

Abstract
Proteolytic cleavage of the tumor necrosis factor (TNF) receptor (TNFR) from the cell surface contributes to anti-inflammatory responses and may be beneficial in reducing the excessive inflammation associated with multiple organ failure and mortality during sepsis. Using a clinically relevant mouse model of polymicrobial abdominal sepsis, we found that the production of inducible nitric oxide synthase (iNOS) in hepatocytes led to the cyclic guanosine monophosphate (cGMP)–dependent activation of the protease TACE (TNF-converting enzyme) and the shedding of TNFR. Furthermore, treating mice with a cGMP analog after the induction of sepsis increased TNFR shedding and decreased systemic inflammation. Similarly, increasing the abundance of cGMP with a clinically approved phosphodiesterase 5 inhibitor (sildenafil) also decreased markers of systemic inflammation, protected against organ injury, and increased circulating amounts of TNFR1 in mice with sepsis. We further confirmed that a similar iNOS-cGMP-TACE pathway was required for TNFR1 shedding by human hepatocytes in response to the bacterial product lipopolysaccharide. Our data suggest that increasing the bioavailability of cGMP might be beneficial in ameliorating the inflammation associated with sepsis.
INTRODUCTION
Tumor necrosis factor (TNF), a proinflammatory cytokine secreted mostly by immune cells, plays an important role in the pathophysiology of sepsis. Appropriate amounts of TNF most likely exert a beneficial effect on host survival to infection, but exaggerated or sustained increases in TNF abundance can lead to toxicity (1, 2), which, at a cellular level, manifests as cell death (3, 4). Plasma TNF concentrations correlate with the severity of sepsis (5), and a meta-analysis of anti-TNF therapies in human sepsis trials indicated that inhibiting TNF or TNF-dependent signaling was on the “side of benefit” (6). Thus, TNF remains a target of interest in studies on the pathobiology of sepsis, as well as a target in new human sepsis trials (7).
TNF initiates cellular inflammatory responses through engagement with two cell surface receptors: TNF receptor 1 (TNFR1) and TNFR2. TNFR1 is found on immune cells, such as macrophages (8), and parenchymal cells, such as hepatocytes (9). Excessive activation of TNFR1 by TNF leads to the apoptosis of hepatocytes, and thus leads to organ damage (9); however, membrane-bound TNFR1 can be cleaved through proteolytic shedding of its ectodomain, which is dependent on activation of TNF-converting enzyme (TACE, also known as ADAM17) (10). Receptor shedding is thought to be protective by reducing cellular responses to TNF and by binding to and sequestering extracellular TNF.
Endotoxemia (10) and sepsis (9, 11) are associated with marked increases in soluble TNFR1 (sTNFR1) concentrations in the circulation. Studies showed that neutralizing TNF with sTNFR1 lessens organ damage (12) and mortality (13) in mice with sepsis. We previously demonstrated that the Toll-like receptor 4 (TLR4)–dependent expression of the gene encoding inducible nitric oxide synthase (iNOS) in hepatocytes leads to nitric oxide (NO) production and activation of the cyclic guanosine monophosphate (cGMP)– and protein kinase G (PKG)–dependent activation of TACE, which cleaves TNFR1 (10). However, the signaling pathways that mediate shedding of hepatocyte TNFR1 (HC-TNFR1) during sepsis are unclear. In polymicrobial sepsis, multiple pathogen-derived ligands of TLRs and cytokines, such as interleukin-1 (IL-1), are potent inducers of iNOS production by hepatocytes (14) and may also be potent inducers of HC-TNFR1 shedding. Therefore, we aimed to elucidate the stimuli and signaling pathways that mediate HC-TNFR1 shedding in polymicrobial sepsis. We provide evidence that multiple TLR ligands and cytokines stimulate HC-TNFR1 shedding in sepsis. Furthermore, we found thatmyeloid differentiation marker 88 (MyD88)–dependent iNOS production in hepatocytes led to the cGMP-dependent activation of TACE and shedding of TNFR1. Finally, strategies that increased cGMP bioavailability enhanced receptor shedding, reduced systemic inflammation, and protected organs from injury in sepsis. These data indicate a link between MyD88, the iNOS-NO-cGMP pathway, and TNF signaling during sepsis. This mechanism for TNFR1 shedding could be exploited therapeutically to limit excessive inflammation during sepsis.
