

Abstract
Human Mesenchymal Stem Cells (hMSCs) have emerged in the last few years as one of the most promising therapeutic cell sources and, in particular, as an important tool for regenerative medicine of skeletal tissues. Although they present a more restricted potency than Embryonic Stem (ES) cells, the use of hMCS in regenerative medicine avoids many of the drawbacks characteristic of ES cells or induced pluripotent stem cells. The challenge in using these cells lies into developing precise protocols for directing cellular differentiation to generate a specific cell lineage. In order to achieve this goal, it is of the upmost importance to be able to control de process of fate decision and lineage commitment. This process requires the coordinate regulation of different molecular layers at transcriptional, posttranscriptional and translational levels. At the transcriptional level, switching on and off different sets of genes is achieved not only through transcriptional regulators, but also through their interplay with epigenetic modifiers. It is now well known that epigenetic changes take place in an orderly way through development and are critical in the determination of lineage-specific differentiation. More importantly, alteration of these epigenetic changes would, in many cases, lead to disease generation and even tumour formation. Therefore, it is crucial to elucidate how epigenetic factors, through their interplay with transcriptional regulators, control lineage commitment in hMSCs.
Keywords: Epigenetics, Bone, Osteoporosis, Mesenchymal stem cells, Histone acetylation, Histone methylation, Histone methylation, DNA methylation.
1. INTRODUCTION
Mesenchymal stem cells (MSCs) are multipotent adult stem cells of mesodermal origin firstly characterized in 1970 [1]. MSCs can be isolated from different adult tissues, including bone marrow and adipose tissue [2-7]. Human MSCs (hMSCs) are thought to be one of the most useful cell sources for clinical application in tissue regeneration due to their ability to produce multiple cell types. Although hMSCs are normally in a quiescent state [8], in response to specific signals (i.e. tissue injury), they are capable of self-renewing to produce more hMSCs but also, of dividing asymmetrically producing lineage-committed progenitors that will differentiate into the required cell types. Although initially it was thought that MSCs were only able to give rise to the cell types found in the tissue they were isolated from, we now know that hMSCs not only are able to differentiate in a variety of cell types of mesodermal germ layer, such as adipocytes and osteocytes, but they can also differentiate into cells types of other germinal layers through a process known as “transdifferentiation” [5, 9-17] (Fig. 1). While hMSCs have a more restricted differentiation potential than embryonic stem (ES) cells, they show other great advantages. hMSCs can be easily sourced from adult tissues such as bone marrow, muscle and fat, and therefore, their use generally implies a lower risk of immune rejection. Additionally, their ability to transdifferentiate helps to create a microenvironment that favours tissue repair [18, 19]. In contrast to the known genomic instability of ES cells, there are few reports of oncogenic transformation in adult derived stem cells, and these cases are normally associated with long-term culture [20]. Moreover, the use of human MSCs is not subjected to the same ethical controversies as the use of human ES cells. Altogether, these unique characteristics have made of the MSCs the best candidates for cell therapy.
Fig. (1).
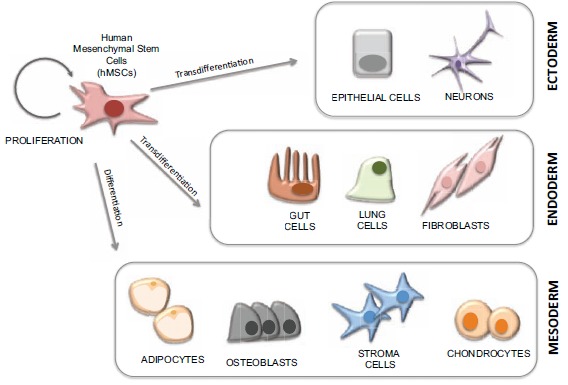
The multipotency of Mesenchymal Stem Cells. Mesenchymal Stem Cells (MSCs) are capable of self-renewing to produce more hMSCs but also, of dividing asymmetrically producing lineage-committed progenitors that will differentiate into the required cell types. hMSCs are able differentiate in a variety of cell types of mesodermal germ layer, such as adipocytes chondrocytes and osteoblasts, but they can also differentiate into endodermal and ectodermal cells types through a process known as “transdifferentiation”.
There are two different approaches for the application of hMSCs-based therapies in regenerative medicine. The first approach would consist in using these cells in their undifferentiated state, allowing the differentiation to take place at the site where the damage needs to be repaired. The downside of this strategy is the possibility of the cells differentiating towards cell types others than the required one at the repair site. To overcome this problem, a second approach is to differentiate the cells in vitro prior to their transplantation [7]. Although his second approach seems to be more appropriate and would involve lower failure risks, it requires a successful in vitro differentiation prior implantation. Thus, in order to attain the required cell identity minimizing the risk of oncogenic transformation we obviously require a deep understanding of all the regulatory mechanisms driving the differentiation process in hMSCs.
Differentiation of stem cells requires the activation of genes involved in the developing of a specific cell type on one hand and the suppression of specific sets of genes responsible for cell stemness on the other hand [21]. Since there are almost no difference between the coding sequences of MSCs and that of the specific cell types that they can give rise to, it seems clear that the differential gene expression that drives the appearance of different cell types is driven by epigenetic factors [22]. Epigenetic changes refer to reversible, heritable changes in gene regulation that occur without a change in DNA sequence. The more studied epigenetic factors include DNA methylation and histone modifications such as methylation, acetylation or ubiquitylation amongst others. Other epigenetic mechanisms are the regulation by non-coding RNAs, such as microRNAs, and mechanisms that control the higher-level organization of chromatin within the nucleus. These epigenetic modifications do not occur randomly but are carefully orchestrated [23]. Once in place, epigenetic changes can direct gene expression by modifying the accessibility of gene promoters and therefore facilitating (or avoiding) the recruitment of additional chromatin modifying enzymes or transcriptional regulators that would drive stem cell differentiation [24-28]. Indeed a recent study has demonstrated that the promoter regions of key genes in osteogenic differentiation such as BMP2 (encoding Bone Morphogenetic protein 2) and ALPL (encoding Alkaline Phosphatase) are epigenetically locked in MSCs to prevent their expression in non-osteogenic cells [29]. These genes have high levels of DNA methylation and low levels of histone acetylation. The alteration of this pattern using demethylation agents or inhibitors of histone deacetylases leads to their transcriptional activation [29]. This idea of an epigenetic control of stem cell differentiation has been supported throughout the years by several studies using chromatin-modifying drugs that can alter the potential of pluripotent and multipotent stem cells to differentiate into several lineages.
Since most of the current therapies are clearly insufficient for repairing damaged bone, to control the ability of hMSCs to differentiate into bone forming cells is of special interest for skeletal tissue engineering. While significant progress has been made in unraveling the molecular regulation of hMSCs differentiation, the comprehensive role of epigenetic factors in the pathophysiology of skeletal diseases, such as osteoporosis, in humans is however mostly unknown at this time.
Osteoblast differentiation can be divided into distinct stages based on the markers expressed at each stage. The first step would consist in the lineage commitment and the formation of osteoprogenitors. This step is regulated by master transcription factors and the correspondent co-regulators, such as RUNX2 (Runt related protein 2) and BMP2. The second step would consist in the proliferation of the osteoprogenitor cells with the expression of growth related genes. This step would be followed by the extracellular matrix maturation stage, where bone matrix proteins, such as Bone Sialoprotein (IBSP), Alkaline phosphatase (ALPL) and Collagen I (COL1A1) would be expressed. After the maturation, the extracellular matrix goes through the mineralization step that would require the expression of Osteocalcin (OCN) and Osteopontin (OPN).
Amongst all the factors involved in osteogenesis RUNX2 seems to be the master regulator since not only is involved in the regulation of most osteblastic specific genes, but also regulates the expression of its targets in response to different signals, incluiding, TGF-b, BMP and WNT signalling pathways, amongst others. Mutations in Runx2 lead to the arrest of osteoblast development and have severe skeletal defects. Subunit b of the core binding factor (CBFb) was the first known RUNX2 binding partner. However, it has been recently discovered that another transcription factor involved in the regulation of OCN, OPN, IBSP and COL1a1 can also cooperate with RUNX2 to for osteoblast-specific gene regulation. This factor is known as Osterix (OSX).
The canonical Wnt and BMP pathways seem to interact together at different steps of osteogenesis. BMP pathway operates early during osteogenesis to drive the differentiation of MSCs into osteoprogenitors. Later on, the function of Wnt signalling is to drive the differentiation of those osteoprogenitors towards osteoblastic fate. RUNX2 seems to have also a key role integrating these pathways.
2. EPIGENETIC CHANGES AND OSTEOPOROSIS
As we previously mentioned, the differentiation of hMSCs is achieved through functional interaction between transcription factors and epigenetic mechanisms. Changes in a specific epigenetic program may directly interfere with the differentiation process, ultimately giving rise to abnormal gene expression and the development of metabolic bone diseases or even, tumour formation [30]. Amid all bone diseases, osteoporosis is, by far, the most prevalent. This disease is suffered by millions of people around the world, affecting 1 in 3 women and 1 in 5 men over the age of 50 [31-33]. Moreover, osteoporotic fractures increase exponentially with advancing age and are a major cause of morbidity in the elderly, highlighting the clinical importance of this illness and its conception as a major problem in public health.
Osteoporosis is characterized by a decrease in bone mineral density (BMD) and the deterioration of bone microarchitecture. In order to maintain a correct bone homeostasis, the processes of bone formation, and bone destruction (known as bone “resorption”) are carefully balanced (Fig. 2). Indeed, the onset of skeletal pathologies such as osteoporosis or osteoarthritis is produced by deregulation of the balance between bone formation, mediated by bone-forming cells (osteoblasts) and bone-resorbing cells (osteoclasts) (Fig. 2). Although it seems clear that the development of osteoporosis is correlated with an increase in bone resorption, this necessarily has to be associated with an insufficient bone formation to replace the destroyed bone. Therefore, we need to contemplate a possible defect in the activity of the osteocytes or bone-forming cells. Indeed, studies in osteoporotic individuals indicate both a deficient activity of the osteoblasts [34] together with an increase in the number of these cells becoming apoptotic [35, 36]. Overall, the latest findings point at the high relevance of epigenetic changes in complex diseases, especially those that, like osteoporosis, develop late in life. The increase of the necessary osteogenic activity to replace the bone being resorpted requires a correct proliferation and differentiation of the osteoblastic precursors from mesenchymal stem cells. It is possible that this inadequate response of the bone-forming cells to the increase in bone resorption could be directly related to alterations in the epigenetic landscape of genes driving the differentiation of hMSCs into osteoblasts.
Fig. (2).
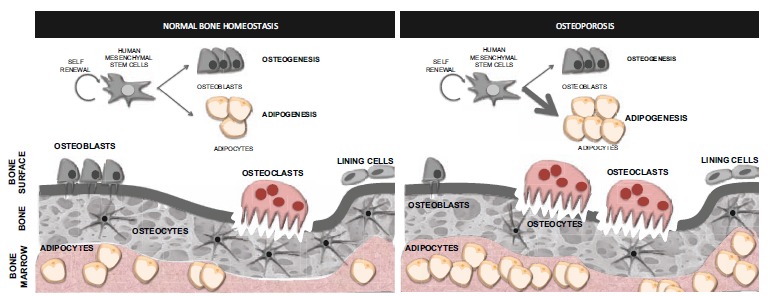
Bone homeostasis and osteoporosis. The maintenance of bone over time requires a balance between bone formation and bone breakdown (resorption). This is accomplished by the bone-forming cells and bone-resorpting cells acting in a coordinated fashion. The onset of osteoporosis is produced by deregulation of the balance between bone-forming cells (osteoblasts) and bone-resorpting cells (osteoclasts). This disease is characterized by an increase of the osteoclastic activity together with a deficient activity of the osteoblast and an increase of the number of these cells suffering apoptosis. The low bone-forming activity of the osteoblasts in osteoporosis could be a result of a defect in the differentiation of the progenitor MSCs that would preferentially differentiate into adipocytes, producing the effect known as “fatty bone marrow” in osteoporotic individuals.
MSCs can differentiate into adipocytes or osteoblasts amongst other cells lineages. In the last few years there has been increasing evidence pointing to the presence of a fatty bone marrow (accumulation of adipocytes) in patients with bone diseases such as osteoporosis (Fig. 2). The fact that these two processes (adipogenesis and osteogenesis) are mutually exclusive in MSCs was highlighted by the fact that inhibition of adipogenesis seems to improve bone development and repair [37, 38]. Since both adipose and bone cells are differentiated from MSCs, it seems clear that this particular disease could be a reflection of an imbalance between adipogenesis and osteoblastogenesis (Fig. 2). Although the molecular mechanisms determining whether a MSCs would differentiate into osteoblast or adipocyte lineage are still unclear, recent works seem to highlight the importance of epigenetic factors in this cell fate decision [39]. Therefore, understanding the epigenetic mechanisms governing this balance seems of the upmost importance in order to target and treat this disease. Since the activity of epigenetic factors is susceptible of being chemically modulated, they constitute perfect targets to control gene expression and thus, the identification of key epigenetic changes together with the prospect of modulate their activity holds great potential for the treatment of osteoporosis and other metabolic bone diseases.
