

Abstract
Optimal healing of damaged tissue following myocardial infarction (MI) requires a coordinated cellular response that can be divided into three phases: inflammatory, proliferative/reparative, and maturation. The inflammatory phase, characterized by rapid influx of cytokines, chemokines, and immune cells, is critical to the removal of damaged tissue. The onset of the proliferative/reparative phase is marked by increased proliferation of myofibroblasts and secretion of collagen to replace dead tissue. Lastly, crosslinking of collagen fibers and apoptosis of immune cells marks the maturation phase. Excessive inflammation or fibrosis has been linked to increased incidence of arrhythmia and other MI-related pathologies. This review describes the roles of inflammation and fibrosis in arrhythmogenesis and prospective therapies for anti-arrhythmic treatment.
Graphical Abstract
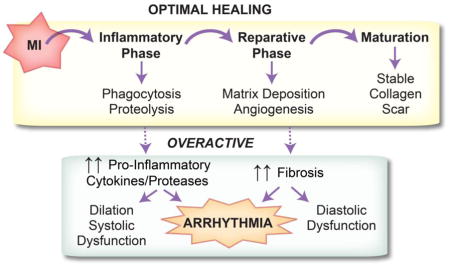
1. Introduction: Post-MI Healing and Repair
Following myocardial infarction (MI), a coordinated cellular response is required for sufficient wound healing and scar formation. This healing process can be classified into three major phases: inflammatory, proliferative/reparative, and maturation (Figure 1) [1]. The inflammatory phase begins with a rapid influx of neutrophils and monocytes that begins within hours of the ischemic event, particularly in the setting of reperfusion. By day 3, the inflammatory phase is dominated by monocyte-derived macrophages, with pro-inflammatory M1 and anti-inflammatory M2 being the major subtypes. Classically activated M1 macrophages clear dead myocyte debris through phagocytosis and proteolysis. M1 macrophages secrete inflammatory cytokines including interleukin (IL)-1β, IL-6, and tumor necrosis factor (TNF)-α as well as proteases including matrix metalloproteinase (MMP)-1, -2, -3, -7, -8, -9, and -12 [2].
Figure 1.

Optimal post-MI healing is comprised of three phases: inflammatory, reparative/proliferative, and maturation. Timely progression and resolution of each phase is required for proper healing. An overactive inflammatory or reparative phase can lead to ventricular arrhythmia.
Alternatively activated anti-inflammatory M2 macrophages, myofibroblasts, and endothelial cells dominate the proliferative/reparative phase [1]. M2 macrophages secrete the anti-inflammatory cytokine IL-10 and growth factors including transforming growth factor (TGF)-β, which in turn recruit and activate reparative myofibroblasts and vascular cells [2]. Myofibroblasts secrete large amounts of extracellular matrix (ECM) in order to replace lost ventricular tissue with a stable scar. The maturation phase is marked by apoptosis of the majority of the inflammatory and reparative cells and scar maturation and remodeling.
Timely progression and resolution of both the inflammatory and reparative phases is necessary for proper infarct healing. If either phase is overactive or incompletely resolved, adverse LV remodeling occurs. A large body of evidence from mouse MI models supports the concept that impaired resolution of inflammation leads to LV dilation and adverse remodeling [3–5]. At the same time, inflammation is required for proper healing, as depleting inflammatory macrophages also leads to impaired healing [6]. An overactive reparative phase is likewise detrimental, promoting fibrosis outside the infarct region and contributing to diastolic dysfunction. A major unanswered question lies in determining which patient populations and which underlying pathologies ultimately contribute to improper resolution of either or both phases.
Importantly, in addition to playing a role in adverse LV remodeling, impaired resolution of either the inflammatory or reparative phase can lead to adverse electrophysiological remodeling, ventricular arrhythmia, and sudden cardiac arrest (Figure 1). Indeed, the mechanisms by which an overactive reparative phase (including interstitial fibrosis and potential myofibroblast-myocyte coupling) may contribute to both triggered and reentrant arrhythmias has been a long-standing area of investigation [7,8]. On the other hand, the mechanisms by which an overactive inflammatory response contributes to ventricular arrhythmias has received less attention. This review focuses on the electrophysiological consequences of both the inflammatory and reparative phases.
2. Post-MI inflammation, electrophysiological remodeling, and arrhythmia
Following ischemia, surviving cardiac myocytes in the infarct border zone (BZ) undergo dramatic electrophysiological remodeling, which, in addition to the fibrotic scar, creates the substrate for ventricular arrhythmia. Some of the most well documented electrophysiological changes in the infarct BZ include a reduction in repolarizing K+ currents that may result in a prolonged action potential duration (APD) [9,10], reduced expression or lateralization of connexin 43 (Cx43) which contributes to slowed conduction [11,12], and intracellular Ca2+ mishandling that may lead to triggered activity [13,14]. Collectively, these changes provide the trigger and substrate for malignant ventricular arrhythmias.
Despite the rigorous characterization of post-MI electrophysiological remodeling, the upstream mechanisms responsible for these changes are not well understood. Importantly, key cytokines and proteases that are elevated in the myocardium following MI (e.g., TNF-α, IL-1β, IL-6, MMPs) produce electrophysiological changes in cardiac myocytes that mirror those found in the infarct BZ, suggesting that inflammation may be an important contributor to electrophysiological remodeling and arrhythmia. Indeed, a growing body of clinical evidence suggests that post-MI patients with arrhythmia have higher circulating levels of inflammatory cytokines compared to post-MI patients who are arrhythmia free [15,16]. Furthermore, even in the absence of MI or structural heart disease, systemic inflammation is associated with a significantly increased risk for ventricular tachyarrhythmias [17]. The studies described below (Table 1) support these clinical observations and demonstrate mechanistic links between the inflammatory phase and post-MI electrophysiological remodeling.
Table 1.
A selection of references demonstrating the impact of inflammatory cytokines and proteases on ionic currents, intracellular Ca2+ handling, and gap junction coupling. Arrows indicate an increase (↑), decrease (↓), or no change (↔) in given parameter.