RESULTS
Multiple TLR ligands and cytokines stimulate HC-TNFR1 shedding during polymicrobial sepsis
We previously showed that HC-TNFR1 shedding is stimulated by the TLR4 ligand lipopolysaccharide (LPS) in vitro and in vivo (10); however, during polymicrobial sepsis, multiple TLR ligands and cytokines are involved in propagating the inflammatory response. To determine the mechanisms involved in a clinically relevant polymicrobial sepsis model, we assessed TNFR1 shedding in mice during sepsis induced by cecal ligation and puncture (CLP). The control mice underwent laparotomy and manipulation of the cecum without puncture. Concentrations of sTNFR1 in the circulation increased over time in both the sublethal and lethal models of CLP (fig. S1A). In the sublethal model, all of the mice survived until 24 hours after CLP. The amounts of sTNFR1 were greatest at 18 hours after CLP and returned to baseline by 24 hours. In the lethal model, all of the mice survived until 18 hours after CLP; however, mortality reached 66% by 24 hours. The amounts of plasma sTNFR1 were substantially greater in the lethal model than in the sublethal model at 18 hours (fig. S1A). Plasma TNF concentrations increased early in both models, with the greatest amounts at 6 hours (fig. S1B). That the amount of sTNFR1 peaked much later than did that of TNF suggests that direct TNF-TNFR1 interactions do not play a major role in sTNFR1 release in sepsis.
We used the lethal model of CLP to assess the mechanisms of TNFR1 shedding in polymicrobial sepsis. We first investigated the extent to which increases in plasma sTNFR1 abundance were TLR4-dependent in CLP. We subjected wild-type and TLR4-deficient (Tlr4−/−) mice to CLP and assessed circulating amounts of sTNFR1 after 8 hours by enzyme-linked immunosorbent assay (ELISA). We found that plasma sTNFR1 amounts were substantially increased in wild-type and Tlr4−/− mice after CLP, with no statistically significant difference between both groups (Fig. 1A). Thus, TNFR1 shedding does not appear to be TLR4-dependent in this polymicrobial sepsis model. We next assessed the capacity of TLR ligands, in addition to LPS, to induce HC-TNFR1 shedding in vitro. Compared to the amount of sTNFR1 shed by untreated control hepatocytes, the amounts of sTNFR1 in the culture media of hepatocytes treated for 24 hours with HKLM (heat-killed Listeria monocytogenes, a TLR2 ligand), LPS (a TLR4 ligand), disulfide HMGB1 (high-mobility group protein 1, a TLR4 ligand), FLA-ST (flagellin from Salmonella typhimurium, a TLR5 ligand), and OD1669 (CpG oligonucleotide 1669, a TLR9 ligand) were markedly increased (Fig. 1B).This observation suggests that multiple TLR ligands stimulate HC-TNFR1 shedding in vitro.
Fig. 1. Multiple TLR ligands and cytokines stimulate HC-TNFR1 shedding in vitro and in vivo.
