

Abstract
Cyclophilin B (CypB) is a member of the immunophilin family and intracellular chaperone. It predominantly localizes to the ER, but also contains a nuclear localization signal and is secreted from cells. CypB has been shown to interact with the Gag protein of human immunodeficiency type 1 (HIV-1). Several proteomic and genetic studies identified it as a potential factor involved in HIV replication. Herein, we show that over-expression of CypB enhances HIV infection by increasing nuclear import of viral DNA. This enhancement was unaffected by cyclosporine treatment and requires the N-terminus of the protein. The N-terminus contains an ER leader sequence, putative nuclear localization signal, and is required for secretion. Deletion of the N-terminus resulted in mislocalization from the ER and suppression of HIV infection. Passive transfer experiments showed that secreted CypB did not impact HIV infection. Combined, these experiments show that intracellular CypB modulates a pathway of HIV nuclear import.
Keywords: HIV-1, cyclophilin b, CypB, nuclear import, virus-host interactions
Introduction
Human Immunodeficiency Virus type 1 (HIV-1) is the causative agent of Acquired Immune Deficiency Syndrome (AIDS) and has remained a global health problem for over thirty years. HIV-1 is retrovirus which primarily targets the CD4+ T-cells of the immune system. The HIV-1 genome consists of 9 open reading frames encoding only 15 viral proteins (Frankel and Young, 1998). As an obligate intracellular parasite, HIV must utilize host proteins and pathways for effective replication while evading the innate and acquired immune responses. Thus far, approximately 3500 human proteins have been found to participate in HIV-1 replication in some capacity (NCBI HIV Interaction Database; (Ako-Adjei et al., 2014)). A major challenge is identifying the subset of these interaction partners that can be targeted as cell-based viral inhibitors. This necessitates characterizing normal protein function as well as deciphering how a factor interacts with HIV and impacts viral replication.
Cyclophilin B (CypB) was identified in a number of proteomic investigations performed by our laboratory, including: a characterization of the interactome of Matrix and Integrase, a study of partially purified preintegration complexes, and a study of the nuclei of HIV-infected T-cells ((DeBoer et al., 2014; Schweitzer et al., 2013; Schweitzer et al., 2012), unpubublished data). Additionally, CypB was shown to be significantly upregulated in the nuclei of HIV-infected monocyte-derived macrophages (Haverland et al., 2014) and identified as an HIV-dependency factor (Brass et al., 2008). CypB, also known as peptidyl prolyl isomerase B (PPIB), is a member of the cyclophilin family of immunophilins. CypA, the initial member of this family, was identified by its ability to bind the immunosuppressant cyclosporine A (CsA). CsA binding to CypA inhibits calcineurin and prevents dephosphorylation and nuclear translocation of the transcription factor NFAT (Friedman and Weissman, 1991; Liu et al., 1991; Schreiber and Crabtree, 1992). Cyclophilins contain a peptidyl prolyl isomerase (PPI) domain that functions to lower the rotational energy barrier in prolyl imide bonds. The core domain of CypB (aa 48–143) has ~80% homology with CypA and contains both the PPI and CsA binding region (Fig. 1). However, the functions of CypA and CypB are distinct. CypA is primarily a cytoplasmic protein, whereas CypB is widely distributed and present in the cytoplasm, nucleus, ER/golgi and plasma membrane (Price et al., 1994). CypB contains a N-terminal hydrophobic leader consistent with an endoplasmic reticulum (ER) signal sequence, as well as a C-terminal ER retention signal (Arber et al., 1992), where it may act as a chaperone for exported proteins (Price et al., 1994). ER localization results in approximately 50% of CypB being secreted from the cell (Caroni et al., 1991). In addition, CypB associates with collagen, is required for its intracellular transport, and correct collagen fibril formation (Choi et al., 2009; Smith et al., 1995). CsA treatment reduces the isomerization activity of CypB (Spik et al., 1991), blocks collagen binding (Smith et al., 1995), and causes an increase in CypB secretion (Price et al., 1994). CypB also acts as an intracellular chaperone for CAML (von Bülow and Bram, 1997), IRF-3 (Obata et al., 2005), and Prolactin (PRL) (Rycyzyn et al., 2000). Knock-down of CypB reduces virus induced phosphorylation of IRF-3 and its subsequent induction of interferon-β. CypB mediates nuclear retrotransport of PRL following receptor interaction and internalization. The intranuclear PRL-CypB complex activates Stat5-mediated gene expression (Rycyzyn and Clevenger, 2002).
Fig. 1.

Domain structure of CypB and the deletion mutants used in this study. The approximate location of previously described domains are shown including the core domain conserved between CypB and CypA.
CypA is an established HIV factor and its interaction with viral cores is necessary for capsid stabilization, proper uncoating, protection of viral cores from cellular restriction factors (Sayah et al., 2004), and efficient HIV replication is inhibited by CsA in vitro (Aiken, 1998; Franke and Luban, 1996; Karpas et al., 1992; Wainberg et al., 1988). Both CypA and CypB have been found to bind HIV-1 Gag (Franke et al., 1994; Luban et al., 1993). The CypA-HIV-1 interaction with Gag occurs at Gag proline 222. An alanine substitution at this position ablates CypA binding and blocks replication. CypB appears to interact in an independent manner with Gag. It binds Gag with stronger affinity due to its hydrophobic N-terminal ER signal sequence and its binding is less sensitive to disruption by CsA compared to CypA (Luban et al., 1993). Moreover, mutation of proline 222 does not block the interaction of CypB and Gag (Franke et al., 1994). Initial studies reported that only CypA is incorporated into virions (Franke et al., 1994; Thali et al., 1994); however, a later study detected CypB in virions by mass spectrometry (Chertova et al., 2006). CypB is also essential for successful infection of multiple viruses, including hepatitis C virus (HCV) (Watashi et al., 2005), Japanese encephalitis virus (JEV) (Kambara et al., 2011), and human papillomavirus type 16 (HPV16) (Bienkowska-Haba et al., 2009).
Given the repeated identification of CypB in HIV proteomic studies, we hypothesized that it played an important role in HIV replication. We were not able to knock-down CypB by RNA interference in 293T cells so alternate methods were explored. We discovered that ectopic expression of CypB enhances HIV infection at the step of nuclear import of the viral DNA. This enhancement is distinct from CypA and the N-terminus of CypB is necessary for the effect. Deletion of the leader sequence of CypB alters localization of the CypB and hinders its secretion from the cell. However, media transfer experiments suggested that secreted CypB does not play a major role in increasing infection. Finally, inhibition of the Stat5 pathway does not interfere with the enhancement. Combined, these results demonstrate that CypB modulates HIV nuclear import.
