

Abstract
Human cytomegalovirus (HCMV) lacking TRS1 and IRS1 (HCMV[ΔI/ΔT]) cannot replicate in cell culture. Although both proteins can block the protein kinase R (PKR) pathway, they have multiple other activities and binding partners. It remains unknown which functions are essential for HCMV replication. To investigate this issue, we first identified a TRS1 mutant that is unable to bind to PKR. Like HCMV[ΔI/ΔT], a recombinant HCMV containing this mutant (HCMV[TRS1-Mut 1]) did not replicate in wild-type cells. However, HCMV[ΔI/ΔT] did replicate in cells in which PKR expression was reduced by RNA interference. Moreover, HCMV[ΔI/ΔT] and HCMV[TRS1-Mut 1] replicated to similar levels as virus containing wild-type TRS1 in cell lines in which PKR expression was knocked out by CRISPR/Cas9-mediated genome editing. These results demonstrate that the sole essential function of TRS1 is to antagonize PKR and that its other activities do not substantially enhance HCMV replication, at least in cultured human fibroblasts.
Keywords: Cytomegalovirus, PKR, translation, double-stranded RNA, TRS1, IRS1, host defense
INTRODUCTION
Host cells express a gauntlet of intrinsic defense mechanisms that sense and impede viral replication. One of these relies on protein kinase R (PKR), which is activated by binding to double-stranded RNA (dsRNA), dimerization, and autophosphorylation. Activated PKR phosphorylates the alpha subunit of eukaryotic initiation factor 2 (eIF2α), which in turn blocks the guanine nucleotide exchange factor eIF2B from replenishing the supply of eIF2α-tRNAMet-GTP ternary complex that is necessary for translation initiation (Dever et al., 2007). The resulting shutdown of protein synthesis is one tactic that host cells use to prevent viruses from replicating.
Many viruses encode at least one antagonist of the PKR pathway (Mohr et al., 2007). Human cytomegalovirus (HCMV) encodes two highly similar proteins, TRS1 and IRS1, each of which is able to inhibit the PKR pathway (Child et al., 2004). Infection of cells with a viral mutant lacking both genes (HCMV[ΔI/ΔT]) shuts off host protein synthesis and causes an increase in the level of phospho-eIF2α, indicating that the PKR pathway has been activated (Marshall et al., 2009). Unlike single mutant viruses that lack either TRS1 or IRS1, HCMV[ΔI/ΔT] does not replicate in cell culture (Blankenship and Shenk, 2002; Dunn et al., 2003; Jones and Muzithras, 1992; Marshall et al., 2009). These observations suggest that PKR pathway inhibition may be the essential role of TRS1 or IRS1 in HCMV replication. However, TRS1 and IRS1 have been reported to have several additional functions, one or more of which might be essential for viral replication. Both proteins have been reported to bind to Beclin 1 and inhibit autophagy (Chaumorcel et al., 2012; Mouna et al., 2015), to block the 2′-5′ oligoadenylate synthetase (OAS)/RNaseL pathway (Child et al., 2004), and to have potential roles as transcriptional activators (Romanowski and Shenk, 1997; Stasiak and Mocarski, 1992). The IRS1 gene has also been reported to encode a second product that acts as a transcriptional repressor (Romanowski and Shenk, 1997). Additional studies focusing on TRS1 have shown that it binds to the DNA polymerase accessory factor UL44 and may play a role in virion assembly (Adamo et al., 2004; Blankenship and Shenk, 2002; Strang et al., 2010). Finally, TRS1 was recently reported to have a role in translation stimulation following mRNA cap binding (Ziehr et al., 2015). Thus, while TRS1 and IRS1 appear to have multiple functions, those that are essential for HCMV replication have not been established.
We undertook a two-pronged approach to investigate the hypothesis that the sole essential function of TRS1 is to block the PKR pathway. First, we identified TRS1 mutants that have lost the ability to bind to and inhibit PKR. A recombinant virus containing a non-PKR binding TRS1 mutant (HCMV[TRS1-Mut 1]) was unable to replicate in wild-type human fibroblasts (HF). Second, we used both RNA silencing and CRISPR gene-editing technology to reduce or eliminate the expression of endogenous PKR in HF. We found that HCMV[ΔI/ΔT] replication was restored in cell lines depleted of PKR. Notably, we did not detect any major differences in the replication of HCMV[ΔI/ΔT] compared to that of a virus expressing TRS1 in PKR knockout cells. These data reveal that PKR antagonism is the only essential function of TRS1 or IRS1, and that, at least for the AD169 strain of HCMV, the additional activities ascribed to TRS1 do not substantially enhance replication in HF.
MATERIALS AND METHODS
Cells
All cells were maintained at 37°C in Dulbecco’s modified Eagle’s medium supplemented with 10% NuSerum. Primary human foreskin fibroblasts (HF) and telomerase-immortalized HF (HF-Tert) were provided by Denise Galloway (Fred Hutchinson Cancer Center). HF transduced with a TRS1-expressing retrovirus (HF-TRS1) were reported previously (Marshall et al., 2009).
PKR knockdown and control knockdown HF were created by transduction with lentiviral vectors expressing either an shRNA targeting PKR or a control shRNA (Open Biosystems, catalog numbers RHS4430-98844125 and RHS4346, respectively) followed by selection in puromycin (1 μg/ml).
PKR knockout A (PKR KO A) cells were constructed by transducing HF-Tert cells with a lentiviral vector that expresses Cas9 (Eckard et al., 2014) and a guide RNA (gRNA) with the genomic target sequence 5′-TCTCTTCCATTGTAGGATA-3′ (kindly provided by Elizabeth Gray and Dan Stetson, University of Washington). The predicted Cas9 cleavage site (between the two underlined nucleotides) is one bp downstream of the conserved AG (bold) of the exon 3 splice-acceptor site and 15 bps upstream of the PKR initiation codon. Following puromycin selection, single-cell clones were isolated and analyzed by PCR-amplifying this region of genomic DNA using primers #2001 and #2030 (Table 1), followed by TOPO-cloning and sequencing of multiple inserts from each of several cell clones. In every case, we detected mutations reflecting nonhomologous end joining resulting from cleavage at the expected site. In some cases, the resulting alleles retained the splice site and did not contain any large deletions. These clones expressed PKR as determined by immunoblot assays and were nonpermissive for VVΔE3L replication (data not shown). Other clones had alleles with mutations that removed the splice acceptor site and/or the PKR initiation codon. Analyses of PKR KO A revealed that this clone had one PKR allele with a 3 bp deletion that inactivated the splice acceptor site and a second allele with both a 66bp deletion and a 7bp duplication that deleted the PKR initiation codon. A control clone (Cas9 Ctrl A) was constructed by transducing HF-Tert with a similar vector that did not contain a gRNA. Sequence analyses of the PKR locus in this clone revealed the wild-type genomic sequence.
Table 1.
