

Abstract
Engineered nanoparticles (ENPs) are increasingly utilized for commercial and medical applications; thus, understanding their potential adverse effects is an important societal issue. Herein, we investigated protein S-glutathionylation (SSG) as an underlying regulatory mechanism by which ENPs may alter macrophage innate immune functions, using a quantitative redox proteomics approach for site-specific measurement of SSG modifications. Three high-volume production ENPs (SiO2, Fe3O4, and CoO) were selected as representatives which induce low, moderate, and high propensity, respectively, to stimulate cellular reactive oxygen species (ROS) and disrupt macrophage function. The SSG modifications identified highlighted a broad set of redox sensitive proteins and specific Cys residues which correlated well with the overall level of cellular redox stress and impairment of macrophage phagocytic function (CoO > Fe3O4 ≫ SiO2). Moreover, our data revealed pathway-specific differences in susceptibility to SSG between ENPs which induce moderate versus high levels of ROS. Pathways regulating protein translation and protein stability indicative of ER stress responses and proteins involved in phagocytosis were among the most sensitive to SSG in response to ENPs that induce subcytoxic levels of redox stress. At higher levels of redox stress, the pattern of SSG modifications displayed reduced specificity and a broader set pathways involving classical stress responses and mitochondrial energetics (e.g., glycolysis) associated with apoptotic mechanisms. An important role for SSG in regulation of macrophage innate immune function was also confirmed by RNA silencing of glutaredoxin, a major enzyme which reverses SSG modifications. Our results provide unique insights into the protein signatures and pathways that serve as ROS sensors and may facilitate cellular adaption to ENPs, versus intracellular targets of ENP-induced oxidative stress that are linked to irreversible cell outcomes.
Keywords: S-glutathionylation, nanotoxicology, macrophage, oxidative stress, redox proteomics, resin-assisted enrichment, immune functions
Graphical Abstract
With the number of nanotechnology-enabled products projected to double every three years involving 6 million nanotechnology workers by 2020,1 concerns over potential detrimental health effects from exposure to engineered nanoparticles (ENPs) are an important societal issue. Unfortunately, our understanding of how ENPs affect regulation of biological systems is inadequate to accurately assess their potential hazard, particularly at a mechanistic level. Macrophages are critical cellular targets for ENPs due to their high efficiency in scavenging foreign particles and their essential role in regulating immune signaling and inflammation. Indeed, a central premise in design of many targeted therapeutic ENPs is to avoid macrophage phagocytic clearance mechanisms and minimize subsequent immune effects. We recently found that exposure to some types of ENPs disrupt macrophage gene activation and inhibit their ability to phagocytize pathogenic bacteria, including Streptococcus pneumonia, the leading cause of community-acquired pneumonia.2 Such observations are important since increased risk of pneumonia is also observed in welders exposed to fumes that are rich in metal oxide nanoparticles, including iron, chromium and manganese oxides.3–5 Furthermore, increased risk of lung infections have been associated with exposure to ultrafine air pollution particulates (<200 nm), particularly in elderly individuals.6 These findings, along with experimental studies in rodents,7,8 suggest suppression of innate immune function is an important adverse effect of some ENPs. However, the underlying mechanisms involved and physicochemical properties of ENPs that cause these effects remain unclear.
The ability of ENPs to stimulate cellular oxidative stress has emerged as one of the leading hypotheses in nanotoxicology,9,10 suggesting that the toxicity potential of ENPs can be predicted from hierarchical analyses of their ability to generate redox stress in cells.11 However, the concept that oxidative stress is the key initiating event broadly involved in the cellular toxicity of ENPs has also been challenged, since some materials have been found to induce adverse effects in the absence of measurable oxidative stress or inflammation.12–14 Furthermore, loss of cellular redox control is a common phenomenon in virtually all processes that lead to cell death, and even noncytotoxic ENPs have been shown to alter the regulation of hundreds of genes.2,15,16 Frequently, nanotoxicity studies focus on general measures of oxidative stress, such as fluorescent reporter dyes (e.g., DCFH) or total glutathione. While these approaches can be rapid to implement, they often lack the sensitivity, specificity, and dynamic range needed to capture biological effects at subcytotoxic exposure levels, and provide little insight into the specific cellular pathways affected. Consequently, the chemical nature and specific molecular targets of oxidative stress, and how it influences regulation of specific biological pathways in cells exposed to ENPs, remains an important question to be addressed.13
Mounting evidence suggests that reversible oxidative post-translational modifications (PTMs) of protein cysteines by reactive oxygen and nitrogen species (ROS and RNS) represent a fundamental mechanism of cell signaling that modulates enzyme activities and protein functions in many cellular activities.17–22 In particular, protein S-glutathionylation (SSG) has emerged as an important type of redox modification that regulates transcription, mitochondrial metabolism, apoptosis, and other critical processes including immune function.19,23 Modification by SSG occurs through multiple mechanisms whereby glutathione reacts with oxidized derivatives of protein cysteines, such as sulfenic acid (–SOH), thiyl radicals (–S•), or S-nitrosyl (–SNO), thereby converting these oxidized forms to a more stable modification.18,22,23 SSG modifications are normally tightly controlled under physiological conditions and can be “repaired” (deglutathionylated) by the glutaredoxin (Grx) enzymes, a highly conserved family of thioltransferases.23,24 We and others have shown SSG modifications are selective toward specific protein subsets, likely depending on the pKa and chemical microenvironment of the cysteinyl residue.24,25 Substrate specificity for deglutathionylation of SSG-modified proteins by Grx has also been demonstrated,26,27 and differences in subcellular localization of Grx isoforms (e.g., Grx1, cytosol; Grx2, mitochondrial) may provide additional levels of substrate specificity. The significance of the SSG/Grx redox axis in immune regulation was also demonstrated recently. For example, Sakai et al. reported that the phagocytic and bactericidal activity of stimulated neutrophils is regulated by SSG modifications of actin.28 Lung alveolar macrophages from Grx1-deficient mice also have attenuated inflammatory cytokine expression responses to lipopolysaccharide (LPS), indicating that Grx1 is necessary for normal macrophage transcriptional activation.20 In fact, nearly a dozen signaling proteins that control activation of the NF-κB transcriptional pathway responsible for immune response have been reported to be regulated by SSG.19,24,26,29,30
We recently developed a quantitative redox proteomics approach that permits site-specific profiling of SSG modifications at a proteome-wide scale.25,31–33 The goal of this study was to apply this new analytical approach to identify the specific molecular targets and pathways in macrophages that are susceptible to redox-dependent regulation following exposure to ENPs. On the basis of our previous work, we selected three high volume commercial ENPs (SiO2, Fe3O4, and CoO) predicted to cause low, moderate, and high redox stress and impacts on macrophage function, respectively.2 Global comparison of the patterns of SSG modifications across treatment groups not only confirmed large differences in overall redox stress induced by these ENPs, but also identified >1100 new SSG sites not previously reported. Through global profiling, cellular pathways under potential redox regulation with increasing levels of cellular oxidative stress were revealed, including pathways associated with ER stress and phagocytosis. Finally, we demonstrated that macrophages with reduced Grx repair capacity have defective phagocytosis, confirming the important role of the SSG/Grx redox axis as an important mediator of the effects of ENPs on innate immune functions.
RESULTS AND DISCUSSION
Characterization of ENPs and ENP-Induced Oxidative Stress
On the basis of our previous work2 and other studies of the toxicity of metal oxide ENPs,11,34 we chose three high volume commercially used ENPs (SiO2, Fe3O4, and CoO) that are predicted to induce low, medium, and high levels of oxidative stress in cells, respectively. All three ENPs are broadly used for industrial applications in food packaging, microelectronics, ceramics, and catalysts, and are also of biomedical importance. SiO2 and Fe3O4 ENPs are also promising mediums for drug delivery,35,36 while nanoparticles composed of cobalt oxides are a potential source of inflammation from degradation of orthopedic implants.37,38 SiO2 and CoO ENPs were obtained commercially, whereas Fe3O4 ENPs were produced in-house, as previously described.2 The physicochemical characteristics of these particles in complete RPMI culture medium, including primary particle size, agglomerate size, effective agglomerate density, and zeta potential, have been extensively characterized and described previously,2,39 and are summarized in Table 1. Transmission electron microscopy (TEM) confirmed the primary size of the nanoparticles that were prepared in house (Figure S5). For cell treatments, the ENPs were first dispersed in fetal bovine serum and subsequently diluted in complete RMPI (with 10% serum) to achieve final concentrations, as previously reported.34 Cells were thereby exposed to ENPs bearing serum protein coronas with net negative surface charges (Table 1). DLS analysis showed some agglomeration occurred when the ENPs were dispersed in complete culture medium, as expected. However, the polydispersity of the particles after dispersion in cell culture medium did not significantly change for at least 24 h.
Table 1.
Characterization of ENPs
| ENP type | source | sizea (nm) | RPMI
|
||
|---|---|---|---|---|---|
| hydrodynamic diameter (nm)b | polydispersity | ζ potential (mV) | |||
| Cobalt oxide(II) (CoO) | Skyspring Nanomaterials Inc. Cat# 2310 SC | <100 | 199 ± 1 | 0.16 | −18.0 ± 3.1 |
| Silicon oxide (SiOx) | Nanostructured and Amorphous Materials Inc. Cat# 4850 MR | 15 | 40 ± 2 | 0.35 | −8.0 ± 3.7 |
| Superparamagnetic iron oxide (Fe3O4) | In House | 13 | 216 ± 3 | 0.16 | −9.0 ± 7.69 |
Manufacturer specified size.
Hydrodynamic diameter obtained by DLS analysis at a concentration of 50 μg/mL in RPMI culture medium.
Quantification of lactate dehydrogenase (LDH) release in RAW 264.7 cells after 24 h exposure revealed that SiO2 and Fe3O4 ENPs were not cytotoxic up to ENP concentrations of 100 μg/mL, whereas CoO induced a dose-dependent increase in cytotoxicity that was significant compared to controls at ≥25 μg/mL concentrations (Figure 1A). The level of cellular oxidative stress induced by the ENPs was also evaluated by measuring the mRNA expression of heme oxygenase-1 (HMOX1), and cellular glutathione levels. HMOX1 is a critical stress response protein that is regulated by redox-sensitive transcription factors.40 Figure 1B shows treatment with Fe3O4 and CoO ENPs caused a dose-dependent increase in HMOX1 expession, with a relatively greater induction caused by CoO ENPs. In contrast, elevations in HMOX1 expression were not observed with SiO2 ENP treatment. Figure 1C,D shows the levels of total glutathione (GSH) and the oxidized glutathione (GSSG)/GSH ratio in RAW 264.7 cells treated by these ENPs. Total GSH levels were unchanged by SiO2 and Fe3O4 ENPs at ENP concentrations of <50 μg/mL, whereas decreases in total GSH content (p < 0.05) were observed with all CoO concentrations tested. This result is in agreement with the previous reported association between cellular GSH levels and nanoparticle-induced cytotocity,41 but also illustrates the limited sensitivity of total GSH as a measure of cellular redox stress.42 A shift to more oxidative cellular conditions was indicated by a significantly increased GSSG/GSH ratio observed for CoO ENP exposures. Fe3O4 ENPs caused only a modest increase in GSSG levels at the highest concentration tested, and no change occurred with SiO2 ENPs. The greater increases in GSSG/GSH ratios caused by CoO compared to Fe3O4 ENPs is in good agreement with the HMOX1 data. The results clearly confirm the differential cellular redox stress induced by these ENPs as expected based on the different physicochemical properties and redox potential of the core metal oxides.
