables in high-dimension data. Noise accumulation occurs when there exist many weak signals that do not contribute to the reduction of modeling errors. The level of accumulated noise is directly proportional to the number of features in a model. These issues increase the statistical complexity of the problem. If not handled correctly, they may lead to false positives and wrong statistical inferences. Dimensionality reduction is typically applied in Big Data settings in which one suspects that all measured variables can be efficiently represented by a smaller number of features (Cunningham and Yu, 2014). Dimensionality reduction methods reduce statistical complexity of the data by discovering/extracting features of interest and discarding some aspect of data as noise. We refer the interested reader to a recent state-of-the-art report on dimensionality reduction techniques with detailed examples in computational neuroscience (Cunningham and Yu, 2014). Here, we briefly mention a few methods that could be useful for psychiatric research.
Dimensionality reduction methods can in general be classified into two types: supervised and unsupervised. In many experimental settings, some variables are labeled as dependent/outcome variables. Supervised approaches aim to select subsets of predictors that preserve differences in the outcome measures as much as possible. Penalized regression methods such as LASSO (least absolute shrinkage and selection operator; (Tibshirani, 1996)) and elastic-net (Zou and Hastie, 2005) are effective supervised dimensionality reduction techniques for handling noise-accumulation issues. In penalized regression methods we assume the model parameters are sparse (i.e., only a few predictors are important to the outcome measure) and selection of the optimal set of important predictors is done by minimizing the goodness of fit of the model (e.g., sum of squares of residuals) plus the sum of a sparsity-inducing penalty on the regression coefficients. As a note, penalized regression methods differ in the penalty functions imposed on regression models. Although the change to the penalty function is subtle, it can have a dramatic impact on the resulting estimator. For example, LASSO employs an L1 penalty (i.e., penalizing the absolute values of the coefficients) to the model, while elastic-net utilizes a combination of L1 and L2 (i.e., penalizing the squares of the coefficients) penalties. The L1 penalty in LASSO introduces shrinkage towards zero thus retaining the most important variables in the model. However, LASSO has some limitations. If the number of predictors is much larger than the sample size n, LASSO can only select up to n predictors. In addition, in genetic studies, genes sharing the same biological ‘pathway’ might form a group effect (i.e., a group of highly correlated genes that are equally important to the outcome measure). In this case, LASSO might not be an ideal method for detecting the grouping effect. In contrast, elastic net, by adding an L2 penalty to the LASSO, removes the limitation on the number of selected variables and encourages grouping effects. LASSO and elastic-net have been widely used in cancer research, but less in psychiatric research. A PubMed search on May 31, 2015 yielded 262 publications using keywords “LASSO” AND “cancer” and 90 publications using “elastic net and cancer”. However, the combination of “LASSO” or “elastic net” with “schizophrenia”, “autism”, or “ADHD” returned less than 10 publications in total. In our opinion, applying LASSO or elastic-net to psychiatric research should be productive in the near future.
Unsupervised dimensionality reduction methods utilize all data aiming to faithfully preserve the statistical properties of the data. Well-known examples include principal component analysis (PCA), independent component analysis (ICA) (Hyvarinen and Oja, 2000), and non-negative matrix factorization (NMF) (Lee and Seung, 1999). Briefly, PCA minimizes covariance of the original data and identifies an ordered set of uncorrelated linear projections (i.e., principal components) that capture the largest portions of variance in the data. ICA linearly transforms the original data into a set of unordered components by maximizing the non-Gaussianity (i.e., normalized kurtosis) so that each component is maximally statistically independent. ICA is frequently used in functional MRI data analysis to extract features that are interpreted as representing neuronal networks or as artifactual signals. NMF is a technique for finding parts-based, linear projections of non-negative data where the loadings of each component are constrained to be positive. These three methods all involve matrix factorization. However, the resulting components from each method have different representational properties due to their different optimization constraints. PCA and ICA extract holistic views of the original data, but NMF decomposes the data into parts-based representations. As illustrated by Lee and Seung (Lee and Seung, 1999), if we were to extract the main features (i.e., basis images) from a set of facial images, PCA generates the components (basis images) accounting for the largest proportions of variance but which may lack intuitive meanings, e.g., low spatial frequency facial features. By contrast, NMF does not provide a global view of the face. Instead, the components are the facial parts themselves, i.e., mouths, noses, etc. All of these methods are being widely applied in psychiatric research. For instance, integrative analysis of MRI, fMRI and phenotypic data via NMF led to the discovery of differential changes in default mode subnetworks in ADHD (Anderson et al., 2014). Parallel ICA was used to explore the genetic underpinnings of white matter abnormalities in SZ (Gupta et al., 2015). Using PCA on polymorphism data, a new ancestral origin was identified for autism cases with a deleterious G34S mutation in the CTNND2 gene (Turner et al., 2015).
In summary, supervised approaches differ from unsupervised approaches in that the former reduce dimensionality while preserving information about outcome measures. Unsupervised approaches, in contrast, find low-dimension representations (e.g., linear or nonlinear combinations of the original data) that capture significant statistical properties (e.g., variance) of all data. Although dimensionality reduction methods all aim to find a lower dimensional representation of high dimensional data, each method differs in the statistical properties it preserves and discards (Cunningham and Yu, 2014). Therefore it is crucial to understand the type of properties which need to be preserved via dimension reduction (Committee on the Analysis of Massive Data et al., 2013) and selection of dimension reduction methods should be project-oriented.
Data analysis/modeling
Statistical models/machine learning methods provide a convenient framework for acquiring knowledge from data. Compared to building models for the analysis of small-scale datasets using traditional statistical procedures, different considerations are needed for building effective models in the Big Data setting. For example, data in massive settings are prone to contamination from multiple sources (Committee on the Analysis of Massive Data et al., 2013). Robust regression models should be developed/used to protect against outliers. The validity of most traditional statistical procedures relies heavily on the assumption that residual noise is uncorrelated with all the predictors in the model (Fan et al., 2014). However, high-throughput technologies allow people to collect as many features as possible. This increases the possibility of incidental correlations between residual noise and predictors (known as incidental endogeneity), leading to model selection inconsistency. Whether incidental correlations exist in the data should be checked. If they do exist, the Focused Generalized Methods of Moments (Fan and Liao, 2014) can be used to improve model selection consistency. Interested readers should refer to the paper for technical details.
Traditional research strategies rely heavily on linear/nonlinear regression models to uncover relationships among variables. Network approaches and clustering analyses have increasingly been recognized as offering promising alternatives for assessing complex systems (Morris and Cuthbert, 2012). Network approaches model biological systems as complex networks of nodes connected through edges. Here nodes represent different units of analysis (e.g., genes, neurophenotypes, behavioral measurements) and edges stand for any conceivable relationship (e.g., correlation, odds ratio, neuron activity) between them. Network modeling conceptualizes disorders as systems of symptoms that interact mutually in a complex network (Borsboom and Cramer, 2013). Analyses using the network approach led to the finding of two SZ genetic networks and an intriguing connection between SZ and ASD, providing insights into the molecular causes of psychiatric disorders (Gilman et al., 2012). The principal goal of clustering analyses is to divide data into clusters based on their similarity, with the hope that the clusters will capture the natural structure of the data. Clustering analysis has been widely used for subtype detection in cancer research. For example, clustering analysis of gene expression patterns discovered tumor subclasses in breast cancer, resulting in better disease prognosis (Sorlie et al., 2001). To cross evidence from multiple genomic data types, an integrative clustering analysis approach was developed for cancer subtype detection (Shen et al., 2009). In psychiatric research, these approaches have become popular for detecting subgroups within patients with ADHD (Clarke et al., 2001), ASD (Hrdlicka et al., 2005), or SZ (Gilbert et al., 2014). As a cautionary note, many clustering techniques including K-means and hierarchical clustering have been developed, but there is little consensus on the definition of a cluster (Rodriguez and Laio, 2014). Different clustering analysis techniques likely result in very different clusters for the same data, and thus the validity of clustering analysis results should be carefully examined. Although clustering analysis remains largely an exploratory data analysis tool, it helps visualize underlying data structure, characterize the study population, and facilitate the generation of novel hypotheses.
