

Abstract
Antimicrobial peptides form part of the first line of defense against pathogens for many organisms. Current treatments for fungal infections are limited by drug toxicity and pathogen resistance. Cm-p5 (SRSELIVHQRLF), a peptide derived from the marine mollusk Cenchritis muricatus peptide Cm-p1, has a significantly increased fungistatic activity against pathogenic Candida albicans (minimal inhibitory concentration, 10 µg/ml; EC50, 1.146 µg/ml) while exhibiting low toxic effects against a cultured mammalian cell line. Cm-p5 as characterized by circular dichroism and nuclear magnetic resonance revealed an α-helical structure in membrane-mimetic conditions and a tendency to random coil folding in aqueous solutions. Additional studies modeling Cm-p5 binding to a phosphatidylserine bilayer in silico and isothermal titration calorimetry using lipid monophases demonstrated that Cm-p5 has a high affinity for the phospholipids of fungal membranes (phosphatidylserine and phosphatidylethanolamine), only moderate interactions with a mammalian membrane phospholipid, low interaction with ergosterol, and no interaction with chitin. Adhesion of Cm-p5 to living C. albicans cells was confirmed by fluorescence microscopy with FITC-labeled peptide. In a systemic candidiasis model in mice, intraperitoneal administration of Cm-p5 was unable to control the fungal kidney burden, although its low amphiphaticity could be modified to generate new derivatives with improved fungicidal activity and stability.—López-Abarrategui, C., McBeth, C., Mandal, S. M., Sun, Z. J., Heffron, G., Alba-Menéndez, A., Migliolo, L., Reyes-Acosta, O., García-Villarino, M., Nolasco, D. O., Falcão, R., Cherobim, M. D., Dias, S. C., Brandt, W., Wessjohann, L., Starnbach, M., Franco, O. L., Otero-González, A. J. Cm-p5: an antifungal hydrophilic peptide derived from the coastal mollusk Cenchritis muricatus (Gastropoda: Littorinidae).
Keywords: antimicrobial peptide, Candida albicans, systemic candidiasis
In the last decade, human fungal infections such as those caused by Aspergillus fumigatus and Candida albicans have been associated with high mortality rates due to the increasing number of immunocompromised patients (1). Unfortunately, antifungal therapies for these infections are limited to only a small number of molecules, such as azoles, echinocandins, and polyenes (2). These classes of molecules typically act on only one target, which enables rapid resistance to develop in the pathogenic population. These resistance mechanisms of pathogenic fungi constitute a critical challenge for current antifungal therapies (3). Additionally, the incomplete efficacy and the toxicity associated with these drugs, especially when used nonspecifically, have further increased the need for novel antifungal agents (4).
Antimicrobial peptides (AMPs) represent a possible alternative solution because they exhibit a broad spectrum of activities against a wide range of microorganisms (5, 6). As a class, AMPs are a conserved defense mechanism across most eukaryotic species (7). Indeed, organisms as genetically diverse as snails and humans both generate AMPs to limit growth of pathogenic microorganisms, including fungi (8–11). Several mechanisms of action have been proposed for these molecules, and current findings suggest that many of them have more than one antimicrobial target at the cellular level (12, 13). Factors that work by modifying the physicochemical properties of the membrane rather than occluding a specific binding site or inhibiting an enzymatic activity are especially difficult for pathogens to counter because they are not related to a single protein target and are thus not amenable to single nucleotide polymorphism–based resistance.
In addition to direct effects, some AMPs may indirectly inhibit microbial growth through activation of the innate immune system (14). By engaging the microbe directly and recruiting additional immune responses, AMPs are substantially less likely to induce pathogen resistance (15). Biophysical characterization of AMPs has revealed the cationic and amphipathic character of AMPs as key determinants for their antimicrobial activity (16–18). Databases linking these physicochemical properties to the functionality of AMPs may aid in designing novel peptides with enhanced activity (19–22). One such metric is the Boman index, which describes the potential of a peptide to bind to other proteins (23). A low hemolytic potential should also be considered when developing these peptides for applications in the clinic. In previous work, we identified Cm-p1, an antifungal peptide derived from the marine mollusk Cenchritis muricatus (24). In order to augment the antimicrobial activity of Cm-p1, we added 2 additional residues to the C terminus. Amino acids were chosen to match the native peptide fragment size and to significantly alter the cationicity, hydrophobicity, and Boman index of the peptide. These indices have been shown to correlate with in vitro antimicrobial activity. Three Cm-p1-derived peptides were synthesized and evaluated for antimicrobial activity against a panel of bacteria and clinically important pathogenic fungi. One of the derivatives, Cm-p5 (SRSELIVHQRLF), had a minimal inhibitory concentration (MIC) against clinically important strains of C. albicans and Cryptococcus neoformans lower than the parental peptide.
To understand the significant effect of the hydrophobic addition to Cm-p5, we examined the behavior of Cm-p5 in both aqueous and membrane-mimetic solutions. Cm-p5 adopted a clear α-helical structure in the membrane-mimetic environment, as determined by circular dichroism (CD) and nuclear magnetic resonance. However, both methods revealed a tendency to relax into a random coil in aqueous environments, suggesting that Cm-p5 interaction with membranes triggers ordering of the peptide. As the phospholipid phosphatidylserine is widely distributed in the plasma membrane of fungi (25), we modeled Cm-p5 in a lipid bilayer of phosphatidylserine and identified multiple specific interactions between the peptide and the phospholipid head group. The affinity of Cm-p5 by the fungal phospholipids phosphatidylserine and phosphatidylethanolamine was demonstrated by isothermal titration calorimetry (ITC). In order to confirm these results, the adhesion of Cm-p5 to living C. albicans cell was evaluated by fluorescence microscopy with FITC-labeled peptide. Furthermore, in a systemic candidiasis model in mice, intraperitoneal administration of Cm-p5 was used to evaluate its therapeutic action.
MATERIALS AND METHODS
Peptide synthesis
All peptides were synthesized by the solid-phase method using 9-fluorenyl-methoxycarbonyl chemistry (26), purified by reverse-phase HPLC to >98% purity using an acetonitrile/H2O-TFA gradient. Purity was confirmed by ion spray mass spectrometry (Micromass, Manchester, United Kingdom).