Oligonucleotide primers.
| Primer # | 5′ to 3′ sequence |
|---|---|
| 622 | CTCGAGACCATGGGCGTGGGCACCCCGCGC |
| 783 | GCCTCTAACATTGAGACAGCATA |
| 841 | GGTTCTGCAGCTTCACCTGGTAACCTTGGTGGTTTCCAATCTTC |
| 876 | GTTACCGAATTCGCGGCCGCCCATCATCACCATCACCATTAACTGCA |
| 877 | GTTAATGGTGATGGTGATGATGGGCGGCCGCGAATTCG |
| 878 | GCGCGGCCGCATCAATTTCCAAACCACATCTC |
| 879 | GCGCGGCCGCACAACCTTTTGGAACAGTTGG |
| 881 | GCGCGGCCGCTTGAGCGTTGTAATGGTAATG |
| 917 | GCGCGGCCGCATCCAAATCATACCAAGAATCTTCACC |
| 918 | GCGCGGCCGCCTTTCTATTAGAACCCAAAACCCAAAAAG |
| 919 | GCGCGGCCGCTTTCTTCCATCTTCTTTGATAAACATCG |
| 1052 | TCGAGCTGGAGCCACCCCCAGTTCGAGAAGGGCGGCGGCAGCGGCGGCGGCAGCGGCGGCGGCAGCTGGAGCCACCCCCAGTTCGAGAAGGGT |
| 1053 | CTAGACCCTTCTCGAACTGGGGGTGGCTCCAGCTGCCGCCGCCGCTGCCGCCGCCGCTGCCGCCGCCCTTCTCGAACTGGGGGTGGCTCCAGC |
| 1096 | ACGTGCTTTGGCCCGGGCCGCCGCCGATTGGAAACCGCCACGTCT |
| 1097 | GTTTCCAATCGGCGGCGGCCCGGGCCAAAGCACGTCCCAAACTGG |
| 1098 | CGACGGGACGCCGCCGATTGGGCCCCGCCACGTCTCCCTGGGGA |
| 1099 | GACGTGGCGGGGCCCAATCGGCGGCGTCCCGTCGCAAAGCACGT |
| 1100 | CTCCCTGGGGCCGCCTCCTGGTACGACTTGGACG |
| 1101 | GTACCAGGAGGCGGCCCCAGGGAGACGTGGCGGT |
| 1102 | TCCTGGTACGCCTTGGCCGCCACTTTCTGGGTTCTGGGGAG |
| 1103 | CCCAGAAAGTGGCGGCCAAGGCGTACCAGGAGTCTTCCCCA |
| 1104 | GGGGAGTAACGCCAAAAACGCCGTGTATCAACGACGTTGGA |
| 1105 | TTGATACACGGCGTTTTTGGCGTTACTCCCCAGAACCCAGA |
| 1106 | AACCGTGTTAGCCTGTGGTTTGGCCATTGATCGTCCCATGCCAAC |
| 1107 | GACGATCAATGGCCAAACCACAGGCTAACACGGTTTTCTTCCAAC |
| 1108 | TGTGGTTTGGCCATTGCCGCCCCCATGCCAACGGTCCCCAA |
| 1109 | TTGGCATGGGGGCGGCAATGGCCAAACCACAGCGTAACACG |
| 1170 | CGAGGCTGATCAGCGGGTTTTACGAGGACAGGCTGGAGC |
| 1171 | TCACCATCACCATTGAGTTTGACGACGACGACAAGTAAGAAG |
| 1185 | TTTCTTGGCTTTATATATCTTGTGGAAAGGACGAAACACCGGTTTTAGAGCTAG |
| 1186 | GACTAGCCTTATTTTAACTTGCTATTTCTAGCTCTAAAACCGGTGTTTCGTCCT |
| 1187 | TTTCTTGGCTTTATATATCTTGTGGAAAGGACGAAACACCGTTCAGGACCTCCACATGAT |
| 1188 | GACTAGCCTTATTTTAACTTGCTATTTCTAGCTCTAAAACATCATGTGGAGGTCCTGAAC |
| 1197 | TAGTTAAGCTTGGTACCGAGCTCGTGACGCGGGTTTGCTTCCTATATAGTGGACGTCGGA |
| 1198 | CTGGGCCATGGCCGCCGGACGGATCCGGGCGCCGGACACCTCCGACGTCCACTATATAGG |
| 1199 | CTAGAAAGTATAGGAACTTCGAATTCACTGGTTTTCTTTTGCAGCTGTCGTTATGTTTCG |
| 2000 | GCTGGAGCCATGGCTGGTGACCTTGTAAAACAAGTTTTCGAAACATAACGACAGCTGCAA |
| 2001 | ATATGTTCTGTGAGCATCACTC |
| 2030 | GACCTCCACATGATAGGAG |
| 2049 | GCTACCACTCCACTTCACTTATT |
| 2050 | AGGCAATCACTCACCTTCTTT |
| 2180 | ATATGAAGCGTCGCGAGTATTA |
| 2183 | GCACGTCGCTGCCTATAAA |
| BGH rev. | TAGAAGGCACAGTCGAGG |
We constructed another PKR KO cell line (PKR KO B) by transducing HF with a construct containing doxycycline-inducible Cas9 and a gRNA targeting the first dsRNA-binding domain of PKR (pEQ1510; target sequence 5′-TTCAGGACCTCCACATGAT-3′; Cas9 is predicted to cut between the two underlined nucleotides). Following puromycin selection (1 μg/ml), Cas9 expression was induced by addition of 1 μg/ml doxycycline for a minimum of two weeks prior to analysis. Screening of this cell line as described above (using primers #2049 and #2050 for PCR amplification) showed only a single bp insertion at the predicted cut site.
Plasmids
We made a series of C-terminal truncations in TRS1 for use in yeast two-hybrid assays. We first introduced a silent mutation to create a BstEII site within TRS1 by PCR-amplifying pEQ1284 (Child et al., 2012) with primers #783 and #841 (Table 1). The amplicon was digested with BglII and PstI and inserted into those same sites in pEQ1284, resulting in pEQ1304. We then annealed primers #876 and #877 and ligated the product into the BstEII and PstI sites of pEQ1304, generating pEQ1331, which added EcoRI and NotI sites and a 6XHis tag to the C-terminus of TRS1[1-667]. For each additional 3′ truncation, we PCR-amplified pEQ1284 with forward primer #783 and the following reverse primers, digested the product with BglII and NotI, and inserted it into the BglII and NotI sites of pEQ1331. Reverse primer #917 was used for pEQ1360 (TRS1[1-679]); primer #918 for pEQ1361 (TRS1[1-690]); primer #919 for pEQ1362 (TRS1[1-700]; Primer #878 for pEQ1340 (TRS1[1-710]); primer #879 for pEQ1341 (TRS1[1-720]); and primer #881 for pEQ1342 (full length TRS1[1-795]).
To make plasmids for use in transfection assays, primers #1052 and #1053, containing sequences encoding a TwinStrep tag, were annealed and ligated into pEQ1180 (Child et al., 2012) that had been digested with XhoI and XbaI to generate pEQ1435, a plasmid that expresses full length TRS1 with TwinStrep and 6XHis tags. The same primers inserted into an EGFP-6XHis expression plasmid derived from pEQ1100 (Child et al., 2006) to generate pEQ1436. TRS1 was excised from pEQ1427, which expresses a triple mutant of TRS1 that does not bind to dsRNA (Bierle et al., 2013) and from pEQ978 (Hakki and Geballe, 2005), which contains a C-terminal truncation (TRS1[1-648]) using HindIII and XhoI and the fragments were cloned into the same sites in pEQ1435 to generate pEQ1442 and pEQ1445, respectively. A plasmid expressing kinase-dead PKR with a biotinylation signal and 6XHis tag (pEQ1232) was constructed by transferring PKR from pEQ1198 (Child et al., 2012) into the pSV2 vector backbone and then cloning in the biotinylation signal from pEQ1068 (Child and Geballe, 2009). To design charged-cluster-to-alanine mutants of TRS1, we aligned TRS1 and IRS1 sequences from HCMV Towne (Accession # FJ616285) and Chimpanzee CMV strain Heberling (Accession # NC_003521) using ClustalX 2.1. To introduce these mutations, TRS1 was amplified in two separate reactions, the first using a common upstream primer (#622) along with a mutant reverse primer (Table 1) and the second using a mutant forward primer and a common reverse primer (BGH). Subsequently, stitch PCR was used to join each set of products using the #622 and BGH reverse primers. The final PCR products were digested with PpuMI and XhoI and cloned into the same sites in pEQ1445. The specific mutant forward and reverse primer pairs were as follows: pEQ1468 (Mut 1) used #1096 and #1097, pEQ1469 (Mut 2) used #1098 and #1099, pEQ1470 (Mut 3) used #1100 and #1101, pEQ1471 (Mut 4) used #1102 and #1103, pEQ1472 (Mut 5) used #1104 and #1105, pEQ1473 (Mut 6) used #1106 and #1107, and pEQ1474 (Mut 7) used #1108 and #1109. The amino acids that were mutated to alanine in each of these constructs are indicated in Fig. 2a.
