

Abstract
Rickettsia conorii is the etiologic agent of Mediterranean spotted fever, a re-emerging infectious disease with significant mortality. This Gram-negative, obligately intracellular pathogen is transmitted via tick bites, resulting in disseminated vascular endothelial cell infection with vascular leakage. In the infected human, Rickettsia conorii infects endothelial cells, stimulating expression of cytokines and pro-coagulant factors. However, the integrated proteomic response of human endothelial cells to R. conorii infection is not known. In this study, we performed quantitative proteomic profiling of primary human umbilical vein endothelial cells (HUVECs) with established R conorii infection versus those stimulated with endotoxin (LPS) alone. We observed differential expression of 55 proteins in HUVEC whole cell lysates. Of these, we observed induction of signal transducer and activator of transcription (STAT)1, MX dynamin-like GTPase (MX1), and ISG15 ubiquitin-like modifier, indicating activation of the JAK-STAT signaling pathway occurs in R. conorii-infected HUVECs. The down-regulated proteins included those involved in the pyrimidine and arginine biosynthetic pathways. A highly specific biotinylated cross-linking enrichment protocol was performed to identify dysregulation of 11 integral plasma membrane proteins that included up-regulated expression of a sodium/potassium transporter and down-regulation of α-actin 1. Analysis of Golgi and soluble Golgi fractions identified up-regulated proteins involved in platelet-endothelial adhesion, phospholipase activity, and IFN activity. Thirty four rickettsial proteins were identified with high confidence in the Golgi, plasma membrane, or secreted protein fractions. The host proteins associated with rickettsial infections indicate activation of interferon-STAT signaling pathways; the disruption of cellular adhesion and alteration of antigen presentation pathways in response to rickettsial infections are distinct from those produced by nonspecific LPS stimulation. These patterns of differentially expressed proteins suggest mechanisms of pathogenesis as well as methods for diagnosis and monitoring Rickettsia infections.
The genus Rickettsia contains non-motile, Gram-negative, obligately intracellular alphaproteobacteria that are of global medical and veterinary health importance due to their endemicity and re-emergence. From the clinical and antigenic perspectives, rickettsial diseases are classified into two groups, spotted fever and typhus. Nearly all of the numerous spotted fever group rickettsiae are transmitted by ticks. The most virulent ones are Rickettsia rickettsii, the agent of Rocky Mountain spotted fever, and Rickettsia conorii, the agent of Mediterranean spotted fever (boutonneuse fever), a disease prevalent throughout the Mediterranean, Africa, the Middle East, and India. In humans, the spotted fevers present as acute fever, headache, maculopapular rash, and vascular leakage that can lead to significant morbidity and mortality due to pulmonary and cerebral edema, particularly if there are delays in diagnosis and treatment (1).
The characteristic leakage of intravascular fluid is a consequence of the specific tropism of rickettsiae for endothelial cells (1). Rickettsial organisms enter endothelial cells through a calcium-dependent zipper-like entry mechanism involving the actin cytoskeleton (2–4). Viable organisms subsequently exit the phagosomes via phospholipase D and hemolysin activities (5, 6), replicate in the cytoplasm, and exhibit early intercellular spread as a consequence of directional actin polymerization without detectable cellular injury (7–9).
Understanding the host response to Rickettsia infection has been advanced by the development of a standardized model of endothelial cell infection using primary human umbilical vein cells (HUVECs)1 (10). In this model, infected endothelial cells have been shown to express cytokines, interferons, cell surface adhesion molecules such as E-selectin, VCAM-1, ICAM-1 (11–13), and αVβ3 integrin (14), and pro-coagulants (tissue factor and von Willebrand factor) (15–17). These endothelial cellular responses explain aspects of the pathobiology of natural infections, including microvascular hemorrhage, endothelial leakage, and multiorgan failure (1).
An integrated understanding of the endothelial cellular response is not yet available. To address this question, we have undertaken a study of the global endothelial cell proteomic response to infection with R. conorii using quantitative proteomic profiling of R. conorii-infected HUVECs, including the analysis of plasma membrane and secreted proteins within the Golgi apparatus. Our experimental design was to use trypsin-mediated exchange of stable isotopes of H2O to quantify differences in protein expression of primary HUVECs infected with R. conorii versus those stimulated with LPS alone to control for nonspecific inflammatory effects. In whole cellular lysates, we observed that R. conorii-infected endothelial cells significantly up-regulated the JAK-STAT signaling pathway. By contrast, analysis of PM and Golgi fractions revealed up-regulation of platelet adhesion proteins and down-regulation of integrin/cadherin components. To identify proteins secreted by R. conorii-infected HUVECs, soluble Golgi fractions were analyzed. Here, we observed significant induction of HLA and β2-microglobulin, providing insights into major histocompatibility complex (MHC)-I-mediated antigen processing, important in the host cytotoxic T cell response. Finally, 34 rickettsial proteins were identified with high confidence in the Golgi apparatus, PM, or secreted protein fractions. These studies advance the understanding of the endothelial response to R. conorii infection through up-regulation of IFN- and MHC class I antigen presentation pathways and implications of the secretome for the host response and diagnostics.
EXPERIMENTAL PROCEDURES
Materials
Link sulfosuccinimidyl-2-(biotinamido) ethyl-1,3-dithiopropionate (Sulfo-NHS-SS-Biotin) was from Pierce (Thermo Scientific, San Jose, CA). NanoLinkTM streptavidin magnetic beads (0.8 μm) were from Solulink, San Diego, CA (catalog no. M-1002); protease inhibitor mixture was from Sigma, St. Louis, MO (catalog no. P8340). LPS was from Escherichia coli 0111:B4 (Sigma).
Cell Cultures
Pools of HUVECs were established from individual human umbilical cords grown in supplemented EGM-2 medium (Lonza). The cells were subcultured when the monolayer became confluent two or three times per week. In this study, the cells were used between passages 3 and 4. For infection, 15 × 106 primary HUVECs in T175 flasks were infected in BSL-3 containment, and subsequently lysates were prepared 10 days later and were inactivated in accordance with University of Texas Medical Branch IBC-approved protocols. Cellular infection was verified by immunofluorescent microscopy using a rabbit polyclonal serum against R. conorii and anti-rabbit IgG conjugated to Alexa 594 (Life Technologies, inc.). HUVECs were stimulated with LPS (50 ng/ml) overnight as controls.
Rickettsia
R. conorii (Malish 7 strain) was obtained from the American Type Culture Collection (ATCC; Manassas, Va.; catalog no. VR-613). The stock, aliquots of a 10% suspension of infected yolk sac containing 4 × 106 pfu per ml, was stored at −70 °C.
Experimental Design and Statistical Rationale
HUVECs were infected with R. conorii and subjected to whole cell lysate, plasma membrane, and Golgi fractionation. Experiments were replicated twice. For each quantitative LC-MS/MS analysis, samples were subjected to label swapping as described below.
