

Abstract
Background and Purpose
Glucose‐dependent insulinotropic polypeptide (GIP) affects lipid, bone and glucose homeostasis. High‐affinity ligands for the GIP receptor are needed to elucidate the physiological functions and pharmacological potential of GIP in vivo. GIP(1–30)NH2 is a naturally occurring truncation of GIP(1–42). Here, we have characterized eight N‐terminal truncations of human GIP(1–30)NH2.
Experimental Approach
COS‐7 cells were transiently transfected with human GIP receptors and assessed for cAMP accumulation upon ligand stimulation or competition binding with 125I‐labelled GIP(1–42), GIP(1–30)NH2, GIP(2–30)NH2 or GIP(3–30)NH2.
Key Results
GIP(1–30)NH2 displaced 125I‐GIP(1–42) as effectively as GIP(1–42) (Ki 0.75 nM), whereas the eight truncations displayed lower affinities (Ki 2.3–347 nM) with highest affinities for GIP(3–30)NH2 and GIP(5–30)NH2 (5–30)NH2. Only GIP(1–30)NH2 (Emax 100% of GIP(1–42)) and GIP(2–30)NH2 (Emax 20%) were agonists. GIP(2‐ to 9–30)NH2 displayed antagonism (IC50 12–450 nM) and Schild plot analyses identified GIP(3–30)NH2 and GIP(5–30)NH2 as competitive antagonists (Ki 15 nM). GIP(3–30) NH2 was a 26‐fold more potent antagonist than GIP(3–42). Binding studies with agonist (125I‐GIP(1–30)NH2), partial agonist (125I‐GIP(2–30)NH2) and competitive antagonist (125I‐GIP(3–30)NH2) revealed distinct receptor conformations for these three ligand classes.
Conclusions and Implications
The N‐terminus is crucial for GIP agonist activity. Removal of the C‐terminus of the endogenous GIP(3–42) creates another naturally occurring, more potent, antagonist GIP(3–30)NH2, which like GIP(5–30)NH2, was a high‐affinity competitive antagonist. These peptides may be suitable tools for basic GIP research and future pharmacological interventions.
Abbreviations
- Bmax
maximal binding capacity
- DPP‐4
dipeptidyl peptidase 4
- DR
dose ratios
- GIP
glucose‐dependent insulinotropic polypeptide
- GLP‐1
glucagon‐like peptide‐1
- MW
molecular weight
- 7TM
seven‐transmembrane
Tables of Links
| TARGETS |
| GPCRs |
| GIP receptor |
| LIGANDS |
| GIP, glucose‐dependent insulinotropic polypeptide |
| GIP(6–30)NH2, GIP‐(6‐30) amide |
| GIP(7–30)NH2, GIP‐(7‐30) |
| GIP(1–30)NH2 |
| GIP(3–42) |
| GIP(3–30) |
| GIP(5–30) |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
Glucose‐dependent insulinotropic polypeptide (GIP) is a 42‐amino acid peptide hormone secreted from the K cells in the proximal small intestine. It appears to influence lipid, bone and glucose homeostasis (Kreymann et al., 1987; Baggio and Drucker, 2007). Together with glucagon‐like peptide‐1 (GLP‐1), GIP is responsible for the incretin effect that accounts for about 60% of the insulin response after ingestion of glucose (Nauck et al., 1986). Loss of the incretin effect is an early characteristic of type 2 diabetes (Holst et al., 2011), and exploitation of GLP‐1 physiology has led to the development of two novel drug classes (GLP‐1 analogues and dipeptidyl peptidase 4, DPP‐4, inhibitors) for the treatment of type 2 diabetes (Knop et al., 2009). The GLP‐1 receptor antagonist exendin(9–39) has been an essential research tool in the elucidation of GLP‐1 biology both in vivo and in vitro (Thorens et al., 1993; Al‐Sabah et al., 2014) and thereby also for the successful development of GLP‐1‐based pharmacological therapy.
Several antagonists of the GIP receptor have been reported with the majority of native GIP(1–42) truncations (Pederson and Brown, 1976; Moroder et al., 1978; Maletti et al., 1986; Sandberg et al., 1986; Rossowski et al., 1992; Gallwitz et al., 1993; Wheeler et al., 1995; Morrow et al., 1996; Tseng et al., 1996; Gelling et al., 1997; Tseng et al., 1999; Hinke et al., 2001; Gault et al., 2002a; Gault et al., 2002b; Hinke et al., 2004; Deacon et al., 2006; Irwin et al., 2006a; Irwin et al., 2006b; Jorgensen et al., 2007; Parthier et al., 2007; Kerr et al., 2011). An overview of the literature of GIP(1–42) truncations is presented in Supporting Information Table 1. Antibodies and a low MW compound with antagonistic properties for the GIP receptor have also been reported (Nakamura et al., 2012; Ravn et al., 2013). Importantly, none of these have been shown to act under human physiological conditions. Thus, an effective GIP receptor antagonist suitable for human studies remains to be found.
Like the GLP‐1 receptor, the GIP receptor belongs to B1 family of seven‐transmembrane (7TM) receptors, signalling through the Gαs pathway (Brubaker and Drucker, 2002), but several other intracellular messengers have also been reported in relation to GIP receptor activation (Kubota et al., 1997; Zhong et al., 2000; Mentlein, 2009; Campbell and Drucker, 2013). The GIP receptor is found in the pancreatic islet cells, osteocytes, cells in the gastrointestinal tract, adipocytes, and in the brain (Usdin et al., 1993). GIP has glucose stabilizing effects, potentiating glucose‐stimulated insulin secretion combined with glucagonotropic properties at low plasma glucose concentrations (Christensen et al., 2011). In humans, GIP may have anabolic effects on adipose tissue under hyperinsulinemic and hyperglycemic conditions (Asmar et al., 2014) and inhibits postprandial bone resorption (Nissen et al., 2014). In agreement with these findings, GIP receptor knockout mice are resistant to diet‐induced obesity, and crossing this mouse with the leptin mutant mouse, which is a known mouse model for obesity due to hyperphagia, led to reduction of weight gain by 23% (Miyawaki et al., 2002). Furthermore, GIP receptor knockout mice showed decreased bone mineral density, decreased bone quality including strength and cortical thickness and increased osteoclast activity (Gaudin‐Audrain et al., 2013; Mieczkowska et al., 2013).
The peptide secreted, GIP(1–42), is a substrate for the enzyme DPP‐4, which is a protease involved in the inactivation of numerous bioactive peptides (Mentlein, 1999). DPP‐4 cleaves the peptide bond following a proline or alanine at the penultimate position of the N‐terminus of peptides and, thus, in the case of GIP(1–42), produces the metabolite GIP(3–42) (Mentlein et al., 1993; Deacon, 2004). This degradation product is a GIP receptor antagonist in supraphysiological concentrations (Gault et al., 2002c, Deacon et al., 2006). Moreover, other truncated GIP variants have been reported to behave as antagonists including GIP(6–30)NH2 and GIP(7–30)NH2, both demonstrating high‐affinity and potent inhibition in vitro and decreasing GIP‐stimulated insulin secretion in vivo in rodents (Tseng et al., 1996; Gelling et al., 1997; Hinke et al., 2001). DPP‐4‐resistant palmitoylated human GIP(3–30), with a C‐terminal 9‐amino acid extension originating from exendin(1–39), was recently presented as a GIP receptor antagonist in both in vitro and mouse studies resulting in weight loss and improved insulin sensitivity (Pathak et al., 2015a). These findings are all consistent with the canonical paradigm for receptor activation in the 7TM B1 family, which includes the GIP receptor, demonstrating a pivotal role for the N‐terminus of the ligands.
The C‐terminally truncated and fully bioactive proGIP product, peptide GIP(1–30)NH2, has been shown to be formed in mice and in human intestine and pancreatic islets (Fujita et al., 2010a; Fujita et al., 2010b). Due to the lack of potent antagonists for the human GIP system and to the promising results in different species with truncated variants of GIP(1–42) (Pederson and Brown, 1976; Moroder et al., 1978; Maletti et al., 1986; Sandberg et al., 1986; Rossowski et al., 1992; Gallwitz et al., 1993; Wheeler et al., 1995; Morrow et al., 1996; Tseng et al., 1996; Gelling et al., 1997; Tseng et al., 1999; Hinke et al., 2001; Gault et al., 2002a; Hinke et al., 2004; Deacon et al., 2006; Irwin et al., 2006a; Parthier et al., 2007; Kerr et al., 2011) summarized in Supporting Information Table 1, we decided to focus on N‐terminally truncated forms of the backbone of the full agonist human GIP(1–30)NH2. GIP(1–30)NH2 and eight N‐terminally truncated variants, including GIP(2–30)NH2, GIP(3–30)NH2, GIP(4–30)NH2, GIP(5–30)NH2, GIP(6–30)NH2, GIP(7–30)NH2, GIP(8–30)NH2 and GIP(9–30)NH2 (Figure 1A), were tested as agonists and antagonists with respect to cAMP accumulation and competitive binding. To further explore the role of the C‐terminus, GIP(3–42) was characterized in parallel with GIP‐(3–30)NH2. Importantly, we found two novel, potent, competitive and high‐affinity antagonists among the GIP(1–30)NH2 truncations, and furthermore, we found that the absence of the C‐terminus is important for potent GIP receptor antagonism.