Determination of peptide concentration
Peptide concentrations were estimated by UV absorption (Abs) at 205, 215, and 225 nm using the following equations (27):
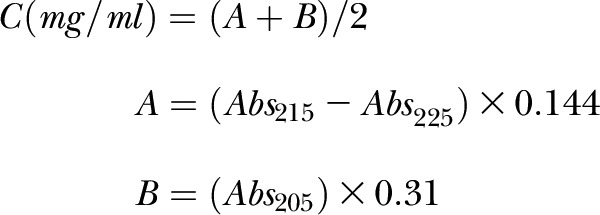 |
Bioassays against bacteria
The antimicrobial bioassays were evaluated for a panel of Bacillus subtilis, Staphylococcus aureus, Enterococcus faecalis, Klebsiella pneumoniae, Pseudomonas aeruginosa, Proteus mirabilis, and Escherichia coli by a colorimetric microreader (BioTek Instruments, Winooski, VT, USA) using 96-well microplates according to Hetru and Bulet’s methodology (28). Bioassay analyses against bacteria were performed in Luria-Bertani medium (pH 7.0). Previously, a growth curve of the original culture was established to determine the relationship between colony-forming units (CFU) and optical density. For antimicrobial activity evaluation, 0.1 ml inoculum was cultured in 4 ml Luria-Bertani medium for 3 h until reaching the midexponential phase. An aliquot corresponding to 5 × 105 CFU ml−1 was then added to Luria-Bertani medium to produce a final volume of 0.1 ml in the microplate wells. Peptides were added to reach a final concentration of 128 µg/ml. Chloramphenicol at a concentration of 40 µg/ml and distilled water were used as positive and negative controls, respectively. Microplates were incubated at 37°C, and bacterial growth was monitored at 620 nm every hour. The percentage of survival was determined as the ratio of optical density of bacterial normal growth (100%) and the growth under peptide action. All bioassays were performed in triplicate.
Antifungal bioassays
Bioassays against fungi were performed using the broth microdilution method according to Clinical and Laboratory Standards Institute guidelines (29). The bioassays against human pathogenic yeast (Candida parapsilosis [ATCC 22019] and clinically isolated strains of C. albicans [01U, 38U], C. neoformans [L26, L30], Trichophyton mentagrophytes [28d, 28e], and Trichophyton rubrum [329] were performed in RPMI 1640 medium (Gibco, Carlsbad, CA, USA). Amphotericin-B (30 µg/ml) and sterile distilled water were used as positive and negative controls, respectively. Microplates were incubated at 37°C, and yeast growth was monitored at 620 nm every hour. The percentage of survival was determined as the ratio of optical density of fungi normal growth (100%) and the growth under peptide action. All tests were conducted in triplicate. The Cm-p5 EC50 value of C. albicans (01U) in the antifungi bioassay was calculated by plotting percent survival as a function of the log of Cm-p5 concentration (0.1–100 µg/ml) and fitting the data to a standard sigmoidal dose–response curve using Microcal Origin, version 8.0 (Microcal Software, Northampton, MA, USA). Each experiment was performed in triplicate.
Cytotoxicity assay
The MTT (3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyltetrazolium bromide; Sigma-Aldrich, St. Louis, MO, USA) assay was performed as previously described (30). RAW 264.7 murine macrophage-like cells (Rio de Janeiro Cell Bank, Rio de Janeiro, Brazil) were plated at a concentration of 1 × 105 cells per well in supplemented DMEM (4 mM glutamine, 10% fetal calf serum, and 100 units/ml penicillin–streptomycin) and incubated with different concentrations of peptides (0–400 µg/ml). After overnight incubation, 10 ml of the MTT solution (5 mg/ml in PBS) was added to each well. Plates were incubated for 4 h in 5% CO2 at 37°C. The generated blue formazan product was dissolved by the addition of 100 µl of 100% DMSO (Mallinckrodt Chemical, USA) per well. The absorbance was monitored at 575 nm in an ELISA plate reader (BioTek).
CD spectroscopy
The CD measurements were made on a J-815 CD spectrometer (Jasco, Tokyo, Japan) at 20°C in a thermally controlled quartz cell with a 0.1 cm path length over 180 to 400 nm. The samples were prepared by dissolving the peptides in water, trifluoroethanol (TFE; 100% w/v), and 60% TFE/water mixture. The final peptide concentration for the CD measurements was 100 µM. Data were collected every 0.1 nm, the bandwidth was set at 1.0 nm, and the sensitivity was 200 mdeg. Data integration time was 1 s, and scanning speed was 200 nm/min. The number of accumulations was 10.
NMR structure determination
Two-dimensional NOESY (nuclear Overhauser effect spectroscopy) and TOCSY (total correlation spectroscopy) data were acquired on an Agilent DD2 600 MHz spectrometer equipped with a cryogenic probe at 25°C (Agilent Technologies, Santa Clara, CA, USA). A 4 mM sample of Cm-p5 was prepared in 40% TFE (d2)/60% D2O. The NOESY data were collected with 100 ms mixing time, 1.5 s recycling delay time, 24 transients per free induction decay, and 2048 and 256 complex points in direct and indirect dimensions, respectively. NMR data were processed by NMRPipe (31). NMR and nuclear Overhauser effect (NOE) peak assignments were completed using CARA (32). Preliminary structures were calculated and NOE distances were calibrated using DYANA (33). Ten lowest energy structures were selected from 20 models calculated using XPLOR-NIH (34). Of these 10 final structures, the average root mean square deviation of backbone atoms to the mean structure is 0.28 ± 0.10 Å. The NMR structures and restraint files are available from Protein Data Bank and BMRB (Biological Magnetic Resonance Data Bank) with Protein Data Bank ID 2MP9 and BMRB accession number 19973.
Interaction of Cm-p5 with a phosphatidylserine bilayer by molecular dynamics
The 3-dimensional structure of the sequence SRSELIVHQRLF (Cm-p5) was constructed with MOE (http://www.chemcomp.com/MOE-Molecular_Operating_Environment.htm) as α-helix as indicated by NMR and CD measurements. The built structure was energy optimized with AMBER12-EHT (a new version of the AMBER force field implemented in MOE). This structure was loaded in YASARA (www.yasara.org/index.html) (35, 36) to perform molecular dynamics simulations to test interactions of the small peptide with a phosphatidylserine bilayer. The corresponding bilayer was automatically created by YASARA with appropriate positioning of the peptide in the bilayer. The membrane bilayer was solvated by a water box on both sides and periodic boundary conditions were applied. The simulation cell was set 20 Å larger than the protein in each direction, and 0.9% mass percent NaCl solution corresponding to a physiologic solution and a pH of 7.0 were applied. The simulation was performed at a temperature of 298 K for 75 ns using the AMBER03 forcefield.
Isothermal titration calorimetry
Three membrane lipids—phosphatidylcholine (PC), phosphatidylserine (PS), and phosphatidylethanolamine (PE)—and other major fungal membrane components such as chitin and ergosterol were used for evaluating interaction with peptide Cm-p5. ITC was performed using the iTC200 Systems (GE Healthcare, Waukesha, WI, USA) coupled with nonreactive Hastelloy cells for chemical resistance. All pure samples were dissolved against phosphate buffer (pH 7.5). The condition and preparation of solution for ITC analysis was previously described by Mahata et al. (37). All samples, including Cm-p5, were initially dissolved in DMSO, and experiments were performed at 25°C. Isothermal interactions among peptide, Cm-p5, and molecules were measured by titrating over 20 injections using 40 µl peptide solution (1 mM) and other molecules in sample cell with a 100 µM concentration of 200 µl. Experiments were repeated 3 times.
