

Abstract
Background and Purpose
In vivo and in vitro studies have demonstrated a protective effect of cannabidiol (CBD) in reducing infarct size in stroke models and against epithelial barrier damage in numerous disease models. We aimed to investigate whether CBD also affects blood–brain barrier (BBB) permeability following ischaemia.
Experimental Approach
Human brain microvascular endothelial cell (HBMEC) and human astrocyte co‐cultures modelled the BBB. Ischaemia was modelled by oxygen–glucose deprivation (OGD) and permeability was measured by transepithelial electrical resistance.
Key Results
CBD (10 μM) prevented the increase in permeability caused by 4 h OGD. CBD was most effective when administered before the OGD, but protective effects were observed up to 2 h into reperfusion. This protective effect was inhibited by a PPARγ antagonist and partly reduced by a 5‐HT1A receptor antagonist, but was unaffected by antagonists of cannabinoid CB1 or CB2 receptors, TRPV1 channels or adenosine A2A receptors. CBD also reduced cell damage, as measured by LDH release and by markers of cellular adhesion, such as the adhesion molecule VCAM‐1. In HBMEC monocultures, CBD decreased VCAM‐1 and increased VEGF levels, effects which were inhibited by PPARγ antagonism.
Conclusions and Implications
These data suggest that preventing permeability changes at the BBB could represent an as yet unrecognized mechanism of CBD‐induced neuroprotection in ischaemic stroke, a mechanism mediated by activation of PPARγ and 5‐HT1A receptors.
Abbreviations
- BBB
blood–brain barrier
- CREB
cAMP response element binding protein
- CBD
cannabidiol
- CBF
cerebral blood flow
- HA
human astrocyte
- HBMEC
human brain microvascular endothelial cell
- OGD
oxygen–glucose deprivation
- TEER
transepithelial electrical resistance
- VCAM‐1
vascular cell adhesion molecule 1
Tables of Links
| TARGETS |
|---|
| Ion channels a |
| TRPV1 channels |
| GPCRs b |
| 5‐HT1A receptors |
| Adenosine A2A receptors |
| CB1 receptors |
| CB2 receptors |
| Nuclear hormone receptors c |
| PPARγ |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a, b, cAlexander et al., 2013a, 2013b, 2013c).
Introduction
The blood–brain barrier (BBB) is formed by brain endothelial cells that line the cerebral microvasculature, the capillary basement membranes and the endfeet of astrocytes. Tight junctions restrict the paracellular pathway for diffusion of hydrophilic solutes, allowing the body to control which substances can gain access to the brain (Abbott et al., 2006). Cerebral reperfusion following ischaemia initiates a cascade of events including inflammation, protease activation and oxidative and nitrosative stress, all of which increase BBB permeability (Lo et al., 2003). Increased BBB permeability aggravates haemorrhagic transformation and vasogenic oedema, and uncontrolled cerebral oedema represents the leading cause of patient mortality within the first week following an ischaemic stroke (Hacke et al., 1996).
Cannabidiol (CBD) is the second most abundant plant‐derived cannabinoid. CBD has multiple pharmacological targets, behaving as an agonist of TRPV1 channels, PPARγ, the adenosine A2A and 5‐HT1A receptors and antagonizing the novel endothelial receptor, GPR55, and μ and δ opioid receptors. CBD is a low‐affinity ligand which can modulate cannabinoid CB1 receptor activity (Laprairie et al., 2015; see also Petitet et al., 1998; McPartland et al., 2015) and has weak antagonistic activity toward signalling mediated at CB2 receptors by the cannabinoid ligands, CP 55,940 and WIN 55212‐2 (Bisogno et al., 2001; McPartland et al., 2015). A CBD/THC combination (1:1 ratio, Sativex/Nabiximols, GW Pharmaceuticals, UK) is currently licensed internationally in more than 20 countries for the treatment of spasticity in multiple sclerosis, and a product containing only CBD (Epidiolex, GW Pharmaceuticals, UK) has entered an expanded access programme in children with intractable epilepsies. CBD has also received orphan designation status in treating newborn children with neonatal hypoxic‐ischaemic encephalopathy.
The protective qualities of CBD in ischaemic stroke using rodent in vivo models have been shown in numerous studies. CBD reduces infarct size (when given either before or after middle cerebral artery occlusion) without the development of tolerance, increases cerebral blood flow (CBF), improves motor behaviour and increases survival (Hayakawa et al., 2010). The mechanism of action includes the ability of CBD to ameliorate glutamate neurotoxicity, behave as an antioxidant and anti‐inflammatory agent and to attenuate adhesion molecule expression, neutrophils and the transendothelial migration of monocytes. The protective effects of CBD in vivo are inhibited by 5‐HT1A receptor antagonists, with no role for TRPV1 channels, CB1 or CB2 receptors (Hayakawa et al., 2010). We recently conducted a meta‐analysis to examine the effects of exogenous application of endocannabinoids, phytocannabinoids and synthetic cannabinoids on infarct volume, functional outcome and survival. In 21 studies, involving 188 animals, treatment with CBD showed a highly significant reduction in infarct volume (P < 0.00001), with a standardized mean difference of −1.20 (England et al., 2015).
At the BBB, in vivo studies have shown that CBD decreased BBB permeability in a mouse model of multiple sclerosis (Mecha et al., 2013) and reduced LPS‐induced BBB disruption in mice (Fernandez‐Ruiz et al., 2013). CBD also modulated the permeability of several other epithelial barriers. For example, blood–retinal barrier permeability is increased in diabetic rats in vivo and this was prevented by regular treatment with CBD (El‐Remessy et al., 2006). CBD also decreased in vitro permeability in human coronary artery endothelial cells exposed to high glucose conditions (Rajesh et al., 2007). In gut epithelial cells, CBD enhanced the speed of recovery of either chemical‐induced or cytokine‐induced permeability activity via activation of CB1 receptors (Alhamoruni et al., 2010; Alhamoruni et al., 2012). A recent study from our group found that endocannabinoid‐like molecules (oleoylethanolamide, palmitoylethanolamide and virodhamine) decreased the oxygen–glucose deprivation (OGD)‐induced increase in permeability using a in vitro model of the human BBB, showing that endocannabinoids can affect BBB permeability in an ischaemic setting (Hind et al., 2015).
The potential effects that CBD has on BBB permeability are yet to be assessed in the context of ischaemia, and we hypothesized that activity at the BBB could represent an as yet unrecognized mechanism of CBD‐mediated protection in ischaemic stroke. To address this, we examined the effects of CBD in an in vitro BBB model, where the potential mechanisms of action were also probed.
Methods
Cell culture and protocols
Human brain microvascular endothelial cells (HBMECs; ScienCell, USA) and human astrocytes (HAs; ScienCell, USA) were cultured in their specialized media. HAs were seeded on the outside of collagen‐coated 0.4 μm pore polytetrafluoroethylene membrane Transwell inserts (12 well type; Corning Costar, USA), and HBMECs were seeded on the inside of the insert. Both cells were grown to confluence to create a contact co‐culture model (Hind et al., 2015). Transepithelial electrical resistance (TEER) was measured as a marker of co‐culture integrity and as a measure of BBB permeability. The resistance across the membrane was measured using STX2 electrodes linked to an epithelial voltohmmeter (World Precision Instruments, UK).