FIG. 2. Binding properties of TRS1 charged-cluster-to-alanine mutants.



(a) HCMV TRS1 and IRS1 (Towne strain) were aligned with Chimpanzee CMV TRS1 and IRS1 (Heberling strain). Clusters of two to five charged residues within five residue windows near the C-terminus were identified (highlighted in yellow). Seven of these clusters were selected for mutation to alanine in human TRS1 (* denotes identical residues; + indicates conserved charge in at least two other genes). (b) For PKR binding assays, COS7 cells were co-transfected with kinase-dead PKR-BH and the indicated TRS1-S2H expression plasmids. At 48 hours post-transfection, lysates were harvested and pulldown assays performed as detailed in Materials and Methods. Input samples representing 5% of each lysate (top panel) were visualized using a His antibody. Following pulldown, bound PKR was detected by immunoblot analysis using the PKR B-10 antibody (bottom panel). (c) For dsRNA binding assays, 293T cells were transfected with the indicated TRS1-S2H constructs or a GFP-S2H control. At 48 hours post-transfection, input samples (I) and proteins that bound to poly[I:C] Sepharose beads (B) or control beads (C) were analyzed by immunoblotting with His antibody.
To construct pEQ1510, the 2kb filler sequences in lentiCRISPR ((Shalem et al., 2014) a gift from Feng Zhang, Addgene plasmid # 49535) were removed by BsmBI digestion, followed by the introduction of a pair of annealed primers that contain an AgeI restriction site (#1185 and #1186) using Gibson Assembly (New England Biolabs). A NotI to XhoI fragment was then excised and cloned into pCW-Cas9 ((Wang et al., 2014), a gift from Eric Lander and David Sabatini, Addgene plasmid # 50661) cut with the same enzymes. This yielded a lentiviral vector expressing tet-inducible Cas9 and containing the U6 promoter, an AgeI site into which different gRNAs can be introduced and the gRNA scaffold. The resulting vector, pEQ1508, was then digested with AgeI, and paired, annealed primers designed to target the first dsRBD of PKR (#1187 and #1188) were introduced by Gibson Assembly.
To construct plasmids for recombination into bacterial artificial chromosomes (BACs), a FRT-Kan-FRT cassette, PCR amplified from pSLFRTkn (Atalay et al., 2002) using primers #1170 and #1171, was cloned into the PmeI site of pcDNA3.1-V5-His-TOPO vectors containing TwinStrep-tagged wild-type TRS1 (pEQ1435) or TRS1-Mut 1 (pEQ1468) by Gibson assembly. The plasmids were digested with BamHI, then paired, annealed oligonucleotides containing the upstream 50 bp homology arm (#1197 and #1198) were introduced by Gibson Assembly. The resulting vectors were then digested with EcoR1 and BstEII, and paired, annealed primers #1199 and #2000 containing the downstream 50 bp homology arm were inserted by Gibson Assembly to yield pEQ1521 (HCMV[TRS1-S2H]) and pEQ1524 (HCMV[TRS1-Mut 1]).
Yeast two-hybrid assay
Yeast two-hybrid assays were conducted as previously described using a yeast codon-optimized variant of TRS1 fused to the GAL4 DNA binding domain and kinase-dead PKR fused to the GAL4 transcriptional activation domain in yeast expression vectors (Child et al., 2012).
Pulldown assays
For the PKR pulldown assays, COS7 cells were transfected with TwinStrep and 6XHis-tagged (-S2H) TRS1 constructs and a Biotin and 6XHis-tagged, kinase-dead form of human PKR (PKR-BH, pEQ1232), using Lipofectamine 2000 (Invitrogen). At 48 hours post-transfection, the cells were pelleted at 2000 × g for 5 minutes at 4°C and washed with cold PBS. The cell pellet was resuspended in 150 μl of lysis buffer (50 mM Tris HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 0.5% NP-40, with freshly added 1 μM each of NaVO4, homoarginine, and benzamidine and 100uM PMSF) at 4°C and incubated on ice for 20 minutes, with periodic vortexing. Nuclei were removed by centrifugation at 10,000 × g for 10 minutes at 4°C. 5 μl per sample was saved on ice as the input and the remaining 145 μl was used for the pulldown. A 1:6.5 mixture of Streptactin beads (IBA Lifesciences; Schmidt 2013) and Sepharose CL-B6 beads (Sigma) was washed twice with lysis buffer and resuspended in 600ul lysis buffer. The lysates were added to the bead mixture and incubated on a rotator at 4°C for 2 hours. The beads were pelleted and washed three times with lysis buffer and twice with wash buffer (lysis buffer without the protease inhibitors and NP-40). Proteins were eluted from the beads by heating to 95°C in loading buffer and separated by SDS-PAGE.
For the dsRNA pulldown, 293T cells were transfected with TRS1-expressing plasmids using Lipofectamine 2000 (Invitrogen) and lysates were harvested at 48 hours post-transfection. The dsRNA-binding assay was performed as previously described (Bierle et al., 2013).
VVΔE3L rescue assays
HeLa cells were transfected in triplicate with expression plasmids using Lipofectamine 2000 (Invitrogen). After 48 hours, the cells were infected (MOI = 0.1) with VVΔE3L, a vaccinia virus mutant in which E3L has been replaced with a LacZ cassette (provided by Bertram Jacobs). At 48 hours post-infection (hpi), VVΔE3L replication was monitored by quantification of β-galactosidase activity by a fluorometric substrate cleavage assay essentially as has been described (Hakki and Geballe, 2005). VVΔE3L replication in PKR knockdown and knockout cell lines was assessed at 48 hpi (MOI = 0.1).
BAC recombineering
After digestion of pEQ1521 (wild-type TRS1) and pEQ1524 (TRS1-Mut 1) with Asp718 and BstEII, the TRS1-FRT-kan-FRT fragments were transformed into Escherichia coli EL250 containing an HCMV BAC lacking both IRS1 and TRS1 (HCMV[ΔI/ΔT] (Marshall et al., 2009)). Following selection of kanamycin-resistant colonies, the kanR gene was removed by arabinose induction of FLP recombinase followed by selection for kan-sensitive colonies. BAC DNAs were analyzed by digestion with EcoRI, BamHI, or HindIII followed by electrophoresis on 0.7% agarose gels and ethidium bromide staining.
To reconstitute viruses, BAC DNA was purified from E. coli using the NucleoBond BAC 100 kit (Clontech) and transfected along with the cre-expression plasmid pPBRepcre into HF-TRS1 using Lipofectamine LTX reagent (Life Technologies). Mutant viruses were propagated and titered in HF-TRS1 or in PKR KO A cells. Viral DNA was harvested from infected PKR KO cells by lysis with 2% SDS, proteinase K digestion, phenol:chloroform extraction and ethanol precipitation. The resulting DNAs were analyzed by PCR with primers located upstream (#2183) and downstream (#2180) of TRS1 and outside of the homologous recombination arms. The PCR products were sequenced with multiple forward and reverse primers, which yielded sequence covering the flanking regions and a minimum of 800 bp within both the 5′ and 3′ ends of TRS1. These results confirmed the identities of the wild-type and mutant TRS1 genes as well as their proper localization in the viral genome.
Immunoblot analyses
With the exception of pulldown assays (above), lysates for immunoblot assays were prepared by washing the monolayer with phosphate-buffered saline and lysing the cells with 2% SDS. After sonication to disrupt the cellular DNA, proteins were separated by 10% SDS-PAGE, transferred to a PVDF membrane (Millipore), and probed with the indicated antibodies using the Western Star chemiluminescent detection system (Applied Biosystems). Antibodies used in these experiments include eIF2α L57A5 (#2103), phospho-eIF2α Ser51 (#3597), and PKR D7F7 (#12297), all from Cell Signaling Technology, PKR B-10 (sc6282, Santa Cruz Biotechnology), Penta-His (Qiagen), phospho-PKR T446 (ab32026, AbCam), and Actin (A2066, Sigma). Rabbit antiserum that recognizes the TRS1/IRS1 dsRNA binding domain (anti-p999) has been described previously (Marshall et al., 2009).
