

Here, Elmore et al. investigate how the Type III-B Cmr complex, which cleaves invader RNAs recognized by the CRISPR RNA (crRNA), functions. The findings demonstrate that the Cmr complex is a novel DNA nuclease activated by invader RNAs containing a crRNA target sequence and a protospacer-adjacent motif (rPAM).
Keywords: CRISPR–Cas, Cmr, PAM, RNAi, DNA nuclease
Abstract
CRISPR–Cas systems eliminate nucleic acid invaders in bacteria and archaea. The effector complex of the Type III-B Cmr system cleaves invader RNAs recognized by the CRISPR RNA (crRNA ) of the complex. Here we show that invader RNAs also activate the Cmr complex to cleave DNA. As has been observed for other Type III systems, Cmr eliminates plasmid invaders in Pyrococcus furiosus by a mechanism that depends on transcription of the crRNA target sequence within the plasmid. Notably, we found that the target RNA per se induces DNA cleavage by the Cmr complex in vitro. DNA cleavage activity does not depend on cleavage of the target RNA but notably does require the presence of a short sequence adjacent to the target sequence within the activating target RNA (rPAM [RNA protospacer-adjacent motif]). The activated complex does not require a target sequence (or a PAM) in the DNA substrate. Plasmid elimination by the P. furiosus Cmr system also does not require the Csx1 (CRISPR-associated Rossman fold [CARF] superfamily) protein. Plasmid silencing depends on the HD nuclease and Palm domains of the Cmr2 (Cas10 superfamily) protein. The results establish the Cmr complex as a novel DNA nuclease activated by invader RNAs containing a crRNA target sequence and a rPAM.
CRISPR–Cas systems eliminate nucleic acid invaders in bacteria and archaea by means of crRNA-guided nuclease effector complexes (Terns and Terns 2013; Barrangou and Marraffini 2014; van der Oost et al. 2014; Jackson and Wiedenheft 2015). The CRISPR RNAs (crRNAs) include target recognition sequences captured from invaders and cached between the repeat elements of the CRISPR locus (Bolotin et al. 2005; Mojica et al. 2005; Pourcel et al. 2005). crRNAs guide Cas proteins to identify and destroy corresponding invader nucleic acid.
There are multiple CRISPR–Cas systems comprised of distinct modules of Cas proteins that effect invader sequence acquisition, crRNA production, and invader destruction. The systems are classified into three broad types (Types I, II, and III), and each includes a signature superfamily protein (Cas3, Cas9, and Cas10, respectively) (Makarova et al. 2011). The characterized Type I and Type II systems cleave DNAs complementary to the crRNA guide sequences (Jinek et al. 2012; Westra et al. 2012). To prevent damage to the host genome (e.g., at the CRISPR where the invader sequences are cached), these systems depend on detection of a second signal to trigger cleavage activity; in addition to the sequence recognized by the crRNA, the Type I and II effector complexes require the presence of a short protospacer-adjacent motif (PAM) adjoining the target sequence in order to activate cleavage. The PAM sequence is found adjacent to the invader target sequences that these systems acquire but not in the CRISPR repeat sequence adjacent to the target sequence integrated in the host genome (Mojica et al. 2009; Shah et al. 2013).
Interestingly, the Type III systems have been found to target RNA and DNA (Hale et al. 2009; Zhang et al. 2012; Deng et al. 2013; Staals et al. 2013; 2014; Tamulaitis et al. 2014; Samai et al. 2015). In particular, the Type III-B Cmr complex from Pyrococcus furiosus cleaves target RNAs at 6-nucleotide (nt) intervals in the region of crRNA complementarity by means of a series of Cmr4 protein subunits that line the region of crRNA–target RNA base pairing (Hale et al. 2009, 2014; Benda et al. 2014; Ramia et al. 2014a; Osawa et al. 2015). At the 5′ end of the crRNA, Cmr3 recognizes the common crRNA tag sequence (derived from the repeat sequence in the CRISPR), and, near the 3′ end of the crRNA, Cmr1 and Cmr6 function in target RNA capture (Spilman et al. 2013; Hale et al. 2014; Osawa et al. 2015). The HD nuclease domain of the signature Cas10 superfamily protein Cmr2, also found at the 5′ end of the crRNA, is not required for RNA cleavage activity (Cocozaki et al. 2012; Spilman et al. 2013; Osawa et al. 2015). Type III-A Csm systems have also been shown to target RNA (Staals et al. 2014; Tamulaitis et al. 2014; Samai et al. 2015).
DNA silencing by the Type III-B Cmr (and Type III-A Csm) systems requires directional transcription of the invader DNA, which has been hypothesized to facilitate access of the crRNA to the target DNA strand (Deng et al. 2013; Goldberg et al. 2014). Csx1, an auxiliary protein associated with Cmr systems (Garrett et al. 2011; Makarova and Koonin 2013), was additionally found to be necessary for plasmid elimination in Sulfolobus islandicus (Deng et al. 2013). In the Csm complex, the Palm domain of the Cas10 superfamily protein Csm1 has been shown to be critical for plasmid silencing (Hatoum-Aslan et al. 2014; Ramia et al. 2014b; Samai et al. 2015). Rather than requiring a PAM sequence in the invader DNA to trigger specific destruction of the invader, it has been proposed that the characterized Type III systems are inhibited from cleaving the host genome CRISPR by the base-pairing potential between the CRISPR repeat sequence and the corresponding crRNA 5′ tag sequence (Marraffini and Sontheimer 2010; Deng et al. 2013).
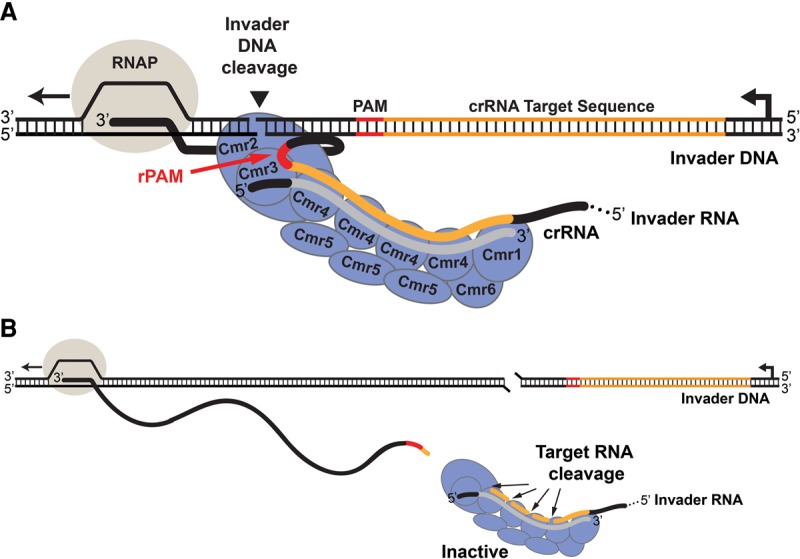
In this study, we investigate the mechanism of DNA silencing by the Cmr system in P. furiosus. Our results reveal a significant new paradigm for CRISPR–Cas silencing (illustrated in Fig. 6A, below): The Cmr complex is a DNA nuclease activated by the presence of an invader RNA containing the crRNA target sequence and a PAM (without a requirement for the crRNA target sequence and PAM in the DNA itself).
Figure 6.

Target RNAs containing a PAM sequence trigger Cmr dsDNA cleavage activity. (A) Model showing the Cmr complex (Cmr1–6 proteins and crRNA with the guide region in gray) activated by a target RNA containing a sequence recognized by crRNA (orange) and an rPAM (PAM sequence in the target RNA) sequence (red). The Cmr2 protein of the activated complex cleaves dsDNA. (B,C) Target RNA binding and cleavage by Cmr4 mutant complexes. 5′ radiolabeled 7.01 target RNA was incubated with Cmr crRNPs containing wild-type (WT) or mutant (D26N) Cmr4, and products were analyzed by native PAGE (B) or denaturing PAGE (C). (D) DNA cleavage activity of the Cmr4 mutant complex. Wild-type and Cmr4 mutant Cmr complexes were incubated with target RNA and 5′ radiolabeled dsDNA containing the crRNA 7.01 target sequence, and products were analyzed by denaturing PAGE. (E–G) Analysis of RNA PAM (rPAM) sequences. Cmr crRNPs were incubated with target RNAs containing the indicated flanking sequences (as well as the crRNA 7.01 target sequence) and with 5′ radiolabeled dsDNA substrates containing the crRNA 7.01 target sequence (E,G, orange) or a mutated target sequence (F, blue). Products were analyzed by denaturing PAGE. For G, reactions were analyzed at 5, 15, and 60 min. Flanking sequences corresponding to PAMs (identified in vivo) are indicated in red. Graphical representations of substrates are shown with the 7.01 target (orange) and mutant 7.01 target (blue) sequences and the radiolabeled strands (asterisks) indicated.
Results
The P. furiosus Type III-B Cmr system silences plasmid DNA in a transcription-dependent manner
The hyperthermophilic archaeon P. furiosus has three CRISPR–Cas systems: a Type III-B Cmr system as well as Type I-A Csa and Type I-G Cst systems (Supplemental Fig. S1). The effector complexes in P. furiosus use crRNAs produced from seven shared CRISPR loci (Majumdar et al. 2015). Using deletion strains containing a single CRISPR–Cas system, we found that the P. furiosus Type I-A Csa and Type I-G Cst systems silence plasmid DNA invaders in a PAM-dependent manner (Elmore et al. 2015). Here we examine the plasmid targeting capability of the P. furiosus Type III-B Cmr system. We generated and infected strains containing various combinations of CRISPR–Cas systems with plasmids containing either no target or a target sequence complementary to endogenous P. furiosus crRNA 7.01 (Fig. 1A). The 7.01 target sequence was included in both orientations relative to the plasmid backbone with or without a constitutive promoter in order to assess the dependence of any observed silencing on transcription and crRNA complementarity (Fig. 1B). Northern analysis confirmed expression of the “target RNA” (complementary to crRNA 7.01) (Fig. 1C, indicated by an asterisk) or the reverse complement of the target RNA (same sequence as crRNA 7.01) (Fig. 1D, indicated by an asterisk) from the respective constructs. The plasmid confers uracil prototrophy on P. furiosus, so successful infection of the plasmid would be observed as colony formation on selective media, and CRISPR–Cas defense would be observed as a reduction in colony formation (Fig. 1A).