Ischaemic conditions were simulated using an OGD protocol (Hind et al., 2015). Cell culture media was replaced with glucose‐free RPMI medium (Invitrogen, UK) and the plates placed into a GasPak EZ Anaerobe Pouch (Beckton Dickinson, UK) with anaerobic conditions being achieved within 20 min. Inserts were left in OGD conditions for 4 h. After OGD, TEER was read, and the RPMI medium was replaced with the cells' normal medium and returned to the incubator (i.e. the reperfusion period). Throughout the course of the experiments, TEER readings were taken at 0, 4, 6, 8, 10, 12, 24 and 32 h.
The effect of CBD treatment (100 nM, 1 μM and 10 μM) on BBB permeability was assessed when given either before or immediately after the OGD protocol. To test the time window of efficacy, CBD (10 μM) was given at 6 and 8 h (i.e. 2 and 4 h into the reperfusion period). A range of receptor antagonists were used to probe the target sites of action for CBD (at 10 μM) at the BBB; for CB1 receptors, AM251 (100 nM); for CB2 receptors, AM630 (100 nM); for TRPV1 channels, capsazepine (1 μM), for PPARγ, GW9662 (100 nM), for adenosine A2A receptors, SCH58261 (100 nM); for 5‐HT1A receptors, WAY100135 (300 nM). In these studies, vehicle, the antagonist alone, and a control response to CBD were conducted at the same time. To confirm the role of PPARγ and 5‐HT1A receptors, the effects of agonists of these receptors, pioglitazone (10 μM) and 8‐OH‐DPAT (1 μM) were measured. In some experiments, HBMECs and HAs were grown in monoculture and treated with CBD and/or antagonist to establish which target site(s) of action were activated by CBD in these two individual cell types.
Biochemical analysis
LDH levels at the end of the experiment (32 h) were measured using a commercially available kit (LDH‐Cytotoxicity Assay Kit II, Biovision, USA). Medium samples were transferred into an optically clear 96‐well plate and Reaction Mix (containing water‐soluble tetrazolium‐1) was added. After 30 min, absorbance was measured at 450 nm, subtracting the 650 nm reading to correct for optical imperfections.
DuoSet elisa kits (R&D Systems, UK) were used to measure the levels of human IL‐6, vascular cell adhesion molecule 1 (VCAM‐1) and VEGF in medium samples from co‐cultures and in medium or cellular samples from monocultures (cellular samples collected in RIPA buffer with phosphatase and protease inhibitors; Sigma‐Aldrich, UK). Data were normalized to protein content as determined using a bicinchoninic acid assay (Sigma‐Aldrich, UK). The medium from co‐culture experiments was analysed for changes in cytokines using a human cytokine/chemokine panel (HCYTOMAG‐60 K, Merck Millipore, UK) that measured levels of IFN‐γ, IL‐1β, IL‐2, IL‐6, IL‐10, chemokines CCL3 and CCL4, TNF‐α and VEGF.
Cell signalling analysis
HBMEC monocultures treated with CBD (100 nM, 1 μM and 10 μM) for 28 h were analysed using human Multi‐Pathway Magnetic Bead 9‐Plex Cell Signalling Multiplex Assay (48‐680MAG, Merck Millipore, UK) to screen for potential changes in the following phosphorylated proteins; PKB (Akt), cAMP response element binding protein (CREB), ERK, JNK, NF‐κB, STAT3, STAT5A/STAT5B, p38 MAPK (p38), and ribosomal protein S6 kinase β‐1.
Materials
Purified CBD was a gift from GW Pharmaceuticals (Cambridge, UK) and was dissolved to 10 mM in ethanol. AM251, AM630, GW9662, SCH58261, (S)‐WAY100135 and capsazepine (Tocris UK) were dissolved in dimethyl sulfoxide to stock solutions of 10 mM,). Serial dilutions were made fresh daily.
Data analysis
All data were tested for normality using the D'Agostino and Pearson omnibus normality test, and AUC was calculated using the trapezoidal method. Time course studies (Figures 1, 2, 3) were analysed by two‐way anova with multiple comparisons at each time point comparing against the vehicle control. Column graphs (Figures 3, 4, 5, 6) were compared by one‐way anova with Dunnett's post hoc test comparing against vehicle controls.
Figure 1.
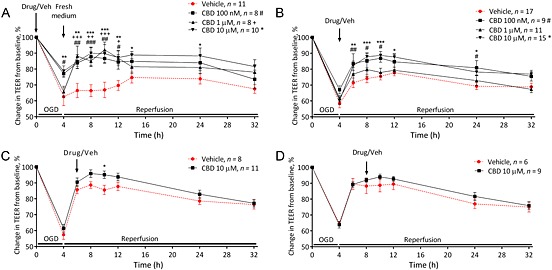
Cannabidiol (CBD) is protective against increased permeability of the BBB associated with oxygen–glucose derivation (OGD). The effects of CBD given before (A), immediately after (B) OGD, or 2 h (C) or 4 h (D) into the reperfusion period on TEER values. Data are given as mean ± SEM (n represents the total number of inserts measured, based on three to four separate experiments). Statistical analysis was conducted by two‐way anova with post hoc analysis at each time point comparing data against the vehicle control. *P < 0.05, **P < 0.01, ***P < 0.001; 10 μM CBD versus vehicle. # P < 0.05, ## P < 0.01, ### P < 0.001, 1 μM CBD versus vehicle, + P < 0.05, ++ P <0.01`, +++ P <, 0.001, 100 nM CBD versus vehicle.
Figure 2.
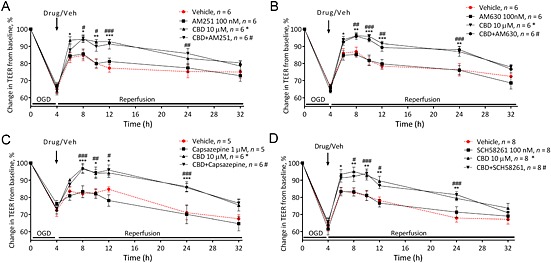
The effects of cannabidiol (CBD) are not mediated by CB1, CB2 receptors or TRPV1 channels. The effect of the receptor antagonists AM251 (A; 100 nM), AM630 (B; 100 nM), capsazepine (C; 1 μM) and SCH58261 (D; 100 nM) on the effects of CBD applied luminally on BBB permeability. Data are given as mean ± SEM (n represents the total number of inserts measured, based on two separate experiments). Statistical analysis was conducted by two‐way anova with post hoc analysis at each time point comparing data against the vehicle control. *P < 0.05, **P < 0.01, ***P < 0.001, vehicle versus CBD. # P < 0.05, ## P < 0.01, ### P < 0.001, vehicle versus CBD + antagonist.
Figure 3.