Interaction of Cm-p5 with C. albicans cells visualized by fluorescence microscopy
C. albicans cells were grown as for the antifungal bioassays described previously. Cm-p5 was labeled with FITC basically using a previously described protocol (38) using Cm-p5 at 1 mg/ml in Na2PO3 0.1 M, pH 9.0, and FITC (Sigma-Aldrich) 1 mg/ml in DMSO. In a darkened laboratory, 100 µl FITC was added by milliliter of peptide. The mixture was maintained for 8 h at 4°C in the dark. The reaction was stopped using ammonium chloride to a final concentration of 50 mM for 2 h. The FITC-labeled peptide was purified by gel filtration using a G-10 resin (GE Healthcare). The antifungal activity of FITC-conjugated Cm-p5 was not affected. The labeled peptide (20 µg/ml) was incubated with C. albicans (105 cells/ml) in RPMI 1640 medium at 30°C. Cells were washed twice in buffered Tris (10 mM) before fluorescence microscopy. Observations were performed with an AxioPhot epifluorescence microscope equipped with an Axiocam MRc camera and complemented with capture software (AxioVision; Carl Zeiss GmbH, Jena, Germany) using an excitation filter (450–490 nm) and fluorescence emission (515 nm). Black-and-white photographs were obtained avoiding fluorescence influence and with phase contrast.
Evaluation of antifungal activity of Cm-p5 in a systemic candidiasis model in mice
Female Balb/c mice, 6–8 wk old, were housed at the Animal Facilities of the Catholic University of Brasilia, Brasilia, Brazil. Groups of 5 animals were infected intravenously (ophthalmic plexus route) with 106 CFU ml−1 of a C. albicans clinical isolate cells suspended in 0.1 ml sterile saline solution for evaluation of fungal clearance. As a negative control of infection, saline solution was used. Infected animals were provided 5, 25, and 50 mg/kg body weight of Cm-p5 peptide intraperitoneally 2, 26, and 50 h after infection. Saline solution and amphotericin B (1 mg/kg body weight) were use as negative and positive treatment controls, respectively. Mice were killed 3 d after infection, and the fungal kidney burden was assessed by plating serial dilutions of kidney homogenates onto Sabouraud agar plates. The numbers of colonies were counted after incubating the plates at 37°C for 24 h. Statistical analysis of the data was performed by GraphPad Prism software, version 6.01, 2012 (GraphPad Software, La Jolla, CA, USA), using a nonparametric statistical analysis of multiple comparison (Dunn’s posttest following a Kruskal-Wallis test). Procedures involving animals and their care were conducted in conformity with the local ethics committee and international recommendations.
RESULTS
Cm-p1-derived peptides
Cm-p1 (SRSELIVHQR) is a hydrophilic peptide that has broad antifungal activity but demonstrated no detectable effect against a panel of bacteria (24). To generate peptides with an improved antimicrobial activity, Cm-p1 was modified by the addition of 2 amino acids to the C terminus. The 2 added residues were selected to match the native peptide fragment size and to modify the cationicity, hydrophobicity, and the Boman index, which are correlated to in vitro antimicrobial activity. By use of these guidelines for our design, 3 new peptides (Cm-p3, Cm-p4, and Cm-p5) were synthesized (Table 1). The C-terminal residue of all Cm-p1-derived peptides was amidated by solid phase synthesis.
TABLE 1.
Characteristics of AMPs derived from C. muricatus
| Sequence | Name | Hydrophobicity (%) | Net charge | Boman index (kcal⋅mol−1) |
|---|---|---|---|---|
| SRSELIVHQR | Cm-p1 | 30 | +2 | 3.97 |
| SRSELIVHQRRC | Cm-p3 | 33 | +3 | 4.45 |
| SRSELIVHQRMK | Cm-p4 | 33 | +3 | 3.58 |
| SRSELIVHQRLF | Cm-p5 | 41 | +2 | 2.65 |
Bioassays against bacteria and fungi
We first tested the antibiotic activity of the 3 derivatives against Bacillus subtilis, Staphylococcus aureus, Enterococcus faecalis, Klebsiella pneumoniae, Pseudomonas aeruginosa, Proteus mirabilis, and Escherichia coli. Unfortunately, each of the peptides tested had similar antibiotic activities to the parental peptide Cm-p1 (data not shown). Because all derivative peptides yielded MICs greater than 128 µg/ml, the determination of individual MICs would likely indicate doses beyond those considered clinically useful and therefore was not pursued further.
In a parallel screening, we also tested the antifungal activity of the 3 derivatives against Candida albicans, Candida parapsilosis, Cryptococcus neoformans, Trichophyton mentagrophytes, and Trichophyton rubrum. Surprisingly, the addition of the dipeptide Leu-Phe to generate Cm-p5 resulted in an improvement in antifungal activity (Table 2). This peptide exhibited an MIC of 10 µg/ml and an EC50 of 1.146 µg/ml against C. albicans. In the same experiment, the peptide LL-37 showed an EC50 of 0.46 µg/ml. To study the fungicidal activity of Cm-p5, different samples of the bioassay were spreading onto agar plates. Unfortunately, the peptide did not show a reduction of CFU per milliliter compared to the starting inoculum or the control group. On the other hand, Cm-p3 and Cm-p4 exhibited very similar MICs as the control Cm-p1 across the panel and were slated for later analysis.
TABLE 2.
Antifungal activity of AMPs derived from C. muricatus
| Microorganism |
MIC (µg/ml) |
|||
|---|---|---|---|---|
| Cm-p1 | Cm-p3 | Cm-p4 | Cm-p5 | |
| Candida albicans (01U) | 64 | 128 | 32 | 10 |
| C. albicans (38U) | 32 | 64 | 32 | 10 |
| Candida parapsilosis | 256 | — | 256 | 32 |
| Cryptococcus neoformans (L26) | 128 | — | — | 64 |
| C. neoformans (L30) | — | — | — | 64 |
| Trichophyton mentagrophytes | — | — | — | 128 |
| Trichophyton rubrum | 32 | 128 | 32 | 10 |
MICs are provided as the most frequently observed value obtained from 5 independent experiments. A dash indicates values higher than 256 µg/ml.
Cytotoxicity assay
One of the major limitations of current antifungal drugs is extensive toxicity toward host cells at therapeutic doses. To evaluate the toxicity of Cm-p5 against mammalian cells, 2 separate concentrations of Cm-p5 at least one order of magnitude above the MIC for C. albicans and C. neoformans were incubated in the presence of the in vitro cultured mouse macrophage line RAW 264.7. Viability and proliferation of the RAW 264.7 cells were monitored using an MTT microtiter assay. Cm-p5 showed no detectable toxicity toward RAW 264.7 cells with concentrations of 400 µg/ml. As in the case of Cm-p1 (24), Cm-p5 was also nontoxic for human erythrocytes at a concentration of 400 µg/ml (data not shown).
Secondary structure elucidation of Cm-p5
The significantly enhanced antifungal activity of Cm-p5 prompted us to pursue the chemical and structural characterization of this AMP. Circular dichroic spectra of Cm-p5 were obtained in 3 conditions: H2O (100%), TFE (40%)/ H2O (60%), and TFE (100%). As shown in Fig. 1, Cm-p5 in aqueous solution exhibited a random coiled structure, whereas in the membrane mimetic environment TFE (40%)/ H2O (60%), the clear tendency is to adopt an α helical structure, as shown by the shift in wavelength to 205 nm; α-helicity is further enhanced in TFE (100%) solutions as shown by a green trace.