RESULTS
Identification of TRS1 mutants that are unable to bind to PKR
In order to determine the role of PKR inhibition by TRS1 in HCMV replication, we first sought to identify TRS1 mutants that are unable to bind to and antagonize PKR. Previous data indicated that a C-terminal region of TRS1 is critical for these functions (Hakki and Geballe, 2005; Hakki et al., 2006). Building on these prior observations, we used yeast two-hybrid assays to more precisely delineate the region of TRS1 required for interaction with PKR. Consistent with our previous report (Child et al., 2012), yeast codon-optimized TRS1 bound to kinase-dead (K296R) human PKR (Fig. 1). Progressive deletions from the TRS1 C-terminus resulted in a gradual decrease in binding. One C-terminal TRS1 truncation (TRS1[1-679]), which was previously reported to not bind detectably to PKR in mammalian cell assays (Hakki et al., 2006), exhibited considerably decreased but measurable PKR binding in this assay. Further deletion of codons following residue 667 eliminated all detectable binding. Although these results did not define a discrete position at which binding is eliminated, they did corroborate the importance of the C-terminus of TRS1 for binding to PKR.
FIG. 1. Yeast two-hybrid analysis of the interaction between PKR and TRS1.
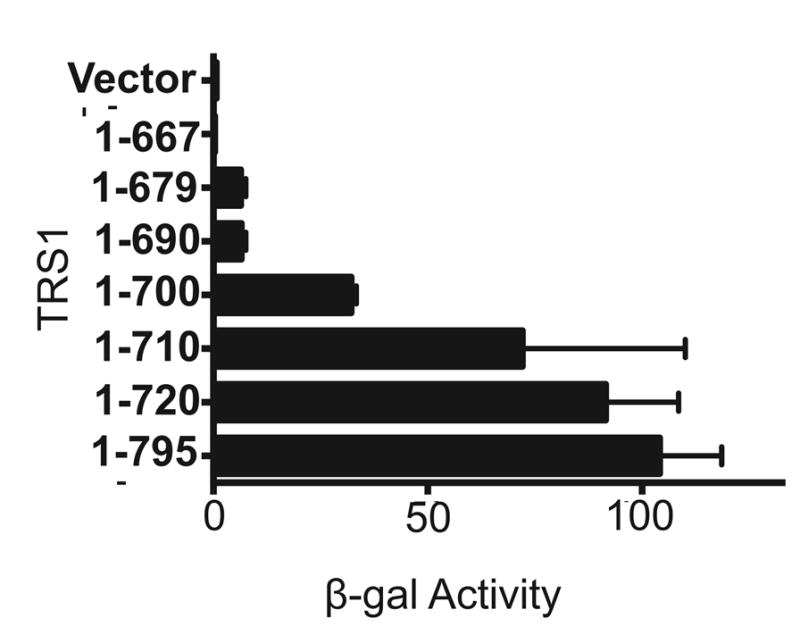
Plasmids expressing kinase-dead PKR fused to the GAL4 activation domain and the indicated regions of TRS1 fused to the GAL4 binding domain or an empty vector control were co-transformed into yeast. The strength of the protein-protein interactions was assessed by β-galactosidase activity assays.
In order to generate a TRS1 mutant that fails to bind to PKR but is expected to retain other functional interactions for which the C-terminus is known to be dispensable (Chaumorcel et al., 2012; Hakki and Geballe, 2005; Strang et al., 2010), we next constructed C-terminal charged-cluster-to-alanine (CCTA) mutations. This method, which has been used in studies of other HCMV proteins and in other systems (Schuessler et al., 2010; Schuessler et al., 2008; Schuessler et al., 2012), capitalizes on the propensity for charged clusters of amino acids to be surface-exposed. Therefore, charged clusters are likely to be mediators of protein-protein interactions and to tolerate mutations without drastically impacting the overall protein structure. An alignment of HCMV TRS1 and IRS1 with closely related chimpanzee CMV homologs reveals several conserved charged amino acid clusters (Fig. 2a). We chose seven clusters that possess at least two conserved residues within a five-residue window and engineered alanine mutations at each of these positions to produce mutants TRS1-Mut 1 through TRS1-Mut 7.
We evaluated PKR binding to these mutants by co-transfecting plasmids that express kinase-dead PKR along with each of the TRS1 variants into 293T cells. The S2H-tagged TRS1 proteins were pulled down using Streptactin beads, after which bound proteins were separated by SDS:PAGE then subjected to immunoblot analysis with an anti-PKR antibody. Expression of the His-tagged transfected genes was also monitored by immunoblot analysis of the cell lysates. As shown previously, wild-type TRS1 and an N-terminal TRS1 mutant that cannot bind to dsRNA (Triple Mut) both bound to PKR, while a C-terminal deletion mutant (TRS1[1-648]) and a GFP control plasmid did not (Fig. 2b, lanes 1–3, 11; (Bierle et al., 2013; Hakki et al., 2006)). Several of the CCTA mutants bound PKR (Fig. 2b, lanes 6, 8, 9, and 10), indicating that these residues are dispensable for this activity. However, TRS1-Mut 1 did not bind to PKR at all and TRS1-Mut 2 bound only weakly (Fig. 2b, lanes 4 and 5). Although TRS1-Mut 4 did not appear to bind to PKR, its expression was lower that the other TRS1 variants in this and other experiments (not shown).
These experiments identified PKR binding deficiencies of TRS1-Mut 1 and TRS1-Mut 2. We expected the CCTA mutants to retain other known TRS1 activities that have been reported to function even when the C-terminal PKR-binding region is deleted (Chaumorcel et al., 2012; Hakki and Geballe, 2005; Strang et al., 2010). To test this prediction, we performed dsRNA-binding assays with each of the CCTA mutants. Immunoblot analysis of proteins bound to poly[I:C] beads revealed that wild-type TRS1 bound dsRNA, while Triple Mut did not (Fig. 2c). As in past experiments, we found considerable variation in the intensity of binding (Bierle et al., 2013). In contrast to the negative controls, Triple Mut and GFP, all of the CCTA mutants bound dsRNA at least to a detectable level. The one exception was TRS1-Mut 4, which again expressed quite poorly. These results suggest that these CCTA mutations, including those that completely disrupt PKR binding, retained some dsRNA-binding activity, although some of the mutants appear to bind to dsRNA less well than the wild-type protein.
PKR binding is required for VVΔE3L rescue by TRS1
Previous data showed that the C-terminal region of TRS1 that is required for PKR binding is also required to rescue replication of VVΔE3L, a vaccinia virus mutant lacking its own PKR antagonist, E3L (Hakki and Geballe, 2005). Therefore, to determine more specifically whether PKR binding is necessary for TRS1 to rescue VVΔE3L, we tested three of our CCTA mutants. HeLa cells were transfected with plasmids expressing TRS1-Mut 1, TRS1-Mut 2, or TRS1-Mut 3, which bind to PKR to varying extents (Fig. 2b). After transfection, cells were infected with VVΔE3L (MOI = 0.1) and viral replication was measured. Wild-type TRS1 rescued VVΔE3L replication, whereas Triple Mut, TRS1[1-648], and GFP did not (Fig. 3), consistent with previous reports (Bierle et al., 2013; Hakki and Geballe, 2005). TRS1-Mut 1 failed to rescue VVΔE3L, while TRS1-Mut 2 rescued VVΔE3L to an intermediate degree and TRS1-Mut 3 rescued to wild-type levels. TRS1-Mut 5, 6, and 7, all of which bound to PKR as well as wild-type TRS1 did, also rescued VVΔE3L to a level similar to wild type TRS1 (data not shown). Thus, the ability of the mutants to rescue VVΔE3L replication correlated well with their ability to bind PKR, supporting the conclusion that PKR binding by TRS1 is likely important for blocking PKR activity and enabling VVΔE3L replication.