Results
CypB is upregulated in activated PBMCs during HIV infection
CypB has been identified in a number of proteomic studies of HIV infected cells and purified virions (Chertova et al., 2006; Haverland et al., 2014; Schweitzer et al., 2013), including a recent study in which we observed that it was increased in the nuclei of HIV infected Jurkat T-cells (DeBoer et al., 2014). To extend those observations we investigated the subcellular expression of CypB in primary peripheral blood mononuclear cells (PBMCs) during HIV infection (Fig. 2). Unstimulated, PHA-stimulated, and HIV-1 infected cells were fractionated into cytosolic, membrane/organelle, nuclear, and cytoskeletal/insoluble fractions using a commercially available kit. Infections were monitored by reverse transcriptase assay of cell supernatants and peaked at 6 dpi (Fig. 2A). In all experiments the integrity of subcellular fractions was monitored by detection of GAPDH (cytosol), ERGIC (membrane/organelle), and TOPO IIa (nuclei) (Fig. 2B, bottom blots). The subcellular distribution of CypB was dynamic. Unstimulated PBMCs expressed CypB primarily in the membrane/organelle fraction. Upon stimulation with PHA, CypB was also detected in the nuclear fractions of cells. Infection with HIV-1 also correlated with the expression of CypB in both the membrane and nuclear fractions. Consistent with a loss of cell viability after the peak of infection there was slightly less CypB detected in both fractions at 7 dpi compared to 3 dpi. These results support the hypothesis that CypB is involved in HIV replication.
Fig 2.

CypB expression in primary PBMCs. (A) Infection of donor-derived lymphocytes. Cells were activated with PHA and infected with NLX virus. Virus replication was monitored by RT assay of cell supernatants using an in vitro [32P]dTTP incorporation assay. Results presented are representative of triplicate experiments. (B) Unstimulated, PHA-stimulated, and PHA-stimulated infected PBMCs were fractionated into cytosolic (Cyt), membrane/organelle (Mem), nuclear (Nu), and cytoskeletal/insoluble (In) fractions. Equivalent amounts of each fraction was separated by SDS-PAGE and immunoblotted for CypB. Example blots of the controls used for fraction integrity and protein concentration are shown in bottom three panels.
CypB enhances HIV-1 infection
To study the potential role of CypB in HIV infection, we initially attempted depletion studies using RNAi. Although previously identified in a siRNA screen (Brass et al., 2008), our efforts to knock-down CypB expression using small interfering or short hairpin RNAs were unsuccessful. Therefore we moved to gain-of-function/over-expression studies to test for its impact on HIV infection. To do this we primed 293T cells with increasing amounts of a FLAG-tagged CypB expression vector 24 h prior to infection with a VSVg-pseudotyped HIV luciferase reporter virus (HIV-Luc). As shown in Fig. 3, increased expression of CypB enhanced the infection of HIV in a dose-dependent manner. Notably, over-expression of FLAG-tagged CypB did not appear to affect endogenous CypB expression (Fig. 3, CypB blot). To assess if this effect was non-specific toward retroviruses, we performed parallel experiments with a VSVg-psuedotyped murine leukemia virus luciferase reporter virus (MLV-Luc). Increased expression of CypB did not alter MLV infection, indicating that CypB did not have a general effect on retrovirus transduction.
Fig. 3.

Over-expression of CypB enhances HIV-1 infection. Cells were transfected with increasing amounts of CypB balanced with empty vector control plasmid. The following day, cells were infected with either a VSVg-pseudotyped HIV-Luc or MLV-Luc reporter virus. At 48 hpi, cells were harvested and infection quantified by Luc assay which was normalized to total protein concentration. Results are normalized and statistics compared to control cells transfected with empty vector alone (* = p<0.005 and ** = p<0.0005 by two-tailed t-test). Cell lysates were blotted for transfected (FLAG) and endogenous CypB expression; and GAPDH was used as loading control (bottom panels). Data shown is representative of three independent experiments with triplicate infections. Error bars in this and all other figures represent standard error of the mean.
The mechanism of CypB infection enhancement is distinct from that of CypA
Many studies have demonstrated a requirement for the interaction of CypA with HIV Gag for efficient HIV infection. Although CypB contains distinct N- and C-termini, both CypA and CypB bind HIV-1 Gag (Luban et al., 1993), and share a core domain that is ~80% homologous (Fig. 1). To investigate if CypB enhancement was mediated through a mechanism analogous to CypA, infection assays were conducted under conditions of increased CypA expression. Over-expression of CypA did not result in any significant enhancement in HIV infection (Fig. 4A). This result suggested that CypB potentiates HIV infection through a mechanism distinct from that of CypA.
Fig 4.

The effect of CypB over-expression is distinct from CypA. (A) Over-expression studies of CypA. Experiments were performed as described in Fig. 1 using a FLAG-tagged CypA expression vector. Results are normalized to results from cells transfected with an empty vector. (B) CsA does not abrogate the ability of CypB to enhance infection. Cells were pretransfected for 24 h with empty vector, CypA, or CypB then pretreated with CsA 2 h prior to infection with HIV-Luc VSVg. Cells were harvested and Luc activity measured to quantify infection (normalized to total protein). Presented results are normalized to control DMSO treated cells; * = p<0.01 by two-tailed t-test. Cell lysates were blotted for CypB and CypA expression using antibodies as indicated. GAPDH was used as loading control (bottom panels). (C) CsA induction of CypB secretion. 293T cells were treated with indicated concentrations of CsA for 48 h. Media and cell lysates were collected and the levels of CypB detected by immunoblot. Actin was detected as a loading control.
CsA binds to and inhibits the peptidyl prolyl isomerase (PPI) activity of both CypA and CypB, and when bound to CypA inhibits calcineurin function (Friedman and Weissman, 1991; Liu et al., 1991; Schreiber and Crabtree, 1992). CsA treatment disrupts the interaction of CypA and Gag leading to inhibition of HIV infection in vitro (Aiken, 1998; Franke and Luban, 1996; Karpas et al., 1992; Wainberg et al., 1988). To further explore the dichotomous results of CypA and CypB over-expression, we tested whether cyclosporine A (CsA) would antagonize the ability of CypB to enhance HIV infection. The over-expression experiments were repeated with cells treated with 5 μM CsA (Fig. 4B). As predicted from previous studies, CsA partially inhibited HIV infection in mock transfected cells. Moreover, this amount of CsA still inhibited infection when CypA was overexpressed. Immunoblot analyses of Cyps revealed that CsA substantially reduced the intracellular expression of both endogenous CypB in 293T cells (Fig. 4B, CypB blot, (+) lanes), although it did not appear to affect CypA expression. The loss of CypB correlated with increased secretion of CypB in to the media as previously reported (Fig. 4C; (Price et al., 1994)). CsA treatment did not affect the ability of exogenous CypB to potentiate HIV infection. Moreover, CsA did not substantially reduce HIV infection in CypB transfected cells despite slightly reducing the intracellular levels of the transfected protein. These results demonstrated that the CsA binding domain of CypB was not required for its ability to enhance HIV infection and confirmed that the ability of CypB to potentiate infection was unrelated to the role of CypA in HIV replication.
