

Abstract
Bifidobacteria are intestinal anaerobes often associated with gut health. Specific bifidobacterial species are particularly common in the gastrointestinal tract of breast-fed infants. Current short read next-generation sequencing approaches to profile fecal microbial ecologies do not discriminate bifidobacteria to the species level. Here we describe a low-cost terminal restriction fragment length polymorphism (TRFLP) procedure to distinguish between the common infant-associated bifidobacterial species. An empirical database of TRF sizes was created from both common reference strains and well-identified isolates from infant feces. Species-specific quantitative PCR validated bifidobacterial-specific TRFLP profiles from infant feces. These results indicate that bifidobacterial-specific TRFLP is a useful method to monitor intestinal bifidobacterial populations from infant fecal samples. When used alongside next generation sequencing methods that detect broader population levels at lower resolution, this high-throughput, low-cost tool can help clarify the role of bifidobacteria in health and disease.
Keywords: TRFLP, Bifidobacteria, Infants, Gut microbiota, NGS
1. Introduction
Bifidobacteria are Gram-positive anaerobic bacteria commonly found in the breast-fed infant intestine [1–5]. The main infant-associated members of this genus include Bifidobacterium longum subsp. infantis, Bifidobacterium longum subsp. longum, Bifidobacterium breve, Bifidobacterium bifidum, Bifidobacterium pseudocatenulatum, and occasionally Bifidobacterium adolescentis and Bifidobacterium animalis [6–8]. The genomic diversity among these species suggests that they have different metabolic capabilities, occupy different niches in the gut, and play different roles within the gut microbiota, especially with regards to their ability to degrade human milk oligosaccharides (HMOs) [9–12]. These differences impact our understanding of the role bifidobacterial colonization plays on infant development. In order to make statistically significant connections between particular bifidobacterial species and environmental factors such as diet, host genetics, the presence of other microbes (including potentially harmful ones), and health outcomes, it is necessary to investigate large numbers of individuals over relatively long periods of time. This requires a high-throughput method for looking at the species-level distribution of bifidobacteria.
Of the methods available for the analysis of microbial community structure, there are few good options for accomplishing the above-mentioned goals in a cost-effective manner. Next-generation sequencing (NGS) is an increasingly common approach for extensive microbial ecology studies; however short read-based NGS technologies are not typically able to provide species-level identification, though with longer read lengths this may be possible in the future. Pyrosequencing can sometimes discriminate between bifidobacteria at a species level [13,14], but remains expensive on a per-sample basis for many applications. Species-specific quantitative PCR (qPCR) is not cost effective for large sample numbers, is time-consuming and does not enumerate non-targeted species. Denaturing gel gradient electrophoresis (DGGE) could provide species-level differentiation [15], however is technically challenging and poorly adapted for high-throughput and routine analyses.
Terminal restriction fragment length polymorphism (TRFLP) is used to evaluate microbial ecology in a culture-independent high-throughput manner [16]. TRFLP is a useful technique for community profiling as it is adaptable, sensitive, rapid, inexpensive, and technologically accessible, enabling comparisons of large, time- and treatment-based sample sets. It is, however, only pseudoquantitative, providing an estimate of the abundance of the taxa present [16]. Here we describe a TRFLP method for evaluating species-level bifidobacterial content (Bif-TRFLP) of feces. This approach enables detailed description of bifidobacterial taxa from infant fecal samples in a rapid, high-throughput, and cost-effective manner. When combined with other measures of microbial ecology, such as short read sequencing and/or genus-specific qPCR, it can be used to fill out the details of emerging stories involving bifidobacteria and the infant gut.
2. Materials and methods
2.1. Sample selection and collection
The study was approved by the Institutional Review Board at UC Davis (protocol # 200715509-4) and informed consent was obtained from the parents of all infants prior to participation. Stool samples were collected at different time points (week of life 0, 23, 29, and 35) after birth from four healthy full-term infants (infant A, B, C, and D, respectively) and stored at −80 °C until processing.
2.2. DNA extractions
Genomic DNA was extracted from pure cell cultures and fecal samples using either the Qiagen Stool Kit (Valencia, California) with the Gram-positive protocol adding an enzymatic lysis and a bead-beating step [17]. The Epicentre Masterpure Gram Positive kit (Madison, Wisconsin) was also used to purify genomic DNA from individual strains in the case of a few of the empirical database samples.
2.3. Illumina sequencing
DNA samples were prepared as previously described [18] with the following modifications. Universal barcoded primers with Illumina sequencing adapters (adapters are italicized and the barcode is highlighted in bold) V4F (5′-AATGATACGGCGACCACCGA GATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT ACTGCTGAGTGTG CCAGCMGCCGCGGTAA-3′) and V4Rev (5′-CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCTCCGGACTACHVGGGTWTCTAAT-3′) were used to PCR amplify the V4 region of the 16S rRNA gene [18]. PCR reactions were carried out in triplicate and contained 12.5 μl 2× GoTaq Green Master Mix (Promega, Madison, WI), 1.0 μl 25 mM MgCl2, 8.5 μl water, 0.5 μl forward and reverse primers (10 μM final concentration), and 2.0 μl genomic DNA. The triplicate reactions were combined and cleaned and DNA concentrations were quantified using the PicoGreen dsDNA Kit (Invitrogen, Carlsbad, California). An equimolar composite sample mixture was made, gel purified, and sequenced at the University of California DNA Technologies Core Facility on an Illumina Genome Analyzer II sequencing platform.