Figure 1.

ENP-induced cytotoxicity and oxidative stress in RAW 264.7 cells. (A) Quantitative lactate dehydrogenase (LDH) assay demonstrated the cytotoxicity induced by different ENPs in macrophages. The final cytotoxicity = 100 × (Total Dead Cells/Total Cells), where Total was the sum of cell counts from supernatants and cells on the plate. (B) Quantitative RT-PCR showed dose-dependent increases in the mRNA levels of HMOX1. SiO2-induced alterations of HMOX1 are marginal, while Fe3O4 and CoO induced significant increases of HMOX1 mRNA. As indicated by the resulting data, the oxidative stress potential of ENPs was assigned as follows: SiO2 ≪ Fe3O4 < CoO. (C) Concentration of total GSH in ENP-treated cells. (D) GSSG/GSH ratios in ENP-treated cells. The concentration of GSH and GSSG in cells was measured by using GSH/GSSG-Glo Assay kit (Promega). The concentrations of total GSH and GSSG in each well were calculated by the intensity of luminescence with 3 biological replicates. Results are displayed as means ± standard error. Note: *, p < 0.05; **, p < 0.01 vs untreated controls.
Site-Specific SSG Modifications
Following confirmation of ENP-induced oxidative stress at a broad level, we investigated whether the pattern of protein SSG modifications reflected the level of cellular redox stress induced by the ENPs. Initial Western blot experiments with anti-SSG antibody indicated that the overall level of SSG increased in a time-dependent manner following CoO ENP treatment (Figure S1). To quantitatively profile site-specific SSG at the proteome-wide level, we applied our recently developed redox proteomic strategy for site-specific identification and quantification of SSG (Figure 2A).25 Briefly, RAW 264.7 macrophages were treated for 24 h with a low concentration (12.5 μg/mL) of the ENPs, which represents a subcytotoxic dose (>90% viability). Cells were lysed in the presence of free thiol blocking reagent N-ethylmaleimide (NEM). After free thiol blocking, SSG-modified proteins were selectively deglutathionylated with a reaction cocktail containing a mutant form of Grx1 (Grx1M, C14S), glutathione reductase (GR), reduced nicotinamide adenine dinucleotide phosphate (NADPH) and GSSG. The reduced proteins were then captured on thiopropyl sepharose 6B resin, followed by on-resin tryptic digestion and isobaric labeling with 6-plex tandem mass tags (TMT). For quantification by LC–MS/MS, we devised a 6-plex experiment with three biological replicates each for both control and each ENP treatment condition (Figure 2B).
Figure 2.

Quantitative site-specific profiling of S-glutathionylation in ENP-treated cells. (A) The overall workflow scheme for enrichment and site-specific identification of SSG-modified Cys-peptides. (Left) the enrichment and quantitative analysis for SSG-modified Cys-peptides; (right) the enrichment and quantitative analysis for total Cys-peptides. (B) Schematic workflow for MS-based multiplexed quantification of SSG in cells. RAW 264.7 cells were treated with SiO2, Fe3O4, and CoO ENPs at a concentration of 12.5 μg/mL for 24 h. Three biological replicates for both untreated and treated conditions were used for TMT6 labeling and LC–MS/MS analysis. (C) Pie graph summary of overlaps between the identified SSG sites and the annotated Cys sites in existing databases, including dbSNO, dbGSH and UniPort disulfide database.
As shown in Table 2, a total of 2494 unique SSG-modified Cys sites from 1276 proteins were identified across all treatment conditions. The number of SSG sites and proteins identified were relatively consistent across each ENP treatment group (Table 2 and Tables S1–S3). Moreover, there was a significant overlap in the SSG sites observed among these ENP-treatment conditions (Figure S2). To further evaluate the SSG sites identified, we compared the results across several available protein databases from Mus musculus. First, we mapped the results against an S-glutathionylation database, dbGSH,43 which contains 2006 SSG-modified Cys sites from 1128 proteins. We found that ~37% of the unique SSG sites (929 out of 2494) were previously annotated as SSG sites (Figure 2C). Since S-nitrosylated (SNO) cysteines can also serve as precursors for SSG, we also compared our results with dbSNO,44 an S-nitrosylation database containing 2646 SNO-modified Cys sites from 1355 proteins. This analysis identified 655 SSG sites that were reported to be also susceptible to SNO. Thus, in total, 47% of the SSG sites matched to either previously SSG or SNO redox-sensitive sites (Figure 2C). Finally, the data was compared to a disulfide database from Uniprot containing 29 782 Cys sites from 2867 proteins. Although the disulfide database is 10 times larger than the dbGSH or dbSNO databases, only ~10% of the SSG sites in our list matched to sites in this database. Among these sites, ~30% were also cross-matched in the dbGSH and dbSNO databases with an additional ~22% annotated as redox active sites by Uniprot. Therefore, only 5.4% of the SSG sites were exclusively annotated as disulfides. The high percentage of the SSG sites matched to the redox databases along with the low percentage matching the disulfide database strongly support that our list of identified SSG sites is specifically enriched for redox-sensitive Cys sites. Importantly, our results revealed over 1100 new SSG sites that have not been previously reported based on the cross-comparison of databases.
Table 2.
Identification of SSG-Modified Proteins and Cys Sites in Macrophages after Exposure by Different ENPs
| ENPs | unique Cys sites | unique proteins | statistically changed sitesa | substantial changed sitesb (proteins) |
|---|---|---|---|---|
| SiO2 | 1686 | 948 | 453 | 238 (182) |
| Fe3O4 | 1521 | 851 | 464 | 235 (178) |
| CoO | 1512 | 845 | 842 | 622 (416) |
| Combined | 2494 | 1276 | - | - |
SSG sites with statistically significant changes, p-value <0.05.
p-value < 0.05, |log2 ratio| > 0.3 vs untreated control.
ENP-Induced Alterations of Site-Specific SSG
Statistical analysis of the SSG levels showed that 27%, 30%, and 56% of the SSG sites were significantly altered following SiO2, Fe3O4, and CoO ENP treatment, respectively, compared to untreated controls (Table 2, p ≤ 0.05, t test). As shown in Figure 3A, CoO induced the highest increase in overall SSG level, followed by Fe3O4, while SiO2 ENPs induced a general pattern of slightly decreased SSG level. After applying a fold change cutoff of ≥0.3 in log2 ratio and a p-value of ≤0.05, we identified 238, 235, and 622 Cys sites with substantial alterations in SSG following SiO2, Fe3O4, and CoO treatment, respectively (Table 2 and Figure 3B). The overall patterns of SSG alterations are in good agreement with the oxidative stress results of Figure 1 and demonstrate that Fe3O4 and CoO ENPs induce a robust oxidative stress while SiO2 ENPs induced a minor cellular reductive stress.
Figure 3.

Alterations of SSG modifications under different ENP conditions. (A) Heat maps of Cys sites with significant SSG alterations under different ENP conditions. (B) Alteration of SSG levels under treatment by different ENPs. Fold change is log2 transformed. Cutoff for significance is p < 0.05 in ANOVA analysis.
Since cellular protein expression may also be altered following the treatment of ENPs, we next attempted to determine whether the changes in the measured SSG ratios might be due to ENP-induced changes in total protein abundance. For this purpose, we performed an quantitative analysis of total protein abundance changes after CoO exposure (Tables S4 and S5 and Figure S2). Total protein abundance levels were quantified based on the reporter ion intensities of the total Cys-containing peptides using the strategy shown in Figure 2A where blocking of free thiols was not applied. In this experiment, the abundances of a total of 1434 proteins were quantified, which covered ~94% of the SSG modified proteins identified under the same treatment condition. With the use of the same filtering threshold (p-value ≤0.05 and |log2 ratio| ≥ 0.3 versus controls), 355 proteins displayed significant changes in abundance after CoO exposure (212 up-regulated and 143 down-regulated). However, only 83 of these proteins were also associated with significant SSG changes (Figure S2). Moreover, more than 40% of these 83 proteins showed a larger increase in SSG level in comparison to their change in protein abundance (Figure S3). The results confirmed that the majority of measured increases in SSG modifications were not due to altered protein abundances. We note that the quantitative protein abundance results also provide an independent insight into the oxidative stress response at the proteome level after ENP treatment. For instance, following CoO ENP treatment, multiple stress-related proteins were found to be substantially altered in their abundances, including multiple chaperones (15 total) and isomerases (14 total) (Table S6). Functional analysis revealed that proteins representing several biological processes related to stress responses, such as protein folding, cell redox homeostasis, and acute inflammatory response, were significantly altered in abundance indicating a compensatory response to oxidative stress at the protein expression level following ENP treatment.
Functional Analyses of Proteins with Significant SSG Alterations
To obtain an overall picture of the biological processes and molecular functions that are potentially regulated by SSG, Gene Ontology (GO) analysis was performed using the DAVID software tool.45,46 A total 497 proteins with substantial SSG changes (|log2 ratio| ≥ 0.3, p < 0.05) in both Fe3O4 ENP- and CoO ENP-induced stress conditions were combined and analyzed. A prominent number of these proteins are enzymes, including kinases, phosphatases, ubiquitin protein ligases, deubiquitinases, peptidases, oxidorductases and acetyl-transferases (Figure S4). Other protein functions identified included transporters, transcription regulators, transmembrane receptors, translation regulators, heat shock proteins, and ion channel proteins. This is in agreement with previous reports that proteins in these functional categories are regulated by SSG.19,22,23,47 As showed in Figure 4A, the SSG-modified proteins were broadly distributed across major cellular compartments and represented diverse biological processes and molecular functions.
Figure 4.

Functional analyses of proteins with significant SSG alterations. (A) Summary of GO analysis. (B) Top canonical pathways from IPA analysis. Proteins with significant elevation of SSG modifications were used for the functional analyses. Threshold of significance: p-value ≤ 0.05 and |log2 ratio| ≥ 0.3 versus controls. Moderate ROS condition: The proteins with significant elevation of SSG modifications under Fe3O4 treatment. High ROS condition: The proteins with significant elevation of SSG modification only under CoO treatment, and |log2 ratio| (CoO) – |log2 ratio|(Fe3O4) ≥ 0.2. Other conditions were the same as Figure 2.
As expected, functional categories related to redox homeostasis and cellular stress response are among the most significantly influenced by SSG. For example, a substantial share of SSG proteins have been identified in organelles related to redox functions, such as endoplasmic reticulum (ER) and mitochondria.48–50 The ER and mitochondria are organelles with well-known redox regulated functions and ENP-induced dysfunction of these organelles can give rise to increased ROS production and oxidative stress.50–52 ER stress-related molecular functions, such as unfolded protein binding and protein disulfide isomerase activity, are also highly enriched. Several biological processes that are associated with cell redox homeostasis, including “response to unfolded protein” and “response to oxidative stress”, are also significant. All of these observations support the idea that S-glutathionylation plays a potential regulatory role in mitochondrial and ER functions as part of the overall oxidative stress response. Several processes associated with macrophage phagocytosis and endocytosis functions, such as actin-binding, cortical cytoskeleton organization, and integrin-mediated signaling pathway, were also significantly enriched with SSG modified proteins. Other enriched processes associated with transcriptional regulation, including translation and nucleotide binding, are consistent with our previous findings that redox-active ENPs can induce global transcriptional reprogramming in mouse macrophages.2 We also observed several processes relevant to protein degradation and cell apoptosis, such as proteasome complex, ubiquitin-protein ligase activity, and apoptotic cell clearance, suggesting a role of SSG in the regulation of apoptosis and cell death.