| TNF-α | Ref. | Sample Type* | |
|---|---|---|---|
| ICaL | Current ↔ | [22,23,36] | Ventricular myocytes from TNF-α overexpressing mice [22]; neonatal ventricular myocytes [36] |
| Ito | Current ↓ Kv4.2, Kv4.3 protein ↓ KCND2, KCNIP2 mRNA ↓ |
[22–24] [22,23] [24,89] |
Ventricular myocytes from TNF-α overexpressing mice [22,89] |
| IKs | Current ↓ during PKA activation | [90] | |
| IKr | Current ↓ hERG protein ↔ |
[25] [25] |
Cardiomyocytes or HERG(+) HEK293 cells |
| IK,slow1, IK,slow2 | Current ↓ Kv1.5 protein ↓ |
[22] [22] |
Ventricular myocytes from TNF-α overexpressing mice |
| IK1 | KCNJ12 mRNA ↓ | [89] | Whole hearts from TNF-α overexpressing mice |
| Systolic [Ca2+]i | ↓ | [21,27] | Whole hearts from TNF-α overexpressing mice [21] |
| Diastolic [Ca2+]i | ↑ | [21] | Whole hearts from TNF-α overexpressing mice |
| [Ca2+]SR | ↓ | [27] | |
| Spontaneous SR Ca2+ release | ↑ synergistically with IL-1β co-application | [29] | |
| SERCA | Slowed Ca2+
reuptake Gene ↓ |
[28] [28] |
HL-1 cells |
| PLB | Gene ↔ | [42] | Neonatal ventricular myocytes |
| Cx43 | Total protein ↔ but
lateralization/internalization ↓ activity of promoter region of Cx43 gene |
[30] [31] |
Whole hearts from TNF-α overexpressing
mice [30]; whole hearts following hepatic ischemia or LPS [31] |
| IL-1β | |||
| ICaL | Current ↓ Current ↑ Suppressed response to β-AR stimulation CACNA1C gene ↓ |
[34–36] [37] [38] [41] |
Neonatal ventricular myocytes [36,41] |
| Systolic [Ca2+]i | ↓ ↓ further with TNF-α co-administration ↔ to β-AR stimulation |
[39] [29] [42] |
Neonatal ventricular myocytes [39,42] |
| [Ca2+]SR | ↓ synergistically with TNF-α co-application | [29] | |
| Spontaneous SR Ca2+ release | ↑ synergistically with TNF-α co-application | [29] | |
| RyR | Gene ↓ | [41] | Neonatal ventricular myocytes |
| SERCA | Protein ↓ Gene ↓ |
[39] [39,41–43] |
Neonatal ventricular myocytes |
| PLB | Protein ↓ Gene ↓ |
[39,42] [39,42] |
Neonatal ventricular myocytes |
| Cx43 | ↓ conductance (dye spread
assay) Protein ↓ |
[45,46] [45,46] |
MDCK cells and neonatal ventricular myocytes [45,46] |
| IL-6 | |||
| ICaL | Current ↑ | [48] | |
| Systolic [Ca2+]i | ↑ | [48] | |
| SERCA | Slowed Ca2+ reuptake Gene ↓ | [28] [28,49] |
HL-1 cells [28]; neonatal ventricular myocytes [49] |
| PLB | ↓ phosphorylation | [50] | |
| MMPs | |||
| Cx43 | Directly cleaved by MMP-7 Protein ↓ by MMP-2 Hypoxia-induced ↓ dependent on MMP-9 MMP-9 activation associated with slow conduction and Cx43 degradation, but MMP-9 found not to cleave Cx43 |
[52] [55] [53] [54] |
Whole hearts from MMP-7-deficient mice with MI [52]; retinal endothelial cells [55]; H9c2 cardiomyocytes [53]; whole hearts from M9PROM mice with myocardial injury [54] |
Unless otherwise indicated, studies were performed in isolated adult ventricular myocytes treated with the cytokine/protease indicated.
2.1 TNF-α
TNF-α is significantly elevated in the post-MI heart and is further increased when MI is superimposed on a background of inflammation including atherosclerosis [18], aging [19], or bacterial gingivitis [20]. The electrophysiological effects of TNF-α are well characterized in isolated cardiac myocytes from normal hearts and from those of cardiac-specific TNF-α over-expressing mice. Over-expression of TNF-α prolongs APD and significantly reduces repolarizing K+ currents (Table 1), whereas L-type Ca2+ current (ICaL) remains unchanged [21,22]. Similar results have been observed in normal myocytes treated with exogenous TNF-α. Fernandez-Velasco et al. reported a dose-dependent decrease in Ito with increasing doses of TNF-α, while ICaL remained unaffected [23]. Decreased Ito was also observed in neonatal rat myocytes treated with TNF-α along with decreased KCND2 (Kv4.2: Ito) mRNA [24]. TNF-α treatment also prolonged APD and caused a dose-dependent reduction in IKr in canine myocytes. Here, the reduction in IKr was not associated with decreased channel (hERG) expression, but rather through increased intracellular reactive oxygen species [25].
TNF-α has negative inotropic effects [26]. Acute treatment of adult rat myocytes with TNF-α leads to rapid decreases in systolic [Ca2+] and reduces the sarcoplasmic reticulum (SR) Ca2+ content ([Ca2+]SR) [27]. Likewise, TNF-α over-expressing mice also display altered intracellular Ca2+ handling, including increased diastolic and suppressed systolic [Ca2+] and prolonged Ca2+ transient duration [21]. Wu et al. showed that TNF-α treatment of HL-1 cardiomyocytes lead to delayed Ca2+ reuptake accompanied by a decrease in SR Ca2+ ATPase (SERCA) gene expression [28]. TNF-α and IL-1β have similar effects on intracellular Ca2+ handling and, when combined, act synergistically to further decrease systolic [Ca2+], decrease [Ca2+]SR, and increase spontaneous SR Ca2+ release events, leading to arrhythmogenic Ca2+ waves [29].
TNF-α also impacts gap junction coupling and cell-cell communication. Optical mapping studies in TNF-α over-expressing mice revealed normal ventricular conduction velocity at baseline, but decreased conduction velocity during premature stimuli [21]. Subsequent studies in the atria of the TNF-α mice revealed no change in total Cx43 expression, but a significant increase in Cx43 internalization and lateralization [30]. In vitro experiments provided direct evidence that TNF-α reduces activity of the promoter region of the Cx43 gene [31].
2.2 IL-1β
IL-1β is a key regulatory cytokine involved in the post-MI inflammatory response and is required for recruitment of immune cells and subsequent production of other pro-inflammatory cytokines [32,33]. Investigations into the electrophysiological effects of IL-1β have centered on changes in Ca2+ handling and cell-cell coupling. Most studies have found a decrease in ICaL by IL-1β [34–36], however Li et al. found an increase in ICaL in guinea pig myocytes [37]. Decreased responsiveness of ICaL to β-adrenergic stimulation has also been observed with IL-1β [38,39].
In addition to the synergistic effects of IL-1β and TNF-α [29,40], IL-1β alone appears to have a significant impact on SR Ca2+ release and reuptake. Several reports indicate that IL-1β decreases expression (at both the gene and protein level) of important Ca2+ handling proteins, including ryanodine receptors (RyR), SERCA, and phospholamban (PLB) (Table 1) [39,41–43].
IL-1β has also been implicated in Cx43 down-regulation. First discovered in astrocytes [44] and later observed in cardiac myocytes from post-MI mouse and canine hearts [45,46], IL-1β produces cell-cell uncoupling, internalization, and reduced expression of Cx43. Our group recently reported similar findings in a mouse model of MI superimposed on atherosclerosis (ApoE−/−), which intensifies the inflammatory phase of healing [18,47]. ApoE+MI hearts had a ~3-fold increase in IL-1β expression, a ~2-fold decrease in Cx43 expression, and significant Cx43 internalization and lateralization (Figure 2). The ApoE+MI hearts also had decreased conduction velocity and a significant increase in inducible ventricular arrhythmias. Importantly, the ApoE+MI phenotype could be completely reproduced by acutely increasing post-MI inflammation with lipopolysaccharide (LPS), suggesting that elevated inflammation during healing can produce deleterious electrophysiological consequences [47]. Ongoing work in our laboratory is further defining the role of IL-1β in this process.
Figure 2.

Inflammation and Cx43. A) Immunofluorescence images of Cx43 (green) and CD-68 (red, macrophage marker) show abundant macrophage infiltration in the infarct region of an ApoE+MI heart (bottom). Myocytes adjacent to macrophages show internalization of Cx43 (Inset). B) Top: Cx43 (green) normally co-localizes with N-cadherin (N-Cad, red), a marker of the intercalated disc. Arrows indicate examples of obvious co-localization of Cx43 and N-Cad. Bottom: In the infarct region of an ApoE+MI heart, Cx43 expression is observed without corresponding N-Cad staining, indicating Cx43 internalization or lateralization to areas outside of the intercalated disc. Arrows indicate obvious examples of co-localization. Dashed boxes indicate examples of internalization (Cx43 without corresponding N-Cad). C) Quantification of Cx43 and N-Cad co-localization, demonstrating increased Cx43 internalization/lateralization in the infarct compared to remote regions, and in ApoE+MI compared to WT+MI hearts. D–F) ApoE+MI hearts have a ~3-fold increase in IL-1β expression and a corresponding ~2-fold decrease in Cx43 expression. Reprinted with permission from [47].