Plasma Membrane (PM) Preparation
LPS-stimulated or R. conorii-infected HUVECs were washed three times with PBS (37 °C) containing calcium and magnesium. PM proteins were cross-linked 15 min in 10 ml of PBS with 10 μl of EZ-Link Sulfo-NHS-SS-Biotin stock solution (100 mg/ml, freshly prepared in DMSO) as described previously (18). Afterward, cross-linker was quenched by addition of 5 ml of lysine solution (1 mg/ml), washed in ice-cold wash buffer (250 mm sucrose, 10 mm Tris, pH 7.4), and resuspended in ice-cold homogenization buffer (250 mm sucrose, 10 mm Tris, pH 7.4, 1:100 dilution of protease inhibitor (Sigma P8340), 1 mm NaF, and 1 mm Na3VO4). The cells were concentrated by centrifugation (10 min at 800 × g at 4 °C), resuspended in homogenization buffer, and Dounce homogenized using 15 strokes of pestle A and 10 strokes of pestle B. The homogenate was then centrifuged (10 min at 1,000 × g, 4 °C), and membranes were captured by addition of suspended streptavidin magnetic beads (15 ml/T165 flask) followed by gentle mixing at 4 °C for 1 h and magnetic capture. The membrane-bound streptavidin beads were then washed with 1 m KCl (high salt wash) three times, followed by washing in 0.1 m Na2CO3, pH 11.5 (high-pH wash), and then ice-cold hypotonic buffer (10 mm HEPES, pH 7.5, 1.5 mm MgCl2, 10 mm KCl, 1:100 dilution of protease inhibitor mixture, 1 mm NaF, and 1 mm Na3VO4). The streptavidin beads were resuspended in twice their volume of 2× SDS sample buffer containing 100 mm dithiothreitol (DTT). After vortexing, beads were removed with a strong magnet, and the supernatant was saved. The proteins in the supernatant were separated by SDS-PAGE and visualized with Colloidal Blue (Life Technologies, Inc.). The gel in each lane was cut into small slices. The proteins were digested with trypsin in-gel as described previously. Briefly, the gel particles were destained in 1 ml of water/methanol solution (50:50, v/v) containing 25 mm NH4HCO3, pH 8.0, three times, changing the solution every 10 min. The destained gel was then washed in 1 ml of an acetic/methanol solution (acetic acid/methanol/water, 10:40:50, v/v/v) for 3 h, with the solution changed every 1 h. The resulting gel was soaked in 1 ml of water for 40 min, changing the solvent twice every 20 min. The gel was then transferred into a 0.5-ml microcentrifuge tube and dehydrated by soaking the gel in 100% acetonitrile (ACN) until it became opaque white. The solution was removed, and the gel was dried in a SpeedVac for 20–30 min. The dried gel was rehydrated with an adequate amount of trypsin digestion solution (10 ng of trypsin/μl in 50 mm NH4HCO3, pH 8.0). The digestion was carried out at 37 °C overnight. To extract tryptic digest, the gel was soaked in 40 μl of extraction solution (ACN/trifluoroacetic acid/water, 50:5:45, v/v/v) for 60 min with vortexing. The extraction solution was then carefully removed with a gel-loading pipette tip, and the extraction was repeated once. The extracts were pooled and dried with a SpeedVac. The tryptic peptides were used for trypsin-catalyzed 18O labeling.
Golgi Preparation
Golgi preparations were performed as described (19). In brief, the flow-through of the magnetic bead separation from above was adjusted to a final concentration of 1.4 m sucrose by addition of 2.0 m sucrose and transferred to SW41 centrifuge tubes. Samples were overlaid with 4 ml of 1.2 m sucrose solution, topped off with 0.8 m sucrose solution, and spun at 38,000 rpm in an SW41Ti rotor (246,000 × g) for 90 min at 4 °C. Crude Golgi preparations were harvested from the 0.8/1.2 m sucrose interface. An aliquot was assayed for total protein concentration by the bicinchoninic acid (BCA) assay (Thermo Scientific).
Golgi Extracts
An equal volume of 1 m KCl was added to the crude Golgi preparation, incubated for 25 min with rotation at 4 °C, and an equal amount of ice-cold Dulbecco's modified PBS (D-PBS) added to each tube. The Golgi preparations were spun at 40,000 rpm (271,000 × g) at 4 °C. The membranes were then resuspended in 0.5 μl/μg of protein of crude extract using a 100 mm ammonium carbonate solution, pH 11.0. Membranes were removed by centrifugation at 40,000 rpm (271,000 × g) for 60 min at 4 °C. The soluble supernatants, representing the secreted proteins, were denatured by addition of an equal volume of 8 m guanidine HCl. Insoluble membranes were subjected to SDS-PAGE, and digested with trypsin in-gel as described above.
Trypsin Digestion of Whole Cell Lysate and Secreted Proteins
50 μg of protein were reduced with 10 mm DTT for 30 min at room temperature. Protein cysteinyl residues were alkylated with 30 mm iodoacetamide for 2 h at 37 °C. Each sample was diluted 1:10 with 100 mm ammonium bicarbonate and digested with 40 μg of trypsin overnight at 37 °C, and each tryptic peptide mixture was desalted with a Sep-Pak® C18 cartridge (Waters, Milford, MA) following the manufacturer's instructions. Peptides were eluted from the cartridge with 80% acetonitrile and completely dried using a Speedvac.
Trypsin-catalyzed 16/18O Labeling
The tryptic digest of each sample was divided into two parts of equal volume. One part was labeled with [18O]H2O, the other part remained unlabeled (16O-H2O). Peptide C-terminal 16O/18O labeling was performed as described previously (20, 21). The dried peptide samples were redissolved with 3 μl of anhydrous acetonitrile, 10 mg of immobilized trypsin (Applied Biosystems, CA), and 200 μl of normal water (H216O) or heavy water H218O containing 50 mm ammonium bicarbonate was added to the Rickettsia-infected and LPS control peptides, respectively, and both samples were incubated for 48 h at 37 °C. Supernatants were collected using a spin column and mixed as follows: 18O-labeled peptides from Rickettsia-treated sample mixed with 16O-labeled peptides from LPS particle-treated sample (forward labeling); 16O-labeled peptides from Rickettsia-treated sample mixed with 18O-labeled peptides from LPS particle-treated sample (reverse labeling). After mixing, the samples were desalted with a SepPak C18 cartridge (Waters). The desalted peptides were stored at −80 °C for LC-MS/MS analysis.
LC-MS/MS Analysis
Dried peptide samples were redissolved in 2 μl of acetonitrile and diluted with 40 μl of 0.1% formic acid. LC-MS/MS analysis was performed with a Q Exactive Orbitrap mass spectrometer (Thermo Scientific, San Jose, CA) equipped with a nanospray source with an on-line Easy-nLC 1000 nano-HPLC system (Thermo Scientific, San Jose, CA). Ten microliters of each peptide solution were injected and separated on a reversed phase nano-HPLC C18 column (75 μm × 150 cm) with a linear gradient of 0–35% mobile phase B (0.1% formic acid, 90% acetonitrile) in mobile phase A (0.1% formic acid) over 120 min at 300 nl/min. The mass spectrometer was operated in the data-dependent acquisition mode with a resolution of 70,000 at full scan mode and 17,500 at MS/MS mode. The 10 most intense ions in each MS survey scan were automatically selected for MS/MS. The acquired MS/MS spectra were analyzed by 1.4 (22) using default parameters (supplemental File 1) in the Swiss-Prot human protein databases (downloaded on February 2013, 20,247 protein entries) using a mass tolerance of ±20 ppm for precursor and product ions and a static mass modification on cysteinyl residues that corresponded to alkylation with iodoacetamide. Differential modifications were defined to be 18O-labeled C-terminal and oxidized methionine with a maximum of two missed cleavages. Protein identification data (accession numbers, peptides observed, sequence coverage) are in supplemental Tables 1–3. Annotated spectra of host proteins identified with single peptides are in supplemental file 2. Annotated spectra of rickettsial proteins identified with single peptides are in supplemental file 3. The FDR cutoff for peptide and protein identification is 0.01.