Prioritizing finding and replication
Big Data approaches can revolutionize our ability to discover and generate interesting, testable large-scale ideas (Lee et al., 2014). Analysis of psychiatric Big Data will likely lead to many statistically significant findings, but it will be critically important to prioritize these findings and test whether they are relevant to psychiatric pathophysiology (Committee on the Analysis of Massive Data et al., 2013).
An example of a Big Data approach for analyzing the genetic architecture of schizophrenia
We acknowledge that the ambitious goal of integrating “broad” and “deep” neuropsychiatric data (i.e., analyzing data collected from multiple sources and covering all five levels – genetic, molecular, cellular, brain imaging and symptomatic) has yet to be carried out to date. However, initial attempts have begun by examining the two extremes of the continuum, genes and symptoms. In a recent example, Arnedo et al. analyzed the genetic architecture of SZ using Molecular Genetics of Schizophrenia (MGS) consortium data (Arnedo et al., 2015). As schematized in Figure 3, the authors used hypothesis-free, data-driven methods to uncover complex genotype-phenotype relationships. Their analyses incorporated various machine-learning techniques including p-value based dimensionality reduction, NMF feature extraction and bi-clustering analysis. They found interactions of sets of SNP and eight classes (subtypes) of SZ by data-driven clustering. Analyses of the interacting SNP sets in relation to the eight SZ subtypes explained at least 70% of the disease risk in their initial sample, which exceeded the average effects of the SNPs in isolation (24%). Importantly, they reported replication of more than 81% of the genotypic-phenotypic relationships in two additional independent samples. We note that skepticism regarding this potential landmark paper was expressed in PubMed Commons shortly after its publication, along with the authors’ detailed responses. We eagerly anticipate further extensions and replications of this effort, as that would signal the potential for Big Data approaches to find the “missing heritability” for many psychiatric diseases and to reveal clues to pathogenesis.
Figure 3.
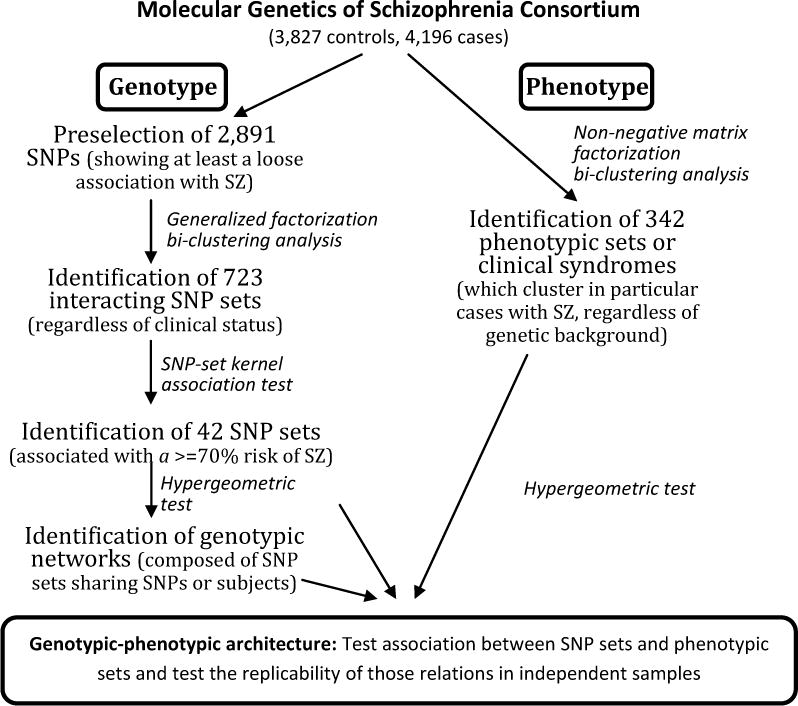
An example of using Big Data approaches to uncover the genotypic-phenotypic architecture of the schizophrenias (SZ) (adapted from Arnedo et al., 2015).
Identification of 8 subtypes of SZ, based on grouping of relationships between single nucleotide polymorphism (SNP) networks and phenotypic sets) 1.
The promise of Big Data
As discussed above, data-driven and discovery-based Big Data approaches have the potential to inform our understanding of the mechanisms underlying psychiatric disease. It would be prohibitively costly to design a large-scale experiment to collect information in every structure and domain proposed by the RDoC matrix. Instead, analyzing available data spanning different levels/units using Big Data approaches can dramatically reduce cost and time requirements. Such a strategy can generate testable specific hypotheses – the principal challenge confronting investigators. In this section, we briefly discuss the potential applications of Big Data approaches in four critical aspects of psychiatric studies.
Disease classification
One of the outstanding questions remains the identification of disease subtypes within psychiatric disorders such as ASD, ADHD and SZ (Clarke et al., 2001, Hrdlicka et al., 2005, Gilbert et al., 2014, Veatch et al., 2014). As in the case of cancer, molecular subtype identification will facilitate genetic, molecular and imaging studies, as revealed by a cross-platform reclassification of 12 cancer types (based on tissue-of-origin) as 11 cancer types based on molecular taxonomy (Hoadley et al., 2014). In breast cancer, molecular subtyping has led to personalized oncological treatment plans for some patients (Barnard et al., 2015). We anticipate that progress in psychiatry will also not apply uniformly across broad diagnostic categories. For example, although variants in L-type voltage-gated calcium channels were implicated as a common mechanism across all five disorders (Cross-Disorder Group of the Psychiatric Genomics, 2013), calcium channel blockers have not been beneficial across psychiatric syndromes (Casamassima et al., 2010). We hold out the possibility that some may be found to be beneficial for some subtypes of psychiatric disorders which can be characterized through genetic, physiological or pharmacological studies. Therefore, one potential application of Big Data approaches is to systematically model behaviors from subgroups representing “rare events,” which can be detected in large samples instead of being treated as outliers in small-scale data (Fan et al., 2014).
Biomarker discovery
Identifying panels of robust biomarkers that can be used to predict psychiatric dysfunctions and assess the effect of treatments remains a high priority. However, initial efforts to identify biomarkers by analyzing brain imaging datasets or genotype/ transcriptome datasets have not met with great success. In part, this may be ascribable to prior efforts having been organized around traditional diagnoses. Neuropsychiatric disorders are frequently transdiagnostic with distinct subtypes likely among all disorders. Therefore, an urgent question remains: Can integrative analysis of datasets from various levels and multiple sources provide a panel of biomarkers for each of the subtypes, domains or structures? We anticipate that Big Data approaches involving clustering and pattern recognition will provide a means to address this crucial question.