3. EPIGENETIC MECHANISMS REGULATING OSTEOGENESIS
The differentiation of osteoblasts from MSCs is accompanied by profound changes in gene expression. It has been shown that two important epigenetic mechanisms, histone modification and DNA methylation have a key role regulating the expression of several genes associated with the osteogenic potential of MSCs as well as with the regulation of different stages of osteoblast differentiation.
High throughput analyses have recently characterized the global changes in gene expression and epigenetic landscape upon differentiation of human MSC towards the osteogenic lineage [40]. Global changes in gene expression during the onset of differentiation were associated with a decrease in proliferation and up-regulation of genes involved in osteoblast function [41, 42]. The differentiation process induces changes in histone methylation and acetylation. It is important to note that although global epigenetic changes were indeed detected during differentiation, these changes were not always linked to changes in gene expression. These epigenetic changes not linked to variations in gene expression were proposed to be part of a pre-patterning mechanism, poising genes for activation or repression in preparation for later parts of the differentiation program.
It has been shown that induction of differentiation is associated with genome-wide loss of histone acetylation, while levels of histone methylation remain practically unchanged [40]. Interestingly, analysis of the methylome of MSC from different origins (adipose tissue, muscle and bone marrow) showed that regardless of their source, MSCs share similar genome-wide methylation patterns and that most of the promoters involved in lineage-specification were hypomethylated in all the different types of MSCs analyzed. The hypomethylated regions harbored a combination of trimethylated H3K4 and H3K27. This could reflect a wide choice of cell fates. On the contrary, early developmental genes were DNA hypermethylated and this could be accompanied or not with H3K27 methylation, another silencing mark [40].
When this genome wide analysis was focused on global DNA methylation, they found altered methylation in a very low percentage of the CpG sites analyzed (1,273 sites of 450,000 sites analyzed) [40]. Therefore, DNA methylation remains relatively constant during MSCs osteogenic differentiation and does not seem an important factor, quantitatively speaking, although these small methylation variations could have great impact in the differentiation process. Interestingly, the differentiation process of MSCs into a specific cell type seems to involve less epigenetic changes, at least regarding DNA methylation, than the differentiation of pluripotent cells into multipotent committed progenitors [43].
3.1. Histone Modifications
Chromatin refers to the state in which DNA and histones are packaged within the eukaryotic nucleus. Chromatin is formed by nucleosomes, a basic repeating unit consisting of 147 bp of DNA wrapped around an octamer of two molecules each of histones H2A, H2B, H3 and H4.
The degree of chromatin packaging greatly determines whether a given DNA sequence is or not available to be targeted by a specific transcription factor. The loosely packaged chromatin, known as “euchromatin”, facilitates the accessibility of transcription factors to the DNA and therefore, gene transcription. On the other hand, tightly packaged chromatin, also called “heterochromatin”, would block gene expression by physically impeding the access of transcriptional regulators to the DNA.
The chromatin conformation would be altered in response to covalent post-translational modification of the flexible N-terminal histone tails that protrude from the nucleosomes. The complexity of these modifications depends not only on the amino acidic residue modified, but also on the type of chemical group added. These modifications include acetylation, methylation, phosphorylation, ubiquitylation and SUMOylation amongst others [21, 44]. In general terms, histone modifications can be classified into those that correlate with transcriptional activation (such as acetylation and phosphorylation) and those related to transcriptional repression (such as methylation, ubiquitilation and SUMOylation). Euchromatin, the more open and relaxed form of the chromatin is, in general, rich in acetylated lysine residues, such as acetylated histone 3 lysine 4 (H3K4Ac) or acetylated lysine 9, (H3K9Ac), while the more compacted form of chromatin, heterochromatin, presents low levels of acetylation and high levels of histone methylation, such as trimethylated H3K9 (H3K9m3) or trymethylated H3K27Me (H3K27m3) [45-47] although methylation of H3K4 is normally related to gene activation. Different modifications would work in a coordinated way to induce structural changes in the chromatin and therefore modify the accessibility of transcription factors to regulatory sequences, allowing the regulation of gene expression in a time and tissue-specific manner. This is known as the “Histone Code Hypothesis” [21].
3.1.1. Histone Acetylation and Deacetylation
One of the most studied epigenetic factors is histone acetylation. The transfer of an acetyl group from an acetyl-CoA molecule to specific lysine residues located at the histone tails is catalysed by histone acetyltransferases (HATs), whereas the opposing effect, the elimination of an acetyl group from a lysine residue is catalysed for the histone deacetylases (HDACs) (Fig. 3A). When this transfer occurs, the acetyl group neutralizes the positive electric charge of the lysine residue and thus relaxes the interaction of the histone tail with the DNA, which is negatively charged, a process defined as “nucleosome opening”. The chromatin structure, once relaxed, makes the DNA accessible for different transcription factors or the transcription machinery. In addition, this acetylated residue could also act as a docking site for regulatory factors that recognise acetylated lysine residues trough a specific domain called bromodomain. The level of acetylation at the histone tails, thus depends on the balance between the activities of HDACs and HATs [21].
Fig. (3).
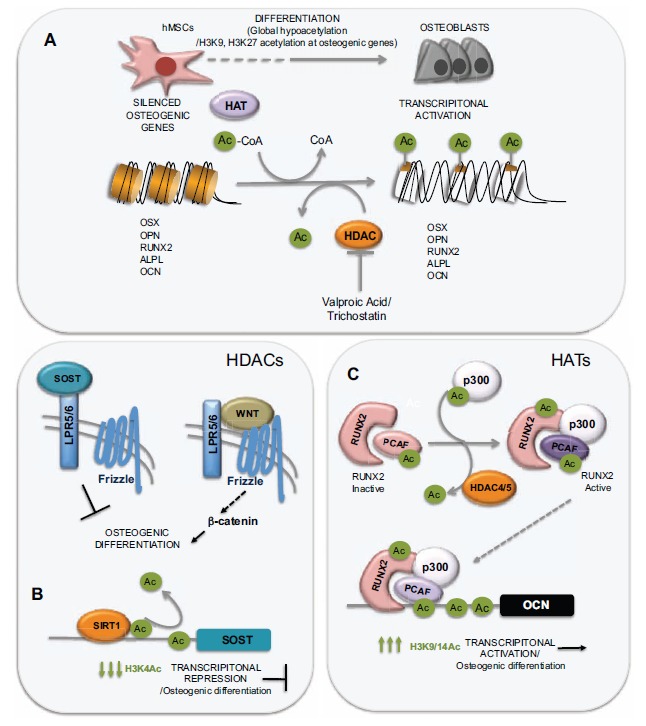
Role of histone acetylation in hMSCs differentiation into osteoblasts. (A) Differentiation of hMSCs into osteoblasts produces a genome-wide reduction of the levels of acetylated H3K9 and H3K27, together with an increase in the histone acetylation at the promoters of osteogenic genes. (B) Sclerostin, the product of the SOST gene, binds to the LPR5/6 receptors inhibiting the activation of the wnt pathway and therefore, osteogenic differentiation. HDAC SIRT1 would activate osteogenesis by deacetylating SOST promoter and thus, inhibiting its expression. (C) Activation of OCN is mediated by an active (acetylated) RUNX2 protein and by the acetylation of histones at its promoter, mediated by the HAT p300.
The analysis of the global epigenetic changes occurring during osteogenic differentiation of hMSCs, revealed that induction of differentiation produces a genome-wide reduction of the levels of acetylated H3K9 and H3K27 [40], although these reduction was considerably less important when the analysis was restricted only to regions surrounding gene promoters (Fig. 3A). Reduction in acetylation occurred at genes involved in gene regulation, cellular survival, growth and proliferation. Other authors have also found a reduction in histone acetylation accompanying the differentiation of hMSCs into myocytes, adipocytes, astrocytes or oligodendrocytes [48-50]. These results suggest that histone deacetylation might be a common step in cellular differentiation and could be related to condensation of chromatin at specific regions to restrict transcription and differentiation potential into other cell lineages. This study also corroborated that the few genes whose transcription was activated during differentiation were enriched in acetylated histone marks [40].
HDACs are classified into four different classes. Class I includes HDACs from 1 to 4. Class II includes HDACs from 5 to 10. Class III includes Sirtuins (Silent Mating Type Information Regulation 2 Homolog) 1 to 7, a special type of HDACs dependent on Nicotinamide adenine dinucleotide (NAD), and finally, only one member, HDAC11, would define Class IV.
Several studies regarding the role of histone acetylation in the control of osteogenesis used inhibitors of HDACs whose effect leads to an important increase in the histone acetylation levels (hyperacetylation).
While in vitro inhibition of HDACs in MSCs derived from umbilical cord or adipose tissue with valproic acid or sodium butyrate, leads to a decrease in chondrogenic or adipogenic differentiation, these treatments seem to have a positive effect on osteogenic differentiation [51]. Similarly, trichostatin, another HDAC inhibitor, was shown to increase osteoblast maturation and calcium deposition in cultures of bone marrow derived MSCs [52]. Interestingly, Cho et al. (2005) showed that treatment with valproic acid, leading to histone hyperacetylation, induced osteogenic differentiation and subsequent up-regulation of osteogeneic genes (OSX, OPN, RUNX2 and BMP-2) in adipose and bone marrow derived MSCs. However, treatment of the same cells with a structural analogue of valproic acid lacking HDAC inhibitor activity did not show this osteogenic induction, clearly indicating that the effect of valproic acid as an inductor of osteogenesis was directly related to its HDAC catalytic activity. This HDAC inhibitor has also been shown to trigger the proliferation of MSCs [53-55]. Similarly to what has been described in human MSCs, in vitro treatment of rat adipose-derived MSCs with trichostatin promotes osteogenic differentiation, an effect that seemed to be mediated by an increase of histone acetylation at the RUNX2 promoter (suggesting RUNX2 up-regulation), and was also dependent on BMP signalling [56]. Overall it seems clear that treatment with HDAC inhibitors, and therefore histone hyperacetylation, promotes osteogenic differentiation in MSCs and that increase in histone acetylation at RUNX2 and its subsequent activation plays a key role in this response.
Although the effect of HDAC inhibitors over MSCs osteogenic differentiation in vitro seems to be consistent, the results using these inhibitors in vivo are controversial and seem, to an extent, determined by the mouse strain used [57, 58] or by the state of the immune system of the subject. Valproate overall was shown to reduce BMD in young rats [57] and mice [58]. While in some cases the treatments led to an increase in osteocalcin in serum, this was not related to any change in osteoblast activity [58]. Mice treated with SAHA (suberanilohydroxamic acid), another HDAC inhibitor, also displayed a decrease in the volume of trabecular bone [59]. Opposing these results, in other studies, mice treated with SAHA showed increased local osteoblast activity and increased mineral apposition and bone formation rate following treatment [51, 55, 60] suggesting that this HDAC inhibitor can cause bone loss even while increasing the activity of mature osteoblasts [59].
In mice, knock down of HDAC4 in chondrocytes leads to an increase in bone formation, an effect correlated with an increase in RUNX2 activity [61]. However, more recently, other results have appeared pointing to a negative effect of some HDACs depletion on bone formation. Conditional knock out of HDAC3 in mice was shown to interfere with bone formation and promote adipogenesis [62]. These results are clearly opposed to those obtained in several in vitro studies [55, 63-65]. Thus, there is a clear discrepancy between the effects of HDAC3 abrogation in vitro in osteoblast cell lines (activation of osteoblastogenesis) and the in vivo deletion of HDAC3 in osteoprogenitor cells expressing OSX (decreased osteogenic activity and bone loss). A possible explanation for these contradictory results is that contrary to the permanent effect of a genetic deletion, HDACs inhibitors have short half-lives in serum and might induce only temporal changes in chromatin structure and gene expression. Besides, whereas non-proliferating cells would be resistant to the toxic effects of HDAC inhibitors they would still be susceptible to HDAC3 deletion [66]. While the different outcomes of HDAC deletion could be explained based on the previous explanations and in the fact that those studies used different cell types, further work needs to be done in order to clarify this issue.
The role of HDACs inhibitors on bone formation in humans is also somehow arguable. Patients suffering for some neurological conditions (epilepsy, mood disorder and bipolar disease) treated with valproic acid showed an important decrease in bone mass [59], whereas in other studies, markers of bone resorption after valproate treatment were reported to decrease, increase or even be unaffected [67-69]. The disagreement between these studies was attributed to the fact that general HDAC inhibitors, such as valproate, could target a wide range of HDACs with different specificities or could have an effect in enzymes others than HDACs. Besides, since this analysis was done in patients suffering from diverse neurological conditions, the authors considered that the results could have also been impacted by the different cohorts of patients analysed [59].