To identify the canonical pathways impacted by SSG, Ingenuity Pathway Analysis (IPA) was applied. Since Fe3O4 and CoO ENPs induced relatively moderate and high levels of oxidative stress in macrophages, respectively, we used these treatment groups to compare and contrast pathways susceptible to modifications at different levels of redox stress. Figure 4B showed the pathways that are significantly enriched with SSG-modified proteins in response to the moderate (Fe3O4) and high ROS (CoO) conditions, respectively. It is interesting that several pathways regulating protein synthesis, protein stability, and phagocytosis were found to be most significantly enriched in SSG-modified proteins in response to the moderate level of ENP-induced redox stress by Fe3O4. For example, the eIF2 signaling pathway, which is a well-known for regulating mRNA translation initiation in response to stress and bacterial invasion,53 ranked as the most significant pathway impacted by SSG modifications associated with Fe3O4 ENP treatment. Other pathways associated with translational regulation and protein turnover (e.g., “Regulation of eIF4 and p70S6K Signaling”, “tRNA Charging”, “RAN Signaling” and “Protein Ubiquitination Pathway”) were also enriched with proteins sensitive to moderate redox stress. These pathways, along with the “mTOR Signaling Pathway”, play important anabolic functions for maintaining translational regulation,54 nucleo-cytoplasmic transport of macromolecules,55 and protein synthesis56 and degradation.57 In addition, the “Fc-gamma Receptor-mediated Phagocytosis in Macrophages and Monocytes” pathway58,59 was also significantly enriched with SSG-modified proteins, which is consistent with our observation that the treatment by Fe3O4 ENPs can remarkably suppress phagocytic activity of macrophages even at a low level of exposure.2
In contrast, at the higher ROS levels associated with CoO ENP exposure, a broader set of pathways associated with intermediary metabolism (Glycolysis I), and signaling pathways associated with cell adhesion (Integrin Signaling, Paxicillin Signaling, PI3K/AKT Signaling), and oxidative stress responses (NRF-2-mediated Oxidative Stress Response) become more prominent, indicating these pathways are triggered only at higher levels of ROS (Figure 4B). Among these, the “PI3K/AKT Signaling” and “ERK/MAPK Signaling” are key signal transduction pathways initiated by extracellular stresses and involved in inflammatory processes.60,61 Whereas pathways that regulate protein translation are more susceptible to lower levels of redox stress, at higher cellular ROS levels associated with CoO ENPs, the “Unfolded Protein Response” signaling network and protein degradation (Ubiquitination Pathway) become more significant. Collectively, the results suggest the SSG modifications induced by ENP exposure initiate responses intended to respond to ER stress.
Potential Regulatory Roles of SSG in ER Stress Response
To explore the potential regulatory roles of SSG, we also examined whether the SSG modifications occur at the active or functional sites of enzymes. By interrogating the Uniprot database, we find that 259 out of 497 proteins with substantial SSG changes have annotated information regarding their active or functional sites (Tables S7 and S8). We identified 112 Cys sites on either active site or functional domains of 60 proteins that were susceptible to SSG modification. For instance, 13 SSG sites matched to the active sites on 9 proteins, including glyceraldehyde 3-phosphate dehydrogenase (GAPDH), ubiquitin-protein ligases and protein disulfide isomerases. Also, 22 SSG sites from 10 proteins were identified as metal binding sites. In addition, 77 of the SSG sites from 53 proteins were found in essential domains for protein functions (Tables S7 and S8). These proteins include allosteric enzymes, ligases and kinases, having functions such as ATP-binding, metal binding, and actin-binding. These data support that SSG serves as a mechanism for directly modulating protein functions.
ER stress is known to play a key role in oxidative stress through the accumulation of misfolded proteins in ER, and the up-regulation of molecular chaperones.62 It has been previously reported that a large number of ER proteins are redox-regulated. In our studies, we observed increases in SSG modifications across a number of ER proteins after both CoO and Fe3O4 ENP treatment, although the majority of these changes were more substantial with CoO ENPs (Figure 5A). For instance, the ER provides a quality-control (QC) system for the correct folding of proteins and for sensing stress, including the calnexin/calreticulin cycle.63,64 Calnexin (CALX) and calreticulin (CALR) are two lectin chaperones that interact with and assist the folding of N-linked monoglucosylated proteins. Protein disulfide isomerases (PDIs), oxidoreductases and other chaperones are also involved in the folding of glycoproteins. Glucosidase II (GANAB) and UDP-glucose:glycoprotein glucosyltranferase (UGGG) are additional ER enzymes responsible for dissociating and reglucosylating of the substrate glycoprotein, respectively. These components collectively comprise a continuous cycling system that promotes correct folding of proteins that enter the secretory pathway and targets misfolded proteins for degradation. We observed significantly increased levels of SSG on all these components, supporting the role of SSG in the QC system of ER. PDIs are major components of the QC system, which promote disulfide interchange activity and facilitate the formation of native disulfide bonds in nascent proteins.62 The inhibition of their enzymatic activity by SNO modification has been previously reported.65 In our work, we identified several PDIs, including PDIA1, PDIA3, PDIA4 and TXND5, with substantially increased levels of SSG modifications after exposure to CoO ENPs. Interestingly, most of the SSG-modified sites on PDIs were localized to their active Cys sites in the redox center of the thioredoxin motif as showed in Figure 5B.66 SSG modification of these Cys sites on PDIs may inhibit their isomerase activity and abrogate the chaperone activity, thus triggering an unfolded protein response.
Figure 5.

SSG modifications in ER stress. (A) Quantitative MS results showing levels of SSG modifications on Cys sites of several ER proteins. (B) Identification of SSG modifications on the active sites of protein disulfide isomerases (PDIs). (C) Western blotting results showing levels of ER stress proteins (p-PERK, PERK, BiP, CHOP) and Caspase-3 proteins in cells treated with 12.5 μg/mL CoO ENPs. (D) Western blotting results of p-PERK, PERK, and caspase-3 proteins in cells were treated with Fe3O4 ENPs and CoO ENPs for 24 h at the indicated concentration.
To validate the predictions from proteomic results that ENP treatment induces ER stress, independent experiments were performed to measure expression of several ER stress marker proteins after CoO ENP exposure by Western blot, including PERK (protein kinase RNA-like ER kinase), p-PERK (phosphorylated form of PERK), BiP (immunoglobulin binding protein), and CHOP (C/EBP homologous protein). In addition, levels of total and activated caspase-3 (CASP-3) were measured to determine if ER-stress induced by CoO ENPs was associated with apoptotic cascades. As showed in Figure 5C, significant increases in PERK phosphorylation along with increased expression of downstream BiP protein were observed at 12–24 h after CoO ENP treatment (12.5 μg/mL). Increases in levels of CHOP protein, a ER stress-induced transcription factor that also functions as a proapoptotic protein,67–69 were also evident at 24 h after CoO ENP treatment. These results, along with quantitative MS data that revealed increased abundances of several general ER stress markers, including PDI, heat shock protein 90 (HSP90) and calnexin (Table S6), confirm that CoO ENP treatment triggers the PERK-dependent ER stress pathway. Western blot data also demonstrated increased activation (cleavage) of CASP-3, directly supporting an apoptotic mode of cell death in CoO ENP-treated cells. Although CHOP induction and prolonged ER stress are associated with apoptotic signaling,67–69 time course studies show that CASP-3 cleavage precedes CHOP induction following CoO ENP treatment, indicating these apoptotic markers may be mediated by independent mechanisms. Increases in markers associated with intrinsic apoptotic cascades in cells treated with CoO ENPs are also consistent with the significantly enhanced SSG modifications of mitochondrial proteins (e.g., Glycolysis) observed by pathway enrichment analysis (Figure 4). Cells treated with Fe3O4 ENPs also displayed increased phosphorylation of PERK, albeit at a lower level of activation compared to CoO ENPs (Figure 5D). These observations are generally consistent with the quantitative MS data of ER proteins (Figure 5A) and suggest the ER stress induced by Fe3O4 is likely a adaptive response to moderate redox stress conditions rather than a pro-apoptotic response. Consistent with the observed differences in cytotoxic potential between these ENPs (Figure 1A), the levels of total and activated CASP-3 in cells treated with Fe3O4 ENPs were also significantly less than compared to CoO ENPs.
Role of SSG in Phagocytosis
In addition to ER stress-related processes, IPA pathway analysis also identified proteins associated with receptor-mediated phagocytosis and phagosome formation as sensitive targets of SSG modification. This observation may be particularly important for understanding how some ENP types modulate macrophage innate immune functions involving phagocytic uptake and clearance of bacterial pathogens.2,8 Actin is an essential component of the cytoskeleton and plays important roles in regulation of immunological responses, tissue remodeling and repair.59 Significant increases in SSG levels were observed on actin binding proteins in macrophages exposed to both Fe3O4 and CoO (Figure 6A), indicating these proteins are particularly susceptible to SSG modifications under even low levels of ENP-induced oxidative stress. SSG alterations of several 14–3–3 proteins (γ/ζ/θ/η) involved in regulating actin dynamics were also observed, consistent with the knowledge that 14–3–3 proteins are redox-regulated.70 Other proteins that play important roles in F-actin formation, such as actin-related proteins (ARC1B and ARP3), adenylyl cyclase-associated protein 1 (CAP1), filamins (FLNA and FLNB), were also modified.71,72 Our results are consistent with recent reports that redox modification of Cys residues occurs on these proteins in response to ROS generation72 or cadmium-induced oxidative stress.73 In addition, plastin-2 (PLSL) and talin-1 (TLN1) are essential proteins associated with phagocytosis and immune defense in macrophages.74,75 Together, our results suggest a close link between SSG modification and the regulation of cytoskeleton remodeling.
Figure 6.

SSG Modifications of Macrophage Phagocytosis Proteins. (A) Alterations of SSG modifications on actin-binding proteins. (B) Inhibitory effects of ENPs on the phagocytic activity of macrophages. The suppression of phagocytic activity is correlated with the oxidative stress induced by ENPs.
The impact of different ENPs on the phagocytic function of RAW 264.7 cells was experimentally assessed by exposing the cells to SiO2, Fe3O4 and CoO ENPs at different concentrations for 24 h followed by challenging the cells for 2 h with fluorescently labeled S. pneumoniae, a model pathogen and the leading cause of community-acquired pneumonia. Flow cytometry analysis (Figure 6B) showed that while macrophages exposed to SiO2 ENPs displayed normal phagocytic activity, the ability to phagocytize S. pneumoniae was dramatically decreased in macrophages exposed to Fe3O4 or CoO ENPs. These inhibitory effects were also observed at the lowest ENP treatment concentration tested (6.25 μg/mL, p < 0.01). The ENP-dependent effects on phygocytic activity are well correlated with the change of SSG levels in cells under treatment of these ENPs (Figure 3B).