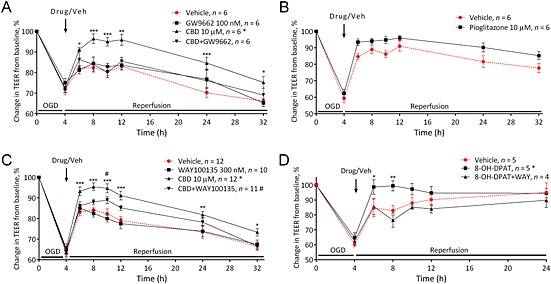
The effects of cannabidiol (CBD) are mediated by PPARγ and 5HT1A receptors. The effect of the receptor antagonists GW9662 (A; 100 nM) and WAY100135 (C; 300 nM) on the effects of CBD on BBB permeability. The effect of the PPARγ agonist pioglitazone (B) and 5HT1A receptor agonist 8‐OH‐DPAT (D) applied luminally on BBB permeability. Data are given as mean ± SEM (n represents the total number of inserts measured, based on two to four separate experiments). Statistical analysis was conducted by two‐way anova with post hoc analysis at each time point comparing data against the vehicle control. *P < 0.05, **P < 0.01, ***P < 0.001,****P < 0.0001; vehicle versus CBD. # P < 0.05; vehicle versus CBD + antagonist.
Figure 4.
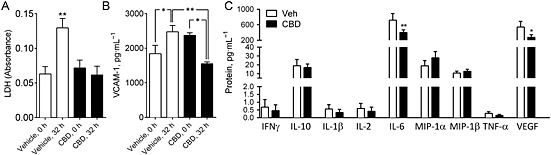
Biochemical analysis of co‐culture medium. Levels of LDH (A) and vascular cell adhesion molecule 1 (VCAM‐1; B) from the luminal (endothelial) chamber of co‐culture inserts at the end of experiment (32 h) (n = 6 inserts). C. Cytokine or chemokine levels from the luminal (endothelial) chamber at 32 h for IFN‐γ, L‐10, IL‐1β, IL‐2, IL‐6, CCL3, CCL4, TNF‐α and VEGF (n = 6 inserts). Data are given as mean ± SEM. Statistical analysis conducted using one‐way anova with Sidak's multiple comparisons comparing the vehicle control against CBD responses (n = 6, *P < 0.05, **P < 0.01).
Figure 5.
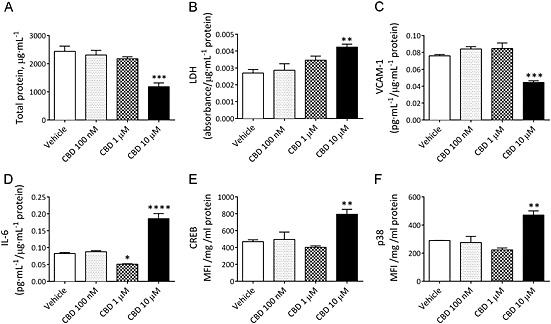
The effects of CBD on HBMECs in monoculture. The total level of protein (A), LDH (B), protein‐normalized medium levels of VCAM (C), IL‐6 (D), and cellular levels of phosphorylated CREB (E) and p38 (F) in HBMEC treated with 100 nM, 1 μM or 10 μM CBD for 28 h in normal culture conditions. MFI, mean fluorescent intensity. Data are given as mean ± SEM (n = 3–6). Statistical analysis conducted using one‐way anova with Dunnett's post hoc test compared with the vehicle control response, *P < 0.05, **P < 0.01, ***P < 0.001, **** P < 0.0001.
Figure 6.
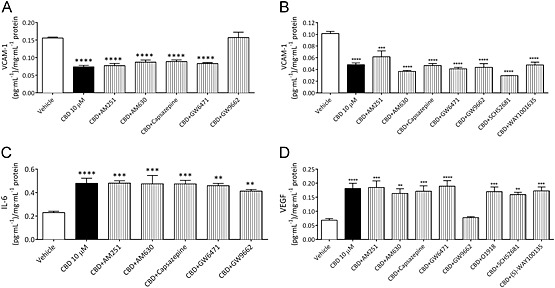
Biochemical analysis of monocultures. The levels of VCAM‐1 in cell culture medium from monocultures of either HBMECs (A) or HAs (B) treated for 28 h with CBD alone or in the presence of receptor antagonists in normal conditions. The levels of IL‐6 (C) and VEGF (D) in HBMEC medium from monocultures treated for 28 h with CBD alone or in the presence of receptor antagonists. All data normalized to total protein (bicinchoninic acid assay). Data are given as mean ± SEM. Statistical analysis conducted using one‐way anova with Dunnett's post hoc test (n = 3–6), **P < 0.01, ***P < 0.001, ****P < 0.0001.
Results
The effects of cannabidiol on BBB permeability after OGD
Exposing the co‐cultures to 4 h OGD caused a significant increase in permeability (P < 0.0001). In some inserts, we showed that 4 h OGD was also accompanied by a significant increase in Evans Blue cleared volume (normoxia 979 ± 23 compared with 4 h OGD 1127 ± 26, n = 3, P < 0.05), an alternative method of measuring increases in cellular permeability (Srivastava et al., 2013). Administering CBD immediately before OGD at 100 nM (P < 0.05), 1 μM (P < 0.05) or 10 μM (P < 0.01) reduced the permeability of the BBB when looking at the effect on the time‐course as a whole (0–32 h, AUC, Table 1). All concentrations of CBD reduced permeability at various time points during reperfusion (P < 0.05–0.001), but only 100 nM (P < 0.05) and 10 μM (P < 0.01) attenuated the initial increase in permeability (Figure 1A). Administering CBD immediately after OGD at 100 nM and 10 μM, but not 1 μM, reduced the permeability of the BBB (Figure 1B). Adding 10 μM CBD 2 h into the reperfusion period reduced permeability at 10 h (Figure 1C). Adding 10 μM CBD 4 h into the reperfusion period did not have any effect on BBB permeability (Figure 1D).
Table 1.