2.4. Sequence analysis
The QIIME software package (version 1.4.0) was used to analyze the results of the Illumina sequencing run [19]. Illumina V4 16S rRNA gene sequences were first demultiplexed and quality filtered. Reads were truncated after a maximum number of 3 consecutive low quality scores. After quality trimming, reads were removed from analysis if they were <60 bp and the number of ambiguous bases were >3. Sequences with a minimum identity of 97% were clustered into operational taxonomic units (OTUs) with UCLUST software [20]. The most abundant sequence was chosen to represent each OTU. Taxonomy was assigned to each OTU with the Ribosomal Database Project (RDP) classifier [21] with a minimum confidence threshold of 80% and the RDP taxonomic nomenclature [22]. OTU representatives were aligned against the Greengenes core set [23] with PyNAST software [24] with a minimum alignment length of 75 bp and a minimum identity of 75%.
2.5. QPCR
SYBR green and TaqMan quantitative PCR (qPCR) assays were performed on a 7500 Fast Real-Time PCR System (Applied Biosystems, Carlsbad, CA) with species-specific primers for B. longum group, B. adolescentis group, B. breve, B. bifidum, and Bifidobacterium catenulatum group [25,26] (see Table S1 for details). SYBR green assays contained 10 μl of 2× Takara Perfect Real Time master mix (TaKaRa, Japan), 6 μl water, 1 μl each forward and reverse primers, and 2 μl genomic DNA. Cycling conditions are described in Table S1. B. longum group TaqMan assays contained 10 μl 2× TaqMan Universal PCR master mix (Applied Biosystems), 1 μl each forward and reverse primers and TaqMan probe, 5 μl water, and 2 μl genomic DNA. Reaction conditions and primer concentrations were as described in supplemental Table S1. All reactions were carried out in triplicate with a nontemplate control.
2.6. Bif-TRFLP
Primers were designed to target a consensus region of the bifidobacterial 16S rRNA gene. The consensus region was determined by a Clustal × 2.0 [27] alignment of all bifidobacteria in the RDP database (release 10) [22,28]. Primer specificity and taxonomic coverage were predicted using PrimerProspector [29] checking against a representative subset of the Greengenes 16S rRNA database filtered at 97% identity [23].
Samples were amplified in 50 μl PCR reactions containing 1 μl of genomic DNA, 25 μl of 2× Promega GoTaq Green PCR master mix, 20 μl of molecular grade water, 5 pmol of each primer (NBIF389 5′[HEX]-GCCTTCGGGTTGTAAAC-3 and NBIF1018REV 5′-GACCATG-CACCACCTGTG-3′), and 1 mM MgCL2. The reaction conditions were 95 °C for 5 min followed by 30 cycles of 95 °C for 1 min, 52 °C for 1 min and 72 °C for 1 min. A final elongation was allowed at the end of the cycles at 72 °C for 5 min, and samples were stored at 4 °C. Successful amplification was confirmed by gel electrophoresis of 5 μl of the finished reaction in a 0.8% agarose gel in 1× TAE stained with Gel Green, and then visualized under UV light. DNA was purified from the reaction using QIAquick PCR purification columns with the manufacturer’s instructions, and eluted in 30 μl of buffer EB.
A portion of the purified DNA was cut with two restriction enzymes (AluI and HaeIII) in separate reactions. Digests involved 1 μl of enzyme in a 10 μl reaction for 3 h at 37 °C. The enzymes were heat inactivated according to supplier’s instructions.
Next, 1.5 μl of the digestion mixture was used for fragment analysis on an ABI 3730 Capillary Electrophoresis Genetic Analyzer at the UC Davis College of Biological Sciences Sequencing Facility. The molecular size markers used were the Gel Company ROX 50–500 size standards (Gel Company Inc., San Francisco, CA).
The resulting electropherograms were read using Peak Scanner software v1.0, (Applied Biosystems). Manual assignment of size standard peaks was performed as necessary. The empirical database was manually constructed by using the Bif-TRFLP method on several Bifidobacterium species from culture collections as well as numerous Bifidobacterium isolates obtained from infant feces. Size predictions were made using the most common TRF size(s) for a given species from a MiCA virtual digest of the RDP bacterial 16S dataset [30].
Peaks from fecal samples were compared to TRF sizes from the empirical database for identification. The data were entered into R software for peak filtering and binning using the scripts of Abdo and coworkers [31] with a filtering cutoff of 1–3 standard deviations. A binning cutoff of 0.1–0.25 bp was found to be optimal. This relatively strict parameter setting is needed due to many of the peaks of different species being found in the same size range. Peak calling and binning were confirmed manually and adjusted as necessary to conform to the electropherograms. In the case of an off-scale peak read as two peaks by the software, the two sizes were averaged together (weighted by peak area) and recorded as one peak. Issues with peak identification in peak calling software have been noted before [32]. Peaks were rounded to the nearest halfbase pair as necessary. Results were calculated as a percentage of the total post-filtering peak area from the electropherograms, excluding primer-dimer and smaller peaks (<35 bp).