Figure 1.

The P. furiosus (Pfu) Cmr system silences plasmid DNA in a transcription-dependent manner. (A) Plasmid challenge assay. In the absence of CRISPR–Cas defense, plasmid infection results in colony formation (growth of cells transformed with a plasmid containing the pyrF gene in the absence of uracil). (B) Target sequence transcription configuration of the various plasmids. The orientation of the 7.01 crRNA target sequence relative to the promoter and plasmid backbone is shown. Plasmids were designed for no transcription (none +/−), transcription of a target RNA complementary to the endogenous 7.01 crRNA (target [tar]), or transcription of an RNA that is not complementary to the 7.01 crRNA (reverse complement of target [rc]). (C,D) Northern analysis of expression of the 7.01 target RNA (C) or the reverse complement RNA (D). The left lane is Decade marker RNA (Life Technologies). All other lanes contain 10 µg of total RNA isolated from P. furiosus TPF20 strains bearing plasmids configured to express the indicated RNA. The primary product is indicated with an asterisk. Blots were also probed for 5S rRNA as a loading control. (E) Colonies produced by infection with five plasmids in wild-type (three endogenous CRISPR–Cas systems; dark gray), ΔCmr (lacking the Cmr system; medium gray), Cmr (Cmr only; blue), and null (no system; light gray) strains. The presence and orientation of crRNA 7.01 target sequence on the plasmids are indicated by an orange arrow above the graph and a dashed line or 7.01 below the graph. The presence of the promoter for target region transcription on the plasmid is indicated by a line arrow above the graph, and the presence of a target region transcript is indicated below the graph as a dashed line, “none,” “target,” and “rc of target.” Colony numbers are plotted with the standard deviation in nine replicates indicated by error bars.
In addition to the wild-type P. furiosus strain with three CRISPR–Cas systems (Supplemental Fig. S1), we tested strains containing only the Cmr system (Cmr), lacking the Cmr system (ΔCmr), and lacking all CRISPR–Cas modules (null) (Fig. 1E). Transformation with the plasmid lacking the target sequence resulted in the formation of 5.81 × 103 to 2.86 × 103 uracil prototroph colonies per 100 ng of plasmid DNA (Fig. 1E, wild-type and Cmr strains, respectively). The Csa and Cst CRISPR–Cas systems in P. furiosus provide orientation-independent and transcription-independent defense against plasmids with crRNA target sequences (Elmore et al. 2015), and thus, as expected, the presence of a target sequence on the plasmid reduced colony formation to <1% of the negative control plasmid in both the wild-type strain and the ΔCmr strain (fewer than two uracil prototroph colonies per 100 ng of plasmid DNA) (Fig. 1E, wild-type [dark gray bars] and ΔCmr [light-gray bars]). In the absence of CRISPR–Cas defense, plasmids containing the target sequence are not silenced and produce colony numbers indistinguishable from the negative control plasmid (Fig. 1E, null, white bars).
Notably, the Cmr strain does not silence plasmids in which the target sequence is not transcribed (RNA: none [+] or none [−]) or in which the transcribed RNA is not complementary to the crRNA (reverse complement of target); however, the Cmr strain effectively silences plasmids that transcribe a target RNA recognized by the crRNA (target RNA) (Fig. 1E, Cmr, blue bars). The results indicate that the P. furiosus Type III-B Cmr system performs transcription-dependent plasmid silencing.
The Type III-B Cmr system in P. furiosus recognizes a PAM in the invader (rather than repeat tag complementarity in the host) to distinguish invader from host
Evidence indicates that the Type III-A Csm system does not target DNA (such as the host's own CRISPR locus) that has complementarity to the 5′ tag sequence of the crRNA adjacent to the crRNA target site (see Supplemental Fig. S2A, gray dashed line; Marraffini and Sontheimer 2010; Samai et al. 2015), while Type I systems specifically target DNA (such as the invader DNA) that has a PAM sequence adjacent to the crRNA target site (see Fig. 2A; Almendros et al. 2012; Fischer et al. 2012; Sinkunas et al. 2013; Westra et al. 2013; Plagens et al. 2014). In order to determine the mechanism of host protection by the P. furiosus Cmr system, we challenged cells with plasmids possessing various sequences 3′ of the crRNA target site (where both tag complementarity and PAMs occur).
Figure 2.
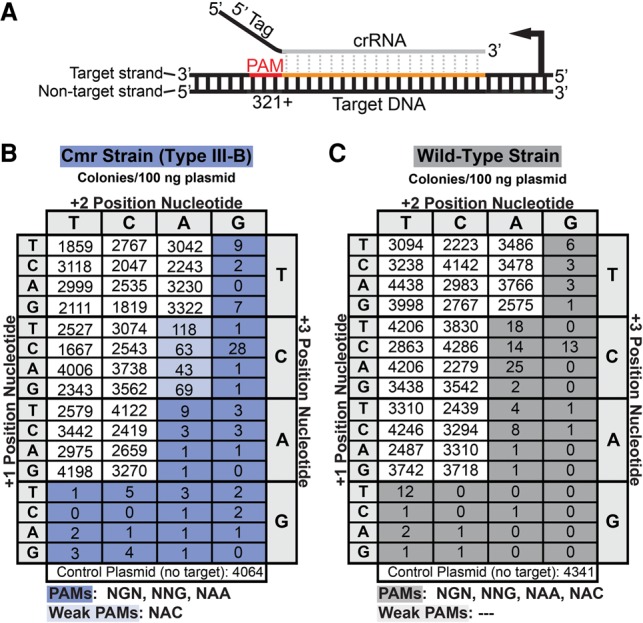
The Cmr system in P. furiosus (Pfu) uses a PAM to distinguish invader from host. (A) The location of tested PAM sequences (red) relative to the target sequence in the DNA (orange) and aligned crRNA. Dotted gray lines indicate complementarity between the crRNA guide region (gray) and DNA. (B,C) Results of plasmid infection assays for Cmr (blue) and wild-type (gray) strains, respectively. The plasmids contain the transcribed 7.01 target sequence and the indicated flanking (PAM) sequence. Colony numbers are the average of at least three replicates. Target-adjacent sequences that activated CRISPR–Cas targeting resulting in >100-fold reduction in colony numbers relative to negative control plasmid are shaded dark blue or gray. Sequences that conferred 30-fold to 100-fold reduction in colony numbers are shaded light blue or gray. The plasmid sequence downstream from the PAM (positions +4 to +8) in all plasmids is 5′-TTCCG-3′.
In Supplemental Figure S2, we tested silencing of several transcribed 7.01 target plasmids containing varying degrees of complementarity to the P. furiosus crRNA 5′ tag sequence. Note that the Csa and Cst systems present in the ΔCmr strain (Supplemental Fig. S2, light-gray bars) require a PAM sequence that is not present in any of the tested plasmids, so the plasmids were not silenced in the control strain containing these two systems (Supplemental Fig. S2B, ΔCmr, light-gray bars). In the Cmr strain (Supplemental Fig. S2B, blue bars), full complementarity of the plasmid sequence to the crRNA tag sequence conferred protection on the plasmid, resulting in colony formation similar to the control plasmid lacking crRNA target sequence (Supplemental Fig. S2B, variant 1 and no target). Moreover, in the absence of 5′ crRNA tag complementarity, the plasmid is not protected (Supplemental Fig. S2B, variant 9). However, analysis of further variants suggests a specific role for the target site-proximal 3 nt of the flanking DNA. Three nucleotides of complementarity proximal to the crRNA target site protected the plasmid (Supplemental Fig. S2B, variant 6); however, modification of these three proximal nucleotides, even in the context of five remaining nucleotides of complementarity, resulted in loss of protection (Supplemental Fig. S2B, variant 10, see also variants 7 and 8), suggesting that the identity of these nucleotides may be more important than tag complementarity. Note that, as expected, plasmids silenced by the Cmr system in the Cmr strain were also silenced in the wild-type strain (Supplemental Fig. S2B, dark-gray bars).
To more clearly determine whether the host protection mechanism was complementarity-driven protection of the host or PAM-driven targeting of the invader and comprehensively delineate the Cmr system-flanking sequence requirement, we tested the complete series of variations in the proximal 3 nt of the 3′ adjacent region (Fig. 2; Supplemental Fig. S3). The negative control plasmid (no target) produced an average of 4.34 × 103 and 4.06 × 103 uracil prototroph colonies per 100 ng of plasmid DNA in the wild-type and Cmr strains, respectively, as expected. We found that plasmids with various trinucleotide sequences completely lacking tag complementarity were not targeted in the Cmr (and wild-type) strain (e.g., TCC, ACC, and GCC) (Fig. 2B), indicating that potential targets are not simply protected by crRNA tag complementarity in this region. On the other hand, plasmids with a contiguous series of 32 trinucleotide sequences were effectively silenced in the Cmr (and wild-type) strain (>100-fold reduction in colony formation relative to negative control plasmid) (Fig. 2B,C, dark-blue and gray shading). (Another four trinucleotide sequences were weakly silenced in the Cmr strain [30-fold to 100-fold reduction in colony formation] [Fig. 2B, light-blue shading].)