Sirtuin 1, a member of HDAC Class III family, encoded by the SIRT1 gene, has been recently shown to be a key regulator of bone mass due to its role as an inhibitor of sclerostin expression. Sclerostin, encoded by the SOST gene, interferes with the Wnt signalling pathway by binding to LPR5/6 receptors and therefore has a negative effect on bone formation. Sirtuin1 protein represses the expression of the SOST gene by means of H3K9 deacetylation at the SOST promoter (Fig. 3B). Cohen-Kfir et al. (2011) showed that mice in which a copy of the SIRT1 gene had been deleted displayed a reduced osteoblast activity leading to a reduced bone mass [70], an effect probably mediated by an increase in the levels of sclerostin.
Several works analysing the epigenetic regulation of osteogenic genes have focused on the regulation of OCN expression. The OCN promoter has, when active, high levels of the acetylated histones H3 and H4 [71, 72]. The activation of this gene depends on an active acetylation of these residues as well as on a decrease in DNA methylation [71, 73, 74]. It has also been shown that vitamin-D dependent activation of OCN is mediated by nucleosome remodelling favouring transcriptional activation [75]. Following these structural changes at the OCN promoter, a protein complex containing RUNX2 as well as other chromatin remodelling factors would bind the OCN promoter to induce its activation [76, 77] (Fig. 3C). Although the transcriptional co-activator p300 has been shown to interact with RUNX2 at the OCN promoter, its HAT catalytic activity was not found to be responsible for the increase in histone acetylation preceding OCN transcriptional activation, as the increase of OCN expression in the presence of p300 was not abolished when a catalytically death version of this HAT is used [72]. However another HAT, PCAF (p300/cyclic AMP receptor element-binding protein associated factor) is able to interact with p300 to promote OCN expression and was suggested to be responsible of the HAT mediated activation of OCN expression [72]. Indeed it has been recently demonstrated, that PCAF can interact and acetylate RUNX2 leading to an increase of osteogenic markers expression and mineralization [78]. It has been suggested that the enhanced RUNX2 activity after its acetylation by PCAF could be due to an increase in its DNA binding capacity.
Many HATs are actually lysine acetyl transferases as they are able to acetylate other non-histone proteins modulating their activity. Jeon et al. (2006) showed that BMP2-mediated induction of osteogenesis requires the acetylation of RUNX2 by p300 and that RUNX2 activity is inhibited by HDAC4/5. This acetylation increases RUNX2 activity and inhibits degradation of RUNX2 by SMURF1 (Fig. 3C). When RUNX2 is deacetylated by the action of HDAC4 and HDAC5, this allows the ubiquilitation of Runx2 by Smurf and its subsequent degradation [79]. Therefore, the activity of RUNX2 is tightly controlled by the equilibrium between the activities of HAT and HDACs.
It is important to highlight that there are other HATs, including MOZ and MORF [80], that have been shown to interact with RUNX2 and could have a role in osteogenic differentiation either by modulating the activity of the ostegenic master regulator RUNX2 or by directly acetylating histones at their target promoters.
3.1.2. Histone Methylation
Histone methylation can occur in lysine or arginine residues located at the histone tails [21]. Depending on the residue methylated, this epigenetic signal could be related to gene activation (methylation of H3K4), or to gene repression (methylation of H3K27 or H3K9). If these two types of modifications take place at the same promoter region (something known as a “bivalent mark”) the gene in question would be in a poised state, a phenomena reported in pluripotent stem cells [81].
The level of methylation at the histone tails is governed by the opposing activities of histone methyltransferases (HMTs) and histone demethylases (HDMTs).
There are two main families of HMTs with opposite activities. The Polycomb (PcG) family, carries out the methylation at lysine 27 of histone H3 (H3K27), a mark, as we previously mentioned, linked to gene silencing. The other group, known as the Trithorax (TrxG) family, methylates H3K4, a mark related to gene activation (Fig. 4B and 4C). Enhancer of Zeste 2 (EZH2) is a member of the PcG family and a component of the Polycomb repressor complex 2 (PRC2). This protein catalyzes the di-and tri- methylation of the lysine 27 at Histone H3 (H3K27me2 and H3K27m3) a modification that would be later recognized by the Polycomb repressor complex 1 (PRC1) leading to the transcriptional repression of the targeted genes. Wei et al. (2011) [82] found that the HMT EZH2 has a key role in the inhibition of MSC differentiation. EZH2 targets the promoter of RUNX2 and promotes its inactivation, by means H3K27 methylation. EZH2 can be inactivated through of CDK1 dependent phosphorylation that disrupts its binding with the rest of components of the PcG complex. CDK1 is one of the major kinases controlling the transition from the G2 to the M phase of the cell cycle and has a key role in regulating proliferation and lineage specification in stem cells [83]. EZH2 inactivation by CDK1 leads to the reduction of the H3K27me3 at the RUNX2 promoter, its subsequent activation, and therefore, the triggering of osteogenic differentiation. Therefore, CDK1 and EZH2 have a key role in osteogenic differentiation of MSCs.
Fig. (4).
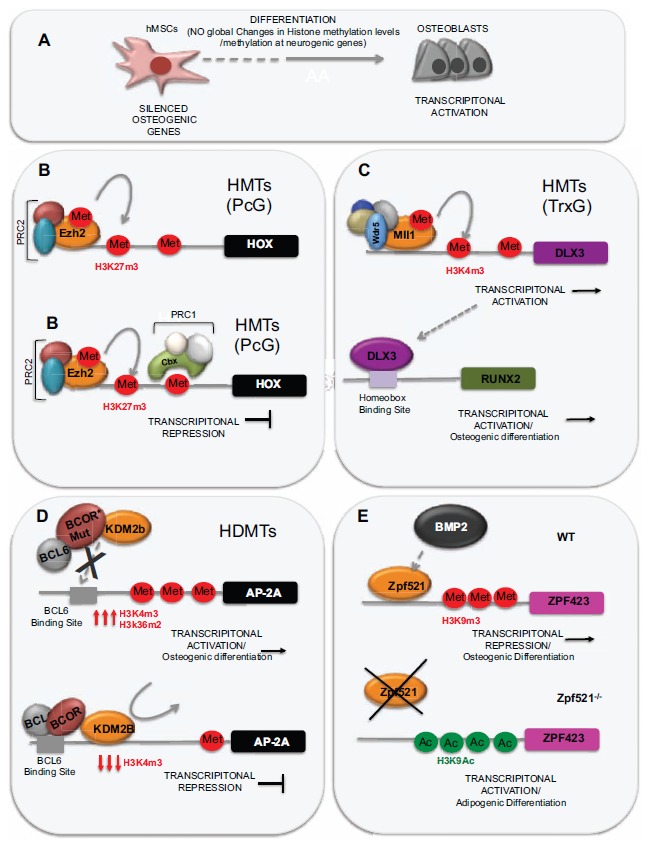
Role of histone methylation in hMSCs differentiation into osteoblasts. (A) Total levels of H3K4m3 and H2K27m3 remain mostly unchanged during differentiation however the silencing mark H3K27m3 was overall enriched at regions surrounding the promoter regions. (B) Polycomb repressor complexes 1 and 2 (PCR1 and PCR2) catalyse the H3K27 methylation leading to gene silencing. (C) BCOR interacts with the KDM2b) demethylase, and inhibits its recruitment to their target promoters and hence the demethylation of lysine residues driven by this enzyme, mainly H3K36 and H3K4. (D) MLL genes, belonging to the Trithorax family, methylate H3K4 leading to gene activation. (E) BMP-induced bone formation is mediated by the binding of Zfp521 to the promoter of Zfp423 and subsequent increase in H3K9 acetylation and decrease of histone H3K9 methylation.
Global analyses of the epigenetic changes undergone during osteogenic differentiation of hMSCs have shown that total levels of H3K4m3 and H2K27m3 remained mostly unchanged during differentiation (Fig. 4A). However, when the analysis was restricted to regions surrounding gene promoters they found that, although no changes were detected again for the H3K4m3 mark, the silencing mark H3K27m3 was overall enriched at these promoter regions, coinciding with the decrease in histone acetylation mentioned in the previous section. Changes in H3K27m3 affected only a few differentially expressed genes. However, this mark was found to be enriched mainly in promoters of genes involved in neurogenesis suggesting that the neurogenic potential of hMSCs is
restricted once osteogenic differentiation has been initiated. Actually, the majority of craniofacial bones are generated during embryogenesis from cranial neural crest cells but this potential is suppressed in more posterior neural crest cells by the action of hometotic (HOX) genes. A recent work highlighting the role of EZH2 in osteogenesis has shown that craniofacial bone is fully prevented in Ezh2 conditional knockout mice due to in an important up-regulation of HOX genes transcription in neural crest cells, (Fig. 4B) showing that the development of craniofacial bones in mammals also depends on epigenetic mechanisms [84].
Members of the TrxG groups, such as the Histone methyltrasnferases MLL1 and MLL2, antagonize the action of PcG proteins, such as EZH2, and therefore work as activators of genes expression. They catalyzed the di and tri- methylation of H3K4, an activation mark. In humans, the TrxG complexes contain one of the MLL proteins and a core module consisting of four proteins (WDR5, RBbp5, Ash2 and DPY-30) (Fig. 4C). All members of the complexes are essential and loss or malfunction of any of the subunits of the complex results in a decrease on H3K4 di- and tri-methylation. Nayak et al., [85] have recently shown that inactivation of
the SUMO-specific isopeptidase SENP3 leads to an important downregulation of genes belonging to a specific subfamily of HOX genes, the DLX genes. One of the members of this family, DLX3 (Distal-less Homeobox 3) was shown in the same work to regulate the differentiation of dental follicle stem cells (DFSCs) into mature osteoblast by controlling RUNX2 expression (Fig. 4C). This group showed that SENP3 catalyzes the de-SUMOylation of RbBP5 one of the core proteins of the TrxG complexes. This step is crucial to have a functional TrxG complex, and therefore, deletion of SNP3 leads to a decrease in the methylated H3K4 levels at the DLX3 promoter and a decrease in the presence of RNA Polymerase II at that promoter. The deficient activation of DLX3 thus translates into a deficient activation of Runx2 and ultimately to a defective osteogenic differentiation [85] (Fig. 4C).
The opposite reaction to histone methylation, that is the removal of methyl groups from specific lysine residues at the histone tails, is carried out by histone demethylases (HDMTs).
Several works have recently pointed to a key role of these enzymes in the decision of cell fate [46, 86-88]. An article from Fan et al. [89] studying the BCOR (BCL6-Co-Repressor) gene was one of the first works to link changes in the histone methylation levels with the ability of hMSCs to initiate osteogenic differentiation. The BCOR gene [90] is frequently mutated in patients with the Oculofacialcardiodental syndrome (OFCD) characterized by craniofacial abnormalities and extremely long teeth roots. The BCOR protein is able to interact with histone deacetylases and demethylases suggesting that this protein might exert its function by means of chromatin modification [91, 92]. Indeed, BCOR interacts with the JHDM1b (KDM2b) demethylase, and seems to inhibit the recruitment of JHDM1b to their target promoters and hence the demethylation of lysine residues driven by this enzyme, mainly H3K36 and H3K4. In OFCD patients, the mutated version of the BCOR gene fails to recruit JHDM1B to the promoter of the AP-2a (Activating Enhancer-Binding Protein 2-Alpha), a transcription factor essential for craniofacial development [93, 94], that mediates the elevated osteogenic capacity of MSCs from OFCD patients, this leads to an increase in H3K4m3 and the subsequent activation of AP-2a transcription, therefore explaining the appearance of some of the characteristic phenotypes of OFCD patients, such as enlarged roots of canine teeth (Fig. 4D).
Another important work from the same group, aimed to further explore the role of these enzymes in cell fate determination, analysed the expression of histone demethylases in MSCs in response to osteogenic induction mediated by BMP4/7. This group found that histone demethylases KDM4B [95] and KDM6B [87] were importantly up-regulated following osteogenic induction and that knockdown of any of these two histone demethylases either in vivo or in vitro blocked on one hand osteogenic differentiation but clearly drives adipogenic differentiation on the other hand [86]. Particularly, knockdown of KDM6B seemed to inhibit expression of key osteogenic proteins, such as IBSP, SPP1 and OCN. To determine whether this activity of KDM6B was directly dependent on its HDM activity, the authors developed a catalytically dead version of the protein. The mutated form of the protein, unlike its wild type version, was unable to rescue osteogenic differentiation of hMSCs in a mouse where KDM6B activity has been silenced, indicating the key role of this catatytic activity in the regulation of osteogenic gene expression. In agreement with this work, Xu et al. [96] recently found that KDM6b has a similar role in dental mesenchymal stem cells. KDM6B was recruited to the promoter of the bone morphogenetic protein 2 (BMP2), leading to the removal of H3K27m3 marks and therefore inducing its activation [86].
Bone morphogenetic proteins (BMPs) are known to drive cell fate towards osteogenesis. Although initial reports led to the idea that BMPs inhibited adipogenic differentiation [97] there is some works reporting some positive effects of BMPs on the adipogenic differentiation under certain conditions [98-101]. The further progression into osteogenesis induced by BMP induction is mediated by SMAD activation, that would, in turn drive the activation of osteogenic transcription factors, such as RUNX2 and Osterix (OSX). On the other hand, the induction of adipogenesis mediated by BMP2 is mediated by the Smad-associated cofactor Schnurri3 [100] and the pre-adipocyte commitment factor zinc finger protein 423 (Zfp423) [102] that would, in turn, activate the peroxisome proliferator-activated receptor (PPAR), the master regulator of adipogenic genes.