Figure 1.
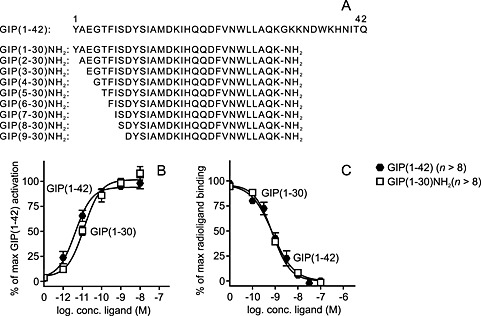
GIP(1–30)NH2 is a high‐affinity full agonist of the GIP receptor. (A) Alignment of the truncated GIP variants. Human native GIP(1–42) sequence was acquired from National Center for Biotechnology Information Protein Database. The GIP receptor was transiently transfected in COS‐7 cells and used for functional (B) and binding studies (C). (B) cAMP accumulation assay with increased concentrations of native GIP(1–42) and GIP(1–30)NH2, mean ± SEM, n = 8. (C) Competitive binding with the 125I‐GIP(1–42) radioligand displaced by GIP(1–42) and GIP(1–30)NH2. Data shown are means ± SEM, n = 13.
Methods
Cell line and transfection
COS‐7 cells were grown in 10% CO2 and at 37°C in DMEM 1885 supplemented with 10% FBS, 2 mM glutamine, 180 units·mL−1 penicillin and 45 g·mL−1 streptomycin. Transfection of COS‐7 cells was performed using the calcium phosphate precipitation method with chloroquine addition as previously described (Kissow et al., 2012).
cAMP assay
COS‐7 cells (30 000 cells per well) were seeded in 96‐well plates 1 day before transfection with human GIP receptor cDNA. 2 days after transfection, the cells were washed once with HEPES buffered saline and incubated with HEPES buffered saline and 0.5 mM IBMX for 30 min at 37°C. The various truncated GIP variants were added to the cells and incubated for 30 min at 37°C in order to test for intrinsic activity. To test for antagonism of a given GIP variant, the cells were preincubated for 10 min at 37°C with the GIP analogue followed by 20 min of incubation with GIP(1–42). The potency of the antagonists was determined from dose–response curves of the antagonist in the presence of a constant concentration of the GIP(1–42) corresponding to 50–80% of the maximal cAMP accumulation response (Emax) of GIP(1–42). For Schild analysis, various antagonist concentrations were added 10 min prior to GIP(1–42) dose–response curves. After ligand incubation, the HitHunter™ cAMP XS assay (an enzyme fragment complementation‐based assay; DiscoveRx, Birmingham, UK) was carried out according to the manufacturer's instructions. All experiments were made in duplicates and repeated at least 3 times. Luminescence was measured by PerkinElmer™ EnVision 2104 Multilabel Reader. In brief, the cells were lysed in the wells, the enzyme fragment‐cAMP‐antibody, an enzyme fragment, and the enzyme substrates were added followed by 1 h incubation at room temperature on shaker tray. The other enzyme fragment was added to the wells and incubated for 4 h on shaker tray followed by measurements of luminescence. The ligand‐induced cAMP competed with the binding of antibody to the first enzyme fragment and left the 2 fragments to fuse. The enzyme complex hydrolyzed the substrates and yielded luminescence. The number ‘n’ refers to individual experiments with separate transfections although from same cell line.
Competitive binding assay
COS‐7 cells were seeded in 96‐well plates 1 day after transfection with human GIP receptor cDNA. The number of cells seeded per well was selected to result in 5–10% specific binding of the added radioactive ligand (1000–5000 cells per well). Two days after transfection, cells were used for competition binding for 3 h at 4°C to inhibit receptor internalization using 6–10 pM per well of 125I‐GIP(1–42), 125I‐GIP(1–30)NH2, 125I‐GIP(2–30)NH2 or 125I‐GIP(3–30)NH2 as well as relevant amounts of unlabelled ligands in 50 mM HEPES buffer and pH 7.4, supplemented with 0.5% (w/v) BSA. After incubation for 3 h at 4°C, the cells were washed twice in ice‐cold binding buffer and lysed using 200 mM NaOH with 1% SDS for 30 min. Non‐specific binding was determined as the binding of radioligand to untransfected cells. All determinations were made in duplicates and all experiments repeated at least 3 times. The samples were analysed for radioactivity using a Wallac Wizard 1470 Gamma Counter (GMI Inc., Ramsey, MN, USA). The number ‘n’ refers to individual experiments with separate transfections although from same cell line.
Data analysis
IC50, EC50 and Kd/Ki values were determined by nonlinear regression. These, as well as maximal binding capacity (Bmax) values and Schild plot analysis, were carried out with the GraphPad Prism 6.0 software (GraphPad, San Diego, CA, USA) and Microsoft Excel™. Statistical analyses of two parameters (unpaired t‐tests) and multiple comparisons (one‐way ANOVAs) were also performed with GraphPad Prism 6.0. The calculations of Bmax and Ki values were based on the formula for one class of binding sites in homologous competition binding studies and the Cheng–Prusoff formula respectively (DeBlasi et al., 1989). Kd is the KD determined by homologous receptor binding. Dose ratios (DR) for the Schild analyses were based on the potency shift of the GIP(1–42) dose–response curve in absence or presence of a fixed antagonist concentration (DR = EC50 in presence of antagonist/EC50 in absence of antagonist). Schild plots were performed with log (DR‐1) (ordinate) and log(antagonist concentration) (abscissa) to estimate the slopes and Ki values (Lazareno and Birdsall, 1993).
Materials
Wild‐type human GIP receptor cDNA was purchased from OrigeneTM, Rockville, MA, USA (SC110906), and cloned into the pCMV‐Script vector. Human native GIP(1–42) was purchased from BachemTM, Bubendorf, Switzerland (H5645). All truncated GIP peptides were synthesized by Caslo™, Lyngby, Denmark, and based on the human GIP sequence. Porcine GIP(3–42) was custom synthesized by PolyPeptide Laboratories (WolfenBüttel, Germany). The peptides had a purity of more than 95% by HPLC analyses and an MS controlled MW. 125I‐labelled native GIP(1–42) was purchased from PerkinElmer Life Sciences, Skovlunde, Denmark (NEX402025UC). Human GIP(1–30)NH2, GIP(2–30)NH2 and GIP(3–30)NH2 were 125I labelled using the standard stoichiometric chloramine T method as described previously (Holst and Bersani, 1991). The labelled peptides were purified by HPLC.
Results
GIP(1–30)NH2 is a full GIP receptor agonist with high affinity equal to native GIP(1–42)
To establish the role of the C‐terminus for agonism in the human GIP system, we first measured cAMP responses to human GIP(1–42) and human GIP(1–30)NH2 in transiently transfected COS‐7 cells expressing the human GIP receptor (Figure 1). GIP(1–30)NH2 was a full agonist on the GIP receptor with a high potency (EC50) of 11.2 pM [logEC50 −10.95 ± 0.11], compared with the 6.0 pM [logEC50 −11.21 ± 0.16] of GIP(1–42), and with the same efficacy as GIP(1–42), consistent with earlier studies (Fujita et al., 2010a; Gault et al., 2011). Binding studies were performed with 125I‐GIP(1–42) as the radioligand in the same cellular background. Truncation of the full length GIP(1–42) peptide at the 30‐position did not change the affinity to the GIP receptor and, thus, resulted in affinities (IC50) of 0.89 and 0.67 nM for GIP(1–30)NH2 and GIP(1–42) respectively (Figure 1). Thus, GIP(1–30)NH2 displayed the same potency, efficacy and affinity for the human GIP receptor as GIP(1–42).