2.3 IL-6
IL-6 is also elevated post-MI and may impact cardiomyocyte Ca2+ handling. Hagiwara et al. reported that IL-6 signaling through gp130 (glycoprotein 130 cytokine receptor) lead to an increase in ICaL, increased systolic [Ca2+] and prolonged APD [48]. IL-6 has also been shown to decrease SERCA gene expression [28,49] and PLB phosphorylation [50], leading to slowed Ca2+ reuptake into the SR [28]. Cytokine signaling through gp130 has also been shown to be a key regulator of remodeling of cardiac sympathetic neurotransmission following MI [51], a known contributor to arrhythmias.
2.4 MMPs
MMPs are important mediators of both the inflammatory and reparative phases of post-MI healing, as they breakdown ECM, a necessary step for proper scar formation. However, these proteinases may also degrade gap junction proteins that are required for proper cell-cell electrical coupling. Lindsey et al. demonstrated one of the first mechanistic links between MMP activity and Cx43. They observed that MMP-7 null mice were protected from Cx43 degradation post-MI, had improved conduction velocity, and reduced propensity to arrhythmia [52]. In vitro studies demonstrated that MMP-7 directly cleaves Cx43 [52]. Similarly, hypoxia-induced Cx43 reduction is MMP-9 dependent [53]. Investigation into the role of MMP-9 and Cx43 following myocardial injury found that MMP-9 activity corresponded to locations of Cx43 degradation and conduction slowing. However, unlike MMP-7, in vitro experiments revealed that Cx43 is not a substrate for MMP-9 [54]. The authors suggest that MMP-9 disrupts normal cell-cell alignment, leading to reduced Cx43 coupling. To date, there are relatively few studies examining the role of MMPs in post-MI Cx43 loss; however, experiments in keratinocytes and epithelial cells support a role for MMPs in Cx43 degradation [55]. Thus, the role of MMP activity and cell-cell electrical communication post-MI remains an important area for future investigation.
3. Post-MI Fibrosis and Arrhythmia
3.1 Mechanisms of Fibrosis Following MI
By days 3–5 post-MI, inflammatory signals start to decline, due to immune cell apoptosis and up-regulation of anti-inflammatory and pro-fibrotic signals, marking the onset of the proliferative/reparative phase of healing (Figure 1). As immune cells retreat, levels of IL-1 and TNF-α decline, while TGF-β and IL-10 increase. TGF-β stimulates the maturation of fibroblasts into myofibroblasts (Mfb), suppresses macrophage activity, and increases expression of both tissue inhibitor of metalloproteinases (TIMPs) and protease inhibitors. Mfbs are characterized by proliferation, increased expression of contractile, focal adhesion, and ECM proteins, and increased collagen synthesis and deposition [56]. Figure 3 shows the time course of collagen deposition and scar maturation following MI wherein the percent of ventricular area and maturity of collagen fibers increases as healing progresses. An overactive reparative phase, however, may lead to fibrosis outside the infarct and contribute to adverse remodeling [1]. When addressing the role of fibrosis and Mfbs in arrhythmogenesis, there are two distinct categories: interstitial fibrosis and Mfb-myocyte coupling.
Figure 3.

The temporal progression of collagen deposition through 28 days post-MI. Sections were stained with 1% picosirius red, which stains collagen red. Reprinted with permission from [85].
3.2 Interstitial Fibrosis and Arrhythmia
Following MI, there are distinct patterns of fibrosis, including the compact fibrotic tissue of the scar and varying degrees of interstitial fibrosis adjacent to and outside the infarct. The compact scar is mostly acellular and is electrically non-excitable, although it may serve as an insulated area where reentrant arrhythmia can anchor and convert to sustained ventricular tachycardia (VT) [57]. We previously showed that the healed compact scar can attract and anchor reentrant VT in the post-MI rabbit heart (Figure 4). In this study, 84% of sustained VTs were found to anchor at the infarct, often stabilizing at the scar following a period of meandering after initiation. Similarly, appropriately timed cardioversion shocks could force detachment and subsequent termination of reentry. These data suggest that compact fibrosis of the scar may be important for maintaining sustained VT and that low-energy methods for detachment of reentry from the scar may be sufficient to terminate VT [57].
Figure 4.

Sustained VT anchored to compact scar. A) Photograph of post-MI rabbit heart. Arrow indicates infarct. B) Masson trichrome staining of short-axis slices. Blue indicates fibrosis. C) 3D histological reconstruction showing entire infarct region in blue. D) Phase map of stable reentrant VT rotating clockwise, anchored to compact scar. E) Location of phase singularities (center of reentry) corresponds to infarct location. F) Lead I ECG during VT. Reprinted with permission from [57].
Interstitial fibrosis in the BZ and non-infarct tissue is in some ways more arrhythmogenic than the compact scar. Here, myocytes are separated to varying degrees by non-conducting collagenous septa. Depending on the severity, this geometry can favor the escape of focal (ectopic) activity, as well as promote slow conduction and unidirectional conduction block that favor reentrant tachycardia [58,59]. These arrhythmogenic situations are created by conditions of source-sink mismatch and are further exacerbated by electrophysiological remodeling of the surviving BZ myocytes.
The source-sink mismatch typically refers to insufficient depolarizing current generated by an area of excited tissue (i.e., the source) compared to that which is necessary to excite the neighboring quiescent tissue (i.e., the sink). Insufficient source current and subsequent conduction block often occur at sites of expansion (e.g., a depolarizing wavefront traveling from a narrow structure into a larger structure) or when small islands of depolarization are surrounded by quiescent myocardium. The source-sink mismatch likely acts to protect the normal myocardium from ectopic activity. However, interstitial fibrosis can uncouple myocytes from one another by creating small, electrically insulated areas between them. This electrical uncoupling reduces the source-sink mismatch and has been shown to promote the escape of ectopic triggers [60,61]. Morita et al. experimentally demonstrated the role of fibrosis in promoting ectopic triggers in hearts of aged rats and rabbits. They showed that the incidence of early afterdepolarizations and triggered activity originated more frequently from the LV base, which had a higher percentage of fibrotic tissue compared to other regions of the heart [62].
Interstitial fibrosis can also have effects on conduction; large areas of fibrosis can force a propagating wavefront to take a ‘zig-zag’ pattern, thus slowing conduction at the macroscopic level. When the activation wavefront is slowed through a fibrotic region, the repolarization wavefront may also be delayed, resulting in an increase in the dispersion of repolarization, another condition that favors reentry. Because interstitial fibrosis often disrupts side-to-side connections between myocytes, the anisotropy of conduction may also be increased. In post-MI swine, Tschabrunn et al. showed that deposition of transmural fibrosis, along with preserved borders of subendocardial tissue, contributed to slowed conduction and non-uniform anisotropy, giving rise to reentrant VT [63].
3.3 Myofibroblast-Cardiomyocyte Coupling
Cardiac arrhythmias stemming from fibrosis have traditionally been attributed to the deposition of ECM in the interstitial space. However, a large population of fibroblasts and Mfbs are present during and after MI and play vital roles in the maintenance of ECM [64]. Mfbs may also play a role in post-MI arrhythmogenesis via Mfb-myocyte coupling. For a more complete discussion of Mfb-myocyte coupling, refer to the accompanying article in this issue by Kohl et al., [JMCC9726] as well as [8].