A recent work by Addison et al. [39] shows that deletion of another zinc finger protein (Zfp521) that regulates lineage progression, impairs BMP-induced bone formation and induces the differentiation of MSCs towards adipose tissue formation. This is mediated by the binding of Zfp521 to the promoter of Zfp423 and subsequent increase in H3K9 acetylation and decrease of histone H3K9 methylation. Although further work would be needed in order to elucidate the particular epigenetic mechanisms driven by the binding of Zpf521 to the Zpf423 promoter these findings support previous evidence that MSCs lineage commitment is epigenetically regulated by histone modifications [86, 103].
3.2. DNA Methylation
The methylation of the cytosine in 5′-position at a CpG dinucleotide is a well-characterized epigenetic modification [104]. While this epigenetic mark was initially considered to mediate stable gene silencing, recent studies have shown that the effect of DNA methylation on gene expression highly depends on the CpG density and that, promoters with low methylated CpG content can be active or inactive [105].
Although there is some controversy [106, 107], DNA methylation is considered by many to be the initiating event leading to the subsequent changes in the epigenome. In this context, it has been suggested that methylated DNA would recruit methyl-CpG binding proteins that would, in turn, recruit histone deacetylases (HDAC) leading to histone deacetylation and therefore gene silencing [73, 108]. On the other hand, more recent discoveries indicate that gene silencing could be initiated by histone deacetylation followed by subsequent histone and DNA methylation of the CpG islands [109]. It has also been proposed that both mechanisms could coexist and work depending on the cell context or genomic locus.
DNA methylation patterns are very dynamic during the development of the organism. After the methylation patterns of the gametes are erased during blastocyst formation, DNA methylation is extensively reprogrammed, up to the post-implantation epiblast, through the activity of the “de novo” DNA methylatransferases DNMT3a and DNMT3b [22, 110]. Mutations in DNMT3 are associated with a rare syndrome called “Immunodeficiency Centromeric Instability and Facial Anomalies type I” (ICF1). A recent work using cells from an individual suffering from this syndrome, has shown that Induced Pluripotent Stem Cells (iPs) derived from fibroblasts of this patient, exhibited global loss of non CG methylation and selective hypomethylation at centromeric regions, specific gene promoters and enhancers. These hypomethylated regions were preserved even when the iPs were differentiated into MSCs [111] and were similar to those found in ICF1 somatic cells, indicating the existence of a specific methylome defects in ICF1 cells. These results also underscore the importance of DNMT3-mediated DNA methylation in the establishment of a correct methylation pattern. A different DNA methyltransferase, DNMT1 would be in charge of maintaining and propagating the methylation patterns through mitosis by reproducing methylation patterns from a hemi-methylated substrate after DNA replication [22]. Inactivation of this enzyme in mice results in loss of genomic imprinting and leads to early embryonic lethality, showing the importance of this protein for development [112, 113]. Although reduced DNMT1 activity might be expected to lead to global demethylation, loss of DNMT1 in vitro only induces demethylation of a subset of key genes [114, 115]. In support of this idea, a recent work has demonstrated that OCT4 and NANOG bind to the DNMT1 promoter to cooperatively induce its expression and to maintain the methylation of genes associated with senescence and developmental regulators [116] (Fig. 5).
Fig. (5).
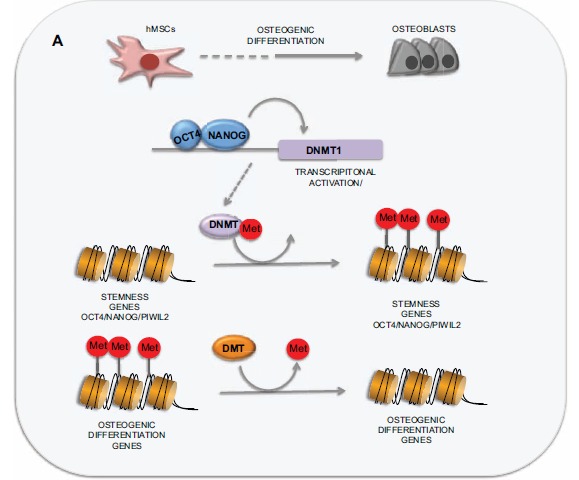
Role of DNA methylation in hMSCs differentiation into osteoblasts. HMSCS differentiation requires both the activation of genes involved in the developing of a specific cell type and the suppression of genes responsible for cell stemness. OCT4 and NANOG bind to the DNMT1 promoter to cooperatively induce its expression and to maintain the methylation of genes associated with senescence and developmental regulators.
High-throughput analysis of the methylome of MSCs derived from different adult tissues has revealed that there is a core of 1755 hypermethylated genes shared by all the hMSCs independently of the source tissue, supporting a common origen of all these cells. This set of hypermethylated genes was enriched in early developmentally regulated genes and probably reflects the functional blocking of older developmental programs that are not longer enabled [117]. Interestingly, it has also been recently shown, that, independently of the developmental lineage they specify, most of the promoters of genes whose expression is related to specific cell types are hypomethylated [43], even though the nature of the MSCs analysed preclude their differentiation into some of those lineages. This clearly suggests the existence of a transcriptionally permissive state in hMSCs that is not necessarily related to their differentiation potential [118, 119].
Many of the evidences regarding the involvement of DNA methylation in the differentiation resulted from studying the effects of demethylating agents. One of them, 5-aza-citydine (AzadC), is incorporated in DNA during cell proliferation instead of endogenous cytosine, inhibiting DNMT1 activity and therefore, reducing DNA methylation [120, 121]. Indeed, it has been demonstrated that treatment of hMSC derived from adipose tissue of aged individuals with AzadC induces proliferation and increases osteogenic potential of those cells [122]. In the same direction, Zhu et al. (2014) recently published a work showing that the osteogenic capacity of hMSCs obtained from umbilical cord was enhanced in absence of the methyltransferase DNMT3b when the cells are exposed to factors inducing osteogenesis [123].
As previously stated, stemness genes such as OCT4 and NANOG are hypomethylated in undifferentiated stem cells but became highly methylated during differentiation [124, 125]. Besides NANOG and OCT4, Berdasco et al. (2012) have described the regulation by DNA methylation of PIWIL2, (Piwi-like RNA-Mediated Gene Silencing 2), another gene involved in stem cell maintenance [126]. In addition to the methylation of stemness genes, the differentiation potential of MSCs seems to be restricted through the methylation of other lineage-specific promoters [118]. Importantly, Boquest et al. (2007) [118] found that the methylation state of adipogenic genes in adipose MSCs was more permissive than that of endothelial genes, such as CD31 or CD144. The methylated state of CD31 and CD144 in undifferentiated adipose-derived hMSCs suggest an important limitation in their endothelial potential. Indeed, when these cells are grown under conditions that promote endothelial differentiation, there is only a minimal decrease in DNA methylation and a marginal up-regulation of CD31 and CD144. Thus, CpG methylation profiles in undifferentiated hMSCs may dictate lineage-specificity of differentiation. This restriction in their differentiation potential would obviously limit their therapeutic applications. In order to establish if these differential potentials of hMSCs are the result of differences in their epigenetic profiles, Aranda et al. (2009) established a relation between the different levels of cell differentiation and their epigenetic signatures using genome-wide approaches [127]. This group analysed the histone modifications and DNA methylation marks at pluripotency and differentiation genes in ES cells, multipotent adult progenitor cells (MAPCs) and MSCs and Multipotent Adult Stem Cells (ADSC). Their analysis showed that the cells with greater differentiation potential (ES cells) showed a profile with higher epigenetic repression of differentiation genes both at the level of DNA methylation and histone modifications. This contrasted with the epigenetic profile of MSCs and ADSC, where genes controlling differentiation showed active epigenetic profiles. Interestingly, MAPC had an intermediate profile with only the presence of repressive histone modifications, but not associated with DNA methylation at the differentiation genes. This interesting work clearly shows that the loss of potential is correlated step by step to the acquisition of epigenetic marks at the differentiation loci.
The significance of promoter methylation in osteoblast-specific gene regulation is underscored by recent studies showing that the expression of RUNX2, DLX5, OSX, OCN, RANK, ALPL, SOST and OPG [74, 128-133] correlates with their promoter methylation status. In agreement with these previous works, Zhou et al. (2009) described that treatment of MSCs with AzadC prior to their culture in osteogenic media significantly promoted osteogenic differentiation, as determined by increased Alkaline Phosphatase (ALPL) activty, and increased mineralization. This was also accompanied by enhanced expression of osteogenic genes (DLX5, RUNX2, COL1a1, OSX, and OCN) and correlated with a decrease in methylation of genomic DNA [134].
DNA methylation is catalized by DNA-methyltrans- ferases and depends on the presence of a methyl donor, normally S-adenosylmethionine (SAM). SAM is a key molecule for mammalian cells since a reduced level of SAM leads to a reduced activity of methyltransferases and the subsequent demethylation of a variety of molecules. Different methyltransferases regulate the incorporation of methyl groups in molecules other than DNA, including proteins [135, 136], lipids [137], mRNA [138], and other small molecules [139]. As described in the previous section, methylation of lysine or arginine residues located at the histone tails can induce or repress gene expression, but also the activity of non-histone proteins can be regulated by methylation. In accordance to previous works, Vaes et al. (2010) [140] described that periodate oxidized adenosine (ADOX), an inhibitor of SAM-dependent methyltransferases, increases osteogenic gene expression under normal culture conditions, similar to the effect described for Aza-dC. However, they showed that under differentiation conditions ADOX inhibits appropriate osteoblast gene expression. In contrast with the effects of AzadC and ADOX on mRNA expression under non-osteogenic conditions, under induction of osteogenic differentiation, ADOX-repressed BMP2-induced mRNA levels of Alp, Osx and Ocn, all genes downstream of Runx2. However, expression of the early inducers of osteogenesis Dlx5 and Msx2 (upstream of Runx2) and mRNA and protein expression of Runx2 itself remained unchanged by ADOX. These results suggest that RUNX2 activity depends on methyltransferase activity. Therefore, global SAM-dependent demethylation leads to impaired osteoblast differentiation despite the fact that DNA hypomethylation can increase mRNA levels.
Overall it seems clear that the differentiation of MSCs into osteoblast requires an active DNA demethylation. Zhang et al. (2011) [130] revealed that the demethylation of CpG regions at the promoters of osteogenic genes and the subsequent increase in their expression levels was associated to a transcriptional up-regulation of the Growth Arrest And DNA-Damage-Inducible, Alpha (GADD45A) protein. Indeed, knockdown of Gadd45a by means of an shRNA inhibits the DNA demethylation of osteogenic gene promoters, leading to an up-regulation of their expression under differentiating culture conditions. Recent data suggests that Nucleotide Excision Repair (NER) is involved in the demethylation of osteogenic gene promoters. According to the mechanism described by Schmith et al. (2009) [141] for the TAF12 promoter, GADD45A can be recruited to a specific promoter DNA and then trigger its demethylation by recruiting the NER complex. The hypomethylated state of the promoter would then allow for the binding of RNA polymerase I, and the transcription of RNA.
4. ENVIRONMENTALLY-INDUCED EPIGENETIC CHANGES
It is well known that skeletal growth can be modified by environmental factors, such as nutrition and physical activity. Recent studies provided mounting evidence that the effect of these environmental factors on bone metabolism is mediated by epigenetic factors.
While nutri-epigenetic studies have been focused on the effect of nutrition during pregnancy and breastfeeding, there is also evidences pointing to an effect of some nutrients on epigenetic programs during adulthood [142]. Of all nutrients influencing bone metabolism, calcium is probably the most studied. The effect of calcium on BMD could be reflecting changes in the epigenetic pattern of genes regulating bone metabolism. Indeed a recent study shows that calcium can act regulating the activity of DNMT3b and DNMT1, and to a lesser extent of DNMT3a [143].
Although nutritional factors have an influence on bone metabolism, probably the most important factor affecting skeletal growth is fluid shear stress tension. Fluid shear stress [144] and compression [145-147] can influence osteogenic differentiation. Pressure and fluid flow-induced shear stress may be the dominant mechanical stimuli for mesenchymal stem cells (MSCs) within the bone marrow [148], while tension and compression are suspected to be more important in the periosteum. There is evidence that bone marrow-derived MSCs increase the expression of Osteopontin (OPN) and OCN twenty-four hours after fluid flow compared to static controls [149]. In the last few years there has been increasing interest in the study of how mechanical signals could alter the epigenetic status of key osteogenic genes and therefore have a key role in mechanotransduction, that is, the process by which cells convert physical stimuli into biochemical responses. Arnsdorf et al. (2010) [150] showed for the first time that the mechanical microenvironment is able to induce epigenetic changes. In this study they designed a novel protocol to promote MSCs differentiation by the application of a mechanical stimulus. They described and increase in OPN expression levels in MSCs subjected to dynamic fluid flow stimulus, and this increase in expression levels was related to a decrease in the DNA methylation at a specific region in the OPN promoter. Importantly, Mechanical stimulation can also induce epigenetic changes in human MSCs through a different epigenetic mechanisms, that is, by altering the histone acetylation levels [151]. MSCs subjected to mechanical strain had a reduced histone deacetylase (HDAC) activity and therefore showed overall higher levels of histone acetylation.