Figure 1.
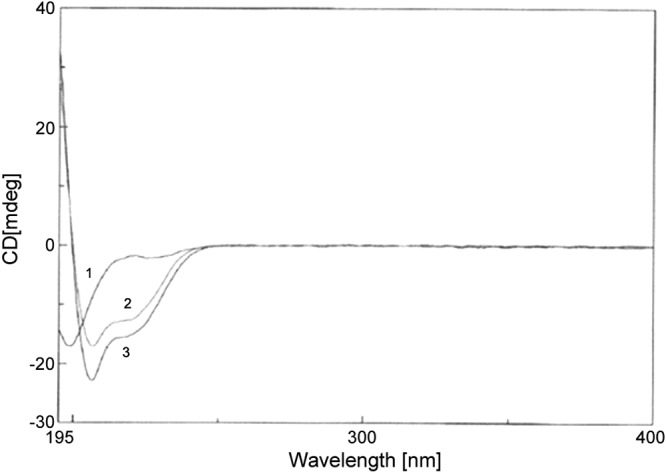
CD spectra of Cm-p5 in water (1) and in membrane mimetic environments, including TFE 40% (2) and TFE 100% (3).
NMR structure of Cm-p5
In a survey of AMP structures, AMPs widely adopt a bipartite surface composed of a cationic face and a hydrophobic face (39, 40). This partition is predicted to enable binding to the lipid bilayer and facilitate either cellular entry or membrane disruption depending on the AMP in question. Threading AMP sequences onto Schiffer-Edmundson wheel projections quickly revealed the division of hydrophobic and cationic residues into the 2 faces. To compare Cm-p5 to other AMPs currently available, the sequence of Cm-p5 was mapped onto a Schiffer-Edmundson wheel projection. Unlike the majority of AMPs currently described, hydrophobic and polar or charged residues were evenly distributed throughout the predicted helix (Fig. 2A). We determined the structure of Cm-p5 by NMR in the presence of TFE (40%)/ D2O (60%) (Supplemental Fig. 1). In this membrane mimetic solvent, Cm-p5 adopts a regular α-helix with a divergent turn at the first 3 residues of the peptide (Fig. 2B). Ser1, Arg2, Ser3, and Glu4 form the first hydrophilic ring at the N terminus. Leu5, Ile6, and Val7 form the first hydrophobic band, whereas His8, Gln9, and Arg10 generate the second hydrophilic ring. Leu11 and Phe12 cap the peptide (Fig. 2C). This finding suggested a potentially divergent mechanism for antifungal activity. However, when we generated the electrostatic map using a Poisson-Boltzman algorithm to account for appropriate dielectric constants, the overall map revealed a typical AMP with both cationic and hydrophobic faces (Fig. 3), albeit ones that did not follow the axis of the peptide.
Figure 2.

A) Schiffer-Edmundson wheel projection of Cm-p5. Hydrophilic residues are shown in white and hydrophobic residues in black. B) NOE distances involving backbone atoms indicating α-helical structure formation of Cm-p5 in 40% TFE (d2)/60% D2O. C) Ten superimposed NMR structures shown with backbones of helices rendered as ribbons.
Figure 3.

Electrostatic map of Cm-p5 as α helix. Red indicates the lowest electrostatic potential energy and blue the highest electrostatic potential energy. Intermediary colors represent intermediary electrostatic potentials.
Interaction of Cm-p5 with a phosphatidylserine bilayer by molecular dynamics
Among the phospholipids found in the plasma membrane of fungi, phosphatidylserine is one of the most abundant. To identify whether or not Cm-p5 is stable within a membrane and could potentially form pores or if it preferentially binds to the external layer of the membrane, we simulated Cm-p5 in a phosphatidylserine bilayer solvated in water at pH 7.0 with 0.9% NaCl. The initial state of the simulation in which the peptide is completely embedded in the hydrophobic portion of the membrane is shown in Fig. 4A. The Cm-p5 orientation is parallel to the hydrophobic tails of each lipid. After approximately 25 ns simulation time, the peptide is expelled from the membrane and binds at the surface (Fig. 4B). It is noteworthy that the α-helical structure of the peptide remained stable during the entire simulation time. Analyzing the molecular simulation with the lipid bilayer in detail reveals key interactions between Cm-p5 and the membrane, as shown in Fig 4C. All hydrophobic side chains along the Cm-p5 hydrophobic face embed within the lipid bilayer and engage lipid side chains. The cationic side chains Arg2 and Arg10 form multiple salt bridges with the C terminus of the serine moiety of several membrane phosphatidylserines. Ser3 and Gln9 form hydrogen bonds with the serine head group, further securing peptide binding to the membrane. In the context of the membrane, the N terminus turn at Glu4 undergoes a slight deformation by engaging in 2 stable intramolecular hydrogen bonds with the backbone amide nitrogen atoms of Arg2 and Ser3.
Figure 4.

A) Starting structure of the orientation of Cm-p5 (green helix) embedded in the hydrophobic area of the phosphatidylserine bilayer. On top and bottom, surrounding water layers are displayed. B) Interaction of the Cm-p5 (green helix) with the surface of the membrane bilayer model. C) Details of the interaction of the Cm-p5 (green helix) with head groups of phosphatidylserine.
Isothermal titration calorimetry
To confirm the direct interaction between Cm-p5 and phosphatidylserine, we performed ITC experiments. The resultant thermograms (Fig. 5) revealed that binding of Cm-p5 to all phospholipids tested progressed exothermically. The rates of binding constant were observed as PS > PE > PC > ergosterol, and chitin did not show any interaction with the peptide. The enthalpic and entropic contributions of each reaction are listed in Table 3.
Figure 5.

Data obtained from ITC binding experiment between membrane lipids and major components of fungal plasma membrane with peptide Cm-p5. Figure representing the integration of raw heat associated thermogram (top) and isotherm (bottom) of each individual set of interaction. A–E) The interaction represents Cm-p5 with PC (A), PS (B), PE (C), ergosterol (D), and chitin (E). Data are corrected by subtraction of appropriate blank experiments and then fitted with nonlinear regression; data derived after analysis are listed in Table 3.
TABLE 3.
Interaction of Cm-p5 with plasma membrane phospholipids by ITC
| Parameter | Phosphatidylcholine | Phosphatidylserine | Phosphatidylethanolamine | Ergosterol | Chitin |
|---|---|---|---|---|---|
| K | 2.34 × 102 M−1 | 4.22 × 105 M−1 | 3.61 × 105 M−1 | 3.8 × 102 M−1 | No interaction |
| ΔH | −1.732 E4 cal/mol | −7.58 E6 cal/mol | −2.01E5 cal/mol | −4.56E2 cal/mol | — |
| ΔS | −356 cal/mol/deg | −6.02E4 cal/mol/deg | −3.580E5 cal/mol/deg | −787 cal/mol/deg | — |
Data derived after fitting the raw heat–associated isotherm with nonlinear regression.