Detection of common and rare genetic variants
The search for the genetic causes responsible for various neuropsychiatric disorders has primarily relied on the use of binary diagnoses. For example, the largest-scale genetic study in any psychiatric disorders to date is on SZ, which led to the identification of 108 loci, many of which had been missed by small-scale studies (Schizophrenia Working Group of the Psychiatric Genomics, 2014). In the first ENIGMA project (ENIGMA1), the intergenic common variant rs7294919 was found to significantly associate with hippocampus volume (Stein et al., 2012), which could not be revealed by any of the individual datasets, illustrating the power of vastly increased sample size. The ENIGMA2 project reported the identification (from genetic and imaging data of 30,717 individuals) of five novel genomic variants influencing the volumes of putamen and caudate nucleus (Hibar et al., 2015). One of these variants is a novel intergenic locus rs945270 which showed evidence of altering KTN1 expression in both brain and blood. Although quantifiable endophenotypes have not yet been used to link genetic variants to psychiatric disorders, it has been argued that integrating endophenotypes into genetic studies will be more powerful for identifying genetic factors associated with psychiatric dysfunction (Glahn et al., 2012, Glahn et al., 2014, Miller and Rockstroh, 2013). However, the data complexity and dimensionality demand better analytical approaches. As most past studies have used univariate or mass-univariate statistical tests, which made it challenging to detect weak effects across multiple variables, multivariate approaches have been proposed to identify aggregate effects, which may still yield robust results with smaller sample sizes. On the other hand, rare variants are hard to detect in Big Data settings using traditional statistical analyses, because of their rarity. However, since rare variants are typically highly penetrant, developing Big Data approaches to detect low frequency events in massive datasets is an active area of research (Morris and Zeggini, 2010, Lee et al., 2014).
Systems view of pathophysiology
Providing a mechanistic view of how genetic contributors and environmental factors intertwine to cause/influence brain dysfunction is not only important for understanding the normal functioning of the brain, but also critical for diagnosing disorders and finding cost-effective and efficient treatments. Genes do not act in isolation, and thus systems or network approaches have allowed the construction of several genetic regulatory networks involved in various psychiatric disorders such as SZ and ASD and in the connections between disorders (Gilman et al., 2012, Alawieh et al., 2012, Gaiteri et al., 2014, Chang et al., 2015). However, to integrate highly complex, heterogeneous and dimensional data (e.g., from (epi)genome to transcriptome, proteome, metabolome, neural networks and brain structure/function) and construct a systems view of pathophysiology in psychiatric disorders is the biggest challenge we face and can only be done in Big Data settings.
Limitations of Big Data approaches
Data sharing
Although data sharing is the norm in molecular genetics and genomics, it is just beginning in neuroscience and psychiatry. To encourage further data sharing, authors have called for appropriately crediting data collectors (Milham, 2012, Poline et al., 2012, Poldrack and Gorgolewski, 2014). With regard to ethical concerns of releasing data, patient or participant authorization to release all data can be achieved as long as ethical policies are strictly followed and re-identification of subjects with or without psychiatric diseases is both prohibited and made difficult.
Data integration and missing data
Once data sharing issues are resolved, the major challenge remains how to integrate data collected from multiple sites. Early unfunded efforts at data sharing have been characterized by poor documentation and variation in experimental approaches across sites which cause problems for data aggregation and utilization (Milham, 2012, Poline et al., 2012). To overcome this limitation, standard ontologies or experimental protocols (such as brain imaging) have been proposed or are currently in place to integrate data from multiple sources. For example, the National Database for Autism Research (NDAR), established in 2008 by NIH and now secured in the Amazon cloud, has developed a data sharing platform. This platform, which has been adopted by the RDoC and National Database of Clinical Trials (NDCT) initiatives, currently hosts a wide range of clinical and behavioral measures and genomic and brain imaging data from 77,000 de-identified human subjects meeting criteria for ASD (Poline et al., 2012). Another issue is missing data. As shown in Table 1, it is obvious that many imaging databases do not have genotyping or molecular (such as gene expression) data and most genetic data sources do not have imaging or behavioral data. Simply put, so far there is not a single database that contains data spanning all four levels (Table 1) or all eight units described in RDoC. We hope and anticipate that collecting data systematically by following the recommended RDoC matrix will eventually bridge this huge data gap.
Possibility of complementing the inaccessibility of the brain
As brain tissues are almost always inaccessible, human postmortem brain tissues have been used for collecting genetic (DNA) and molecular (RNA, protein, metabolites) data and have provided key insights into brain function and psychopathology. While genotypes can be preserved in those tissues, the DNA methylation, gene expression and metabolite profiles can be overwhelmed by artifacts. To overcome this limitation, methods such as profiling blood and in particular brain cerebrospinal fluid have been proposed to complement postmortem brain tissues. Some studies find that some molecular signatures such as DNA methylation or gene expression are similarly regulated in blood or brain cerebrospinal fluid as in postmortem tissues or the brain in case of animal model systems. For example, the marker rs945270, which was previously shown to strongly associate with KTN1 expression in blood (Westra et al., 2013), was recently revealed in a large-scale GWAS study to also strongly associate with KTN1 expression in putamen and explain 0.52% of the variance of putamen volume (Hibar et al., 2015). Clearly, more large-scale studies using Big Data approaches are needed to determine the utility of peripheral biomarkers.
Integrative analysis
Datasets for each level (such as genotyping, expression and imaging) are complex and a big challenge in typical meta-analysis; however, linking different levels and various domains/structures from various data sources for integrative analysis is a much bigger challenge (Craddock et al., 2015). It is possible that the current methods discussed in the previous sections may not be powerful or robust enough to identify the genetic factors and the mechanisms underlying psychiatric disorders. Therefore, investigators in neuroscience and psychiatry, mathematicians and statisticians, and computer science experts must work together to modify existing approaches or develop new approaches to analyze Big Data in psychiatry.
Validation of robustness of results
It is anticipated that discovery-based Big Data analysis will lead to new findings that cannot be revealed from analyses of small-scale data. The major concern will be whether findings are robust and validation of results will become the biggest challenge. While results can be statistically validated using independent datasets, ultimate validation depends on tests of functionality. Therefore, collaborative efforts, including investigation using animal model systems such as rodents and monkeys, will be critical.
Conclusion
Understanding how genetic and environmental interactions impact brain structure and function leading to a continuum of psychiatric function and dysfunction is now the central question in neuropsychiatry. Advances in genomics and imaging have brought an enormous explosion in (epi)genomic, transcriptomic and proteomic data and a dramatic increase in the quantities (and more modest improvements in quality) of brain structural and functional imaging data (Lichtman et al., 2014). Many examples of success have been reported using meta-analyses. However, rapidly accumulated, psychiatry-related data share several key features: large-scale, high heterogeneity and high dimensionality. Basically, these data are generated from numerous studies with dynamic measurements at various levels (from genes to molecules/cells and neural circuits and to behavior and symptoms) and thus can be considered deep data, or are derived from different platforms and collected in different labs/institutions/consortia using different standards and thus also represent broad data. Such data are increasingly being made available in major databases for use by psychiatric investigators or data scientists, seeking to use Big Data approaches for integrated analyses. However, we cannot underestimate the enormity of the challenge, and the striking lack of useful biological markers in psychiatry to date. In response, we concur that we “need to cultivate a new generation of computationally trained researchers who are aware of the richness of data and can draw on knowledge from many laboratories, courageous enough to make judicious simplifications and to have their ideas tested, and imaginative enough to generate interesting, testable large-scale ideas” (Sejnowski et al., 2014).
Key Points.
A transition from traditional symptom-based categorical diagnoses to a domain/structure-based nosology should lead to pathophysiological understanding of neuropsychiatric disorders.
A paradigm shift from linear causal chains to complex causal network models of brain-behavior relationship is emerging.
Increasing complexity, dimensionality and heterogeneity of large-scale high-throughput neuropsychiatric data collected from multiple sources (“broad” data) at multiple levels of analysis (“deep” data) demands powerful Big Data approaches for data mining.
Big Data pipelines from study design, data cleaning, and dimensionality reduction to data analysis and modeling are presented.
We discuss the promise and limitations of Big Data approaches that aim to increase the power of mining neuropsychiatric data.