2.7. Limit of detection
A genomic DNA dilution series of 6 infant bifidobacterial species (B. breve ATCC15700, B. longum ssp. longum DJO10A, B. longum ssp. infantis ATCC15697, B. adolescentis ATCC15703, B. animalis UCD VEN 316, and B. bifidum DSM20456) in the presence of ~14 ng/μl non-target (Escherichia coli) DNA was created. The initial stock DNA concentration of each species was between 10 and 100 ng/μl and 1 ml of each stock was added to form a multi-species mix. One ml of this mix was added to the PCR master mix (1 μl was also used in each subsequent dilution). In calculating the detection limit we used 1.023 × 10−9 pg as the weight of one base pair and 2.5 Mbp as the average bifidobacterial genome size.
3. Results
3.1. Primer coverage
For Bif-TRFLP we designed primers to amplify a portion of the 16S gene of members of the genus Bifidobacterium. Taxonomic specificity and coverage of the Bif-TRFLP primers (as tested by PrimerProspector [29]) revealed that Bifidobacteriales was the only order significantly covered (see Fig. 1). One other phylum (MVP-15) was partially covered, however that represented only a few species. In addition, a few lesser-known Actinobacteria families related to Bifidobacteriaceae received very high coverage, however they also represent a small absolute number of species. An in silico database created from a MiCA virtual digest [30] showed that all the non-target species picked up by PrimerProspector are easily differentiated from the target bifidobacteria by the size of their TRFs. Fig. 1 also shows the high coverage of bifidobacterial species achieved by these primers.
Fig. 1.

Taxonomic coverage of primers used according to PrimerProspector. The x-axis represents percent coverage within a given taxon. The numbers next to the bar for each taxon give the number of species contained within that taxon.
3.2. Empirical database
To verify that the MiCA virtual digest was representative of actual results from the Bif-TRFLP method, we created an empirical database of TRF sizes from pure strains. As shown in Table 1, the TRF sizes showed good consistency within each species. There was one discrepancy, namely the TRF sizes of B. breve SC559, but this is understandable in light of the high diversity present between B. breve species [33]. The main peaks also corresponded well with the predicted TRF size once variability in electrophoretic mobility due to fluorophore conjugation is taken into account [34,35]. One interesting phenomenon shown in Table 1 is the presence of multiple peaks for each species. These peaks are consistent within a species and generally distinct between species, and thus do not impact species assignment in most scenarios. Smaller-than-predicted TRFs may be the result of truncated PCR products and are discussed below.
Table 1.
TRF size database. “Predicted” columns refer to the MiCA virtual digest; “empirical” columns refer to the TRF size shown by our methods.
| Strain | AluI predicted | AluI empirical | HaeIII predicted | HaeIII empirical |
|---|---|---|---|---|
| B. adolescentis ATCC15703 | 628.5 | 621, 425, 144, 140 | 311 | 310, 306, 286, 464 |
| B. adolescentis SC590 | 628.5 | 623, 570, 144 | 311 | 310, 287, 464 |
| B. adolescentis SC656 | 628.5 | 623, 570.5, 144 | 311 | 286, 310, 464 |
| B. adolescentis SC69 | 628.5 | 621, 570, 144 | 311 | 310, 287, 464 |
| B. animalis subsp. animalis ATCC27672 | 637.5 | 631, 225, 576, 152 | 49 or 320 | 41.5, 46, 319 |
| B. animalis subsp. animalis JCM11658 | 637.5 | 632, 225, 152, | 49 or 320 | 42, 46, 319 |
| B. animalis subsp. lactis 358 | 637.5 | 630, 225 | 49 or 320 | 41, 45, 319 |
| B. animalis subsp. lactis DSMZ10140 | 637.5 | 631, 225, 152 | 49 or 320 | 40, 45, 319 |
| B. animalis subsp. lactis S149 | 637.5 | 575, 225 | 49 or 320 | 40, 45, 319, 296 |
| B. animalis subsp. lactis S306 | 637.5 | 629, 574, 225 | 49 or 320 | 41, 45, 319 |
| B. animalis subsp. lactis S332 | 637.5 | 632, 225, 575, 152, | 49 or 320 | 40, 45, 319 |
| B. animalis subsp. lactis UCD316 | 637.5 | 632, 225 | 49 or 320 | 40, 45, 319 |
| B. bifidum DSMZ20456 | 429 | 426, 620, 214, 142 | 309 | 284, 307, 462 |
| B. bifidum JCM1209 | 429 | 425, 620, 393, 214 | 309 | 284, 308, 462 |
| B. bifidum JCM1254 | 429 | 425, 620, 393, 214, 142 | 309 | 284, 308, 462 |
| B. bifidum JCM7003 | 429 | 425, 622, 214, 142 | 309 | 285, 308 |