To further validate the regulatory role of SSG in phagocytosis, RNA silencing was performed to knock down the expression of Grx1 in macrophages. Grx1 plays an important role in the maintenance of redox balance by catalyzing deglutathionylation of SSG-modified proteins and restoring their normal functions. Knockout or down-regulation of Grx1 expression can cause augmentation of SSG-modified proteins and inhibition of their activities in cells, eventually leading to cellular dysfunction.23,28–30 Using siRNA, we were able to selectively suppress expression of Grx1 mRNA and protein (Figure 7). Control experiments which included scrambled siRNA showed the Grx1 siRNA had no effect on the related Grx2 enzyme expression or expression of a housekeeping gene (Chpa). Consistent with the expected increase in SSG modifications due to reduced Grx1 activity,30,76 knockdown of Grx1 also resulted in ~60% inhibition in macrophage phagocytic function compared to control cells (Figure 7C). Collectively, the results indicate that even relatively low (subcytotoxic) levels of oxidative stress, such as that induced by Fe3O4 ENPs, are sufficient to alter macrophage function through SSG modifications.
Figure 7.

Regulatory role of Grx1 in phagocytosis of macrophages. (A) Western blot analysis of Grx1 expression after RNA silencing. (B) qRT-PCR analysis of Grx1 expression at mRNA level. (C) Effects of Grx1 knockdown on the phagocytic activity of macrophages.
CONCLUSIONS
In this study we investigated protein S-glutathionylation, a major reversible oxidative modification that regulates multiple aspects of inflammation, innate immunity and cell death, as a potential mechanism underlying ENP-induced oxidative stress. Our results demonstrated that the levels of SSG modifications correlate well with the overall level of cellular oxidative stress induced by three types of ENPs (CoO > Fe3O4 ≫ SiO2), as determined by generalized measures of cellular redox state, such as cellular GSH content and GSSG/GSH ratio. However, a major advantage of our quantitative redox proteomic approach is the ability to discern differential sensitivities among specific proteins and cellular pathway targets impacted by different physicochemical types of ENPs. For instance, the large differences in SSG modifications in cells exposed to SiO2 and Fe3O4 ENPs extend our previous findings that Fe3O4 ENPs cause dramatic transcriptional reprogramming of macrophages, whereas SiO2 ENPs have relatively little impact on gene regulation, despite being internalized by macrophages at similar levels.2 These observations demonstrate that the cytotoxic potential of a particle, which is often used as a surrogate for measuring biocompatibility, is a poor determinant of the bioactivity of an ENP.
To our knowledge, our results provide the first demonstration at a proteome-wide scale of selectivity in the specific Cys sites of proteins and pathways impacted by ENP-induced redox modifications. Our findings are also consistent with previous studies that show SSG modifications of proteins occur in a nonstochastic manner.25 In particular, we found that proteins associated with pathways involved in protein synthesis and protein stability, and pathways associated with regulation of receptor-mediated phagocytosis, were statistically most enriched with SSG modifications at lower (subcytotoxic) levels of ROS, as shown with Fe3O4 ENPs. On the other hand, at elevated levels of redox stress induced by CoO ENPs, classical stress response and mitochondrial energetic pathways are more prominently impacted by SSG modification. These results provide insights into which protein signatures (Table 3) and pathways may serve as sensitive ROS sensors and facilitate cellular adaption to redox stress induced by ENPs, versus those that are linked to irreversible cell outcomes at primarily high oxidative stress loads.
Table 3.
Selected SSG-Sites as Potential Signatures of Redox Stress Induced by ENPs
| Groupa | UniProt ID | SSG-modified Site | Log2 Ratio (SiO2) | Log2 Ratio (Fe3O4) | Log2 Ratio (CoO) |
|---|---|---|---|---|---|
| 1 | SMHD1_MOUSE | 897 | 1.397 | 1.465 | 1.218 |
| ALDOA_MOUSE | 178 | 0.263 | 0.974 | 2.320 | |
| VIME_MOUSE | 328 | 0.790 | 0.908 | 0.804 | |
|
| |||||
| 2 | RS29_MOUSE | 39 | 0.041 | 0.864 | 0.792 |
| UROK_MOUSE | 169 | 0.122 | 0.699 | 0.702 | |
| RL37_MOUSE | 19 | 0.103 | 0.534 | 0.767 | |
| ERP29_MOUSE | 159 | −0.050 | 0.377 | 0.801 | |
| QCR6_MOUSE | 35 | −0.150 | 0.292 | 1.137 | |
|
| |||||
| 3 | MT2_MOUSE | 33, 34 | −0.575 | 0.195 | 0.933 |
| MT1_MOUSE | 33, 34 | −0.588 | 0.138 | 0.917 | |
| LDHA_MOUSE | 163 | −0.226 | 0.124 | 0.858 | |
| PDIA1_MOUSE | 55, 58 | −0.234 | −0.183 | 1.293 | |
| GILT_MOUSE | 69, 72 | −0.733 | −0.726 | 1.061 | |
The patterns of increases in SSG at assigned Cys sites indicate their distinct susceptibility to different levels of oxidative stress: mild ROS (Group 1), moderate ROS (Group 2), and high ROS (Group 3), respectively.
The cell trafficking events that lead to SSG modifications after ENP exposure have yet to be determined, but our results suggest regulatory roles of SSG in stress response pathways, which at elevated ROS levels may lead to loss of cell regulation and cell death (Figure 8). We and others have shown that many metal oxide ENPs, including SiO2 and Fe3O4, are internalized in macrophages via clathrin-dependent endocytosis involving macrophage scavenger receptors, such as SR-A.77–80 Most endocytic routes of ENP uptake converge within the lysosomal compartment, where acidic conditions can promote dissolution of ENPs and release of metal ions that exacerbate production of ROS. For many biopersistent ENPs, disruption of lysosome function can also trigger inflammasome activation and cell death through “protective” mechanisms, including autophagy and apoptosis.81–83 Although studies with Co II (CoO) are limited, the cytotoxicity of even poorly soluble cobalt oxide ENPs have been attributed entirely to release of cobalt ions within the lysosome following endocytic internalization.84 The lower solubility and redox potential of Fe3O4 versus CoO ENPs may partially explain differences in the cytoxic potential of these ENPs. The GSH/Grx system serves as a major protective mechanism that prevents protein degradation resulting from thiol oxidation, by masking critical thiols from irreversible oxidation through reversible and site-specific modification with glutathione.85 Our results provide multiple lines of evidence that proteins within the ER quality control system are highly sensitive to SSG modification following ENP exposure. First, the identification of SSG modifications on active sites or functionally essential regions of many ER enzymes suggests direct modulation of enzyme activities and protein functions by SSG. In addition, protein disulfide isomerases, which play critical roles in catalyzing disulfide formation necessary for normal protein folding and maturation in the ER,86 were among the most robustly modified proteins identified in our analyses across all ENP types. The inhibition of enzyme activities by SSG modification of protein active sites may cause dysfunction of the QC system and accumulation of misfolded proteins, leading to a state of ER stress. The potential role of these ER pathways as sensitive redox sensors is also supported by our findings that increased SSG modifications are clearly detected at oxidative stress levels that are well below those which alter cellular glutathione content, as shown with Fe3O4 ENPs. In this respect, ER stress triggered by selective SSG modifications may serve as a protective response to low levels of cellular redox stress, perhaps functioning to limit protein translation and prevent accumulation of unfolded proteins that can lead to autophagy and cell death.87
Figure 8.
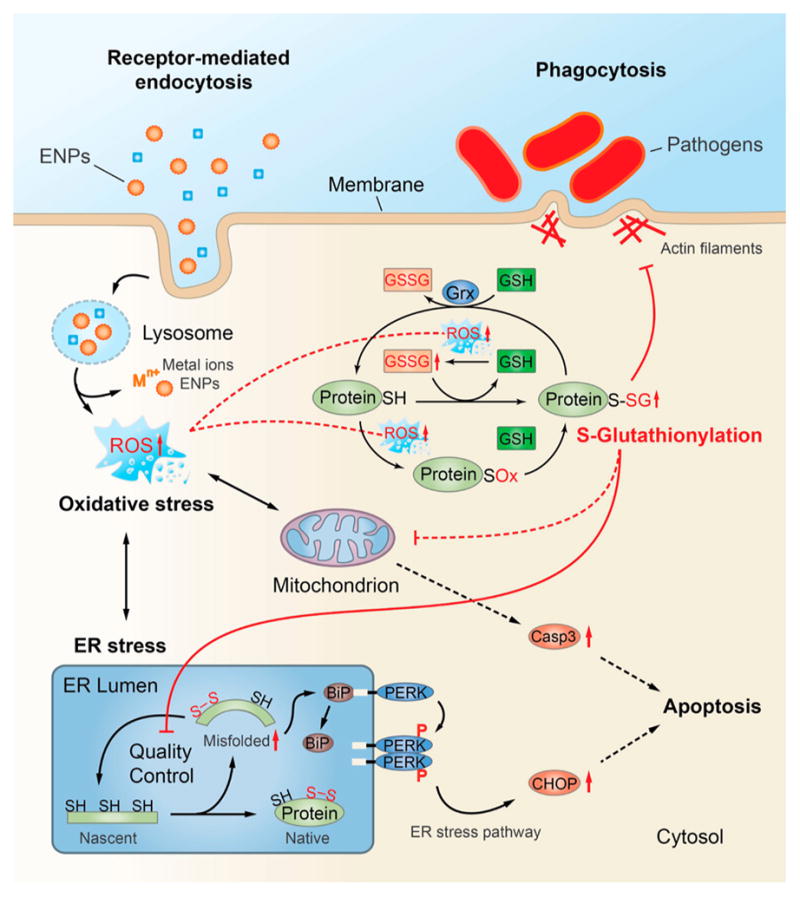
Schematic representation of the proposed pathways and subcellular organelles involved in ENP-induced oxidative stress and S-glutathionylation of proteins and potential influence on ER-stress, phagosome function, and cell survival.
In contrast to either Fe3O4 or SiO2 ENP treatments, elevated levels of oxidative stress associated with CoO ENP treatment result in diminished cellular GSH and increased cellular GSSG levels, and were associated with a much broader accumulation of SSG-modified proteins, indicating less specificity in the proteins and pathways susceptible to SSG modification when the Grx system is overwhelmed. This reduced specificity may simply reflect a broader availability of oxidized protein thiols that can serve as efficient substates for SSG modification when cellular antioxidant levels such as GSH are reduced. We found that under these conditions proteins involved in mitochondrial energetics pathways, which are known to be tightly coupled to intrinsic apoptotic cascades,88 are among the pathways most significantly targeted by SSG modifications. These findings and the strong induction of CHOP protein and increased activation of CASP-3 following CoO ENP treatment support the concept that when critical levels of GSSG/GSH are reached, the ER stress response, which is normally a pro-survival response, is exacerbated and intrinsic mechanisms of apoptosis ensue. For example, it is well established that CASP-3 activation is coupled to mitochondrial permeability changes and is subject to direct redox regulation by SSG.24 Induction of CHOP can also downregulate of the antiapoptotic Bcl-2 regulator, further enhance ROS production in the ER, and is involved in induction of caspases required for cleavage and secretion of IL-1β, a marker of inflammasome activation.67 Thus, under redox conditions that overwhelm the Grx and GSH systems, multiple mechanisms that switch the ER stress pathway from a pro-survival to pro-apoptotic outcome may be operative.