Acknowledgments
Funding from NIAAA R21-AA023800 (to Y.Z.), R01MH094639, U01MH099059 and UL1TR000038 (F.X.C.) is gratefully acknowledged. Y.Z. is partly supported by a neuroscience fellowship from the Leon Levy Foundation. This review was invited by the Editors of this journal (who offered a small honorarium to the second author to cover expenses) and has been subjected to full external review.
Appendix 1: Glossary
Genetics/epigenetics/imaging-related terms
Common variant: a genetic variant frequently present in all human populations (note that the frequency boundaries used in the literature vary, but in this review it refers to having a minor allele frequency of >5%). Each variant at each gene influencing a complex disease usually has low penetrance and may have a small additive or multiplicative effect on the disease phenotype.
Copy number variant (CNV): the DNA alteration resulting in the cell having a variation in the number of copies of one or more DNA sections.
Endophenotype ranking value (ERV): an index developed for assessing the genetic utility of endophenotypes by quantifying the similarity between the endophenotype and the disease of interest (between 0 and 1). It is defined as . Here and represent heritability of the disease (i) and endophenotype (e), respectively, and ρg is their genetic correlation.
Exome sequencing: a technique for sequencing all the protein-coding genes in a genome (known as the exome) of an organism or individual.
Epigenetics: the study of the processes that ensure the inheritance of variation (“-genetic”) above and beyond (“epi”) changes in the DNA sequence.
Genome-wide association studies (GWAS): examination of genetic variants (typically single nucleotide polymorphism or SNP) through the whole genome in different individuals (usually groups of controls vs. cases) to see if any variant is associated with a trait or disease. Does not include determining whether the identified variants are causal.
Methylome: the DNA methylation profiles in the whole genome of an organism or individual.
Pleiotropy: a situation when one gene controls two or more seemingly unrelated phenotypes.
Rare variant: a genetic variant which occurs at low frequency in a population (typically with a minor allele frequency of <5%) but which may be responsible for a portion of the missing heritability of complex diseases in a population specific manner. Detectable rare variants associated with complex phenotypes have higher penetrance and larger effect sizes than common variants.
Single nucleotide polymorphism (SNP): a common DNA sequence variant (with a minor allele frequency ≥ 1% within the population) in which a base (i.e., A, T, C or G) differs among individuals.
Discovery science/Big Data-related terms
Big Data: refers to highly complex, heterogeneous and high-dimensional large-scale datasets. Big Data approaches are hypothesis-generating and discovery-oriented, with a goal of revealing the hidden patterns or information behind complex data via integrating computer science, statistical learning and psychiatry/neuroscience.
“Broad” data: data created by linking massive amounts of data collected from multiple sources (labs/institutions/consortia) using different standards. Typically, the linked data are highly heterogeneous but can add value for understanding the pathophysiology of psychiatric disorders.
Causal chain: an ordered sequence of events in which any one event in the chain causes the next one, leading to the final event.
Causal network: a directed acyclic graph (or network) which consists of a set of variables (also called nodes) and a set of directed links (also called edges) between variables. In a causal network, each node is the direct causal effect of its parental nodes.
Clustering analysis: aims to group objects into subsets/clusters such that objects within each cluster are more similar (i.e., more closely related to each other than objects assigned to other clusters. Many metrics (e.g., Euclidean distance, probabilistic distance, mutual information) can be used to measure the degree of similarity. Although clustering analysis techniques can be useful for patient subgroup detection, it should be noted that the choice of similarity measure is of critical importance in obtaining meaningful clustering results and depends largely on subject matter considerations.
Data mining: the process of exploring/analyzing data from different perspectives so that useful information (e.g., consistent patterns in data, systematic relationship between variables of interest) can be discovered. This is sometimes called data/knowledge discovery.
“Deep” data: deeply phenotyped data (i.e., detailed biological and clinical data collected for each individual). “Deep” data are critically important to personalized medicine, as they entail multiple levels including genetic alteration, epigenetic modification, clinical symptoms, and environmental factors.
Dimensionality reduction: the process of reducing the number of random variables under consideration. Sometimes called feature selection or feature extraction.
Independent component analysis (ICA): a variant of PCA that assumes the components are statistically independent and follow non-gaussian distribution. ICA is used in functional MRI data analysis to extract features that are linearly mixed.
Non-negative matrix factorization (NMF): aims to find a low-dimensional approximation to the original non-negative data. Unlike PCA and ICA which generate holistic representation of the data, NMF extracts part-based, localized features from the data.
Principal component analysis (PCA): the primary interest of PCA is to find a set of linear combinations (i.e., principal components) of the original data such that the components explain as much variation in the data as possible.
Supervised dimensionality reduction methods: seeks to find a low dimensional representation of the original data via regression or classification models. The choice of the low-dimensional space is influenced by a target variable called the response variable.
Systems analysis: a holistic approach that analyzes the interactions between the components (e.g., genes, proteins, metabolites) of complex biological systems, and how these interactions within the network affect the function and behavior of that system.
Unsupervised dimensionality reduction methods: seeks to preserve significant statistical properties of the data in a low-dimensional space. By contrast with supervised methods, no response variable is available for helping select the low-dimensional space. Well-known examples include principal component analysis and its many variants.
Footnotes
Conflict of interest statements: No conflicts declared.
The authors have declared that they have no competing or potential conflicts of interest.
References
- Aberg KA, Mcclay JL, Nerella S, Clark S, Kumar G, Chen W, Van Den Oord EJ. Methylome-wide association study of schizophrenia: identifying blood biomarker signatures of environmental insults. JAMA Psychiatry. 2014;71:255–264. doi: 10.1001/jamapsychiatry.2013.3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alawieh A, Zaraket FA, Li JL, Mondello S, Nokkari A, Razafsha M, Kobeissy FH. Systems biology, bioinformatics, and biomarkers in neuropsychiatry. Front Neurosci. 2012;6:187. doi: 10.3389/fnins.2012.00187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson A, Douglas PK, Kerr WT, Haynes VS, Yuille AL, Xie J, Cohen MS. Non-negative matrix factorization of multimodal MRI, fMRI and phenotypic data reveals differential changes in default mode subnetworks in ADHD. Neuroimage. 2014;102Pt 1:207–219. doi: 10.1016/j.neuroimage.2013.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arcos-Burgos M, Jain M, Acosta MT, Shively S, Stanescu H, Wallis D, Muenke M. A common variant of the latrophilin 3 gene, LPHN3, confers susceptibility to ADHD and predicts effectiveness of stimulant medication. Mol Psychiatry. 2010;15:1053–1066. doi: 10.1038/mp.2010.6. [DOI] [PubMed] [Google Scholar]