FIG. 3. PKR binding is required for VVΔE3L rescue.

HeLa cells were transfected with the indicated TRS1 constructs or a GFP control and infected with VVΔE3L (MOI = 0.1) at 48 hours post-transfection in triplicate. At 48 hpi, VVΔE3L replication was measured by β-galactosidase activity assay (top panel). Expression of TRS1 (and actin as a loading control) was monitored by immunoblot assay (bottom panel).
PKR binding by TRS1 is necessary for HCMV replication
Although these data suggest that the ability of TRS1 to bind PKR is required for rescue of VVΔE3L, the importance of PKR binding and inhibition by TRS1 during HCMV infection has not yet been tested. Therefore, we constructed recombinant HCMVs expressing wild-type TRS1 or TRS1-Mut 1 as described in Materials and Methods. The expected structures of the BAC DNAs were verified by restriction enzyme digestion (Fig. 4a). We reconstituted each virus by transfection into HF-TRS1. The correct location and identity of the TRS1 gene in each of the reconstituted viruses was confirmed by PCR using primers external to the TRS1 locus (Fig. 4b) and sequencing (data not shown). TRS1 protein expression following infection of HF-TRS1 cells (which express very low amounts of TRS1) was detectable from all viruses, with the exception of HCMV[ΔI/ΔT] (Fig. 4c).
FIG. 4. Analyses of the BACs and recombinant viruses.



(a) BAC DNAs corresponding to the indicated recombinant viruses were digested with restriction enzymes and separated on agarose gels. Arrowheads point to bands expected to differ between the BACs. (b) Primers external to the homology arms used for recombination into the HCMV[ΔI/ΔT] BAC were used to PCR-amplify this region from the indicated viruses. The HCMV[TRS1-S2H] and HCMV[TRS1-Mut1] products are ~ 0.23 kb larger than TRS1 from AD169 due to sequences encoding the epitope tags. Amplification of HCMV[ΔI/ΔT] DNA revealed the expected TRS1 deletion. (c) HF-TRS1 cell lysates were made at 72 hpi with the indicated viruses (MOI = 3). Immunoblot assays were performed with an antibody to the dsRNA-binding domain shared by TRS1 and IRS1. * indicates IRS1 expressed from AD169.
To examine whether PKR binding by TRS1 is necessary for HCMV replication, we infected HF with each virus and measured the amount of virus produced in cell supernatants by titering on HF-TRS1 (Fig. 5a). AD169 and HCMV[TRS1-S2H] replicated in HF, while HCMV[ΔI/ΔT] did not, consistent with previous results (Marshall et al., 2009). We did not detect any HCMV[TRS1-Mut 1] replication in HF (Fig. 5a). A second HCMV mutant containing TRS1-Mut 1 also failed to replicate at all in HF (data not shown), supporting the conclusion that the replication defect in HCMV[TRS1-Mut 1] is due to the TRS1-Mut 1 and not to another mutation elsewhere in the genome. Although these data do not exclude the possibility that the mutations in TRS1-Mut 1 also weaken another TRS1 function for which the C-terminal region is not absolutely required, they do suggest that PKR binding by TRS1 is most likely necessary for HCMV replication.
FIG. 5. HCMV[TRS1-Mut 1] fails to replicate in HF.


(a) HF were infected in triplicate with the indicated viruses (MOI = 0.1). Virus present in the cell culture medium was collected at two-day intervals and viral titers were determined on HF-TRS1 cells. Mean titers (and standard deviations) are shown. (b) Lysates from HF that were mock-infected or infected with each virus (MOI = 3) were collected at 48 hpi and the levels of total and phosphorylated PKR and eIF2α were examined by immunoblot analysis using the indicated antisera.
TRS1-Mut 1 fails to block PKR activation
During infection of HF with HCMV[ΔI/ΔT], the abundance of phosphorylated eIF2α increases and protein synthesis levels are reduced, suggesting that the failure of HCMV[ΔI/ΔT] to replicate is due to the inability of this virus to block PKR activation (Marshall et al., 2009). To evaluate the impact of the inability of HCMV[TRS1-Mut 1] to bind to PKR on activation of the PKR pathway, we infected HF with HCMV[TRS1-Mut 1] or control viruses at an MOI of 3, collected lysates at 48 hpi, and evaluated the levels of phosphorylated and total PKR and eIF2α by immunoblot assays.
While infection with AD169 resulted in a very low amount of phosphorylated PKR, similar to the level detected in mock-infected cells, infection with either of two viruses that express TRS1 but not IRS1 (HCMV[TRS1-HA] or HCMV[TRS1-S2H]) resulted in a small increase of phospho-PKR compared to mock- and AD169-infected cells. In contrast, considerably more phospho-PKR accumulated after infection with either HCMV[TRS1-Mut 1] or HCMV[ΔI/ΔT] (Fig. 5b). In addition, the amount of total PKR was markedly reduced after infection with HCMV[TRS1-Mut 1] or HCMV[ΔI/ΔT]. This observation might reflect a reduction in PKR expression following HCMV[TRS1-Mut 1] and HCMV[ΔI/ΔT] infection due to the shutoff of protein synthesis that occurs following PKR activation, as has been observed in other systems (Bierle et al., 2012; Child et al., 2012; Rothenburg et al., 2009).
Analyses of eIF2α in these same lysates revealed an increase in phosphorylated eIF2α during infection with HCMV[TRS1-Mut 1] or HCMV[ΔI/ΔT] compared to mock infection or infection with AD169, HCMV[TRS1-HA] or HCMV[TRS1-S2H] (Fig. 5b, compare lanes 4 and 5 to lanes 1 and 6). We also detected a decrease in the amount of total eIF2α, similar to that observed for total PKR, again suggesting an effect of shutoff of translation in cells in which TRS1 was absent or unable to block the PKR pathway. Cells infected with either of the two viruses that express TRS1 but not IRS1 caused an intermediate level of phosphorylated eIF2α to accumulate, but did not show any reduction in total eIF2α. These results are consistent with moderate but incomplete inhibition of PKR pathway activation by these viruses.
Most importantly, these data reveal that the PKR pathway is activated during infection with HCMV[TRS1-Mut 1] just as it is following infection with HCMV[ΔI/ΔT]. Therefore, TRS1-Mut 1 appears to be unable to prevent PKR activation during HCMV infection, supporting the hypothesis that blocking PKR activation is an essential function of TRS1.
PKR knockdown restores HCMV[ΔI/ΔT] replication
The above data confirm that PKR antagonism is likely critical for HCMV replication, but do not exclude the possibility that additional TRS1 functions are also essential. To explore this possibility, we next tested the ability of HCMV[ΔI/ΔT] to replicate in cells deficient in PKR expression.
First, we used an shRNA directed against PKR to knock down expression in primary HF. This PKR-kd cell line expressed much less PKR than cells containing a control shRNA as can be seen by immunoblot analysis (Fig. 6a, lanes 1 and 2). As well, VVΔE3L replication was significantly increased in the knockdown cells, indicating a reduction in PKR-mediated inhibition (Fig. 6b), as has been reported in other cell types (Child et al., 2012; Zhang et al., 2008).
FIG. 6. PKR expression in knockdown and knockout cells.


(a) The abundance of total PKR in PKR knockdown and knockout cell lines compared to that in control cells was measured by immunoblot assay. VVΔE3L replication in triplicate wells of (b) PKR knockdown cells or (c) PKR knockout cells was quantified by measuring β-galactosidase activity at 48 hpi as described in Materials and Methods.