5. FUTURE PERSPECTIVES
It seems clear that owed to their ease of isolation and characteristics, the future treatment of bone diseases relies on the use of hMSCs. To be able to use these cells successfully, it is necessary to be able to define and tightly control the conditions that promote their differentiation, including the epigenetic changes involved. The investigations overtaken during the last decade have been crucial to make a progress in this direction. Understanding the epigenetic mechanisms governing MSCs differentiation is crucial to maintain stemness. In addition, understanding these epigenetic changes would ultimately allow us to use chemical agents to promote or avoid specific epigenetic modifications thus, controlling the differentiation process.
It seems now widely accepted that the use of in vitro differentiated MSCs has more advantages for their therapeutic use than the use of MSCs in their undifferentiated form. However, there are increasing evidences indicating that MSCs might experiment some loss of potential or premature senescence during in vitro culture, which would seriously hamper their use for therapeutic porpoises. The changes taking place in culture might be a reflection of epigenetic deregulation and therefore are also susceptible to be controlled. Although some techniques like the use of 3D spheroid culture instead of monolayer culture seems to somehow ameliorate this loss of potential during in vitro culture, future efforts should be aimed to unravel the cause of these changes and develop techniques in order to preserve MSCs properties during in vitro culture.
ACKNOWLEDGEMENTS
Suppoted by a grant from Instituto de Salud Carlos III-European Union FEDER funds (PI 12/615).
CONFLICT OF INTEREST
The author(s) confirm that this article content has no conflict of interest.
REFERENCES
- 1.Friedenstein A.J., Chailakhjan R.K., Lalykina K.S. The development of fibroblast colonies in monolayer cultures of guinea-pig bone marrow and spleen cells. Cell Tissue Kinet. 1970;3(4):393–403. doi: 10.1111/j.1365-2184.1970.tb00347.x. [DOI] [PubMed] [Google Scholar]
- 2.Alison M., Sarraf C. Hepatic stem cells. J. Hepatol. 1998;29(4):676–682. doi: 10.1016/S0168-8278(98)80165-7. [DOI] [PubMed] [Google Scholar]
- 3.Conrad S., Renninger M., Hennenlotter J., Wiesner T., Just L., Bonin M., Aicher W., Bühring H.J., Mattheus U., Mack A., Wagner H.J., Minger S., Matzkies M., Reppel M., Hescheler J., Sievert K.D., Stenzl A., Skutella T. Generation of pluripotent stem cells from adult human testis. Nature. 2008;456(7220):344–349. doi: 10.1038/nature07404. [DOI] [PubMed] [Google Scholar]
- 4.Margolis J., Spradling A. Identification and behavior of epithelial stem cells in the Drosophila ovary. Development. 1995;121(11):3797–3807. doi: 10.1242/dev.121.11.3797. [DOI] [PubMed] [Google Scholar]
- 5.Pittenger M.F., Mackay A.M., Beck S.C., Jaiswal R.K., Douglas R., Mosca J.D., Moorman M.A., Simonetti D.W., Craig S., Marshak D.R. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284(5411):143–147. doi: 10.1126/science.284.5411.143. [DOI] [PubMed] [Google Scholar]
- 6.Potten C.S. Stem cells in gastrointestinal epithelium: numbers, characteristics and death. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1998;353(1370):821–830. doi: 10.1098/rstb.1998.0246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weissman I.L. Translating stem and progenitor cell biology to the clinic: barriers and opportunities. Science. 2000;287(5457):1442–1446. doi: 10.1126/science.287.5457.1442. [DOI] [PubMed] [Google Scholar]
- 8.Peiffer I., Eid P., Barbet R., Li M.L., Oostendorp R.A., Haydont V., Monier M.N., Milon L., Fortunel N., Charbord P., Tovey M., Hatzfeld J., Hatzfeld A. A sub-population of high proliferative potential-quiescent human mesenchymal stem cells is under the reversible control of interferon alpha/beta. Leukemia. 2007;21(4):714–724. doi: 10.1038/sj.leu.2404589. [DOI] [PubMed] [Google Scholar]
- 9.Anjos-Afonso F., Siapati E.K., Bonnet D. In vivo contribution of murine mesenchymal stem cells into multiple cell-types under minimal damage conditions. J. Cell Sci. 2004;117(Pt 23):5655–5664. doi: 10.1242/jcs.01488. [DOI] [PubMed] [Google Scholar]
- 10.Hsiao S.H., Lee K.D., Hsu C.C., Tseng M.J., Jin V.X., Sun W.S., Hung Y.C., Yeh K.T., Yan P.S., Lai Y.Y., Sun H.S., Lu Y.J., Chang Y.S., Tsai S.J., Huang T.H., Leu Y.W. DNA methylation of the Trip10 promoter accelerates mesenchymal stem cell lineage determination. Biochem. Biophys. Res. Commun. 2010;400(3):305–312. doi: 10.1016/j.bbrc.2010.08.048. [DOI] [PubMed] [Google Scholar]
- 11.Koç O.N., Lazarus H.M. Mesenchymal stem cells: heading into the clinic. Bone Marrow Transplant. 2001;27(3):235–239. doi: 10.1038/sj.bmt.1702791. [DOI] [PubMed] [Google Scholar]
- 12.Lee K.D., Kuo T.K., Whang-Peng J., Chung Y.F., Lin C.T., Chou S.H., Chen J.R., Chen Y.P., Lee O.K. In vitro hepatic differentiation of human mesenchymal stem cells. Hepatology. 2004;40(6):1275–1284. doi: 10.1002/hep.20469. [DOI] [PubMed] [Google Scholar]
- 13.Lee O.K., Kuo T.K., Chen W.M., Lee K.D., Hsieh S.L., Chen T.H. Isolation of multipotent mesenchymal stem cells from umbilical cord blood. Blood. 2004;103(5):1669–1675. doi: 10.1182/blood-2003-05-1670. [DOI] [PubMed] [Google Scholar]
- 14.Halvorsen Y.D., Bond A., Sen A., Franklin D.M., Lea-Currie Y.R., Sujkowski D., Ellis P.N., Wilkison W.O., Gimble J.M. Thiazolidinediones and glucocorticoids synergistically induce differentiation of human adipose tissue stromal cells: biochemical, cellular, and molecular analysis. Metabolism. 2001;50(4):407–413. doi: 10.1053/meta.2001.21690. [DOI] [PubMed] [Google Scholar]
- 15.Zuk P.A., Zhu M., Mizuno H., Huang J., Futrell J.W., Katz A.J., Benhaim P., Lorenz H.P., Hedrick M.H. Multilineage cells from human adipose tissue: implications for cell-based therapies. Tissue Eng. 2001;7(2):211–228. doi: 10.1089/107632701300062859. [DOI] [PubMed] [Google Scholar]
- 16.Erickson G.R., Gimble J.M., Franklin D.M., Rice H.E., Awad H., Guilak F. Chondrogenic potential of adipose tissue-derived stromal cells in vitro and in vivo. Biochem. Biophys. Res. Commun. 2002;290(2):763–769. doi: 10.1006/bbrc.2001.6270. [DOI] [PubMed] [Google Scholar]
- 17.Kern S., Eichler H., Stoeve J., Klüter H., Bieback K. Comparative analysis of mesenchymal stem cells from bone marrow, umbilical cord blood, or adipose tissue. Stem Cells. 2006;24(5):1294–1301. doi: 10.1634/stemcells.2005-0342. [DOI] [PubMed] [Google Scholar]
- 18.Kolf C.M., Cho E., Tuan R.S. Mesenchymal stromal cells. Biology of adult mesenchymal stem cells: regulation of niche, self-renewal and differentiation. Arthritis Res. Ther. 2007;9(1):204. doi: 10.1186/ar2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Phinney D.G., Prockop D.J. Concise review: mesenchymal stem/multipotent stromal cells: the state of transdifferentiation and modes of tissue repair--current views. Stem Cells. 2007;25(11):2896–2902. doi: 10.1634/stemcells.2007-0637. [DOI] [PubMed] [Google Scholar]
- 20.Tolar J., Nauta A.J., Osborn M.J., Panoskaltsis Mortari A., McElmurry R.T., Bell S., Xia L., Zhou N., Riddle M., Schroeder T.M., Westendorf J.J., McIvor R.S., Hogendoorn P.C., Szuhai K., Oseth L., Hirsch B., Yant S.R., Kay M.A., Peister A., Prockop D.J., Fibbe W.E., Blazar B.R. Sarcoma derived from cultured mesenchymal stem cells. Stem Cells. 2007;25(2):371–379. doi: 10.1634/stemcells.2005-0620. [DOI] [PubMed] [Google Scholar]
- 21.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 22.Jaenisch R., Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat. Genet. 2003;33(Suppl.):245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 23.Vaissière T., Sawan C., Herceg Z. Epigenetic interplay between histone modifications and DNA methylation in gene silencing. Mutat. Res. 2008;659(1-2):40–48. doi: 10.1016/j.mrrev.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 24.Bibikova M., Chudin E., Wu B., Zhou L., Garcia E.W., Liu Y., Shin S., Plaia T.W., Auerbach J.M., Arking D.E., Gonzalez R., Crook J., Davidson B., Schulz T.C., Robins A., Khanna A., Sartipy P., Hyllner J., Vanguri P., Savant-Bhonsale S., Smith A.K., Chakravarti A., Maitra A., Rao M., Barker D.L., Loring J.F., Fan J.B. Human embryonic stem cells have a unique epigenetic signature. Genome Res. 2006;16(9):1075–1083. doi: 10.1101/gr.5319906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Calvanese V., Horrillo A., Hmadcha A., Suarez-Alvarez B., Fernandez A.F., Lara E., Casado S., Menendez P., Bueno C., Garcia-Castro J., Rubio R., Lapunzina P., Alaminos M., Borghese L., Terstegge S., Harrison N.J., Moore H.D., Brüstle O., Lopez-Larrea C., Andrews P.W., Soria B., Esteller M., Fraga M.F. Cancer genes hypermethylated in human embryonic stem cells. PLoS One. 2008;3(9):e3294. doi: 10.1371/journal.pone.0003294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mohn F., Weber M., Rebhan M., Roloff T.C., Richter J., Stadler M.B., Bibel M., Schübeler D. Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol. Cell. 2008;30(6):755–766. doi: 10.1016/j.molcel.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 27.Qiu J. Epigenetics: unfinished symphony. Nature. 2006;441(7090):143–145. doi: 10.1038/441143a. [DOI] [PubMed] [Google Scholar]
- 28.Vincent A., Van Seuningen I. Epigenetics, stem cells and epithelial cell fate. Differentiation. 2009;78(2-3):99–107. doi: 10.1016/j.diff.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 29.Cho Y.D., Yoon W.J., Kim W.J., Woo K.M., Baek J.H., Lee G., Ku Y., van Wijnen A.J., Ryoo H.M. Epigenetic modifications and canonical wingless/int-1 class (WNT) signaling enable trans-differentiation of nonosteogenic cells into osteoblasts. J. Biol. Chem. 2014;289(29):20120–20128. doi: 10.1074/jbc.M114.558064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wagner E.R., Luther G., Zhu G., Luo Q., Shi Q., Kim S.H., Gao J.L., Huang E., Gao Y., Yang K., Wang L., Teven C., Luo X., Liu X., Li M., Hu N., Su Y., Bi Y., He B.C., Tang N., Luo J., Chen L., Zuo G., Rames R., Haydon R.C., Luu H.H., He T.C. Defective osteogenic differentiation in the development of osteosarcoma. 2011. Sarcoma. 2011 doi: 10.1155/2011/325238. 325238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kanis J.A., Johnell O., Oden A., Sembo I., Redlund-Johnell I., Dawson A., De Laet C., Jonsson B. Long-term risk of osteoporotic fracture in Malmö. Osteoporos. Int. 2000;11(8):669–674. doi: 10.1007/s001980070064. [DOI] [PubMed] [Google Scholar]
- 32.Melton L.J., III, Atkinson E.J., O’Connor M.K., O’Fallon W.M., Riggs B.L. Bone density and fracture risk in men. J. Bone Miner. Res. 1998;13(12):1915–1923. doi: 10.1359/jbmr.1998.13.12.1915. [DOI] [PubMed] [Google Scholar]
- 33.Melton L.J., III, Chrischilles E.A., Cooper C., Lane A.W., Riggs B.L. Perspective. How many women have osteoporosis? J. Bone Miner. Res. 1992;7(9):1005–1010. doi: 10.1002/jbmr.5650070902. [DOI] [PubMed] [Google Scholar]