| B. bifidum PRL2010 | 429 | 425, 393, 622, 606, 214, 399 | 309 | 285, 308, 462, 451 |
| B. bifidum SC128 | 429 | 427, 392, 142, 621, 214 | 309 | 284, 308 |
| B. bifidum SC555 | 429 | 427, 393, 142, 214, 620 | 309 | 284, 308 |
| B. bifidum SC572 | 429 | 393, 427, 622, 214, 142 | 309 | 284, 308 |
| B. bifidum SC583 | 429 | 427, 620, 392, 196, 77 | 309 | 284, 308, 195 |
| B. bifidum SC721 | 429 | 427, 392, 214, 142, 619 | 309 | 284, 308 |
| B. breve ATCC15700 | 432 | 396, 430, 145, 71 | 312 | 288, 311 |
| B. breve JCM1273 | 432 | 396, 430, 145 | 312 | 287, 311 |
| B. breve JCM7016 | 432 | 397, 430.5, 145 | 312 | 288, 311 |
| B. breve JCM7017 | 432 | 396, 430, 145, 71 | 312 | 287, 311 |
| B. breve JCM7019 | 432 | 396, 430, 145, 625 | 312 | 287, 311 |
| B. breve JCM7020 | 432 | 396, 430, 400, 145, 71 | 312 | 287, 311 |
| B. breve KA179 | 432 | 430, 396, 145, 623, 71 | 312 | 287, 311 |
| B. breve S17c | 432 | 396, 430, 145, 71, 622 | 312 | 287, 311 |
| B. breve S46 | 432 | 430, 396, 145, 71, 624 | 312 | 287, 311 |
| B. breve SC139 | 432 | 396, 430, 145, 625 | 312 | 288, 311 |
| B. breve SC152 | 432 | 430, 396, 145, 624, 346, 70 | 312 | 311, 287, 405 |
| B. breve SC154 | 432 | 397, 430, 404, 145, 148, 71 | 312 | 287, 311 |
| B. breve SC500 | 432 | 397, 430, 624, 145 | 312 | 288, 311 |
| B. breve SC506 | 432 | 396, 430, 145, 71, 624 | 312 | 287, 311 |
| B. breve SC508 | 432 | 398, 431, 625, 146, 75 | 312 | 289, 312 |
| B. breve SC519 | 432 | 430, 397, 623, 145 | 312 | 288, 311 |
| B. breve SC522 | 432 | 397, 430, 409, 145, 71 | 312 | 287, 311 |
| B. breve SC559 | 432 | 394, 427, 31, 142, 620 | 312 | 285, 308 |
| B. breve SC567 | 432 | 431, 402, 397, 146, 72 | 312 | 288, 312 |
| B. breve SC568 | 432 | 397, 430, 145 | 312 | 288, 311 |
| B. breve SC573 | 432 | 397, 431, 146 | 312 | 289, 312 |
| B. breve SC580 | 432 | 430, 395.5, 145, 624, 71 | 312 | 287.5, 311 |
| B. breve SC670 | 432 | 431, 396, 146, 626, 72 | 312 | 288, 312 |
| B. breve SC81 | 432 | 396, 430, 145, 71, 624 | 312 | 287, 311 |
| B. breve SC95 | 432 | 398, 431, 146 | 312 | 289, 312 |
| B. breve UCC2003 | 432 | 396, 430, 624, 145 | 312 | 287.5, 311 |
| B. longum subsp. infantis ATCC15697 | 427 | 425, 618.5, 391, 140 | 307 | 306, 283, 400 |
| B. longum subsp. infantis ATCC15702 | 427 | 425, 391, 140, 619, 66 | 307 | 282, 306 |
| B. longum subsp. infantis ATCC17930 | 427 | 425, 391, 140, 66, 620 | 307 | 282, 306 |
| B. longum subsp. infantis ATCC25962 | 427 | 391, 425, 140, 619 | 307 | 282, 306 |
| B. longum subsp. infantis JCM7007 | 427 | 392, 425, 619, 140 | 307 | 283, 306 |
| B. longum subsp. infantis SC143 | 427 | 425, 391, 619, 140 | 307 | 282, 306 |
| B. longum subsp. infantis SC145 | 427 | 425, 391, 140, 66, 618.5 | 307 | 283, 306 |
| B. longum subsp. infantis SC30 | 427 | 425, 391, 140, 620, 66 | 307 | 282, 306 |
| B. longum subsp. infantis SC523 | 427 | 425, 391, 621, 140 | 307 | 282, 306 |
| B. longum subsp. infantis SC569 | 427 | 392, 425, 618, 140 | 307 | 283, 306 |
| B. longum subsp. longum 3L-1 | 427 | 391, 425, 140, 620, 66 | 307 | 282, 306 |
| B. longum subsp. longum ATCC55813 | 427 | 425, 391, 618, 140 | 307 | 282, 306 |
| B. longum subsp. longum SC116 | 427 | 425, 391, 623, 140 | 307 | 282, 306 |
| B. longum subsp. longum SC513 | 427 | 425, 391, 140, 618, 66 | 307 | 282, 306 |
| B. longum subsp. longum SC536 | 427 | 425, 391, 140, 619, 66 | 307 | 283, 306 |
| B. longum subsp. longum SC592 | 427 | 425, 391, 620, 140 | 307 | 283, 306 |
| B. longum subsp. longum SC596 | 427 | 391, 425, 140, 619 | 307 | 283, 306 |
| B. longum subsp. longum SC657 | 427 | 391, 425, 620, 140 | 307 | 283, 306 |
| B. longum subsp. longum UCC35624 | 427 | 425, 391, 140, 66, 619 | 307 | 283, 306 |
| B. pseudocatenulatum SC11 | 429 | 393, 424, 621, 604, 215 | 309 | 308.5, 285.5, 399, 403 |
| B. pseudocatenulatum SC564 | 429 | 427.5, 623.5, 392, 215, 142 | 309 | 308.5, 284, 403 |
| B. pseudocatenulatum SC585 | 429 | 427.5, 620, 393, 215, 142 | 309 | 284, 308.5, 403 |
| B. pseudocatenulatum SC665 | 429 | 427.5, 621, 215, 142, 392 | 309 | 284, 308.5, 403 |
In all cases, TRF size differences between species are sufficient to enable discrimination of mixed populations of bifidobacteria. Importantly, the major bifidobacterial species found in the infant gastrointestinal tract are consistently distinguishable from each other based on these two restriction cuts.