CypB increases nuclear import of HIV DNA
To determine how CypB mechanistically enhances HIV infection, we analyzed the early steps of HIV infection using real-time PCR to quantify viral DNA species. In addition to over-expressing CypB, we also included the following controls: Mock transfected cells, cells infected with heat-inactivated virus, and cells transfected with CypB Δ40, a mutant that does not potentiate infection (see below). Late RT product accumulation was analyzed at 24 hpi using gag-specific primers and normalized to the level of mitochondrial DNA as an internal control (Fig. 5A). Over-expression of CypB did not alter the total amount of late RT products in comparison with either mock-transfected cells or those over-expressing the Δ40 mutant. A similar result was found when we quantified early RT products (− strand, strong stop DNA; data not shown). This indicated that CypB did not enhance steps prior to, and through reverse transcription. The import of viral DNA into the nuclei of cells was assessed by quantifying 2-LTR circle accumulation. The levels of 2-LTR circles were normalized to the levels of late RT products to account for differences in the magnitude of infection. As shown in Fig. 5B, the level of 2-LTR circles was higher in cells over-expressing CypB compared to the mock-transfected cells. The amount of 2-LTR circles was also slightly decreased in the Δ40 transfected cells. To confirm the increase in nuclear import in CypB expressing cells, we also quantified the levels of viral DNA integrated in the genome of cells using an established alu-based nested PCR assay (Brussel and Sonigo, 2003). Consistent with the nuclear import/2-LTR data, the amount of integrated viral DNA was higher in cells over-expressing CypB (Fig. 5C). Moreover, cells expressing the Δ40 mutant had reduced levels of integrated viral DNA. Combined, the 2-LTR and integrated viral DNA data suggest that the over-expression of CypB enhances infection by increasing the nuclear import of the viral DNA.
Fig 5.
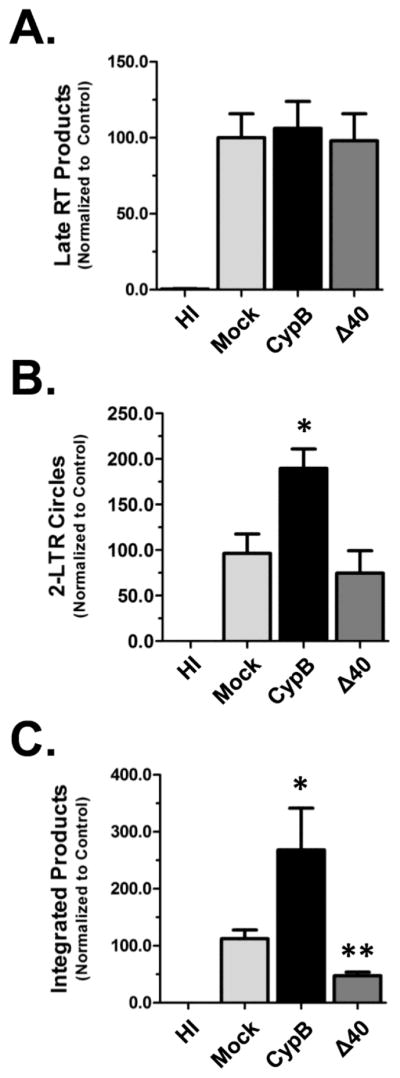
CypB increases nuclear import of HIV. In all experiments 293T cells were transfected with either CypB or the Δ40 mutant and infected the following day with NLX+VSVg. (A) For Late RT experiments extrachromosomal DNA was isolated at 24 hpi. Late RT products quantified by qPCR using gag-specific primers. Levels were normalized for DNA isolation efficiency by quantification of mitochondrial DNA (mtDNA) levels by qPCR. (B) 2-LTR circles were quantified from samples harvested at 48hpi. To account for infection efficiency, the levels of 2-LTR circles were normalized to the level of late RT DNA in each sample (which was normalized to mtDNA level). (C) Measurement of integrated viral DNA. Chromosomal DNA was isolated at 48 hpi and integrated viral DNA measured by an alu-based nested qPCR as outlined in Methods. Mock transfected and cells infected with heat inactivated (HI) virus were used as controls in all experiments. Data presented in A–C is normalized to the mock transfected group and combined from three independent experiments. Statistical significance was analyzed by two-tailed t-test: * = p<0.05 and ** = p<0.005.
The N-terminus of CypB is necessary to enhance HIV-1 infection
The CypA data suggested that a region outside the core domain of CypB was responsible for promoting HIV infection. To map the domain(s) of CypB that were required for the enhancement effect, we constructed a series of deletion mutants, as well as a PPI-inactive mutant (PPIm; Fig. 1). Similar to the parental construct, all the mutants were created with a C-terminal FLAG epitope to support uniform detection. The ability of each mutant to affect HIV infection was assessed under conditions of over-expression as in previous experiments (Fig. 6A). Consistent with the CsA experiments, disruption of the PPI motif did not affect the ability of CypB to augment HIV infection. Deletion of the C-terminus (Δ206) was also indistinguishable from WT CypB. In contrast, N-terminal deletions reduced or eliminated the ability of CypB to potentiate infection. Only the Δ5 mutant retained partial capacity to increase infection. Consistent with the viral DNA analysis, the expression of two larger N-terminal deletion mutants (Δ33 or Δ40) caused a slight reduction of infection compared to mock-transfected cells. Control immunoblots showed that all the mutants expressed (lower panels), although mutation of the PPI domain or deletion of the C-terminus at amino acid 206 appeared to enhance the expression of a larger isoform of the protein, which may be the uncleaved or glycosylated form of CypB (Spik et al., 1991). This isoform was not present in cells transfected with the Δ33 or Δ40 mutants. Finally, MTT assays showed that over-expression of CypB and the derivative mutants did not impact overall health of the cells (Fig. 6B).
Fig. 6.

The N-terminus of CypB is required for the enhancement of HIV infection. (A) Infection experiments were performed as described in legend of FIG 1 using indicated deletion mutants and a PPI inactive mutant. Results shown were normalized to cells transfected with empty vector control and are representative of a minimum of three replicate experiments. Asterisks indicated significant difference in infection compared to cells transfected with wild-type CypB (*= p<0.001 by two-tailed t-test). At the end of each experiment the expression of each mutant was verified by FLAG immunoblot of cell lysates (bottom panels). (B) Overall cell viability following expression of CypB and each mutant was monitored by MTT assay.
Deletion of the N-terminus alters CypB localization
The N-terminus of CypB contains an ER signal sequence, a putative nuclear localization signal (NLS), and is necessary for the secretion of CypB (Rycyzyn et al., 2000). To assess the effect the deletions had on the intracellular localization of CypB we performed immunofluorescence assays on cells transfected with the different CypB deletion mutants (Fig. 7). Wild-type CypB showed strong perinuclear localization typically with one distinct foci of fluorescence. The Δ5 mutant exhibited less perinuclear staining than full-length CypB, but maintained distinct foci of fluorescence near the nuclei of cells. In contrast, the Δ10 mutant showed strong perinuclear staining, but less distinct foci. Both the Δ33 and Δ40 mutants displayed diffuse cellular localization losing both the perinuclear localization and distinct foci. The Δ206 C-terminal mutant show staining similar to wild-type CypB and the PPI mutant maintained generally perinuclear staining but with more cytoplasmic punctae compared to wild-type CypB. Overall, all the N-terminal deletions except Δ5 appear to distinctly perturb the localization of CypB correlating with a loss of enhancement. Combined, these results show that the mutants that lost the ability to enhance HIV infection (Δ10, Δ33, and Δ40) also failed to form perinuclear foci.
