

Abstract
Centrioles are ancient organelles that build centrosomes, the major microtubule-organizing centers of animal cells. Extra centrosomes are a common feature of cancer cells. To investigate the importance of centrosomes in the proliferation of normal and cancer cells, we developed centrinone, a reversible inhibitor of Polo-like kinase 4 (Plk4), a serine-threonine protein kinase that initiates centriole assembly. Centrinone treatment caused centrosome depletion in human and other vertebrate cells. Centrosome loss irreversibly arrested normal cells in a senescence-like G1 state by a p53-dependent mechanism that was independent of DNA damage, stress, Hippo signaling, extended mitotic duration, or segregation errors. In contrast, cancer cell lines with normal or amplified centrosome numbers could proliferate indefinitely after centrosome loss. Upon centrinone washout, each cancer cell line returned to an intrinsic centrosome number “set point.” Thus, cells with cancer-associated mutations fundamentally differ from normal cells in their response to centrosome loss.
Centrioles template assembly of cilia and recruit pericentriolar material to form centrosomes (1, 2). Centriole duplication is tightly controlled, so that mitotic cells have precisely two centrosomes (3, 4). Supernumerary centrosomes are prevalent in cancer and have been postulated to contribute to tumorigenesis (5–7), perhaps by promoting chromosomal instability (8, 9) or increasing cellular invasiveness (10). However, whether cancer cells become de- pendent upon extra centrosomes for proliferation is unknown.
Centriole assembly is controlled by the serine- threonine protein kinase Polo-like kinase 4 (Plk4) (11–15). Of all the compounds previously reported to bind Plk4, only CFI-400945 and related analogs exhibit any in vitro Plk4 selectivity (16–20), and none prevent centrosome assembly in cells. CFI-400945 also induces centrosome amplification and phenotypes associated with Aurora B inhibition (fig. S1) (18). Therefore, to develop a selective Plk4 inhibitor with in vivo efficacy, we chose the pan-Aurora kinase inhibitor VX-680, which also inhibits Plk4 (16, 17, 20), as a template (fig. S2, A and B). Guided by modeling, we introduced a methoxy substituent at the VX-680 C5 position (magenta shading in Fig. 1A) to target the rare hinge-region methionine in Plk4 (Met91) (fig. S2B) and generated a compound with ∼15-fold in vitro preference for Plk4 over Aurora A. Out of an additional 390 analogs synthesized and characterized, 133 (34%) had half-maximal inhibitory concentration (IC50) values ≤100 nM for Plk4 in vitro, but only one, LCR-015 (in which the VX-680 cyclopropylamide was replaced with a benzyl sulfone) (orange shading in Fig. 1A), depleted centrosomes in NIH/3T3 mouse embryonic fibroblasts and HCT-116 human colon carcinoma cells at concentrations <10 mM (fig. S2A). Optimization of LCR-015 produced two highly selective Plk4 inhibitors with robust cellular activity: centrinone [LCR-263; inhibition constant (Ki) = 0.16 nM in vitro; centrosome depletion at 100 nM] and centrinone-B (LCR-323; Ki = 0.6 nM in vitro; centrosome depletion at 500 nM) (Fig. 1A). A 2.65 Å centrinone-Plk4 kinase domain cocrystal structure (Fig. 1, B and C, and table S1) revealed that the benzyl sulfone moiety required for cellular activity (orange in Fig. 1, A and C) wraps around the catalytic lysine (Lys41) and forms hydrophobic contacts with Asp154 of the Asp-Phe- Gly (DFG) motif (Fig. 1C and fig. S2C), which should disfavor transition to the active state. Both centrinones exhibited >1000-fold selectivity for Plk4 over Aurora A/B (Fig. 1A and table S2) in vitro and did not affect cellular Aurora A or B substrate phosphorylation at concentrations that deplete centrosomes (fig. S2D). In vitro screening against 442 human kinases (16) at ∼500 × Ki and subsequent dose-response analysis indicated high selectivity (tables S3 and S4), particularly against mitotic kinases. Although we report data obtained with centrinone, key results were replicated with centrinone-B.
Fig. 1.