Integrated responses over time (0–32 h) to CBD in combination with an oxygen/glucose deprivation protocol
| Pre‐hypoxia | AUC |
| Vehicle (n = 8) | 1518 ± 85 |
| CBD 100 nM (n = 6) | 1144 ± 104 * |
| CBD 1 μM (n = 6) | 1185 ± 101 * |
| CBD 10 μM (n = 8) | 1033 ± 82 ** |
| Immediately after OGD | |
| Vehicle (n = 17) | 1151 ± 54 |
| CBD 100 nM (n = 9) | 930 ± 89 |
| CBD 1 μM (n = 11) | 1075 ± 68 |
| CBD 10 μM (n = 15) | 880 ± 53 ** |
| 2 h post‐OGD | |
| Vehicle (n = 8) | 931 ± 63 |
| CBD 10 μM (n = 11) | 788 ± 62 |
| 4 h post‐OGD | |
| Vehicle (n = 6) | 778 ± 84 |
| CBD 10 μM (n = 9) | 689 ± 39 |
| CB1 receptor studies | |
| Vehicle (n = 6) | 798 ± 79 |
| AM251 alone (n = 6) | 750 ± 60 |
| CBD (n = 6) | 531 ± 49 * |
| CBD and AM251 (n = 6) | 532 ± 62 * |
| CB2 receptor studies | |
| Vehicle (n = 6) | 831 ± 68 |
| AM630 alone (n = 6) | 847 ± 58 |
| CBD (n = 6) | 551 ± 37 ** |
| CBD and AM630 (n = 6) | 573 ± 42 ** |
| TRPV1 receptor studies | |
| Vehicle (n = 5) | 858 ± 66 |
| Capsazepine alone (n = 5) | 918 ± 107 |
| CBD (n = 5) | 543 ± 46 * |
| CBD and capsazepine (n = 5) | 535 ± 32 * |
| PPARγ receptor studies | |
| Vehicle (n = 6) | 889 ± 28 |
| GW9662 alone (n = 6) | 805 ± 73 |
| CBD (n = 6) | 574 ± 54 * |
| CBD and GW9662 (n = 6) | 780 ± 102 |
| Further PPARγ studies | |
| Vehicle (n = 6) | 587 ± 71 |
| Pioglitazone (n = 6) | 381 ± 50 * |
| A2A adenosine receptor studies | |
| Vehicle (n = 8) | 976 ± 63 |
| SCH58261 alone (n = 8) | 949 ± 73 |
| CBD (n = 8) | 683 ± 64 ** |
| CBD and SCH58261 (n = 8) | 697 ± 50 * |
| 5‐HT1A receptor studies | |
| Vehicle (n = 12) | 1033 ± 75 |
| WAY100135 alone (n = 10) | 1082 ± 56 |
| CBD (n = 12) | 768 ± 67 * |
| CBD and WAY100135 (n = 11) | 936.2 ± 44 |
AUC values are presented as mean ± SEM. Data were compared by one‐way anova with Dunnett's post hoc test, comparing against the vehicle response in experiments that did not contain antagonist. In experiments that did contain an antagonist, data were compared by one‐way anova with Tukey's post hoc test. Variability in the vehicle controls reflects differences in BBB permeability responses from experiment to experiment. In every experiment, vehicle controls and CBD alone positive controls were carried out to account for this.* P < 0.05.** P < 0.01.
The permeability lowering effects of 10 μM CBD given immediately after OGD were not inhibited by AM251, AM630, capsazepine or SCH58261 (Figure 2 and Table 1). The permeability lowering effects of 10 μM CBD were inhibited in the presence of GW9662 (Figure 3A), and the PPARγ agonist pioglitazone also reduced BBB permeability after OGD in a similar manner (P < 0.05; Table 1 and Figure 3B). In the presence of WAY100135 (a 5‐HT1A antagonist), the effects of CBD were no longer significantly different to vehicle except at 10 h (Figure 3C). The 5‐HT1A agonist 8‐OH‐DPAT also reduced BBB permeability after OGD in a similar manner to CBD, which was antagonized by WAY100135 (Figure 3D and Table 1).
Combining all experiments that were conducted with CBD (10 μM) together (n = 45), a highly significant reduction in permeability (P < 0.0001) was observed at all time points following the addition of CBD after OGD (Supporting Information Fig. S1).
Co‐culture biochemical assays
In medium from the luminal chamber of co‐culture inserts, LDH levels were significantly increased following reperfusion (P < 0.01), but LDH levels were not increased in medium from the inserts treated with 10 μM CBD, which had significantly lower LDH levels than vehicle‐treated samples (P < 0.05; Figure 4A). VCAM‐1 was increased in vehicle‐treated samples following OGD + reperfusion in medium from luminal chambers (P < 0.05; Figure 4B). Levels of VCAM‐1 were significantly reduced after treatment with 10 μM CBD (P < 0.05; Figure 4B).
Detectable levels of IL‐6, IL‐10, CCL3, CCL4, VEGF, INFγ, IL‐1β, IL‐2 and TNF‐α were seen in medium samples at 32 h from the luminal chambers of the co‐culture, (Figure 4C). A similar pattern was observed in the abluminal chamber (Supporting Information Fig. S2). There was a significant reduction in IL‐6 (P < 0.01) and VEGF (P < 0.01) levels in the medium of inserts treated with CBD (Figure 4C).
Effects of CBD on HBMECs and HAs in monoculture
The amount of total protein in HBMEC monocultures was lower in samples treated with 10 μM (but not 100 nM or 1 μM) CBD for 28 h in normal conditions (P < 0.001; Figure 5A). In medium from HBMEC monocultures, the levels of LDH were significantly higher in samples that were treated with 10 μM CBD (but not 100 nM or 1 μM) for 28 h in normal conditions (P < 0.01; Figure 5B). These increases in levels of cell damage were reflected in the morphology of HBMECs (Supporting Information Fig. S3). These observations were in contrast to the situation in co‐cultures, where 10 μM CBD inhibited the OGD‐induced increase in LDH and visual examination of the cells under the microscope at various time points throughout the 32 h procedure did not reveal any damaging effects of CBD.
In HBMEC monocultures, CBD at 10 μM, but not 100 nM or 1 μM, decreased the levels of VCAM‐1 (data normalized to total protein) compared with vehicle in samples that were treated for 28 h in normal conditions (P < 0.001; Figure 5C). This effect was inhibited by the PPARγ antagonist GW9662 (Figure 6A). There was a concentration‐dependent effect of CBD on IL‐6 in normal conditions, where 1 μM decreased (P < 0.05), but 10 μM increased (P < 0.0001) IL‐6 levels (data normalized to total protein; Figure 5D). Receptor involvement in the 10 μM response was probed; however, none of the antagonists tested altered the effect on IL‐6 levels (Figure 6C). VEGF levels were increased in HBMEC monocultures treated with 10 μM CBD in normal conditions (P < 0.001; Figure 6D) (data normalized to total protein). This effect was inhibited by the PPARγ antagonist GW9662 (Figure 6D).
HBMECs that were treated with 10 μM CBD (but not 100 nM or 1 μM) for 28 h had increased levels of phosphorylated CREB (P < 0.01; Figure 5E) and p38 (P < 0.01; Figure 5F). The levels of JNK, NF‐κB, ERK 1/2, Akt, p70s6K, STAT3 and STAT5 were not altered (Supporting Information Fig. S4).
HA monocultures treated with 10 μM CBD showed similar results to HBMEC monocultures with decreases in protein levels (P < 0.001), increases in LDH (P < 0.001) and decreases in secreted levels of IL‐6 (P < 0.001) and VCAM (P < 0.001) in cells exposed to 10 μM CBD (all data normalized to total protein count; Supporting Information Fig. S5). Unlike the endothelial cells, the decrease in VCAM in human astrocytes was not inhibited by GW9662 (Figure 6B) or any other antagonist tested.