Fig. 7.

The intracellular localization of the CypB deletion mutants. HeLa cells were transfected with CypB or indicated mutant expression vectors. After 24 h the cells were fixed, permeabilized and protein localization detected by immunofluorescence using a FITC conjugated anti-FLAG antibody. Nuclei were stained with DAPI. Panels are representative images of the localization of each protein as observed in two independent experiments.
We also looked at the localization of the CypB mutants by cell fractionation and immunoblotting (Fig. 8). 293T cells expressing the tagged CypB mutants were fractionated into cytoplasmic, membrane/organelle, nuclear and insoluble/cytoskeletal fractions. Fraction consistency was again monitored by detection of various subcellular markers (Fig. 8, bottom blots). Wild-type CypB was observed in the cytoplasmic, membrane/organelle, as nuclear fractions consistent with our immunofluorescence data and previous reports (Price et al., 1994). Several isoforms of CypB were detected in the membrane fractions. The mature form of CypB in cells lacks the N-terminal leader sequence with removal of the first 32 amino acids and migrates at approximately 18–20 kDa (Spik et al., 1991). The mature form of the FLAG-tagged protein migrates at approximately 23–24 kDa The larger band likely represented either the full length “pro” form of CypB or the mature form with a modification (consistent with this idea, WT, Δ5, Δ10, and Δ33 all migrate at a similar size (Fig. 6)). Interestingly, only the mature form of CypB was detected in the cytoplasm of cells and conversely, only larger isoforms of WT CypB were detected in nuclei. Both the Δ5 and Δ10 mutants fractionated similar to WT CypB with the notable exception that the mature isoforms were detected in nuclei instead of the larger forms. Consistent with the IFA data as well as the removal of the ER leader sequences, both Δ33 and Δ40 were detected in the cytoplasmic fractions only. The Δ206 mutant was present in the cytoplasmic, organelle, and nuclear fractions but as expected with the deletion of the C-terminal ER retention signal (Arber et al., 1992), it exhibited a higher proportion of cytoplasmic to membrane localization compared to WT CypB. Notably, three isoforms of Δ206 were observed, confirming that any modification, if present, was not in the C-terminus of CypB. Finally, the PPIm showed a localization distribution similar to Δ206, with a higher proportion in the cytoplasmic fractions of cells. Overall, these results show that the N-terminal truncation of CypB caused aberrant localization and expression of the CypB isoforms. Thus, the CypB mutants that lost the ability to enhance HIV infection showed changes in their localization compared to wild-type CypB.
Fig. 8.

The subcellular distribution of the CypB deletion mutants. 293T cells transfected with CypB or indicated mutant were fractionated into cytosolic (Cyt), membrane/organelle (Mem), nuclear (Nu), and cytoskeletal/insoluble (In) subcellular fractions. Fractions were separated by SDS-PAGE and immunoblotted with anti-FLAG antibodies. Bottom three panels show control immunoblots for cell fractionation. Results shown are representative of two independent experiments.
Secreted CypB does not enhance HIV infection
CypB is secreted from cells through the ER pathway and found in milk and blood (Mariller et al., 1996). Therefore we surmised that deletion of the N-terminus of CypB would block secretion of the protein. Since all the enhancement-null mutants displayed changes in their intracellular localization, we next tested if the secretion of CypB was necessary for the enhancement of HIV infection. First we assessed the secretion of each mutant by immunoblot (Fig. 9A). Only the PPI mutant was detected in supernatants at a level equivalent to wild-type CypB. Importantly, the loss of secretion of the Δ206 mutant suggested that the secretion of CypB was not critical for its ability to enhance infection as it potentiated infection as well as WT CypB. To further test if extracellular CypB was involved in the enhancement of infection, we performed media transfer experiments in which cells were infected with virus pre-mixed 1:1 with media from cells over-expressing CypB, CypA, or mock transfected cells. Both CypB and CypA could be detected in the media from transfected cells (Fig. 9B, top blot). Trans-addition of the media from CypB over-expressing cells to target cells only slightly enhanced HIV infection, and no effect was seen with the CypA media (Fig. 9B, graph). Notably, we did not observe any uptake of either CypB or CypA into the recipient cells (Fig. 9B, bottom blot, 2° lanes). We concluded from these results that secreted CypB does not play a principal role in the enhancement of HIV infection.
Fig. 9.

Secreted CypB does not enhance HIV infection in trans. (A) Secretion of CypB mutants. Cells were transfected with CypB or indicated mutants. After 24 h supernatants and cell lysates were collected and analyzed for protein expression by immmunoblot. (B) Media transfer experiments. Donor 293T cells were transfected with indicated expression constructs. At 24 h post-transfection the media was changed to eliminate excess transfection complexes. After an additional 24 h supernatants and cell lysates were collected. NLX-Luc virus stock was mixed 1:1 with donor supernatant and used to infected untransfected recipient cells. At 48 hpi recipient cells were harvested and infection levels measured by Luc assay. The secretion of CypB and CypA was monitored by immunoblot of donor supernatants (top blot) and the ectopic expression of both CypB and CypA (1°) as well as uptake into recipient cells (2°) was monitored by immunoblotting with anti-FLAG antibodies (bottom blots). Graphed data represents the compilation of two independent experiments and blots are representative images.
CypB potentiation does not involve the STAT5 pathway
Previous reports suggest that CypB is a chaperone for the nuclear localization of IRF-3 and prolactin (Obata et al., 2005; Rycyzyn et al., 2000), suggesting it interacts with a nuclear import pathway. In the nucleus, the CypB-prolactin complex interacts with and activates the Stat5 transcription factor (Rycyzyn and Clevenger, 2002). A single report previously indicated that ectopic expression of Stat5 increases HIV-1 expression in primary T-cells (Selliah et al., 2006). To determine if Stat5 activation was necessary for CypB potentiation, we tested whether over-expression of CypB would enhance HIV infection in the presence of the commercially available Stat5 inhibitor N′-((4-Oxo-4H-chromen-3-yl)methylene)nicotinohydrazide. At odds with the findings of the previous report, we observed that treatment with the inhibitor increased HIV infection in a dose-dependent manner (Fig. 10A). Importantly for our studies, the over-expression of CypB still potentiated infection at all treatment levels. Stat5 did not appear to alter cell metabolism as measured by MTT assay (Fig. 10B), but did cause a decrease in DNA synthesis as evidenced by few cells in S and G2 phases of the cell cycle (Fig. 10C). These results coupled with the fact that 1) deletion of the 40 N-terminal amino acids of CypB retains prolactin binding (Rycyzyn et al., 2000), but blocks nuclear import of prolactin; and 2) mutation of the PPI domain of CypB, which still enhances HIV infection, does not activate Stat5 (Rycyzyn and Clevenger, 2002), lead us to conclude that the enhancement effect of CypB is independent of Stat5 activation and involves a novel mechanism.