- 34.Misof B.M., Gamsjaeger S., Cohen A., Hofstetter B., Roschger P., Stein E., Nickolas T.L., Rogers H.F., Dempster D., Zhou H., Recker R., Lappe J., McMahon D., Paschalis E.P., Fratzl P., Shane E., Klaushofer K. Bone material properties in premenopausal women with idiopathic osteoporosis. J. Bone Miner. Res. 2012;27(12):2551–2561. doi: 10.1002/jbmr.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jilka R.L. Biology of the basic multicellular unit and the pathophysiology of osteoporosis. Med. Pediatr. Oncol. 2003;41(3):182–185. doi: 10.1002/mpo.10334. [DOI] [PubMed] [Google Scholar]
- 36.Kousteni S., Bellido T., Plotkin L.I., O’Brien C.A., Bodenner D.L., Han L., Han K., DiGregorio G.B., Katzenellenbogen J.A., Katzenellenbogen B.S., Roberson P.K., Weinstein R.S., Jilka R.L., Manolagas S.C. Nongenotropic, sex-nonspecific signaling through the estrogen or androgen receptors: dissociation from transcriptional activity. Cell. 2001;104(5):719–730. [PubMed] [Google Scholar]
- 37.Kawai M., Rosen C.J. PPARγ: a circadian transcription factor in adipogenesis and osteogenesis. Nat. Rev. Endocrinol. 2010;6(11):629–636. doi: 10.1038/nrendo.2010.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McCauley L.K. c-Maf and you won’t see fat. J. Clin. Invest. 2010;120(10):3440–3442. doi: 10.1172/JCI44786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Addison W.N., Fu M.M., Yang H.X., Lin Z., Nagano K., Gori F., Baron R. Direct transcriptional repression of Zfp423 by Zfp521 mediates a bone morphogenic protein-dependent osteoblast versus adipocyte lineage commitment switch. Mol. Cell. Biol. 2014;34(16):3076–3085. doi: 10.1128/MCB.00185-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Håkelien A.M., Bryne J.C., Harstad K.G., Lorenz S., Paulsen J., Sun J., Mikkelsen T.S., Myklebost O., Meza-Zepeda L.A. The regulatory landscape of osteogenic differentiation. Stem Cells. 2014;32(10):2780–2793. doi: 10.1002/stem.1759. [DOI] [PubMed] [Google Scholar]
- 41.Qi H., Aguiar D.J., Williams S.M., La Pean A., Pan W., Verfaillie C.M. Identification of genes responsible for osteoblast differentiation from human mesodermal progenitor cells. Proc. Natl. Acad. Sci. USA. 2003;100(6):3305–3310. doi: 10.1073/pnas.0532693100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kulterer B., Friedl G., Jandrositz A., Sanchez-Cabo F., Prokesch A., Paar C., Scheideler M., Windhager R., Preisegger K.H., Trajanoski Z. Gene expression profiling of human mesenchymal stem cells derived from bone marrow during expansion and osteoblast differentiation. BMC Genomics. 2007;8:70. doi: 10.1186/1471-2164-8-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sørensen A.L., Jacobsen B.M., Reiner A.H., Andersen I.S., Collas P. Promoter DNA methylation patterns of differentiated cells are largely programmed at the progenitor stage. Mol. Biol. Cell. 2010;21(12):2066–2077. doi: 10.1091/mbc.E10-01-0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gibney E.R., Nolan C.M. Epigenetics and gene expression. Heredity (Edinb) 2010;105(1):4–13. doi: 10.1038/hdy.2010.54. [DOI] [PubMed] [Google Scholar]
- 45.Portela A., Esteller M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010;28(10):1057–1068. doi: 10.1038/nbt.1685. [DOI] [PubMed] [Google Scholar]
- 46.Agger K., Christensen J., Cloos P.A., Helin K. The emerging functions of histone demethylases. Curr. Opin. Genet. Dev. 2008;18(2):159–168. doi: 10.1016/j.gde.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 47.Shi Y., Whetstine J.R. Dynamic regulation of histone lysine methylation by demethylases. Mol. Cell. 2007;25(1):1–14. doi: 10.1016/j.molcel.2006.12.010. [DOI] [PubMed] [Google Scholar]
- 48.Asp P., Blum R., Vethantham V., Parisi F., Micsinai M., Cheng J., Bowman C., Kluger Y., Dynlacht B.D. Genome-wide remodeling of the epigenetic landscape during myogenic differentiation. Proc. Natl. Acad. Sci. USA. 2011;108(22):E149–E158. doi: 10.1073/pnas.1102223108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hsieh J., Nakashima K., Kuwabara T., Mejia E., Gage F.H. Histone deacetylase inhibition-mediated neuronal differentiation of multipotent adult neural progenitor cells. Proc. Natl. Acad. Sci. USA. 2004;101(47):16659–16664. doi: 10.1073/pnas.0407643101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Takanashi M., Oikawa K., Fujita K., Kudo M., Kinoshita M., Kuroda M. Heterochromatin protein 1gamma epigenetically regulates cell differentiation and exhibits potential as a therapeutic target for various types of cancers. Am. J. Pathol. 2009;174(1):309–316. doi: 10.2353/ajpath.2009.080148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee S., Park J.R., Seo M.S., Roh K.H., Park S.B., Hwang J.W., Sun B., Seo K., Lee Y.S., Kang S.K., Jung J.W., Kang K.S. Histone deacetylase inhibitors decrease proliferation potential and multilineage differentiation capability of human mesenchymal stem cells. Cell Prolif. 2009;42(6):711–720. doi: 10.1111/j.1365-2184.2009.00633.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.de Boer J., Licht R., Bongers M., van der Klundert T., Arends R., van Blitterswijk C. Inhibition of histone acetylation as a tool in bone tissue engineering. Tissue Eng. 2006;12(10):2927–2937. doi: 10.1089/ten.2006.12.2927. [DOI] [PubMed] [Google Scholar]
- 53.Cho H.H., Park H.T., Kim Y.J., Bae Y.C., Suh K.T., Jung J.S. Induction of osteogenic differentiation of human mesenchymal stem cells by histone deacetylase inhibitors. J. Cell. Biochem. 2005;96(3):533–542. doi: 10.1002/jcb.20544. [DOI] [PubMed] [Google Scholar]
- 54.Schroeder T.M., Nair A.K., Staggs R., Lamblin A.F., Westendorf J.J. Gene profile analysis of osteoblast genes differentially regulated by histone deacetylase inhibitors. BMC Genomics. 2007;8:362. doi: 10.1186/1471-2164-8-362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schroeder T.M., Westendorf J.J. Histone deacetylase inhibitors promote osteoblast maturation. J. Bone Miner. Res. 2005;20(12):2254–2263. doi: 10.1359/JBMR.050813. [DOI] [PubMed] [Google Scholar]
- 56.Hu X., Zhang X., Dai L., Zhu J., Jia Z., Wang W., Zhou C., Ao Y. Histone deacetylase inhibitor trichostatin A promotes the osteogenic differentiation of rat adipose-derived stem cells by altering the epigenetic modifications on Runx2 promoter in a BMP signaling-dependent manner. Stem Cells Dev. 2013;22(2):248–255. doi: 10.1089/scd.2012.0105. [DOI] [PubMed] [Google Scholar]
- 57.Nissen-Meyer L.S., Svalheim S., Taubøll E., Reppe S., Lekva T., Solberg L.B., Melhus G., Reinholt F.P., Gjerstad L., Jemtland R. Levetiracetam, phenytoin, and valproate act differently on rat bone mass, structure, and metabolism. Epilepsia. 2007;48(10):1850–1860. doi: 10.1111/j.1528-1167.2007.01176.x. [DOI] [PubMed] [Google Scholar]
- 58.Senn S.M., Kantor S., Poulton I.J., Morris M.J., Sims N.A., O’Brien T.J., Wark J.D. Adverse effects of valproate on bone: defining a model to investigate the pathophysiology. Epilepsia. 2010;51(6):984–993. doi: 10.1111/j.1528-1167.2009.02516.x. [DOI] [PubMed] [Google Scholar]
- 59.McGee-Lawrence M.E., Westendorf J.J. Histone deacetylases in skeletal development and bone mass maintenance. Gene. 2011;474(1-2):1–11. doi: 10.1016/j.gene.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Di Bernardo G., Squillaro T., Dell’Aversana C., Miceli M., Cipollaro M., Cascino A., Altucci L., Galderisi U. Histone deacetylase inhibitors promote apoptosis and senescence in human mesenchymal stem cells. Stem Cells Dev. 2009;18(4):573–581. doi: 10.1089/scd.2008.0172. [DOI] [PubMed] [Google Scholar]
- 61.Vega R.B., Matsuda K., Oh J., Barbosa A.C., Yang X., Meadows E., McAnally J., Pomajzl C., Shelton J.M., Richardson J.A., Karsenty G., Olson E.N. Histone deacetylase 4 controls chondrocyte hypertrophy during skeletogenesis. Cell. 2004;119(4):555–566. doi: 10.1016/j.cell.2004.10.024. [DOI] [PubMed] [Google Scholar]
- 62.Razidlo D.F., Whitney T.J., Casper M.E., McGee-Lawrence M.E., Stensgard B.A., Li X., Secreto F.J., Knutson S.K., Hiebert S.W., Westendorf J.J. Histone deacetylase 3 depletion in osteo/chondroprogenitor cells decreases bone density and increases marrow fat. PLoS One. 2010;5(7):e11492. doi: 10.1371/journal.pone.0011492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Choo M.K., Yeo H., Zayzafoon M. NFATc1 mediates HDAC-dependent transcriptional repression of osteocalcin expression during osteoblast differentiation. Bone. 2009;45(3):579–589. doi: 10.1016/j.bone.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Iwami K., Moriyama T. Effects of short chain fatty acid, sodium butyrate, on osteoblastic cells and osteoclastic cells. Int. J. Biochem. 1993;25(11):1631–1635. doi: 10.1016/0020-711X(93)90522-G. [DOI] [PubMed] [Google Scholar]
- 65.Lee H.W., Suh J.H., Kim A.Y., Lee Y.S., Park S.Y., Kim J.B. Histone deacetylase 1-mediated histone modification regulates osteoblast differentiation. Mol. Endocrinol. 2006;20(10):2432–2443. doi: 10.1210/me.2006-0061. [DOI] [PubMed] [Google Scholar]
- 66.Bhaskara S., Chyla B.J., Amann J.M., Knutson S.K., Cortez D., Sun Z.W., Hiebert S.W. Deletion of histone deacetylase 3 reveals critical roles in S phase progression and DNA damage control. Mol. Cell. 2008;30(1):61–72. doi: 10.1016/j.molcel.2008.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kim S.H., Lee J.W., Choi K.G., Chung H.W., Lee H.W. A 6-month longitudinal study of bone mineral density with antiepileptic drug monotherapy. Epilepsy Behav. 2007;10(2):291–295. doi: 10.1016/j.yebeh.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 68.Sato Y., Kondo I., Ishida S., Motooka H., Takayama K., Tomita Y., Maeda H., Satoh K. Decreased bone mass and increased bone turnover with valproate therapy in adults with epilepsy. Neurology. 2001;57(3):445–449. doi: 10.1212/WNL.57.3.445. [DOI] [PubMed] [Google Scholar]
- 69.Tsukahara H., Kimura K., Todoroki Y., Ohshima Y., Hiraoka M., Shigematsu Y., Tsukahara Y., Miura M., Mayumi M. Bone mineral status in ambulatory pediatric patients on long-term anti-epileptic drug therapy. Pediatr. Int. 2002;44(3):247–253. doi: 10.1046/j.1442-200X.2002.01561.x. [DOI] [PubMed] [Google Scholar]
- 70.Cohen-Kfir E., Artsi H., Levin A., Abramowitz E., Bajayo A., Gurt I., Zhong L., D’Urso A., Toiber D., Mostoslavsky R., Dresner-Pollak R. Sirt1 is a regulator of bone mass and a repressor of Sost encoding for sclerostin, a bone formation inhibitor. Endocrinology. 2011;152(12):4514–4524. doi: 10.1210/en.2011-1128. [DOI] [PubMed] [Google Scholar]
- 71.Shen J., Hovhannisyan H., Lian J.B., Montecino M.A., Stein G.S., Stein J.L., Van Wijnen A.J. Transcriptional induction of the osteocalcin gene during osteoblast differentiation involves acetylation of histones h3 and h4. Mol. Endocrinol. 2003;17(4):743–756. doi: 10.1210/me.2002-0122. [DOI] [PubMed] [Google Scholar]
- 72.Sierra J., Villagra A., Paredes R., Cruzat F., Gutierrez S., Javed A., Arriagada G., Olate J., Imschenetzky M., Van Wijnen A.J., Lian J.B., Stein G.S., Stein J.L., Montecino M. Regulation of the bone-specific osteocalcin gene by p300 requires Runx2/Cbfa1 and the vitamin D3 receptor but not p300 intrinsic histone acetyltransferase activity. Mol. Cell. Biol. 2003;23(9):3339–3351. doi: 10.1128/MCB.23.9.3339-3351.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bird A.P., Wolffe A.P. Methylation-induced repression--belts, braces, and chromatin. Cell. 1999;99(5):451–454. doi: 10.1016/S0092-8674(00)81532-9. [DOI] [PubMed] [Google Scholar]