To confirm the generality of these observations, we tested for targeting of plasmids by P. furiosus crRNAs 2.01 and 6.01 (Supplemental Fig. S4). As expected, plasmids containing the 2.01 or 6.01 target sequences were silenced when the plasmids included the PAMs identified by crRNA 7.01 targeting (GGG or AAA) and not non-PAM sequences (CCC or TTT) (Supplemental Fig. S4).
The results indicate that the P. furiosus Cmr system requires a PAM in addition to the crRNA target sequence in order to target DNA for silencing. Functional Cmr PAM sequences include NGN, NNG, and NAA (and NAC) (Fig. 2C). The crRNA target sequences stored in the seven P. furiosus CRISPR loci do not contain a functional PAM in their adjacent CRISPR repeats (CTT). The range of PAM sequences recognized by the Cmr system in P. furiosus fully encompasses and significantly extends beyond the combined set recognized by the P. furiosus Type I-A Csa and Type I-G Cst systems (NGG, NGA, NAG, and HCG) (Elmore et al. 2015). Our findings indicate that the Type III-B Cmr system in P. furiosus depends on a PAM sequence to activate silencing (rather than on complementarity to inhibit silencing) in order to protect against self-destruction at CRISPR loci.
Csx1 is not essential for transcription-dependent DNA silencing by the Type III-B Cmr system in P. furiosus
Csx1 and Csm6 are CARF (CRISPR-associated Rossman fold) domain proteins often associated with Type III CRISPR–Cas systems, which were recently identified as important for transcription-dependent silencing by the S. islandicus Type III-B Cmr and Staphylococcus epidermidis Type III-A Csm systems, respectively (Deng et al. 2013; Goldberg et al. 2014; Hatoum-Aslan et al. 2014; Makarova et al. 2014). P. furiosus harbors a csx1 gene within the Cmr module (Supplemental Fig. 1). To test whether Csx1 is required for P. furiosus Cmr transcription-dependent DNA silencing, we deleted csx1 in the Cmr strain (Cmr ΔCsx1). The csx1 deletion strain was infected with plasmids that produce 7.01 target RNA, its reverse complement, or no target RNA; however, we observed no change in plasmid silencing in the absence of Csx1 (Supplemental Fig. S5A). We confirmed the absence of Csx1 in the deletion strain by Western analysis (Supplemental Fig. S5B). The protein composition and RNA cleavage activity of immunopurified Cmr complexes were also not disrupted by deletion of csx1 (Supplemental Fig. S5C,D). Furthermore, crRNA maturation and accumulation were not observably affected by the absence of Csx1 (Northern analysis) (data not shown). Our results indicate that Csx1 is not essential for Cmr crRNP formation, RNA cleavage activity, or, unlike Csx1/Csm6 in other characterized Type III systems (Deng et al. 2013; Goldberg et al. 2014; Hatoum-Aslan et al. 2014), transcription-dependent plasmid silencing activity by the Cmr system in P. furiosus in our assay.
The Cmr2 protein cleaves ssDNA via its HD nuclease domain
Cmr2 is a member of the Cas10 superfamily—the signature protein family of the Type III CRISPR–Cas systems—with characteristic Palm and N-terminal HD nuclease domains (Makarova et al. 2011). Csm1, the Cas10 protein of the Type III-A Csm complex, is essential for plasmid elimination (Hatoum-Aslan et al. 2014) and has been shown to have 3′–5′ ssDNA exonuclease activity via the Palm domain in S. epidermidis and ssDNA endonuclease activity via the HD domain in Thermococcus onnurineus (Ramia et al. 2014b; Jung et al. 2015). Cmr2 is an essential component of the P. furiosus Cmr RNA targeting complex; however, the HD domain is not required for the RNA cleavage activity of the complex in vitro (Hale et al. 2009; Cocozaki et al. 2012).
To test whether Cmr2 is a DNA nuclease, we incubated purified P. furiosus Cmr2 with linear ssDNA and dsDNA, a “bubble” dsDNA (having a ssDNA internal sequence), and circular ssDNA. Cmr2 efficiently cleaved linear 5′ radiolabeled ssDNA (Fig. 3A) but not dsDNA (Fig. 3B). Cleavage of linear ssDNA by Cmr2 is metal-dependent: Cleavage was most efficient in the presence of Ni2+ (included in assays shown in Fig. 3) and Co2+, less efficient with Mn2+, and blocked by addition of EDTA (Supplemental Fig. S6). Cmr2 also cleaves circular single-stranded M13 (ssM13) phage DNA (Fig. 3C) and within the single-stranded region of a bubble DNA substrate (Fig. 3D), revealing that Cmr2 can act endonucleolytically to cleave ssDNA.
Figure 3.
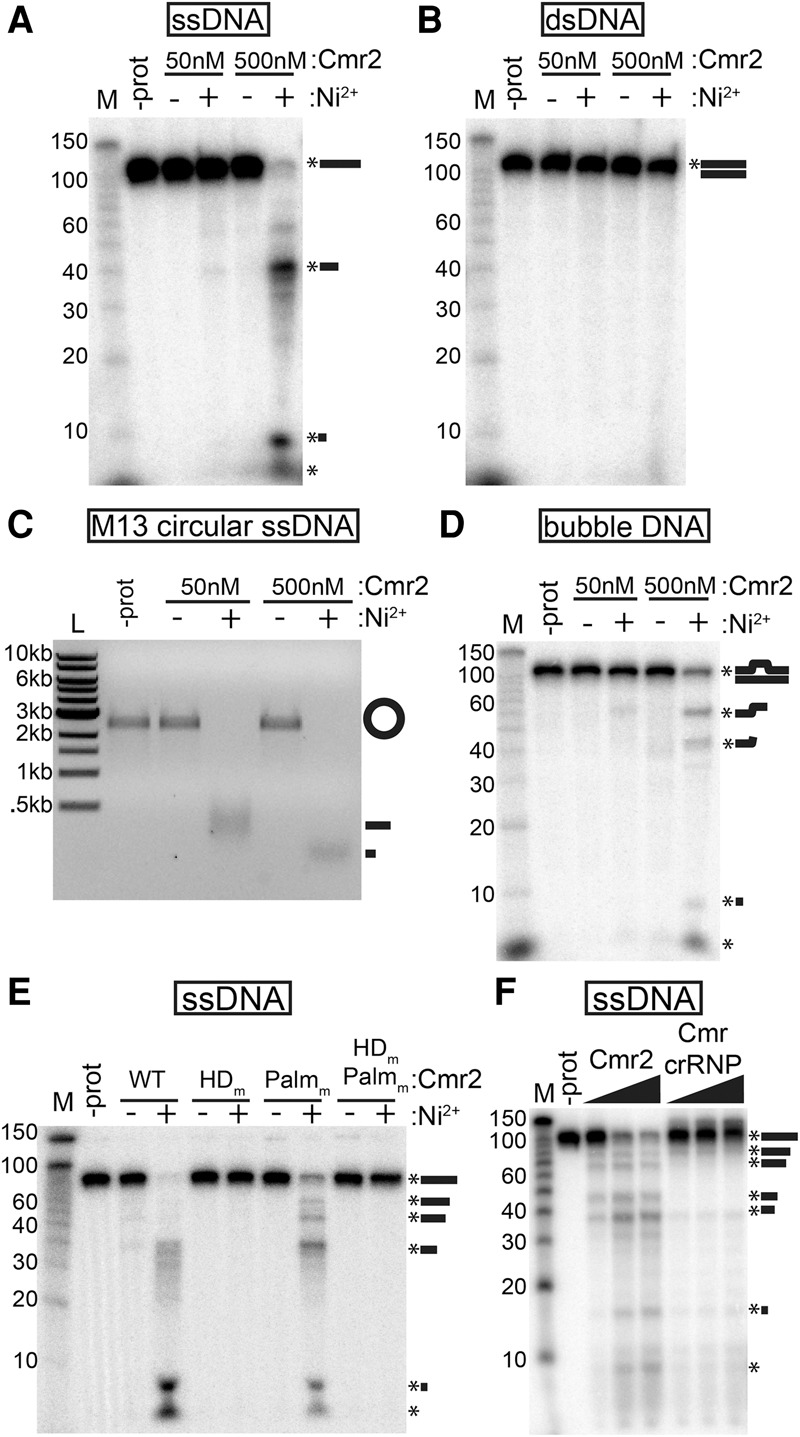
Cmr2 is a ssDNA endonuclease. (A–D) Substrate analysis. Recombinant Cmr2 (50 or 500 nM) was incubated with (+) or without (−) added NiCl2 (200 µM) in the presence of a 5′ radiolabeled ssDNA oligo (A), a 5′ radiolabeled dsDNA annealed from oligos (B), unlabeled circular ssM13 phage DNA (C), or a 5′ radiolabeled “bubble” DNA substrate (with a central single-stranded region) annealed from two partially complementary ssDNA oligos (D). (E) Mutant analysis. Wild-type (WT), HD domain putative active site mutant (HDm), Palm domain GGDD motif mutant (Palmm), or double-mutant (HDm,Palmm) Cmr2 proteins were incubated with 5′ radiolabeled ssDNA with (+) or without (−) added NiCl2. (F) Activity in the context of the Cmr complex. Increasing amounts (50–200 nM) of either recombinant Cmr2 or preformed Cmr crRNP complexes (Cmr1–6 + crRNA) were incubated with 5′ radiolabeled ssDNA oligo. Products were analyzed by denaturing PAGE (A,B,D–F) or agarose gel electrophoresis (C). Radiolabeled Decade markers (M) or New England Biolabs 1-kb ladder (L) were used for size estimation.
Mutation of the HD domain active site abolishes the ssDNA cleavage activity of Cmr2 (alanine substitution of residues H13 and D14) (Fig. 3E, HDm). Mutation of the GGDD motif in the Palm domain has no effect on activity (alanine substitution of residues D673 and D674) (Fig. 3E, Palmm). Additional assays indicate that deletion of the HD domain also abolishes ssDNA cleavage activity and that the HD domain alone is insufficient for ssDNA cleavage (data not shown).