Fig. 10.

CypB enhancement does not require Stat5 activity. (A) Infection assays. 293T cells were transfected with either pcDNA3.1 empty vector or CypB. The following day media was changed and the cells pre-treated with indicated concentrations of STAT5 inhibitor for 4 h prior to transduction with NLX-Luc. At 48hpi cells were lysed and assayed for Luc activity, which was normalized to protein total protein concentration. Results are presented relative to infected control cells treated with DMSO and statistical differences analyzed by two-tailed t-test (** = p<0.0001 and * = p<0.05). (B) MTT assay of cells treated with Stat5 inhibitor. (C) Cell cycle analysis of cells treated with 5, 50, or 200 μM Stat5 inhibitor.
Discussion
The results from these studies show that the over-expression of CypB enhances the infection of HIV-1 by increasing nuclear import of viral DNA and suggests that CypB supports or activates a pathway of HIV nuclear import. Overall, the over-expression of CypB significantly enhanced infection at a magnitude comparable to other HIV cell factors (Hoque et al., 2011; Maddon et al., 1986; Pushkarsky et al., 2007), and the effect occurred in the presence of endogenous CypB. Unfortunately, we were unable to assess the necessity of CypB for HIV infection as we could not knock-down CypB in 293T cells without cytopathic effects (data not shown). Another limitation of these studies was that the functional experiments were limited to fibroblasts. Ongoing studies seek to advance these observations in immune cells. However, previous proteomic studies indicate that CypB is upregulated in HIV infected cells (DeBoer et al., 2014; Haverland et al., 2014). Moreover, on global siRNA study identified CypB as an HIV dependency factor (Brass et al., 2008). Combined, these observations suggested strongly that CypB plays a role in HIV infection.
CypB is a multifunctional protein involved in collagen transport and fibril formation (Choi et al., 2009; Smith et al., 1995), prolactin signaling (Rycyzyn and Clevenger, 2002; Rycyzyn et al., 2000), and immunosuppression (Swanson et al., 1992). Although shown previously to bind Gag, the role of CypB in HIV replication has not been extensively studied, possibly because of its homology to CypA and an assumption that it must function analogously. Indeed, both CypA and CypB are inhibited by CsA and both bind Gag; albeit, CypB at higher affinity due to its hydrophobic N-terminus (Franke et al., 1994). The enhancement of HIV-1 infection by CypB was clearly distinct from CypA as over-expression of CypA did not increase infection, and CsA treatment, which blocks CypA-Gag binding and increases secretion of CypB, did not impact the ability of intracellular CypB to potentiate infection.
Consistent with the concept that the mechanism of CypB enhancement is distinct from that of CypA, the deletion mutant studies demonstrated that the N-terminus of CypB is required for the effect. Moreover, the Δ33 and Δ40 mutants modestly inhibited HIV infection. Deletion of this region of CypB resulted in a loss of perinuclear localization and extracellular secretion. However, the media transfer experiments showed that CypB did not substantially enhance infection when provided in trans, indicating that secreted CypB was not involved in potentiating HIV infection. Moreover, CypB was not efficiently taken up by the recipient cells, nor could we detect it binding to virions in immunoprecipitation assays (data not shown). Finally, CsA treatment increased the secretion of CypB (Price et al., 1994), but in our experiments it did not substantially affect (either positively or negatively) the ability of CypB to boost infection when overexpressed.
Combined, the conclusion we draw from these results is that the intracellular CypB is responsible for the enhancement of infection. The localization of CypB is dynamic, consistent with its role as a chaperon for several proteins. The IFA and cell fractionation data of the deletion mutants show that the perinuclear localization of CypB correlates with its ability to potentiate HIV infection. The non-enhancing Δ33 and Δ40 mutants showed a loss of perinuclear localization by IFA and a loss of detectable membrane/organelle and nuclear localization in the immunoblot experiments of fractionated cells. The Δ10 N-terminal mutant lost the ability to potentiate infection and showed less concentrated perinuclear staining in cells, although the immunoblot analyses showed it still could enter the nuclear compartment. Combined, the data suggests that perinuclear localization of CypB is necessary for increasing HIV infection.
We theorize two possible mechanisms of CypB enhancement. First, that CypB may interact with the viral capsid, RTC, or PIC and facilitate interactions with cellular pathways to increase the nuclear import of the vDNA. Alternatively, the over-expression of CypB may activate one or more cellular pathways that boost import of the vDNA. CypB has been shown to contribute to the nuclear localization of IRF-3 and prolactin (Obata et al., 2005; Rycyzyn et al., 2000), but the mechanism of CypB nuclear import and its interactions with the nuclear transport machinery have yet to be defined. The protein contains a lysine rich region at amino acids 36–45 suggestive of a “classical”-type NLS, and deletion of this region ablates prolactin nuclear import. We did not mutate this region alone, and cannot rule out that both the NLS and the N-terminus are needed to support HIV nuclear import. Indeed, since deletions N-terminal to the NLS ablated the ability of CypB to increase infection, it may be that CypB interacts with HIV or a cellular factor in its N-terminus. Another possibility is that CypB facilitates infection through an interaction in the perinuclear region. One previous report suggested that the ER-associated SET DNA repair complex was crucial for HIV nuclear import by limiting auto-integration of viral DNA prior to nuclear import (Yan et al., 2009). In addition to high resolution studies of CypB localization, further study of the interactions of CypB with the HIV RTC and PIC as well as the nuclear transport machinery are needed to clarify the role of CypB as a nuclear chaperon. Delineating the mechanism through which CypB modulates HIV infection will further our understanding of HIV replication and may provide a target for novel therapeutics.
Materials and Methods
Plasmids
The C-terminal FLAG-tagged CypB and CypA ORFs were obtained from Origene Technologies (Rockville, MD). The CypB ORF was moved by PCR into the pTarget eukaryotic expression vector (Promega Corp., Madison WI USA), the backbone used for the deletion mutant ORFs which were constructed and inserted by PCR mutagenesis. The PPI mutant was constructed in pTarget by PCR-overlap mutagenesis and contained the amino acid substitutions R96 to A96 and F101 to A101. The primers used for mutagenesis were designed using NCBI Reference Sequence # NM000942.4; sequences are available upon request.