- Arnedo J, Svrakic DM, Del Val C, Romero-Zaliz R, Hernandez-Cuervo H, Molecular Genetics of Schizophrenia, C. Zwir I. Uncovering the hidden risk architecture of the schizophrenias: confirmation in three independent genome-wide association studies. Am J Psychiatry. 2015;172:139–153. doi: 10.1176/appi.ajp.2014.14040435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey A, Le Couteur A, Gottesman I, Bolton P, Simonoff E, Yuzda E, Rutter M. Autism as a strongly genetic disorder: evidence from a British twin study. Psychol Med. 1995;25:63–77. doi: 10.1017/s0033291700028099. [DOI] [PubMed] [Google Scholar]
- Barnard ME, Boeke CE, Tamimi RM. Established breast cancer risk factors and risk of intrinsic tumor subtypes. Biochim Biophys Acta. 2015;1856:73–85. doi: 10.1016/j.bbcan.2015.06.002. [DOI] [PubMed] [Google Scholar]
- Bassett AS, Scherer SW, Brzustowicz LM. Copy number variations in schizophrenia: critical review and new perspectives on concepts of genetics and disease. Am J Psychiatry. 2010;167:899–914. doi: 10.1176/appi.ajp.2009.09071016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigos KL, Mattay VS, Callicott JH, Straub RE, Vakkalanka R, Kolachana B, Weinberger DR. Genetic variation in CACNA1C affects brain circuitries related to mental illness. Arch Gen Psychiatry. 2010;67:939–945. doi: 10.1001/archgenpsychiatry.2010.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bis JC, Decarli C, Smith AV, Van Der Lijn F, Crivello F, Fornage M, Aging Research in Genomic Epidemiology, C Common variants at 12q14 and 12q24 are associated with hippocampal volume. Nat Genet. 2012;44:545–551. doi: 10.1038/ng.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobadilla JL, Macek M, Jr, Fine JP, Farrell PM. Cystic fibrosis: a worldwide analysis of CFTR mutations–correlation with incidence data and application to screening. Hum Mutat. 2002;19:575–606. doi: 10.1002/humu.10041. [DOI] [PubMed] [Google Scholar]
- Borsboom D, Cramer AO. Network analysis: an integrative approach to the structure of psychopathology. Annu Rev Clin Psychol. 2013;9:91–121. doi: 10.1146/annurev-clinpsy-050212-185608. [DOI] [PubMed] [Google Scholar]
- Cannon TD, Keller MC. Endophenotypes in the genetic analyses of mental disorders. Annu Rev Clin Psychol. 2006;2:267–290. doi: 10.1146/annurev.clinpsy.2.022305.095232. [DOI] [PubMed] [Google Scholar]
- Casamassima F, Hay AC, Benedetti A, Lattanzi L, Cassano GB, Perlis RH. L-type calcium channels and psychiatric disorders: A brief review. Am J Med Genet B Neuropsychiatr Genet. 2010;153B:1373–1390. doi: 10.1002/ajmg.b.31122. [DOI] [PubMed] [Google Scholar]
- Casey BJ, Craddock N, Cuthbert BN, Hyman SE, Lee FS, Ressler KJ. DSM-5 and RDoC: progress in psychiatry research? Nat Rev Neurosci. 2013;14:810–814. doi: 10.1038/nrn3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellanos FX, Tannock R. Neuroscience of attention-deficit/hyperactivity disorder: the search for endophenotypes. Nat Rev Neurosci. 2002;3:617–628. doi: 10.1038/nrn896. [DOI] [PubMed] [Google Scholar]
- Catterall WA, Few AP. Calcium channel regulation and presynaptic plasticity. Neuron. 2008;59:882–901. doi: 10.1016/j.neuron.2008.09.005. [DOI] [PubMed] [Google Scholar]
- Chan RC, Gottesman II. Neurological soft signs as candidate endophenotypes for schizophrenia: a shooting star or a Northern star? Neurosci Biobehav Rev. 2008;32:957–971. doi: 10.1016/j.neubiorev.2008.01.005. [DOI] [PubMed] [Google Scholar]
- Chang J, Gilman SR, Chiang AH, Sanders SJ, Vitkup D. Genotype to phenotype relationships in autism spectrum disorders. Nat Neurosci. 2015;18:191–198. doi: 10.1038/nn.3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke AR, Barry RJ, Mccarthy R, Selikowitz M. EEG-defined subtypes of children with attention-deficit/hyperactivity disorder. Clin Neurophysiol. 2001;112:2098–2105. doi: 10.1016/s1388-2457(01)00668-x. [DOI] [PubMed] [Google Scholar]
- Coghill D. Editorial: Acknowledging complexity and heterogeneity in causality–implications of recent insights into neuropsychology of childhood disorders for clinical practice. J Child Psychol Psychiatry. 2014;55:737–740. doi: 10.1111/jcpp.12284. [DOI] [PubMed] [Google Scholar]
- Coghill D. Commentary: We’ve only just begun: unravelling the underlying genetics of neurodevelopmental disorders–a commentary on Kiser et al. (2015) J Child Psychol Psychiatry. 2015;56:296–298. doi: 10.1111/jcpp.12399. [DOI] [PubMed] [Google Scholar]
- Committee on the Analysis of Massive Data, Committee on Applied and Theoretical Statistics, Board on Mathematical Sciences and Their Applications, Division on Engineering and Physical Sciences & National Research Council. Frontiers in Massive Data Analysis. Washington, D.C.: National Academies Press; 2013. [Google Scholar]
- Craddock RC, Tungaraza RL, Milham MP. Connectomics and new approaches for analyzing human brain functional connectivity. Gigascience. 2015;4:13. doi: 10.1186/s13742-015-0045-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross-Disorder Group of the Psychiatric Genomics, C. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet. 2013;381:1371–1379. doi: 10.1016/S0140-6736(12)62129-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross-Disorder Group of the Psychiatric Genomics, C. Lee SH, Ripke S, Neale BM, Faraone SV, Purcell SM, Wray NR. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat Genet. 2013;45:984–994. doi: 10.1038/ng.2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham JP, Yu BM. Dimensionality reduction for large-scale neural recordings. Nat Neurosci. 2014;17:1500–1509. doi: 10.1038/nn.3776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuthbert BN, Insel TR. Toward the future of psychiatric diagnosis: the seven pillars of RDoC. BMC Med. 2013;11:126. doi: 10.1186/1741-7015-11-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasu T, Johnson T. Data Quality, in Exploratory Data Mining and Data Cleaning. Hoboken, NJ, USA: John Wiley & Sons, Inc; 2013. [Google Scholar]
- Davies MN, Volta M, Pidsley R, Lunnon K, Dixit A, Lovestone S, Mill J. Functional annotation of the human brain methylome identifies tissue-specific epigenetic variation across brain and blood. Genome Biol. 2012;13:R43. doi: 10.1186/gb-2012-13-6-r43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deco G, Kringelbach ML. Great expectations: using whole-brain computational connectomics for understanding neuropsychiatric disorders. Neuron. 2014;84:892–905. doi: 10.1016/j.neuron.2014.08.034. [DOI] [PubMed] [Google Scholar]
- Doyle AE, Willcutt EG, Seidman LJ, Biederman J, Chouinard VA, Silva J, Faraone SV. Attention-deficit/hyperactivity disorder endophenotypes. Biol Psychiatry. 2005;57:1324–1335. doi: 10.1016/j.biopsych.2005.03.015. [DOI] [PubMed] [Google Scholar]