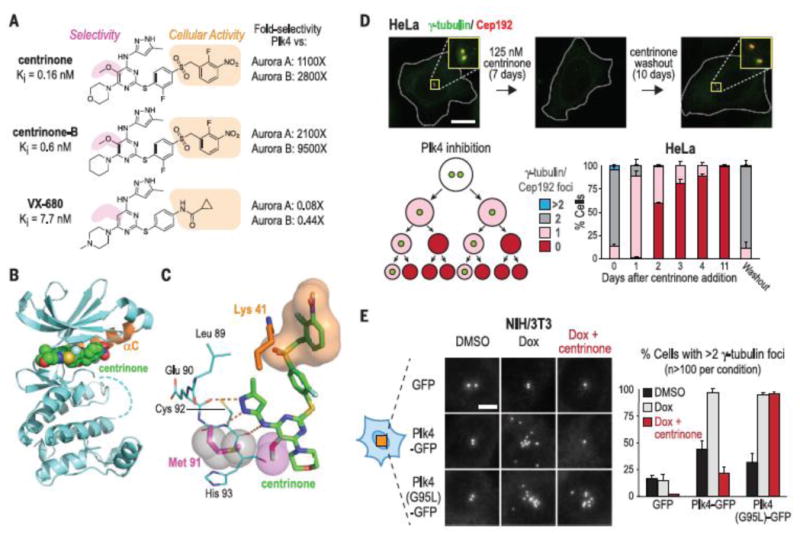
Centrinone is a selective Plk4 inhibitor that reversibly depletes centrioles from cells. (A) Chemical structures, Ki values, and selectivities [Plk4 versus Aurora A/B; Ki (kinase)/Ki (Plk4)] of the centrinones and VX-680 (B) Crystal structure of the centrinone-Plk4 kinase domain complex (orange αC helix). (C) Close-up of centrinone in the Plk4 active site. The aminopyrazole moiety of centrinone hydrogen bonds (orange dashes) with the main chain carbonyl of Glu90 and the carbonyl and amide nitrogen of Cys92. The 5-methoxy substituent (magenta spheres) packs against the Met91 side chain (magenta stick and gray spheres). The benzyl sulfone moiety (orange surface) wraps around Lys41 (orange). (D) HeLa cells 7 days after centrinone addition and 10 days after centrinone washout. (Insets: 3.3× magnified). Scale bar, 10 mm. Schematic shows progressive centrosome depletion after Plk4 inhibition. Bar graph shows the centrosome number distribution after centrinone addition and washout. (E) g-Tubulin foci in NIH/3T3 cells induced to overexpress wild-type or centrinone-resistant (G95L) Plk4-GFP. Scale bar, 5 mm. Data in (D) and (E) are means T SD; number of experiments (N) = 3.
Plk4 inhibition prevents new centriole assembly without disassembling preexisting centrioles (11, 12, 14). Consistent with this, centrinone treatment of HeLa human cervical carcinoma cells led to a progressive reduction in foci containing centriolar and pericentriolar material markers at each round of cell division, until most cells lacked centrioles and centrosomes (Fig. 1D and fig. S2E). Centriole loss prevented formation of primary cilia and resulted in the absence of focal microtubule organization during recovery from nocodazole treatment (fig. S3, A and B). Golgi organization was unaffected (fig. S3C), consistent with its ability to nucleate microtubules independently of centrosomes (21). Centriole loss was fully reversible; 10 days after centrinone washout, all cells exhibited normal centrosome numbers (Fig. 1D). Treatment with centrinone reduced centriole number in multiciliated Xenopus epithelial cells, which indicated that Plk4 also controls centriole amplification in differentiated cells (fig. S4). To confirm that these effects were due to Plk4 inhibition, we generated a Plk4 mutant [in which Gly95 is replaced by Leu (G95L)] with wild-type biochemical activity that sterically hindered centrinone binding [Ki (mutant)/Ki (wild type) > 400] (table S2 and fig. S2C). Treatment with cen- trinone blocked centriole amplification in cells over- expressing wild-type but not G95L Plk4 (Fig. 1E), which confirmed that centrinone prevents centriole assembly by inhibiting Plk4.