We next infected the PKR-kd cells with AD169, HCMV[TRS1-S2H], and HCMV[ΔI/ΔT] to determine whether reduced PKR expression was sufficient to rescue HCMV lacking both TRS1 and IRS1. We quantified the amount of virus present in the medium after infection by titering the viruses on HF-TRS1. Replication of AD169 and HCMV[TRS1-S2H] in the PKR-kd cells was ~10-fold lower than in Ctrl-kd cells, but this modest difference was not always observed in other experiments and in other PKR deficient cells (see below). However, HCMV[ΔI/ΔT] was consistently able to replicate only in the PKR-kd cells (Fig. 7). At 6 days post-infection, HCMV[ΔI/ΔT] titers in the PKR-kd cells were one to two logs lower than those of AD169 or HCMV[TRS1-S2H]. This could indicate that TRS1 might provide one or more auxiliary functions that aid replication. Alternatively, this reduction in HCMV[ΔI/ΔT] titer might simply be due to an inhibitory effect of the small amount of residual PKR remaining in these cells. Regardless, these data demonstrate that PKR antagonism is the only function of TRS1 that is essential for HCMV replication in HF.
FIG. 7. Knockdown of PKR partially restores HCMV[ΔI/ΔT] replication.

Triplicate wells of PKR knockdown and control HF were infected with AD169, HCMV[TRS1-S2H] or HCMV[ΔI/ΔT] (MOI = 0.1) Virus present in the cell culture medium was collected at two-day intervals and viral titers determined on HF-TRS1 cells. Mean titers (and standard deviations) are shown.
PKR knockout restores replication of HCMV[ΔI/ΔT] and HCMV[TRS1-Mut 1]
To determine whether the reduced replication of HCMV[ΔI/ΔT] compared to AD169 and HCMV[TRS1-S2H] in the PKR knockdown cells might be due to the presence of residual PKR, we generated two independent PKR knockout cell lines using CRISPR/Cas9 technology. The first cell line (PKR KO A) contained disruptive mutations adjacent to a splice acceptor site in both alleles, as described in Materials and Methods. The second cell line (PKR KO B) targeted the first double-stranded RNA binding domain in PKR. In this case, the cells contained a single nucleotide insertion in both alleles that resulted in a frameshift mutation.
PKR was expressed in vector-matched control cells but was undetectable in PKR KO A or PKR KO B cells (Fig. 6a, lanes 3–6). Additionally, when these cells were infected with VVΔE3L, the virus replicated to a much higher level in both KO lines than in control cells (Fig. 6b), and we were not able to detect any phospho-PKR (data not shown), confirming the reduction in PKR expression.
We next evaluated the impact of PKR knockout on the replication of AD169 and the HCMV recombinants. Both PKR KO lines and their paired control cells were infected with HCMV[TRS1-S2H], HCMV[TRS1-Mut 1], or HCMV[ΔI/ΔT] at an MOI of 0.1 and the amount of progeny virus in supernatants was determined by titering on HF-TRS1. As expected, HCMV[TRS1-S2H] replicated well and both HCMV[TRS1-Mut 1] and HCMV[ΔI/ΔT] failed to replicate in control cells (Fig. 8). In contrast, both of these viruses replicated as well as HCMV[TRS1-S2H] in each knockout cell line, again confirming that TRS1 is required to block the PKR pathway. This result also suggests that any ancillary functions performed by TRS1 are unnecessary and do not contribute substantially to viral replication in this system (Fig. 8).
FIG. 8. PKR knockout fully restores HCMV[TRS1-Mut 1] and HCMV[ΔI/ΔT] replication.

Triplicate wells of PKR KO A and PKR KO B cells, along with paired control cells, were infected with HCMV[TRS1-S2H], HCMV[TRS1-Mut 1], or HCMV[ΔI/ΔT] (MOI = 0.1). Virus accumulating in the medium from days 6 to 8 post-infection was titered on HF-TRS1 cells. Mean titers (and standard deviations) are shown.
DISCUSSION
HCMV replication depends on either TRS1 or IRS1 (Marshall et al., 2009). While each of these proteins have been reported to have multiple functions, which of these are essential for replication of HCMV remained unknown prior to this study. TRS1 and IRS1 are partially encoded within the repeats surrounding the unique short region of the genome and thus their N-terminal two-thirds are identical. In addition, their C-termini are ~50% conserved. Thus, it is not surprising that they share several reported activities, including inhibition of autophagy and of the PKR and RNase L pathways (Chaumorcel et al., 2012; Child et al., 2004; Mouna et al., 2015). Both TRS1 and IRS1 are able to bind to dsRNA, PKR, and Beclin-1 (Chaumorcel et al., 2012; Hakki and Geballe, 2005; Hakki et al., 2006; Mouna et al., 2015). Also, TRS1 has been reported to play a role in virion assembly and to bind the HCMV DNA processivity factor UL44 as well as binding to the 7-methylguanosine mRNA cap (Adamo et al., 2004; Blankenship and Shenk, 2002; Strang et al., 2010; Ziehr et al., 2015). IRS1 encodes a second smaller protein, IRS1263, which was reported to be a transcriptional repressor (Romanowski and Shenk, 1997). Moreover, these genes may have additional, as yet unknown, activities that could be necessary for replication.
In this study we provide strong evidence, based on analyses of mutant viruses and cell lines, that the sole essential function of TRS1 and IRS1 during infection of cultured HF is the ability to inhibit the PKR pathway. We generated a TRS1 mutant, TRS1-Mut 1, that is unable to bind to PKR. This mutant could not rescue VVΔE3L replication and, importantly, when inserted into HCMV[ΔI/ΔT], was incapable of rescuing HCMV replication. In contrast, either of two wild-type TRS1 genes (TRS1-HA or TRS1-S2H) or the major PKR antagonist from vaccinia virus (E3L, which also binds to PKR), expressed from the same virus backbone could each rescue viral replication (Marshall et al., 2009). These results suggest that TRS1 needs to interact directly with PKR to inhibit PKR activation. However, in a prior study, we identified a mutant that does not bind to dsRNA or rescue VVΔE3L replication, yet does bind to PKR ((Bierle et al., 2013) and Fig. 2b and c). Thus, PKR binding appears to be necessary but not sufficient for TRS1 to block PKR activation. These results suggest a model in which TRS1 heterodimerizes with PKR in a complex that depends on the ability of TRS1 to bind to both PKR and dsRNA, thereby preventing PKR homodimerization and activation.
Although these results are consistent with the conclusion that the essential role of TRS1 is to block the PKR pathway, it remained possible that another function of TRS1 that might be necessary for HCMV replication could have been disrupted by the mutations introduced into TRS1-Mut 1. In order to address this issue more directly, we constructed PKR knockdown and knockout cell lines. Our finding that HCMV[ΔI/ΔT] was able to replicate in each of these PKR deficient cell lines but not in control lines demonstrates that the only essential function of TRS1 (or IRS1) is PKR antagonism.
Like HCMV, many other viruses contain one or more genes that target the PKR pathway in order to enable viral replication (Mohr et al., 2007). Mouse cytomegalovirus (MCMV) m142 and m143 act together to block PKR (Child et al., 2006; Valchanova et al., 2006). Deletion of either gene abolishes MCMV replication and results in PKR pathway activation in wild type MEFs, but MCMV lacking both genes replicates as well as wild-type MCMV in PKR-null MEFs (Budt et al., 2009). Moreover, viruses lacking either m142 or m143 fail to replicate in wild-type mice but replicate to levels similar to wild-type MCMV in PKR-null mice (Ostermann et al., 2015), indicating that the only apparent role for m142 and m143 is to block the PKR pathway. VVΔE3L is incapable of replicating in many cell types and is avirulent in wild-type mice, but its replication is at least partially restored in PKR-deficient cells and mice (Brandt and Jacobs, 2001; Rice et al., 2011). Similarly, the reduced neurovirulence of herpes simplex virus 1 lacking ICP34.5 in wild-type mice is restored in PKR-null mice (Leib et al., 2000). Vaccinia virus and herpes simplex virus 1 each contain at least one additional PKR antagonist, K3L and US11, respectively, that might account for the ability of the ΔE3L and ΔICP34.5 viruses to replicate in some settings (Langland and Jacobs, 2002; Mulvey et al., 2004). These examples highlight the critical contribution of PKR antagonism to viral replication in multiple viral systems.