Discussion
In this study we have shown that CBD decreases BBB permeability when given either before or after OGD, involving activation of PPARγ and 5‐HT1A receptors and associated with reductions in cell damage and VCAM‐1, and increases in VEGF. These data increase the case for further investigations into CBD as a future treatment in ischaemic stroke.
Given before OGD, CBD reduced permeability at all concentrations tested. This is consistent with data from in vitro and in vivo models showing CBD is effective at reducing the permeability of the BBB in multiple sclerosis (Mecha et al., 2013) and sepsis‐related encephalitis (Fernandez‐Ruiz et al., 2013) and other epithelial barriers in various diseases (El‐Remessy et al., 2006; Rajesh et al., 2007; Alhamoruni et al., 2010, 2012). Most strokes occur without warning and a treatment that protects the brain when administered post ictus is more clinically relevant; this makes CBD an attractive candidate as we also showed that 10 μM CBD decreased BBB permeability when given immediately after OGD and also 2 h after reperfusion (i.e. 4 and 6 h from initial onset). Indeed, administration of CBD at 1 or even 3 days (but not 5 days) following middle cerebral artery occlusion increases survival in vivo in mice (Hayakawa et al., 2010). CBD afforded protection to the BBB at all concentrations tested when given pre OGD, but only at 10 μM when given post‐OGD. The reasons for this may be complex as OGD induces many cellular changes which could influence CBD efficacy such as changes in signalling proteins, Ca2+ levels, receptor expression and function, enzymic activities and mRNA translation. CBD decreased the levels of cell damage that were elevated following OGD +reperfusion in co‐cultures, contributing to a less disrupted BBB. In vivo, CBD decreases the level of cell death in the hippocampus, the liver and the retina following ischaemic stroke in mice (Shiavon et al., 2014), hepatic ischaemia + reperfusion in mice (Fernandez‐Ruiz et al., 2013) and in diabetic rats (El‐Remessy et al., 2006) respectively. CBD also decreased VCAM‐1 after OGD + reperfusion. VCAM‐1 is mainly expressed by endothelial cells and selectively binds to the very late antigen‐4 expressed on monocytes and lymphocytes (Yusuf‐Makagiansar et al., 2002), thus increasing their transmigration across the BBB. VCAM‐1 levels are raised during cerebral ischaemia, and it seems likely that they play an important role during the inflammatory response to stroke (Jander et al., 1996). This is consistent with previous studies where CBD reduced VCAM‐1 levels using in vitro human coronary artery endothelial cells (CB1/2 independent; Rajesh et al., 2007) and using in vivo mouse models of multiple sclerosis (partly mediated by adenosine A2A receptors; Mecha et al., 2013). In our monoculture studies, CBD reduced VCAM secretion in both HBMECs and HAs. Etymologically, it may seem paradoxical that VCAM‐1 is produced by astrocytes, but this finding has also been observed in human astroglial cells (Song et al., 2011).
CBD has a rich pharmacology with known activity at multiple sites. In the present study, we found no role for CB1, CB2 or adenosine A2A receptors or TRPV1 channels. We did however find that permeability reducing properties of CBD were mediated via activity at PPARγ. The concentration of CBD (10 μM) required to affect BBB permeability when given after OGD is in line with CBD's micromolar affinity for PPARγ (O'Sullivan et al., 2009; Fernandez‐Ruiz et al., 2013). Furthermore, pioglitazone (a PPARγ agonist) mimicked CBD's permeability‐reducing properties. We also found that the CBD‐induced decrease in VCAM‐1 in HBMEC monocultures was attenuated by PPARγ antagonism, consistent with a previous study showing activation of PPARγ by the CB1/CB2 agonist WIN55,212‐2 decreases VCAM‐1 levels in mouse brain endothelial cells in vitro (Mestre et al., 2009). PPARγ activation by rosiglitazone and pioglitazone decreases BBB permeability induced by inflammation (Ramirez et al., 2008). Our results provide evidence to suggest that activation of PPARγ may play a role in reducing permeability following ischaemia. Other groups have shown that PPARγ activation is protective in rat models of ischaemic stroke; thiazolidinediones (TZDs) such rosiglitazone, as are PPARγ agonists that reduce infarct size and improve functional recovery from ischaemic stroke in rats (Luo et al., 2006). Therefore, PPARγ activity at the BBB in ischaemic stroke could represent an as yet unrecognized mechanism of TZD‐mediated protection.
The effects of CBD were also partly inhibited by a 5‐HT1A receptor antagonist. We also showed that a 5‐HT1A receptor agonist was capable of reducing the increased permeability induced by OGD, and this was inhibited by a 5‐HT1A receptor antagonist. Again, the concentration of CBD (10 μM) required to affect BBB permeability is consistent with CBD's micromolar affinity for 5‐HT1A receptors (Fernandez‐Ruiz et al., 2013). This is the first study to investigate the potential role that 5‐HT1A plays in regulating the permeability of the BBB or any epithelial barrier. In mouse models of ischaemic stroke, CBD activity at the 5‐HT1A receptor was shown to be responsible, at least in part, for the beneficial effects of reducing infarct volume and increasing CBF (Hayakawa et al., 2010). Therefore, CBD activation of the 5‐HT1A receptor at the BBB could potentially contribute to the protective effects reported in the published literature.
The expression of VEGF is potentiated by hypoxia, and it is an important pathogenic factor for induction of vascular leakage in the brain (Schoch et al., 2002), and inhibition of VEGF reduces the permeability of the BBB in vivo in the rat (Zhang et al., 2002). In HBMEC monocultures, we found that VEGF levels were increased following treatment with 10 μM CBD, sensitive to PPARγ antagonism. The PPARγ agonist troglitazone increased VEGF levels in OP9 mouse preadipocytes in a GW9662‐sensitive manner (Kotake and Hirasawa, 2013). Our findings are in contrast to a study by El‐Remessy et al. (2006, where they showed that CBD reduced VEGF levels in the blood–retinal barrier following experimental diabetes in the rat. However, it should also be noted that in the co‐culture experiments (Figure 4C), a trend for a reduction in VEGF levels was observed with CBD, indicating a difference in the cellular response to CBD between monoculture and co‐cultures.
In HA and HBMEC monocultures, we found that CBD‐reduced IL‐6 levels at 10 and 1 μM, respectively; and there was a trend for reduced levels of IL‐6 in medium from the luminal chamber of our co‐cultures that were treated with 10 μM CBD (P = 0.11; Figure 4C). CBD decreases IL‐6 levels in vitro following ischaemic damage to the mouse forebrain (Castillo et al., 2010), which is associated with CBD's ability to reduce infarct volume. IL‐6 levels are increased following experimental and human ischaemic stroke (Miao and Liao, 2014), with increased IL‐6 levels being shown to interrupt the BBB by disrupting endothelial TJs (Yenari et al., 2006). Indeed, in clinical studies, IL‐6 is often suggested as a good predictive marker of stroke severity (Miao and Liao, 2014). However, IL‐6‐deficient mice do not exhibit improved outcome from stroke (Ceulemans et al., 2010), and production of IL‐6 promotes post‐stroke angiogenesis, thus promoting long‐term histological and functional benefits (Gertz et al., 2012).