For the first 2 days after centrinone addition, when cells retained two or one centrosomes, the proliferation of HeLa and NIH/3T3 cells was identical to controls; this was followed by a de- crease in proliferation rate coincident with the appearance of centrosome-less cells (Fig. 2A, Fig. 1D, and fig. S5). Cells treated long-term continued to proliferate at the slower rate and returned to the control rate after washout-mediated centrosome recovery (Fig. 2B). Measurement by single-cell imaging in cells coexpressing green fluorescent protein–proliferating cell nuclear antigen (GFP- PCNA) and histone 2B–red fluorescent protein (H2B-RFP) revealed that G1+S and G2 durations were not substantially different in centrosome- less cells compared with controls (Fig. 2D and fig. S6). Imaging of cells coexpressing centrin-GFP and H2B-RFP revealed that mitotic duration was increased by ∼20 min in centrosome-less NIH/ 3T3 cells and by ∼1 hour in HeLa cells (fig. S7). Consistent with prior work (22, 23), centrosome loss increased the frequency of mitotic errors (Fig. 2E and fig. S7), which resulted in cell death (Fig. 2F and fig. S6C) that quantitatively explained the reduced proliferation after centro- some removal (fig. S6D). Centrosome-less NIH/ 3T3 and HeLa cells arrested in response to DNA damage and also retained the ability to bypass this arrest when treated with caffeine (fig. S8).
Fig. 2.
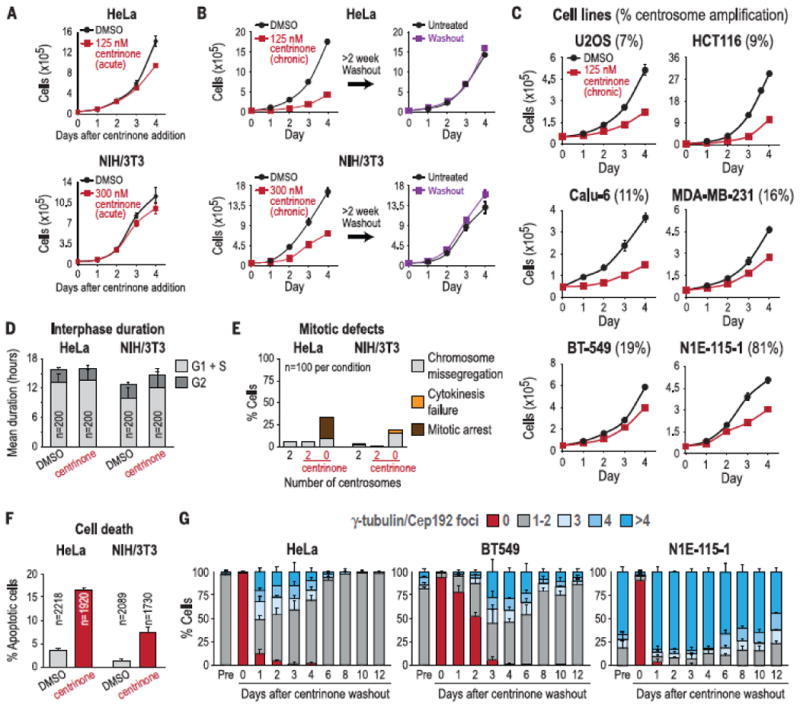
Transformed cells proliferate indefinitely in the absence of centrosomes. (A) Proliferation curves of HeLa and NIH/3T3 cells immediately after addition of centrinone or DMSO (control). (B) Proliferation curves after chronic (>2 weeks) centrinone treatment (left), or after chronic centrinone treatment followed by drug washout for >2 weeks (right). (C) Proliferation curves after chronic (>2 weeks) centrinone treatment in cell lines with varying degrees of centrosome amplification. Numbers in parentheses are percentages of cells exhibiting centrosome amplification in untreated population. Data in (A) to (C) are means T SEM (N = 3). (D) G1+S and G2 durations measured in HeLa and NIH/3T3 cells coexpressing GFP-PCNA and H2B-RFP (see fig. S6). Data are means T SD. (E) Percentage of cells exhibiting mitotic defects measured in HeLa and NIH/3T3 cells coexpressing centrin-GFP and H2B-RFP (see fig. S7). (F) Percentage of cells undergoing cell death in HeLa and NIH/3T3 cells, measured by using a fluorescent caspase substrate. Data are means T SD (N = 2). (G) Graphs showing centrosome number distribution over time after centrinone washout from HeLa, BT-549, and N1E-115-1 cells treated long-term (>2 weeks). The centrosome number distribution in untreated cells (“Pre” bars) is also shown for each cell line. Data are means T SD (N = 3).