An equally important finding from our work is that proteins which control the phagocytic efficiency of macrophages toward bacterial pathogens are among the most susceptible to SSG modifications, even at relatively low levels of oxidative stress. The downregulation of macrophage phagocytic processes may have evolved as a normal feedback control mechanism triggered by protein redox modifications in response to bacterial stimuli (“phagocytic burst”), functioning to prevent uncontrolled macrophage activation and tissue damage. This study and our previous transcriptomics analyses2 suggest these feedback control mechanisms are also triggered by cellular uptake of redox-active ENPs, resulting in an altered macrophage activation phenotype and reduced capacity to phagocytose pathogenic bacteria such as S. pneumonia. The impairment of cytoskeletal assembly and macrophage phagocytosis via reversible protein redox modifications is consistent with earlier observations that low levels of intracellular ROS are a contributing risk factor for pneumonia, and more recent experimental evidence demonstrating inhalation exposure to copper ENPs reduces bacterial lung clearance mechanisms in mice.7,89 Our studies are also in agreement with a previous study that showed SSG modification of actin impaired cytoskeletal assembly necessary for efficient phagosome formation in neutrophils.28 Most important, redox-dependent dysregulation of macrophage innate immune function may provide a mechanistic explanation for the increased susceptibility to pneumonia observed in epidemiological studies of welders exposed to metal oxide nanoparticles and individuals exposed to ultrafine urban air particulates.3,4,6 While the critical SSG sites that influence macrophage functions warrants further investigation, our redox proteomic analyses and gene silencing studies implicate the SSG/Grx axis as an important regulator of these processes. The use of the RAW 264.7 cell model, which is readily amenable to genetic manipulation with siRNA, allowed us to established an important functional role for the Grx/SSG axis in regulation of phagocytosis of S. pneumonia. Future studies will be focused on extending these findings to alveolar macrophages in vivo, as well as in human macrophages.
The SSG/Grx redox axis is implicated in many diseases associated with inflammation and aging.18,19,24 Furthermore, prevalent polymorphisms in the human glutaredoxin gene have been linked to redox stress,90 yet their potential role in modulating susceptibility to particulates, including engineered materials, is not understood. The unique quantitative site-specific redox modification data reported here provides valuable insight not only for understanding biological impacts of ENPs, but also in stimulating new hypotheses of redox-dependent signaling regulation across a broader research community.
METHODS
Nanoparticle Preparation and Physicochemical Characterization
CoO and amorphous SiO2 ENPs were acquired from commercial sources, whereas Fe3O4 ENPs were synthesized in house as previously described (Table 1).2,34 Extensive physicochemical characterization of these ENPs has been previously described.2,39 TEM and SEM analysis of the Fe3O4 particles synthesized in house demonstrated a primary size of 12.9 nm (see Figure S5 and ref34). SEM images for CoO and SiO2 ENPs are also found in manufacturer’s Web sites (http://www.nanoamor.com; http://www.ssnano.com). To minimize agglomeration due to zwitterionic interactions with media components, the stock ENPs in water (5 mg/mL) were initially dispersed directly into fetal bovine serum (FBS) (Atlanta Biologicals, Lawrenceville, GA) and sonicated using a cup sonicator for 3 min. The ENPs were subsequently diluted with an appropriate volume of RPMI media to achieve a final working concentration of ENPs in complete RPMI medium with 10% FBS. We previously demonstrated this method of suspension minimizes agglomeration and stabilize the agglomerate sizes in culture medium.34 Dynamic light scattering (DLS) and ζ-potentiometric measurements were conducted in complete RPMI media with 10% serum at a particle concentration of 50 μg/mL using a BI 90 particle sizer (Brookhaven Instruments Corp., Holtsville, NY). Before DLS measurement or cell treatment, ENPs were further suspended in media by sonication. The experimental nanoparticle agglomeration densities of these ENPs in complete RPMI were previously reported.39,91
Cell Culture and Nanoparticle Treatment
Raw 264.7 (ATCC # TIB 71) cells were cultured in RPMI 1640 supplemented with 10% FBS, 2 mM L-glutamine (Fisher Scientific, Rockford, IL), and 1% penicillin–streptomycin (Fisher Scientific, Rockford, IL). The cells were maintained in a humid incubator at 37 °C with 5% CO2. Prior to treatment, RAW 264.7 cells were seeded into 100 mm culture plates and grown until 60% confluent. For treatment, original growth media was removed and replaced with media containing ENPs at the desired concentration. Cells were incubated with ENPs media for 24 h.
ENP-Induced Cytotoxicity
The cytotoxic potential of the ENPs was determined by measuring the release of intracellular lactate dehydrogenase (LDH) using the CytoTox-ONE membrane integrity assay per manufacturer’s instructions (Promega, G7890). For the cytotoxicity experiments, cells were plated in a 96-well plate at 1 × 104 cells per well in 100 μL of media. To determine cytotoxicity, cells were incubated at 37 °C with 1.5 mM propidium iodide (PI) (Sigma, Cat# P-4170) and 16.7 μM Hoechst (Anaspec, Inc., Cat#83218) for 15 min then washed once with PBS. The media, staining solution, and washes were collected and spun down, and the cell pellet was resuspended in equal parts of Trypan blue (Sigma, Cat#T8154) and PBS. The number of live and dead cells in the supernatants were quantified using a hematocytometer. The number of live and dead cells remaining on the plates were visualized using a Nikon Eclipse TE300 microscope and further quantified using ImageJ software (http://imagej.nih.gov/). The final cytotoxicity = 100 × (Total Dead Cells/Total Cells), where Total is the sum of cell counts from supernatants and cells remaining on the plate.
Quantification of Cellular GSH and GSSG
GSH and GSSG content in cells was quantified using a luminescence-based GSH/GSSG-Glo Assay per manufacturer’s instructions (Promega Cat#V6611).
Cell Lysis and Enrichment of Protein SSG
After ENP treatment, cells were rinsed twice briefly with RPMI-1640 media containing no supplements and harvested in lysis buffer (250 mM HEPES, 1% Triton X-100, pH 7.0) containing freshly prepared 100 mM N-ethylmaleimide (NEM) (Fisher Scientific, Rockford, IL). Cell lysates were centrifuged at 14 000 rpm, for 10 min at 4 °C and the soluble protein fraction was retained. Alkylation reaction was carried out at 55 °C in dark for 30 min with existence of 2% SDS, followed by acetone precipitation to remove unreacted reagents. Purified proteins were resuspended in 250 mM HEPES containing 8 M urea (pH 7.5) and 0.1% SDS, followed by buffer change to 25 mM HEPES containing 1 M urea (pH 7.5) by using 0.5 mL Amicon Ultra 10 K filter units (EMD Millipore, MA). Protein concentration was determined using the bicinchoninic acid assay (BCA).
For the reduction of SSG-modified proteins, 480 μg of the alkylated samples were prepared at a final concentration of 1 μg/μL in 25 mM HEPES containing 1 M urea (pH 7.5) followed by the addition of 2.5 μg/mL Grx1M (C14S mutant from Escherichia coli, IMCO Corp. Ltd. AB), 0.25 mM GSSG, 1 mM NADPH, and 4 U/mL GR. Samples were incubated at 37 °C for 10 min, immediately placed on ice and transferred to a 0.5 mL Amicon Ultra 10K filter. Excess reagents were removed by buffer exchanged with 3× 8 M urea (pH 7.0) resulting in a final volume of 30–40 μL. Protein concentration of the deglutathionylated samples was measured by the BCA assay before enrichment.
For enrichment of SSG-modifed proteins, 350 μg of the reduced samples was resuspended in 120 μL of 25 mM HEPES buffer containing 0.2% SDS and loaded to Handee Mini-Spin columns containing 30 mg of preconditioned thiopropyl sepharose 6B resin. Enrichment was carried out in a thermomixer at room temperature with shaking at 850 rpm for 2 h. The experimental conditions for resin washing, on-beads tryptic digestion, TMT-labeling, DTT eluting and C18 cleanup were performed as previously described.25,31,32,92,93 The enriched peptides were dissolved in a final volume of 30 μL of water containing 20 mM DTT prior to LC–MS/MS analysis.
LC–MS/MS and Data Analysis
LC–MS/MS was performed as previously described.32 MS analysis was performed on a Thermo Scientific LTQ-Orbitrap Velos mass spectrometer (Thermo Scientific, San Jose, CA) coupled with an electrospray ionization interface using a homemade 150 μm o.d. × 20 μm i.d. chemically etched electrospray emitter. The heated capillary temperature and spray voltage were 350 °C and 2.2 kV, respectively. Full MS spectra were recorded at resolution of 60 K over the range of m/z 300–2000 with an automated gain control (AGC) value of 1 × 106. MS/MS was performed in the data-dependent mode at a resolution of 7.5 K with an AGC target value of 3 × 104. The most abundant 10 parent ions were selected for MS/MS using high-energy collision dissociation (HCD) with a normalized collision energy setting of 45. Precursor ion activation was performed with an isolation width of 2.5 Da, a minimal intensity of 1000 counts, and an activation time of 0.1 s. A dynamic exclusion time of 60 s was used.
LC–MS/MS raw data were converted into dta files using Bioworks Cluster 3.2 (Thermo Fisher Scientific, Cambridge, MA), and MSGF plus algorithm94 (v9979, released in March 2014) was used to search MS/MS spectra against the mouse protein sequence database (UniProt, released in September 2013). The key search parameters used were 20 ppm tolerance for precursor ion masses, 0.5 Da tolerance for fragment ions, partial tryptic search with up to 2 missed cleavages, dynamic oxidation of methionine (15.9949 Da), dynamic NEM modification of Cys (125.0477 Da), and static 6-plex TMT modification of lysine and N-termini of peptides (229.1629 Da). Peptides were identified from database searching results applying the following criteria: MSGF E-value <10–10, Q-value <0.01, and mass measurement error <10 ppm (±5 ppm). The decoy database searching methodology (38) was used to confirm the final false discovery rate at the unique peptide level to be ~0.9%. Since NEM blocked original free Cys sites, all glutathionylated Cys residues were identified as unmodified Cys.