To gain a better understanding of the mechanisms through which CBD was having its effects on the HBMEC and HA monocultures, the levels of several well‐known signalling proteins involved in cell survival and inflammation were measured, and an increase in the levels of phosphorylated p38 and CREB were observed at 10 μM. p38 is a stress‐activated serine/threonine protein kinase, which is a downstream target of proinflammatory cytokines and oxidative stress, and hence causes cellular damage. Increased levels of p38 contribute to transcriptional activation of the IL‐6 promoter by modulating the transactivation capacity of the NF‐κB subunit (Vanden Berghe et al., 2000). Therefore, the increased levels of p38 may be partly responsible for the increased levels of IL‐6 seen in HBMEC monocultures treated with 10 μM CBD.
CBD also increased phosphorylated CREB levels in HBMEC monocultures. Cerebral ischaemia increases glutamate levels, which results in elevated levels of neuronal CREB activation. CREB is known to lead to the expression of certain genes that encode neuroprotective molecules, such as the anti‐apoptotic protein Bcl‐2, and contributes to neuronal survival following ischaemia (Kitagawa, 2007). However, CREB has also been shown to induce transcription of immune‐related genes that possess a cAMP responsive element, such as IL‐6, so this could be partially responsible for an increase in IL‐6 levels (Wen et al., 2010). The CREB signalling pathway is linked to p38, whereby an increase in p38 raises CREB phosphorylation through interaction with ERK‐1/ERK‐2 and then mitogen and stress‐activated kinases (MSK)‐1/MSK‐2. CREB then initiates transcription of the anti‐inflammatory protein IL‐10 and also induces dual specificity protein phosphatase 1, which feeds back to inhibit p38 (Wen et al., 2010).
The effects of CBD on cells in monocultures did not always mirror the data obtained from the co‐cultures. For example, in co‐culture, CBD decreased LDH levels, but in HBMEC and HA monocultures, CBD (10 μM) increased the levels of cell damage. A potential explanation for why 10 μM CBD was cytotoxic in monocultures but cytoprotective in co‐cultures is that in the co‐culture, the HBMECs and HAs communicate with each other, and thus is more representative of the BBB in vivo. Communication between these two cell types is vital for the formation of tight junctions; furthermore, paracrine signalling occurs between these two cells. Given the large repertoire of astrocyte‐released agents, many of which have matching receptors on brain endothelium, a range of distinct and complex responses of the endothelium can be influenced by astrocyte activity (Abbott et al., 2006). For example, there is an up‐regulation of numerous specific transport systems in brain endothelia when they are in contact with glia, an effect that has been observed with GLUT‐1, the L‐system and A‐system amino acid carriers and P‐glycoprotein. Several molecules, including glial cell line‐derived neurotrophic factor and various steroids, are known to be capable of mimicking aspects of glial‐mediated barrier induction of brain endothelium. In the opposite direction, there is also evidence that endothelial cells induce the astrocytic phenotype at the BBB, with leukaemia inhibitory factor, secreted from brain endothelia, being shown to induce astrocytic differentiation (Abbott, 2002). Another potential explanation for the distinct effect of CBD in co‐culture and monoculture may be that in co‐culture, the cells were grown on a collagen‐coated membrane, which may provide a more stable environment for the cells compared with monoculture. Of the two different types of cellular environment employed in this study (i.e. monoculture and co‐culture), the co‐culture is considered to be the more physiologically relevant environment. Whilst our co‐culture reproduced many of the important characteristics of the BBB, regulation of the BBB is highly complex and involves input from an array of different cell types and molecules (Abbott et al., 2006), implying that the actual in vivo physiology of the BBB can never be fully reproduced in vitro. Our data demonstrate the limitations of performing in vitro cell work in a single cell type only, in the absence of other cells that will influence cellular responses.
In conclusion, this study demonstrated that CBD decreased the permeability of the BBB following OGD‐reperfusion and has identified a role for PPARγ and 5‐HT1A receptors. CBD may achieve this decrease in permeability by attenuating the OGD + reperfusion‐associated increases in cell damage and by decreasing VCAM‐1. The results presented here (in addition to the existing literature; where CBD has shown neuroprotection, CBF increasing properties and improved motor performance following ischaemic stroke in vivo and in vitro; Hayakawa et al., 2010) increases the case for further investigations into CBD as a future treatment in ischaemic stroke and potential clinical application. Many neuroprotective drugs have shown preclinical promise, but all those that have reached clinical development failed to demonstrate efficacy or had unacceptable adverse effects (Green and Shuaib, 2006). CBD has an excellent tolerability profile (Bergamaschi et al., 2011) and is currently licensed to treat spasticity in multiple sclerosis (in combination with THC, as the medicine Sativex; GW Pharmaceuticals, Cambridge UK) in over 20 countries. The sequelae of ischaemic stroke are multifaceted; therefore, a drug that targets multiple systems such as CBD may have an increased chance of success compared with compounds aimed at a single‐target (O'Collins et al., 2006).
Author contributions
W. H. H. performed the research. T. J. E. and S. O. S. designed the research study. W. H. H. and S. O. S. analysed the data. W. H. H., T. J. E. and S. O. S. wrote the paper.
Conflict of interest
None.
Supporting information
Figure S1 The effect of 10 μM cannabidiol (CBD) applied after 4 hours OGD on BBB permeability from all experiments (n = 45) presented as a time course (A) and as the area under the curve of the whole experiment (B). Data are given as mean ± SEM. Statistical analysis conducted using two‐way ANOVA (A) or Student's t‐test (B). *** P<0.001, **** P<0.0001.
Figure S2 Cytokine/chemokine levels in the abluminal medium of BBB co‐culture inserts at 32 hours for IFNγ, IL‐10, IL‐1β, IL‐2, IL‐6, macrophage inflammatory protein (MIP)‐1α, MIP‐1β, TNF‐α and VEGF. Data are given as mean ± SEM. Statistical analysis conducted using one‐way ANOVA with post hoc analysis of selected pairs (vehicle versus CBD).
Figure S3 The effect of 28h treatment with increasing concentrations of cannabidiol (CBD) on HBMEC cell morphology. At the highest concentration, CBD appeared to cause cell damage, which was associated with a significant decrease in protein levels and significant increase in LDH release (see Figure 5).
Figure S4 No differences were observed after CBD treatment in the levels of phosphorylated JNK (A), ERK (B), Akt (c), NFκB (D), p70s6K (E), STAT3 (F) or STAT5 (G) in HBMECs treated for 28 h with CBD. Data are given as mean ± SEM. Statistical analysis conducted using one‐way ANOVA with Dunnett's post hoc test.
Figure S5 A. Light microscope images of human brain astrocytes treated by vehicle or increasing concentrations of CBD for 28 h in normal conditions. At the highest concentration, CBD appeared to cause cell damage, which was associated with a significant decrease in protein levels (B) and significant increase in LDH release (C). The levels of IL‐6 (D) and VCAM (E) in medium from astrocyte monocultures treated for 28 h with CBD. All data was normalised to total protein (BCA assay). Data are given as mean ± SEM. Statistical analysis conducted using one‐way ANOVA with Dunnett's post hoc test, n = 3‐6, ** P<0.01, *** P<0.001.