Interestingly, the observed ssDNA nuclease activity of the Cmr2 protein is significantly attenuated within the Cmr crRNP complex. The reconstituted Cmr complex (including Cmr1–6 and crRNA) displays very little ssDNA nuclease activity in comparison with approximately equimolar concentrations of Cmr2 alone (Fig. 3F). Our results indicate that Cmr2 is a latent DNA nuclease present within the Cmr effector complex.
Binding of a target RNA activates generic DNA cleavage activity of the Type III-B Cmr complex
Plasmid targeting by the Cmr complex depends on directional transcription of the target region of the plasmid in vivo (Fig. 1), suggesting that activity of the complex may depend on transcription-induced access to a ssDNA target site and/or on the RNA product of transcription of the target site. To test for stimulation of DNA cleavage activity by the target RNA, we assayed DNA cleavage in the presence of the 7.01 target RNA (complementary to the 7.01 crRNA guide region) or the reverse complement of the target RNA (Fig. 4).
Figure 4.
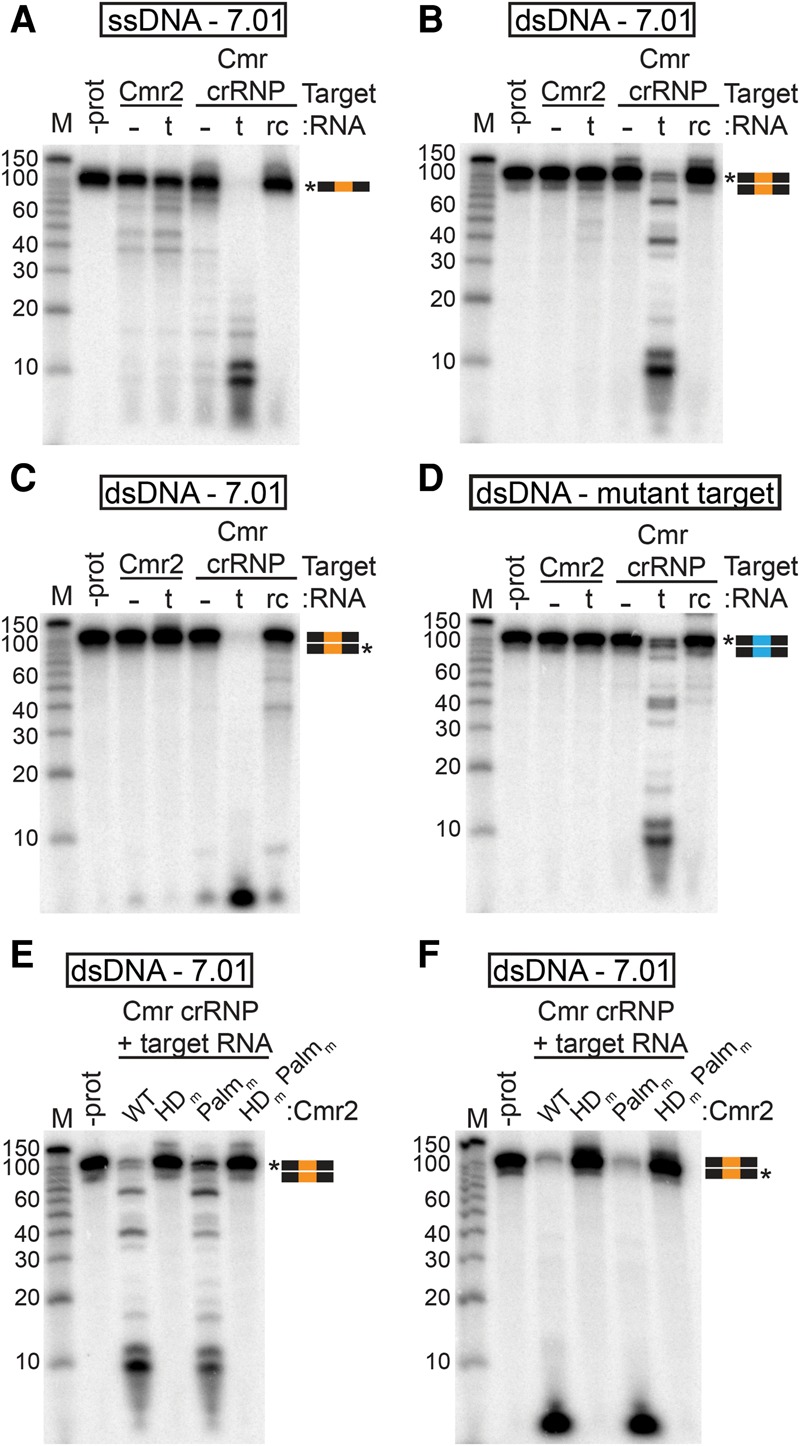
The Cmr complex cleaves dsDNA in the presence of target RNA. (A–D) Substrate analysis. Cmr2 (50 nM) and Cmr crRNP complexes (∼50 nM) were analyzed with (t) or without (−) the target RNA (complementary to the 7.01 crRNA) or with the reverse complement of the target RNA (rc). The substrates used were 5′ radiolabeled ssDNA complementary to crRNA 7.01 (A), dsDNA with a crRNA 7.01 target sequence 5′ radiolabeled on the target (B) or nontarget (C) strand, and dsDNA with a mutant 7.01 target sequence (D). (E,F) Mutant analysis. Cmr crRNP complexes containing wild-type (WT), HD domain putative active site mutant (HDm), Palm domain GGDD motif mutant (Palmm), or double-mutant (HDm,Palmm) Cmr2 were incubated with dsDNA with the crRNA 7.01 target sequence 5′ radiolabeled on the target (E) or nontarget (F) strand. Graphical representations of substrates are shown with the 7.01 target (orange) and mutant 7.01 target (blue) sequences and radiolabeled strands (asterisks) indicated. Products were analyzed by denaturing PAGE. Decade marker RNA (M) were included for size estimations.
We found that the Cmr complex cleaves both ssDNA and dsDNA specifically upon addition of the target RNA (complementary to the crRNA guide sequence) (Fig. 4A,B, Cmr crRNP, t lanes). Activity was not stimulated by the reverse complement of the target RNA (Fig. 4A,B, rc lanes). Notably, while stimulation of the complex is specifically dependent on the complementary target RNA (both in vivo and in vitro) (Figs. 1, 4), cleavage by the complex is independent of complementarity of the DNA to the crRNA: dsDNA lacking the crRNA target sequence is also efficiently cleaved by the activated complex (dsDNA mutant target) (Fig. 4D, Cmr crRNP, t lane). Both strands of the dsDNA are cleaved (individual strands 5′ radiolabeled in Fig. 4B,C). The ssDNA cleavage activity of the Cmr complex stimulated by the target RNA was greater than the ssDNA cleavage activity of an approximately equimolar concentration of Cmr2 alone (Fig. 4A).
Cmr2 is the DNA nuclease of the target RNA-activated Cmr complex
The importance of Cmr2 nuclease activity in the observed dsDNA cleavage by the activated Cmr complex was determined using complexes reconstituted with Cmr2 active site mutants. As with the free Cmr2 protein, mutation of the Palm domain GGDD motif (Palmm) did not significantly affect cleavage of either strand of the DNA, but mutation of the HD domain of Cmr2 (HDm) abolishes cleavage of both strands of dsDNA by the activated Cmr complex in vitro (Fig. 4E,F). The results indicate that Cmr2 mediates DNA cleavage by the Cmr complex. Furthermore, Cmr2 is inactive in the Cmr complex except in the presence of a target RNA complementary to the crRNA (Fig. 4A–D).
To test the importance of Cmr2 in plasmid silencing in vivo, we generated P. furiosus Cmr strains with various Cmr2 mutations. Each strain was assayed for transcription-dependent plasmid silencing activity (using plasmids that produce 7.01 target RNA, its reverse complement, or no target RNA) (Fig. 5A). While mutation of the HD domain abolishes DNA cleavage activity of the activated Cmr complex and the Cmr2 protein in vitro (Figs. 3,4), deletion of the HD domain did not disrupt plasmid silencing in vivo (Fig. 5A, ΔHD). Mutation of the Palm domain also did not disrupt silencing (Fig. 5A, Palmm). However, simultaneous mutation of both the Palm domain and the HD domain (either deletion or active site mutation) abolished Cmr plasmid silencing (Fig. 5A, ΔHD-Palmm and HDm-Palmm), suggesting that both the HD and Palm domains can mediate plasmid silencing in vivo. The silencing mediated by the HD and Palm domains in vivo was dependent on transcription of the target sequence (Fig. 5A, ΔHD and Palmm), consistent with the requirement for the target RNA for DNA cleavage in vitro (Fig. 4). The lack of DNA cleavage activity supported by the Palm domain in vitro (Fig. 4) suggests that an additional requirement for Palm domain activity is provided in vivo (which could be related to other aspects of transcription or numerous other differences). Analysis of complexes immunoprecipitated from each strain indicates that the Cmr2 mutations did not prevent formation of the Cmr effector complex (Fig. 5B) or target RNA cleavage by the Cmr complex (Fig. 5C). The results indicate that Cmr2 plays an essential function in DNA cleavage activity of the Cmr complex that can be mediated by its Palm or HD nuclease domain in vivo.
Figure 5.