Data Processing
For each subcellular fraction, the experiments were repeated after swapping the 18O-labeling between Rickettsia-infected cells and LPS-treated cells. This label swapping strategy allows for detection of irreproducible ratios that might arise due to interference in precursor quantification provided there is a minimum of two biological replicates. Each dataset was first centered by subtracting the most frequent value in that dataset and then subjected to MaxQuant Significant A analysis (22). Next, the forward and reverse datasets were plotted with forward log2 heavy/light (H/L) ratio (x axis) against reverse log2 H/L ratio (y axis). Only the proteins that have a Significant A p value below 0.05 and also located in either upper-left or lower-right quadrant are considered to be the significantly expressed proteins.
The normalized spectral abundance factor (NSAF) value for each protein was calculated as described (23) in Equation 1,
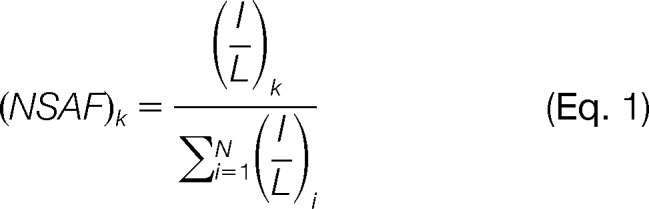 |
where the total MS intensity (I) of the matching peptides from protein k was divided by the protein length (L) and then divided by the sum of I/L for all uniquely identified proteins in the dataset.
Stable Isotope Dilution (SID)-selected Reaction Monitoring (SRM)-MS
The SID-SRM-MS assays were developed as described previously (24, 25). For each targeted protein, two or three peptides were initially selected and then the sensitivity and selectivity of these were experimentally evaluated as described previously (24, 25). The peptide with the best sensitivity and selectivity was selected as the surrogate for that protein. For each peptide, 3–5 SRM transitions were monitored. The signature peptides and SRM parameters are listed in Table VI. The peptides were chemically synthesized incorporating isotopically labeled [13C615N4]arginine or [13C615N2]lysine to a 99% isotopic enrichment (Thermo Scientific). The amount of stable isotope standard (SIS) peptides was determined by amino acid analysis. The tryptic digests were then reconstituted in 30 μl of 5% formic acid, 0.01% TFA. An aliquot of 10 μl of 50 fmol/μl diluted SIS peptides was added to each tryptic digest. These samples were desalted with a ZipTip C18 cartridge. The peptides were eluted with 80% ACN and dried. The peptides were reconstituted in 30 μl of 5% formic acid, 0.01% TFA and were directly analyzed by LC-SRM-MS. LC-SRM-MS analysis was performed with a TSQ Vantage triple quadrupole mass spectrometer equipped with nanospray source (Thermo Scientific, San Jose, CA). 8–10 targeted proteins were analyzed in a single LC-SRM run. The on-line chromatography was performed using an Eksigent NanoLC-2D HPLC system (AB SCIEX, Dublin, CA). An aliquot of 10 μl of each of the tryptic digests was injected on a C18 reverse-phase nano-HPLC column (PicoFritTM, 75 μm × 10 cm; tip inner diameter of 15 μm) at a flow rate of 500 nl/min over 20 min in 98% buffer A (0.1% formic acid), followed by a 15-min linear gradient from 2 to 30% mobile buffer B (0.1% formic acid, 90% acetonitrile). The TSQ Vantage was operated in high resolution SRM mode with Q1 and Q3 set to 0.2 and 0.7-Da full width half-maximum. All acquisition methods used the following parameters: 2100 V ion spray voltage, a 275 °C ion-transferring tube temperature, and a collision-activated dissociation pressure at 1.5 millitorr. The S-lens voltage used corresponded to the value in S-lens table generated during MS calibration.
Table VI. SID-SRM-MS assays for human and rickettsial proteins.
For each protein, the proteotypic peptide (sequence) is shown, along with the mass to charge ratio (m/z) for the first quadrupole (Q1) and third quadrupole (Q3) measurement, the ion type, and optimized collision energy.

All SRM data were manually inspected to ensure peak detection and accurate integration. The chromatographic retention time and the relative product ion intensities of the analyte peptides were compared with those of the SIS peptides. The variation of the retention time between the analyte peptides and their SIS counterparts should be within 0.05 min, and the difference in the relative product ion intensities of the analyte peptides and SIS peptides were below 20%. The peak areas in the extract ion chromatography of the native and SIS version of each signature peptide were integrated using Xcalibur® 2.1. The default values for noise percentage and baseline subtraction window were used. The ratio between the peak area of native and SIS version of each peptide was calculated.
Bioinformatics Analysis
High confidence protein identifications were subjected to pathway enrichment analysis using the Protein ANalysis THrough Evolutionary Relationship (Panther) pathway classification system (26). Pathways are rank-ordered based on statistically significant enrichment of the number of proteins in the dataset relative to the total number of proteins in the pathway. Ingenuity Pathway Analysis (IPA) was performed to identify relevant networks, rank-ordered by the number of proteins in the dataset relative to the pathway.
RESULTS
Experimental Design
To obtain a global understanding of the endothelial proteomics response to Rickettsia infections, we applied a standardized model developed by us using R. conorii-infected HUVECs, cells selected because they represent the primary target of R. conorii infection in vivo. Whole cell lysates (WCLs), PM, and Golgi fractions were prepared from uniformly R. conorii-infected primary HUVECs (Fig. 1A). In this experiment, conditions were established such that the cells were uniformly infected at the time of harvest (Fig. 1B). Quantitative stable isotopic labeling LC-MS/MS analysis was performed using trypsin-mediated 18O exchange and was compared with LPS-stimulated HUVECs to control for inflammatory responses that are not specific to rickettsial infection.
Fig. 1.

Quantitative proteomics study of R. conorii infection in HUVECs. A, experimental strategy. Shown is a schematic diagram of the experimental work flow for the identification of differential protein expression control (LPS-stimulated) or R. conorii-infected HUVECs. B, immunofluorescence assay. Immunofluorescence assay for rickettsial antigen (red) and nuclear DNA (blue) in HUVECs smeared on a glass slide to determine rickettsial growth. Original objective magnification was ×40. The primary antibody was a rabbit anti-R. conorii immune serum. The secondary antibody was a donkey anti-rabbit labeled with Alexa 546. C, 18O-labeling efficiency. MS spectra of two 18O-labeled peptides (HSPB1, LATQSNEITIPVTFESR, and ANXA2, GVDEVTIVNILTNR) are shown.