Our studies also reveal insights into why HCMV might have retained two seemingly redundant PKR antagonists. Although other reports have indicated that deletion of just IRS1 does not impact replication, we have observed a mild reduction in the replication of two different viruses that lack IRS1, HCMV[TRS1-HA] and HCMV[TRS1-S2H] ((Marshall et al., 2009) and Fig. 5a). Moreover, infection with these viruses results in an increased level of phospho-PKR compared to cells infected with wild-type HCMV (Fig. 5b). Although it is possible that the epitope tags weaken TRS1’s ability to block PKR in these viruses, our data suggest the possibility that TRS1 alone is not as efficient at blocking PKR as are TRS1 and IRS1 in combination. In studies involving experimental evolution of vaccinia virus recombinants, we found that gene amplification of a weak PKR antagonist led to improved viral replication (Brennan et al., 2014; Elde et al., 2012). In the same way, it is possible that deletion of TRS1 or IRS1 leads to a reduction in the total level of PKR antagonism during HCMV replication. Some data suggest that wild-type HCMV expresses more TRS1 than IRS1 (Bierle et al., 2013; Mouna et al., 2015), which may explain the replication defect observed in single deletion mutants of TRS1 but not IRS1 (Blankenship and Shenk, 2002; Dunn et al., 2003; Jones and Muzithras, 1992). It is possible that simultaneous expression of both proteins might be necessary in order to generate an optimal concentration of TRS1 and IRS1 to block PKR.
Our finding that PKR antagonism is the only essential function of TRS1 and IRS1 raises questions about the importance of the other activities that have been attributed to these factors. Some of those functions may be indirect consequences of their ability to block PKR activation. For example, the described activation of reporter gene expression might simply be due to maintenance of protein synthesis as a result of TRS1 or IRS1 blocking PKR. Consistent with this interpretation, we found that TRS1 did not activate reporter gene expression in PKR-deficient cells (Bierle et al., 2012). Ziehr et al found that most but not all of the increase in reporter gene expression by TRS1 was eliminated by PKR depletion (Ziehr et al., 2015). Their study implicated a PKR-independent effect of TRS1 on translation that might be mediated by the ability of TRS1 to bind to the mRNA cap. Likewise, TRS1’s reported role in virion formation might be a secondary consequence of the effects of TRS1 on translation of factors needed for assembly. We previously reported that TRS1 and IRS1 are able to prevent activation of the OAS/RNaseL pathway in the setting of VVΔE3L infection (Child et al., 2004). However, infection with HCMV[ΔI/ΔT] did not activate this pathway (Marshall et al., 2009), possibly because PKR activation represses viral replication prior to accumulation of sufficiently high levels of dsRNA to activate OAS/RNaseL. Regardless, the finding that HCMV[ΔI/ΔT] replicates in PKR–deficient cells demonstrates that antagonism of the RNaseL pathway is not an essential function of TRS1 or IRS1.
In contrast to reported activities that might simply result from maintenance of translation, the inhibition of autophagy by TRS1 and IRS1 cannot be explained by antagonism of PKR (Chaumorcel et al., 2012; Mouna et al., 2015). These activities map to separate domains of the proteins. While PKR inhibition requires the C-terminus of TRS1 and IRS1, this domain is not necessary for Beclin-1 binding or blocking autophagy. Conversely, the N-terminal 44 codons of both proteins are required to block autophagy efficiently but not to inhibit PKR. In addition, TRS1 blocks autophagy even in PKR-null cells. Finally, some data show that autophagy induction may aid, rather than inhibit, HCMV replication (Mouna et al., 2015; Zhao et al., 2013), but another report suggests the reverse effect (Belzile et al., 2015). Regardless, our data indicate that inhibition of autophagy is not an essential function of TRS1 or IRS1.
Our results reveal that inhibition of PKR is the single essential function of TRS1 or IRS1, at least in cultured HF infected with HCMV AD169. Indeed, the observation that HCMV[ΔI/ΔT] replicates as well as wild-type virus in PKR-null cells suggests that TRS1’s other activities contribute little to productive HCMV replication in cell culture. Although it remains possible that other functions of TRS1 and IRS1 might have an important or even essential role in other cell types or in natural HCMV infection, these studies highlight the importance of PKR as a strong viral restriction factor that has compelled many viruses to evolve effective evasion strategies.
HIGHLIGHTS.
TRS1 must bind to PKR in order to inhibit its activity.
Inhibition of PKR is the only essential function of TRS1.
The other functions performed by TRS1 do not augment HCMV replication in cell culture.
Acknowledgments
We thank Denise Galloway and Harmit Malik (Fred Hutchinson Cancer Research Center [FHCRC]), Elizabeth Gray and Daniel Stetson (University of Washington), and Bertram Jacobs (Arizona State University) for reagents. We also thank the Genomics Core facility (FHCRC) for technical assistance.
This work was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health NIH grant under award number RO1AI027762 to A.P.G. J.E.B. was supported in part by a grant from the National Institute of General Medical Sciences, Public Health Service, National Research Service Award, T32 GM007270. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Jacquelyn E. Braggin, Email: jbraggin@uw.edu.
Stephanie J. Child, Email: schild@fhcrc.org.
Adam P. Geballe, Email: ageballe@fhcrc.org.
References
- Adamo JE, Schroer J, Shenk T. Human cytomegalovirus TRS1 protein is required for efficient assembly of DNA-containing capsids. J Virol. 2004;78:10221–10229. doi: 10.1128/JVI.78.19.10221-10229.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atalay R, Zimmermann A, Wagner M, Borst E, Benz C, Messerle M, Hengel H. Identification and expression of human cytomegalovirus transcription units coding for two distinct Fcgamma receptor homologs. J Virol. 2002;76:8596–8608. doi: 10.1128/JVI.76.17.8596-8608.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belzile JP, Sabalza M, Craig M, Clark E, Morello CS, Spector DH. Trehalose, an mTOR-Independent Inducer of Autophagy, Inhibits Human Cytomegalovirus Infection in Multiple Cell Types. J Virol. 2015 doi: 10.1128/JVI.02651-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierle CJ, Schleiss MR, Geballe AP. Antagonism of the protein kinase R pathway by the guinea pig cytomegalovirus US22-family gene gp145. Virology. 2012;433:157–166. doi: 10.1016/j.virol.2012.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierle CJ, Semmens KM, Geballe AP. Double-stranded RNA binding by the human cytomegalovirus PKR antagonist TRS1. Virology. 2013;442:28–37. doi: 10.1016/j.virol.2013.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankenship CA, Shenk T. Mutant human cytomegalovirus lacking the immediate-early TRS1 coding region exhibits a late defect. J Virol. 2002;76:12290–12299. doi: 10.1128/JVI.76.23.12290-12299.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt TA, Jacobs BL. Both carboxy- and amino-terminal domains of the vaccinia virus interferon resistance gene, E3L, are required for pathogenesis in a mouse model. J Virol. 2001;75:850–856. doi: 10.1128/JVI.75.2.850-856.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan G, Kitzman JO, Rothenburg S, Shendure J, Geballe AP. Adaptive gene amplification as an intermediate step in the expansion of virus host range. PLoS Pathog. 2014;10:e1004002. doi: 10.1371/journal.ppat.1004002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budt M, Niederstadt L, Valchanova RS, Jonjic S, Brune W. Specific inhibition of the PKR-mediated antiviral response by the murine cytomegalovirus proteins m142 and m143. J Virol. 2009;83:1260–1270. doi: 10.1128/JVI.01558-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaumorcel M, Lussignol M, Mouna L, Cavignac Y, Fahie K, Cotte-Laffitte J, Geballe A, Brune W, Beau I, Codogno P, Esclatine A. The human cytomegalovirus protein TRS1 inhibits autophagy via its interaction with Beclin 1. J Virol. 