The N‐terminus is essential for high‐affinity binding
To study the role of the N‐terminus of human GIP(1–30)NH2, the affinity of the eight N‐terminally truncated peptides was compared with that of GIP(1–30)NH2 in transiently transfected COS‐7 cells using 125I‐GIP(1–42) as radioligand (Figure 2). Truncation resulted in decreased affinity with a tendency towards length dependency, with a span from 2.3‐fold to 347‐fold decrease in affinity compared with GIP(1–30)NH2. GIP(3–30)NH2 followed by GIP(5–30)NH2 displayed the highest affinities, while GIP(9–30)NH2 and GIP(6–30)NH2 had more than 300‐fold lower affinities compared with GIP(1–30)NH2. Taken together, this emphasizes the importance of the N‐terminus for receptor binding.
Figure 2.
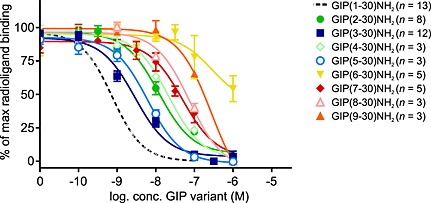
GIP(3–30)NH2 and GIP(5–30)NH2 display the highest affinity among the eight truncated GIP variants. The binding of 125I‐GIP(1–42) to transiently transfected COS‐7 cells with the GIP receptor was tested in the presence of increasing amounts of GIP(1–30)NH2, GIP(3–30)NH2, GIP(5–30)NH2, GIP(2–30)NH2, GIP(4–30)NH2, GIP(7–30)NH2, GIP(8–30)NH2, GIP(9–30)NH2 or GIP(6–30)NH2. Data shown are means ± SEM.
GIP(2–30)NH2 is a partial agonist, and GIP(3‐ to 9–30)NH2 are antagonists of the GIP receptor
We measured cAMP accumulation in COS‐7 cells, transiently transfected with the human GIP receptor, after incubation with each of the GIP variants (Figure 3). Removal of the first amino acid from GIP(1–30)NH2 created GIP(2–30)NH2, which is a weak partial agonist with an efficacy of 20% compared with GIP(1–30)NH2 and a potency of 3.7 nM [logEC50 −8.43 ± 0.33, n = 8], which is >3000‐fold lower than GIP(1–30)NH2. Removal of the second amino acid completely eliminated intrinsic activity (Figure 3A), a pattern that was also seen for the remaining truncations (Figure 3B). To determine whether the inactive forms had antagonistic properties, increasing concentrations of the GIP variants were added to a submaximal (50–80%) activation by GIP(1–42). All were able to inhibit the cAMP response induced by GIP(1–42) (Figure 3C and D). The most potent antagonists were GIP(3–30)NH2 and GIP(5–30)NH2 with IC50 of 11.8 and 11.9 nM, respectively (Table 1), in agreement with their high binding affinities. Similar to the binding studies, the shortest GIP variant, GIP(9–30)NH2, had the lowest antagonistic potency with a 38‐fold right shift compared with GIP‐(3–30)NH2.
Figure 3.
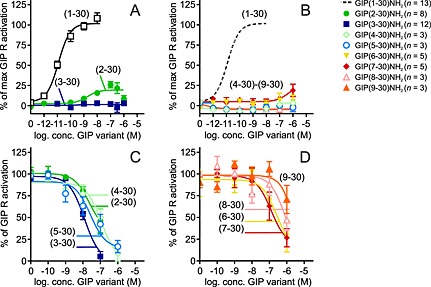
GIP(3–30) and GIP(5–30) are the most potent GIP receptor antagonists. cAMP accumulation in transiently transfected COS‐7 cells with GIP receptor. (A, B) Ligand dose–response stimulated cAMP accumulation. Data shown are means ± SEM. (C, D) Dose–response curves of antagonists inhibited a constant amount of native GIP(1–42) corresponding to 50–80% of max receptor activation. Data shown are means ± SEM.
Table 1.
Affinity and inhibitory potencies of the GIP variants
| Competitive binding | cAMP accumulation | ||||||
|---|---|---|---|---|---|---|---|
| logIC50 ± SEM | Ki (nM) | Fold | n | logIC50 ± SEM | IC50 (nM) | n | |
| GIP(1–30)NH2 | −9.05 ± 0.02 | 0.89 | 1.0 | 13 | — | — | — |
| GIP(2–30)NH2 | −7.85 ± 0.04 | 14.3 | 16 | 10 | −7.66 ± 0.1 | 21.7 | 4 |
| GIP(3–30)NH2 | −8.63 ± 0.04 | 2.3 | 2.6 | 12 | −7.93 ± 0.04 | 11.8 | 6 |
| GIP(4–30)NH2 | −7.67 ± 0.02 | 21.5 | 24 | 3 | −6.97 ± 0.4 | 108 | 4 |
| GIP(5–30)NH2 | −8.23 ± 0.05 | 5.9 | 6.6 | 3 | −7.92 ± 0.4 | 11.9 | 4 |
| GIP(6–30)NH2 | −6.46 ± 0.09 | 347 | 391 | 10 | −6.47 ± 0.6 | 342 | 4 |
| GIP(7–30)NH2 | −7.58 ± 0.08 | 26 | 30 | 9 | −6.86 ± 0.4 | 137 | 7 |
| GIP(8–30)NH2 | −7.10 ± 0.04 | 79 | 89 | 3 | −6.88 ± 0.5 | 133 | 5 |
| GIP(9–30)NH2 | −6.51 ± 0.08 | 307 | 345 | 3 | −6.35 ± 0.6 | 450 | 4 |
GIP(3–30)NH2 and GIP(5–30)NH2 are competitive antagonists
A Schild analysis was performed for the four most potent antagonists, in addition to the previously described antagonists GIP(6–30)NH2 and GIP(7–30)NH2 (Gelling et al., 1997; Tseng et al., 1999; Hinke et al., 2001). This analysis determines whether an antagonist acts competitively and is illustrated by the Schild plot. A straight line with a Hill slope of 1.0 indicates competitive antagonism. The antagonists were added in various constant concentrations to the dose–response curves of GIP(1–42) (Figure 4). All six antagonists were able to right shift the GIP(1–42) dose–response curve with no changes in efficacy. However, only GIP(3–30)NH2 and GIP(5–30)NH2 act as pure competitive antagonists judged by a straight line with a slope of 1 (inserts in Figure 4). These 2 ligands displayed slopes of 0.93 ± 0.02 and 1.1 ± 0.04, respectively, while the slopes for GIP(2–30)NH2, GIP(4–30)NH2, GIP(6–30)NH2 and GIP(7–30)NH2 were 0.49 ± 0.14, 0.75 ± 0.02, 0.38 ± 0.13 and 0.17 ± 0.03 respectively (Figure 4). The lack of ability to compete equally with the agonist could indicate an allosteric component in the antagonistic properties of these ligands. The X‐intercept or pA2‐value of the Schild plot corresponds to the affinity constant of the antagonist if the Hill slope equals 1. For the 2 competitive antagonists GIP(3–30)NH2 and GIP(5–30)NH2, the pA2‐values were 14.9 and 15.2 nM, respectively, thus in the same range as the Ki determined from the binding studies (2.3 and 5.9 nM respectively). In summary, this analysis identified GIP(3–30)NH2 and GIP(5–30)NH2 as high‐affinity competitive GIP receptor antagonists.
Figure 4.
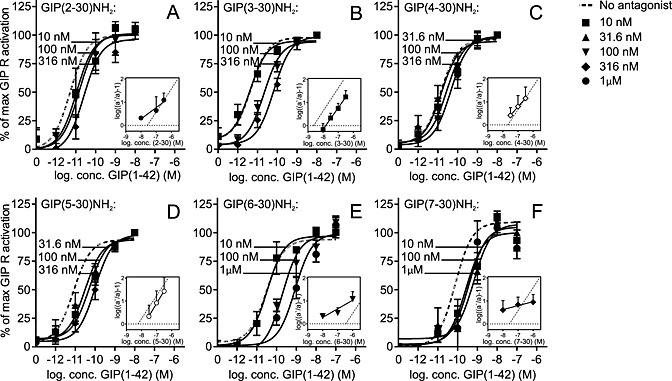
Of the six antagonists, only GIP(3–30)NH2 and GIP(5–30)NH2 are the competitive antagonists. GIP(1–42)‐mediated cAMP accumulation assayed for transiently transfected COS‐7 cells with the GIP receptor in the absence of and with increasing concentrations of either GIP(2–30)NH2, GIP(3–30)NH2, GIP(4–30)NH2, GIP(5–30)NH2, GIP(6–30)NH2 or GIP(7–30)NH2. The corresponding Schild plot is presented with a comparison with a linear regression with a slope of 1.0 and the X‐intercept of Ki for the antagonist. GIP(2–30)NH2 (A, n = 4), GIP(3–30)NH2 (B, n = 6), GIP(4–30)NH2 (C, n = 3), GIP(5–30)NH2 (D, n = 4), GIP(6–30)NH2 (E, n = 3) and GIP(7–30)NH2 (F, n = 4). Data shown are means ± SEM.