- 74.Villagra A., Gutiérrez J., Paredes R., Sierra J., Puchi M., Imschenetzky M., Wijnen Av Av., Lian J., Stein G., Stein J., Montecino M. Reduced CpG methylation is associated with transcriptional activation of the bone-specific rat osteocalcin gene in osteoblasts. J. Cell. Biochem. 2002;85(1):112–122. doi: 10.1002/jcb.10113. [DOI] [PubMed] [Google Scholar]
- 75.Paredes R., Gutiérrez J., Gutierrez S., Allison L., Puchi M., Imschenetzky M., van Wijnen A., Lian J., Stein G., Stein J., Montecino M. Interaction of the 1alpha,25-dihydroxyvitamin D3 receptor at the distal promoter region of the bone-specific osteocalcin gene requires nucleosomal remodelling. Biochem. J. 2002;363(Pt 3):667–676. doi: 10.1042/bj3630667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gutierrez S., Javed A., Tennant D.K., van Rees M., Montecino M., Stein G.S., Stein J.L., Lian J.B. CCAAT/enhancer-binding proteins (C/EBP) beta and delta activate osteocalcin gene transcription and synergize with Runx2 at the C/EBP element to regulate bone-specific expression. J. Biol. Chem. 2002;277(2):1316–1323. doi: 10.1074/jbc.M106611200. [DOI] [PubMed] [Google Scholar]
- 77.Javed A., Gutierrez S., Montecino M., van Wijnen A.J., Stein J.L., Stein G.S., Lian J.B. Multiple Cbfa/AML sites in the rat osteocalcin promoter are required for basal and vitamin D-responsive transcription and contribute to chromatin organization. Mol. Cell. Biol. 1999;19(11):7491–7500. doi: 10.1128/MCB.19.11.7491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang C.Y., Yang S.F., Wang Z., Tan J.M., Xing S.M., Chen D.C., Xu S.M., Yuan W. PCAF acetylates Runx2 and promotes osteoblast differentiation. J. Bone Miner. Metab. 2013;31(4):381–389. doi: 10.1007/s00774-013-0428-y. [DOI] [PubMed] [Google Scholar]
- 79.Jeon E.J., Lee K.Y., Choi N.S., Lee M.H., Kim H.N., Jin Y.H., Ryoo H.M., Choi J.Y., Yoshida M., Nishino N., Oh B.C., Lee K.S., Lee Y.H., Bae S.C. Bone morphogenetic protein-2 stimulates Runx2 acetylation. J. Biol. Chem. 2006;281(24):16502–16511. doi: 10.1074/jbc.M512494200. [DOI] [PubMed] [Google Scholar]
- 80.Pelletier N., Champagne N., Stifani S., Yang X.J. MOZ and MORF histone acetyltransferases interact with the Runt-domain transcription factor Runx2. Oncogene. 2002;21(17):2729–2740. doi: 10.1038/sj.onc.1205367. [DOI] [PubMed] [Google Scholar]
- 81.Bernstein B.E., Mikkelsen T.S., Xie X., Kamal M., Huebert D.J., Cuff J., Fry B., Meissner A., Wernig M., Plath K., Jaenisch R., Wagschal A., Feil R., Schreiber S.L., Lander E.S. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125(2):315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 82.Wei Y., Chen Y.H., Li L.Y., Lang J., Yeh S.P., Shi B., Yang C.C., Yang J.Y., Lin C.Y., Lai C.C., Hung M.C. CDK1-dependent phosphorylation of EZH2 suppresses methylation of H3K27 and promotes osteogenic differentiation of human mesenchymal stem cells. Nat. Cell Biol. 2011;13(1):87–94. doi: 10.1038/ncb2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Van Hoof D., Muñoz J., Braam S.R., Pinkse M.W., Linding R., Heck A.J., Mummery C.L., Krijgsveld J. Phosphorylation dynamics during early differentiation of human embryonic stem cells. Cell Stem Cell. 2009;5(2):214–226. doi: 10.1016/j.stem.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 84.Schwarz D., Varum S., Zemke M., Schöler A., Baggiolini A., Draganova K., Koseki H., Schübeler D., Sommer L. Ezh2 is required for neural crest-derived cartilage and bone formation. Development. 2014;141(4):867–877. doi: 10.1242/dev.094342. [DOI] [PubMed] [Google Scholar]
- 85.Nayak A., Viale-Bouroncle S., Morsczeck C., Muller S. The SUMO-specific isopeptidase SENP3 regulates MLL1/MLL2 methyltransferase complexes and controls osteogenic differentiation. Mol. Cell. 2014;55(1):47–58. doi: 10.1016/j.molcel.2014.05.011. [DOI] [PubMed] [Google Scholar]
- 86.Ye L., Fan Z., Yu B., Chang J., Al Hezaimi K., Zhou X., Park N.H., Wang C.Y. Histone demethylases KDM4B and KDM6B promotes osteogenic differentiation of human MSCs. Cell Stem Cell. 2012;11(1):50–61. doi: 10.1016/j.stem.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lan F., Bayliss P.E., Rinn J.L., Whetstine J.R., Wang J.K., Chen S., Iwase S., Alpatov R., Issaeva I., Canaani E., Roberts T.M., Chang H.Y., Shi Y. A histone H3 lysine 27 demethylase regulates animal posterior development. Nature. 2007;449(7163):689–694. doi: 10.1038/nature06192. [DOI] [PubMed] [Google Scholar]
- 88.Shi Y. Histone lysine demethylases: emerging roles in development, physiology and disease. Nat. Rev. Genet. 2007;8(11):829–833. doi: 10.1038/nrg2218. [DOI] [PubMed] [Google Scholar]
- 89.Fan Z., Yamaza T., Lee J.S., Yu J., Wang S., Fan G., Shi S., Wang C.Y. BCOR regulates mesenchymal stem cell function by epigenetic mechanisms. Nat. Cell Biol. 2009;11(8):1002–1009. doi: 10.1038/ncb1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Huynh K.D., Fischle W., Verdin E., Bardwell V.J. BCoR, a novel corepressor involved in BCL-6 repression. Genes Dev. 2000;14(14):1810–1823. [PMC free article] [PubMed] [Google Scholar]
- 91.Gearhart M.D., Corcoran C.M., Wamstad J.A., Bardwell V.J. Polycomb group and SCF ubiquitin ligases are found in a novel BCOR complex that is recruited to BCL6 targets. Mol. Cell. Biol. 2006;26(18):6880–6889. doi: 10.1128/MCB.00630-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sánchez C., Sánchez I., Demmers J.A., Rodriguez P., Strouboulis J., Vidal M. Proteomics analysis of Ring1B/Rnf2 interactors identifies a novel complex with the Fbxl10/Jhdm1B histone demethylase and the Bcl6 interacting corepressor. Mol. Cell. Proteomics. 2007;6(5):820–834. doi: 10.1074/mcp.M600275-MCP200. [DOI] [PubMed] [Google Scholar]
- 93.Brewer S., Feng W., Huang J., Sullivan S., Williams T. Wnt1-Cre-mediated deletion of AP-2alpha causes multiple neural crest-related defects. Dev. Biol. 2004;267(1):135–152. doi: 10.1016/j.ydbio.2003.10.039. [DOI] [PubMed] [Google Scholar]
- 94.Schorle H., Meier P., Buchert M., Jaenisch R., Mitchell P.J. Transcription factor AP-2 essential for cranial closure and craniofacial development. Nature. 1996;381(6579):235–238. doi: 10.1038/381235a0. [DOI] [PubMed] [Google Scholar]
- 95.Fodor B.D., Kubicek S., Yonezawa M., O’Sullivan R.J., Sengupta R., Perez-Burgos L., Opravil S., Mechtler K., Schotta G., Jenuwein T. Jmjd2b antagonizes H3K9 trimethylation at pericentric heterochromatin in mammalian cells. Genes Dev. 2006;20(12):1557–1562. doi: 10.1101/gad.388206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Xu J., Yu B., Hong C., Wang C.Y. KDM6B epigenetically regulates odontogenic differentiation of dental mesenchymal stem cells. Int. J. Oral Sci. 2013;5(4):200–205. doi: 10.1038/ijos.2013.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gimble F.S., Stephens B.W. Substitutions in conserved dodecapeptide motifs that uncouple the DNA binding and DNA cleavage activities of PI-SceI endonuclease. J. Biol. Chem. 1995;270(11):5849–5856. doi: 10.1074/jbc.270.11.5849. [DOI] [PubMed] [Google Scholar]
- 98.Hata K., Nishimura R., Ikeda F., Yamashita K., Matsubara T., Nokubi T., Yoneda T. Differential roles of Smad1 and p38 kinase in regulation of peroxisome proliferator-activating receptor gamma during bone morphogenetic protein 2-induced adipogenesis. Mol. Biol. Cell. 2003;14(2):545–555. doi: 10.1091/mbc.E02-06-0356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Huang H., Song T.J., Li X., Hu L., He Q., Liu M., Lane M.D., Tang Q.Q. BMP signaling pathway is required for commitment of C3H10T1/2 pluripotent stem cells to the adipocyte lineage. Proc. Natl. Acad. Sci. USA. 2009;106(31):12670–12675. doi: 10.1073/pnas.0906266106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jin W., Takagi T., Kanesashi S.N., Kurahashi T., Nomura T., Harada J., Ishii S. Schnurri-2 controls BMP-dependent adipogenesis via interaction with Smad proteins. Dev. Cell. 2006;10(4):461–471. doi: 10.1016/j.devcel.2006.02.016. [DOI] [PubMed] [Google Scholar]
- 101.Tseng Y.H., Kokkotou E., Schulz T.J., Huang T.L., Winnay J.N., Taniguchi C.M., Tran T.T., Suzuki R., Espinoza D.O., Yamamoto Y., Ahrens M.J., Dudley A.T., Norris A.W., Kulkarni R.N., Kahn C.R. New role of bone morphogenetic protein 7 in brown adipogenesis and energy expenditure. Nature. 2008;454(7207):1000–1004. doi: 10.1038/nature07221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gupta R.K., Arany Z., Seale P., Mepani R.J., Ye L., Conroe H.M., Roby Y.A., Kulaga H., Reed R.R., Spiegelman B.M. Transcriptional control of preadipocyte determination by Zfp423. Nature. 2010;464(7288):619–623. doi: 10.1038/nature08816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wang L., Xu S., Lee J.E., Baldridge A., Grullon S., Peng W., Ge K. Histone H3K9 methyltransferase G9a represses PPARγ expression and adipogenesis. EMBO J. 2013;32(1):45–59. doi: 10.1038/emboj.2012.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Klose R.J., Bird A.P. Genomic DNA methylation: the mark and its mediators. Trends Biochem. Sci. 2006;31(2):89–97. doi: 10.1016/j.tibs.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 105.Weber M., Hellmann I., Stadler M.B., Ramos L., Pääbo S., Rebhan M., Schübeler D. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat. Genet. 2007;39(4):457–466. doi: 10.1038/ng1990. [DOI] [PubMed] [Google Scholar]
- 106.Ben-Porath I., Cedar H. Epigenetic crosstalk. Mol. Cell. 2001;8(5):933–935. doi: 10.1016/S1097-2765(01)00399-9. [DOI] [PubMed] [Google Scholar]
- 107.Richards E.J., Elgin S.C. Epigenetic codes for heterochromatin formation and silencing: rounding up the usual suspects. Cell. 2002;108(4):489–500. doi: 10.1016/S0092-8674(02)00644-X. [DOI] [PubMed] [Google Scholar]
- 108.Jones P.L., Veenstra G.J., Wade P.A., Vermaak D., Kass S.U., Landsberger N., Strouboulis J., Wolffe A.P. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet. 1998;19(2):187–191. doi: 10.1038/561. [DOI] [PubMed] [Google Scholar]
- 109.Mutskov V., Felsenfeld G. Silencing of transgene transcription precedes methylation of promoter DNA and histone H3 lysine 9. EMBO J. 2004;23(1):138–149. doi: 10.1038/sj.emboj.7600013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Borgel J., Guibert S., Li Y., Chiba H., Schübeler D., Sasaki H., Forné T., Weber M. Targets and dynamics of promoter DNA methylation during early mouse development. Nat. Genet. 2010;42(12):1093–1100. doi: 10.1038/ng.708. [DOI] [PubMed] [Google Scholar]
- 111.Huang K., Wu Z., Liu Z., Hu G., Yu J., Chang K.H., Kim K.P., Le T., Faull K.F., Rao N., Gennery A., Xue Z., Wang C.Y., Pellegrini M., Fan G. Selective demethylation and altered gene expression are associated with ICF syndrome in human-induced pluripotent stem cells and mesenchymal stem cells. Hum. Mol. Genet. 2014;23(24):6448–6457. doi: 10.1093/hmg/ddu365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Biniszkiewicz D., Gribnau J., Ramsahoye B., Gaudet F., Eggan K., Humpherys D., Mastrangelo M.A., Jun Z., Walter J., Jaenisch R. Dnmt1 overexpression causes genomic hypermethylation, loss of imprinting, and embryonic lethality. Mol. Cell. Biol. 2002;22(7):2124–2135. doi: 10.1128/MCB.22.7.2124-2135.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chan M.F., van Amerongen R., Nijjar T., Cuppen E., Jones P.A., Laird P.W. Reduced rates of gene loss, gene silencing, and gene mutation in Dnmt1-deficient embryonic stem cells. Mol. Cell. Biol. 2001;21(22):7587–7600. doi: 10.1128/MCB.21.22.7587-7600.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Jackson-Grusby L., Beard C., Possemato R., Tudor M., Fambrough D., Csankovszki G., Dausman J., Lee P., Wilson C., Lander E., Jaenisch R. Loss of genomic methylation causes p53-dependent apoptosis and epigenetic deregulation. Nat. Genet. 2001;27(1):31–39. doi: 10.1038/83730. [DOI] [PubMed] [Google Scholar]