- Engert F. The big data problem: turning maps into knowledge. Neuron. 2014;83:1246–1248. doi: 10.1016/j.neuron.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escudero I, Johnstone M. Genetics of schizophrenia. Curr Psychiatry Rep. 2014;16:502. doi: 10.1007/s11920-014-0502-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Han F, Liu H. Challenges of Big Data Analysis. Natl Sci Rev. 2014;1:293–314. doi: 10.1093/nsr/nwt032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Liao Y. Endogeneity in High Dimensions. Ann Stat. 2014;42:872–917. doi: 10.1214/13-AOS1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faraone SV, Perlis RH, Doyle AE, Smoller JW, Goralnick JJ, Holmgren MA, Sklar P. Molecular genetics of attention-deficit/hyperactivity disorder. Biol Psychiatry. 2005;57:1313–1323. doi: 10.1016/j.biopsych.2004.11.024. [DOI] [PubMed] [Google Scholar]
- Ferguson AR, Nielson JL, Cragin MH, Bandrowski AE, Martone ME. Big data from small data: data-sharing in the ‘long tail’ of neuroscience. Nat Neurosci. 2014;17:1442–1447. doi: 10.1038/nn.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint J, Munafo M. Schizophrenia: genesis of a complex disease. Nature. 2014;511:412–413. doi: 10.1038/nature13645. [DOI] [PubMed] [Google Scholar]
- Flint J, Munafo MR. The endophenotype concept in psychiatric genetics. Psychol Med. 2007;37:163–180. doi: 10.1017/S0033291706008750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint J, Timpson N, Munafo M. Assessing the utility of intermediate phenotypes for genetic mapping of psychiatric disease. Trends Neurosci. 2014;37:733–741. doi: 10.1016/j.tins.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromer M, Pocklington AJ, Kavanagh DH, Williams HJ, Dwyer S, Gormley P, O’donovan MC. De novo mutations in schizophrenia implicate synaptic networks. Nature. 2014;506:179–184. doi: 10.1038/nature12929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaiteri C, Ding Y, French B, Tseng GC, Sibille E. Beyond modules and hubs: the potential of gene coexpression networks for investigating molecular mechanisms of complex brain disorders. Genes Brain Behav. 2014;13:13–24. doi: 10.1111/gbb.12106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gershon ES, Alliey-Rodriguez N, Liu C. After GWAS: searching for genetic risk for schizophrenia and bipolar disorder. Am J Psychiatry. 2011;168:253–256. doi: 10.1176/appi.ajp.2010.10091340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geschwind DH, Konopka G. Neuroscience in the era of functional genomics and systems biology. Nature. 2009;461:908–915. doi: 10.1038/nature08537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert E, Merette C, Jomphe V, Emond C, Rouleau N, Bouchard RH, Maziade M. Cluster analysis of cognitive deficits may mark heterogeneity in schizophrenia in terms of outcome and response to treatment. Eur Arch Psychiatry Clin Neurosci. 2014;264:333–343. doi: 10.1007/s00406-013-0463-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilman SR, Chang J, Xu B, Bawa TS, Gogos JA, Karayiorgou M, Vitkup D. Diverse types of genetic variation converge on functional gene networks involved in schizophrenia. Nat Neurosci. 2012;15:1723–1728. doi: 10.1038/nn.3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glahn DC, Curran JE, Winkler AM, Carless MA, Kent JW, Jr, Charlesworth JC, Blangero J. High dimensional endophenotype ranking in the search for major depression risk genes. Biol Psychiatry. 2012;71:6–14. doi: 10.1016/j.biopsych.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glahn DC, Knowles EE, Mckay DR, Sprooten E, Raventos H, Blangero J, Almasy L. Arguments for the sake of endophenotypes: examining common misconceptions about the use of endophenotypes in psychiatric genetics. Am J Med Genet B Neuropsychiatr Genet. 2014;165B:122–130. doi: 10.1002/ajmg.b.32221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesman Ii, Gould TD. The endophenotype concept in psychiatry: etymology and strategic intentions. Am J Psychiatry. 2003;160:636–645. doi: 10.1176/appi.ajp.160.4.636. [DOI] [PubMed] [Google Scholar]
- Guintivano J, Kaminsky ZA. Role of epigenetic factors in the development of mental illness throughout life. Neurosci Res. 2014 doi: 10.1016/j.neures.2014.08.003. [DOI] [PubMed] [Google Scholar]
- Guo JU, Ma DK, Mo H, Ball MP, Jang MH, Bonaguidi MA, Song H. Neuronal activity modifies the DNA methylation landscape in the adult brain. Nat Neurosci. 2011;14:1345–1351. doi: 10.1038/nn.2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JU, Su Y, Shin JH, Shin J, Li H, Xie B, Song H. Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat Neurosci. 2014;17:215–222. doi: 10.1038/nn.3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta CN, Chen J, Liu J, Damaraju E, Wright C, Perrone-Bizzozero NI, Calhoun VD. Genetic markers of white matter integrity in schizophrenia revealed by parallel ICA. Front Hum Neurosci. 2015;9:100. doi: 10.3389/fnhum.2015.00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasler G. Evaluating endophenotypes for psychiatric disorders. Rev Bras Psiquiatr. 2006;28:91–92. doi: 10.1590/s1516-44462006000200003. [DOI] [PubMed] [Google Scholar]
- Hawi Z, Cummins TD, Tong J, Johnson B, Lau R, Samarrai W, Bellgrove MA. The molecular genetic architecture of attention deficit hyperactivity disorder. Mol Psychiatry. 2015;20:289–297. doi: 10.1038/mp.2014.183. [DOI] [PubMed] [Google Scholar]
- Hibar DP, Stein JL, Renteria ME, Arias-Vasquez A, Desrivieres S, Jahanshad N, Medland SE. Common genetic variants influence human subcortical brain structures. Nature. 2015;520:224–229. doi: 10.1038/nature14101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoadley KA, Yau C, Wolf DM, Cherniack AD, Tamborero D, Ng S, Stuart JM. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell. 2014;158:929–944. doi: 10.1016/j.cell.2014.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrdlicka M, Dudova I, Beranova I, Lisy J, Belsan T, Neuwirth J, Urbanek T. Subtypes of autism by cluster analysis based on structural MRI data. Eur Child Adolesc Psychiatry. 2005;14:138–144. doi: 10.1007/s00787-005-0453-z. [DOI] [PubMed] [Google Scholar]
- Hunt KA, Mistry V, Bockett NA, Ahmad T, Ban M, Barker JN, Van Heel DA. Negligible impact of rare autoimmune-locus coding-region variants on missing heritability. Nature. 2013;498:232–235. doi: 10.1038/nature12170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussong AM, Curran PJ, Bauer DJ. Integrative data analysis in clinical psychology research. Annu Rev Clin Psychol. 2013;9:61–89. doi: 10.1146/annurev-clinpsy-050212-185522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyvarinen A, Oja E. Independent component analysis: algorithms and applications. Neural Netw. 2000;13:411–430. doi: 10.1016/s0893-6080(00)00026-5. [DOI] [PubMed] [Google Scholar]
- Ikram MA, Fornage M, Smith AV, Seshadri S, Schmidt R, Debette S, Aging Research in Genomic Epidemiology, C Common variants at 6q22 and 17q21 are associated with intracranial volume. Nat Genet. 2012;44:539–544. doi: 10.1038/ng.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insel TR. The NIMH Research Domain Criteria (RDoC) Project: precision medicine for psychiatry. Am J Psychiatry. 2014;171:395–397. doi: 10.1176/appi.ajp.2014.14020138. [DOI] [PubMed] [Google Scholar]
- International Schizophrenia, C. Purcell SM, Wray NR, Stone JL, Visscher PM, O’donovan MC, Sklar P. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeste SS, Geschwind DH. Disentangling the heterogeneity of autism spectrum disorder through genetic findings. Nat Rev Neurol. 2014;10:74–81. doi: 10.1038/nrneurol.2013.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jogia J, Ruberto G, Lelli-Chiesa G, Vassos E, Maieru M, Tatarelli R, Frangou S. The impact of the CACNA1C gene polymorphism on frontolimbic function in bipolar disorder. Mol Psychiatry. 2011;16:1070–1071. doi: 10.1038/mp.2011.49. [DOI] [PubMed] [Google Scholar]