The functionalities of the ligands reflect the binding properties
The N‐terminal truncations of GIP(1–30)NH2 had a span in affinities (Ki) from 1 to 350 nM (Figure 2 and Table 1) and, concomitantly, displayed different pharmacodynamics with both competitive and non‐competitive antagonistic properties (Figures 3, 4). To further analyse the receptor interaction of these variants, we performed homologous competitive binding studies with 125I‐GIP(1–30)NH2, 125I‐GIP(2–30)NH2 and 125I‐GIP(3–30)NH2 as radioligands (representing a full agonist, a partial agonist and a competitive antagonist respectively). The Kd values for GIP(1–30)NH2, GIP(2–30)NH2 and GIP(3–30)NH2 obtained from the homologous binding experiments (Figure 5 and Table 2) were in the same range as the Ki values obtained in the heterologous binding experiments using 125I‐GIP(1–42) as radioligand (Table 1). However, minor, yet significant, changes were observed upon a closer look at the affinities, as higher affinities were observed when GIP(1–30)NH2 and GIP(2–30)NH2 competed with their own iodinated versions (homologous binding), compared with when they competed with 125I‐GIP(1–42) (heterologous binding) (P = 0.012 and P = 0.0031, respectively; Figure 5). Thus, the lack of C‐terminus decreased the ability of GIP(1–30)NH2 and GIP‐(2–30)NH2 to compete with the full‐length agonist GIP(1–42) for the GIP receptor. In contrast, the N‐terminally truncated antagonist GIP(3–30)NH2 was able to displace the homologous radioligand with the same affinity as the full agonist 125I‐GIP‐(1–42) radioligands (P = 0.45; Figure 5). The Bmax was calculated from the homologous binding studies (DeBlasi et al., 1989) and uncovered significantly more binding sites for the antagonists compared with the two agonists (Figure 5), which illustrates the general property of antagonists to stabilize several inactive receptor confirmations, while agonists preferentially bind to the active confirmation(s) (Rosenkilde et al., 1994).
Figure 5.
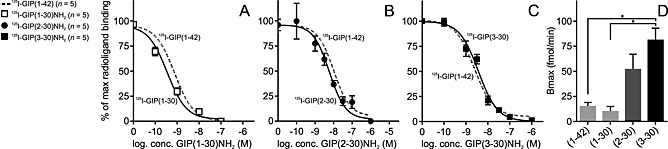
The homologous binding curves are equivalent to the heterologous binding studies with native 125I‐GIP(1–42) radioligand. (A–C) Transiently transfected COS‐7 cells with the GIP receptor were used in homolog competitive binding studies with 125I‐GIP(1–30)NH2, 125I‐GIP(2–30)NH2 and 125I‐GIP(3–30)NH2 and heterologous binding studies with 125I‐GIP(1–42), mean ± SEM. (D) Bmax values calculated from the homologous binding curves for GIP(1–42), GIP(1–30)NH2, GIP(2–30)NH2 and GIP(3–30)NH2. Data shown are means ± SEM, n = 5. Significance determined by multiple comparisons (one‐way ANOVA).
Table 2.
Homologous and heterologous binding studies
| 125I‐GIP(1–30)NH2 | 125I‐GIP(2–30)NH2 | 125I‐GIP(3–30)NH2 | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| log (IC50) | ±SEM | IC50 (nM) | Fold change GIP(1–30)NH2 | n | log (IC50) | ±SEM | IC50 (nM) | Fold change GIP(1–30)NH2 | n | log (IC50) | ±SEM | IC50 (nM) | Fold change GIP(1–30)NH2 | n | |
| GIP(1–42)NH2 | −9.24 | 0.19 | 0.58 | 1.9 | 3 | −9.36 | 0.087 | 0.43 | 0.9 | 3 | −8.97 | 0.0015 | 1.07 | 0.6 | 3 |
| GIP(1–30)NH2 | −9.52 | 0.16 | 0.30 | 1.0 | 5 | −9.32 | 0.482 | 0.48 | 1.0 | 3 | −8.78 | 0.063 | 1.7 | 1.0 | 3 |
| GIP(2–30)NH2 | −7.59 | 0.18 | 26 | 84.3 | 4 | −8.57 | 0.28 | 2.7 | 10.5 | 5 | −8.11 | 0.065 | 7.7 | 4.6 | 4 |
| GIP(3–30)NH2 | −8.35 | 0.071 | 4.4 | 14.5 | 4 | −9.12 | 0.20 | 0.76 | 1.6 | 3 | −8.47 | 0.12 | 3.4 | 2.0 | 5 |
| GIP(6–30)NH2 | −5.97 | 0.066 | 1065 | 3502 | 5 | −6.47 | 0.28 | 340 | 707 | 4 | −6.43 | 0.26 | 370 | 223 | 4 |
| GIP(7–30)NH2 | −7.43 | 0.25 | 37 | 120.9 | 5 | −7.54 | 0.23 | 29 | 60.6 | 5 | −7.68 | 0.16 | 21 | 12.7 | 5 |
The binding properties were further elucidated through heterologous binding studies with 125I‐GIP(1–30)NH2, 125I‐GIP(2–30)NH2 and 125I‐GIP(3–30)NH2 displaced by GIP(1–42), GIP(1–30)NH2, GIP(2–30)NH2 and GIP(3–30)NH2 and the previously described GIP(6–30)NH2 and GIP(7–30)NH2 (Supporting Information Figure 1 and Table 2). Again, the agonists GIP(1–30)NH2 and GIP(1–42) displaced the agonist radioligand 125I‐GIP(1–30)NH2 most efficiently, while their affinities decreased in competition with the radiolabeled antagonists. The opposite was observed for the antagonists that displaced the partial agonist 125I‐GIP(2–30)NH2 and the antagonist 125I‐GIP(3–30)NH2 radioligand with highest affinities. Thereby, when looking at apparent affinities, the largest effects of increased truncation of GIP(1–30)NH2 were observed with the agonist as radioligand with >3000‐fold decrease in affinity of GIP(7–30)NH2 compared with GIP(1–30)NH2 measured with agonist radioligand and only 223‐fold decrease when measured with 125I‐GIP(3–30)NH2 as radioligand. This pattern was observed for all four antagonists (Table 2).
The C‐terminal part of GIP acts as a negative regulator of the antagonist action of GIP(3–42)
The identification of GIP(3–30)NH2 as the most potent antagonist prompted us to compare it with GIP(3–42) in order to directly determine the impact of the C‐terminal amino acids 31 through 42. We also included the porcine GIP(3–42), representing a low‐potent antagonist on the human GIP receptor in vitro, with no ability to antagonize porcine GIP(1–42)‐mediated insulin secretion in pigs at physiological concentrations (Deacon et al., 2006). Porcine GIP(3–42) has arginine in position 18 and serine in position 34, whereas the human sequence has histidine and asparagine respectively (Figure 6). Like GIP(3–30)NH2 (Figure 3), neither of the GIP(3–42) variants had any intrinsic agonistic activity in cAMP accumulation assay (data not shown, n = 3), but both were able to antagonize submaximal (50–80%) human GIP(1–42)‐induced activation (Figure 6). Importantly, human GIP(3–42) was remarkably less potent than human GIP(3–30)NH2 (26‐fold lower potency; Figure 6), and 1 μM of this resulted in only 4.9‐fold shift in the dose–response curve of human GIP(1–42) compared with 247‐fold for human GIP‐(3–30)NH2 (Figure 6). The porcine variant displayed higher potency compared with human GIP(3–42), yet not as high as human GIP(3–30)NH2. Thus, the C‐terminus has a functional role as its absences improve the antagonistic properties in GIP(3–30)NH2 compared with GIP(3–42).
Figure 6.