Supporting info item
Hind, W. H. , England, T. J. , and O'Sullivan, S. E. (2016) Cannabidiol protects an in vitro model of the blood–brain barrier from oxygen‐glucose deprivation via PPARγ and 5‐HT1A receptors. British Journal of Pharmacology, 173: 815–825. doi: 10.1111/bph.13368.
References
- Abbott NJ (2002). Astrocyte–endothelial interactions and blood–brain barrier permeability. J Anat 200: 629–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbott NJ, Ronnback L, Hansson E (2006). Astrocyte–endothelial interactions at the blood–brain barrier. Nat Rev Neurosci 7: 41–53. [DOI] [PubMed] [Google Scholar]
- Alexander SP, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA et al (2013a). The Concise Guide to PHARMACOLOGY 2013/14:Ion channels. Br J Pharmacol 170: 1607–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al (2013b). The Concise Guide to PHARMACOLOGY 2013/14: G protein‐coupled receptors. Br J Pharmacol 170: 1459–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al (2013c). The Concise Guide to PHARMACOLOGY 2013/14: Nuclear hormone receptors. Br J Pharmacol 170: 1652–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alhamoruni A, Lee AC, Wright KL, Larvin M, O'Sullivan SE (2010). Pharmacological effects of cannabinoids on the Caco‐2 cell culture model of intestinal permeability. J Pharmacol Exp Ther 335: 92–102. [DOI] [PubMed] [Google Scholar]
- Alhamoruni A, Wright KL, Larvin M, O'Sullivan SE (2012). Cannabinoids mediate opposing effects on inflammation‐induced intestinal permeability. Br J Pharmacol 165: 2598–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen CL, Bayraktutan U (2009). Antioxidants attenuate hyperglycaemia‐mediated brain endothelial cell dysfunction and blood–brain barrier hyperpermeability. Diabetes Obes Metab 11: 480–490. [DOI] [PubMed] [Google Scholar]
- Bergamaschi MM, Queiroz RH, Zuardi AW, Crippa JA (2011). Safety and side effects of cannabidiol, a Cannabis sativa constituent. Curr Drug Saf 6: 237–249. [DOI] [PubMed] [Google Scholar]
- Bisogno T, Hanus L, De Petrocellis L, Tchilibon S, Ponde DE, Brandi I et al (2001). Molecular targets for cannabidiol and its synthetic analogues: effect on vanilloid VR1 receptors and on the cellular uptake and enzymatic hydrolysis of anandamide. Br J Pharmacol 134: 845–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo A, Tolon MR, Fernandez‐Ruiz J, Romero J, Martinez‐Orgado J (2010). The neuroprotective effect of cannabidiol in an in vitro model of newborn hypoxic‐ischemic brain damage in mice is mediated by CB(2) and adenosine receptors. Neurobiol Dis 37: 434–440. [DOI] [PubMed] [Google Scholar]
- Ceulemans AG, Zgavc T, Kooijman R, Hachimi‐Idrissi S, Sarre S, Michotte Y (2010). The dual role of the neuroinflammatory response after ischemic stroke: modulatory effects of hypothermia. J Neuroinflammation 7: 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El‐Remessy AB, Al‐Shabrawey M, Khalifa Y, Tsai NT, Caldwell RB, Liou GI (2006). Neuroprotective and blood‐retinal barrier‐preserving effects of cannabidiol in experimental diabetes. Am J Pathol 168: 235–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- England TJ, Hind WH, Rasid NA, O'Sullivan SE (2015). Cannabinoids in experimental stroke: a systematic review and meta‐analysis. J Cereb Blood Flow Metab 35: 348–358. Epub 2014 Dec 10. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez‐Ruiz J, Sagredo O, Pazos MR, Garcia C, Pertwee R, Mechoulam R et al (2013). Cannabidiol for neurodegenerative disorders: important new clinical applications for this phytocannabinoid? Br J Clin Pharmacol 75: 323–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertz K, Kronenberg G, Kalin RE, Baldinger T, Werner C, Balkaya M et al (2012). Essential role of interleukin‐6 in post‐stroke angiogenesis. Brain 135: 1964–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green AR, Shuaib A (2006). Therapeutic strategies for the treatment of stroke. Drug Discov Today 11: 681–693. [DOI] [PubMed] [Google Scholar]
- Hacke W, Schwab S, Horn M, Spranger M, De Georgia M, von Kummer R (1996). ‘Malignant’ middle cerebral artery territory infarction: clinical course and prognostic signs. Arch Neurol 53: 309–315. [DOI] [PubMed] [Google Scholar]
- Hayakawa K, Mishima K, Fujiwara M (2010). Therapeutic potential of non‐psychotropic cannabidiol in ischemic stroke. Pharmaceuticals 3: 2197–2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hind WH, Tufarelli C, Neophytou M, Anderson SI, England TJ, O'Sullivan SE (2015). Endocannabinoids modulate human blood–brain barrier permeability in vitro. Br J Pharmacol 172: 3015–3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jander S, Pohl J, Gillen C, Schroeter M, Stoll G (1996). Vascular cell adhesion molecule‐1 mRNA is expressed in immune‐mediated and ischemic injury of the rat nervous system. J Neuroimmunol 70: 75–80. [DOI] [PubMed] [Google Scholar]
- Kitagawa K (2007). CREB and cAMP response element‐mediated gene expression in the ischemic brain. FEBS J 274: 3210–3217. [DOI] [PubMed] [Google Scholar]
- Kotake D, Hirasawa N (2013). Activation of a retinoic acid receptor pathway by thiazolidinediones induces production of vascular endothelial growth factor/vascular permeability factor in OP9 adipocytes. Eur J Pharmacol 707: 95–103. [DOI] [PubMed] [Google Scholar]
- Laprairie RB, Bagher AM, Kelly ME, Denovan‐Wright EM (2015) Cannabidiol is a negative allosteric modulator of the type 1 cannabinoid receptor. Br J Pharmacol. doi: 10.1111/bph.13250 [Epub ahead of print] PubMed PMID: 26218440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo EH, Dalkara T, Moskowitz MA (2003). Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci 4: 399–415. [DOI] [PubMed] [Google Scholar]
- Luo Y, Yin W, Signore AP, Zhang F, Hong Z, Wang S et al (2006). Neuroprotection against focal ischemic brain injury by the peroxisome proliferator‐activated receptor‐gamma agonist rosiglitazone. J Neurochem 97: 435–448. [DOI] [PubMed] [Google Scholar]
- McPartland JM, Duncan M, Di Marzo V, Pertwee RG (2015). Are cannabidiol and Δ(9)‐tetrahydrocannabivarin negative modulators of the endocannabinoid system? A systematic review. Br J Pharmacol 172: 737–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mecha M, Feliu A, Inigo PM, Mestre L, Carrillo‐Salinas FJ, Guaza C (2013). Cannabidiol provides long‐lasting protection against the deleterious effects of inflammation in a viral model of multiple sclerosis: a role for A2A receptors. Neurobiol Dis 59: 141–150. [DOI] [PubMed] [Google Scholar]