- Johnstone M, Thomson PA, Hall J, Mcintosh AM, Lawrie SM, Porteous DJ. DISC1 in schizophrenia: genetic mouse models and human genomic imaging. Schizophr Bull. 2011;37:14–20. doi: 10.1093/schbul/sbq135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavanagh DH, Tansey KE, O’donovan MC, Owen MJ. Schizophrenia genetics: emerging themes for a complex disorder. Mol Psychiatry. 2015;20:72–76. doi: 10.1038/mp.2014.148. [DOI] [PubMed] [Google Scholar]
- Khadka S, Meda SA, Stevens MC, Glahn DC, Calhoun VD, Sweeney JA, Pearlson GD. Is aberrant functional connectivity a psychosis endophenotype? A resting state functional magnetic resonance imaging study. Biol Psychiatry. 2013;74:458–466. doi: 10.1016/j.biopsych.2013.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiser DP, Rivero O, Lesch KP. Annual research review: The (epi)genetics of neurodevelopmental disorders in the era of whole-genome sequencing–unveiling the dark matter. J Child Psychol Psychiatry. 2015;56:278–295. doi: 10.1111/jcpp.12392. [DOI] [PubMed] [Google Scholar]
- Kitchen RR, Rozowsky JS, Gerstein MB, Nairn AC. Decoding neuroproteomics: integrating the genome, translatome and functional anatomy. Nat Neurosci. 2014;17:1491–1499. doi: 10.1038/nn.3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofink D, Boks MP, Timmers HT, Kas MJ. Epigenetic dynamics in psychiatric disorders: environmental programming of neurodevelopmental processes. Neurosci Biobehav Rev. 2013;37:831–845. doi: 10.1016/j.neubiorev.2013.03.020. [DOI] [PubMed] [Google Scholar]
- Lee DD, Seung HS. Learning the parts of objects by non-negative matrix factorization. Nature. 1999;401:788–791. doi: 10.1038/44565. [DOI] [PubMed] [Google Scholar]
- Lee S, Abecasis GR, Boehnke M, Lin X. Rare-variant association analysis: study designs and statistical tests. Am J Hum Genet. 2014;95:5–23. doi: 10.1016/j.ajhg.2014.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy F, Hay DA, Mcstephen M, Wood C, Waldman I. Attention-deficit hyperactivity disorder: a category or a continuum? Genetic analysis of a large-scale twin study. J Am Acad Child Adolesc Psychiatry. 1997;36:737–744. doi: 10.1097/00004583-199706000-00009. [DOI] [PubMed] [Google Scholar]
- Lichtenstein P, Yip BH, Bjork C, Pawitan Y, Cannon TD, Sullivan PF, Hultman CM. Common genetic determinants of schizophrenia and bipolar disorder in Swedish families: a population-based study. Lancet. 2009;373:234–239. doi: 10.1016/S0140-6736(09)60072-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtman JW, Pfister H, Shavit N. The big data challenges of connectomics. Nat Neurosci. 2014;17:1448–1454. doi: 10.1038/nn.3837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lionel AC, Crosbie J, Barbosa N, Goodale T, Thiruvahindrapuram B, Rickaby J, Scherer SW. Rare copy number variation discovery and cross-disorder comparisons identify risk genes for ADHD. Sci Transl Med. 2011;3:95ra75. doi: 10.1126/scitranslmed.3002464. [DOI] [PubMed] [Google Scholar]
- Lister R, Mukamel EA, Nery JR, Urich M, Puddifoot CA, Johnson ND, Ecker JR. Global epigenomic reconfiguration during mammalian brain development. Science. 2013;341:1237905. doi: 10.1126/science.1237905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mcintyre RS, Cha DS, Jerrell JM, Swardfager W, Kim RD, Costa LG, Alsuwaidan M. Advancing biomarker research: utilizing ‘Big Data’ approaches for the characterization and prevention of bipolar disorder. Bipolar Disord. 2014;16:531–547. doi: 10.1111/bdi.12162. [DOI] [PubMed] [Google Scholar]
- Meda SA, Ruano G, Windemuth A, O’neil K, Berwise C, Dunn SM, Pearlson GD. Multivariate analysis reveals genetic associations of the resting default mode network in psychotic bipolar disorder and schizophrenia. Proc Natl Acad Sci U S A. 2014;111:E2066–2075. doi: 10.1073/pnas.1313093111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medland SE, Jahanshad N, Neale BM, Thompson PM. Whole-genome analyses of whole-brain data: working within an expanded search space. Nat Neurosci. 2014;17:791–800. doi: 10.1038/nn.3718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milham MP. Open neuroscience solutions for the connectome-wide association era. Neuron. 2012;73:214–218. doi: 10.1016/j.neuron.2011.11.004. [DOI] [PubMed] [Google Scholar]
- Miller GA, Rockstroh B. Endophenotypes in psychopathology research: where do we stand? Annu Rev Clin Psychol. 2013;9:177–213. doi: 10.1146/annurev-clinpsy-050212-185540. [DOI] [PubMed] [Google Scholar]
- Montano CM, Irizarry RA, Kaufmann WE, Talbot K, Gur RE, Feinberg AP, Taub MA. Measuring cell-type specific differential methylation in human brain tissue. Genome Biol. 2013;14:R94. doi: 10.1186/gb-2013-14-8-r94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-De-Luca A, Myers SM, Challman TD, Moreno-De-Luca D, Evans DW, Ledbetter DH. Developmental brain dysfunction: revival and expansion of old concepts based on new genetic evidence. Lancet Neurol. 2013;12:406–414. doi: 10.1016/S1474-4422(13)70011-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris AP, Zeggini E. An evaluation of statistical approaches to rare variant analysis in genetic association studies. Genet Epidemiol. 2010;34:188–193. doi: 10.1002/gepi.20450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris SE, Cuthbert BN. Research Domain Criteria: cognitive systems, neural circuits, and dimensions of behavior. Dialogues Clin Neurosci. 2012;14:29–37. doi: 10.31887/DCNS.2012.14.1/smorris. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munafo MR, Flint J. Dissecting the genetic architecture of human personality. Trends Cogn Sci. 2011;15:395–400. doi: 10.1016/j.tics.2011.07.007. [DOI] [PubMed] [Google Scholar]
- Munafo MR, Flint J. The genetic architecture of psychophysiological phenotypes. Psychophysiology. 2014;51:1331–1332. doi: 10.1111/psyp.12355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munafo MR, Zammit S, Flint J. Practitioner review: A critical perspective on gene-environment interaction models–what impact should they have on clinical perceptions and practice? J Child Psychol Psychiatry. 2014;55:1092–1101. doi: 10.1111/jcpp.12261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Research Council. Frontiers in Massive Data Analysis. Washington, DC: The National Academies Press; 2013. [Google Scholar]
- Numata S, Ye T, Herman M, Lipska BK. DNA methylation changes in the postmortem dorsolateral prefrontal cortex of patients with schizophrenia. Front Genet. 2014;5:280. doi: 10.3389/fgene.2014.00280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’roak BJ, State MW. Autism genetics: strategies, challenges, and opportunities. Autism Res. 2008;1:4–17. doi: 10.1002/aur.3. [DOI] [PubMed] [Google Scholar]
- Ongur D, Lundy M, Greenhouse I, Shinn AK, Menon V, Cohen BM, Renshaw PF. Default mode network abnormalities in bipolar disorder and schizophrenia. Psychiatry Res. 2010;183:59–68. doi: 10.1016/j.pscychresns.2010.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersson E, Anckarsater H, Gillberg C, Lichtenstein P. Different neurodevelopmental symptoms have a common genetic etiology. J Child Psychol Psychiatry. 2013;54:1356–1365. doi: 10.1111/jcpp.12113. [DOI] [PubMed] [Google Scholar]