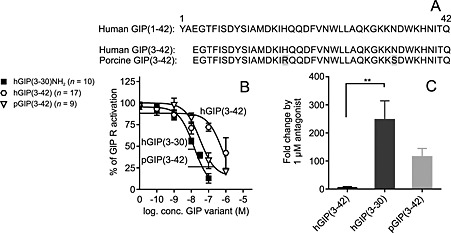
Human GIP(3–42) is a low‐potent antagonist on the human GIP receptor compared with human GIP(3–30)NH2 and porcine GIP(3–42). (A) Alignment of the truncated GIP variants. Human and porcine GIP(1–42) sequence was acquired from National Center for Biotechnology Information Protein Database. The human GIP receptor transiently transfected in COS‐7 cells was used in cAMP accumulation assay (B and C). (B) Dose–response curves of antagonists inhibited a constant amount of native GIP(1–42) corresponding to 50–80% of max receptor activation. Data shown are means ± SEM. (C) Fold change in potency of human GIP(1–42) by 1 μM antagonist. The bars display the mean fold change ± SEM, n = 4. Significance determined by multiple comparisons (one‐way ANOVA).
Discussion and conclusion
In this pharmacological study of truncated human GIP variants, we evaluated GIP(2‐ to 9–30)NH2 as antagonists of the human GIP receptor. GIP(1–30)NH2 was a full agonist with affinity equal to the full‐length native GIP(1–42), and GIP(2–30)NH2 acted as a low‐potent partial agonist and an antagonist. Both the affinities and the antagonistic properties appeared to decrease by the truncation length. GIP(3–30)NH2 and GIP(5–30)NH2 were high‐affinity competitive antagonists of the human GIP receptor, whereas the rest displayed lower affinities and more complex antagonism. Additionally, when comparing human GIP(3–30)NH2 with GIP(3–42), the C‐terminus (position 31–42) was found to dampen the antagonist properties of GIP(3–42).
Many truncated GIP variants have been presented, but GIP(3–30)NH2 and GIP(5–30)NH2 seem superior
GIP has been truncated into numerous variants and characterized in various species (Figure 7 and Supporting Information Table 1). None of the previous studies have focused on the pure human system, and recently, the species differences have been shown to influence the pharmacology fundamentally (Sparre‐Ulrich et al., 2015). Several studies have established GIP(1–30)NH2 as a full agonist with equal potency to GIP(1–42) primarily using the rat system (Rossowski et al., 1992; Gallwitz et al., 1993; Morrow et al., 1996; Tseng et al., 1996; Hinke et al., 2001). In line with this, we find GIP(1–30)NH2 to be a high‐affinity full agonist of the human GIP receptor. Eight N‐terminally truncated GIP(1–30)NH2 variants have previously been published: GIP(6–30)NH2, GIP(7–30)NH2, GIP(10–30), GIP(15–30)NH2, GIP(16–30)NH2, GIP(17–30)‐NH2, GIP(19–30)NH2 and GIP(21–30)NH2 (Maletti et al., 1986; Wheeler et al., 1995; Morrow et al., 1996; Tseng et al., 1996; Gelling et al., 1997; Tseng et al., 1999; Hinke et al., 2001; Gault et al., 2002a). The porcine GIP(6–30)NH2 has been presented as a high‐affinity and potent rat GIP receptor antagonist [IC50 of 3.1 nM in competition with porcine 125I‐GIP(1–42)] with 58% reduction in human GIP(1–42)‐induced cAMP response by 100 nM porcine GIP(6–30)NH2 (Gelling et al., 1997). In a recent study, GIP(6–30)Cex‐K40[Pal] inhibited human GIP(1–42)‐induced cAMP production from the human GIP receptor (IC50 3.1 nM) and insulin release (IC50 49 pM) in a rat beta cell line and enhanced insulin sensitivity and improved glucose tolerance in diabetic mice (Pathak et al., 2015b). On the human GIP receptor, we observed GIP(6–30)NH2 to have the lowest affinity of the truncated GIP(1–30)NH2 variants and poor antagonistic properties. This is not in line with previous studies, but the pharmacological differences between rodent and human GIP systems are not yet clarified (Sparre‐Ulrich et al., 2015). In contrast to the findings for GIP(6–30)NH2, our results of the human GIP(7–30)NH2 are in line with previous studies using the porcine GIP(7–30)NH2 on the rat GIP receptor (Tseng et al., 1996; Gelling et al., 1997; Tseng et al., 1999; Hinke et al., 2001). We found GIP(3–30)NH2 and GIP(5–30)NH2 to be superior to GIP(6–30)NH2 and GIP‐(7–30)NH2 and to be the only competitive antagonists. Recently, palmitoylated human GIP(3–30)NH2 extended with the nine last amino acids from exendin(1–39) was published as a GIP receptor antagonist in mice with significant effects on weight loss and improved glycemic control and insulin sensitivity (Pathak et al., 2015a). Our results from the human receptor are consistent with this. Our systematic truncation approach revealed that the potency and affinity of the antagonists decreased with the length of the truncations (Figures 1, 2, 3, 4, 8) and that amino acids 2–5 in human GIP(1–30)NH2 are important for potent inhibition. A possible explanation for the decreased antagonist properties of GIP(6‐ to 9–30)NH2 could be found in changes of the secondary structure (Figure 8). GIP(1–42) has an α‐helix initiated by amino acid Thr‐5 as the helix‐capping residue (Parthier et al., 2007). Removal of this threonine in GIP(6–30)NH2 decreases the antagonism (IC50) by 29‐fold and the affinity (Ki) by 59‐fold, which could be due to an impaired ability to attain this important secondary structure. The fact that GIP(6–30)‐NH2 displays the lowest affinity and potency indicates that not only the disruption of the α‐helix initiation by the lack of position 5 [which is also missing in the truncations GIP(7‐ to 9–30)NH2] but also the exact position of the truncation and N‐terminally exposed amino acids is essential for proper receptor binding.
Figure 7.
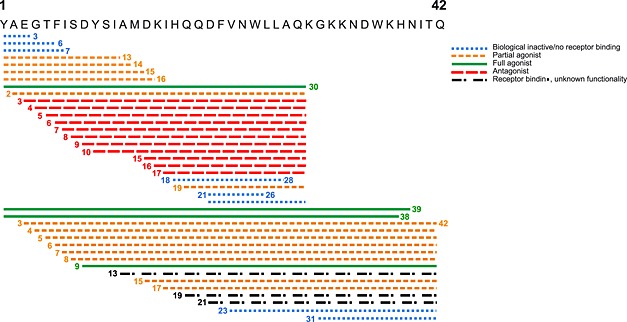
Overview of truncated GIP variants studied in vitro or in vivo in rodents. The truncated GIP variants of various species are biological inactive/no receptor binding, partial agonist, full agonist, antagonist or receptor binding but unknown functionality. This figure is based on the studies referred in supplementary Table 1.
Figure 8.
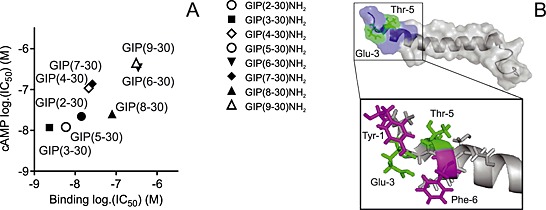
Correlation of affinity and antagonistic potency (A) and structure of the N‐terminus of GIP (B). (A) The correlation of calculated affinities (binding log.IC50) and antagonistic potencies (cAMP log.IC50) plotted for the eight GIP receptor antagonists (data from Table 1). (B) The published structure (Parthier et al., 2007) of the native GIP(1–42) peptide with amino acids 1–9 in blue, Glu3 and Thr5 in green and Tyr1 and Phe6 in pink.