When fibroblasts are cultured, they differentiate into Mfbs and upregulate expression of Cx43 and Cx45 [64]. The most compelling evidence for Mfb-myocyte electrical coupling comes from several in vitro co-culture studies where coupling was confirmed via direct observation of wavefront propagation through Mfbs, or by changes to myocyte resting membrane potential [65–67]. When cardiomyocytes and Mfbs are well-coupled they are difficult to differentiate electrically, thus evidence of Mfb-myocyte coupling in intact tissue has yet to be unequivocally demonstrated. Immunohistochemical staining of Cx43 in infarcted hearts has, however, demonstrated the potential for coupling [68]. Given the experimental difficulties with differentiating and confirming Mfb-myocyte coupling in the intact post-MI heart, computational modeling has become an important investigational tool in this area.
Using a 2D computational model, Xie et al. demonstrated the arrhythmogenic consequences of Mfb-myocyte coupling [69]. Mfbs within the sheet, but not electrically coupled to myocytes act similarly to interstitial fibrosis in that they lead to zig-zag (slowed) conduction. When Mfbs are electrically coupled to myocytes, however, they significantly reduce the conduction velocity (CV) as the ratio of Mfbs to cardiomyocytes increases. The authors attribute this finding to increased electrotonic loading of the myocytes (i.e., increased current sink), reduced myocyte-myocyte coupling, and to differences in resting membrane potential between myocytes and Mfbs (~-80 mV versus ~-50 to -20 mV, respectively) [67]. As Mfb density increases, the resting membrane potential of myocytes becomes more depolarized, inactivating voltage-gated Na+ channels, which can slow conduction and lead to post-repolarization refractoriness. In this study, only in instances of Mfb-myocyte coupling could reentry be induced by programmed stimulation [69].
McDowell et al. demonstrated similar results in the infarct region and peri-infarct zone (PZ) using a 3-dimensional, anatomically accurate model of an infarct rabbit heart. They found that Mfb-myocyte coupling in the PZ only affects reentry at intermediate Mfb:myocyte densities. At higher Mfb densities, reentry is suppressed due to depolarization of myocytes and increased refractoriness; however, this state is also pro-arrhythmic since diastolic depolarization can lower the threshold for ectopic activity. Higher rates of reentry were observed at intermediate Mfb densities wherein coupling of Mfbs to myocytes shortened the APD (because Mfbs act as a current sink when myocytes are depolarized), which lead to increased APD dispersion and enhanced reentry potential [70].
4. Post-MI Arrhythmia Prevention
Prevention of ventricular arrhythmias is primarily achieved through use of implantable devices or anti-arrhythmic drugs. Implantable cardioverter defibrillators (ICDs) are lifesaving, but are highly invasive and impermanent fixes [71]. Ion channel blockers, including flecainide, encainide, and d-sotalol, have been used to prevent arrhythmias in the post-MI setting but were discontinued due to dangerous pro-arrhythmic effects. While these drugs decreased incidence of premature ventricular contractions, they increased incidence of reentrant and fatal arrhythmias [72]. Current therapeutics, such as beta blockers and angiotensin-converting enzyme inhibitors, avoid ion channels and may act indirectly to prevent arrhythmias by altering neurohumoral tone. However, most of these approaches treat the symptom (arrhythmia), rather than the underlying cause (electrophysiological and structural remodeling). Targeting the post-MI inflammatory or reparative phase at the outset might represent a more effective anti-arrhythmic approach.
4.1 Targeting Inflammation
Treatment with non-specific anti-inflammatory steroids following acute MI has resulted in compromised infarct healing, scar tissue thinning, and increased risk of ventricular rupture [73]. However, other studies have reported decreased mortality and no change in rupture risk [74]. Use of non-steroidal anti-inflammatory drugs is associated with worse clinical outcomes and current guidelines advise against their use in MI patients [75]. Thus, more specific targeting of inflammatory signaling is likely required to avoid suppression of beneficial functions.
Given its potentially important role in electrophysiological remodeling (Table 1), TNF-α may represent a novel anti-arrhythmic target. However, anti-TNF-α therapies have been tested for the prevention of heart failure following MI and, although results are somewhat contradictory, the overall consensus is that inhibition of TNF-α yields more detrimental outcomes and should not be used to treat MI [76]. These results underscore the difficulties in targeting post-MI inflammation and are likely due to abatement of the beneficial functions of TNF-α [77].
IL-1 is rapidly upregulated following MI and may play a role in post-MI arrhythmogenesis [45,47]. In the Virginia Commonwealth University Anakinra Remodeling Trial (VCU-ART), Abbate et al. demonstrated that administration of anakinra, a human recombinant IL-1 receptor antagonist, following acute MI attenuated LV remodeling without compromising infarct healing [78]. Other therapeutic strategies for modulating IL-1 activity have also been shown to improve LV remodeling and preserve cardiac function [79,80]. Furthermore, the first large, multi-center trial evaluating the efficacy of IL-1 inhibition for limiting the progression of atherosclerosis (CANTOS) is currently ongoing [81]. These studies, combined with the role of IL-1 in post-MI Ca2+ mishandling and gap junction uncoupling (Table 1), make IL-1 an attractive therapeutic target that warrants further investigation.
4.2 Targeting Fibrosis
TGF-β is central to post-MI scar formation; therefore, TGF-β inhibition may attenuate interstitial fibrosis. Dobaczewski et al. demonstrated in mice subject to MI that knockdown of Smad3, a downstream transcription factor for TGF-β, markedly impaired Mfb differentiation, migration, and contractility [82]. Consistent with previous reports [83], they also demonstrated that despite increased Mfb numbers and unchanged infarct size, Mfb inhibition attenuated LV remodeling and ventricular dysfunction [82].
Pirfenidone, a TGF-β inhibitor, was also shown to attenuate TGF-β-mediated fibrosis and reduce arrhythmia following MI [84]. In an ischemia-reperfusion model, Nguyen et al. showed that pirfenidone treatment improved LV ejection fraction, lowered incidence of inducible VT, and preserved CV by reducing total LV fibrosis and infarct size [84].
Therefore, directly inhibiting fibroblast/Mfb activity reduces total fibrosis, improves structural remodeling, and reduces propensity to arrhythmias by preventing fibrosis-induced slowing of conduction. Importantly, Mfbs are also a source of pro-inflammatory cytokines [45], therefore, suppression of Mfb activation may also be anti-arrhythmic by inhibiting cytokine production. Considering Mfb-myocyte coupling, however, treatments that increase Mfb numbers [82,83] may be pro-arrhythmic by increasing electrotonic loading on myocytes. Thus, the pro- or anti-arrhythmic effects of Mfb inhibition require further study.
The activity of several MMPs, including MMP-1, -2, -3, -7, -8, -9, -13, and -14 is upregulated following MI [85]. ECM fragments produced by MMPs are bioactive and act to promote fibrosis, ECM turnover, and LV remodeling. Indeed, several mouse models of MMP over-expression have shown adverse LV remodeling and increased mortality [86,87]. As such, modulation of MMPs, or their inhibitors—mainly TIMPs 1 and 2—is a possible avenue for post-MI therapy [88] and may reduce arrhythmogenesis by limiting interstitial fibrosis. Furthermore, MMPs degrade connexins [52], thus MMP inhibition may improve cell-cell coupling and conduction. As a note of caution, MMPs are widespread and expressed in most tissues and broad-spectrum MMP inhibitors have been unsuccessful in clinical trials due to musculoskeletal toxicity. Thus, therapeutics with affinities for specific MMPs may prove more beneficial [88]. For more discussion on MMP roles in post-MI remodeling, see Lindsey et al. in this issue [JMCC9601].