Interaction of Cm-p5 with C. albicans cells visualized by fluorescence microscopy
To demonstrate the interaction of Cm-p5 with C. albicans, fluorescence microscopy was used. The interaction of Cm-p5 with the surface of the living yeast was observed (Fig. 6). Interestingly, the interaction of Cm-p5 with C. albicans is almost exclusively to the yeast surface, both at 10 min and at 4 h incubation (Fig. 6B, D).
Figure 6.

Microscopic analyses of C. albicans cultured cells with FITC-conjugated Cm-p5. A, B) C. albicans was evaluated by phase contrast (A) and epifluorescence conditions (B) with excitation filter of 450–490 nm and fluorescence emission of 515 nm. Fungal cells were exposed to FITC-Cm-p5 at 20 µg/ml for 10 min at 30°C. C, D) Identical experimental conditions were also analyzed after 4 h.
Evaluation of antifungal activity of Cm-p5 in a systemic candidiasis model in mice
The therapeutic effect of different concentrations of Cm-p5 was evaluated in a systemic candidiasis in Balb/c mice. The peptide effectiveness was measured by the fungal kidney burden. Neither of the groups treated with Cm-p5 diminished the CFU per kidneys compared to the placebo group. In fact, the groups treated with peptide concentrations of 5 and 25 mg/kg showed a tendency to augment the colonization of kidneys by C. albicans. In contrast, treatment with amphotericin B resulted in lower levels of colonies in kidney, although no significant differences existed with respect to the placebo and the higher doses used with Cm-p5 (data not shown).
DISCUSSION
The reverse-phase chromatography fragment derived from the coastal snail C. muricatus had a molecular mass of 1485 Da (24), suggesting that it would be reasonable to add 2 amino acids to the C terminus of Cm-p1 in order to approximate the original in vivo fragment size. The goal of generating these derivatives was to increase the antimicrobial activity of Cm-p1. Keeping in mind the pharmaceutical properties and technical requirements for synthesis, we designed 3 derivatives of Cm-p1 by the addition of a dipeptide to the C terminus. These 3 sequences—Cm-p3, Cm-p4, and Cm-p5—were also designed by taking into account the cationicity, hydrophobicity, and Boman index.
The increase in cationicity of AMPs has been related with an increase in antibacterial activity due to interaction with the negatively charged phospholipid head groups of the bacterial plasma membrane. This interaction is predicted to facilitate either pore formation or disruption of the membrane by surface-level separation of phospholipids (carpet model) (41, 42). In the work presented here, Cm-p3 and Cm-p4 showed a modest increase in cationic charge, yet were largely similar in activities against the panel of bacteria described compared to the parental Cm-p1. Although bacterial activity of the derivatives did not increase compared to Cm-p1, the antifungal activity of Cm-p5 was significantly enhanced against different fungi of clinical importance. In fact, both MIC and EC50 values of Cm-p5 were equivalent to other antifungal peptides. The increase in hydrophobicity and the lower Boman index of Cm-p5 may describe the biophysical characteristics that lead to its higher antifungal activity.
The NMR structure determined here reveals that the C-terminal addition of LF to generate Cm-p5 creates a broad hydrophobic face. This hydrophobic face is extended by amidation of the terminal carboxy group and likely significantly contributes to its antifungal activity, although we did not test this hypothesis by generating a native carboxy group for this derivative. The importance of the hydrophobic motif at the C terminus is evident in the case of the peptide PAF26, where the triple alanine substitution of the C-terminal WFW motif substantially eliminated most of the antifungal activity (18). The structure–activity relationship studies of pleurocidin also demonstrate that hydrophobicity is an important factor for its antifungal activity compared to other parameters such as helicity and cationicity (43).
CD analysis confirmed the unfolded configuration of Cm-p5 in a hydrophilic environment and a tendency to adopt an α-helix as the molecule transitions to a more nonpolar environment with the addition of TFE. Our findings match previous work in the AMP field that demonstrates that many AMPs can adopt multiple conformations depending on the nature of the environment (44–46).
Structural studies of Cm-p5, conducted with an NMR analysis, showed that along the length of the α-helix, hydrophilic and hydrophobic residues alternate in succeeding bands. Electrostatic mapping of the surface demonstrated a division of hydrophobic and cationic surfaces similar to other AMPs, albeit not along the helical axis. Such a banded pattern was noted for dermaseptin S9 in the presence of 30% TFE, where the cationic termini flank a central hydrophobic core (47). The dual arginines of Cm-p5 at positions 2 and 10, in conjunction with the N terminus, are sufficient to generate a cationic shield across the intervening hydrophobic Leu5 and Ile6 residues. The opposing hydrophobic face is dominated by Val7, Leu11, and Phe12, with the charge on the C terminus dampened by the amide group. The amide terminus also plays a role in the stability of the α-helix by participating in a hydrogen bonding network with the carbonyl oxygens of both His8 and Gln9.
This hydrophobic/cationic duality is likely what enables efficient killing by AMPs through membrane disruption. We note that even with atypical distribution of amino acids, as seen here in Cm-p5, the folding of the peptide and rotamer selection by individual residues generates these same 2 distinct faces. On the other hand, the simulations also suggest a strong potential for Cm-p5 to specifically bind to fungal membranes at the surface rather than form membrane spanning pores. However, the behavior of transport through membranes, the effect of metabolic cation gradients, or several molecules of Cm-p5 working together need to be studied. We currently have no evidence that the peptide will destroy or substantially deform the plasma membrane. On the other hand, we have evidence by in vitro experiments that Cm-p5 has not shown a fungicidal behavior, so a destructive action on plasma membrane should be less probable. Another clue about the membranolytic capacity of Cm-p5 could be better explored by using the Eisenberg’s hydrophobicity plot (48). Such analysis was performed similarly to the data of Helmerhorst et al. (49) developed with the antifungal peptide histatin-5. Despite the limitation of the hydrophobicity plot, this analysis has made many successful predictions (50). The representation of Cm-p5 directly compared to other α-helical AMPs (Fig. 7) indicates that plotting of Cm-p5 and histatin-5 are located outside the surface-seeking region in the hydrophobicity plot, whereas classic pore formers are located inside this region. Furthermore, the dependency of the antifungal activity of histatin-5 to different intracellular targets have been exhaustively confirmed (51). All data discussed above suggest that Cm-p5 could be a non-pore-forming peptide.
Figure 7.

Hydrophobicity plot of 7 pore-forming and non-pore-forming AMPs. The vertical axis represents the amphipathicity (µ) of the most amphipathic 11-residue sequence of the peptide and the horizontal axis the hydrophobicity (H) of the same 11-residue sequence. Histatin-5, AKRHHGYKRKF; LL-37, FKRIVQRIKDF; buforin-2, PVGRVHRLLRK; melittin, GLPALISWIKR; chromofungin A, QNLLKELQDLA; pleurocidin, WGSFFKKAAHV; Cm-p5, RSELIVHQRLF.