3.3. Limits of detection/co-detection
A series of experiments were performed to evaluate the sensitivity of Bif-TRFLP under various community conditions. In a genomic DNA dilution series of 6 bifidobacterial species in the presence of non-target DNA all the corresponding TRF peaks were still visible at a dilution of 1:10,000, or ~65–650 genome equivalents (data not shown). This is comparable to the sensitivity of other PCR-based assays [36] and is sufficient to track the major populations of bifidobacteria in infants.
In artificial communities containing only two species of Bifidobacterium, both were detected if there was a 10-fold or fewer difference in their relative abundances (data not shown). The less abundant species was not consistently visible with a 100-fold or greater difference. With more complex artificial bifidobacterial communities containing 4 species however, there were instances where only the 2–3 most abundant species were detectable even though all species were within an order of magnitude of each other in abundance (Table S2). This type of bias is not unprecedented in TRFLP, and may be related to the fact that the detection of any given species by TRFLP is dependent on its rank abundance [16,37,38]. Given the strong similarity between the rrn templates of different bifidobacterial species this problem may be caused by chimera formation [39,40]. However, troubleshooting steps appropriate for reducing chimera formation, such as lowering the number of cycles, increasing primer concentration, and increasing elongation time, did not ameliorate this phenomenon (data not shown).
3.4. qPCR validation
In order to validate that the results of Bif-TRFLP were representative of community profiles, both Bif-TRFLP and species-specific qPCR were performed on several infant fecal samples. Fig. 2 compares qPCR and Bif-TRFLP data from four representative samples. Bifidobacterial species and relative amounts detected by both methods correlate very closely. However, as indicated above, when one species is dominant, Bif-TRFLP occasionally does not detect the lesser abundant species (such as B. breve and B. adolescentis in the HaeIII digest of infant B, week 29). The pseudoquantitative nature of TRFLP is also seen throughout the data, as Bif-TRFLP does not always precisely reflect the exact percentages of each species detected by qPCR. Relatively little of the total peak area is contained in “unknown” peaks (TRF sizes not associated with any species in the database), generally less than ~10%.
Fig. 2.
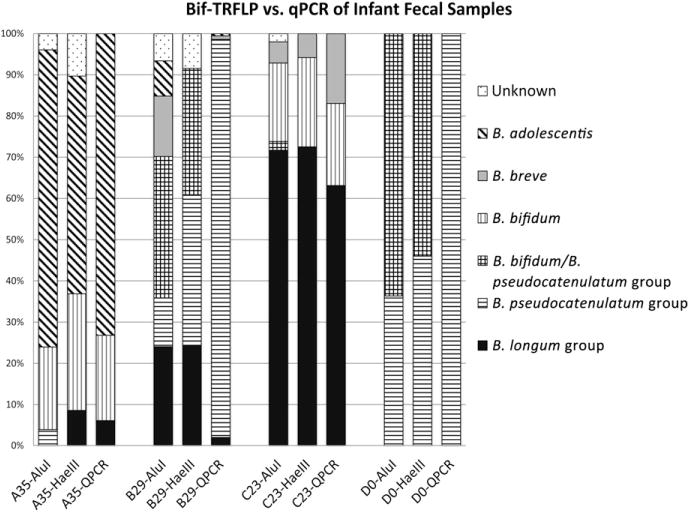
Comparison of Bif-TRFLP to bifidobacterial species-specific qPCR. The percentage of the total bifidobacterial content contributed by each species is shown for four infant fecal samples (A35, B29, C23, D0). The results of both restriction cuts of the Bif-TRFLP are shown, as well as the combined data for all species measured by qPCR. The letter in the bar label indicates the specific infant, while the number indicates the week of life when the sample was collected.
The species-specific qPCR data validates that Bif-TRFLP is reflective of the dominant bifidobacterial species content of the samples. Fig. 2 shows examples where Bif-TRFLP agrees with qPCR data whether there is one dominant species (infant D) or four prevalent species (infant B). The only ambiguity in TRF identification was a peak prevalent in infants B and D that represents either B. pseudocatenulatum or B. bifidum but could not be clearly distinguished. The discovery of B. pseudocatenulatum group species in these infants was somewhat unexpected due to the relative paucity of isolates of these species in culture-based work from these samples (data not shown). The prevalence of the B. pseudocatenulatum group in the Bif-TRFLP data prompted the investigation of these samples with B. catenulatum group-specific qPCR, which confirmed the high abundance of B. catenulatum in these infants.
3.5. NGS analysis
To illustrate how Bif-TRFLP can augment the taxonomic discrimination that Illumina methods are able to provide, the microbial diversity for one infant fecal sample (infant B, week of life 29) was characterized by Illumina sequencing (V4 region) (Figure S1) and Bif-TRFLP. Illumina sequencing yielded 9913 partial 16S rRNA gene sequence reads with a mean length of 120 bp. Figure S1 shows the diversity described by Illumina sequencing. Illumina sequencing identified much of the infant gut community, but achieved resolution only to the family level. Bif-TRFLP, in contrast, was able to separate the Bifidobacterium in this sample into species-level data, including proportions of B. longum group, B. breve, B. adolescentis, B pseudocatenulatum group, and B. pseudocatenulatum/B. bifidum group (see Fig. 2).