- Plomin R, Owen MJ, Mcguffin P. The genetic basis of complex human behaviors. Science. 1994;264:1733–1739. doi: 10.1126/science.8209254. [DOI] [PubMed] [Google Scholar]
- Poldrack RA, Gorgolewski KJ. Making big data open: data sharing in neuroimaging. Nat Neurosci. 2014;17:1510–1517. doi: 10.1038/nn.3818. [DOI] [PubMed] [Google Scholar]
- Poline JB, Breeze JL, Ghosh S, Gorgolewski K, Halchenko YO, Hanke M, Kennedy DN. Data sharing in neuroimaging research. Front Neuroinform. 2012;6:9. doi: 10.3389/fninf.2012.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Port RG, Gandal MJ, Roberts TP, Siegel SJ, Carlson GC. Convergence of circuit dysfunction in ASD: a common bridge between diverse genetic and environmental risk factors and common clinical electrophysiology. Front Cell Neurosci. 2014;8:414. doi: 10.3389/fncel.2014.00414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell SM, Moran JL, Fromer M, Ruderfer D, Solovieff N, Roussos P, Sklar P. A polygenic burden of rare disruptive mutations in schizophrenia. Nature. 2014;506:185–190. doi: 10.1038/nature12975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribases M, Ramos-Quiroga JA, Sanchez-Mora C, Bosch R, Richarte V, Palomar G, Casas M. Contribution of LPHN3 to the genetic susceptibility to ADHD in adulthood: a replication study. Genes Brain Behav. 2011;10:149–157. doi: 10.1111/j.1601-183X.2010.00649.x. [DOI] [PubMed] [Google Scholar]
- Rodriguez A, Laio A. Machine learning. Clustering by fast search and find of density peaks. Science. 2014;344:1492–1496. doi: 10.1126/science.1242072. [DOI] [PubMed] [Google Scholar]
- Rommelse NN, Franke B, Geurts HM, Hartman CA, Buitelaar JK. Shared heritability of attention-deficit/hyperactivity disorder and autism spectrum disorder. Eur Child Adolesc Psychiatry. 2010;19:281–295. doi: 10.1007/s00787-010-0092-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzicka WB, Subburaju S, Benes FM. Circuit- and Diagnosis-Specific DNA Methylation Changes at gamma-Aminobutyric Acid-Related Genes in Postmortem Human Hippocampus in Schizophrenia and Bipolar Disorder. JAMA Psychiatry. 2015 doi: 10.1001/jamapsychiatry.2015.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schizophrenia Working Group of the Psychiatric Genomics, C. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–427. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sejnowski TJ, Churchland PS, Movshon JA. Putting big data to good use in neuroscience. Nat Neurosci. 2014;17:1440–1441. doi: 10.1038/nn.3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serretti A, Fabbri C. Shared genetics among major psychiatric disorders. Lancet. 2013;381:1339–1341. doi: 10.1016/S0140-6736(13)60223-8. [DOI] [PubMed] [Google Scholar]
- Shen R, Olshen AB, Ladanyi M. Integrative clustering of multiple genomic data types using a joint latent variable model with application to breast and lung cancer subtype analysis. Bioinformatics. 2009;25:2906–2912. doi: 10.1093/bioinformatics/btp543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons JM, Quinn KJ. The NIMH Research Domain Criteria (RDoC) Project: implications for genetics research. Mamm Genome. 2014;25:23–31. doi: 10.1007/s00335-013-9476-9. [DOI] [PubMed] [Google Scholar]
- Sivakumaran S, Agakov F, Theodoratou E, Prendergast JG, Zgaga L, Manolio T, Campbell H. Abundant pleiotropy in human complex diseases and traits. Am J Hum Genet. 2011;89:607–618. doi: 10.1016/j.ajhg.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Borresen-Dale AL. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98:10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein JL, Medland SE, Vasquez AA, Hibar DP, Senstad RE, Winkler AM, Enhancing Neuro Imaging Genetics through Meta-Analysis, C Identification of common variants associated with human hippocampal and intracranial volumes. Nat Genet. 2012;44:552–561. doi: 10.1038/ng.2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taal HR, St Pourcain B, Thiering E, Das S, Mook-Kanamori DO, Warrington NM, Early Growth Genetics, C Common variants at 12q15 and 12q24 are associated with infant head circumference. Nat Genet. 2012;44:532–538. doi: 10.1038/ng.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thimm M, Kircher T, Kellermann T, Markov V, Krach S, Jansen A, Krug A. Effects of a CACNA1C genotype on attention networks in healthy individuals. Psychol Med. 2011;41:1551–1561. doi: 10.1017/S0033291710002217. [DOI] [PubMed] [Google Scholar]
- Thompson PM, Stein JL, Medland SE, Hibar DP, Vasquez AA, Renteria ME, Alzheimer’s Disease Neuroimaging Initiative, E. C. I. C. S. Y. S. G The ENIGMA Consortium: large-scale collaborative analyses of neuroimaging and genetic data. Brain Imaging Behav. 2014;8:153–182. doi: 10.1007/s11682-013-9269-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson PA, Parla JS, Mcrae AF, Kramer M, Ramakrishnan K, Yao J, Porteous DJ. 708 Common and 2010 rare DISC1 locus variants identified in 1542 subjects: analysis for association with psychiatric disorder and cognitive traits. Mol Psychiatry. 2014;19:668–675. doi: 10.1038/mp.2013.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibshirani R. Regression shrinkage and selection via the lasso. J Royal Stat Soc B. 1996;58:267–288. [Google Scholar]
- Tong JH, Cummins TD, Johnson BP, Mckinley LA, Pickering HE, Fanning P, Bellgrove MA. An association between a dopamine transporter gene (SLC6A3) haplotype and ADHD symptom measures in nonclinical adults. Am J Med Genet B Neuropsychiatr Genet. 2015;168:89–96. doi: 10.1002/ajmg.b.32283. [DOI] [PubMed] [Google Scholar]
- Tordjman S, Somogyi E, Coulon N, Kermarrec S, Cohen D, Bronsard G, Xavier J. Gene x Environment interactions in autism spectrum disorders: role of epigenetic mechanisms. Front Psychiatry. 2014;5:53. doi: 10.3389/fpsyt.2014.00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner TN, Sharma K, Oh EC, Liu YP, Collins RL, Sosa MX, Chakravarti A. Loss of delta-catenin function in severe autism. Nature. 2015;520:51–56. doi: 10.1038/nature14186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Horn JD, Gazzaniga MS. Opinion: Databasing fMRI studies towards a ‘discovery science’ of brain function. Nat Rev Neurosci. 2002;3:314–318. doi: 10.1038/nrn788. [DOI] [PubMed] [Google Scholar]
- Veatch OJ, Veenstra-Vanderweele J, Potter M, Pericak-Vance MA, Haines JL. Genetically meaningful phenotypic subgroups in autism spectrum disorders. Genes Brain Behav. 2014;13:276–285. doi: 10.1111/gbb.12117. [DOI] [PubMed] [Google Scholar]
- Viding E, Blakemore SJ. Endophenotype approach to developmental psychopathology: implications for autism research. Behav Genet. 2007;37:51–60. doi: 10.1007/s10519-006-9105-4. [DOI] [PubMed] [Google Scholar]
- Wessa M, Linke J, Witt SH, Nieratschker V, Esslinger C, Kirsch P, Rietschel M. The CACNA1C risk variant for bipolar disorder influences limbic activity. Mol Psychiatry. 2010;15:1126–1127. doi: 10.1038/mp.2009.103. [DOI] [PubMed] [Google Scholar]
- Westra HJ, Peters MJ, Esko T, Yaghootkar H, Schurmann C, Kettunen J, Franke L. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat Genet. 2013;45:1238–1243. doi: 10.1038/ng.2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Camarillo C, Ping Y, Arana TB, Zhao H, Thompson PM, Xu C. The DNA methylome and transcriptome of different brain regions in schizophrenia and bipolar disorder. PLoS One. 2014;9:e95875. doi: 10.1371/journal.pone.0095875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou H, Hastie T. Regularization and variable selection via the elastic net. J Royal Stat Soc B. 2005;67:301–320. [Google Scholar]