ITC is a sensitive technique that provides information about the thermodynamics of the process of peptide–lipid interactions (52, 53). Because the ∆H and ∆S are both negative, complex formation is enthalpically driven by van der Waals interactions and H bonding facilitated by charged lipid head groups. Additionally, as the hydrophobicity of the lipid itself increases, the affinity for Cm-p5 also increases, as can be seen by comparing the set of binding constants. Ordering the phospholipids tested by increasing hydrophobicity also partitions fungal membrane components from those found in mammalian cells, highlighting a trend that may represent a rough mechanism for targeting pathogen membranes. The rates of binding constant are PS ≥ PE > PC > ergosterol in favor of fungal components of plasma membrane compared to a phospholipid from mammalian plasma membranes. Indeed, the specificity showed by Cm-p5 is in accordance with its condition to be nontoxic for mammalian cells. It is noteworthy that the antibacterial peptide dermaseptin S9 (similar nonamphipatic pattern regarding Cm-p5) inserts less efficiently in phosphatidylglycerol than in phosphatidylcholine monolayers (54). This behavior is in contrast with the specificity showed by Cm-p5. Moreover, the interaction of AMPs with specific phospholipids has been widely demonstrated. In this sense, the antifungal plant defensin NaD1 interacts specifically with phospholipid phosphatidylinositol 4,5-bisphosphate, disrupting the fungal cell membrane (55). The ability of Cm-p5 to interact with the C. albicans cell surface could be driven by their affinity by the phospholipids above mentioned. Otherwise, Cm-p5 interaction with other molecules such as proteoglycans and/or sphingolipids could not be discarded. In fact, the interaction of different plant defensins with the fungal glycosphingolipid glucosylceramide has been demonstrated (56). It is noteworthy that at least during the first 4 h of interaction between Cm-p5 and C. albicans, the peptide remains almost exclusively at the cellular surface. Apparently Cm-p5 does not enter the cell. Indeed, antifungal mechanisms that inhibit the cell wall biosynthesis through the binding to lipid II (57) or the inhibition of the β-(1,3) glucan synthase (58) have been described. Nevertheless, it cannot be excluded the possibility that Cm-p5 could enter the cell following slow kinetics. For example, the uptake of the antifungal peptide MMGP1 to C. albicans occurs in this way. The uptake of MMGP1 to the yeast after 2 h incubation was only 1.5% but after 6 h reached 99% (59).
Finally, to demonstrate the therapeutic effects of Cm-p5, we used a model of systemic candidiasis in Balb/c mice that, in general, has been previously described (60–63). None of the concentrations assayed of Cm-p5 significantly reduced the CFU per kidney in this model. Nevertheless, a similar study conducted with the antifungal peptide N10K did not show a reduction of CFU per milliliter compared to the placebo at day 7 after infection, but significant differences were observed at day 14 (62). In this case, the treatment started 1 h after the infection. On the other hand, the antifungal peptide RsAFP2 showed a significant reduction in the CFU per gram at a concentration of 14 mg/kg in a similar murine model of candidiasis, although the peptide was administrated 1 h after infection (61). Starting therapy soon after infection simulates preventative therapy. The longer the time between infection and treatment, the greater the fungal growth in animals and the more difficult the therapy. A 2 h delay between infection and therapy allows the yeast to enter the log phase of growth (64). On the other hand, factors like the possible instability of Cm-p5, biodistribution, and fungicidal activity need to be improved to augment their therapeutic potential. In this sense, the development of nanoparticles for entrapment and delivery of AMPs could represent an alternative (65, 66).
In summary, it is possible to conclude that Cm-p5, a short peptide derived from the sea snail C. muricatus, exhibits an antifungal activity against C. albicans and other clinically important fungi. Importantly, this peptide is nontoxic for human cells. On the other hand, the low amphipathicity of the peptide could be modified to generate new derivatives with improved therapeutic potential.
Supplementary Material
Acknowledgments
The authors thank CNPq, FAPDF, FUNDECT, CAPES-MES, Brazil; International Foundation of Science (IFS), Sweden (F 4614 and F/5199 projects); and David Rockefeller Center for Latin American Studies (DRCLAS), Harvard University. It was also relevant the support of the Organization for the Prohibition of Chemical Weapons (OPCW), The Netherlands. Furthermore, they also appreciate the efforts of Dr. A. Porcel from the Leibniz Institute of Plant Biochemistry, Department of Bio-organic Chemistry, Halle (Saale), Germany, for her assistance with CD experiments. They also extend their sincerest thanks and appreciation to Dr. J. David, Richard Pearson Strong Professor of Tropical Public Health, Emeritus, Harvard School of Public Health, for his critical review and advice.
Glossary
- AMP
antimicrobial peptide
- CD
circular dichroism
- CFU
colony-forming unit
- ITC
isothermal titration calorimetry
- MIC
minimal inhibitory concentration
- MTT
(3-[4,5-dimethylthlazol-2-yl]-2,5-diphenyltetrazolium bromide
- NOE
nuclear Overhauser effect
- NOESY
NOE spectroscopy
- PC
phosphatidy‐lcholine
- PE
phosphatidylethanolamine
- PS
phosphatidylserine
- TFE
trifluoroethanol
- TOCSY
total correlation spectroscopy
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
REFERENCES
- 1.Drew R. H., Townsend M. L., Pound M. W., Johnson S. W., Perfect J. R. (2013) Recent advances in the treatment of life-threatening, invasive fungal infections. Expert Opin. Pharmacother. 14, 2361–2374 [DOI] [PubMed] [Google Scholar]
- 2.Arnold T. M., Dotson E., Sarosi G. A., Hage C. A. (2010) Traditional and emerging antifungal therapies. Proc. Am. Thorac. Soc. 7, 222–228 [DOI] [PubMed] [Google Scholar]
- 3.Pemán J., Cantón E., Espinel-Ingroff A. (2009) Antifungal drug resistance mechanisms. Expert Rev. Anti Infect. Ther. 7, 453–460 [DOI] [PubMed] [Google Scholar]
- 4.Matejuk A., Leng Q., Begum M. D., Woodle M. C., Scaria P., Chou S. T., Mixson A. J. (2010) Peptide-based antifungal therapies against emerging infections. Drugs Future 35, 197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jenssen H., Hamill P., Hancock R. E. (2006) Peptide antimicrobial agents. Clin. Microbiol. Rev. 19, 491–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aoki W., Ueda M. (2013) Characterization of antimicrobial peptides toward the development of novel antibiotics. Pharmaceuticals (Basel) 6, 1055–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Phoenix, D. A., Dennison, S. R., Harris, F. (2013) Antimicrobial Peptides. Wiley-VCH, Weinheim, Germany [Google Scholar]