4. Discussion
The establishment of the intestinal microbiota in the first few months of life appears to have short and long-term clinical significance. Since bifidobacteria are often the dominant member of the gut microbial community [5] and differ in their metabolic abilities in significant ways [9–12], there is a pressing need for rapid, inexpensive species-level identification of bifidobacteria from fecal samples. The ability to profile microbial communities has advanced greatly with the advent of short-read NGS technologies, however the level of taxonomic discrimination does not always enable precise genus and species level discrimination. This problem necessitated the development of Bif-TRFLP for future use in studying our infant fecal samples. Similar targeted TRFLP-based methods have been successfully used by our group before to complement short read sequencing in studying the microbial ecology of beverage fermentations [41,42]. A TRFLP-based method of studying bifidobacteria had been previously developed by Sakamoto and coworkers, who used TRFLP with Bifidobacterium genus-specific primers to look at the bifidobacterial content of adults [43]. Our method adds the ability to distinguish between important infant-borne bifidobacterial species such as B. bifidum and B. longum, and the support of a more extensive empirical database of TRF sizes.
Bif-TRFLP is a rapid and inexpensive way to obtain bifidobacterial species-level data on large numbers of samples when compared to alternatives such as species-specific qPCR. The strengths of the method lie in its robustness, specificity for bifidobacteria, discriminatory power at the species level, and lack of a priori assumptions as required by qPCR (as demonstrated with the discovery of B. pseudocatenulatum group species in infants B and D). We have shown that this method provides community profiles closely matching those obtained by qPCR at a lower cost. This method is a particularly good tool for studies evaluating large numbers of bifidobacterial communities from the infant gut, as it has been shown to accurately distinguish the most common species found in that environment.
One of the possible drawbacks to this method may be a limited ability to detect multiple species at similar population levels. This effect in our artificial communities was less prevalent in fecal samples, suggesting that it may be caused by the paucity of diverse templates in the artificial communities. Another possible limitation is the decreased detection by Bif-TRFLP of lower abundance species when a sample is heavily dominated by one species. Natural variation in the bifidobacterial rrn gene leading to variant TRF sizes within species (such as in B. breve, as noted earlier) may be a concern, but this seems to be an uncommon occurrence according to our empirical database. There are also complementary methods for detecting when rrn variants do occur, such as performing Bif-TRFLP on identified isolates or comparing Bif-TRFLP data with bifidobacterial species-specific qPCR data. None of these limitations prevent Bif-TRFLP from providing useful inexpensive data on the most prevalent species in an infant fecal sample.
As noted earlier, when constructing the empirical database, there was more than one TRF that corresponded to each species; a result that was not predicted. The possibility of some smaller-than predicted peaks being the result of truncated chimeras was noted earlier. Other possible explanations for these unpredicted TRFs include star activity of the restriction enzyme recognizing non-target sequences, differences in rrn sequence between multiple copies that generate different TRF sizes, and incomplete sample digestion (for larger size peaks around 615–635 bp, as this is the size range of the undigested target amplicons based on sequence gazing). Incomplete digestion has been seen in TRFLP before, and can occur even when time and enzyme amount are not limiting [44]. The TRFs of >600 bps were principally found in the electropherograms of pure culture samples and mixed samples where one strain dominates. As these TRFs are consistent within a species, they do not present an identification problem.
Overall, we propose Bif-TRFLP as a useful tool for tracking the major populations of bifidobacteria across a large number of samples. It could be applied to target a specific subset of samples for study by other methods such as qPCR or NGS. When combined with methods for evaluating the overall composition of gut microbiota, such as NGS, it can also be useful for detecting correlations between the presence of specific bifidobacterial species and changes in the microbiota. While it could be applied to other types of samples, such as bifidobacteria-containing probiotic products, we anticipate using it principally to study infant fecal samples. Bif-TRFLP represents a new, improved tool for looking at the species-level composition of an important group of human-associated bacteria.
Supplementary Material
Acknowledgments
This work was supported by grants from the University of California Discovery Grant Program, the California Dairy Research Foundation, USDA NRICSREES award 2008-35200-18776, and National Institutes of Health NICHD awards R01HD059127, and R01HD061923
Appendix A. Supplementary data
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.anaerobe.2012.12.005.