To determine whether centriole depletion is preferentially deleterious to cell lines with supernumerary centrosomes, we analyzed the basal level of centrosome amplification across a panel of 21 cell lines that continued to proliferate after centrinone treatment (table S5). Nine lines span- ning a range of amplification levels (HeLa, 4%; NIH/3T3, 6%; U2OS, 7%; HCT116, 9%; Calu-6, 11%; MDA-MB-231, 16%; BT-549, 19%; and N1E-115-1, 81%) were depleted of centrosomes, and their proliferation rate was compared with dimethyl sulfoxide (DMSO)–treated controls (Fig. 2, A and C). We observed no correlation between basal centrosome amplification state and proliferation after centrosome depletion, which indicated that cells with multiple centrosomes are not addicted to them.
To study the origins of centrosome amplification within cancer cell lines, we depleted centrosomes from three cell lines that normally exhibit low (HeLa, 4%); medium (BT-549, 19%); or high (N1E-115-1, 81%) amplification (Fig. 2G). We then washed out centrinone and counted centrosomes at regular intervals. In all three lines, centrinone washout triggered an initial wave of centrosome overduplication (Fig. 2G and fig. S9, A and C), owing to the lack of copy number control during de novo assembly and elevated Plk4 levels that resulted from inhibition of autophosphorylation-mediated degradation (figs. S1C and S9D). This wave of overduplication was followed by a gradual return to a centrosome number distribution similar to that before depletion (Fig. 2G). Live imaging of HeLa cells revealed that recovery of the original distribution occurred by removal of cells with supernumerary centrosomes via multipolar mitoses with death of the resulting progeny (fig. S9B). Thus, each cancer cell line has an intrinsic centrosome number distribution, or “set point,” that is independent of preexisting centrosomes and reflects a dynamic equilibrium between ongoing overduplication and selection against cells with extra centrosomes.
To determine whether centrosomes are required for the proliferation of normal human cells, we analyzed the effect of centrosome depletion in three cell lines and three primary cell cultures. Prior work with RPE1 human retinal pigment epithelial cells showed that transient centrosome removal did not block passage through the sub- sequent G1-S transition (24), but the effect of multigenerational centrosome removal could not be analyzed because S-phase entry triggered de novo centriole assembly. Using centrinone to persistently block centriole assembly in RPE1 cells, we found that centrosome loss coincided with a plateau in cell number (Fig. 3A). A 12-day passaging assay and flow cytometry showed that centrosome depletion led to cell cycle arrest in G1 (Fig. 3E and fig. S10A); an identical arrest was observed after centrosome depletion in three primary cell cultures and two other lines lacking cancer- associated mutations (table S6 and fig. S10, B and D). Centrinone treatment did not lead to centrosome loss or proliferation arrest in RPE1 cells where both endogenous PLK4 alleles were engineered to express the centrinone-resistant G95L mutant (Fig. 3B and fig. S11), which indicated that the arrest is triggered by centrosome loss due to Plk4 inhibition.
Fig. 3.
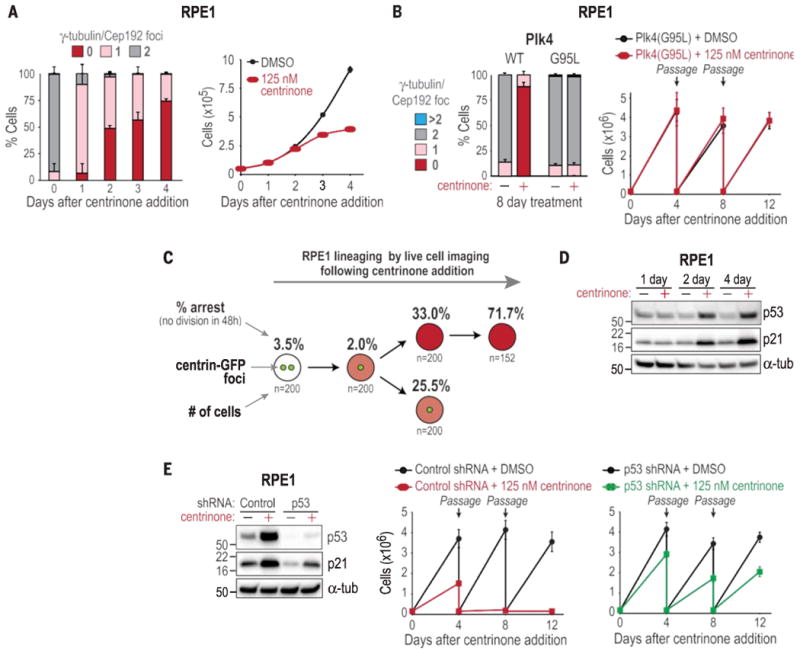