Cell culture and virus preparation
Lymphocytes from seronegative human donors were purchased from the Elutriation Core facility at the University of Nebraska Medical Center. Cells were cultured in RPMI media supplemented with 10% fetal bovine serum, 50 U/ml IL-2 (NIH AIDS Research and Reference Reagent Program, Germantown, MD), 8 mM L-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. Cells were activated by the addition of 10 μg/ml PHA for 48 h. 293T and HeLa cells were maintained in Dulbecco’s modified eagle medium (DMEM) supplemented with 10% fetal clone 3 (Hyclone, Logan, UT), 8 mM L-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. Viruses were produced by transient transfection of 293T cells using PolyJet reagent following the manufacturer’s instructions (SignaGen Laboratories, Gaithersburg, MD). Briefly, cells were seeded into 10 cm dishes at 70% confluency one day prior to transfection. NLX virus was produced by transfecting 5 μg of pNLX molecular clone (Brown et al., 1999); NLXLuc-VSVg virus was produced by transfecting 3.5 μg of viral molecular clone (pNLX-luc) DNA and 1.5 μg of pMD2.G vesicular stomatitis virus glycoprotein G (VSVg) expression vector (Addgene Plasmid Repository, Cambridge, MA). Media was collected at 24 and 48 h post transduction and clarified by centrifugation at 4000 xg for 5 min. MLV-luc viruses were produced following the same protocol using 2.5 μg viral molecular clone (pFB-luc), 1.5 μg packaging vector (pCG-gag-pol), and 1 μg pMD2.G. NLX virus was tittered on HeLa T4.β-gal cells (NIH AIDS Repository) and VSVg-pseudotyped viruses were titered on 293T cells. A TCID50 used for all infection experiments.
Infection assays
For lymphocyte infections, 2x107 cells were infected for 6 h, washed and maintained in IL-2 containing media. Virus replication was monitored by sampling supernatants and measuring exogenous reverse transcriptase activity as described previously (Belshan et al., 2009). For 293T transduction assays, 2x105 cells were seeded in triplicate wells of a 6-well plate. The following day each well was transfected with a total of 500 ng plasmid using PolyJet transfection reagent. For experiments showing a range of transfection, expression plasmids were supplemented with empty vector control plasmids to a total 500 ng. The next day, wells were inoculated with 100 μl of HIV-luc or MLV-luc and incubated for 48 h at 37°C. In indicated experiments, CsA was added 2 h prior to infection. Cells were lysed in 300 μl M-PER solution (Pierce Biotechnology, Rockford, IL) and clarified by centrifugation. Luciferase activity was determined using One-glo luciferase reagent (Promega, Madison, WI) and measured using a Spectramax L (Molecular Devices, Sunnyvale, CA). Protein concentrations were determined by BCA protein assay (Thermo Scientific, Rockford, IL) and used to normalize the amount of luciferase activity in each sample. Subcellular compartments (cytosol, membrane/organelle, nuclear, and cytoskeleton/insoluble) were isolated using the Qproteome Cell Compartment kit as described by the manufacturer (Qiagen, Valencia, CA). MTT assays were performed on transfected cells using the CellTiter 96 non-radioactive cell proliferation assay according to the manufacturer’s specifications (Promega, Madison, WI).
Immunoblotting
Samples were normalized by protein concentration with PBS, mixed 1:1 with 2x SDS-PAGE loading buffer, boiled for 10 min., separated by SDS-PAGE, and transferred to PVDF. Proteins were detected by western blot using the following primary antibodies: Anti-CypA (C-14), anti-CypB (k2E2), anti-ERGIC (H-245), anti-actin (C4), and anti-GAPDH (6C5) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA); and anti-Topo IIa was from BD Biosciences (San Jose, CA). The HRP-conjugated anti-Flag (M2) antibody was from Sigma-Aldrich (St Louis, MO). HRP-conjugated anti-rabbit and anti-mouse IgG secondary antibodies were also from Santa Cruz Biotechnology. Antibody binding was detected and visualized by West Pico chemiluminescent staining (Thermo Scientific, Rockford, IL). Images were captured by exposure to radiographic film, scanned to computer, adjusted for brightness and contrast if necessary, and cropped for size.
Indirect immunofluorescence
HeLa cells were plated on sterile coverslips in a six-well plate at 50% confluence. The following day, cells were transfected with CypB expression plasmids and incubated overnight. Cells were washed with PBS and fixed for 5 min by addition of 2 mL 4% paraformaldehyde (w/v in PBS). Cells were permeabilized using 2 mL of 0.2% Triton X-100 in PBS for 5 min. CypB proteins were detected with FITC conjugated anti-Flag (M2) antibody diluted 1:1000 in PBS+1% FBS; 2 mL was added to cells at 37° C for 60 min. Cells were washed 3x with PBS+1% FBS and coverslips mounted onto slides with ProLong® Gold antifade reagent with DAPI (Life Technologies, Eugene, OR). Slides were dried overnight and imaged using a Nikon Eclipse Ci microscope (Nikon USA, Melville, NY) and images captured using Infinity Analyze software (Lumenera, Ottawa, ON).
Quantification of viral DNA species
293T cells were seeded into 6 well plates to achieve 50% confluence the following day. Cells were transfected with empty control vector, CypB, or Δ40 and incubated overnight. NLX+VSVg stocks were treated with 2 U/ml Turbo DNase (Ambion, Austin TX) for 1 h at 37°C prior to inoculation. Heat inactivated virus (30 min at 65°C) was also used as a negative control for each experiment. Extra-chromosomal DNA was isolated at indicated times using the modified HIRT protocol (Arad, 1998; Belshan et al., 2009). In all experiments DNA was quantified by real-time PCR using a CFX Connect real-time PCR detection system (Bio-Rad) with Bullseye EvaGreen qPCR Master Mix (Midsci, Valley Park MO). Late reverse transcription (LRT) viral DNA was quantified using gag-specific primers NL919 (5′-TTCGCAGTTAATCCTGGACTT-3′) and NL1054 (5′-GCACACAATAGAGGACTGCTATTGTA). LRT was normalized to detection of mitochondrial DNA, which was quantified using primers MitoFor (5′-ACCCACTCCCTCTTAGCCAATATT-3′) and MitoRev (5′-GTAGGGCTAGGCCCACCG-3′). 2-LTR circles were quantified using primers NL500 (5′-AACTAGGGAACCCACTGCTTAAG-3′) and NL9126 (5′-TCCACAGATCAAGGATATCTTGTC-3′). These values were normalized to the level of LRT viral DNA to account for variance in infection. Chromosomal DNA was isolated using the EZNA Tissue DNA Kit (Omega Biotek, Frederick CO) and integration was assessed by nested Alu-PCR, using primers cNL658 (5′-TTTCAGGTCCCTGTTCGGGCGCCAC-3′), Alu1 (TCCCAGCTACTGGGGAGGCTGAGG-3′), and Alu2 (GCCTCCCAAAGTGCTGGGATTACAG-3′) for the initial PCR and primers NL493 (5′-TCTGGCTAACTAGGGAACCCAC-3′) and cNL616 (5′-CTGACTAAAAGGGTCTGAGG-3′) for the second PCR (Brussel and Sonigo, 2003). Integrated DNA was normalized to detection of β-globin intron DNA, which was quantified using primers BGlobinFor (5′-GCAAGAAAGTGCTCGGTGC-3′) and BGlobinRev (5′- CTACTCAGTGTGGCAAAG-3′).