5. Conclusions
We have briefly reviewed the inflammatory and reparative/proliferative phases of post-MI healing and the mechanisms by which overactivity of either phase may contribute to malignant ventricular arrhythmias. Pharmacological therapies that target the post-MI electrophysiological substrate (i.e., ion channel blockers) have been largely unsuccessful in treating ventricular arrhythmias. Thus, by shifting the focus upstream of electrophysiological remodeling (i.e., to the inflammatory and fibrotic mechanisms that ultimately potentiate arrhythmia), novel anti-arrhythmic targets may be uncovered. An improved understanding of these mechanisms in arrhythmogenesis in the post-MI heart will provide more individualized treatment options for patients with MI.
Highlights.
Post-MI healing is divided into 3 phases: inflammatory, reparative, and maturation
Timely progression through each phase is required for optimal healing
Over-activity of either the inflammatory or reparative phase may lead to arrhythmia
Targeting inflammation or fibrotic repair may lead to new anti-arrhythmics
Acknowledgments
This work was supported by the National Institutes of Health HL075360 and GM114833 (to MLL) and HL111600 (to CMR), the American Heart Association 12SDG9010015 (to CMR), and the Biomedical Laboratory Research and Development Service of the Veterans Affairs Office of Research and Development Award 5I01BX000505 (to MLL).
Footnotes
Disclosures
None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Frangogiannis NG. The inflammatory response in myocardial injury, repair, and remodelling. Nat Rev Cardiol. 2014;11:255–65. doi: 10.1038/nrcardio.2014.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lambert JM, Lopez EF, Lindsey ML. Macrophage roles following myocardial infarction. Int J Cardiol. 2008;130:147–58. doi: 10.1016/j.ijcard.2008.04.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dobaczewski M, Xia Y, Bujak M, Gonzalez-Quesada C, Frangogiannis NG. CCR5 signaling suppresses inflammation and reduces adverse remodeling of the infarcted heart, mediating recruitment of regulatory T cells. Am J Pathol. 2010;176:2177–87. doi: 10.2353/ajpath.2010.090759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cochain C, Auvynet C, Poupel L, Vilar J, Dumeau E, Richart A, et al. The chemokine decoy receptor D6 prevents excessive inflammation and adverse ventricular remodeling after myocardial infarction. Arterioscler Thromb Vasc Biol. 2012;32:2206–13. doi: 10.1161/ATVBAHA.112.254409. [DOI] [PubMed] [Google Scholar]
- 5.Iyer RP, Patterson NL, Zouein FA, Ma Y, Dive V, de Castro Brás LE, et al. Early matrix metalloproteinase-12 inhibition worsens post-myocardial infarction cardiac dysfunction by delaying inflammation resolution. Int J Cardiol. 2015;185:198–208. doi: 10.1016/j.ijcard.2015.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van Amerongen MJ, Harmsen MC, van Rooijen N, Petersen AH, van Luyn MJA. Macrophage depletion impairs wound healing and increases left ventricular remodeling after myocardial injury in mice. Am J Pathol. 2007;170:818–29. doi: 10.2353/ajpath.2007.060547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karagueuzian HS. Targeting cardiac fibrosis: a new frontier in antiarrhythmic therapy? Am J Cardiovasc Dis. 2011;1:101–9. [PMC free article] [PubMed] [Google Scholar]
- 8.Kohl P, Gourdie RG. Fibroblast-myocyte electrotonic coupling: does it occur in native cardiac tissue? J Mol Cell Cardiol. 2014;70:37–46. doi: 10.1016/j.yjmcc.2013.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lue WM, Boyden PA. Abnormal electrical properties of myocytes from chronically infarcted canine heart. Alterations in Vmax and the transient outward current. Circulation. 1992;85:1175–88. doi: 10.1161/01.cir.85.3.1175. [DOI] [PubMed] [Google Scholar]
- 10.Pinto JM, Boyden PA. Electrical remodeling in ischemia and infarction. Cardiovasc Res. 1999;42:284–97. doi: 10.1016/s0008-6363(99)00013-9. [DOI] [PubMed] [Google Scholar]
- 11.Peters NS, Coromilas J, Severs NJ, Wit AL. Disturbed connexin43 gap junction distribution correlates with the location of reentrant circuits in the epicardial border zone of healing canine infarcts that cause ventricular tachycardia. Circulation. 1997;95:988–96. doi: 10.1161/01.cir.95.4.988. [DOI] [PubMed] [Google Scholar]
- 12.Yao J-A, Hussain W, Patel P, Peters NS, Boyden PA, Wit AL. Remodeling of gap junctional channel function in epicardial border zone of healing canine infarcts. Circ Res. 2003;92:437–43. doi: 10.1161/01.RES.0000059301.81035.06. [DOI] [PubMed] [Google Scholar]
- 13.Pu J, Robinson RB, Boyden PA. Abnormalities in Ca(i)handling in myocytes that survive in the infarcted heart are not just due to alterations in repolarization. J Mol Cell Cardiol. 2000;32:1509–23. doi: 10.1006/jmcc.2000.1184. [DOI] [PubMed] [Google Scholar]
- 14.Dun W, Keurs ter H, Boyden PA. In: The Role of Intracellular Ca2+ in Arrhythmias in the Postmyocardial Infarction Heart. Tripathi ON, Ravens U, Sanguinetti MC, editors. Berlin, Heidelberg: Springer Berlin Heidelberg; 2011. pp. 305–22. [DOI] [Google Scholar]
- 15.Streitner F, Kuschyk J, Veltmann C, Brueckmann M, Streitner I, Brade J, et al. Prospective study of interleukin-6 and the risk of malignant ventricular tachyarrhythmia in ICD-recipients--a pilot study. Cytokine. 2007;40:30–4. doi: 10.1016/j.cyto.2007.07.187. [DOI] [PubMed] [Google Scholar]
- 16.Streitner F, Kuschyk J, Veltmann C, Ratay D, Schoene N, Streitner I, et al. Role of proinflammatory markers and NT-proBNP in patients with an implantable cardioverter-defibrillator and an electrical storm. Cytokine. 2009;47:166–72. doi: 10.1016/j.cyto.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 17.Maradit-Kremers H, Crowson CS, Nicola PJ, Ballman KV, Roger VL, Jacobsen SJ, et al. Increased unrecognized coronary heart disease and sudden deaths in rheumatoid arthritis: a population-based cohort study. Arthritis Rheum. 2005;52:402–11. doi: 10.1002/art.20853. [DOI] [PubMed] [Google Scholar]
- 18.Panizzi P, Swirski FK, Figueiredo J-L, Waterman P, Sosnovik DE, Aikawa E, et al. Impaired infarct healing in atherosclerotic mice with Ly-6C(hi) monocytosis. J Am Coll Cardiol. 2010;55:1629–38. doi: 10.1016/j.jacc.2009.08.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yabluchanskiy A, Ma Y, DeLeon-Pennell KY, Altara R, Halade GV, Voorhees AP, et al. Myocardial Infarction Superimposed on Aging: MMP-9 Deletion Promotes M2 Macrophage Polarization. J Gerontol a Biol Sci Med Sci. 2015 doi: 10.1093/gerona/glv034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DeLeon-Pennell KY, de Castro Brás LE, Iyer RP, Bratton DR, Jin Y-F, Ripplinger CM, et al. P. gingivalis lipopolysaccharide intensifies inflammation post-myocardial infarction through matrix metalloproteinase-9. J Mol Cell Cardiol. 2014;76C:218–26. doi: 10.1016/j.yjmcc.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.London B, Baker LC, Lee JS, Shusterman V, Choi B-R, Kubota T, et al. Calcium-dependent arrhythmias in transgenic mice with heart failure. Am J Physiol Heart Circ Physiol. 2003;284:H431–41. doi: 10.1152/ajpheart.00431.2002. [DOI] [PubMed] [Google Scholar]