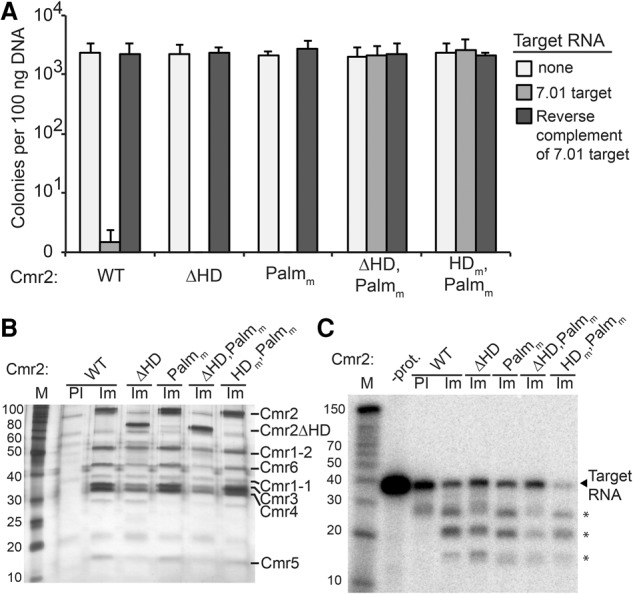
Mutation of both the HD and Palm domains of Cmr2 is required to abolish Type III-B plasmid silencing in vivo. (A) Plasmid infection of strains with Cmr2 mutants. Cmr strains expressing the indicated Cmr2 proteins (wild type [WT], ΔHD, Palmm, ΔHD,Palmm, and HDm,Palmm) were infected with plasmids expressing no target RNA (light gray), crRNA 7.01 target RNA (gray) or the reverse complement RNA (dark gray). Colony numbers are plotted with error bars indicating the standard deviation in three replicates. (B) Cmr complexes from Cmr2 mutant strains. Proteins immunoprecipitated with preimmune (PI; wild type only) or immune (Im) antibodies against Cmr2 from Cmr strains expressing the indicated Cmr2 proteins (wild type, ΔHD, Palmm, ΔHD,Palmm, and HDm,Palmm) were analyzed by SDS-PAGE and silver staining. Cmr protein identities are indicated based on predicted molecular weights and mass spectrometry. (C) RNA cleavage activity of Cmr complexes in Cmr2 mutant strains. Complexes immunopurified from Cmr strains expressing the indicated Cmr2 proteins (wild type, ΔHD, Palmm, ΔHD,Palmm, and HDm,Palmm) were incubated with 5′ end-labeled crRNA 7.01 target RNA. Products were analyzed by denaturing PAGE. Decade marker RNAs (M) were included for size estimations. Asterisks mark primary RNA cleavage products. Note that the top product migrates distinctly from the band in observed in preimmune sample in multiple experiments.
Cleavage of the target RNA is not required for DNA cleavage
The Cmr complex cleaves complementary target RNAs in vitro and in vivo (Hale et al. 2009, 2012). To determine whether target RNA cleavage is required to stimulate Cmr DNA cleavage activity, we disrupted RNA cleavage with a Cmr4 active site mutation. Cmr complexes containing either wild-type or mutant Cmr4 (RNA cleavage active site mutant D26N) were incubated with radiolabeled target RNA (complementary to crRNA guide sequence). Target RNA binding (assessed by native PAGE) was not affected by the Cmr4 mutations (Fig. 6B), but target RNA cleavage was abolished (Fig. 6C). To examine DNA cleavage activity, we incubated the Cmr complexes containing the Cmr4 mutant with the target RNA and radiolabeled dsDNA. The Cmr4 mutations had no observable effect on DNA cleavage activity of the complex (Fig. 6D), indicating that Cmr DNA cleavage activity does not require cleavage of the target RNA.
A PAM sequence in the target RNA (rPAM) is required to activate the Type III-B Cmr complex DNA cleavage activity
Many Type I and Type II CRISPR–Cas effector complexes depend on the bipartite PAM sequence/crRNA recognition sequence in the target DNA to activate target DNA cleavage (Jinek et al. 2012; Westra et al. 2012; Sinkunas et al. 2013; Plagens et al. 2014). We identified a series of PAM sequences required in vivo for transcription-dependent plasmid silencing by the Cmr system (Fig. 2; Supplemental Fig. S3). However, we found that the target RNA-activated Cmr complex does not depend on recognition of the crRNA target site in the target DNA (Fig. 4D), suggesting that the identified PAM sequences may also not be recognized in the invader DNA.
As the PAM sequences are also found adjacent to the crRNA recognition site in the target RNAs in vivo, we tested the ability of target RNAs with and without identified PAMs (Fig. 2) to stimulate DNA cleavage. We found that target RNAs containing a PAM 3′ of the 7.01 crRNA recognition sequence (GGG and AAA) (see Fig. 6A) effectively activated DNA cleavage, but, strikingly, RNAs lacking PAMs (CCC and UUU) did not (Fig. 6E). Figure 6G shows the time course of DNA cleavage stimulated by a further series of target RNAs. Again, the target RNAs containing Cmr PAMs defined by in vivo plasmid silencing assays (Fig. 2) stimulated DNA cleavage (Fig. 6G, UGG, UGU, and UUG). RNAs lacking PAMs failed to activate DNA cleavage despite having the crRNA recognition sequence and, notably, despite the presence of an identified PAM and crRNA recognition sequence in the DNA substrate included in the assay (Fig. 6G [CUU, UCU, and UUC], E [CCC and UUU]).
DNA cleavage activity was only stimulated by target RNAs containing both the crRNA recognition sequence and a PAM (Fig. 6A). RNAs with a GGG PAM but the reverse complement (rc-GGG) or a mutated version (mut-GGG) of the 7.01 crRNA recognition sequence failed to activate DNA cleavage (Fig. 6E). Also consistent with our previous findings (Fig. 4D), DNA cleavage stimulated by the 7.01-GGG and 7.01-AAA target RNAs is independent of a crRNA recognition site in the DNA: Cmr complexes activated by both target RNAs cleave DNA with and without the crRNA recognition sequence (Fig. 6E, F, respectively).
Discussion
The findings presented here reveal novel mechanisms for CRISPR–Cas effector complex activation and invader specificity: The Cmr effector complex is activated to cleave DNA by RNAs that contain an rPAM sequence and crRNA target sequence (Fig. 6A).
Transcription-dependent invader silencing: DNA nuclease activity is triggered by the target RNA
Plasmid silencing by the Cmr system in P. furiosus requires transcription of the crRNA target region (Fig. 1; Supplemental Fig. S4). Invader silencing had also previously been observed to be transcription-dependent for the Type III-A Csm system in S. epidermidis (Goldberg et al. 2014) and the Type III-B Cmr system in S. islandicus (Deng et al. 2013). A leading hypothesis is that transcription is important to allow access of the CRISPR–Cas DNA nuclease to the target DNA sequence; however, our findings indicate that the product of transcription, rather than the physical process of transcription, is essential for function. We found that exogenously supplied target RNA is sufficient to activate Cmr DNA nuclease activity in vitro (Figs. 4, 6). The S. epidermidis Csm crRNP was observed to nick target strand DNA in vitro when coupled with target transcription (Samai et al. 2015). Notably, the Csm in vitro DNA-nicking activity and the previously observed in vivo invader silencing by both the Csm and Cmr systems (like the Cmr system in P. furiosus) (Fig. 1; Supplemental Fig. S4; Deng et al. 2013; Goldberg et al. 2014) specifically require transcription of the target strand, consistent with a requirement for the target RNA product. Invader silencing likely involves a complex biochemical mechanism; our findings indicate that target RNA detection is one essential aspect.
rPAM: Recognition of the PAM sequence in the target RNA licenses DNA cleavage
CRISPR–Cas systems use mechanisms to distinguish invader targets from host CRISPR loci. To date, two mechanisms have been described: deactivation of the DNA nuclease by complementarity of the crRNA 5′ tag region with the potential CRISPR target for the Type III-A Csm system (see Supplemental Fig. S2; Marraffini and Sontheimer 2010) and requisite activation of the nuclease by a PAM sequence (in addition to the target sequence) in the invader DNA for Type I and II systems (see Fig. 2; Mojica et al. 2009; Shah et al. 2013). Notably, we found that the Type III-B Cmr system in P. furiosus is activated by PAMs rather than deactivated by 5′ tag complementarity (Fig. 2; Supplemental Figs. S3, S4). Note that plasmids with the 5′ tag complementary sequence are not targeted, but neither are plasmids with many flanking sequences lacking 5′ tag complementarity; i.e., GCC (Fig. 2; Supplemental Figs. S2, S3). (Although there is no discernable pattern to suggest interaction between the crRNA tag sequence and non-PAM sequences, we cannot exclude a role for miscellaneous non-Watson-Crick interactions in function.) The PAM that activates the P. furiosus Cmr complex is very flexible, requiring only a single G (or two As) in one of two critical positions (Fig. 2) to induce silencing, and includes the PAM that was bioinformatically predicted (5′-NGG-3′) for the CRISPR repeat sequence found in P. furiosus (Kunin et al. 2007; Mojica et al. 2009).
Remarkably, the DNA cleavage activity of the P. furiosus Cmr complex depends on the presence of the discriminating PAM sequence within the target RNA molecule. This contrasts the well-characterized Type I and II CRISPR–Cas systems, in which the PAM sequence is recognized in the DNA substrate (Jinek et al. 2012; Westra et al. 2012; Sinkunas et al. 2013; Rollins et al. 2015). We expect that further analysis will reveal that other Type III systems use an rPAM to effect invader silencing. Based on the orientation of the target RNA by the crRNA within the Cmr complex (Spilman et al. 2013; Osawa et al. 2015), we predict that Cmr2 and/or Cmr3 are involved in rPAM recognition (see Fig. 6). Electron microscopic studies of the architecturally similar Type I and Type III CRISPR–Cas complexes have demonstrated that interaction with a target DNA or RNA can effect concerted structural changes across multiple subunits of these effector complexes (Wiedenheft et al. 2011; Hochstrasser et al. 2014; Taylor et al. 2015). Our findings indicate that bipartite recognition of an invader—both the crRNA target sequence and the discriminating PAM sequence within the invader RNA—activates DNA cleavage activity of the Type III-B CRISPR–Cas complex.