For TMT-based relative quantification of SSG-modified sites, all MS/MS spectra were grouped based on individual Cys-sites. The intensities of TMT reporter ion were summed from all spectra corresponding to a given Cys-site and transformed to log2 values for normalization by using the central tendency approach. Biological triplicates in ENP-treated samples and untreated controls were normalized at unique Cys-site level, separately. The SSG level was considered substantially different when p-value is less than 0.05 (ANOVA) with at least 1.23-fold of change between ENP-treated sample and untreated controls (log2 ratio < 0.3). For quantification of protein expression with total Cys enrichment, the intensities of TMT reporter ion were summed from all spectra corresponding to a given unique protein and transformed to log2 values for normalization by using the central tendency approach. ENP-treated samples and controls were normalized together at protein level. The protein expression was considered substantially different when p-value < 0.05 (ANOVA) with at least 1.23-fold of change between ENP-treated sample and untreated controls (log2 ratio < 0.3). Biological function categorization of the identified proteins was clustered based on GO with DAVID tools. Protein interaction network and pathway analysis was performed with Ingenuity Pathway Analysis (www.ingenuity.com). Protein motif analysis was carried out by using MOTIF tools (www.genome.jp/tools/motif). Several PTM databases were used for data comparison and function annotation, including dbGSH43 (Cys S-glutathionylation database with 2006 SSG-modified Cys sites from 1128 proteins), dbSNO44 (Cys S-nitrosylatioin database with 2646 SNO-modified Cys sites from 1355 proteins) and protein disulfide database from UniProt (http://www.uniprot.org, annotation for 29 782 Cys sites from 2867 proteins).
RT-PCR Analysis
Isolation of RNA, generation of cDNA and real-time PCR amplification were performed as previously described.2 The primers used included (5′ to 3′) CPHA sense, GAG CTG TTT GCA GAC AAA GTT C; CPHA antisense, CCC TGG CAC ATG AAT CCT GG; Grx1 sense, TGG AGC TCT GCA GTT ATA AAA GG; Grx1 antisense, GCC ATC AGC ATG GTT AGA CA; HMOX sense, AGG CTA AGA CCG CCT TCC T; HMOX antisense, TGT GTT CCT CTG TCA GCA TCA.
RNA Silencing Knockdown of Grx1 in RAW 264.7
To knock down expression of Grx1, 5 × 105 cells were plated in six-well plates. After the cells adhered for 6 h, the regular media was exchanged with 600 μL of Opti-MEM (Invitrogen cat # 31985070) containing siRNA for mouse Grx1 (Invitrogen) (sense, CCU ACU GCA GAA AGA CCC AAG AAA U; antisense, AUU UCU UGG GUC UUU CUG CAG UAG G) or scrambled RNA was applied at 125 nM using Lipofectamine 2000 (Invitrogen). After 6 h of incubation, RPMI media containing 10% FBS without antibiotics was added to the wells. Cells were used for functional assays after 48 h.
Gel Electrophoresis and Western Blot
For Western blot analysis, cells were lysed in PBS with 1% Triton X-100 containing protease and phosphatase inhibitors. Equal amounts of isolated proteins (i.e., 20 μg) were diluted in NuPAGE LDS sample buffer (Thermo Fisher Scientific) and separated electrophoretically under reducing conditions on a 4–12% Bis-Tris polyacrylamide gel in MES SDS running buffer (Invitrogen, Carlsbad, CA). Separated proteins were then subjected to electrophoretic transfer to a polyvinylidene difluoride (PVDF) membrane (Invitrogen, Carlsbad, CA). Membranes were blocked for 1 h at room temperature in PBS solution containing 5% (w/v) nonfat milk and 0.1% (w/v) Tween-20 prior to the addition of primary antibody at 1:1000 dilution in PBS containing 1% milk and 0.1% Tween-20. Membranes were incubated with primary antibodies overnight at 4 °C followed by incubation for 1 h at room temperature with HRP-conjugated secondary antibody at 1:2000 dilution in PBS containing 1% milk and 0.1% Tween-20. The membranes were then washed in PBS with 1% Tween-20 prior to the addition of SuperSignal West Pico or Femto chemiluminescent substrate for visualization (Thermo Scientific, Rockford, IL) according to the manufacturer’s instructions. Primary antibodies against BiP (Cat# 3177), PERK (Cat# 3192), p-PERK (Cat# 3179), CHOP (Cat# 5554), caspase-3 (Cat# 9662), and GAPDH (Cat# 2118) were purchased from Cell Signaling Technology, Inc.
Supplementary Material
Acknowledgments
This research was supported by in part by NIH grants DP2OD006668 (W.-J.Q.), UC4 DK104167 (W.-J.Q.), U19 ES019544 (B.D.T.), P41 GM103493 (R.D.S.), U24-CA-160019 (R.D.S), and a DOE Early Career Research Award (W.-J.Q.). This work was performed in the Environmental Molecular Sciences Laboratory, a national scientific user facility sponsored by DOE/BER and located at Pacific Northwest National Laboratory, which is operated by Battelle Memorial Institute for the DOE under Contract DE-AC05-76RL0 1830. The authors would like to thank Prof. Tzong-Yi Lee at Yuan Ze University, Academia Sinica, Taiwan, China for providing the database of dbGSH 1.0.
Footnotes
Notes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsnano.5b05524. The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the data set identifier PXD003356 and 10.6019/PXD003356.
Figures S1–S4, described in the text, with accompanying figure legends (PDF) Tables S1–S12 (XLSX)
References
- 1.Roco MC, Mirkin CA, Hersam MC. Nanotechnology Research Directions for Societal Needs in 2020. Retrospective and Outlook. National Science Foundation (Sponsor); Washington, D.C: 2010. [Google Scholar]
- 2.Kodali V, Littke MH, Tilton SC, Teeguarden JG, Shi L, Frevert CW, Wang W, Pounds JG, Thrall BD. Dysregulation of Macrophage Activation Profiles by Engineered Nanoparticles. ACS Nano. 2013;7:6997–7010. doi: 10.1021/nn402145t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coggon D, Inskip H, Winter P, Pannett B. Lobar Pneumonia: An Occupational Disease in Welders. Lancet. 1994;344:41–43. doi: 10.1016/s0140-6736(94)91056-1. [DOI] [PubMed] [Google Scholar]
- 4.Palmer KT, Poole J, Ayres JG, Mann J, Burge PS, Coggon D. Exposure to Metal Fume and Infectious Pneumonia. Am J Epidemiol. 2003;157:227–233. doi: 10.1093/aje/kwf188. [DOI] [PubMed] [Google Scholar]
- 5.Andujar P, Simon-Deckers A, Galateau-Salle F, Fayard B, Beaune G, Clin B, Billon-Galland MA, Durupthy O, Pairon JC, Doucet J, et al. Role of Metal Oxide Nanoparticles in Histopathological Changes Observed in the Lung of Welders. Part Fibre Toxicol. 2014;11:23. doi: 10.1186/1743-8977-11-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neupane B, Jerrett M, Burnett RT, Marrie T, Arain A, Loeb M. Long-Term Exposure to Ambient Air Pollution and Risk of Hospitalization with Community-Acquired Pneumonia in Older Adults. Am J Respir Crit Care Med. 2010;181:47–53. doi: 10.1164/rccm.200901-0160OC. [DOI] [PubMed] [Google Scholar]
- 7.Kim JS, Adamcakova-Dodd A, O’Shaughnessy PT, Grassian VH, Thorne PS. Effects of Copper Nanoparticle Exposure on Host Defense in a Murine Pulmonary Infection Model. Part Fibre Toxicol. 2011;8:29. doi: 10.1186/1743-8977-8-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Braydich-Stolle LK, Speshock JL, Castle A, Smith M, Murdock RC, Hussain SM. Nanosized Aluminum Altered Immune Function. ACS Nano. 2010;4:3661–3670. doi: 10.1021/nn9016789. [DOI] [PubMed] [Google Scholar]
- 9.Meng H, Xia T, George S, Nel AE. A Predictive Toxicological Paradigm for the Safety Assessment of Nanomaterials. ACS Nano. 2009;3:1620–1627. doi: 10.1021/nn9005973. [DOI] [PubMed] [Google Scholar]
- 10.Xia T, Kovochich M, Brant J, Hotze M, Sempf J, Oberley T, Sioutas C, Yeh JI, Wiesner MR, Nel AE. Comparison of the Abilities of Ambient and Manufactured Nanoparticles to Induce Cellular Toxicity According to an Oxidative Stress Paradigm. Nano Lett. 2006;6:1794–1807. doi: 10.1021/nl061025k. [DOI] [PubMed] [Google Scholar]
- 11.Zhang H, Ji Z, Xia T, Meng H, Low-Kam C, Liu R, Pokhrel S, Lin S, Wang X, Liao YP, et al. Use of Metal Oxide Nanoparticle Band Gap to Develop a Predictive Paradigm for Oxidative Stress and Acute Pulmonary Inflammation. ACS Nano. 2012;6:4349–4368. doi: 10.1021/nn3010087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Manke A, Wang L, Rojanasakul Y. Mechanisms of Nanoparticle-Induced Oxidative Stress and Toxicity. BioMed Res Int. 2013;2013:942916. doi: 10.1155/2013/942916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Donaldson K, Borm PJ, Castranova V, Gulumian M. The Limits of Testing Particle-Mediated Oxidative Stress in Vitro in Predicting Diverse Pathologies; Relevance for Testing of Nano-particles. Part Fibre Toxicol. 2009;6:13. doi: 10.1186/1743-8977-6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shvedova AA, Pietroiusti A, Fadeel B, Kagan VE. Mechanisms of Carbon Nanotube-Induced Toxicity: Focus on Oxidative Stress. Toxicol Appl Pharmacol. 2012;261:121–133. doi: 10.1016/j.taap.2012.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Perkins TN, Shukla A, Peeters PM, Steinbacher JL, Landry CC, Lathrop SA, Steele C, Reynaert NL, Wouters EF, Mossman BT. Differences in Gene Expression and Cytokine Production by Crystalline Vs. Amorphous Silica in Human Lung Epithelial Cells. Part Fibre Toxicol. 2012;9:6. doi: 10.1186/1743-8977-9-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Waters KM, Masiello LM, Zangar RC, Tarasevich BJ, Karin NJ, Quesenberry RD, Bandyopadhyay S, Teeguarden JG, Pounds JG, Thrall BD. Macrophage Responses to Silica Nanoparticles Are Highly Conserved across Particle Sizes. Toxicol Sci. 2009;107:553–569. doi: 10.1093/toxsci/kfn250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Janssen-Heininger YMW, Mossman BT, Heintz NH, Forman HJ, Kalyanaraman B, Finkel T, Stamler JS, Rhee SG, van der Vliet A. Redox-Based Regulation of Signal Transduction: Principles, Pitfalls, and Promises. Free Radical Biol Med. 2008;45:1–17. doi: 10.1016/j.freeradbiomed.2008.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mieyal JJ, Gallogly MM, Qanungo S, Sabens EA, Shelton MD. Molecular Mechanisms and Clinical Implications of Reversible Protein S-Glutathionylation. Antioxid Redox Signaling. 2008;10:1941–1988. doi: 10.1089/ars.2008.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shelton MD, Mieyal JJ. Regulation by Reversible S-Glutathionylation: Molecular Targets Implicated in Inflammatory Diseases. Mol Cells. 2008;25:332–346. [PMC free article] [PubMed] [Google Scholar]