References
- 1.Grześkowiak Ł, Collado MC, Mangani C, Maleta K, Laitinen KA, Isolauri E, et al. Distinct gut microbiota in south eastern African and northern European infants. Journal of Pediatric Gastroenterology and Nutrition. 2012;54:812–6. doi: 10.1097/MPG.0b013e318249039c. [DOI] [PubMed] [Google Scholar]
- 2.Arboleya S, Binetti A, Salazar N, Fernández N, Solís G, Hernández-Barranco A, et al. Establishment and development of intestinal microbiota in preterm neonates. FEMS Microbiology Ecology. 2011;79:763–72. doi: 10.1111/j.1574-6941.2011.01261.x. [DOI] [PubMed] [Google Scholar]
- 3.Yap GC, Chee KK, Hong P-Y, Lay C, Satria CD, Sumadiono, et al. Evaluation of stool microbiota signatures in two cohorts of Asian (Singapore and Indonesia) newborns at risk of atopy. BMC Microbiology. 2011;11:193. doi: 10.1186/1471-2180-11-193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fallani M, Amarri S, Uusijarvi A, Adam R, Khanna S, Aguilera M, et al. Determinants of the human infant intestinal microbiota after the introduction of first complementary foods in infant samples from five European centres. Microbiology (Reading, England) 2011;157:1385–92. doi: 10.1099/mic.0.042143-0. [DOI] [PubMed] [Google Scholar]
- 5.Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, et al. Human gut microbiome viewed across age and geography. Nature. 2012;486:222. doi: 10.1038/nature11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sakata S, Tonooka T, Ishizeki S, Takada M, Sakamoto M, Fukuyama M, et al. Culture-independent analysis of fecal microbiota in infants, with special reference to Bifidobacterium species. FEMS Microbiology Letters. 2005;243:417–23. doi: 10.1016/j.femsle.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 7.Donovan SM. Promoting bifidobacteria in the human infant intestine: why, how, and which one? Journal of Pediatric Gastroenterology and Nutrition. 2011;52:648–9. doi: 10.1097/MPG.0b013e31821e2799. [DOI] [PubMed] [Google Scholar]
- 8.Turroni F, Foroni E, Pizzetti P, Giubellini V, Ribbera A, Merusi P, et al. Exploring the diversity of the bifidobacterial population in the human intestinal tract. Applied and Environmental Microbiology. 2009;75:1534–45. doi: 10.1128/AEM.02216-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Turroni F, van Sinderen D, Ventura M. Genomics and ecological overview of the genus Bifidobacterium. International Journal of Food Microbiology. 2011;149:37–44. doi: 10.1016/j.ijfoodmicro.2010.12.010. [DOI] [PubMed] [Google Scholar]
- 10.Bottacini F, Medini D, Pavesi A, Turroni F, Foroni E, Riley D, et al. Comparative genomics of the genus Bifidobacterium. Microbiology (Reading, England) 2010;156:3243–54. doi: 10.1099/mic.0.039545-0. [DOI] [PubMed] [Google Scholar]
- 11.Ventura M, Canchaya C, Fitzgerald GF, Gupta RS, van Sinderen D. Genomics as a means to understand bacterial phylogeny and ecological adaptation: the case of bifidobacteria. Antonie Van Leeuwenhoek. 2007;91:351–72. doi: 10.1007/s10482-006-9122-6. [DOI] [PubMed] [Google Scholar]
- 12.Sela DA. Bifidobacterial utilization of human milk oligosaccharides. International Journal of Food Microbiology. 2011;149:58–64. doi: 10.1016/j.ijfoodmicro.2011.01.025. [DOI] [PubMed] [Google Scholar]
- 13.Turroni F, Peano C, Pass DA, Foroni E, Severgnini M, Claesson MJ, et al. Diversity of bifidobacteria within the infant gut microbiota. PLoS One. 2012;7:e36957. doi: 10.1371/journal.pone.0036957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davis LMG, Martínez I, Walter J, Goin C, Hutkins RW. Barcoded pyrosequencing reveals that consumption of galactooligosaccharides results in a highly specific bifidogenic response in humans. PloS One. 2011;6:e25200. doi: 10.1371/journal.pone.0025200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hong W-S, Chen M-J. Rapid identification of bifidobacteria in dairy products by gene-targeted species-specific PCR technique and DGGE. Asian-Australasian Journal Of Animal Sciences. 2007;20:1887–94. [Google Scholar]
- 16.Liu W, Marsh TL, Cheng H. Characterization of microbial diversity by determining terminal restriction fragment length polymorphisms of genes encoding 16S rRNA. Applied and Environmental Microbiology. 1997;63:4516–22. doi: 10.1128/aem.63.11.4516-4522.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martínez I, Kim J, Duffy PR, Schlegel VL, Walter J. Resistant starches types 2 and 4 have differential effects on the composition of the fecal microbiota in human subjects. PloS One. 2010;5:e15046. doi: 10.1371/journal.pone.0015046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. PNAS. 2011;108:4516–22. doi: 10.1073/pnas.1000080107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nature Methods. 2010;7:335–6. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics (Oxford, England) 2010;26:2460–1. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 21.Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Applied and Environmental Microbiology. 2007;73:5261–7. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, et al. The Ribosomal database project: improved alignments and new tools for rRNA analysis. Nucleic Acids Research. 2009;37:D141–5. doi: 10.1093/nar/gkn879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Applied and Environmental Microbiology. 2006;72:5069–72. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Andersen GL, Knight R. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics (Oxford, England) 2010;26:266–7. doi: 10.1093/bioinformatics/btp636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matsuki T, Watanabe K, Fujimoto J, Takada T, Matsumoto K, Kado Y, et al. Quantitative PCR with 16S primers for analysis of human intestinal bifidobacteria. Applied and Environmental Microbiology. 2004;70:167–73. doi: 10.1128/AEM.70.1.167-173.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Malinen E, Rinttilä T, Kajander K, Mättö J, Kassinen A, Krogius L, et al. Analysis of the fecal microbiota of irritable bowel syndrome patients and healthy controls with real-time PCR. The American Journal of Gastroenterology. 2005;100:373–82. doi: 10.1111/j.1572-0241.2005.40312.x. [DOI] [PubMed] [Google Scholar]