For each pairwise comparison, the experiment was repeated after swapping the 18O labeling between R. conorii-infected and LPS-stimulated HUVECs. Before the heavy- and light-labeled peptides were mixed, a small fraction of the 18O-labeled sample was tested for 18O-labeling efficiency with LC-MS/MS. Based on the analysis, the 18O-labeling efficiency is higher than 95% based on the abundance of two 18O-labeled peptides and their 16O-labeled counterparts. For example, in the whole cell lysate 18O quantification experiment, a total of 3,242 peptides were identified with 1% FDR by forward labeling. Among these, 3,127 (96.5%) were peptides with double-incorporated 18O labels. Out of 5,938 peptides in the reverse labeling experiment, 5,845 (98.4%) were double-incorporated with 18O labels. We further inspected the spectra manually. As shown in Fig. 1C, the 18O-labeling efficiency was higher than 95% based on the abundance of two 18O-labeled peptides and their 16O-labeled counterparts.
Differentially Expressed Proteins Identified from WCLs
In WCLs, a total of 1,082 proteins were quantified in both the forward and reversed 18O-labeling experiments, resulting in 784 high confidence proteins quantified (Fig. 2). A total of 55 proteins was differentially expressed (Fig. 2A), with 31 being up-regulated (Table I). The Pearson correlation of the quantitation of these significantly differentially expressed proteins between the two replicates is 0.68.
Fig. 2.

Differential protein expression in R. conorii-infected HUVEC WCLs. A, quantification of regulation. The plots are log2-transformed forward and reverse heavy/light ratios of individual proteins quantitated in WCLs from each replicate. Up-regulated proteins identified from the R. conorii-infected HUVECs are located in the bottom-right quadrant (red circles), and the proteins down-regulated in Rickettsia-infected HUVECs are located in the upper-left quadrant (green squares). B, Panther pathway analysis. Top panel, top pathways for proteins enriched in HUVEC WCL identified by the Panther classification system. x axis is the percentage of the pathway represented in the identified proteins. Bottom panel is pathway analysis for proteins depleted in HUVEC WCLs. C, network by IPA. Shown is the top-ranked network of differentially expressed WCL proteins identified in the Ingenuity Knowledge base. For each node, red indicates up-regulation in Rickettsia-infected cells; green, down-regulation. For abbreviations, see Table I.
Table I. Proteins with altered abundances in HUVEC WLCs.
For each significantly regulated protein is shown the accession number (Acc #), protein name, common gene name, and mean heavy (H)/light (L) enrichment ratio for the stable isotopic quantification in the replicate measurements. The L/H ratio measured after the label swap experiment is also shown.

To understand the biological pathways affected by R. conorii infection, we subjected the data to pathway enrichment analysis using the Panther pathway classification system (see under “Experimental Procedures”). This classification is a simplified functional ontology of functional protein groups based on curated data linked by Hidden Markov Models, allowing for more accurate functional inferences (26). In this representation, pathways enriched in the data set are presented as a rank-ordered list based on the percentage of the proteins within the pathway that are represented in the observed data set. We noted that the 31 up-regulated proteins are found within 13 pathways (Fig. 2B, top panel); Parkinson disease, FGF signaling, JAK-STAT pathways, interleukin signaling, and chemokine/cytokine inflammation pathways are enriched. The pathways represented by the down-regulated proteins were RNA polymerase I, arginine biosynthesis, and de novo pyrimidine biosynthesis (Fig. 2B, bottom panel), suggesting that R. conorii infection significantly perturbs metabolic pathways in HUVECs.
IPA was performed on the 55 differentially expressed protein WCLs. One of the top-ranked networks enriched in this fraction was Molecular Transport and Protein Trafficking, containing a cluster of IFN-signaling proteins, including MX1, ISG3, STAT1, and ISG15 (Fig. 2C), consistent with the Panther pathway analysis.
Differentially Expressed PM Proteins
Previous work has shown that R. conorii induces significant changes in the expression of cell surface proteins, including tissue factor (16) and adhesion molecules (11–14), promoting a pro-thrombotic phenotype. Because PM proteins are under-represented in total cellular lysates, we separately analyzed the integral membrane proteome of R. conorii-infected HUVECs. For this purpose, we applied a biotin-directed affinity purification method developed by us for the preparation of integral PM proteins (18). A total of 286 plasma membrane proteins were quantified (Fig. 3). We used NSAF to confirm the enrichment of the plasma membrane proteins. NSAF is based on spectral counting, which has been widely used in label-free proteomics quantitation (27–29). In spectral counting, larger proteins usually generate more peptides and therefore more spectral counts than smaller proteins. Therefore, the number of spectral counts for each protein is first divided by the mass or protein length, which defines the spectral abundance factor (SAF). Furthermore, to accurately account for sample to sample variation, individual SAF values are normalized to one by dividing by the sum of all SAFs for proteins identified in the sample, resulting in the NSAF value (23). In this manner, NSAFs values are standardized across distinct samples, allowing direct comparisons to be made between individual samples. The NSAF value of one protein is positively correlated with the relative abundance of the protein in this sample, where a highly abundant protein would have a higher NSAF value. We calculated the NSAF value for each protein identified in the PM fraction and whole cell lysate and then we compared their NSAF scores in the PM fraction and WCLs. As shown in Fig. 3A, compared with WCLs, plasma membrane proteins were highly enriched in the PM fraction, whereas the highly abundant cytosolic proteins HSP90AB1, HSPB1, and ENO1 were almost completely removed from our PM fraction. Proteins specific to the mitochondria (LDHA and LDHB), endoplasmic reticulum (VCP), and nucleus (NPM1 and NCL) were also significantly depleted from the PM fraction, indicating that we have successfully enriched the plasma membrane fraction.
Fig. 3.

Differential protein expression in R. conorii-infected HUVEC PM fractions. A, enrichment analysis. Top, spectral count measurements (NSAF) for representative plasma membrane proteins in the PM and WCL fractions. Note the enrichment of plasma membrane in the PM fractions. Bottom left panel, NSAF for cytosolic proteins, which are reduced (depleted) in PM fractions. Bottom right panel, NSAF for representative mitochondrial, endoplasmic reticulum, and nuclear proteins, also depleted in PM fractions. B, quantification of regulation. Shown are up-regulated proteins in PM fractions from Rickettsia-infected HUVECs in the bottom-right quadrant (red), and the proteins down-regulated in Rickettsia-infected HUVECs are located in the top-left quadrant (green). C, Panther pathway analysis. Top panel, highest ranked pathways identified by the Panther classification system for proteins enriched in HUVEC PMs. Bottom panel, pathways associated with down-regulated PM proteins. D, IPA network. Shown is the top-ranked network (“organismal disease”) of PM proteins in the IPA Knowledge Base. Abbreviations are shown in Table II.
Among 286 quantified PM proteins, a total of 11 differentially expressed proteins was identified (Fig. 3B). The Pearson correlation of the quantitation of these significantly differentially expressed proteins between the two replicates is 0.80. Four of these PM proteins were up-regulated by R. conorii infection (Table II). These proteins included hyperpolarization-activated potassium channel (HCN1) and 5′-nucleotidase. We noted that the most down-regulated PM protein was α-actin 1 (ACTA1), suggesting that R. conorii infection depletes PM ACTA1 as part of its effect on adherens junction formation and stress fiber formation Rickettsia (2, 8, 9, 30).
Table II. Differentially expressed PM proteins.