The N‐terminus has a pivotal role in GIP receptor activation
We have previously demonstrated that porcine GIP(3–42) was unable to induce cAMP accumulation (Deacon et al., 2006), underlining the necessity of the first two amino acids of GIP(1–42) for agonistic properties. Many 7TM receptors of family A (for instance, the chemokine and C5a receptors) and most B1 receptors are thought to be activated in (at least) two steps. Firstly, the ligand binds to the extracellular domain of the receptor, and secondly, the N‐terminus of the ligand interacts with transmembrane domains, resulting in receptor activation (Clark‐Lewis et al., 1995; Hjorth and Schwartz, 1996; Vilardaga et al., 2011; Thiele and Rosenkilde, 2014). Previously, alanine screening of human GIP identified Tyr1 to be crucial for GIP receptor‐mediated insulin secretion in vitro (Alaña et al., 2006). In line with this, NMR and molecular modelling described the interaction of Tyr1 with multiple amino acids of the transmembrane domains of the GIP receptor (Malde et al., 2007; Yaqub et al., 2010). Ala2 is thought to both interact with residues of TM3 and participate in hydrogen bonding with Thr5, which is thought to be important for receptor activation (Yaqub et al., 2010). Accordingly, our study demonstrates that Tyr1 and Ala2 are pivotal for GIP receptor activation by the binding to and stabilization of the active conformations as seen by the low efficacy of GIP‐(2–30)NH2 and no intrinsic activity of GIP(3–30)NH2 (and of subsequent truncations). The differences in Bmax values and the radioligand‐dependent affinities (agonists compete best with agonists and antagonists with antagonists; Supporting Information Figure 1) underline the structural alterations in the recognition and conformational constraining of active and inactive GIP receptor conformations. A similar phenomenon with changes in apparent affinities determined by the choice of radioligand has previously been described, for example, in the neurokinin system (Rosenkilde et al., 1994) and in the chemokine system (Jensen et al., 2008). Apart from a surprising finding for GIP(19–30)NH2, which seems to be a weak partial agonist in vivo and in vitro (Figure 7) (Maletti et al., 1986; Morrow et al., 1996; Hinke et al., 2001), the N‐terminal truncations of GIP(1–30)NH2 beyond the first two amino acids do not activate the GIP receptor, emphasizing the essential role of the Tyr1 and Ala2 for GIP receptor activation in the GIP(1–30)NH2 scaffold (absence of amino acid 31 to 42).
The effects of the C‐terminus for receptor activation are unmasked by N‐terminal GIP truncations
In a previous study, consecutive N‐terminal truncations of human GIP(1–42) were characterized in vitro in the rat GIP receptor and in leptin mutant mice (Kerr et al., 2011). In contrast to our findings for the similar truncations in the human GIP(1–30)NH2 scaffold, GIP(3‐ to 8–42) displayed partial agonism, and GIP(9–42) acted as a full GIP receptor agonist in vitro. Only GIP(8–42), with a Emax of 10% in terms of cAMP release, was able to antagonize the action of GIP(1–42) in vivo with decreased insulin secretion and increased plasma glucose in an acute setting. In line with our results with GIP(5–30)NH2, GIP(5–42) was the most potent antagonist in vitro; however, the length‐dependent functional changes described here for the human GIP(1–30)NH2 variants were not observed in the truncated GIP(1–42) variants (Kerr et al., 2011). All together, this indicates that the C‐terminus is important for intrinsic activity, and even though the activities of GIP(1–30)NH2 and GIP(1–42) are similar, the N‐terminally truncated GIP variants reveal that the C‐terminus (position 31–42) improves intrinsic activity. It is, however, also possible that species differences may play a role as the rat system was used for the GIP(1–42) truncations and we used the human system for the GIP(1–30)NH2 truncations.
Improved antagonistic properties in the absence of the C‐terminus of GIP
Our study demonstrates that although GIP(1–30)NH2 and GIP(1–42) were equally potent and efficient agonists, GIP(1–30)NH2 displayed a threefold lower apparent affinity in competition with 125I‐GIP(1–42) as compared with 125I‐GIP(1–30)NH2. In contrast, GIP(1–42) displaced both radioligands with the same affinity, which together with the same amount of binding sites (Bmax) for the two agonists indicate largely overlapping binding sites. Interestingly, in terms of (antagonistic) functionality of the N‐terminal truncated variants, the C‐terminus had a huge negative impact, as GIP(3–30)NH2 was a 26‐fold more potent antagonist compared with GIP(3–42). Taken together, our study combined with previous studies (Gallwitz et al., 1993; Wheeler et al., 1995; Morrow et al., 1996; Tseng et al., 1996; Hinke et al., 2001; Kerr et al., 2011) indicates that the C‐terminus (position 31–42) has only a minute impact on the binding and no impact on the agonistic properties of GIP (with preserved N‐terminus), whereas it affects the functionality of the N‐terminally truncated GIP variants. Thus, the presence of the C‐terminus enhances the intrinsic activity (confers agonism) to GIP(3‐ to 8–42) (Kerr et al., 2011), while the absence of it in our GIP(3‐ to 9–30)NH2 variants improves the antagonism. In other words, the best antagonism was obtained in the absence of the C‐terminus.
GIP(3–30)NH2 may be a suitable research tool for the human GIP system
We identified GIP(3–30)NH2 and GIP(5–30)NH2 as the most potent and efficacious antagonists for the human GIP receptor (Figure 7). They were also the only truncated GIP variants with true competitive properties. As GIP(1–30)NH2 is expressed in human pancreatic α‐cells and in specific enteroendocrine cells (Fujita et al., 2010b), the presence of GIP(1–30)NH2 in human plasma is highly likely. Due to the efficient DPP‐4 degradation of N‐terminally intact GIP, GIP(3–30)NH2 should therefore also be present, similar to what has been observed for GIP(3–42) (Deacon et al., 2006). Given the high and competitive antagonistic potency of GIP(3–30)NH2 and its putative presence in the body, which would decrease the risk of immune reactions, GIP(3–30)NH2 has the potential to be a safe and efficient GIP receptor antagonist suitable for human studies. Our discovery could therefore contribute to a better elucidation of the human GIP system including the determination of GIP's contribution to the human incretin effect and extrapancreatic functions of GIP. Ultimately, the promising characteristics of GIP(3–30)NH2 and GIP(5–30)NH2 can lead to the development of GIP receptor antagonists as a future therapeutic possibility.
Author contributions
L. S. H. contributed to the study design, conducted the experiments and wrote and edited the manuscript. A. H. S.‐U. conducted the experiments and reviewed and edited the manuscript. M. C., F. K. K., B. H. and J. J. H. contributed to the study design and reviewed and edited the manuscript. M. M. R. contributed to the study design and wrote and edited the manuscript.
Conflict of interest
Authors declare that they have no conflict of interest.
Supporting information
Supporting info item
Supporting info item
Hansen, L. S. , Sparre‐Ulrich, A. H. , Christensen, M. , Knop, F. K. , Hartmann, B. , Holst, J. J. , and Rosenkilde, M. M. (2016) N‐terminally and C‐terminally truncated forms of glucose‐dependent insulinotropic polypeptide are high‐affinity competitive antagonists of the human GIP receptor. British Journal of Pharmacology, 173: 826–838. doi: 10.1111/bph.13384.