- 27.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, et al. Clustal W and Clustal X version 2.0. Bioinformatics (Oxford, England) 2007;23:2947–8. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 28.Cole JR, Chai B, Farris RJ, Wang Q, Kulam-Syed-Mohideen AS, McGarrell DM, et al. The ribosomal database project (RDP-II): introducing myRDP space and quality controlled public data. Nucleic Acids Research. 2007;35:D169–72. doi: 10.1093/nar/gkl889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Walters WA, Caporaso JG, Lauber CL, Berg-Lyons D, Fierer N, Knight R. PrimerProspector: de novo design and taxonomic analysis of barcoded polymerase chain reaction primers. Bioinformatics (Oxford, England) 2011;27:1159–61. doi: 10.1093/bioinformatics/btr087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shyu C, Soule T, Bent SJ, Foster JA, Forney LJ. MiCA: a web-based tool for the analysis of microbial communities based on terminal-restriction fragment length polymorphisms of 16S and 18S rRNA genes. Microbial Ecology. 2007;53:562–70. doi: 10.1007/s00248-006-9106-0. [DOI] [PubMed] [Google Scholar]
- 31.Abdo Z, Schüette UME, Bent SJ, Williams CJ, Forney LJ, Joyce P. Statistical methods for characterizing diversity of microbial communities by analysis of terminal restriction fragment length polymorphisms of 16S rRNA genes. Environmental Microbiology. 2006;8:929–38. doi: 10.1111/j.1462-2920.2005.00959.x. [DOI] [PubMed] [Google Scholar]
- 32.Clement BG, Kehl LE, DeBord KL, Kitts CL. Terminal restriction fragment patterns (TRFPs), a rapid, PCR-based method for the comparison of complex bacterial communities. Journal of Microbiological Methods. 1998;31:135–42. [Google Scholar]
- 33.Boesten R, Schuren F, Wind RD, Knol J, de Vos WM. Analysis of infant isolates of Bifidobacterium breve by comparative genome hybridization indicates the existence of new subspecies with marked infant specificity. Research in Microbiology. 2011;162:664–70. doi: 10.1016/j.resmic.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 34.Marsh T. Culture-independent microbial community analysis with terminal restriction fragment length polymorphism. Methods in Enzymology. 2005;397:308–29. doi: 10.1016/S0076-6879(05)97018-3. [DOI] [PubMed] [Google Scholar]
- 35.Kaplan CW, Kitts CL. Variation between observed and true Terminal Restriction Fragment length is dependent on true TRF length and purine content. Journal of Microbiological Methods. 2003;54:121–5. doi: 10.1016/s0167-7012(03)00003-4. [DOI] [PubMed] [Google Scholar]
- 36.Bastien P, Procop GW, Reischl U. Quantitative real-time PCR is not more sensitive than “conventional” PCR. Journal of Clinical Microbiology. 2008;46:1897–900. doi: 10.1128/JCM.02258-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lueders T, Friedrich MW. Evaluation of PCR amplification bias by terminal restriction fragment length polymorphism analysis of small-subunit rRNA and mcrA genes by using defined template mixtures of methanogenic pure cultures and soil DNA extracts. Applied and Environmental Microbiology. 2003;69:320–6. doi: 10.1128/AEM.69.1.320-326.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hiraishi A, Iwasaki M, Shinjo H. Terminal restriction pattern analysis of 16S rRNA genes for the characterization of bacterial communities of activated sludge. Journal of Bioscience and Bioengineering. 2000;90:148–56. doi: 10.1016/s1389-1723(00)80102-4. [DOI] [PubMed] [Google Scholar]
- 39.Qiu X, Wu L, Huang H, Mcdonel PE, Palumbo AV, Tiedje JM, et al. Evaluation of PCR-generated chimeras, mutations, and heteroduplexes with 16S rRNA gene-based cloning. Applied and Environmental Microbiology. 2001;67:880–7. doi: 10.1128/AEM.67.2.880-887.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smyth RP, Schlub TE, Grimm A, Venturi V, Chopra A, Mallal S, et al. Reducing chimera formation during PCR amplification to ensure accurate genotyping. Gene. 2010;469:45–51. doi: 10.1016/j.gene.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 41.Bokulich NA, Bamforth CW, Mills DA. Brewhouse-resident microbiota are responsible for multi-stage fermentation of American coolship ale. PloS One. 2012;7:e35507. doi: 10.1371/journal.pone.0035507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bokulich NA, Joseph CML, Allen G, Benson AK, Mills DA. Next-generation sequencing reveals significant bacterial diversity of botrytized wine. PloS One. 2012;7:e36357. doi: 10.1371/journal.pone.0036357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sakamoto M, Hayashi H, Benno Y. Terminal restriction fragment length polymorphism analysis for human fecal microbiota and its application for analysis of complex bifidobacterial communities. Microbiology and Immunology. 2003;47:133–42. doi: 10.1111/j.1348-0421.2003.tb02796.x. [DOI] [PubMed] [Google Scholar]
- 44.Li F, Hullar MAJ, Lampe JW. Optimization of terminal restriction fragment polymorphism (TRFLP) analysis of human gut microbiota. Journal of Microbiological Methods. 2007;68:303–11. doi: 10.1016/j.mimet.2006.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.