To more globally understand the functions controlled by this coordinated up-regulation of cell surface proteins, we subjected the differentially expressed proteins to enrichment analysis. Interestingly, proteins involved in purine and pyrimidine metabolism pathways were present in the up-regulated protein data set (Fig. 3C, top panel). Importantly, proteins controlling Wnt signaling, endothelin signaling, and cadherin signaling were in the down-regulated data set, among others (Fig. 3C, bottom panel). The effect of R. conorii on cadherin signaling may explain previously observed vascular leak phenomena and the effect on endothelial cell adherens junctions (1, 30). An IPA analysis showed a single network, “organismal disease” populated by down-regulated ACTA1 and the up-regulated HCN1 (Fig. 3D).
Differentially Expressed Golgi Proteins
Endothelial cells infected with R. conorii inducibly secrete a variety of soluble mediators. To better understand these, we profiled Golgi-enriched fractions containing proteins being processed for the secretory pathway. In this analysis, both total and soluble Golgi fractions were subjected to quantitative proteomic profiling. A total of 499 Golgi proteins were quantified; of these, 336 proteins were quantified in both experiments (supplemental Fig. 3). We used NSAF values to evaluate the enrichment of Golgi proteins and the proteins regulating the secretory pathways. As shown in Fig. 4A, Golgi apparatus protein 1 (GLG1) and several proteins of Golgi-derived retrograde transport vesicles such as SEC22B and three members of p24 family (TMED10, TMED3, and TMED7) were enriched in the Golgi fraction. Transmembrane emp24 domain-containing proteins are a widely conserved family of transmembrane proteins that play a functional role in protein transport within the early secretory pathway.
Fig. 4.

Differential protein expression in R. conorii-infected HUVEC Golgi fractions. A, enrichment analysis. NSAF for selected Golgi proteins in HUVEC Golgi and WCL fractions. Note the high spectral counts in the Golgi fractions relative to that in WCL fractions. B, quantification of regulation. Golgi proteins up-regulated in Rickettsia-stimulated HUVECs are located in the bottom-right quadrant (red), the Golgi proteins down-regulated in Rickettsia stimulated cells are located in the upper left (green). C, Panther pathway analysis. Top panel, top ranked pathways for proteins enriched in HUVEC Golgi fractions. Bottom panel, pathway analysis for proteins depleted in HUVEC Golgi fractions. D, IPA network analysis. Shown is the top-ranked network (“cell-cell signaling and compromise”) of Golgi proteins in IPA. Abbreviations are shown in Table III.
Among the 336 quantified proteins, a total of 52 differentially expressed proteins were identified (Fig. 4B), including 25 up-regulated proteins. The Pearson correlation of the quantitation of these significantly differentially expressed proteins between the two replicates is 0.88. These proteins included cell surface proteins PECAM, HLA-C, annexin, and others (Table III). We also noted interferon-induced transmembrane protein (IFITM)-3, consistent with the activation of the IFN-JAK-STAT signaling pathway observed in the WCL fractions. Enriched pathways of the up-regulated proteins in the Golgi membranes included GABA-type B protein receptor signaling and endogenous cannabinoid signaling (Fig. 4C, top panel).
Table III. Golgi proteins with altered abundances in response to R. conorii infection and LPS stimulation.

The down-regulated proteins in the Golgi fractions represented Huntington disease, Rho GTPase, and cadherin-signaling pathways (Fig. 4C, bottom panel). The depletion of cadherins in the Golgi pathway is consistent with the reduction of cadherins in the PM fractions noted earlier. An IPA analysis showed a network linked to cell-cell signaling and cellular compromise, including von Willebrand factor, flotillin (FLOT1), annexin A2 (ANXA2), phospholipase D3 (PLD3), HLA-C, and others (Fig. 4D).
Differentially Expressed Soluble Golgi Proteins
To identify secreted proteins processed by the canonical secretory pathway, we separately analyzed changes in soluble Golgi protein proteomes; 371 proteins were identified in soluble Golgi extracts, with 216 proteins being quantified in both experiments (supplemental Fig. 4). We used NSAF values to evaluate the enrichment of secreted proteins in this fraction. As shown in Fig. 5A, extracellular proteins such as secreted protein, acidic, cysteine-rich (SPARC), fibronectin (FN1), Intercellular Adhesion Molecule (ICAM)-1, Connective Tissue Growth Factor (CTGF), Prosaposin (PSAP), and Golgi Glycoprotein 1 (GLG1) were enriched in this fraction. These proteins included HLA class I, PECAM, and β2-microglobulin (Table IV).
Fig. 5.

Differential expression of secreted proteins in R. conorii infection. A, enrichment analysis. NSAF for selected secreted proteins in soluble Golgi fraction relative to WCL fractions. Note the high spectral counts in the soluble fractions relative to that in WCLs. B, quantification of regulation. Secreted proteins identified in soluble Golgi fraction. Proteins up-regulated in Rickettsia-infected HUVECs are located in the bottom-right quadrant (red), and down-regulated proteins are located in the upper left (green). C, Panther protein classification. Shown are the protein classifications for the secreted proteins in the R. conorii-infected HUVECs. D, IPA network. Shown is the top-ranked network (“immunological disease”) of soluble Golgi proteins from IPA. Abbreviations are shown in Table IV.
Table IV. Soluble Golgi proteins.

In the Golgi fraction, a total of 13 differentially expressed proteins were identified (Fig. 5B); of these, six were up-regulated by Rickettsia infection (Table V). The Pearson correlation of the quantitation of these significantly differentially expressed proteins between the two replicates is 0.86. To provide some biological insight into the activities of these secreted proteins, the up-regulated proteins were subjected to a protein class analysis. The most abundant protein activities of the secreted proteins were those encoding classes of cell adhesion/cell junction activity and hydrolase and protease activity (Fig. 5C). IPA analysis identified a pathway dominated by β2-microglobulin MHC class I and HLA isoforms (Fig. 5D).
Table V. R. conorii proteins identified.
Each protein is identified with FDR of <1%. For each protein, the subcellular fraction in which it was identified is shown.

Intracellular Distribution of Rickettsial Proteins
Rickettsia invade eukaryotic cells through an induced phagocytosis mechanism. The bacteria then escape from the phagosome and utilize actin filaments to spread (even to the nucleus) and replicate in the host cell cytoplasm and nucleus. However, the subcellular distribution of Rickettsia proteins has not yet been determined to our knowledge. Unmatched spectra from our proteomics study were searched against the Rickettsia proteome database (downloaded from SwissProt protein database on February 20, 2013, 4,189 entries); 34 proteins were identified with a false discovery rate estimation of 1% or less in the PM, Golgi, and secreted protein fractions (Table V). These proteins show a characteristic non-random distribution. For example, the Rickettsia chaperone proteins, HtpG, DnaK, NADPH reductase, and cytosolic aminopeptidase, were enriched in the Golgi fractions, consistent with either contamination of Golgi fraction with Rickettsia organisms or biological processing of rickettsial proteins by the host Golgi pathway. The cytochrome c oxidase and NADH-quinone oxidoreductases were observed in the Golgi preparations, perhaps suggesting that these molecules may be a source of enhanced superoxide and lipid peroxidation observed in Rickettsia-infected endothelial cells (31).