- 22.Petkova-Kirova PS, Gursoy E, Mehdi H, McTiernan CF, London B, Salama G. Electrical remodeling of cardiac myocytes from mice with heart failure due to the overexpression of tumor necrosis factor-alpha. Am J Physiol Heart Circ Physiol. 2006;290:H2098–107. doi: 10.1152/ajpheart.00097.2005. [DOI] [PubMed] [Google Scholar]
- 23.Fernández-Velasco M, Ruiz-Hurtado G, Hurtado O, Moro MA, Delgado C. TNF-alpha downregulates transient outward potassium current in rat ventricular myocytes through iNOS overexpression and oxidant species generation. Am J Physiol Heart Circ Physiol. 2007;293:H238–45. doi: 10.1152/ajpheart.01122.2006. [DOI] [PubMed] [Google Scholar]
- 24.Kawada H, Niwano S, Niwano H, Yumoto Y, Wakisaka Y, Yuge M, et al. Tumor necrosis factor-alpha downregulates the voltage gated outward K+ current in cultured neonatal rat cardiomyocytes: a possible cause of electrical remodeling in diseased hearts. Circ J. 2006;70:605–9. doi: 10.1253/circj.70.605. [DOI] [PubMed] [Google Scholar]
- 25.Wang J, Wang H, Zhang Y, Gao H, Nattel S, Wang Z. Impairment of HERG K(+) channel function by tumor necrosis factor-alpha: role of reactive oxygen species as a mediator. J Biol Chem. 2004;279:13289–92. doi: 10.1074/jbc.C400025200. [DOI] [PubMed] [Google Scholar]
- 26.Finkel MS, Oddis CV, Jacob TD, Watkins SC, Hattler BG, Simmons RL. Negative inotropic effects of cytokines on the heart mediated by nitric oxide. Science. 1992;257:387–9. doi: 10.1126/science.1631560. [DOI] [PubMed] [Google Scholar]
- 27.Greensmith DJ, Nirmalan M. The effects of tumor necrosis factor-alpha on systolic and diastolic function in rat ventricular myocytes. Physiol Rep. 2013;1:e00093. doi: 10.1002/phy2.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu C-K, Lee J-K, Chiang F-T, Yang C-H, Huang S-W, Hwang J-J, et al. Plasma levels of tumor necrosis factor-α and interleukin-6 are associated with diastolic heart failure through downregulation of sarcoplasmic reticulum Ca2+ ATPase. Crit Care Med. 2011;39:984–92. doi: 10.1097/CCM.0b013e31820a91b9. [DOI] [PubMed] [Google Scholar]
- 29.Duncan DJ, Yang Z, Hopkins PM, Steele DS, Harrison SM. TNF-alpha and IL-1beta increase Ca2+ leak from the sarcoplasmic reticulum and susceptibility to arrhythmia in rat ventricular myocytes. Cell Calcium. 2010;47:378–86. doi: 10.1016/j.ceca.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sawaya SE, Rajawat YS, Rami TG, Szalai G, Price RL, Sivasubramanian N, et al. Downregulation of connexin40 and increased prevalence of atrial arrhythmias in transgenic mice with cardiac-restricted overexpression of tumor necrosis factor. Am J Physiol Heart Circ Physiol. 2007;292:H1561–7. doi: 10.1152/ajpheart.00285.2006. [DOI] [PubMed] [Google Scholar]
- 31.Fernandez-Cobo M, Gingalewski C, Drujan D, De Maio A. Downregulation of connexin 43 gene expression in rat heart during inflammation. The role of tumour necrosis factor. Cytokine. 1999;11:216–24. doi: 10.1006/cyto.1998.0422. [DOI] [PubMed] [Google Scholar]
- 32.Saxena A, Chen W, Su Y, Rai V, Uche OU, Li N, et al. IL-1 induces proinflammatory leukocyte infiltration and regulates fibroblast phenotype in the infarcted myocardium. J Immunol. 2013;191:4838–48. doi: 10.4049/jimmunol.1300725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guillén I, Blanes M, Gómez-Lechón MJ, Castell JV. Cytokine signaling during myocardial infarction: sequential appearance of IL-1 beta and IL-6. Am J Physiol. 1995;269:R229–35. doi: 10.1152/ajpregu.1995.269.2.R229. [DOI] [PubMed] [Google Scholar]
- 34.Liu S, Schreur KD. G protein-mediated suppression of L-type Ca2+ current by interleukin-1 beta in cultured rat ventricular myocytes. Am J Physiol. 1995;268:C339–49. doi: 10.1152/ajpcell.1995.268.2.C339. [DOI] [PubMed] [Google Scholar]
- 35.Schreur KD, Liu S. Involvement of ceramide in inhibitory effect of IL-1 beta on L-type Ca2+ current in adult rat ventricular myocytes. Am J Physiol. 1997;272:H2591–8. doi: 10.1152/ajpheart.1997.272.6.H2591. [DOI] [PubMed] [Google Scholar]
- 36.Khoury El N, Mathieu S, Fiset C. Interleukin-1β reduces L-type Ca2+ current through protein kinase Cε activation in mouse heart. Journal of Biological Chemistry. 2014;289:21896–908. doi: 10.1074/jbc.M114.549642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li YH, Rozanski GJ. Effects of human recombinant interleukin-1 on electrical properties of guinea pig ventricular cells. Cardiovasc Res. 1993;27:525–30. doi: 10.1093/cvr/27.3.525. [DOI] [PubMed] [Google Scholar]
- 38.Liu SJ, Zhou W, Kennedy RH. Suppression of beta-adrenergic responsiveness of L-type Ca2+ current by IL-1beta in rat ventricular myocytes. Am J Physiol. 1999;276:H141–8. doi: 10.1152/ajpheart.1999.276.1.H141. [DOI] [PubMed] [Google Scholar]
- 39.Combes A, Frye CS, Lemster BH, Brooks SS, Watkins SC, Feldman AM, et al. Chronic exposure to interleukin 1beta induces a delayed and reversible alteration in excitation-contraction coupling of cultured cardiomyocytes. Pflugers Arch. 2002;445:246–56. doi: 10.1007/s00424-002-0921-y. [DOI] [PubMed] [Google Scholar]
- 40.Duncan DJ, Hopkins PM, Harrison SM. Negative inotropic effects of tumour necrosis factor-alpha and interleukin-1beta are ameliorated by alfentanil in rat ventricular myocytes. Br J Pharmacol. 2007;150:720–6. doi: 10.1038/sj.bjp.0707147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thaik CM, Calderone A, Takahashi N, Colucci WS. Interleukin-1 beta modulates the growth and phenotype of neonatal rat cardiac myocytes. J Clin Invest. 1995;96:1093–9. doi: 10.1172/JCI118095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McTiernan CF, Lemster BH, Frye C, Brooks S, Combes A, Feldman AM. Interleukin-1 beta inhibits phospholamban gene expression in cultured cardiomyocytes. Circ Res. 1997;81:493–503. doi: 10.1161/01.res.81.4.493. [DOI] [PubMed] [Google Scholar]
- 43.Patten M, Hartogensis WE, Long CS. Interleukin-1beta is a negative transcriptional regulator of alpha1-adrenergic induced gene expression in cultured cardiac myocytes. J Biol Chem. 1996;271:21134–41. doi: 10.1074/jbc.271.35.21134. [DOI] [PubMed] [Google Scholar]