- 8.Aerts A. M., François I. E., Cammue B. P., Thevissen K. (2008) The mode of antifungal action of plant, insect and human defensins. Cell. Mol. Life Sci. 65, 2069–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Belguesmia Y., Choiset Y., Rabesona H., Baudy-Floc’h M., Le Blay G., Haertlé T., Chobert J. M. (2013) Antifungal properties of durancins isolated from Enterococcus durans A5-11 and of its synthetic fragments. Lett. Appl. Microbiol. 56, 237–244 [DOI] [PubMed] [Google Scholar]
- 10.Stoumieux-Garzon D., Saulnier D., Garnier J., Jouffrey C., Bulet P., Bachere E. (2001) Crustacean immunity. Antifungal peptides are generated from the C terminus of shrimp hemocyanin in response to microbial challenge. J. Biol. Chem. 276, 47070–47077 [DOI] [PubMed] [Google Scholar]
- 11.Pál T., Abraham B., Sonnevend A., Jumaa P., Conlon J. M. (2006) Brevinin-1BYa: a naturally occurring peptide from frog skin with broad-spectrum antibacterial and antifungal properties. Int. J. Antimicrob. Agents 27, 525–529 [DOI] [PubMed] [Google Scholar]
- 12.Brogden K. A. (2005) Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 3, 238–250 [DOI] [PubMed] [Google Scholar]
- 13.van der Weerden N. L., Bleackley M. R., Anderson M. A. (2013) Properties and mechanisms of action of naturally occurring antifungal peptides. Cell. Mol. Life Sci. 70, 3545–3570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Steinstraesser L., Kraneburg U., Jacobsen F., Al-Benna S. (2011) Host defense peptides and their antimicrobial–immunomodulatory duality. Immunobiology 216, 322–333 [DOI] [PubMed] [Google Scholar]
- 15.Yeung A. T., Gellatly S. L., Hancock R. E. (2011) Multifunctional cationic host defence peptides and their clinical applications. Cell. Mol. Life Sci. 68, 2161–2176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Su Y., Li S., Hong M. (2013) Cationic membrane peptides: atomic-level insight of structure–activity relationships from solid-state NMR. Amino Acids 44, 821–833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cho J., Choi H., Lee D. G. (2012) Influence of the N- and C-terminal regions of antimicrobial peptide pleurocidin on antibacterial activity. J. Microbiol. Biotechnol. 22, 1367–1374 [DOI] [PubMed] [Google Scholar]
- 18.Muñoz A., Harries E., Contreras-Valenzuela A., Carmona L., Read N. D., Marcos J. F. (2013) Two functional motifs define the interaction, internalization and toxicity of the cell-penetrating antifungal peptide PAF26 on fungal cells. PLoS ONE 8, e54813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang G., Li X., Wang Z. (2009) APD2: the updated antimicrobial peptide database and its application in peptide design. Nucleic Acids Res. 37, D933–D937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Waghu F. H., Gopi L., Barai R. S., Ramteke P., Nizami B., Idicula-Thomas S. (2014) CAMP: collection of sequences and structures of antimicrobial peptides. Nucleic Acids Res. 42, D1154–D1158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao X., Wu H., Lu H., Li G., Huang Q. (2013) LAMP: a database linking antimicrobial peptides. PLoS ONE 8, e66557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seshadri Sundararajan V., Gabere M. N., Pretorius A., Adam S., Christoffels A., Lehväslaiho M., Archer J. A., Bajic V. B. (2012) DAMPD: a manually curated antimicrobial peptide database. Nucleic Acids Res. 40, D1108–D1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boman H. G. (2003) Antibacterial peptides: basic facts and emerging concepts. J. Intern. Med. 254, 197–215 [DOI] [PubMed] [Google Scholar]
- 24.López-Abarrategui C., Alba A., Silva O. N., Reyes-Acosta O., Vasconcelos I. M., Oliveira J. T., Migliolo L., Costa M. P., Costa C. R., Silva M. R., Garay H. E., Dias S. C., Franco O. L., Otero-González A. J. (2012) Functional characterization of a synthetic hydrophilic antifungal peptide derived from the marine snail Cenchritis muricatus. Biochimie 94, 968–974 [DOI] [PubMed] [Google Scholar]
- 25.Ratledge C., Wilkinson S. G. (1988) Microbial Lipids, Vol. 1 Academic Press, London [Google Scholar]
- 26.Merrifield B. (1986) Solid phase synthesis. Science 232, 341–347 [DOI] [PubMed] [Google Scholar]
- 27.Murphy J., Kies M. (1960) Note on the spectrophotometric determination of proteins in dilute solutions. Biochim. Biophys. Acta 45, 382–384 [Google Scholar]
- 28.Hetru C., Bulet P. (1997) Strategies for the isolation and characterization of antimicrobial peptides of invertebrates. Methods Mol. Biol. 78, 35–49 [DOI] [PubMed] [Google Scholar]
- 29.CLSI (2008) Reference method for broth dilution antifungal susceptibility testing of yeasts. In Approved Standard, 3rd EdCLSI document M27-A3, pages 3–12, Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 30.Pasupuleti M., Schmidtchen A., Chalupka A., Ringstad L., Malmsten M. (2009) End-tagging of ultra-short antimicrobial peptides by W/F stretches to facilitate bacterial killing. PLoS ONE 4, e5285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 32.Keller R. L. (2004) Computer Aided Resonance Assignment Tutorial. Cantina Verlag, Goldau, Switzerland [Google Scholar]
- 33.Güntert P., Mumenthaler C., Wüthrich K. (1997) Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol. 273, 283–298 [DOI] [PubMed] [Google Scholar]
- 34.Schwieters C. D., Kuszewski J. J., Tjandra N., Clore G. M. (2003) The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 160, 65–73 [DOI] [PubMed] [Google Scholar]
- 35.Krieger E., Darden T., Nabuurs S. B., Finkelstein A., Vriend G. (2004) Making optimal use of empirical energy functions: force-field parameterization in crystal space. Proteins 57, 678–683 [DOI] [PubMed] [Google Scholar]
- 36.Krieger E., Joo K., Lee J., Lee J., Raman S., Thompson J., Tyka M., Baker D., Karplus K. (2009) Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: four approaches that performed well in CASP8. Proteins 77(Suppl 9), 114–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mahata D., Mandal S. M., Bharti R., Gupta V. K., Mandal M., Nag A., Nando G. B. (2014) Self-assembled cardanol azo derivatives as antifungal agent with chitin-binding ability. Int. J. Biol. Macromol. 69, 5–11 [DOI] [PubMed] [Google Scholar]
- 38.Hermanson G. T. (2013) Bioconjugate Techniques. Academic Press, San Diego, USA [Google Scholar]
- 39.Bhattacharjya S., Ramamoorthy A. (2009) Multifunctional host defense peptides: functional and mechanistic insights from NMR structures of potent antimicrobial peptides. FEBS J. 276, 6465–6473 [DOI] [PubMed] [Google Scholar]
- 40.Haney E. F., Hunter H. N., Matsuzaki K., Vogel H. J. (2009) Solution NMR studies of amphibian antimicrobial peptides: linking structure to function? Biochim. Biophys. Acta 1788, 1639–1655 [DOI] [PubMed] [Google Scholar]