A large number of R. conorii proteins was identified in the soluble fraction of the Golgi preparation and some of these included ferredoxin, heme biosynthetic enzymes, peptide chain releasing factors, protein translocases, and others (Table V). In the soluble Golgi fractions, we also identified the cell surface antigen Sca2, a formin mimic responsible for interacting with the host actin cytoskeleton (32). Finally, a distinct group of R. conorii proteins was identified in HUVEC plasma membrane fractions; these proteins were putative ligases, dehydrogenases, glycoprotein transferases, and lipoprotein-metabolizing proteins (Table V).
Verification of Host- and Rickettsial Proteins
To qualify the differential expression of endothelial innate response proteins, we developed quantitative high throughput SRM assays to selectively measure each protein. The proteins, gene names, and accession numbers are shown in Table VI. The optimal empirically determined collision energy is also tabulated. Compared with LPS-stimulated cells, significant induction of STAT1 and ISG15 was observed in the WCL (Fig. 6A).
Fig. 6.

Qualification of innate response proteins in R. conorii-infected HUVECs. A, qualification of IFN pathway. Shown are SID-selected reaction-monitoring (SRM)-MS measurements of STAT1 and ISG15 (bottom) for HUVEC stimulated with LPS (left) or infected with R. conorii (right). y axis is ratio of protein relative to internal SIS peptide (native/SIS) peptide (native/aqua). B, qualification of rickettsial proteins in Golgi membranes. SID-SRM-MS measurements for UvrABC system protein C are shown; putative ankyrin repeat protein RBE; and chaperone protein HtpG. Compared with LPS-stimulated cells, significant induction of each was observed. C, qualification of rickettsial proteins in soluble Golgi fraction. SID-SRM-MS measurements for human HLA proteins and the rickettsial proteins UVRVC and HPTG in the soluble Golgi fraction.
We also selected candidate rickettsial proteins identified in the Golgi and PM fractions as follows: the putative UvrABC system protein C; putative ankyrin repeat protein RBE; and chaperone protein HtpG. Compared with LPS-stimulated cells, significant induction of each was observed (Fig. 6B). The up-regulation of HLA proteins and the rickettsial proteins UVRVC and HPTG in the soluble Golgi fraction was also confirmed with SID-SRM-MS (Fig. 6C).
DISCUSSION
Rickettsiae are non-motile, Gram-negative, and obligately intracellular bacterial pathogens of global medical and veterinary health importance. During transmission, an infected hematophagous arthropod vector introduces rickettsiae into the dermis, where the organism disseminates to vascular endothelial cells throughout the body. Here, dividing Rickettsia induce cellular stress leading to cell detachment. Detached endothelial cells, which are heavily infected, lodge into downstream capillaries and initiate new foci of vascular infection. In our approach, we harvested infected endothelial cells once they were homogeneously infected, which mimic these foci of vascular infection. Such multifocal lesions are found throughout the course of the disease, even at early stages. We applied subcellular fractionation and quantitative proteomics to develop an integrated understanding of the human endothelial cellular response and subcellular distribution of Rickettsia proteins in this model of established infection. We deduced the activation of the JAK-STAT-ISG15 signaling pathway along with significant perturbations of cell surface enzymatic activities of infected endothelial cells. We think it important that R. conorii-infected endothelial cells show significant down-regulation of cadherin components whose putative role in pathogenesis is discussed below.
Previous work by us has shown that the IFN response is a major determinant limiting severity of rickettsial disease in vivo (33, 34). Although the pattern recognition receptors for rickettsial infection are not fully understood, replication of Rickettsia is a stimulus for production of type I IFN (IFNβ) (35). The type I IFN response has been observed as a common response to Rickettsia infections for all endothelial cell types examined (36). Downstream of IFN, activation of the STAT1 signaling pathway triggers production of ISG, MX1, SOCS, and other proteins important in host defense (37). ISG15 is a small ubiquitin-like modifier induced at the transcriptional level by IFN signaling known to covalently modify target proteins by ISGylation, inducing changes in signaling, chemotaxis, and translation. In human microvascular endothelial cells infected with R. conorii, ISG15 is up-regulated through an IFN-β-dependent pathway, and it controls intracellular rickettsial replication (35). Our data extend the Rickettsia-induced IFN pathway to include intracellular MX1 and cell-surface associated IFITM3 expression. More study will be required to understand their role, if any, in restricting rickettsial infection.
Previous work has shown that E-selectin (11), VCAM-1, ICAM-1 (11–13), and αVβ3 integrin (14) are plasma membrane proteins up-regulated in Rickettsia-infected cells to mediate neutrophil attachment, as well as vascular integrity. Because of their size, hydrophobic characteristics, and insolubility, the analysis of PM proteins is typically challenging. In this study, we applied a cell surface-directed biotin cross-linking method to affinity-enrich PM proteins in Rickettsia-infected cells. This method enables the enrichment and quantitation of integral PM proteins (18). Our data suggest that rickettsial infection up-regulates voltage channels in endothelial cell and depletes plasma membrane-associated ACTA1. Such changes could underlie vascular reactivity and the phenomenon of endothelial cell detachment (38), which could be a source of new foci of infection once they lodge in distal capillaries.
Previous studies provided evidence that T lymphocytes are critical in the development of immune protection in rickettsial disease; CD4 and CD8 T lymphocytes protect mice against lethal disseminated endothelial infection with R. conorii (39), but CD8 T cells are the most critical effectors (40). We note that up-regulation of HLA-I antigen transporter is observed, along with the presence of β2-microglobulin, hydrolases, and Rickettsia OmpB (Sca5) and Sca2 in the secretory pathway (soluble Golgi fraction). Importantly, OmpB is an immunodominant antigen for CD8 T lymphocytes in a mouse model of R. conorii infection (41). These data may explain how this immunodominant rickettsial antigen is processed and presented to the PM.
von Willebrand factor (vWF), an adhesive glycoprotein involved in primary hemostasis, is primarily stored in endothelial secretory granules, Weibel-Palade bodies, from which it is released during rickettsial infection (42). We observe increased abundance of vWF in R. conorii-infected endothelial cells. These data may suggest that newly synthesized vWF is processed by the Golgi apparatus prior to its packaging within the Weibel-Palade body.
Analysis of the soluble fraction of Golgi-enriched organelles provides a number of insights into the host response of endothelial cells. Endothelial host response proteins are highly enriched in MHC class I activity, including HLA-A, -C, and β2-microglobulin. Interestingly, Rickettsia cell surface antigens, Sca-5 and -2, are found in the secretory pathway, which may be a reflection of antigenic processing for presentation through the MHC class I pathway. Previous work by us has shown that MHC I knockout mice are highly susceptible to rickettsial infection due to defects in mobilizing an efficient cytotoxic T cell response (40). Interestingly, we have observed that cathepsin is an abundant cysteine protease enriched in the secretory fraction. Cathepsin has recently been shown to be involved in proteolytic processing of chemerin to trigger migration of human blood-derived plasmacytoid dendritic cells (43). Whether cathepsin plays a role in host response to Rickettsia infection may be an important direction for future research.
Our experimental design was intended to identify the major responses of human endothelial cells to established rickettsial infection. In our design, we harvested infected endothelial cells once they were homogeneously infected, a model that mimics foci of vascular infection from detached endothelial cells important in rickettsial dissemination. Other mechanistic studies have shown rapid activation of STAT3 signaling within hours of rickettsial infection, followed by a later activation of STAT1 (44). More work will be required to understand the early patterns of host response in rickettsial infection.