Target RNA cleavage by Type III complexes: possible role in the limitation of DNA nuclease activity
Cleavage of invader RNAs (complementary to the crRNA) has been observed for numerous Type III systems (Hale et al. 2009; Zhang et al. 2012; Staals et al. 2013, 2014; Tamulaitis et al. 2014; Samai et al. 2015) and, prior to detection of DNA nuclease activity, was thought to be the mechanism of invader resistance by Cmr systems. Indeed, RNA cleavage can silence RNA viruses (Tamulaitis et al. 2014). DNA and RNA cleavage activities of the Type III CRISPR–Cas systems may function in parallel to eliminate DNA invaders and their transcripts.
Cleavage of the target RNA is not required for transcription-dependent DNA interference associated with Type III systems (Fig. 6D; Samai et al. 2015). Nonetheless, RNA cleavage may play an important role in the regulation of the function of Type III DNA nucleases in vivo. As outlined below, cleavage of the target RNA may be important for deactivation or turnover of complexes activated by the target RNA, which will be an interesting area of future investigation.
Cmr2 is the DNA nuclease of the Type III-B CRISPR–Cas system
In this study, we determined that the Cas10 superfamily protein of the P. furiosus Cmr effector complex, Cmr2 (Makarova et al. 2011), is a latent DNA endonuclease (Figs. 3, 4, 6). Our results revealed that the activity of Cmr2 is curbed in the context of the Cmr crRNP (Fig. 3F) and indicate that it is activated by interaction of the complex with target RNAs containing rPAMs (Fig. 6). Although other silencing mechanisms are possible, Cmr2-mediated DNA cleavage likely effects plasmid silencing by the Type III-B CRISPR–Cas system, which is target RNA transcription-dependent and PAM-dependent and was not observed when the HD and Palm domains of the Cmr2 nuclease were inactivated (Fig. 5).
The Cas10 proteins of the Type III CRISPR–Cas systems are characterized by the presence of two Palm domains (one of which is typically predicted to be inactive) (Cocozaki et al. 2012) and often contain a fused HD nuclease domain (Makarova et al. 2006). The evidence regarding the roles of the HD and Palm domains of Cas10 proteins in DNA targeting is complex.
Our findings indicate that both the Palm domain and the HD domain of Cmr2 can mediate silencing in the P. furiosus Type III-B Cmr complex. We found that mutation of the HD domain (but not the Palm domain) of Cmr2 disrupts cleavage of both DNA strands by the complex in vitro (Fig. 4). While the Palm domain does not function in the in vitro assay, mutation of both domains is required to disrupt silencing in vivo (Fig. 5), indicating that both domains can mediate silencing in vivo.
At the same time, current evidence implicates one or the other of the domains in Type III-A Csm systems from different organisms. For the Type III-A Csm1 protein from S. epidermidis, mutation of the Palm domain GGDD motif disrupts DNA cleavage by the complex (and by the isolated protein) in vitro (Ramia et al. 2014b; Samai et al. 2015). (The HD domain mutant was not tested in Samai et al. 2015.) In addition, mutation of the Palm domain alone (and not of the HD domain alone) of S. epidermidis Csm1 disrupts silencing in vivo (Hatoum-Aslan et al. 2014). However, mutation of the HD domain alone of T. onnurineus Csm1 disrupts DNA cleavage in vitro (Jung et al. 2015). (The Palm domain mutant was not tested in Jung et al. 2015.)
Cmr2 crystal structures indicate that both domains coordinate divalent metals in their active sites (Cocozaki et al. 2012; Osawa et al. 2015). However, some Cas10 proteins appear to contain only one or the other of the two nuclease domains (Makarova et al. 2006; Vestergaard et al. 2014). Collectively, the data suggest that either or both the Palm and HD domains of Cas10 superfamily nucleases may catalyze DNA cleavage within Type III CRISPR–Cas effector complexes.
Models for function of DNA nuclease in invader silencing by Type III-B Cmr systems
A DNA nuclease activity that does not require a crRNA target sequence within the DNA is a novel mechanism in CRISPR–Cas invader silencing that presents challenges in understanding its function. We propose two models to describe how the Cmr system may function in invader defense, in which the DNA nuclease acts locally in cis or more broadly in trans.
The DNA nuclease activity of the Cmr complex depends on the target RNA, and interaction with the nascent invader RNA may physically tether the nuclease and limit its activity to the invader DNA (illustrated in Fig. 7A). Regulation of the DNA nuclease activity by cleavage of the target RNA could further function to limit nuclease activity to the invader DNA. For example, cleavage of the target RNA may terminate the activity of the DNA nuclease as it drifts away from the site of transcription (illustrated in Fig. 7B). Consistent with this model, disruption of target RNA cleavage by the S. epidermidis Csm complex results in a hyperactive DNA silencing activity phenotype in vivo (Samai et al. 2015).
Figure 7.
Model for limitation of Cmr nuclease activity to invader DNA. (A) The Cmr complex DNA nuclease is activated and tethered by the nascent target RNA. The transcribed invader RNA containing the crRNA target sequence (orange) and rPAM (red) is recognized by the Cmr complex containing the crRNA with the guide sequence (gray). The activated nuclease is tethered to and cleaves the invader DNA. (B) As the length of the tether increases, the Cmr complex DNA nuclease is deactivated by cleavage of the target RNA.
Alternatively, the Cmr system may act as a sophisticated abortive infection system that, when activated by invader RNA, destroys host cell DNA to curb the spread of the infection: the altruistic sacrifice of the individual cell for the benefit of the population. Such an immune response would be analogous to bacterial abortive infection systems (Labrie et al. 2010; Samson et al. 2013) or the hypersensitive response in plants (Spoel and Dong 2012). For a CRISPR–Cas immune system, this mode of action (destruction of the cell in which the system is activated) might be expected to eliminate the relevant CRISPR-acquired invader targeting sequence from the population, which might be an acceptable cost for survival of the population. However, notably, Cmr systems are rarely found in the absence of another CRISPR–Cas system (Haft et al. 2005) that may act as the first line of defense and, when successful, preserve the invader-directed spacer for rapid elimination of infection in future generations. The Cmr system may be activated, and the cell may be eliminated, when infection progresses to the point where invader sequence transcription is rampant.
Materials and methods
Strains and growth conditions
The strains and plasmids used in this study are listed in Supplemental Table S1. P. furiosus strains were grown under strict anaerobic conditions at 90°C in defined medium or complex medium (cell extract preparation only). Cultures and media were prepared as described previously (Lipscomb et al. 2011), with medium pH adjusted to ∼6.5. Cultures were inoculated with 1%–2% inoculum or a single colony and grown anaerobically. Medium was supplemented with 20 µM uracil and/or 2.75 mM 5-FOA as needed for selection.
Escherichia coli strains TOP10 and BL21-CodonPlus(DE3)-RIPL were used for plasmid DNA manipulation and protein expression, respectively. Cultures were grown at 37°C or 25°C in Luria broth (Millers) or Terrific broth supplemented with 50 µg/mL apramycin sulfate, 50 µg/mL kanamycin sulfate, or 100 µg/mL ampicillin.
Northern analysis
Northern analysis was carried out as previously described (Hale et al. 2008). Following initial probing, membranes were reprobed for the 5S rRNA loading control. The probe sequences used are listed in Supplemental Table S2.
General DNA manipulation and target plasmid construction
Plasmids were isolated from Top10 with QIAprep Spin Miniprep (Qiagen) for routine analysis and Zyppy Plasmid Maxiprep kit (Zymo Research) for plasmid construction and assays. Plasmids were isopropanol-precipitated following initial isolation. Phusion polymerase (New England Biolabs) was used for all PCR cloning. P. furiosus gDNA was isolated from 1-mL overnight cultures with Zymo Quick gDNA kit (Zymo Research).
The expression cassette in pJE47 was constructed by amplifying the Pcsg promoter and ChiA terminator of pLC64-ChiA. Products were spliced by SOE-PCR and cloned into NotI/EcoRV sites of pJFW18 (Farkas et al. 2011).
Plasmids pJE65–85, pJE275–276, pJE294, and pJE299–306 were generated by ligation of annealed 5′ phosphorylated oligos with NdeI/BamHI-linearized pJE47. Plasmids pJE186–249 were constructed by a combination of two methods. The majority of the plasmid inserts was constructed by extension of 701_NNN_F, containing degenerate PAM nucleotides, with primer 701_NNN_R. The remainder was generated with annealed oligos. Products were cloned into NdeI/BamHI sites of pJE47. Plasmids pJE271/272 were constructed by NotI/NdeI digestion of pJE65/66 (respectively) to remove the Pcsg promoter and treated with Quick Blunting kit (New England Biolabs). Oligo sequences are listed in Supplemental Table S2, with “+” oligos annealed with cognate “−” oligos to generate inserts.
P. furiosus strain construction
P. furiosus strains were constructed using a variant of the previously described pop-in/pop-out marker replacement technique (Supplemental Fig. 7; Lipscomb et al. 2011; Farkas et al. 2012).
Plasmid transformation assay in P. furiosus
Incubations were performed anaerobically at 90°C with defined P. furiosus medium. Liquid cultures were grown to mid to late log phase, and 33.3 μL of culture was mixed with 66.7 ng of plasmid DNA (in 1.66 μL) and incubated briefly (5–60 min) at room temperature during plating. The mixtures were spread on solid defined medium and incubated for ∼64 h. Following incubation, colonies per plate were enumerated. All assays were carried out with a minimum of three replicates.
P. furiosus cell extract preparation and coimmunoprecipitation (co-IP) reactions
Cultures were grown to late log phase, harvested by centrifugation, and weighed. Lysis buffer (50 mM Tris-Cl at pH 8, 30 U/mL SUPERase• In [Life Technologies], 1× Complete-mini EDTA-free protease inhibitor [Roche]) was added (3 mL per 1 g of cells). Cells were lysed by sonication and centrifuged at 20,000g for 30 min at 4°C. The soluble fraction was collected as S20 cell extract, with protein quantified by Qubit assay (Life Technologies).