Centrosome loss triggers a p53-dependent arrest in normal cells. (A) Centro- some number distribution (left; data are means T SD; N = 3) and proliferation (right, data are means T SEM; N = 3) of RPE1 cells after centrinone addition. (B) (left) Centro- some number distribution in RPE1 cells expressing WT or centrinone-resistant G95L (homozygous knock-in at the endogenous locus) Plk4. (Right) Passaging assay on RPE1 Plk4 G95L cells. Data are means T SD (N = 2). (C) Lineage analysis showing the percentage of RPE1 cells with the indicated number of centrosomes arresting in each generation after centrinone addition (N = 2). Cells coexpressing centrin-GFP and H2B-RFP were initially filmed in both GFP and RFP channels to count centrosomes and monitor mother cell mitosis. Daughter cell fate was subsequently tracked by using RFP only. Arrest was the inability to enter mitosis within 48 hours of cell birth. (D) Western blot of p53 and p21 in RPE1 cells. a-tub, a-tubulin. (E) Western blot of RPE1 cells expressing control or p53 shRNA, and passaging assay of RPE1 cells expressing control or p53 shRNA after centrinone addition. Data are means T SD (N = 2).
The potent G1 arrest in the absence of centrosomes was in contrast to the normal progression through G1-S observed after transient centrosome removal (24). To address this difference, we used live-cell imaging to establish the lineage of RPE1 cells coexpressing centrin-GFP and H2B- RFP after acute centrinone treatment (Fig. 3C). Pioneer one-centrosome mothers divided at normal frequency, but a significant fraction of their one- and zero-centrosome progeny arrested (25.5% and 33.0%, respectively). The majority (70%) of the progeny of pioneer zero-centrosome mothers arrested. That one-centrosome progeny of one- centrosome mothers arrested indicates that cells detect loss of even a single centrosome. Thus, penetrant G1 arrest requires one to two cell cycles after centrosome removal, which explains why it was not observed after transient centrosome ablation. We speculate that progressive arrest, rather than an immediate block when one or both centrosomes are absent, allows for rescue by the de novo centriole assembly pathway.
Of the cell lines we identified that continue to proliferate in the absence of centrosomes, 12 have mutations in or suppress expression of p53 (table S5), which suggests that the arrest is p53-dependent. Consistent with this, immunoblot analysis showed increased levels of p53 and its downstream effector p21 after centrosome depletion (Fig. 3D and fig. S10C). Fixed analysis in RPE1 cells and primary fibroblasts revealed that this p53 increase paralleled the arrest observed in the lineage analysis (fig. S10, E and F), and small hairpin RNA (shRNA)–mediated p53 depletion in RPE1 cells allowed indefinite proliferation in the absence of centrosomes (Fig. 3E). Three lines of evidence indicate that the p53-dependent arrest was not a consequence of DNA damage. First, no posttranslational modifications were observed at eight p53 residues associated with DNA damage signaling (Ser9, Ser15, Ser20, Ser33, Ser37, Ser315, and Ser392 phosphorylation and Lys382 acetylation) (Fig. 4A and fig. S12A) (25, 26). Second, g-H2A.X foci, which mark sites of double-stranded DNA breaks, were not more abundant in centrinone-treated cells (Fig. 4B and fig. S12B). Third, chemical inhibition of the DNA damage– response kinases ATM, ATR, Chk1, and DNA-PK had no effect on the proliferation arrest induced by centrosome loss (fig. S12C). The G1 arrest induced by centrosome loss was also not due to stress signaling; p38 stress kinase, activated by both doxorubicin-induced DNA damage and chromosome missegregation (induced by Mps1 inhibition), was not activated by centrosome loss (Fig. 4C), and a p38 inhibitor had no effect on the G1 arrest (fig. S12, D and E). Knockdown of LATS2 or expression of constitutively active YAP—both recently shown to bypass a Hippo pathway–mediated arrest resulting from cytokinesis failure (27)—also did not bypass the arrest caused by centrosome loss (Fig. 4D and fig. S13).