- 20.Aesif SW, Kuipers I, van der Velden J, Tully JE, Guala AS, Anathy V, Sheely JI, Reynaert NL, Wouters EFM, van der Vliet A, et al. Activation of the Glutaredoxin-1 Gene by Nuclear Factor Kappa B Enhances Signaling. Free Radical Biol Med. 2011;51:1249–1257. doi: 10.1016/j.freeradbiomed.2011.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Anathy V, Roberson EC, Guala AS, Godburn KE, Budd RC, Janssen-Heininger YMW. Redox-Based Regulation of Apoptosis: S-Glutathionylation as a Regulatory Mechanism to Control Cell Death. Antioxid Redox Signaling. 2012;16:496–505. doi: 10.1089/ars.2011.4281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dalle-Donne I, Rossi R, Colombo G, Giustarini D, Milzani A. Protein S-Glutathionylation: A Regulatory Device from Bacteria to Humans. Trends Biochem Sci. 2009;34:85–96. doi: 10.1016/j.tibs.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 23.Shelton MD, Chock PB, Mieyal JJ. Glutaredoxin: Role in Reversible Protein S-Glutathionylation and Regulation of Redox Signal Transduction and Protein Translocation. Antioxid Redox Signaling. 2005;7:348–366. doi: 10.1089/ars.2005.7.348. [DOI] [PubMed] [Google Scholar]
- 24.Allen EM, Mieyal JJ. Protein-Thiol Oxidation and Cell Death: Regulatory Role of Glutaredoxins. Antioxid Redox Signaling. 2012;17:1748–1763. doi: 10.1089/ars.2012.4644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Su D, Gaffrey MJ, Guo J, Hatchell KE, Chu RK, Clauss TR, Aldrich JT, Wu S, Purvine S, Camp DG, et al. Proteomic Identification and Quantification of S-Glutathionylation in Mouse Macrophages Using Resin-Assisted Enrichment and Isobaric Labeling. Free Radical Biol Med. 2014;67:460–470. doi: 10.1016/j.freeradbiomed.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gravina SA, Mieyal JJ. Thioltransferase Is a Specific Glutathionyl Mixed Disulfide Oxidoreductase. Biochemistry. 1993;32:3368–3376. doi: 10.1021/bi00064a021. [DOI] [PubMed] [Google Scholar]
- 27.Fratelli M, Demol H, Puype M, Casagrande S, Eberini I, Salmona M, Bonetto V, Mengozzi M, Duffieux F, Miclet E, et al. Identification by Redox Proteomics of Glutathionylated Proteins in Oxidatively Stressed Human T Lymphocytes. Proc Natl Acad Sci U S A. 2002;99:3505–3510. doi: 10.1073/pnas.052592699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sakai J, Li J, Subramanian KK, Mondal S, Bajrami B, Hattori H, Jia Y, Dickinson BC, Zhong J, Ye K, et al. Reactive Oxygen Species-Induced Actin Glutathionylation Controls Actin Dynamics in Neutrophils. Immunity. 2012;37:1037–1049. doi: 10.1016/j.immuni.2012.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aesif SW, Anathy V, Kuipers I, Guala AS, Reiss JN, Ho YS, Janssen-Heininger YMW. Ablation of Glutaredoxin-1 Attenuates Lipopolysaccharide-Induced Lung Inflammation and Alveolar Macrophage Activation. Am J Respir Cell Mol Biol. 2011;44:491–499. doi: 10.1165/rcmb.2009-0136OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reynaert NL, van der Vliet A, Guala AS, McGovern T, Hristova M, Pantano C, Heintz NH, Heim J, Ho YS, Matthews DE, et al. Dynamic Redox Control of Nf-Kappa B through Glutaredoxin-Regulated S-Glutathionylation of Inhibitory Kappa B Kinase Beta. Proc Natl Acad Sci U S A. 2006;103:13086–13091. doi: 10.1073/pnas.0603290103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guo J, Gaffrey MJ, Su D, Liu T, Camp DG, II, Smith RD, Qian WJ. Resin-Assisted Enrichment of Thiols as a General Strategy for Proteomic Profiling of Cysteine-Based Reversible Modifications. Nat Protoc. 2014;9:64–75. doi: 10.1038/nprot.2013.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guo J, Nguyen AY, Dai Z, Su D, Gaffrey MJ, Moore RJ, Jacobs JM, Monroe ME, Smith RD, Koppenaal DW, et al. Proteome-Wide Light/Dark Modulation of Thiol Oxidation in Cyanobacteria Revealed by Quantitative Site-Specific Redox Proteomics. Mol Cell Proteomics. 2014;13:3270–3285. doi: 10.1074/mcp.M114.041160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Su D, Shukla AK, Chen B, Kim JS, Nakayasu E, Qu Y, Aryal U, Weitz K, Clauss TRW, Monroe ME, et al. Quantitative Site-Specific Reactivity Profiling of S-Nitrosylation in Mouse Skeletal Muscle Using Cysteinyl Peptide Enrichment Coupled with Mass Spectrometry. Free Radical Biol Med. 2013;57:68–78. doi: 10.1016/j.freeradbiomed.2012.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sharma G, Kodali V, Gaffrey M, Wang W, Minard KR, Karin NJ, Teeguarden JG, Thrall BD. Iron Oxide Nanoparticle Agglomeration Influences Dose Rates and Modulates Oxidative Stress-Mediated Dose-Response Profiles. Nanotoxicology. 2014;8:663–675. doi: 10.3109/17435390.2013.822115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu G, Gao J, Ai H, Chen X. Applications and Potential Toxicity of Magnetic Iron Oxide Nanoparticles. Small. 2013;9:1533–1545. doi: 10.1002/smll.201201531. [DOI] [PubMed] [Google Scholar]
- 36.Tang L, Cheng J. Nonporous Silica Nanoparticles for Nanomedicine Application. Nano Today. 2013;8:290–312. doi: 10.1016/j.nantod.2013.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dalal A, Pawar V, McAllister K, Weaver C, Hallab NJ. Orthopedic Implant Cobalt-Alloy Particles Produce Greater Toxicity and Inflammatory Cytokines Than Titanium Alloy and Zirconium Alloy-Based Particles in Vitro, in Human Osteoblasts, Fibroblasts, and Macrophages. J Biomed Mater Res, Part A. 2012;100A:2147–2158. doi: 10.1002/jbm.a.34122. [DOI] [PubMed] [Google Scholar]
- 38.Sansone V, Pagani D, Melato M. The Effects on Bone Cells of Metal Ions Released from Orthopaedic Implants. A Review. Clin Cases Miner Bone Metab. 2013;10:34–40. doi: 10.11138/ccmbm/2013.10.1.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cohen JM, Teeguarden JG, Demokritou P. An Integrated Approach for the in Vitro Dosimetry of Engineered Nanomaterials. Part Fibre Toxicol. 2014;11:20. doi: 10.1186/1743-8977-11-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Choi AMK, Alam J. Heme Oxygenase-1: Function, Regulation, and Implication of a Novel Stress-Inducible Protein in Oxidant-Induced Lung Injury. Am J Respir Cell Mol Biol. 1996;15:9–19. doi: 10.1165/ajrcmb.15.1.8679227. [DOI] [PubMed] [Google Scholar]
- 41.Zhao Y, Gu X, Ma H, He X, Liu M, Ding Y. Association of Glutathione Level and Cytotoxicity of Gold Nanoparticles in Lung Cancer Cells. J Phys Chem C. 2011;115:12797–12802. [Google Scholar]
- 42.Schafer FQ, Buettner GR. Redox Environment of the Cell as Viewed through the Redox State of the Glutathione Disulfide/Glutathione Couple. Free Radical Biol Med. 2001;30:1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 43.Chen YJ, Lu CT, Lee TY, Chen YJ. Dbgsh: A Database of S-Glutathionylation. Bioinformatics. 2014;30:2386–2388. doi: 10.1093/bioinformatics/btu301. [DOI] [PubMed] [Google Scholar]
- 44.Lee TY, Chen YJ, Lu CT, Ching WC, Teng YC, Huang HD, Chen YJ. Dbsno: A Database of Cysteine S-Nitrosylation. Bioinformatics. 2012;28:2293–2295. doi: 10.1093/bioinformatics/bts436. [DOI] [PubMed] [Google Scholar]
- 45.Huang DW, Sherman BT, Lempicki RA. Systematic and Integrative Analysis of Large Gene Lists Using David Bioinformatics Resources. Nat Protoc. 2008;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 46.Huang DW, Sherman BT, Tan Q, Kir J, Liu D, Bryant D, Guo Y, Stephens R, Baseler MW, Lane HC, et al. David Bioinformatics Resources: Expanded Annotation Database and Novel Algorithms to Better Extract Biology from Large Gene Lists. Nucleic Acids Res. 2007;35:W169–W175. doi: 10.1093/nar/gkm415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Klatt P, Lamas S. Regulation of Protein Function by S-Glutathiolation in Response to Oxidative and Nitrosative Stress. Eur J Biochem. 2000;267:4928–4944. doi: 10.1046/j.1432-1327.2000.01601.x. [DOI] [PubMed] [Google Scholar]
- 48.Figueira TR, Barros MH, Camargo AA, Castilho RF, Ferreira JCB, Kowaltowski AJ, Sluse FE, Souza-Pinto NC, Vercesi AE. Mitochondria as a Source of Reactive Oxygen and Nitrogen Species: From Molecular Mechanisms to Human Health. Antioxid Redox Signaling. 2013;18:2029–2074. doi: 10.1089/ars.2012.4729. [DOI] [PubMed] [Google Scholar]
- 49.Bhandary B, Marahatta A, Kim HR, Chae HJ. An Involvement of Oxidative Stress in Endoplasmic Reticulum Stress and Its Associated Diseases. Int J Mol Sci. 2013;14:434–456. doi: 10.3390/ijms14010434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fruehauf JP, Trapp V. Reactive Oxygen Species: An Achilles’ Heel of Melanoma? Expert Rev Anticancer Ther. 2008;8:1751–1757. doi: 10.1586/14737140.8.11.1751. [DOI] [PubMed] [Google Scholar]
- 51.Sun L, Li Y, Liu X, Jin M, Zhang L, Du Z, Guo C, Huang P, Sun Z. Cytotoxicity and Mitochondrial Damage Caused by Silica Nanoparticles. Toxicol In Vitro. 2011;25:1619–1629. doi: 10.1016/j.tiv.2011.06.012. [DOI] [PubMed] [Google Scholar]
- 52.Tsai YY, Huang YH, Chao YL, Hu KY, Chin LT, Chou SH, Hour AL, Yao YD, Tu CS, Liang YJ, et al. Identification of the Nanogold Particle-Induced Endoplasmic Reticulum Stress by Omic Techniques and Systems Biology Analysis. ACS Nano. 2011;5:9354–9369. doi: 10.1021/nn2027775. [DOI] [PubMed] [Google Scholar]