References
- Al‐Sabah S, Al‐Fulaij M, Ahmed HA (2014). Selectivity of peptide ligands for the human incretin receptors expressed in HEK‐293 cells. Eur J Pharmacol 741: 311–315. [DOI] [PubMed] [Google Scholar]
- Alaña I, Parker JC, Gault VA, Flatt PR, O'harte FPM, Malthouse JP et al (2006). NMR and alanine scan studies of glucose‐dependent insulinotropic polypeptide in water. Journal of Biological Chemistry 281: 16370–16376. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al (2013). The Concise Guide to PHARMACOLOGY 2013/14: G Protein‐Coupled Receptors. Br J Pharmacol 170: 1459–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asmar M, Simonsen L, Arngrim N, Holst JJ, Dela F, Bulow J (2014). Glucose‐dependent insulinotropic polypeptide has impaired effect on abdominal, subcutaneous adipose tissue metabolism in obese subjects. Int J Obes 38: 259–265. [DOI] [PubMed] [Google Scholar]
- Baggio LL, Drucker DJ (2007). Biology of incretins: GLP‐1 and GIP. Gastroenterology 132: 2131–2157. [DOI] [PubMed] [Google Scholar]
- Brubaker PL, Drucker DJ (2002). Structure‐function of the glucagon receptor family of G protein‐coupled receptors: the glucagon, GIP, GLP‐1, and GLP‐2 receptors. Recept Channel 8: 179–188. [PubMed] [Google Scholar]
- Campbell JE, Drucker DJ (2013). Pharmacology, physiology, and mechanisms of incretin hormone action. Cell Metab 17: 819–837. [DOI] [PubMed] [Google Scholar]
- Christensen M, Vedtofte L, Holst JJ, Vilsboell T, Knop FK (2011). Glucose‐dependent insulinotropic polypeptide: a bifunctional glucose‐dependent regulator of glucagon and insulin secretion in humans. Diabetes 60: 3103–3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark‐Lewis I, Kim KS, Rajarathnam K, Gong JH, Dewald B, Moser B et al (1995). Structure–activity relationships of chemokines. J Leukoc Biol 57: 703–711. [DOI] [PubMed] [Google Scholar]
- Deacon CF (2004). Circulation and degradation of GIP and GLP‐1. Horm Metab Res 36: 761–765. [DOI] [PubMed] [Google Scholar]
- Deacon CF, Plamboeck A, Rosenkilde MM, De HJ, Holst JJ (2006). GIP‐(3–42) does not antagonize insulinotropic effects of GIP at physiological concentrations. Am J Physiol Endocrinol Metab 291: E468–E475. [DOI] [PubMed] [Google Scholar]
- Deblasi A, O'reilly K, Motulsky HJ (1989). Calculating receptor number from binding experiments using same compound as radioligand and competitor. Trends Pharmacol Sci 10: 227–229. [DOI] [PubMed] [Google Scholar]
- Fujita Y, Asadi A, Yang GK, Kwok YN, Kieffer TJ (2010a). Differential processing of pro‐glucose‐dependent insulinotropic polypeptide in gut. Am J Physiol Gastrointest Liver Physiol 298: G608–G614. [DOI] [PubMed] [Google Scholar]
- Fujita Y, Wideman RD, Asadi A, Yang GK, Baker R, Webber T et al (2010b). Glucose‐dependent insulinotropic polypeptide is expressed in pancreatic islet alpha‐cells and promotes insulin secretion. Gastroenterology 138: 1966–1975. [DOI] [PubMed] [Google Scholar]
- Gallwitz B, Witt M, Folsch UR, Creutzfeldt W, Schmidt WE (1993). Binding specificity and signal transduction of receptors for glucagon‐like peptide‐1(7–36)amide and gastric inhibitory polypeptide on RINm5F insulinoma cells. J Mol Endocrinol 10: 259–268. [DOI] [PubMed] [Google Scholar]
- Gaudin‐Audrain C, Irwin N, Mansur S, Flatt PR, Thorens B, Baslé M et al (2013). Glucose‐dependent insulinotropic polypeptide receptor deficiency leads to modifications of trabecular bone volume and quality in mice. Bone 53: 221–230. [DOI] [PubMed] [Google Scholar]
- Gault VA, Harriott P, Flatt PR, O'harte FP (2002a). Cyclic AMP production and insulin releasing activity of synthetic fragment peptides of glucose‐dependent insulinotropic polypeptide. Biosci Rep 22: 523–528. [DOI] [PubMed] [Google Scholar]
- Gault VA, O'harte FPM, Harriott P, Flatt PR (2002b). Characterization of the cellular and metabolic effects of a novel enzyme‐resistant antagonist of glucose‐dependent insulinotropic polypeptide. Biochem Biophys Res Comm 290: 1420–1426. [DOI] [PubMed] [Google Scholar]
- Gault VA, Parker JC, Harriott P, Flatt PR, O'harte FP (2002c). Evidence that the major degradation product of glucose‐dependent insulinotropic polypeptide, GIP(3–42), is a GIP receptor antagonist in vivo. J Endocrinol 175: 525–533. [DOI] [PubMed] [Google Scholar]
- Gault VA, Porter DW, Irwin N, Flatt PR (2011). Comparison of sub‐chronic metabolic effects of stable forms of naturally occurring GIP(1–30) and GIP(1–42) in high‐fat fed mice. J Endocrinol 208: 265–271. [DOI] [PubMed] [Google Scholar]
- Gelling RW, Coy DH, Pederson RA, Wheeler MB, Hinke S, Kwan T et al (1997). GIP(6‐30amide) contains the high affinity binding region of GIP and is a potent inhibitor of GIP1‐42 action in vitro. Regul Pept 69: 151–154. [DOI] [PubMed] [Google Scholar]
- Hinke SA, Manhart S, Pamir N, Demuth HU, Gelling W, Pederson RA et al (2001). Identification of a bioactive domain in the amino‐terminus of glucose‐dependent insulinotropic polypeptide (GIP). Biochimica et Biophysica Acta (BBA) ‐ Protein Struct Mol Enzymol 1547: 143–155. [DOI] [PubMed] [Google Scholar]
- Hinke SA, Manhart S, Speck M, Pederson RA, Demuth HU, Mcintosh CHS (2004). In depth analysis of the N‐terminal bioactive domain of gastric inhibitory polypeptide. Life Sci 75: 1857–1870. [DOI] [PubMed] [Google Scholar]
- Hjorth SA, Schwartz TW (1996). Glucagon and GLP‐1 receptors: lessons from chimeric ligands and receptors. Acta Physiologica Scandinavica 157: 343–345. [DOI] [PubMed] [Google Scholar]
- Holst JJ, Bersani M (1991). 1 ‐ assays for peptide products of somatostatin gene expression In: Conn PM. (ed.). Methods in Neurosciences. Academic Press. [Google Scholar]
- Holst JJ, Knop FK, Vilsboll T, Krarup T, Madsbad S (2011). Loss of incretin effect is a specific, important, and early characteristic of type 2 diabetes. Diabetes Care 34 (Suppl 2): S251–S257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin N, Green BD, Parker JC, Gault VA, O'harte FPM, Flatt PR (2006a). Biological activity and antidiabetic potential of synthetic fragment peptides of glucose‐dependent insulinotropic polypeptide, GIP(1–16) and (Pro3)GIP(1–16). Regul Pept 135: 45–53. [DOI] [PubMed] [Google Scholar]
- Irwin N, O'harte FP, Gault VA, Green BD, Greer B, Harriott P et al (2006b). GIP(Lys16PAL) and GIP(Lys37PAL): novel long‐acting acylated analogues of glucose‐dependent insulinotropic polypeptide with improved antidiabetic potential. J Med Chem 49: 1047–1054. [DOI] [PubMed] [Google Scholar]
- Jensen PC, Thiele S, Ulven T, Schwartz TW, Rosenkilde MM (2008). Positive versus negative modulation of different endogenous chemokines for CC‐chemokine receptor 1 by small molecule agonists through allosteric versus orthosteric binding. J Biol Chem 283: 23121–23128. [DOI] [PubMed] [Google Scholar]
- Jorgensen R, Kubale V, Vrecl M, Schwartz TW, Elling CE (2007). Oxyntomodulin differentially affects glucagon‐like peptide‐1 receptor +Ý‐arrestin recruitment and signaling through G+Ý. J Pharmacol Exp Therapeut 322: 148–154. [DOI] [PubMed] [Google Scholar]
- Kerr BD, Flatt AJS, Flatt PR, Gault VA (2011). Characterization and biological actions of N‐terminal truncated forms of glucose‐dependent insulinotropic polypeptide. Biochem Biophys Res Comm 404: 870–876. [DOI] [PubMed] [Google Scholar]
- Kissow H, Hartmann B, Holst JJ, Viby N‐E, Hansen LS, Rosenkilde MM et al (2012). Glucagon‐like peptide‐1 (GLP‐1) receptor agonism or DPP‐4 inhibition does not accelerate neoplasia in carcinogen treated mice. Regul Pept 179: 91–100. [DOI] [PubMed] [Google Scholar]
- Knop FK, Vilsboll T, Holst JJ (2009). Incretin‐based therapy of type 2 diabetes mellitus. Curr Protein Pept Sci 10: 46–55. [DOI] [PubMed] [Google Scholar]
- Kreymann B, Williams G, Ghatei MA, Bloom SR (1987). Glucagon‐like peptide‐1 7–36: a physiological incretin in man. Lancet 2: 1300–1304. [DOI] [PubMed] [Google Scholar]
- Kubota A, Yamada Y, Yasuda K, Someya Y, Ihara Y, Kagimoto S et al (1997). Gastric inhibitory polypeptide activates MAP kinase through the wortmannin‐sensitive and ‐insensitive pathways. Biochem Biophys Res Commun 235: 171–175. [DOI] [PubMed] [Google Scholar]