- Mestre L, Docagne F, Correa F, Loria F, Hernangomez M, Borrell J et al (2009). A cannabinoid agonist interferes with the progression of a chronic model of multiple sclerosis by downregulating adhesion molecules. Mol Cell Neurosci 40: 258–266. [DOI] [PubMed] [Google Scholar]
- Miao Y, Liao JK (2014). Potential serum biomarkers in the pathophysiological processes of stroke. Expert Rev Neurother 14: 173–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Collins VE, Macleod MR, Donnan GA, Horky LL, van der Worp BH, Howells DW (2006). 1,026 experimental treatments in acute stroke. Ann Neurol 59: 467–477. [DOI] [PubMed] [Google Scholar]
- O'Sullivan SE, Kendall DA, Randall MD (2009). Time‐dependent vascular effects of endocannabinoids mediated by peroxisome proliferator‐activated receptor gamma (PPARgamma). PPAR Res 2009: 425289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP et al (2014). The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledge base of drug targets and their ligands. Nucl Acids Res 42 (Database Issue): D1098–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petitet F, Jeantaud B, Reibaud M, Imperato A, Dubroeucq MC (1998). Complex pharmacology of natural cannabinoids: evidence for partial agonist activity of delta9‐tetrahydrocannabinol and antagonist activity of cannabidiol on rat brain cannabinoid receptors. Life Sci 63: PL1–PL6. [DOI] [PubMed] [Google Scholar]
- Rajesh M, Mukhopadhyay P, Batkai S, Hasko G, Liaudet L, Drel VR et al (2007). Cannabidiol attenuates high glucose‐induced endothelial cell inflammatory response and barrier disruption. Am J Physiol Heart Circ Physiol 293: H610–H619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez SH, Heilman D, Morsey B, Potula R, Haorah J, Persidsky Y (2008). Activation of peroxisome proliferator‐activated receptor gamma (PPARgamma) suppresses Rho GTPases in human brain microvascular endothelial cells and inhibits adhesion and transendothelial migration of HIV‐1 infected monocytes. J Immunol 180: 1854–1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoch HJ, Fischer S, Marti HH (2002). Hypoxia‐induced vascular endothelial growth factor expression causes vascular leakage in the brain. Brain 125: 2549–2557. [DOI] [PubMed] [Google Scholar]
- Shiavon AP, Soares LM, Bonato JM, Milani H, Guimaraes FS, Weffort de Oliveira RM (2014). Protective Effects of Cannabidiol against hippocampal cell death and cognitive impairment induced by bilateral common carotid artery occlusion in mice. Neurotox Res 26: 307–316. [DOI] [PubMed] [Google Scholar]
- Song HY, Ju SM, Seo WY, Goh AR, Lee JK, Bae YS et al (2011). Nox2‐based NADPH oxidase mediates HIV‐1 Tat‐induced up‐regulation of VCAM‐1/ICAM‐1 and subsequent monocyte adhesion in human astrocytes. Free Radic Biol Med 50: 576–584. [DOI] [PubMed] [Google Scholar]
- Srivastava K, Shao B, Bayraktutan U (2013). PKC‐beta exacerbates in vitro brain barrier damage in hyperglycemic settings via regulation of RhoA/Rho‐kinase/MLC2 pathway. J Cereb Blood Flow Metab 33: 1928–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanden Berghe W, Vermeulen L, De Wilde G, De Bosscher K, Boone E, Haegeman G (2000). Signal transduction by tumor necrosis factor and gene regulation of the inflammatory cytokine interleukin‐6. Biochem Pharmacol 60: 1185–1195. [DOI] [PubMed] [Google Scholar]
- Wen AY, Sakamoto KM, Miller LS (2010). The role of the transcription factor CREB in immune function. J Immunol 185: 6413–6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yenari MA, Xu L, Tang XN, Qiao Y, Giffard RG (2006). Microglia potentiate damage to blood–brain barrier constituents: improvement by minocycline in vivo and in vitro. Stroke 37: 1087–1093. [DOI] [PubMed] [Google Scholar]
- Yusuf‐Makagiansar H, Anderson ME, Yakovleva TV, Murray JS, Siahaan TJ (2002). Inhibition of LFA‐1/ICAM‐1 and VLA‐4/VCAM‐1 as a therapeutic approach to inflammation and autoimmune diseases. Med Res Rev 22: 146–167. [DOI] [PubMed] [Google Scholar]
- Zhang ZG, Zhang L, Tsang W, Soltanian‐Zadeh H, Morris D, Zhang R et al (2002). Correlation of VEGF and angiopoietin expression with disruption of blood–brain barrier and angiogenesis after focal cerebral ischemia. J Cereb Blood Flow Metab 22: 379–392. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 The effect of 10 μM cannabidiol (CBD) applied after 4 hours OGD on BBB permeability from all experiments (n = 45) presented as a time course (A) and as the area under the curve of the whole experiment (B). Data are given as mean ± SEM. Statistical analysis conducted using two‐way ANOVA (A) or Student's t‐test (B). *** P<0.001, **** P<0.0001.
Figure S2 Cytokine/chemokine levels in the abluminal medium of BBB co‐culture inserts at 32 hours for IFNγ, IL‐10, IL‐1β, IL‐2, IL‐6, macrophage inflammatory protein (MIP)‐1α, MIP‐1β, TNF‐α and VEGF. Data are given as mean ± SEM. Statistical analysis conducted using one‐way ANOVA with post hoc analysis of selected pairs (vehicle versus CBD).
Figure S3 The effect of 28h treatment with increasing concentrations of cannabidiol (CBD) on HBMEC cell morphology. At the highest concentration, CBD appeared to cause cell damage, which was associated with a significant decrease in protein levels and significant increase in LDH release (see Figure 5).
Figure S4 No differences were observed after CBD treatment in the levels of phosphorylated JNK (A), ERK (B), Akt (c), NFκB (D), p70s6K (E), STAT3 (F) or STAT5 (G) in HBMECs treated for 28 h with CBD. Data are given as mean ± SEM. Statistical analysis conducted using one‐way ANOVA with Dunnett's post hoc test.
Figure S5 A. Light microscope images of human brain astrocytes treated by vehicle or increasing concentrations of CBD for 28 h in normal conditions. At the highest concentration, CBD appeared to cause cell damage, which was associated with a significant decrease in protein levels (B) and significant increase in LDH release (C). The levels of IL‐6 (D) and VCAM (E) in medium from astrocyte monocultures treated for 28 h with CBD. All data was normalised to total protein (BCA assay). Data are given as mean ± SEM. Statistical analysis conducted using one‐way ANOVA with Dunnett's post hoc test, n = 3‐6, ** P<0.01, *** P<0.001.
Supporting info item