- 53.Shrestha N, Bahnan W, Wiley DJ, Barber G, Fields KA, Schesser K. Eukaryotic Initiation Factor 2 (Eif2) Signaling Regulates Proinflammatory Cytokine Expression and Bacterial Invasion. J Biol Chem. 2012;287:28738–28744. doi: 10.1074/jbc.M112.375915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Clemens MJ. Translational Regulation in Cell Stress and Apoptosis. Roles of the Eif4e Binding Proteins. J Cell Mol Med. 2001;5:221–239. doi: 10.1111/j.1582-4934.2001.tb00157.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Moore MS, Blobel G. A G-Protein Involved in Nucleocytoplasmic Transport - the Role of Ran. Trends Biochem Sci. 1994;19:211–216. doi: 10.1016/0968-0004(94)90024-8. [DOI] [PubMed] [Google Scholar]
- 56.Phizicky EM, Hopper AK. Trna Biology Charges to the Front. Genes Dev. 2010;24:1832–1860. doi: 10.1101/gad.1956510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ciechanover A. The Ubiquitin-Proteasome Pathway: On Protein Death and Cell Life. EMBO J. 1998;17:7151–7160. doi: 10.1093/emboj/17.24.7151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pricop L, Salmon JE. Redox Regulation of Fc Gamma Receptor-Mediated Phagocytosis: Implications for Host Defense and Tissue Injury. Antioxid Redox Signaling. 2002;4:85–95. doi: 10.1089/152308602753625889. [DOI] [PubMed] [Google Scholar]
- 59.Cox D, Greenberg S. Phagocytic Signaling Strategies: Fc(Gamma) Receptor-Mediated Phagocytosis as a Model System. Semin Immunol. 2001;13:339–345. doi: 10.1006/smim.2001.0330. [DOI] [PubMed] [Google Scholar]
- 60.Vara JAF, Casado E, de Castro J, Cejas P, Belda-Iniesta C, Gonzalez-Baron M. Pi3k/Akt Signalling Pathway and Cancer. Cancer Treat Rev. 2004;30:193–204. doi: 10.1016/j.ctrv.2003.07.007. [DOI] [PubMed] [Google Scholar]
- 61.Whelan JT, Hollis SE, Cha DS, Asch AS, Lee MH. Post-Transcriptional Regulation of the Ras-Erk/Mapk Signaling Pathway. J Cell Physiol. 2012;227:1235–1241. doi: 10.1002/jcp.22899. [DOI] [PubMed] [Google Scholar]
- 62.Halloran M, Parakh S, Atkin JD. The Role of S-Nitrosylation and S-Glutathionylation of Protein Disulphide Isomerase in Protein Misfolding and Neurodegeneration. Int J Cell Biol. 2013;2013:797914. doi: 10.1155/2013/797914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mekahli D, Bultynck G, Parys JB, De Smedt H, Missiaen L. Endoplasmic-Reticulum Calcium Depletion and Disease. Cold Spring Harbor Perspect Biol. 2011;3:a004317. doi: 10.1101/cshperspect.a004317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ellgaard L, Helenius A. Quality Control in the Endoplasmic Reticulum. Nat Rev Mol Cell Biol. 2003;4:181–191. doi: 10.1038/nrm1052. [DOI] [PubMed] [Google Scholar]
- 65.Nakamura T, Tu S, Akhtar MW, Sunico CR, Okamoto S-i, Lipton SA. Aberrant Protein S-Nitrosylation in Neuro-degenerative Diseases. Neuron. 2013;78:596–614. doi: 10.1016/j.neuron.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ellgaard L, Ruddock LW. The Human Protein Disulphide Isomerase Family: Substrate Interactions and Functional Properties. EMBO Rep. 2005;6:28–32. doi: 10.1038/sj.embor.7400311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nishitoh H. Chop Is a Multifunctional Transcription Factor in the Er Stress Response. J Biochem. 2012;151:217–219. doi: 10.1093/jb/mvr143. [DOI] [PubMed] [Google Scholar]
- 68.Gotoh T, Oyadomari S, Mori K, Mori M. Nitric Oxide-Induced Apoptosis in Raw 264.7 Macrophages Is Mediated by Endoplasmic Reticulum Stress Pathway Involving Atf6 and Chop. J Biol Chem. 2002;277:12343–12350. doi: 10.1074/jbc.M107988200. [DOI] [PubMed] [Google Scholar]
- 69.Tagawa Y, Hiramatsu N, Kasai A, Hayakawa K, Okamura M, Yao J, Kitamura M. Induction of Apoptosis by Cigarette Smoke via Ros-Dependent Endoplasmic Reticulum Stress and Ccaat/Enhancer-Binding Protein-Homologous Protein (Chop) Free Radical Biol Med. 2008;45:50–59. doi: 10.1016/j.freeradbiomed.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 70.Rouhier N, Villarejo A, Srivastava M, Gelhaye E, Keech O, Droux M, Finkemeier I, Samuelsson G, Dietz KJ, Jacquot JP, et al. Identification of Plant Glutaredoxin Targets. Antioxid Redox Signaling. 2005;7:919–929. doi: 10.1089/ars.2005.7.919. [DOI] [PubMed] [Google Scholar]
- 71.Schafer DA, Schroer TA. Actin-Related Proteins. Annu Rev Cell Dev Biol. 1999;15:341–363. doi: 10.1146/annurev.cellbio.15.1.341. [DOI] [PubMed] [Google Scholar]
- 72.Kim JS, Huang TY, Bokoch GM. Reactive Oxygen Species Regulate a Slingshot-Cofilin Activation Pathway. Mol Biol Cell. 2009;20:2650–2660. doi: 10.1091/mbc.E09-02-0131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Go YM, Orr M, Jones DP. Actin Cytoskeleton Redox Proteome Oxidation by Cadmium. Am J Physiol-Lung C. 2013;305:L831–L843. doi: 10.1152/ajplung.00203.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lim J, Wiedemann A, Tzircotis G, Monkley SJ, Critchley DR, Caron E. An Essential Role for Talin During Alpha(M)Beta(2)-Mediated Phagocytosis. Mol Biol Cell. 2007;18:976–985. doi: 10.1091/mbc.E06-09-0813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shinomiya H. Plastin Family of Actin-Bundling Proteins: Its Functions in Leukocytes, Neurons, Intestines, and Cancer. Int J Cell Biol. 2012;2012:213492. doi: 10.1155/2012/213492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kuipers I, Bracke KR, Brusselle GG, Aesif SW, Krijgsman R, Arts IC, Wouters EFM, Reynaert NL. Altered Cigarette Smoke-Induced Lung Inflammation Due to Ablation of Grx1. PLoS One. 2012;7:e38984. doi: 10.1371/journal.pone.0038984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Orr GA, Chrisler WB, Cassens KJ, Tan R, Tarasevich BJ, Markillie LM, Zangar RC, Thrall BD. Cellular Recognition and Trafficking of Amorphous Silica Nanoparticles by Macrophage Scavenger Receptor A. Nanotoxicology. 2011;5:296–311. doi: 10.3109/17435390.2010.513836. [DOI] [PubMed] [Google Scholar]
- 78.Minard KR, Littke MH, Wang W, Xiong Y, Teeguarden JG, Thrall BD. Magnetic Particle Detection (Mpd) for in-Vitro Dosimetry. Biosens Bioelectron. 2013;43:88–93. doi: 10.1016/j.bios.2012.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chao Y, Makale M, Karmali PP, Sharikov Y, Tsigelny I, Merkulov S, Kesari S, Wrasidlo W, Ruoslahti E, Simberg D. Recognition of Dextran-Superparamagnetic Iron Oxide Nanoparticle Conjugates (Feridex) via Macrophage Scavenger Receptor Charged Domains. Bioconjugate Chem. 2012;23:1003–1009. doi: 10.1021/bc200685a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chao Y, Karmali PP, Mukthavaram R, Kesari S, Kouznetsova VL, Tsigelny IF, Simberg D. Direct Recognition of Superparamagnetic Nanocrystals by Macrophage Scavenger Receptor Sr-Ai. ACS Nano. 2013;7:4289–4298. doi: 10.1021/nn400769e. [DOI] [PubMed] [Google Scholar]
- 81.Andon FT, Fadeel B. Programmed Cell Death: Molecular Mechanisms and Implications for Safety Assessment of Nanomaterials. Acc Chem Res. 2013;46:733–742. doi: 10.1021/ar300020b. [DOI] [PubMed] [Google Scholar]
- 82.Stern ST, Adiseshaiah PP, Crist RM. Autophagy and Lysosomal Dysfunction as Emerging Mechanisms of Nanomaterial Toxicity. Part Fibre Toxicol. 2012;9:20. doi: 10.1186/1743-8977-9-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.De Stefano D, Carnuccio R, Maiuri MC. Nanomaterials Toxicity and Cell Death Modalities. J Drug Delivery. 2012;2012:167896. doi: 10.1155/2012/167896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ortega R, Bresson C, Darolles C, Gautier C, Roudeau S, Perrin L, Janin M, Floriani M, Aloin V, Carmona A, et al. Low-Solubility Particles and a Trojan-Horse Type Mechanism of Toxicity: The Case of Cobalt Oxide on Human Lung Cells. Part Fibre Toxicol. 2014;11:14. doi: 10.1186/1743-8977-11-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Grek CL, Zhang J, Manevich Y, Townsend DM, Tew KD. Causes and Consequences of Cysteine S-Glutathionylation. J Biol Chem. 2013;288:26497–26504. doi: 10.1074/jbc.R113.461368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wilkinson B, Gilbert HF. Protein Disulfide Isomerase. Biochim Biophys Acta, Proteins Proteomics. 2004;1699:35–44. doi: 10.1016/j.bbapap.2004.02.017. [DOI] [PubMed] [Google Scholar]
- 87.Han J, Backa SH, Hur J, Lin YH, Gildersleeve R, Shan J, Yuan CL, Krokowski D, Wang S, Hatzoglou M, et al. Er-Stress-Induced Transcriptional Regulation Increases Protein Synthesis Leading to Cell Death. Nat Cell Biol. 2013;15:481–90. doi: 10.1038/ncb2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.El Kasmi KC, Stenmark KR. Contribution of Metabolic Reprogramming to Macrophage Plasticity and Function. Semin Immunol. 2015;27:267–275. doi: 10.1016/j.smim.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.O’Reilly PJ, Hickman-Davis JM, Davis IC, Matalon S. Hyperoxia Impairs Antibacterial Function of Macrophages through Effects on Actin. Am J Respir Cell Mol Biol. 2003;28:443–450. doi: 10.1165/rcmb.2002-0153OC. [DOI] [PubMed] [Google Scholar]
- 90.Schlawicke Engstrom K, Nermell B, Concha G, Stromberg U, Vahter M, Broberg K. Arsenic Metabolism Is Influenced by Polymorphisms in Genes Involved in One-Carbon Metabolism and Reduction Reactions. Mutat Res, Fundam Mol Mech Mutagen. 2009;667:4–14. doi: 10.1016/j.mrfmmm.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 91.DeLoid G, Cohen JM, Darrah T, Derk R, Rojanasakul L, Pyrgiotakis G, Wohlleben W, Demokritou P. Estimating the Effective Density of Engineered Nanomaterials for in Vitro Dosimetry. Nat Commun. 2014;5:3514. doi: 10.1038/ncomms4514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Benham AM, van Lith M, Sitia R, Braakman I. Ero1-Pdi Interactions, the Response to Redox Flux and the Implications for Disulfide Bond Formation in the Mammalian Endoplasmic Reticulum. Philos Trans R Soc, B. 2013;368:20110403. doi: 10.1098/rstb.2011.0403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Aitken RJ, Chaudhry MQ, Boxall ABA, Hull M. Manufacture and Use of Nanomaterials: Current Status in the Uk and Global Trends. Occup Med. 2006;56:300–306. doi: 10.1093/occmed/kql051. [DOI] [PubMed] [Google Scholar]
- 94.Kim S, Pevzner PA. Ms-Gf Plus Makes Progress Towards a Universal Database Search Tool for Proteomics. Nat Commun. 2014;5:5277. doi: 10.1038/ncomms6277. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.