- 41.Jiang Z., Kullberg B. J., van der Lee H., Vasil A. I., Hale J. D., Mant C. T., Hancock R. E., Vasil M. L., Netea M. G., Hodges R. S. (2008) Effects of hydrophobicity on the antifungal activity of alpha-helical antimicrobial peptides. Chem. Biol. Drug Des. 72, 483–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Powers J. P., Hancock R. E. (2003) The relationship between peptide structure and antibacterial activity. Peptides 24, 1681–1691 [DOI] [PubMed] [Google Scholar]
- 43.Lee J., Lee D. G. (2010) Influence of the hydrophobic amino acids in the N- and C-terminal regions of pleurocidin on antifungal activity. J. Microbiol. Biotechnol. 20, 1192–1195 [DOI] [PubMed] [Google Scholar]
- 44.Mahalka A. K., Kinnunen P. K. (2009) Binding of amphipathic alpha-helical antimicrobial peptides to lipid membranes: lessons from temporins B and L. Biochim. Biophys. Acta 1788, 1600–1609 [DOI] [PubMed] [Google Scholar]
- 45.Sato H., Feix J. B. (2006) Peptide-membrane interactions and mechanisms of membrane destruction by amphipathic alpha-helical antimicrobial peptides. Biochim. Biophys. Acta 1758, 1245–1256 [DOI] [PubMed] [Google Scholar]
- 46.Lequin O., Bruston F., Convert O., Chassaing G., Nicolas P. (2003) Helical structure of dermaseptin B2 in a membrane-mimetic environment. Biochemistry 42, 10311–10323 [DOI] [PubMed] [Google Scholar]
- 47.Lequin O., Ladram A., Chabbert L., Bruston F., Convert O., Vanhoye D., Chassaing G., Nicolas P., Amiche M. (2006) Dermaseptin S9, an alpha-helical antimicrobial peptide with a hydrophobic core and cationic termini. Biochemistry 45, 468–480 [DOI] [PubMed] [Google Scholar]
- 48.Eisenberg D. (1984) Three-dimensional structure of membrane and surface proteins. Annu. Rev. Biochem. 53, 595–623 [DOI] [PubMed] [Google Scholar]
- 49.Helmerhorst E. J., van’t Hof W., Breeuwer P., Veerman E. C., Abee T., Troxler R. F., Amerongen A. V., Oppenheim F. G. (2001) Characterization of histatin 5 with respect to amphipathicity, hydrophobicity, and effects on cell and mitochondrial membrane integrity excludes a candidacidal mechanism of pore formation. J. Biol. Chem. 276, 5643–5649 [DOI] [PubMed] [Google Scholar]
- 50.Phoenix D. A., Harris F. (2002) The hydrophobic moment and its use in the classification of amphiphilic structures (review). Mol. Membr. Biol. 19, 1–10 [DOI] [PubMed] [Google Scholar]
- 51.Puri S., Edgerton M. (2014) How does it kill? Understanding the candidacidal mechanism of salivary histatin 5. Eukaryot. Cell 13, 958–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sikorska E., Dawgul M., Greber K., Iłowska E., Pogorzelska A., Kamysz W. (2014) Self-assembly and interactions of short antimicrobial cationic lipopeptides with membrane lipids: ITC, FTIR and molecular dynamics studies. Biochim. Biophys. Acta 1838, 2625–2634 [DOI] [PubMed] [Google Scholar]
- 53.Domingues T. M., Mattei B., Seelig J., Perez K. R., Miranda A., Riske K. A. (2013) Interaction of the antimicrobial peptide gomesin with model membranes: a calorimetric study. Langmuir 29, 8609–8618 [DOI] [PubMed] [Google Scholar]
- 54.Caillon L., Killian J. A., Lequin O., Khemtémourian L. (2013) Biophysical investigation of the membrane-disrupting mechanism of the antimicrobial and amyloid-like peptide dermaseptin S9. PLoS ONE 8, e75528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Poon I. Kh., Baxter A. A., Lay F. T., Mills G. D., Adda C. G., Payne J. A., Phan T. K., Ryan G. F., White J. A., Veneer P. K., van der Weerden N. L., Anderson M. A., Kvansakul M., Hulett M. D. (2014) Phosphoinositide-mediated oligomerization of a defensin induces cell lysis. eLife 3, e01808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vriens K., Cammue B. P., Thevissen K. (2014) Antifungal plant defensins: mechanisms of action and production. Molecules 19, 12280–12303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schneider T., Kruse T., Wimmer R., Wiedemann I., Sass V., Pag U., Jansen A., Nielsen A. K., Mygind P. H., Raventós D. S., Neve S., Ravn B., Bonvin A. M., De Maria L., Andersen A. S., Gammelgaard L. K., Sahl H. G., Kristensen H. H. (2010) Plectasin, a fungal defensin, targets the bacterial cell wall precursor lipid II. Science 328, 1168–1172 [DOI] [PubMed] [Google Scholar]
- 58.Martins I. M., Cortés J. C., Muñoz J., Moreno M. B., Ramos M., Clemente-Ramos J. A., Durán A., Ribas J. C. (2011) Differential activities of three families of specific beta(1,3)glucan synthase inhibitors in wild-type and resistant strains of fission yeast. J. Biol. Chem. 286, 3484–3496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pushpanathan M., Rajendhran J., Jayashree S., Sundarakrishnan B., Jayachandran S., Gunasekaran P. (2012) Direct cell penetration of the antifungal peptide, MMGP1, in Candida albicans. J. Pept. Sci. 18, 657–660 [DOI] [PubMed] [Google Scholar]
- 60.Ribeiro P. D., Medina-Acosta E. (2003) Prevention of lethal murine candidiasis using HP (2-20), an antimicrobial peptide derived from the N-terminus of Helicobacter pylori ribosomal protein L1. Peptides 24, 1807–1814 [DOI] [PubMed] [Google Scholar]
- 61.Tavares P. M., Thevissen K., Cammue B. P., François I. E., Barreto-Bergter E., Taborda C. P., Marques A. F., Rodrigues M. L., Nimrichter L. (2008) In vitro activity of the antifungal plant defensin RsAFP2 against Candida isolates and its in vivo efficacy in prophylactic murine models of candidiasis. Antimicrob. Agents Chemother. 52, 4522–4525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Polonelli L., Ciociola T., Magliani W., Zanello P. P., D’Adda T., Galati S., De Bernardis F., Arancia S., Gabrielli E., Pericolini E., Vecchiarelli A., Arruda D. C., Pinto M. R., Travassos L. R., Pertinhez T. A., Spisni A., Conti S. (2012) Peptides of the constant region of antibodies display fungicidal activity. PLoS ONE 7, e34105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lupetti A., Brouwer C. P., Bogaards S. J., Welling M. M., de Heer E., Campa M., van Dissel J. T., Friesen R. H., Nibbering P. H. (2007) Human lactoferrin-derived peptide’s antifungal activities against disseminated Candida albicans infection. J. Infect. Dis. 196, 1416–1424 [DOI] [PubMed] [Google Scholar]
- 64.Andes D. (2005) Use of an animal model of disseminated candidiasis in the evaluation of antifungal therapy. Methods Mol. Med. 118, 111–128 [DOI] [PubMed] [Google Scholar]
- 65.Brandelli A. (2012) Nanostructures as promising tools for delivery of antimicrobial peptides. Mini Rev. Med. Chem. 12, 731–741 [DOI] [PubMed] [Google Scholar]
- 66.López-Abarrategui C., Figueroa-Espí V., Reyes-Acosta O., Reguera E., Otero-Gonzalez A. J. (2013) Magnetic nanoparticles: new players in antimicrobial peptide therapeutics. Curr. Protein Pept. Sci. 14, 595–606 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.