STAT5 inhibition assays
The STAT5 inhibitor (N′-((4-Oxo-4H-chromen-3-yl)methylene)nicotinohydrazide) was obtained from Santa Cruz Biotechnology and resuspended in DMSO to a concentration of 10mg/ml. 293T cells were seeded into 6 well plates to achieve 50–60% confluence the following day. Cells were pre-treated with STAT5 inhibitor for 4 hours prior to transduction with NLX-Luc VSVg virus. At 24 hours, virus was removed and new media containing inhibitor added. At 48 hours post-transduction, cells were lysed with MPER and infection levels measured as described above. For the both the MTT and cell cycle analyses, 293T cells were pretreated with inhibitor for 24 h. For cell cycle analysis, cells were harvested by centrifugation and resuspended in Vindelov’s reagent (10mM Tris (pH 7.6), 10 μg/ml RNase A, 75 μM propidium iodide, and 0.1% Igepal CA-630). Cells were analyzed at the Creighton University flow cytometry core using a FACSAria flow cytometer (BD Biosciences, San Jose, CA) and Flowjo software (Treestar Inc., Ashland, OR).
HIGHLIGHTS.
CypB has been identified in several proteomic studies of HIV-1 infection.
CypB expression is upregulated in activated and infected T-cells.
Over-expression of CypB enhances HIV nuclear import and infection.
The N-terminus of CypB is necessary for these effects.
Acknowledgments
This work was supported by Public Health Service grant AI080348 from the National Institute of Allergy and Infectious Diseases. Jason DeBoer is funded by the US Army’s Medical Service Corp Long Term Health Education Program. The following reagent was obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: Human rIL-2 from Dr. Maurice Gately, Hoffmann - La Roche Inc. The authors also wish to thank John West for his critique of the manuscript and the Creighton University Molecular Biology Core Facility for support with the microscopy.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aiken C. Mechanistic independence of Nef and cyclophilin A enhancement of human immunodeficiency virus type 1 infectivity. Virology. 1998;248:139–147. doi: 10.1006/viro.1998.9254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ako-Adjei D, Fu W, Wallin C, Katz KS, Song G, Darji D, Brister JR, Ptak RG, Pruitt KD. HIV-1, human interaction database: current status and new features. Nucleic Acids Research. 2014;43:D566–570. doi: 10.1093/nar/gku1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arad U. Modified Hirt procedure for rapid purification of extrachromosomal DNA from mammalian cells. Biotechniques. 1998;24:760–762. doi: 10.2144/98245bm14. [DOI] [PubMed] [Google Scholar]
- Arber S, Krause KH, Caroni P. s-cyclophilin is retained intracellularly via a unique COOH-terminal sequence and colocalizes with the calcium storage protein calreticulin. The Journal of cell biology. 1992;116:113–125. doi: 10.1083/jcb.116.1.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belshan M, Schweitzer CJ, Donnellan MD, Lu R, Engelman A. In vivo biotinylation and capture of HIV-1 matrix and integrase proteins. J Virol Methods. 2009;159:178–184. doi: 10.1016/j.jviromet.2009.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienkowska-Haba M, Patel HD, Sapp M. Target Cell Cyclophilins Facilitate Human Papillomavirus Type 16 Infection. PLoS Pathog. 2009;5:e1000524. doi: 10.1371/journal.ppat.1000524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brass AL, Dykxhoorn DM, Benita Y, Yan N, Engelman A, Xavier RJ, Lieberman J, Elledge SJ. Identification of host proteins required for HIV infection through a functional genomic screen. Science. 2008;319:921–926. doi: 10.1126/science.1152725. [DOI] [PubMed] [Google Scholar]
- Brown HE, Chen H, Engelman A. Structure-based mutagenesis of the human immunodeficiency virus type 1 DNA attachment site: effects on integration and cDNA synthesis. J Virol. 1999;73:9011–9020. doi: 10.1128/jvi.73.11.9011-9020.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brussel A, Sonigo P. Analysis of Early Human Immunodeficiency Virus Type 1 DNA Synthesis by Use of a New Sensitive Assay for Quantifying Integrated Provirus. J Virol. 2003;77:10119–10124. doi: 10.1128/JVI.77.18.10119-10124.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caroni P, Rothenfluh A, McGlynn E, Schneider C. S-cyclophilin. New member of the cyclophilin family associated with the secretory pathway. Journal of Biological Chemistry. 1991;266:10739–10742. [PubMed] [Google Scholar]
- Chertova E, Chertov O, Coren LV, Roser JD, Trubey CM, Bess JW, Jr, Sowder RC, 2nd, Barsov E, Hood BL, Fisher RJ, Nagashima K, Conrads TP, Veenstra TD, Lifson JD, Ott DE. Proteomic and biochemical analysis of purified human immunodeficiency virus type 1 produced from infected monocyte-derived macrophages. J Virol. 2006;80:9039–9052. doi: 10.1128/JVI.01013-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JW, Sutor SL, Lindquist L, Evans GL, Madden BJ, Bergen HR, 3rd, Hefferan TE, Yaszemski MJ, Bram RJ. Severe osteogenesis imperfecta in cyclophilin B-deficient mice. PLoS Genet. 2009;5:e1000750. doi: 10.1371/journal.pgen.1000750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBoer J, Jagadish T, Haverland NA, Madson CJ, Ciborowski P, Belshan M. Alterations in the nuclear proteome of HIV-1 infected T-cells. Virology. 2014:468–470. 409–420. doi: 10.1016/j.virol.2014.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke EK, Luban J. Inhibition of HIV-1 replication by cyclosporine A or related compounds correlates with the ability to disrupt the Gag-cyclophilin A interaction. Virology. 1996;222:279–282. doi: 10.1006/viro.1996.0421. [DOI] [PubMed] [Google Scholar]
- Franke EK, Yuan HE, Luban J. Specific incorporation of cyclophilin A into HIV-1 virions. Nature. 1994;372:359–362. doi: 10.1038/372359a0. [DOI] [PubMed] [Google Scholar]
- Frankel AD, Young JAT. HIV-1: Fifteen Proteins and an RNA. Annual Review of Biochemistry. 1998;67:1–25. doi: 10.1146/annurev.biochem.67.1.1. [DOI] [PubMed] [Google Scholar]
- Friedman J, Weissman I. Two cytoplasmic candidates for immunophilin action are revealed by affinity for a new cyclophilin: one in the presence and one in the absence of CsA. Cell. 1991;66:799–806. doi: 10.1016/0092-8674(91)90123-g. [DOI] [PubMed] [Google Scholar]