- Lazareno S, Birdsall NJ (1993). Estimation of competitive antagonist affinity from functional inhibition curves using the Gaddum, Schild and Cheng‐Prusoff equations. Br J Pharmacol 109: 1110–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malde AK, Srivastava SS, Coutinho EC (2007). Understanding interactions of gastric inhibitory polypeptide (GIP) with its G‐protein coupled receptor through NMR and molecular modeling. J Pept Sci 13: 287–300. [DOI] [PubMed] [Google Scholar]
- Maletti M, Carlquist M, Portha B, Kergoat M, Mutt V, Rosselin G (1986). Structural requirements for gastric inhibitory polypeptide (GIP) receptor binding and stimulation of insulin release. Peptides 7 (Supplement 1): 75–78. [DOI] [PubMed] [Google Scholar]
- Mentlein R (1999). Dipeptidyl‐peptidase IV (CD26)‐role in the inactivation of regulatory peptides. Regul Pept 85: 9–24. [DOI] [PubMed] [Google Scholar]
- Mentlein R (2009). Mechanisms underlying the rapid degradation and elimination of the incretin hormones GLP‐1 and GIP. Best Pract Res Clin Endocrinol Metab 23: 443–452. [DOI] [PubMed] [Google Scholar]
- Mentlein R, Gallwitz B, Schmidt WE (1993). Dipeptidyl‐peptidase IV hydrolyses gastric inhibitory polypeptide, glucagon‐like peptide‐1(7–36)amide, peptide histidine methionine and is responsible for their degradation in human serum. Eur J Biochem 214: 829–835. [DOI] [PubMed] [Google Scholar]
- Mieczkowska A, Irwin N, Flatt PR, Chappard D, Mabilleau G (2013). Glucose‐dependent insulinotropic polypeptide (GIP) receptor deletion leads to reduced bone strength and quality. Bone 56: 337–342. [DOI] [PubMed] [Google Scholar]
- Miyawaki K, Yamada Y, Ban N, Ihara Y, Tsukiyama K, Zhou H et al (2002). Inhibition of gastric inhibitory polypeptide signaling prevents obesity. Nat Med 8: 738–742. [DOI] [PubMed] [Google Scholar]
- Moroder LHA, Thamm P, Wilschowitz L, Brown JC, Wünsch E (1978). Studies on gastric inhibitory polypeptide: synthesis of the octatricontapeptide GIP(1–38) with full insulinotropic activity. Scand J Gastroenterol 13 suppl. 49: p.129. [Google Scholar]
- Morrow GW, Kieffer TJ, Mcintosh CH, Macgillivray RT, Brown JC, St Pierre S et al (1996). The insulinotropic region of gastric inhibitory polypeptide; fragment analysis suggests the bioactive site lies between residues 19 and 30. Can J Physiol Pharmacol 74: 65–72. [PubMed] [Google Scholar]
- Nakamura T, Tanimoto H, Mizuno Y, Tsubamoto Y, Noda H (2012). Biological and functional characteristics of a novel low‐molecular weight antagonist of glucose‐dependent insulinotropic polypeptide receptor, SKL‐14959, in vitro and in vivo. Diabetes Obes Metab 14: 511–517. [DOI] [PubMed] [Google Scholar]
- Nauck MA, Homberger E, Siegel EG, Allen RC, Eaton RP, Ebert R et al (1986). Incretin effects of increasing glucose loads in man calculated from venous insulin and C‐peptide responses. J Clin Endocrinol Metab 63: 492–498. [DOI] [PubMed] [Google Scholar]
- Nissen A, Christensen M, Knop FK, Vilsbøll T, Holst JJ, Hartmann B (2014). Glucose‐dependent insulinotropic polypeptide inhibits bone resorption in humans. J Clin Endocrinol Metabol 99: E2325–E2329. [DOI] [PubMed] [Google Scholar]
- Parthier C, Kleinschmidt M, Neumann P, Rudolph R, Manhart S, Schlenzig D et al (2007). Crystal structure of the incretin‐bound extracellular domain of a G protein‐coupled receptor. Proc Natl Acad Sci U S A 104: 13942–13947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak V, Gault VA, Flatt PR, Irwin N (2015a). Antagonism of gastric inhibitory polypeptide (GIP) by palmitoylation of GIP analogues with N‐ and C‐terminal modifications improves obesity and metabolic control in high fat fed mice. Mol Cell Endocrinol 401: 120–129. [DOI] [PubMed] [Google Scholar]
- Pathak V, Vasu S, Gault VA, Flatt PR, Irwin N (2015b). Sequential induction of beta cell rest and stimulation using stable GIP inhibitor and GLP‐1 mimetic peptides improves metabolic control in C57BL/KsJ db/db mice. Diabetologia 58: 2144–2153. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP et al (2014). The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledge base of drug targets and their ligands. Nucl Acids Res 42 (Database Issue): D1098–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pederson RA, Brown JC (1976). The insulinotropic action of gastric inhibitory polypeptide in the perfused isolated rat pancreas. Endocrinology 99: 780–785. [DOI] [PubMed] [Google Scholar]
- Ravn P, Madhurantakam C, Kunze S, Matthews E, Priest C, O'brien S et al (2013). Structural and pharmacological characterization of novel potent and selective monoclonal antibody antagonists of glucose‐dependent insulinotropic polypeptide receptor. J Biol Chem 288: 19760–19772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenkilde MM, Cahir M, Gether U, Hjorth SA, Schwartz TW (1994). Mutations along transmembrane segment II of the NK‐1 receptor affect substance P competition with non‐peptide antagonists but not substance P binding. J Biol Chem 269: 28160–28164. [PubMed] [Google Scholar]
- Rossowski WJ, Zacharia S, Mungan Z, Ozmen V, Ertan A, Baylor LM et al (1992). Reduced gastric acid inhibitory effect of a pGIP(1–30)NH2 fragment with potent pancreatic amylase inhibitory activity. Regul Pept 39: 9–17. [DOI] [PubMed] [Google Scholar]
- Sandberg E, Ahren B, Tendler D, Carlquist M, Efendic S (1986). Potentiation of glucose‐induced insulin secretion in the perfused rat pancreas by porcine GIP (gastric inhibitory polypeptide), bovine GIP, and bovine GIP(1–39). Acta Physiol Scand 127: 323–326. [DOI] [PubMed] [Google Scholar]
- Sparre‐Ulrich AH, Hansen LS, Svendsen B, Christensen M, Knop FK, Hartmann B et al (2015). Species‐specific action of (Pro3)GIP ‐ an efficacious agonist on human GIP receptor, but partial agonist and competitive antagonist on rat and mouse GIP receptors. Br J Pharmacol. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiele S, Rosenkilde MM (2014). Interaction of chemokines with their receptors–from initial chemokine binding to receptor activating steps. Curr Med Chem 21: 3594–3614. [DOI] [PubMed] [Google Scholar]
- Thorens B, Porret A, Buhler L, Deng SP, Morel P, Widmann C (1993). Cloning and functional expression of the human islet GLP‐1 receptor. Demonstration that exendin‐4 is an agonist and exendin‐(9–39) an antagonist of the receptor. Diabetes 42: 1678–1682. [DOI] [PubMed] [Google Scholar]
- Tseng CC, Kieffer TJ, Jarboe LA, Usdin TB, Wolfe MM (1996). Postprandial stimulation of insulin release by glucose‐dependent insulinotropic polypeptide (GIP). Effect of a specific glucose‐dependent insulinotropic polypeptide receptor antagonist in the rat. J Clin Invest 98: 2440–2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng CC, Zhang XY, Wolfe MM (1999). Effect of GIP and GLP‐1 antagonists on insulin release in the rat. Am J Physiol 276: E1049–E1054. [DOI] [PubMed] [Google Scholar]
- Usdin TB, Mezey E, Button DC, Brownstein MJ, Bonner TI (1993). Gastric inhibitory polypeptide receptor, a member of the secretin‐vasoactive intestinal peptide receptor family, is widely distributed in peripheral organs and the brain. Endocrinology 133: 2861–2870. [DOI] [PubMed] [Google Scholar]
- Vilardaga JP, Romero G, Friedman PA, Gardella TJ (2011). Molecular basis of parathyroid hormone receptor signaling and trafficking: a family B GPCR paradigm. Cell Mol Life Sci 68: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler MB, Gelling RW, Mcintosh CH, Georgiou J, Brown JC, Pederson RA (1995). Functional expression of the rat pancreatic islet glucose‐dependent insulinotropic polypeptide receptor: ligand binding and intracellular signaling properties. Endocrinology 136: 4629–4639. [DOI] [PubMed] [Google Scholar]
- Yaqub T, Tikhonova IG, Lättig J, Magnan R, Laval M, Escrieut C et al (2010). Identification of determinants of glucose‐dependent insulinotropic polypeptide receptor that interact with N‐terminal biologically active region of the natural ligand. Mol Pharmacol 77: 547–558. [DOI] [PubMed] [Google Scholar]
- Zhong Q, Bollag RJ, Dransfield DT, Gasalla‐Herraiz J, Ding KH, Min L et al (2000). Glucose‐dependent insulinotropic peptide signaling pathways in endothelial cells. Peptides 21: 1427–1432. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting info item
Supporting info item