IgY antibodies previously raised against recombinant Cmr2 (Hale et al. 2012) were used for co-IPs. Co-IPs were carried out as previously described (Hale et al. 2012) with the following modifications: Each immunoprecipitation (30 µL of resin) used S20 cell extract containing 2 mg of protein. UltraLink hydrazide (Pierce) resin was used for silver stain analysis of immunoprecipitations, and anti-IgY agarose resin (Gallus Immunotech) was used for RNA cleavage assays.
Co-IP RNA cleavage activity assays
Co-IP RNA cleavage assays were carried out as described (Hale et al. 2012).
Co-IP silver staining
Co-IP samples (30 µL resin) were resuspended in 60 µL of nonreducing Laemmli buffer and heated for 5 min at 60°C to elute samples from UltraLink beads. An equal fraction of each elution (one-half of a co-IP) was separated on 11% SDS–polyacrylamide gels, and subjected to silver staining.
Protein expression and purification
Cmr2 mutants HDm (H13A, D14A) and Palmm (D673A, D674A) were generated by PCR mutagenesis (primers in Supplemental Table S2). Expression and purification of both recombinant wild type and mutants, including Cmr4-D26N (Ramia et al. 2014a), was performed as described (Hale et al. 2009, 2014) with the following modifications: Cells were lysed, and protein was purified in buffer containing 40 mM Tris-Cl (pH 7.5), 500 mM NaCl, and 0.2 mM PMSF. Purified proteins were dialyzed into 40 mM Tris-Cl (pH 7.5) and 500 mM NaCl and quantified by Qubit assay (Life Technologies).
Preparation of DNA and RNA substrates
DNA oligos were purchased from IDT (DNA 1) (Supplemental Fig. S6) or Operon (all others). RNAs for in vitro Cmr crRNP formation (45-mer 7.01 crRNA) or cleavage assay substrate (37-mer 7.01 target RNA) were purchased from IDT. DNA and RNA substrates used in cleavage assays were 5′ end labeled with 32P using T4 polynucleotide kinase (New England Biolabs). Annealing 5′ radiolabeled DNA oligos with 2× molar excess of gel-purified, unlabeled oligos generated radiolabeled dsDNA substrates. To form a bubble substrate, oligos 2397 and 3124 (Supplemental Table S2) were annealed for Cmr2 assays (Fig. 3). Annealing oligos 2397 and 2398 (7.01 target) or 2765 and 2766 (mutant nontarget) generated full-length dsDNA substrates. All substrates were gel-purified prior to use on nondenaturing (dsDNA) or denaturing (ssDNA/RNA) PAGE.
Cmr complex assembly and in vitro Cmr2/Cmr complex assays
Cmr complex (Cmr1–6 + crRNA) assembly was performed as described (Hale et al. 2009, 2012, 2014) with modifications. The reconstitution buffer used was 20 mM Tris-Cl (pH 7.5 at 25°C), 250 mM NaCl, 10% glycerol, 2.5 mM MgCl2, and 200 µM NiCl2; the protein concentrations were 500 nM with the exception of 50 nM Cmr2, and the crRNA concentration was 50 nM.
Assays were performed with either Cmr2 or assembled Cmr complex. All samples were preincubated in reconstitution buffer (-crRNA for Cmr2 alone) prior to the addition of substrates. Cleavage reactions were initiated by addition of either 5000 cpm (∼0.05 pmol) 5′ radiolabeled DNA/RNA or 0.21 pmol of M13 circular ssDNA (New England Biolabs). Unless otherwise indicated, substrates were incubated with 500 nM (Fig. 3) or 50 nM (Fig. 4) Cmr2 or ∼50 nM Cmr complex (Figs. 4, 6). Unless indicated, assays were incubated for 1 h at 70°C. Following incubation (cleavage assays), reactions were treated with proteinase K (New England Biolabs) for 15 min at 37°C, denatured in gel loading buffer II (ThermoFisher) at 95°C, and separated on either 15% TBE-UREA PAGE or 0.7% TAE-agarose. For target binding, following 70°C incubation, half of the reaction was separated on 6% TBE-PAGE with 4% glycerol. Agarose gels were visualized by SYBR Gold (Thermofisher) stain. PAGE gels were dried and imaged by phosphorimaging.
The substrates (asterisks indicate labeled oligos) used for Figures 3, 4, 6 were as follows: oligo *2397 in Figure 3, A, E, and F; annealed *2397/2398 in Figure 3B; M13 in Figure 3C; annealed *2397/3124 in Figure 3D; *2397 in Figure 4A; *2397/2398 in Figure 4, B and F; 2397/*2398 in Figure 4, C and G; *2765/2766 in Figure 4D; RNA oligo *2 in Figure 6, B and C; *2397/2398 in Figure 6, D, E, and G; and *2765/2766 in Figure 6F.
Assay-specific details are as follows: Figure 3F assays used 50, 100, or 200 nM Cmr2 or assembled Cmr2 complex as indicated. For RNA target-dependent DNA cleavage assays (Figs. 4, 6), following preincubation, 22.5 nM of target RNA was added immediately before DNA substrates.
In vitro transcription (IVT) of target RNAs
Target RNAs were generated using MEGAshortscript T7 kit (ThermoFisher) with PCR templates. Products were gel-purified and quantified by Qubit analysis (ThermoFisher). IVT templates for Figures 4 and 6C target RNAs were made by PCR of the annealed oligos 2397/2398 with primers 3110/3112 (t) or 3115/3114 (rc). For Figure 6E–G IVT templates, target plasmids were amplified with primers 2798/2801. Plasmids pJE65 (GGG), pJE66 (rc-GGG), pJE67 (AAA), pJE69 (CCC), pJE71 (UUU), pJE294 (mut-GGG), pJE201 (UGG), pJE189 (UUG), pJE198 (UGU), pJE202 (CUU), pJE190 (UCU), and pJE187 (UUC) were used as PCR templates.
Supplementary Material
Acknowledgments
We thank Claiborne V.C. Glover III for critical review of the manuscript and helpful discussions throughout the course of this work. We also thank members of the Terns laboratory—Caryn Hale, Erica Figueroa, and Janet Figueroa—for help with strain construction. We thank University of Georgia colleagues Barney Whitman and Felipe Sarmiento for technical advice with anaerobic culturing, Farris Poole for assistance with anaerobic culturing equipment, and Joel Farkas and Jan Westpheling for helpful advice regarding working with P. furiosus. This work was funded by the National Institutes of Health (R01 GM54682 to M.P.T. and R.M.T.).
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.272153.115.
References
- Almendros C, Guzman NM, Diez-Villasenor C, Garcia-Martinez J, Mojica FJ. 2012. Target motifs affecting natural immunity by a constitutive CRISPR–Cas system in Escherichia coli. PLoS One 7: e50797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrangou R, Marraffini LA. 2014. CRISPR–Cas systems: prokaryotes upgrade to adaptive immunity. Mol Cell 54: 234–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benda C, Ebert J, Scheltema RA, Schiller HB, Baumgartner M, Bonneau F, Mann M, Conti E. 2014. Structural model of a CRISPR RNA-silencing complex reveals the RNA-target cleavage activity in Cmr4. Mol Cell 56: 43–54. [DOI] [PubMed] [Google Scholar]
- Bolotin A, Quinquis B, Sorokin A, Ehrlich SD. 2005. Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology 151: 2551–2561. [DOI] [PubMed] [Google Scholar]
- Cocozaki AI, Ramia NF, Shao Y, Hale CR, Terns RM, Terns MP, Li H. 2012. Structure of the Cmr2 subunit of the CRISPR–Cas RNA silencing complex. Structure 20: 545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L, Garrett RA, Shah SA, Peng X, She Q. 2013. A novel interference mechanism by a Type IIIB CRISPR–Cmr module in Sulfolobus. Mol Microbiol 87: 1088–1099. [DOI] [PubMed] [Google Scholar]
- Elmore J, Deighan T, Westpheling J, Terns RM, Terns MP. 2015. DNA targeting by the Type I-G and Type I-A CRISPR–Cas systems of Pyrococcus furiosus. Nucleic Acids Res 43: 10353–10363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farkas J, Chung D, DeBarry M, Adams MW, Westpheling J. 2011. Defining components of the chromosomal origin of replication of the hyperthermophilic archaeon Pyrococcus furiosus needed for construction of a stable replicating shuttle vector. Appl Environ Microbiol 77: 6343–6349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farkas J, Stirrett K, Lipscomb GL, Nixon W, Scott RA, Adams MW, Westpheling J. 2012. Recombinogenic properties of Pyrococcus furiosus strain COM1 enable rapid selection of targeted mutants. Appl Environ Microbiol 78: 4669–4676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer S, Maier LK, Stoll B, Brendel J, Fischer E, Pfeiffer F, Dyall-Smith M, Marchfelder A. 2012. An archaeal immune system can detect multiple protospacer adjacent motifs (PAMs) to target invader DNA. J Biol Chem 287: 33351–33363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett RA, Vestergaard G, Shah SA. 2011. Archaeal CRISPR-based immune systems: exchangeable functional modules. Trends Microbiol 19: 549–556. [DOI] [PubMed] [Google Scholar]