- Haverland NA, Fox HS, Ciborowski P. Quantitative proteomics by SWATH-MS reveals altered expression of nucleic acid binding and regulatory proteins in HIV-1-infected macrophages. Journal of Proteome Research. 2014;13:2109–2119. doi: 10.1021/pr4012602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoque M, Shamanna RA, Guan D, Pe’ery T, Mathews MB. HIV-1 replication and latency are regulated by translational control of cyclin T1. Journal of Molecular Biology. 2011;410:917–932. doi: 10.1016/j.jmb.2011.03.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kambara H, Tani H, Mori Y, Abe T, Katoh H, Fukuhara T, Taguwa S, Moriishi K, Matsuura Y. Involvement of cyclophilin B in the replication of Japanese encephalitis virus. Virology. 2011;412:211–219. doi: 10.1016/j.virol.2011.01.011. [DOI] [PubMed] [Google Scholar]
- Karpas A, Lowdell M, Jacobson SK, Hill F. Inhibition of human immunodeficiency virus and growth of infected T cells by the immunosuppressive drugs cyclosporin A and FK 506. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:8351–8355. doi: 10.1073/pnas.89.17.8351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Farmer JD, Jr, Lane WS, Friedman J, Weissman I, Schreiber SL. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell. 1991;66:807–815. doi: 10.1016/0092-8674(91)90124-h. [DOI] [PubMed] [Google Scholar]
- Luban J, Bossolt KL, Franke EK, Kalpana GV, Goff SP. Human immunodeficiency virus type 1 Gag protein binds to cyclophilins A and B. Cell. 1993;73:1067–1078. doi: 10.1016/0092-8674(93)90637-6. [DOI] [PubMed] [Google Scholar]
- Maddon PJ, Dalgleish AG, McDougal JS, Clapham PR, Weiss RA, Axel R. The T4 gene encodes the AIDS virus receptor and is expressed in the immune system and the brain. Cell. 1986;47:333–348. doi: 10.1016/0092-8674(86)90590-8. [DOI] [PubMed] [Google Scholar]
- Mariller C, Allain F, Kouach M, Spik G. Evidence that human milk isolated cyclophilin B corresponds to a truncated form. Biochimica et biophysica acta. 1996;1293:31–38. doi: 10.1016/0167-4838(95)00230-8. [DOI] [PubMed] [Google Scholar]
- Obata Y, Yamamoto K, Miyazaki M, Shimotohno K, Kohno S, Matsuyama T. Role of cyclophilin B in activation of interferon regulatory factor-3. J Biol Chem. 2005;280:18355–18360. doi: 10.1074/jbc.M501684200. [DOI] [PubMed] [Google Scholar]
- Price ER, Jin M, Lim D, Pati S, Walsh CT, McKeon FD. Cyclophilin B trafficking through the secretory pathway is altered by binding of cyclosporin A. Proc Natl Acad Sci U S A. 1994;91:3931–3935. doi: 10.1073/pnas.91.9.3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pushkarsky T, Yurchenko V, Laborico A, Bukrinsky M. CD147 stimulates HIV-1 infection in a signal-independent fashion. Biochemical and Biophysical Research Communications. 2007;363:495–499. doi: 10.1016/j.bbrc.2007.08.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rycyzyn MA, Clevenger CV. The intranuclear prolactin/cyclophilin B complex as a transcriptional inducer. Proceedings of the National Academy of Sciences. 2002;99:6790–6795. doi: 10.1073/pnas.092160699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rycyzyn MA, Reilly SC, O’Malley K, Clevenger CV. Role of cyclophilin B in prolactin signal transduction and nuclear retrotranslocation. Mol Endocrinol. 2000;14:1175–1186. doi: 10.1210/mend.14.8.0508. [DOI] [PubMed] [Google Scholar]
- Sayah DM, Sokolskaja E, Berthoux L, Luban J. Cyclophilin A retrotransposition into TRIM5 explains owl monkey resistance to HIV-1. Nature. 2004;430:569–573. doi: 10.1038/nature02777. [DOI] [PubMed] [Google Scholar]
- Schreiber SL, Crabtree GR. The mechanism of action of cyclosporin A and FK506. Immunol Today. 1992;13:136–142. doi: 10.1016/0167-5699(92)90111-J. [DOI] [PubMed] [Google Scholar]
- Schweitzer CJ, Jagadish T, Haverland N, Ciborowski P, Belshan M. Proteomic analysis of early HIV-1 nucleoprotein complexes. Journal of Proteome Research. 2013;12:559–572. doi: 10.1021/pr300869h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweitzer CJ, Matthews JM, Madson CJ, Donnellan MR, Cerny RL, Belshan M. Knockdown of the Cellular Protein LRPPRC Attenuates HIV-1 Infection. PLoS One. 2012;7:e40537. doi: 10.1371/journal.pone.0040537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selliah N, Zhang M, DeSimone D, Kim H, Brunner M, Ittenbach RF, Rui H, Cron RQ, Finkel TH. The γc-cytokine regulated transcription factor, STAT5, increases HIV-1 production in primary CD4 T cells. Virology. 2006;344:283–291. doi: 10.1016/j.virol.2005.09.063. [DOI] [PubMed] [Google Scholar]
- Smith T, Ferreira LR, Hebert C, Norris K, Sauk JJ. Hsp47 and Cyclophilin B Traverse the Endoplasmic Reticulum with Procollagen into Pre-Golgi Intermediate Vesicles: A ROLE FOR Hsp47 AND CYCLOPHILIN B IN THE EXPORT OF PROCOLLAGEN FROM THE ENDOPLASMIC RETICULUM. Journal of Biological Chemistry. 1995;270:18323–18328. doi: 10.1074/jbc.270.31.18323. [DOI] [PubMed] [Google Scholar]
- Spik G, Haendler B, Delmas O, Mariller C, Chamoux M, Maes P, Tartar A, Montreuil J, Stedman K, Kocher HP. A novel secreted cyclophilin-like protein (SCYLP) Journal of Biological Chemistry. 1991;266:10735–10738. [PubMed] [Google Scholar]
- Swanson SK, Born T, Zydowsky LD, Cho H, Chang HY, Walsh CT, Rusnak F. Cyclosporin-mediated inhibition of bovine calcineurin by cyclophilins A and B. Proc Natl Acad Sci U S A. 1992;89:3741–3745. doi: 10.1073/pnas.89.9.3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thali M, Bukovsky A, Kondo E, Rosenwirth B, Walsh CT, Sodroski J, Gottlinger HG. Functional association of cyclophilin A with HIV-1 virions. Nature. 1994;372:363–365. doi: 10.1038/372363a0. [DOI] [PubMed] [Google Scholar]
- von Bülow GU, Bram RJ. NF-AT Activation Induced by a CAML-Interacting Member of the Tumor Necrosis Factor Receptor Superfamily. Science. 1997;278:138–141. doi: 10.1126/science.278.5335.138. [DOI] [PubMed] [Google Scholar]
- Wainberg MA, Dascal A, Blain N, Fitz-Gibbon L, Boulerice F, Numazaki K, Tremblay M. The effect of cyclosporine A on infection of susceptible cells by human immunodeficiency virus type 1. Blood. 1988;72:1904–1910. [PubMed] [Google Scholar]
- Watashi K, Ishii N, Hijikata M, Inoue D, Murata T, Miyanari Y, Shimotohno K. Cyclophilin B is a functional regulator of hepatitis C virus RNA polymerase. Molecular Cell. 2005;19:111–122. doi: 10.1016/j.molcel.2005.05.014. [DOI] [PubMed] [Google Scholar]
- Yan N, Cherepanov P, Daigle JE, Engelman A, Lieberman J. The SET complex acts as a barrier to autointegration of HIV-1. PLoS Pathog. 2009;5:e1000327. doi: 10.1371/journal.ppat.1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]