2012;86:2571–2584. doi: 10.1128/JVI.05746-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Child SJ, Brennan G, Braggin JE, Geballe AP. Species specificity of protein kinase r antagonism by cytomegalovirus TRS1 genes. J Virol. 2012;86:3880–3889. doi: 10.1128/JVI.06158-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Child SJ, Geballe AP. Binding and relocalization of protein kinase R by murine cytomegalovirus. J Virol. 2009;83:1790–1799. doi: 10.1128/JVI.01484-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Child SJ, Hakki M, De Niro KL, Geballe AP. Evasion of cellular antiviral responses by human cytomegalovirus TRS1 and IRS1. J Virol. 2004;78:197–205. doi: 10.1128/JVI.78.1.197-205.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Child SJ, Hanson LK, Brown CE, Janzen DM, Geballe AP. Double-stranded RNA binding by a heterodimeric complex of murine cytomegalovirus m142 and m143 proteins. J Virol. 2006;80:10173–10180. doi: 10.1128/JVI.00905-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dever TE, Dar AC, Sicheri F. The eIF2alpha kinases. In: Matthews MB, Sonenberg N, Hershey JWB, editors. Translational Control in Biology and Medicine. Cold Spring Harbor Press; 2007. pp. 319–344. [Google Scholar]
- Dunn W, Chou C, Li H, Hai R, Patterson D, Stolc V, Zhu H, Liu F. Functional profiling of a human cytomegalovirus genome. Proc Natl Acad Sci U S A. 2003;100:14223–14228. doi: 10.1073/pnas.2334032100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckard SC, Rice GI, Fabre A, Badens C, Gray EE, Hartley JL, Crow YJ, Stetson DB. The SKIV2L RNA exosome limits activation of the RIG-I-like receptors. Nat Immunol. 2014;15:839–845. doi: 10.1038/ni.2948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elde NC, Child SJ, Eickbush MT, Kitzman JO, Rogers KS, Shendure J, Geballe AP, Malik HS. Poxviruses deploy genomic accordions to adapt rapidly against host antiviral defenses. Cell. 2012;150:831–841. doi: 10.1016/j.cell.2012.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakki M, Geballe AP. Double-stranded RNA binding by human cytomegalovirus pTRS1. J Virol. 2005;79:7311–7318. doi: 10.1128/JVI.79.12.7311-7318.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakki M, Marshall EE, De Niro KL, Geballe AP. Binding and nuclear relocalization of protein kinase R by human cytomegalovirus TRS1. J Virol. 2006;80:11817–11826. doi: 10.1128/JVI.00957-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TR, Muzithras VP. A cluster of dispensable genes within the human cytomegalovirus genome short component: IRS1, US1 through US5, and the US6 family. J Virol. 1992;66:2541–2546. doi: 10.1128/jvi.66.4.2541-2546.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langland JO, Jacobs BL. The role of the PKR-inhibitory genes, E3L and K3L, in determining vaccinia virus host range. Virology. 2002;299:133–141. doi: 10.1006/viro.2002.1479. [DOI] [PubMed] [Google Scholar]
- Leib DA, Machalek MA, Williams BR, Silverman RH, Virgin HW. Specific phenotypic restoration of an attenuated virus by knockout of a host resistance gene. Proc Natl Acad Sci U S A. 2000;97:6097–6101. doi: 10.1073/pnas.100415697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall EE, Bierle CJ, Brune W, Geballe AP. Essential role for either TRS1 or IRS1 in human cytomegalovirus replication. J Virol. 2009;83:4112–4120. doi: 10.1128/JVI.02489-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr IJ, Pe’ery T, Mathews MB. Protein Synthesis and Translational Control during Viral Infection. In: Matthews MB, Sonenberg N, Hershey JWB, editors. Translational Control in Biology and Medicine. Cold Spring Harbor Press; 2007. pp. 545–599. [Google Scholar]
- Mouna L, Hernadez E, Bonte D, Brost R, Amazit L, Delgui LR, Brune W, Geballe AP, Beau I, Esclatine A. Analysis of the role of autophagy inhibition by two complementary human cytomegalovirus BECN1/Beclin 1-binding proteins. Autophagy. 2015 doi: 10.1080/15548627.2015.1125071. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulvey M, Camarena V, Mohr I. Full resistance of herpes simplex virus type 1-infected primary human cells to alpha interferon requires both the Us11 and gamma(1)34.5 gene products. J Virol. 2004;78:10193–10196. doi: 10.1128/JVI.78.18.10193-10196.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostermann E, Warnecke G, Waibler Z, Brune W. Knockout of the host resistance gene Pkr fully restores replication of murine cytomegalovirus (MCMV) m142 and m143 mutants in vivo. J Virol. 2015 doi: 10.1128/JVI.02003-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice AD, Turner PC, Embury JE, Moldawer LL, Baker HV, Moyer RW. Roles of vaccinia virus genes E3L and K3L and host genes PKR and RNase L during intratracheal infection of C57BL/6 mice. J Virol. 2011;85:550–567. doi: 10.1128/JVI.00254-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanowski MJ, Shenk T. Characterization of the human cytomegalovirus irs1 and trs1 genes: a second immediate-early transcription unit within irs1 whose product antagonizes transcriptional activation. J Virol. 1997;71:1485–1496. doi: 10.1128/jvi.71.2.1485-1496.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothenburg S, Seo EJ, Gibbs JS, Dever TE, Dittmar K. Rapid evolution of protein kinase PKR alters sensitivity to viral inhibitors. Nat Struct Mol Biol. 2009;16:63–70. doi: 10.1038/nsmb.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuessler A, Sampaio KL, Scrivano L, Sinzger C. Mutational mapping of UL130 of human cytomegalovirus defines peptide motifs within the C-terminal third as essential for endothelial cell infection. J Virol. 2010;84:9019–9026. doi: 10.1128/JVI.00572-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuessler A, Sampaio KL, Sinzger C. Charge cluster-to-alanine scanning of UL128 for fine tuning of the endothelial cell tropism of human cytomegalovirus. J Virol. 2008;82:11239–11246. doi: 10.1128/JVI.01069-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuessler A, Sampaio KL, Straschewski S, Sinzger C. Mutational mapping of pUL131A of human cytomegalovirus emphasizes its central role for endothelial cell tropism. J Virol. 2012;86:504–512. doi: 10.1128/JVI.05354-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG, Zhang F. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stasiak PC, Mocarski ES. Transactivation of the cytomegalovirus ICP36 gene promoter requires the alpha gene product TRS1 in addition to IE1 and IE2. J Virol. 1992;66:1050–1058. doi: 10.1128/jvi.66.2.1050-1058.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strang BL, Geballe AP, Coen DM. Association of human cytomegalovirus proteins IRS1 and TRS1 with the viral DNA polymerase accessory subunit UL44. The Journal of general virology. 2010;91:2167–2175. doi: 10.1099/vir.0.022640-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valchanova RS, Picard-Maureau M, Budt M, Brune W. Murine cytomegalovirus m142 and m143 are both required to block protein kinase R-mediated shutdown of protein synthesis. J Virol. 2006;80:10181–10190. doi: 10.1128/JVI.00908-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Wei JJ, Sabatini DM, Lander ES. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343:80–84. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Jacobs BL, Samuel CE. Loss of protein kinase PKR expression in human HeLa cells complements the vaccinia virus E3L deletion mutant phenotype by restoration of viral protein synthesis. J Virol. 2008;82:840–848. doi: 10.1128/JVI.01891-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Li Z, Wang M, Zhang Z, Ma H, Chang J, Gao D, Wang S. Manipulation of autophagy by HCMV infection is involved in mTOR and influences the replication of virus. Acta Biochim Biophys Sin (Shanghai) 2013;45:979–981. doi: 10.1093/abbs/gmt102. [DOI] [PubMed] [Google Scholar]
- Ziehr B, Lenarcic E, Vincent HA, Cecil C, Garcia B, Shenk T, Moorman NJ. Human cytomegalovirus TRS1 protein associates with the 7-methylguanosine mRNA cap and facilitates translation. Proteomics. 2015;15:1983–1994. doi: 10.1002/pmic.201400616. [DOI] [PMC free article] [PubMed] [Google Scholar]