In summary, our study provides an integrated host and bacterial proteomics analysis of the infection of primary human endothelial cells with the etiologic agent of human Mediterranean spotted fever, R. conorii. Our study identifies the STAT1-ISG15 and HLA antigen production as the major components of the innate and adaptive immune response triggered by endothelial cells. We observe significant reprogramming of the plasma membrane proteome and induction of adhesion molecules, with down-regulation of endothelial cell cadherins. These observations generate insights into how rickettsiae induce the endothelial stress response.
Supplementary Material
Footnotes
Author contributions: Y.Z., G.V., D.H.W., M.H., R.D., J.A.O., and A.R.B. designed the research; Y.Z., G.V., M.G., and Y.G. performed the research; Y.Z., G.V., M.H., R.D., and A.R.B. contributed new reagents or analytic tools; Y.Z., G.V., D.H.W., and A.R.B. analyzed the data; and Y.Z., G.V., D.H.W., J.A.O., and A.R.B. wrote the paper.
* This work was supported by NIAID Clinical Proteomics Center Grant HHSN272200800048C (to A.R.B.), University of Texas Medical Branch Clinical and Translational Science Award (CTSA) UL1TR000071 (to A.R.B.), and NIEHS Grant P30 ES006676 (to A.R.B.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
 This article contains supplemental Files 1–3, Tables 1–3, and Figs. 1–4.
This article contains supplemental Files 1–3, Tables 1–3, and Figs. 1–4.
Data deposition Repository: ProteomeXchange Consortium; Project Name: Endothelial cell proteomic response to Rickettsia conorii infection; Project accession: PXD002650 Reviewer account details: Username: reviewer72296@ebi.ac.uk Password: PSZLXnUC.
1 The abbreviations used are:
- HUVEC
- human umbilical vein endothelial cell
- FDR
- false discovery rate
- IPA
- Ingenuity Pathway Analysis
- PM
- plasma membrane
- SID
- stable isotopic dilution
- SRM
- selected reaction monitoring
- WCL
- whole cell lysate
- STAT
- signal transducer and activator of transcription
- NSAF
- normalized spectral abundance factor
- SAF
- spectral abundance factor
- ACN
- acetonitrile
- SIS
- stable isotope standard.
REFERENCES
- 1.Walker D. H., Valbuena G. A., and Olano J. P. (2003) Pathogenic mechanisms of diseases caused by Rickettsia. Ann. N.Y. Acad. Sci. 990, 1–11 [DOI] [PubMed] [Google Scholar]
- 2.Walker T. S., and Winkler H. H. (1978) Penetration of cultured mouse fibroblasts (L cells) by Rickettsia prowazeki. Infect. Immun. 22, 200–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martinez J. J., and Cossart P. (2004) Early signaling events involved in the entry of Rickettsia conorii into mammalian cells. J. Cell Sci. 117, 5097–5106 [DOI] [PubMed] [Google Scholar]
- 4.Chan Y. G., Cardwell M. M., Hermanas T. M., Uchiyama T., and Martinez J. J. (2009) Rickettsial outer-membrane protein B (rOmpB) mediates bacterial invasion through Ku70 in an actin, c-Cbl, clathrin, and caveolin 2-dependent manner. Cell. Microbiol. 11, 629–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Renesto P., Dehoux P., Gouin E., Touqui L., Cossart P., and Raoult D. (2003) Identification and characterization of a phospholipase D-superfamily gene in rickettsiae. J. Infect. Dis. 188, 1276–1283 [DOI] [PubMed] [Google Scholar]
- 6.Whitworth T., Popov V. L., Yu X. J., Walker D. H., and Bouyer D. H. (2005) Expression of the Rickettsia prowazekii pld or tlyC gene in Salmonella enterica serovar Typhimurium mediates phagosomal escape. Infect. Immun. 73, 6668–6673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schaechter M., Bozeman F. M., and Smadel J. E. (1957) Study on the growth of Rickettsiae. II. Morphologic observations of living Rickettsiae in tissue culture cells. Virology 3, 160–172 [DOI] [PubMed] [Google Scholar]
- 8.Teysseire N., Chiche-Portiche C., and Raoult D. (1992) Intracellular movements of Rickettsia conorii and R. typhi based on actin polymerization. Res. Microbiol. 143, 821–829 [DOI] [PubMed] [Google Scholar]
- 9.Heinzen R. A., Hayes S. F., Peacock M. G., and Hackstadt T. (1993) Directional actin polymerization associated with spotted fever group Rickettsia infection of Vero cells. Infect. Immun. 61, 1926–1935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Walker D. H., Firth W. T., and Edgell C. J. (1982) Human endothelial cell culture plaques induced by Rickettsia rickettsii. Infect. Immun. 37, 301–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sporn L. A., Lawrence S. O., Silverman D. J., and Marder V. J. (1993) E-selectin-dependent neutrophil adhesion to Rickettsia rickettsii-infected endothelial cells. Blood 81, 2406–2412 [PubMed] [Google Scholar]
- 12.Dignat-George F., Teysseire N., Mutin M., Bardin N., Lesaule G., Raoult D., and Sampol J. (1997) Rickettsia conorii infection enhances vascular cell adhesion molecule-1- and intercellular adhesion molecule-1-dependent mononuclear cell adherence to endothelial cells. J. Infect. Dis. 175, 1142–1152 [DOI] [PubMed] [Google Scholar]
- 13.Damås J. K., Davi G., Jensenius M., Santilli F., Otterdal K., Ueland T., Flo T. H., Lien E., Espevik T., Frøland S. S., Vitale G., Raoult D., and Aukrust P. (2009) Relative chemokine and adhesion molecule expression in Mediterranean spotted fever and African tick bite fever. J. Infect. 58, 68–75 [DOI] [PubMed] [Google Scholar]
- 14.Bechah Y., Capo C., Grau G., Raoult D., and Mege J. L. (2009) Rickettsia prowazekii infection of endothelial cells increases leukocyte adhesion through αvβ3 integrin engagement. Clin. Microbiol. Infect. 15, 249–250 [DOI] [PubMed] [Google Scholar]
- 15.Clifton D. R., Rydkina E., Huyck H., Pryhuber G., Freeman R. S., Silverman D. J., and Sahni S. K. (2005) Expression and secretion of chemotactic cytokines IL-8 and MCP-1 by human endothelial cells after Rickettsia rickettsii infection: regulation by nuclear transcription factor NF-κB. Int. J. Med. Microbiol. 295, 267–278 [DOI] [PubMed] [Google Scholar]
- 16.Sporn L. A., Haidaris P. J., Shi R. J., Nemerson Y., Silverman D. J., and Marder V. J. (1994) Rickettsia rickettsii infection of cultured human endothelial cells induces tissue factor expression. Blood 83, 1527–1534 [PubMed] [Google Scholar]
- 17.Sporn L. A., and Marder V. J. (1996) Interleukin-1α production during Rickettsia rickettsii infection of cultured endothelial cells: potential role in autocrine cell stimulation. Infect. Immun. 64, 1609–1613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao Y., Zhang W., Kho Y., and Zhao Y. (2004) Proteomic analysis of integral plasma membrane proteins. Anal. Chem. 76, 1817–1823 [DOI] [PubMed] [Google Scholar]
- 19.Bell A. W., Ward M. A., Blackstock W. P., Freeman H. N., Choudhary J. S., Lewis A. P., Chotai D., Fazel A., Gushue J. N., Paiement J., Palcy S., Chevet E., Lafrenière-Roula M., Solari R., Thomas D. Y, Rowley A., and Bergeron J. J. (2001) Proteomics characterization of abundant Golgi membrane proteins. J. Biol. Chem. 276, 5152–5165 [DOI] [PubMed] [Google Scholar]