- 44.John GR, Scemes E, Suadicani SO, Liu JS, Charles PC, Lee SC, et al. IL-1beta differentially regulates calcium wave propagation between primary human fetal astrocytes via pathways involving P2 receptors and gap junction channels. P Natl Acad Sci Usa. 1999;96:11613–8. doi: 10.1073/pnas.96.20.11613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Baum JR, Long B, Cabo C, Duffy HS. Myofibroblasts cause heterogeneous Cx43 reduction and are unlikely to be coupled to myocytes in the healing canine infarct. AJP: Heart and Circulatory Physiology. 2012;302:H790–800. doi: 10.1152/ajpheart.00498.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baum JR, Dolmatova E, Tan A, Duffy HS. Omega 3 fatty acid inhibition of inflammatory cytokine-mediated Connexin43 regulation in the heart. Front Physiol. 2012;3:272. doi: 10.3389/fphys.2012.00272/abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.De Jesus NM, Wang L, Herren AW, Wang J, Shenasa F, Bers DM, et al. Atherosclerosis exacerbates arrhythmia following myocardial infarction: Role of myocardial inflammation. Heart Rhythm. 2015;12:169–78. doi: 10.1016/j.hrthm.2014.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hagiwara Y, Miyoshi S, Fukuda K, Nishiyama N, Ikegami Y, Tanimoto K, et al. SHP2-mediated signaling cascade through gp130 is essential for LIF-dependent I CaL, [Ca2+]i transient, and APD increase in cardiomyocytes. J Mol Cell Cardiol. 2007;43:710–6. doi: 10.1016/j.yjmcc.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 49.Tanaka T, Kanda T, Takahashi T, Saegusa S, Moriya J, Kurabayashi M. Interleukin-6-induced reciprocal expression of SERCA and natriuretic peptides mRNA in cultured rat ventricular myocytes. J Int Med Res. 2004;32:57–61. doi: 10.1177/147323000403200109. [DOI] [PubMed] [Google Scholar]
- 50.Yu X-W, Chen Q, Kennedy RH, Liu SJ. Inhibition of sarcoplasmic reticular function by chronic interleukin-6 exposure via iNOS in adult ventricular myocytes. J Physiol (Lond) 2005;566:327–40. doi: 10.1113/jphysiol.2005.086686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Parrish DC, Alston EN, Rohrer H, Hermes SM, Aicher SA, Nkadi P, et al. Absence of gp130 in dopamine beta-hydroxylase-expressing neurons leads to autonomic imbalance and increased reperfusion arrhythmias. AJP: Heart and Circulatory Physiology. 2009;297:H960–7. doi: 10.1152/ajpheart.00409.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lindsey ML, Escobar GP, Mukherjee R, Goshorn DK, Sheats NJ, Bruce JA, et al. Matrix metalloproteinase-7 affects connexin-43 levels, electrical conduction, and survival after myocardial infarction. Circulation. 2006;113:2919–28. doi: 10.1161/CIRCULATIONAHA.106.612960. [DOI] [PubMed] [Google Scholar]
- 53.Wu X, Huang W, Luo G, Alain LA. Hypoxia induces connexin 43 dysregulation by modulating matrix metalloproteinases via MAPK signaling. Mol Cell Biochem. 2013;384:155–62. doi: 10.1007/s11010-013-1793-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mukherjee R, Colbath GP, Justus CD, Bruce JA, Allen CM, Hewett KW, et al. Spatiotemporal induction of matrix metalloproteinase-9 transcription after discrete myocardial injury. Faseb J. 2010;24:3819–28. doi: 10.1096/fj.10-155531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mohammad G, Kowluru RA. Novel role of mitochondrial matrix metalloproteinase-2 in the development of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2011;52:3832–41. doi: 10.1167/iovs.10-6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bujak M, Frangogiannis NG. The role of TGF-beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc Res. 2007;74:184–95. doi: 10.1016/j.cardiores.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ripplinger CM, Lou Q, Li W, Hadley J, Efimov IR. Panoramic imaging reveals basic mechanisms of induction and termination of ventricular tachycardia in rabbit heart with chronic infarction: implications for low-voltage cardioversion. Heart Rhythm. 2009;6:87–97. doi: 10.1016/j.hrthm.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nguyen TP, Qu Z, Weiss JN. Cardiac fibrosis and arrhythmogenesis: the road to repair is paved with perils. J Mol Cell Cardiol. 2014;70:83–91. doi: 10.1016/j.yjmcc.2013.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dhein S, Seidel T, Salameh A, Jozwiak J, Hagen A, Kostelka M, et al. Remodeling of cardiac passive electrical properties and susceptibility to ventricular and atrial arrhythmias. Front Physiol. 2014;5:424. doi: 10.3389/fphys.2014.00424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xie Y, Sato D, Garfinkel A, Qu Z, Weiss JN. So little source, so much sink: requirements for afterdepolarizations to propagate in tissue. Biophys J. 2010;99:1408–15. doi: 10.1016/j.bpj.2010.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Myles RC, Wang L, Kang C, Bers DM, Ripplinger CM. Local β-adrenergic stimulation overcomes source-sink mismatch to generate focal arrhythmia. Circ Res. 2012;110:1454–64. doi: 10.1161/CIRCRESAHA.111.262345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Morita N, Sovari AA, Xie Y, Fishbein MC, Mandel WJ, Garfinkel A, et al. Increased susceptibility of aged hearts to ventricular fibrillation during oxidative stress. Am J Physiol Heart Circ Physiol. 2009;297:H1594–605. doi: 10.1152/ajpheart.00579.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tschabrunn CM, Roujol S, Nezafat R, Faulkner-Jones B, Buxton AE, Josephson ME, et al. A swine model of infarct-related reentrant ventricular tachycardia: Electroanatomic, magnetic resonance, and histopathological characterization. Heart Rhythm. 2015 doi: 10.1016/j.hrthm.2015.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.van den Borne SWM, Díez J, Blankesteijn WM, Verjans J, Hofstra L, Narula J. Myocardial remodeling after infarction: the role of myofibroblasts. Nat Rev Cardiol. 2010;7:30–7. doi: 10.1038/nrcardio.2009.199. [DOI] [PubMed] [Google Scholar]
- 65.Gaudesius G, Miragoli M, Thomas SP, Rohr S. Coupling of cardiac electrical activity over extended distances by fibroblasts of cardiac origin. Circ Res. 2003;93:421–8. doi: 10.1161/01.RES.0000089258.40661.0C. [DOI] [PubMed] [Google Scholar]
- 66.Chilton L, Giles WR, Smith GL. Evidence of intercellular coupling between co-cultured adult rabbit ventricular myocytes and myofibroblasts. J Physiol (Lond) 2007;583:225–36. doi: 10.1113/jphysiol.2007.135038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Miragoli M, Gaudesius G, Rohr S. Electrotonic modulation of cardiac impulse conduction by myofibroblasts. Circ Res. 2006;98:801–10. doi: 10.1161/01.RES.0000214537.44195.a3. [DOI] [PubMed] [Google Scholar]