- Goldberg GW, Jiang W, Bikard D, Marraffini LA. 2014. Conditional tolerance of temperate phages via transcription-dependent CRISPR–Cas targeting. Nature 514: 633–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haft DH, Selengut J, Mongodin EF, Nelson KE. 2005. A guild of 45 CRISPR-associated (Cas) protein families and multiple CRISPR/Cas subtypes exist in prokaryotic genomes. PLoS Comput Biol 1: e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale C, Kleppe K, Terns RM, Terns MP. 2008. Prokaryotic silencing (psi)RNAs in Pyrococcus furiosus. RNA 14: 2572–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale CR, Zhao P, Olson S, Duff MO, Graveley BR, Wells L, Terns RM, Terns MP. 2009. RNA-guided RNA cleavage by a CRISPR RNA–Cas protein complex. Cell 139: 945–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale CR, Majumdar S, Elmore J, Pfister N, Compton M, Olson S, Resch AM, Glover CV III, Graveley BR, Terns RM, et al. 2012. Essential features and rational design of CRISPR RNAs that function with the Cas RAMP module complex to cleave RNAs. Mol Cell 45: 292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale CR, Cocozaki A, Li H, Terns RM, Terns MP. 2014. Target RNA capture and cleavage by the Cmr Type III-B CRISPR–Cas effector complex. Genes Dev 28: 2432–2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatoum-Aslan A, Maniv I, Samai P, Marraffini LA. 2014. Genetic characterization of antiplasmid immunity through a Type III-A CRISPR–Cas system. J Bacteriol 196: 310–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochstrasser ML, Taylor DW, Bhat P, Guegler CK, Sternberg SH, Nogales E, Doudna JA. 2014. CasA mediates Cas3-catalyzed target degradation during CRISPR RNA-guided interference. Proc Natl Acad Sci 111: 6618–6623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson RN, Wiedenheft B. 2015. A conserved structural chassis for mounting versatile CRISPR RNA-guided immune responses. Mol Cell 58: 722–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. 2012. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337: 816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung TY, An Y, Park KH, Lee MH, Oh BH, Woo E. 2015. Crystal structure of the Csm1 subunit of the Csm complex and its single-stranded DNA-specific nuclease activity. Structure 23: 782–790. [DOI] [PubMed] [Google Scholar]
- Kunin V, Sorek R, Hugenholtz P. 2007. Evolutionary conservation of sequence and secondary structures in CRISPR repeats. Genome Biol 8: R61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrie SJ, Samson JE, Moineau S. 2010. Bacteriophage resistance mechanisms. Nat Rev Microbiol 8: 317–327. [DOI] [PubMed] [Google Scholar]
- Lipscomb GL, Stirrett K, Schut GJ, Yang F, Jenney FE Jr, Scott RA, Adams MW, Westpheling J. 2011. Natural competence in the hyperthermophilic archaeon Pyrococcus furiosus facilitates genetic manipulation: construction of markerless deletions of genes encoding the two cytoplasmic hydrogenases. Appl Environ Microbiol 77: 2232–2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar S, Zhao P, Pfister NT, Compton M, Olson S, Glover CV III, Wells L, Graveley BR, Terns RM, Terns MP. 2015. Three CRISPR–Cas immune effector complexes coexist in Pyrococcus furiosus. RNA 21: 1147–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Koonin EV. 2013. Evolution and classification of CRISPR–Cas systems and Cas protein families. In CRISPR–Cas systems (ed. Barrangou R, van der Oost J), pp. 61–91. Springer, Berlin. [Google Scholar]
- Makarova KS, Grishin NV, Shabalina SA, Wolf YI, Koonin EV. 2006. A putative RNA-interference-based immune system in prokaryotes: computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol Direct 1: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Haft DH, Barrangou R, Brouns SJ, Charpentier E, Horvath P, Moineau S, Mojica FJ, Wolf YI, Yakunin AF, et al. 2011. Evolution and classification of the CRISPR–Cas systems. Nat Rev Microbiol 9: 467–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Anantharaman V, Grishin NV, Koonin EV, Aravind L. 2014. CARF and WYL domains: ligand-binding regulators of prokaryotic defense systems. Front Genet 5: 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marraffini LA, Sontheimer EJ. 2010. Self versus non-self discrimination during CRISPR RNA-directed immunity. Nature 463: 568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mojica FJ, Diez-Villasenor C, Garcia-Martinez J, Soria E. 2005. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J Mol Evol 60: 174–182. [DOI] [PubMed] [Google Scholar]
- Mojica FJ, Diez-Villasenor C, Garcia-Martinez J, Almendros C. 2009. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology 155: 733–740. [DOI] [PubMed] [Google Scholar]
- Osawa T, Inanaga H, Sato C, Numata T. 2015. Crystal structure of the CRISPR–Cas RNA silencing Cmr complex bound to a target analog. Mol Cell 58: 418–430. [DOI] [PubMed] [Google Scholar]
- Plagens A, Tripp V, Daume M, Sharma K, Klingl A, Hrle A, Conti E, Urlaub H, Randau L. 2014. In vitro assembly and activity of an archaeal CRISPR–Cas Type I-A cascade interference complex. Nucleic Acids Res 42: 5125–5138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pourcel C, Salvignol G, Vergnaud G. 2005. CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology 151: 653–663. [DOI] [PubMed] [Google Scholar]
- Ramia NF, Spilman M, Tang L, Shao Y, Elmore J, Hale C, Cocozaki A, Bhattacharya N, Terns RM, Terns MP, et al. 2014a. Essential structural and functional roles of the Cmr4 subunit in RNA cleavage by the Cmr CRISPR–Cas complex. Cell Rep 9: 1610–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramia NF, Tang L, Cocozaki AI, Li H. 2014b. Staphylococcus epidermidis Csm1 is a 3′–5′ exonuclease. Nucleic Acids Res 42: 1129–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollins MF, Schuman JT, Paulus K, Bukhari HS, Wiedenheft B. 2015. Mechanism of foreign DNA recognition by a CRISPR RNA-guided surveillance complex from Pseudomonas aeruginosa. Nucleic Acids Res 43: 2216–2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samai P, Pyenson N, Jiang W, Goldberg GW, Hatoum-Aslan A, Marraffini LA. 2015. Co-transcriptional DNA and RNA cleavage during Type III CRISPR–Cas immunity. Cell 161: 1164–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samson JE, Magadan AH, Sabri M, Moineau S. 2013. Revenge of the phages: defeating bacterial defences. Nat Rev Microbiol 11: 675–687. [DOI] [PubMed] [Google Scholar]
- Shah SA, Erdmann S, Mojica FJ, Garrett RA. 2013. Protospacer recognition motifs: mixed identities and functional diversity. RNA Biol 10: 891–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinkunas T, Gasiunas G, Waghmare SP, Dickman MJ, Barrangou R, Horvath P, Siksnys V. 2013. In vitro reconstitution of cascade-mediated CRISPR immunity in Streptococcus thermophilus. EMBO J 32: 385–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spilman M, Cocozaki A, Hale C, Shao Y, Ramia N, Terns R, Terns M, Li H, Stagg S. 2013. Structure of an RNA silencing complex of the CRISPR–Cas immune system. Mol Cell 52: 146–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spoel SH, Dong X. 2012. How do plants achieve immunity? Defence without specialized immune cells. Nat Rev Immunol 12: 89–100. [DOI] [PubMed] [Google Scholar]
- Staals RH, Agari Y, Maki-Yonekura S, Zhu Y, Taylor DW, van Duijn E, Barendregt A, Vlot M, Koehorst JJ, Sakamoto K, et al. 2013. Structure and activity of the RNA-targeting Type III-B CRISPR–Cas complex of Thermus thermophilus. Mol Cell 52: 135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staals RH, Zhu Y, Taylor DW, Kornfeld JE, Sharma K, Barendregt A, Koehorst JJ, Vlot M, Neupane N, Varossieau K, et al. 2014. RNA targeting by the Type III-A CRISPR–Cas Csm complex of Thermus thermophilus. Mol Cell 56: 518–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamulaitis G, Kazlauskiene M, Manakova E, Venclovas C, Nwokeoji AO, Dickman MJ, Horvath P, Siksnys V. 2014. Programmable RNA shredding by the Type III-A CRISPR–Cas system of Streptococcus thermophilus. Mol Cell 56: 506–517. [DOI] [PubMed] [Google Scholar]
- Taylor DW, Zhu Y, Staals RH, Kornfeld JE, Shinkai A, van der Oost J, Nogales E, Doudna JA. 2015. Structures of the CRISPR–Cmr complex reveal mode of RNA target positioning. Science 348: 581–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terns RM, Terns MP. 2013. The RNA- and DNA-targeting CRISPR–Cas immune systems of Pyrococcus furiosus. Biochem Soc Trans 41: 1416–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Oost J, Westra ER, Jackson RN, Wiedenheft B. 2014. Unravelling the structural and mechanistic basis of CRISPR–Cas systems. Nat Rev Microbiol 12: 479–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vestergaard G, Garrett RA, Shah SA. 2014. CRISPR adaptive immune systems of archaea. RNA Biol 11: 156–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westra ER, van Erp PB, Kunne T, Wong SP, Staals RH, Seegers CL, Bollen S, Jore MM, Semenova E, Severinov K, et al. 2012. CRISPR immunity relies on the consecutive binding and degradation of negatively supercoiled invader DNA by cascade and Cas3. Mol Cell 46: 595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westra ER, Semenova E, Datsenko KA, Jackson RN, Wiedenheft B, Severinov K, Brouns SJ. 2013. Type I-E CRISPR–Cas systems discriminate target from non-target DNA through base pairing-independent PAM recognition. PLoS Genet 9: e1003742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedenheft B, Lander GC, Zhou K, Jore MM, Brouns SJ, van der Oost J, Doudna JA, Nogales E. 2011. Structures of the RNA-guided surveillance complex from a bacterial immune system. Nature 477: 486–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Rouillon C, Kerou M, Reeks J, Brugger K, Graham S, Reimann J, Cannone G, Liu H, Albers SV, et al. 2012. Structure and mechanism of the CMR complex for CRISPR-mediated antiviral immunity. Mol Cell 45: 303–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.