Fig. 4.
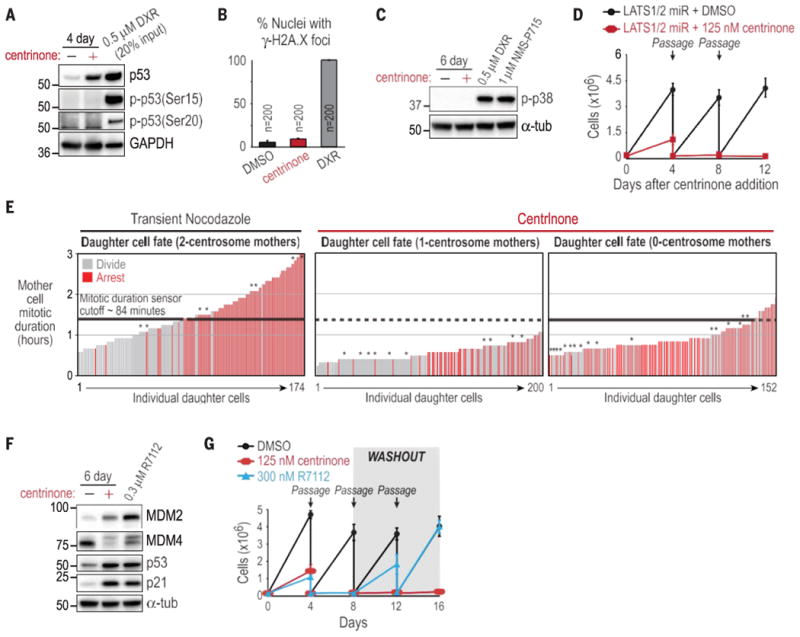
The irreversible G1 arrest after centrosome loss occurs via an unidentified mechanism. (A) Western blot for p53 phosphoepitopes associated with DNA damage in RPE1 cells treated with centrinone or doxorubicin as a positive control. (B) Quantification of g-H2A.X foci in RPE1 nuclei. Data are means T SD (N = 3). (C) Western blot of activated p38 in RPE1 cells. (D) Passaging assay of RPE1 cells expressing LATS1/2 microRNA, after addition of centrinone. (E) Daughter cell fate in RPE1 cells coexpressing centrin-GFP and H2B-RFP. Vertical bars represent measurements from individual daughter cells. Bar height is the time their mother spent in mitosis, and color indicates arrest (red) or division (gray). Asterisks indicate chromosome missegregation in the mother cell. Daughter cell fate after nocodazole treatment of mother cells with a normal two-centrosome complement (left) confirms the existence of a mitotic duration sensor that arrests daughter cells if the mother cell spends more than ∼84 min (black dashed lines on all plots) in mitosis. (F) Western blot of RPE1 cells treated with centrinone or R7112. (G) Passaging assay of RPE1 cells after addition (day 0) and washout (day 8) of centrinone or R7112. Data in (D) and (G) are means T SD (N = 2).
When mitosis is artificially prolonged beyond ∼90 min in RPE1 cells (unperturbed duration is ∼20 min), a mitotic duration sensor arrests the resulting progeny in G1 in a p53-dependent manner (28) (Fig. 4E, left). In a study of Sas4–/– mouse embryos, it was proposed that centriole loss delays mitosis and activates the sensor, which triggers p53-dependent apoptosis (29, 30). To test this idea, we correlated the mitotic duration of mother cells with daughter cell fate during the course of centrosome depletion (Fig. 4E and fig. S14A). All one-centrosome mothers and 87% of zero-centrosome mothers spent less time in mitosis than the duration sensor timing cutoff (dashed black line in Fig. 4E), with most completing this step in significantly less time. There was no correlation between mitotic duration in the mother cell and daughter cell fate. Thus, the G1 arrest triggered by centrosome loss is not a result of extended mitotic duration. In addition, chromosome missegregation was observed only in a minority of cells (asterisks in Fig. 4E), which suggested that aneuploidy resulting from centrosome loss was not the cause of the arrest. Consistent with this, after deliberate induction of chromosome missegregation via Mps1 inhibition, only 11% of the progeny of mothers with visible missegregation arrested in G1 (fig. S14B).