- 68.Camelliti P, Devlin GP, Matthews KG, Kohl P, Green CR. Spatially and temporally distinct expression of fibroblast connexins after sheep ventricular infarction. Cardiovasc Res. 2004;62:415–25. doi: 10.1016/j.cardiores.2004.01.027. [DOI] [PubMed] [Google Scholar]
- 69.Xie Y, Garfinkel A, Camelliti P, Kohl P, Weiss JN, Qu Z. Effects of fibroblast-myocyte coupling on cardiac conduction and vulnerability to reentry: A computational study. Heart Rhythm. 2009;6:1641–9. doi: 10.1016/j.hrthm.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McDowell KS, Arevalo HJ, Maleckar MM, Trayanova NA. Susceptibility to arrhythmia in the infarcted heart depends on myofibroblast density. Biophys J. 2011;101:1307–15. doi: 10.1016/j.bpj.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nattel S, Andrade J, Macle L, Rivard L, Dyrda K, Mondesert B, et al. New directions in cardiac arrhythmia management: present challenges and future solutions. Can J Cardiol. 2014;30:S420–30. doi: 10.1016/j.cjca.2014.09.027. [DOI] [PubMed] [Google Scholar]
- 72.Waldo AL, Camm AJ, de Ruyter H, Friedman PL, MacNeil DJ, Pauls JF, et al. Effect of d-sotalol on mortality in patients with left ventricular dysfunction after recent and remote myocardial infarction. The SWORD Investigators. Survival With Oral d-Sotalol. Lancet. 1996;348:7–12. doi: 10.1016/s0140-6736(96)02149-6. [DOI] [PubMed] [Google Scholar]
- 73.Hammerman H, Kloner RA, Schoen FJ, Brown EJ, Hale S, Braunwald E. Indomethacin-induced scar thinning after experimental myocardial infarction. Circulation. 1983;67:1290–5. doi: 10.1161/01.cir.67.6.1290. [DOI] [PubMed] [Google Scholar]
- 74.Giugliano GR, Giugliano RP, Gibson CM, Kuntz RE. Meta-analysis of corticosteroid treatment in acute myocardial infarction. Am J Cardiol. 2003;91:1055–9. doi: 10.1016/S0002-9149(03)00148-6. [DOI] [PubMed] [Google Scholar]
- 75.American College of Emergency Physicians, Society for Cardiovascular Angiography and Interventions. O’Gara PT, Kushner FG, Ascheim DD, Casey DE, et al. 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2013;61:e78–140. doi: 10.1016/j.jacc.2012.11.019. [DOI] [PubMed] [Google Scholar]
- 76.Moliterno DJ, Smyth SS. Tissue necrosis factor α and targeting its receptor in ischaemic heart disease. Heart. 2013;99:1305–6. doi: 10.1136/heartjnl-2013-303960. [DOI] [PubMed] [Google Scholar]
- 77.Kurrelmeyer KM, Michael LH, Baumgarten G, Taffet GE, Peschon JJ, Sivasubramanian N, et al. Endogenous tumor necrosis factor protects the adult cardiac myocyte against ischemic-induced apoptosis in a murine model of acute myocardial infarction. P Natl Acad Sci Usa. 2000;97:5456–61. doi: 10.1073/pnas.070036297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Abbate A, Kontos MC, Grizzard JD, Biondi-Zoccai GGL, Van Tassell BW, Robati R, et al. Interleukin-1 blockade with anakinra to prevent adverse cardiac remodeling after acute myocardial infarction (Virginia Commonwealth University Anakinra Remodeling Trial [VCU-ART] Pilot study) Am J Cardiol. 2010;105:1371–1. doi: 10.1016/j.amjcard.2009.12.059. [DOI] [PubMed] [Google Scholar]
- 79.Abbate A, Van Tassell BW, Seropian IM, Toldo S, Robati R, Varma A, et al. Interleukin-1beta modulation using a genetically engineered antibody prevents adverse cardiac remodelling following acute myocardial infarction in the mouse. Eur J Heart Fail. 2010;12:319–22. doi: 10.1093/eurjhf/hfq017. [DOI] [PubMed] [Google Scholar]
- 80.Van Tassell BW, Varma A, Salloum FN, Das A, Seropian IM, Toldo S, et al. Interleukin-1 trap attenuates cardiac remodeling after experimental acute myocardial infarction in mice. J Cardiovasc Pharmacol. 2010;55:117–22. doi: 10.1097/FJC.0b013e3181c87e53. [DOI] [PubMed] [Google Scholar]
- 81.Ridker PM, Thuren T, Zalewski A, Libby P. Interleukin-1β inhibition and the prevention of recurrent cardiovascular events: rationale and design of the Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS) Am Heart J. 2011;162:597–605. doi: 10.1016/j.ahj.2011.06.012. [DOI] [PubMed] [Google Scholar]
- 82.Dobaczewski M, Bujak M, Li N, Gonzalez-Quesada C, Mendoza LH, Wang X-F, et al. Smad3 signaling critically regulates fibroblast phenotype and function in healing myocardial infarction. Circ Res. 2010;107:418–28. doi: 10.1161/CIRCRESAHA.109.216101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bujak M, Ren G, Kweon HJ, Dobaczewski M, Reddy A, Taffet G, et al. Essential role of Smad3 in infarct healing and in the pathogenesis of cardiac remodeling. Circulation. 2007;116:2127–38. doi: 10.1161/CIRCULATIONAHA.107.704197. [DOI] [PubMed] [Google Scholar]
- 84.Nguyen DT, Ding C, Wilson E, Marcus GM, Olgin JE. Pirfenidone mitigates left ventricular fibrosis and dysfunction after myocardial infarction and reduces arrhythmias. Heart Rhythm. 2010;7:1438–45. doi: 10.1016/j.hrthm.2010.04.030. [DOI] [PubMed] [Google Scholar]
- 85.Lindsey ML, Zamilpa R. Temporal and spatial expression of matrix metalloproteinases and tissue inhibitors of metalloproteinases following myocardial infarction. Cardiovasc Ther. 2012;30:31–41. doi: 10.1111/j.1755-5922.2010.00207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang G-Y, Bergman MR, Nguyen AP, Turcato S, Swigart PM, Rodrigo MC, et al. Cardiac transgenic matrix metalloproteinase-2 expression directly induces impaired contractility. Cardiovasc Res. 2006;69:688–96. doi: 10.1016/j.cardiores.2005.08.023. [DOI] [PubMed] [Google Scholar]
- 87.Lemaître V, O’Byrne TK, Dalal SS, Tall AR, D’Armiento JM. Macrophage-specific expression of human collagenase (MMP-1) in transgenic mice. Annals of the New York Academy of Sciences. 1999;878:736–9. doi: 10.1111/j.1749-6632.1999.tb07776.x. [DOI] [PubMed] [Google Scholar]
- 88.Yabluchanskiy A, Li Y, Chilton RJ, Lindsey ML. Matrix metalloproteinases: drug targets for myocardial infarction. Curr Drug Targets. 2013;14:276–86. [PMC free article] [PubMed] [Google Scholar]
- 89.Tang Z, McGowan BS, Huber SA, McTiernan CF, Addya S, Surrey S, et al. Gene expression profiling during the transition to failure in TNF-alpha over-expressing mice demonstrates the development of autoimmune myocarditis. J Mol Cell Cardiol. 2004;36:515–30. doi: 10.1016/j.yjmcc.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 90.Hatada K, Washizuka T, Horie M, Watanabe H, Yamashita F, Chinushi M, et al. Tumor necrosis factor-alpha inhibits the cardiac delayed rectifier K current via the asphingomyelin pathway. Biochem Biophys Res Commun. 2006;344:189–93. doi: 10.1016/j.bbrc.2006.03.115. [DOI] [PubMed] [Google Scholar]