- 115.Rhee I., Jair K.W., Yen R.W., Lengauer C., Herman J.G., Kinzler K.W., Vogelstein B., Baylin S.B., Schuebel K.E. CpG methylation is maintained in human cancer cells lacking DNMT1. Nature. 2000;404(6781):1003–1007. doi: 10.1038/35010000. [DOI] [PubMed] [Google Scholar]
- 116.Tsai C.C., Su P.F., Huang Y.F., Yew T.L., Hung S.C. Oct4 and Nanog directly regulate Dnmt1 to maintain self-renewal and undifferentiated state in mesenchymal stem cells. Mol. Cell. 2012;47(2):169–182. doi: 10.1016/j.molcel.2012.06.020. [DOI] [PubMed] [Google Scholar]
- 117.Collas P. Programming differentiation potential in mesenchymal stem cells. Epigenetics. 2010;5(6):476–482. doi: 10.4161/epi.5.6.12517. [DOI] [PubMed] [Google Scholar]
- 118.Boquest A.C., Noer A., Sørensen A.L., Vekterud K., Collas P. CpG methylation profiles of endothelial cell-specific gene promoter regions in adipose tissue stem cells suggest limited differentiation potential toward the endothelial cell lineage. Stem Cells. 2007;25(4):852–861. doi: 10.1634/stemcells.2006-0428. [DOI] [PubMed] [Google Scholar]
- 119.Sørensen A.L., Timoskainen S., West F.D., Vekterud K., Boquest A.C., Ahrlund-Richter L., Stice S.L., Collas P. Lineage-specific promoter DNA methylation patterns segregate adult progenitor cell types. Stem Cells Dev. 2010;19(8):1257–1266. doi: 10.1089/scd.2009.0309. [DOI] [PubMed] [Google Scholar]
- 120.Jüttermann R., Li E., Jaenisch R. Toxicity of 5-aza-2′-deoxycytidine to mammalian cells is mediated primarily by covalent trapping of DNA methyltransferase rather than DNA demethylation. Proc. Natl. Acad. Sci. USA. 1994;91(25):11797–11801. doi: 10.1073/pnas.91.25.11797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.El-Serafi A.T., Oreffo R.O., Roach H.I. Epigenetic modifiers influence lineage commitment of human bone marrow stromal cells: Differential effects of 5-aza-deoxycytidine and trichostatin A. Differentiation. 2011;81(1):35–41. doi: 10.1016/j.diff.2010.09.183. [DOI] [PubMed] [Google Scholar]
- 122.Yan X., Ehnert S., Culmes M., Bachmann A., Seeliger C., Schyschka L., Wang Z., Rahmanian-Schwarz A., Stöckle U., De Sousa P.A., Pelisek J., Nussler A.K. 5-azacytidine improves the osteogenic differentiation potential of aged human adipose-derived mesenchymal stem cells by DNA demethylation. PLoS One. 2014;9(6):e90846. doi: 10.1371/journal.pone.0090846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zhu X.W., Zuo J.L., Liu Y.H., Zang R., Li Y.K., Wang X., Li J.M. Osteogenesis of umbilical mesenchymal stem cells is enhanced in absence of DNA methyltransferase 3B (DNMT3B) through upregulating Runx2 expression. Eur. Rev. Med. Pharmacol. Sci. 2014;18(20):3004–3009. [PubMed] [Google Scholar]
- 124.Fouse S.D., Shen Y., Pellegrini M., Cole S., Meissner A., Van Neste L., Jaenisch R., Fan G. Promoter CpG methylation contributes to ES cell gene regulation in parallel with Oct4/Nanog, PcG complex, and histone H3 K4/K27 trimethylation. Cell Stem Cell. 2008;2(2):160–169. doi: 10.1016/j.stem.2007.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lagarkova M.A., Volchkov P.Y., Lyakisheva A.V., Philonenko E.S., Kiselev S.L. Diverse epigenetic profile of novel human embryonic stem cell lines. Cell Cycle. 2006;5(4):416–420. doi: 10.4161/cc.5.4.2440. [DOI] [PubMed] [Google Scholar]
- 126.Berdasco M., Melguizo C., Prados J., Gómez A., Alaminos M., Pujana M.A., Lopez M., Setien F., Ortiz R., Zafra I., Aranega A., Esteller M. DNA methylation plasticity of human adipose-derived stem cells in lineage commitment. Am. J. Pathol. 2012;181(6):2079–2093. doi: 10.1016/j.ajpath.2012.08.016. [DOI] [PubMed] [Google Scholar]
- 127.Aranda P., Agirre X., Ballestar E., Andreu E.J., Román-Gómez J., Prieto I., Martín-Subero J.I., Cigudosa J.C., Siebert R., Esteller M., Prosper F. Epigenetic signatures associated with different levels of differentiation potential in human stem cells. PLoS One. 2009;4(11):e7809. doi: 10.1371/journal.pone.0007809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kang M.I., Kim H.S., Jung Y.C., Kim Y.H., Hong S.J., Kim M.K., Baek K.H., Kim C.C., Rhyu M.G. Transitional CpG methylation between promoters and retroelements of tissue-specific genes during human mesenchymal cell differentiation. J. Cell. Biochem. 2007;102(1):224–239. doi: 10.1002/jcb.21291. [DOI] [PubMed] [Google Scholar]
- 129.Lee J.Y., Lee Y.M., Kim M.J., Choi J.Y., Park E.K., Kim S.Y., Lee S.P., Yang J.S., Kim D.S. Methylation of the mouse DIx5 and Osx gene promoters regulates cell type-specific gene expression. Mol. Cells. 2006;22(2):182–188. [PubMed] [Google Scholar]
- 130.Zhang R.P., Shao J.Z., Xiang L.X. GADD45A protein plays an essential role in active DNA demethylation during terminal osteogenic differentiation of adipose-derived mesenchymal stem cells. J. Biol. Chem. 2011;286(47):41083–41094. doi: 10.1074/jbc.M111.258715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Delgado-Calle J., Sañudo C., Bolado A., Fernández A.F., Arozamena J., Pascual-Carra M.A., Rodriguez-Rey J.C., Fraga M.F., Bonewald L., Riancho J.A. DNA methylation contributes to the regulation of sclerostin expression in human osteocytes. J. Bone Miner. Res. 2012;27(4):926–937. doi: 10.1002/jbmr.1491. [DOI] [PubMed] [Google Scholar]
- 132.Delgado-Calle J., Sañudo C., Fernández A.F., García-Renedo R., Fraga M.F., Riancho J.A. Role of DNA methylation in the regulation of the RANKL-OPG system in human bone. Epigenetics. 2012;7(1):83–91. doi: 10.4161/epi.7.1.18753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Delgado-Calle J., Sañudo C., Sánchez-Verde L., García-Renedo R.J., Arozamena J., Riancho J.A. Epigenetic regulation of alkaline phosphatase in human cells of the osteoblastic lineage. Bone. 2011;49(4):830–838. doi: 10.1016/j.bone.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 134.Zhou G.S., Zhang X.L., Wu J.P., Zhang R.P., Xiang L.X., Dai L.C., Shao J.Z. 5-Azacytidine facilitates osteogenic gene expression and differentiation of mesenchymal stem cells by alteration in DNA methylation. Cytotechnology. 2009 doi: 10.1007/s10616-009-9203-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Bedford M.T., Richard S. Arginine methylation an emerging regulator of protein function. Mol. Cell. 2005;18(3):263–272. doi: 10.1016/j.molcel.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 136.Huang J., Berger S.L. The emerging field of dynamic lysine methylation of non-histone proteins. Curr. Opin. Genet. Dev. 2008;18(2):152–158. doi: 10.1016/j.gde.2008.01.012. [DOI] [PubMed] [Google Scholar]
- 137.Bartel R.L., Borchardt R.T. Effects of adenosine dialdehyde on S-adenosylhomocysteine hydrolase and S-adenosylmethionine-dependent transmethylations in mouse L929 cells. Mol. Pharmacol. 1984;25(3):418–424. [PubMed] [Google Scholar]
- 138.Hermes M., Osswald H., Mattar J., Kloor D. Influence of an altered methylation potential on mRNA methylation and gene expression in HepG2 cells. Exp. Cell Res. 2004;294(2):325–334. doi: 10.1016/j.yexcr.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 139.Fontecave M., Atta M., Mulliez E. S-adenosylmethionine: nothing goes to waste. Trends Biochem. Sci. 2004;29(5):243–249. doi: 10.1016/j.tibs.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 140.Vaes B.L., Lute C., van der Woning S.P., Piek E., Vermeer J., Blom H.J., Mathers J.C., Müller M., de Groot L.C., Steegenga W.T. Inhibition of methylation decreases osteoblast differentiation via a non-DNA-dependent methylation mechanism. Bone. 2010;46(2):514–523. doi: 10.1016/j.bone.2009.09.033. [DOI] [PubMed] [Google Scholar]
- 141.Schmitz K.M., Schmitt N., Hoffmann-Rohrer U., Schäfer A., Grummt I., Mayer C. TAF12 recruits Gadd45a and the nucleotide excision repair complex to the promoter of rRNA genes leading to active DNA demethylation. Mol. Cell. 2009;33(3):344–353. doi: 10.1016/j.molcel.2009.01.015. [DOI] [PubMed] [Google Scholar]
- 142.Davis C.D., Ross S.A. Dietary components impact histone modifications and cancer risk. Nutr. Rev. 2007;65(2):88–94. doi: 10.1111/j.1753-4887.2007.tb00285.x. [DOI] [PubMed] [Google Scholar]
- 143.Chen C.C., Wang K.Y., Shen C.K. DNA 5-methylcytosine demethylation activities of the mammalian DNA methyltransferases. J. Biol. Chem. 2013;288(13):9084–9091. doi: 10.1074/jbc.M112.445585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Haasper C., Jagodzinski M., Drescher M., Meller R., Wehmeier M., Krettek C., Hesse E. Cyclic strain induces FosB and initiates osteogenic differentiation of mesenchymal cells. Exp. Toxicol. Pathol. 2008;59(6):355–363. doi: 10.1016/j.etp.2007.11.013. [DOI] [PubMed] [Google Scholar]
- 145.Haudenschild A.K., Hsieh A.H., Kapila S., Lotz J.C. Pressure and distortion regulate human mesenchymal stem cell gene expression. Ann. Biomed. Eng. 2009;37(3):492–502. doi: 10.1007/s10439-008-9629-2. [DOI] [PubMed] [Google Scholar]
- 146.Jagodzinski M., Breitbart A., Wehmeier M., Hesse E., Haasper C., Krettek C., Zeichen J., Hankemeier S. Influence of perfusion and cyclic compression on proliferation and differentiation of bone marrow stromal cells in 3-dimensional culture. J. Biomech. 2008;41(9):1885–1891. doi: 10.1016/j.jbiomech.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 147.Pelaez D., Arita N., Cheung H.S. Extracellular signal-regulated kinase (ERK) dictates osteogenic and/or chondrogenic lineage commitment of mesenchymal stem cells under dynamic compression. Biochem. Biophys. Res. Commun. 2012;417(4):1286–1291. doi: 10.1016/j.bbrc.2011.12.131. [DOI] [PubMed] [Google Scholar]
- 148.Gurkan U.A., Akkus O. The mechanical environment of bone marrow: a review. Ann. Biomed. Eng. 2008;36(12):1978–1991. doi: 10.1007/s10439-008-9577-x. [DOI] [PubMed] [Google Scholar]
- 149.Li Y.J., Batra N.N., You L., Meier S.C., Coe I.A., Yellowley C.E., Jacobs C.R. Oscillatory fluid flow affects human marrow stromal cell proliferation and differentiation. J. Orthop. Res. 2004;22(6):1283–1289. doi: 10.1016/j.orthres.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 150.Arnsdorf E.J., Tummala P., Castillo A.B., Zhang F., Jacobs C.R. The epigenetic mechanism of mechanically induced osteogenic differentiation. J. Biomech. 2010;43(15):2881–2886. doi: 10.1016/j.jbiomech.2010.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Li Y., Chu J.S., Kurpinski K., Li X., Bautista D.M., Yang L., Sung K.L., Li S. Biophysical regulation of histone acetylation in mesenchymal stem cells. Biophys. J. 2011;100(8):1902–1909. doi: 10.1016/j.bpj.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]