Our results indicate that the p53-mediated G1 arrest after centrosome loss is not due to any previously described signaling mechanism. Instead, centrosome loss resembles the effect of chemically blocking the interaction between p53 and MDM2, the E3 ubiquitin ligase that targets p53 for degradation (31). Both centrosome loss and treatment with the Mdm2 inhibitor R7112 raised p53 levels without genotoxic stress and led to increased MDM2 and decreased MDM4 levels (Fig. 4F). However, whereas R7112 washout led to resumption of proliferation, centrinone washout did not (Fig. 4G), even though arrested cells remained viable for >3 weeks. This difference could result from centrosomes being necessary to suppress p53 levels and new centriole assembly requiring S-phase entry. Effectively, this would trap centrosome-less G1-arrested cells in a “Catch-22” situation—unable to reduce p53 levels and enter S-phase because they lack centrosomes and, at the same time, unable to form new centrosomes because they cannot enter S-phase.
In summary, the Plk4 inhibitor centrinone permits reversible knockout of centrioles and centrosomes and may prove broadly useful for analysis of these organelles. Centrinone treatment revealed that centrosomes are essential for the proliferation of normal human cells, settling a long debate and highlighting an important difference from Drosophila (32). In their absence, a centrosome loss sensor arrests cells in G1 in a p53-dependent manner distinct from previously described signaling mechanisms. In addition to preventing the proliferation of centrosome-less cells, the centrosome loss sensor may also serve a physiological function. As centrosome inactivation is coincident with differentiation in many contexts (33–35), we speculate that it may not only be important to form specialized micro- tubule arrays, but may also function as a barrier restricting cell cycle reentry. Cancer-derived cell lines, irrespective of their basal amplification state, continue to proliferate without centrosomes, albeit with substantially reduced mitotic fidelity. The differential effect of centrosome removal on normal cells and cells with cancer-associated mutations suggests the possibility of combining centrosome depletion with other perturbations to selectively target dividing cancer cells.
Supplementary Material
Acknowledgments
We thank Z. Li for leading the Sundia Meditech chemistry team, Advanced Photon Source NE-CAT (supported by NIH GM103403) for assistance with x-ray data collection, Q. Zhu and I. Verma for the p53 and Glu4 shRNA lentiviruses, A. Holland and D. Cleveland for the Plk4-YFP DLD-1 cell line, R. Gassmann for help with initial cell-based assays, M. Kaulich for technical advice on CRISPR/Cas9, and A. Dammermann for generation of the Sas6 and Cep192 antibodies. J.V.A, R.L.D., M.Y., A.M., C.P.S., J.E.H., A.D., T.C.G., A.K.S., and K.O. receive salary and other support from the Ludwig Institute for Cancer Research. This work was supported by NIH grants to K.O. (GM074207) and B.J.M. (GM089970), and with funds from the Hilton Ludwig Cancer Prevention Initiative to A.K.S. and T.C.G. The data described here are tabulated in the main paper and Supplementary Materials. The structure of the centrinone-bound Plk4 kinase domain complex has been deposited in the Protein Data Bank (4YUR). The Ludwig Institute for Cancer Research has filed a patent application (62/149,292) related to the structures, syntheses, and uses of centrinone, centrinone B, and chemically related Plk4 inhibitors. Requests for the centrinones should be directed to A.K.S. (ashiau@ucsd.edu).