- 20.Starkey J. M., Zhao Y., Sadygov R. G., Haidacher S. J., Lejeune W. S., Dey N., Luxon B. A., Kane M. A., Napoli J. L., Denner L., and Tilton R. G. (2010) Altered retinoic acid metabolism in diabetic mouse kidney identified by O isotopic labeling and 2D mass spectrometry. PLoS One 5, e11095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sadygov R. G., Zhao Y., Haidacher S. J., Starkey J. M., Tilton R. G., and Denner L. (2010) Using power spectrum analysis to evaluate 18O-water labeling data acquired from low resolution mass spectrometers. J. Proteome Res. 9, 4306–4312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cox J., and Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 23.Zybailov B., Mosley A. L., Sardiu M. E., Coleman M. K., Florens L., and Washburn M. P. (2006) Statistical analysis of membrane proteome expression changes in Saccharomyces cerevisiae. J. Proteome Res. 5, 2339–2347 [DOI] [PubMed] [Google Scholar]
- 24.Zhao Y., and Brasier A. R. (2013) Applications of selected reaction monitoring (SRM)-mass spectrometry (MS) for quantitative measurement of signaling pathways. Methods 61, 313–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao Y., Tian B., Edeh C. B., and Brasier A. R. (2013) Quantitation of the dynamic profiles of the innate immune response using multiplex selected reaction monitoring-mass spectrometry. Mol. Cell. Proteomics 12, 1513–1529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mi H., Lazareva-Ulitsky B., Loo R., Kejariwal A., Vandergriff J., Rabkin S., Guo N., Muruganujan A., Doremieux O., Campbell M. J., Kitano H., and Thomas P. D. (2005) The PANTHER database of protein families, subfamilies, functions and pathways. Nucleic Acids Res. 33, D284–D288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu H., Sadygov R. G., and Yates J. R. 3rd (2004) A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal. Chem. 76, 4193–4201 [DOI] [PubMed] [Google Scholar]
- 28.Old W. M., Meyer-Arendt K., Aveline-Wolf L., Pierce K. G., Mendoza A., Sevinsky J. R., Resing K. A., and Ahn N. G. (2005) Comparison of label-free methods for quantifying human proteins by shotgun proteomics. Mol. Cell. Proteomics 4, 1487–1502 [DOI] [PubMed] [Google Scholar]
- 29.Paoletti A. C., Parmely T. J., Tomomori-Sato C., Sato S., Zhu D., Conaway R. C., Conaway J. W., Florens L., and Washburn M. P. (2006) Quantitative proteomic analysis of distinct mammalian Mediator complexes using normalized spectral abundance factors. Proc. Natl. Acad. Sci. U.S.A. 103, 18928–18933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Valbuena G., and Walker D. H. (2005) Changes in the adherens junctions of human endothelial cells infected with spotted fever group rickettsiae. Virchows Arch. 446, 379–382 [DOI] [PubMed] [Google Scholar]
- 31.Santucci L. A., Gutierrez P. L., and Silverman D. J. (1992) Rickettsia rickettsii induces superoxide radical and superoxide dismutase in human endothelial cells. Infect. Immun. 60, 5113–5118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kleba B., Clark T. R., Lutter E. I., Ellison D. W., and Hackstadt T. (2010) Disruption of the Rickettsia rickettsii Sca2 autotransporter inhibits actin-based motility. Infect. Immun. 78, 2240–2247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jerrells T. R., Li H., and Walker D. H. (1988) In vivo and in vitro role of γ interferon in immune clearance of Rickettsia species. Adv. Exp. Med. Biol. 239, 193–200 [DOI] [PubMed] [Google Scholar]
- 34.Li H., Jerrells T. R., Spitalny G. L., and Walker D. H. (1987) γ interferon as a crucial host defense against Rickettsia conorii in vivo. Infect. Immun. 55, 1252–1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Colonne P. M., Eremeeva M. E., and Sahni S. K. (2011) β interferon-mediated activation of signal transducer and activator of transcription protein 1 interferes with Rickettsia conorii replication in human endothelial cells. Infect. Immun. 79, 3733–3743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rydkina E., Turpin L. C., and Sahni S. K. (2010) Rickettsia rickettsii infection of human macrovascular and microvascular endothelial cells reveals activation of both common and cell type-specific host response mechanisms. Infect. Immun. 78, 2599–2606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Colonne P. M., Sahni A., and Sahni S. K. (2013) Suppressor of cytokine signalling protein SOCS1 and UBP43 regulate the expression of type I interferon-stimulated genes in human microvascular endothelial cells infected with Rickettsia conorii. J. Med. Microbiol. 62, 968–979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.George F., Brouqui P., Boffa M. C., Mutin M., Drancourt M., Brisson C., Raoult D., and Sampol J. (1993) Demonstration of Rickettsia conorii-induced endothelial injury in vivo by measuring circulating endothelial cells, thrombomodulin, and von Willebrand factor in patients with Mediterranean spotted fever. Blood 82, 2109–2116 [PubMed] [Google Scholar]
- 39.Feng H., Popov V. L., Yuoh G., and Walker D. H. (1997) Role of T lymphocyte subsets in immunity to spotted fever group rickettsiae. J. Immunol. 158, 5314–5320 [PubMed] [Google Scholar]
- 40.Walker D. H., Olano J. P., and Feng H. M. (2001) Critical role of cytotoxic T lymphocytes in immune clearance of rickettsial infection. Infect. Immun. 69, 1841–1846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li Z., Díaz-Montero C. M., Valbuena G., Yu X.-J., Olano J. P., Feng H.-M., and Walker D. H. (2003) Identification of CD8 T-lymphocyte epitopes in OmpB of Rickettsia conorii. Infect. Immun. 71, 3920–3926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sporn L. A., Shi R. J., Lawrence S. O., Silverman D. J., and Marder V. J. (1991) Rickettsia rickettsii infection of cultured endothelial cells induces release of large von Willebrand factor multimers from Weibel-Palade bodies. Blood 78, 2595–2602 [PubMed] [Google Scholar]
- 43.Kulig P., Kantyka T., Zabel B. A., Banas M., Chyra A., Stefanska A., Tu H., Allen S. J., Handel T. M., Kozik A., Potempa J., Butcher E. C., and Cichy J. (2011) Regulation of chemerin chemoattractant and antibacterial activity by human cysteine cathepsins. J. Immunol. 187, 1403–1410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sahni S. K., Kiriakidi S., Colonne M. P., Sahni A., and Silverman D. J. (2009) Selective activation of signal transducer and activator of transcription (STAT) proteins STAT1 and STAT3 in human endothelial cells infected with Rickettsia rickettsii. Clin. Microbiol. Infect. 15, 303–304 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.