Footnotes
Supplementary materials: www.sciencemag.org/content/348/6239/1155/suppl/DC1
References and Notes
- 1.Drummond IA. Curr Opin Cell Biol. 2012;24:24–30. doi: 10.1016/j.ceb.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schatten H. Histochem Cell Biol. 2008;129:667–686. doi: 10.1007/s00418-008-0427-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brito DA, Gouveia SM, Bettencourt-Dias M. Curr Opin Cell Biol. 2012;24:4–13. doi: 10.1016/j.ceb.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 4.Färat-Karalar EN, Stearns T. Philos Trans R Soc Lond B Biol Sci. 2014;369:20130460. doi: 10.1098/rstb.2013.0460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boveri T. Zur Frage der Entstehung maligner Tumoren. Gustav Fischer Verlag; Jena, Germany: 1914. [Google Scholar]
- 6.Godinho SA, Pellman D. Philos Trans R Soc Lond B Biol Sci. 2014;369:20130467. doi: 10.1098/rstb.2013.0467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nigg EA, Čajánek L, Arquint C. FEBS Lett. 2014;588:2366–2372. doi: 10.1016/j.febslet.2014.06.030. [DOI] [PubMed] [Google Scholar]
- 8.Ganem NJ, Godinho SA, Pellman D. Nature. 2009;460:278–282. doi: 10.1038/nature08136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Silkworth WT, Nardi IK, Scholl LM, Cimini D. PLOS ONE. 2009;4:e6564. doi: 10.1371/journal.pone.0006564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Godinho SA, et al. Nature. 2014;510:167–171. doi: 10.1038/nature13277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bettencourt-Dias M, et al. Curr Biol. 2005;15:2199–2207. doi: 10.1016/j.cub.2005.11.042. [DOI] [PubMed] [Google Scholar]
- 12.Habedanck R, Stierhof YD, Wilkinson CJ, Nigg EA. Nat Cell Biol. 2005;7:1140–1146. doi: 10.1038/ncb1320. [DOI] [PubMed] [Google Scholar]
- 13.Kleylein-Sohn J, et al. Dev Cell. 2007;13:190–202. doi: 10.1016/j.devcel.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 14.O'Connell KF, et al. Cell. 2001;105:547–558. doi: 10.1016/s0092-8674(01)00338-5. [DOI] [PubMed] [Google Scholar]
- 15.Peel N, Stevens NR, Basto R, Raff JW. Curr Biol. 2007;17:834–843. doi: 10.1016/j.cub.2007.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davis MI, et al. Nat Biotechnol. 2011;29:1046–1051. doi: 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
- 17.Johnson EF, Stewart KD, Woods KW, Giranda VL, Luo Y. Biochemistry. 2007;46:9551–9563. doi: 10.1021/bi7008745. [DOI] [PubMed] [Google Scholar]
- 18.Mason JM, et al. Cancer Cell. 2014;26:163–176. doi: 10.1016/j.ccr.2014.05.006. [DOI] [PubMed] [Google Scholar]
- 19.Sampson PB, et al. J Med Chem. 2015;58:147–169. [PubMed] [Google Scholar]
- 20.Sloane DA, et al. ACS Chem Biol. 2010;5:563–576. doi: 10.1021/cb100053q. [DOI] [PubMed] [Google Scholar]
- 21.Rios RM. Philos Trans R Soc Lond B Biol Sci. 2014;369:20130462. doi: 10.1098/rstb.2013.0462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khodjakov A, Rieder CL. J Cell Biol. 2001;153:237–242. doi: 10.1083/jcb.153.1.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sir JH, et al. J Cell Biol. 2013;203:747–756. doi: 10.1083/jcb.201309038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Uetake Y, et al. J Cell Biol. 2007;176:173–182. doi: 10.1083/jcb.200607073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jenkins LM, Durell SR, Mazur SJ, Appella E. Carcinogenesis. 2012;33:1441–1449. doi: 10.1093/carcin/bgs145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lakin ND, Jackson SP. Oncogene. 1999;18:7644–7655. doi: 10.1038/sj.onc.1203015. [DOI] [PubMed] [Google Scholar]
- 27.Ganem NJ, et al. Cell. 2014;158:833–848. doi: 10.1016/j.cell.2014.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Uetake Y, Sluder G. Curr Biol. 2010;20:1666–1671. doi: 10.1016/j.cub.2010.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bazzi H, Anderson KV. Proc Natl Acad Sci USA. 2014;111:E1491–E1500. doi: 10.1073/pnas.1400568111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Izquierdo D, Wang WJ, Uryu K, Tsou MF. Cell Reports. 2014;8:957–965. doi: 10.1016/j.celrep.2014.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shen H, Maki CG. Curr Pharm Des. 2011;17:560–568. doi: 10.2174/138161211795222603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Basto R, et al. Cell. 2006;125:1375–1386. doi: 10.1016/j.cell.2006.05.025. [DOI] [PubMed] [Google Scholar]
- 33.Bartolini F, Gundersen GG. J Cell Sci. 2006;119:4155–4163. doi: 10.1242/jcs.03227. [DOI] [PubMed] [Google Scholar]
- 34.Stiess M, et al. Science. 2010;327:704–707. doi: 10.1126/science.1182179. [DOI] [PubMed] [Google Scholar]
- 35.Sumigray KD, Lechler T. BioArchitecture. 2011;1:221–224. doi: 10.4161/bioa.18403. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.