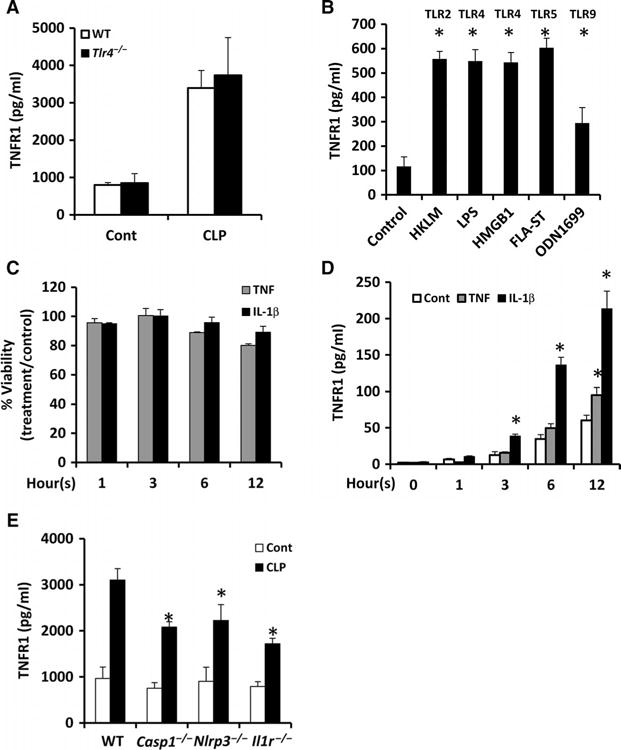
(A) Wild-type (WT) and Tlr4−/− mice were subjected to CLP. In the control mice, laparotomy and cecal manipulation were performed without ligation and puncture. Eight hours later, plasma sTNFR1 concentrations were measured by ELISA. Data are means ± SD from 8 to 10 mice per group from two separate experiments. (B) Mouse hepatocytes were left untreated (Control) or were stimulated for 24 hours with the TLR ligands HKLM (1 × 108 cells/ml), LPS (100 ng/ml), HMGB1 (5 μg/ml), FLA-ST (1 μg/ml), and ODN1669 (5 μg/ml). The concentrations of TNFR1 in the culture media were measured by ELISA. Data are means ± SD from three experiments. *P < 0.05 versus control by two-tailed unpaired t test. (C and D) Mouse hepatocytes were left untreated or were incubated with TNF (500 U/ml) or IL-1β (100 U/ml) for the indicated times. At each time point, (C) cell viability and (D) the concentration of TNFR1 in the culture media were determined. Data are means ± SD from three experiments. *P < 0.05 versus control by two-tailed unpaired t test. (E) Mice of the indicated genotypes were subjected to CLP. Eight hours later, the plasma concentrations of TNFR1 were determined by ELISA. Data are means ± SD from six mice per group of each genotype from two experiments. *P < 0.05 versus control by two-tailed unpaired t test.
Cytokines, such as TNF and IL-1β, are important early inflammatory mediators during sepsis. To investigate whether these cytokines also stimulated HC-TNFR1 shedding, we treated primary hepatocytes with TNF and IL-1β in culture. Treatment with either cytokine did not result in substantial cell death (Fig. 1C). We found that hepatocytes spontaneously released low amounts of sTNFR1 over time (Fig. 1D). The amounts of sTNFR1 in the media of cells treated with IL-1β increased in a time-dependent manner compared to that in the media of untreated cells (Fig. 1D), whereas sTNFR1 amounts in the media were only slightly increased after 12 hours of treatment with TNF. These results suggest that IL-1β stimulates HC-TNFR1 shedding in vitro. Consistent with these results, knockout of caspase-1 or the inflammasone component NLRP3 (NACHT, LRR, and PYD domains–containing protein 3), both of which result in abrogation of IL-1β processing (fig. S2), was associated with lower circulating concentrations of sTNFR1 at 8 hours after CLP (Fig. 1E). Loss of IL-1 signaling using Il1r−/− mice was also associated with substantially decreased circulating concentrations of sTNFR1 after CLP compared to those of wild-type mice (Fig. 1E). These results suggest that IL-1β also stimulates TNFR1 shedding in vivo.
TNFR1 shedding from hepatocytes and myeloid cells during sepsis is dependent on MyD88
Because TLR ligands and IL-1β initiate signaling mediated by the adaptor proteins MyD88 and TRIF (TIR domain–containing adaptor–inducing interferon-β), we hypothesized that TNFR1 shedding was mediated by signaling pathways requiring one or both molecules. When wild-type, Myd88−/−, or Trif−/− mice were subjected to CLP for 8 hours, their plasma concentrations of sTNFR1 increased substantially (Fig. 2A); however, the amounts of plasma sTNFR1 in Myd88−/− mice subjected to CLP were markedly less than those in similarly treated wild-type or Trif−/− mice (Fig. 2A). The abundance of Tnfr1 mRNA increased twofold in the livers of all mice after CLP, with no statistically significant difference between the strains (Fig. 2B). These data suggest that MyD88 does not regulate Tnfr1 expression in mouse livers after CLP but that it contributes to the signaling that leads to TNFR1 shedding.
Fig. 2. TNFR1 shedding from hepatocytes and myeloid cells during sepsis depends on Myd88.
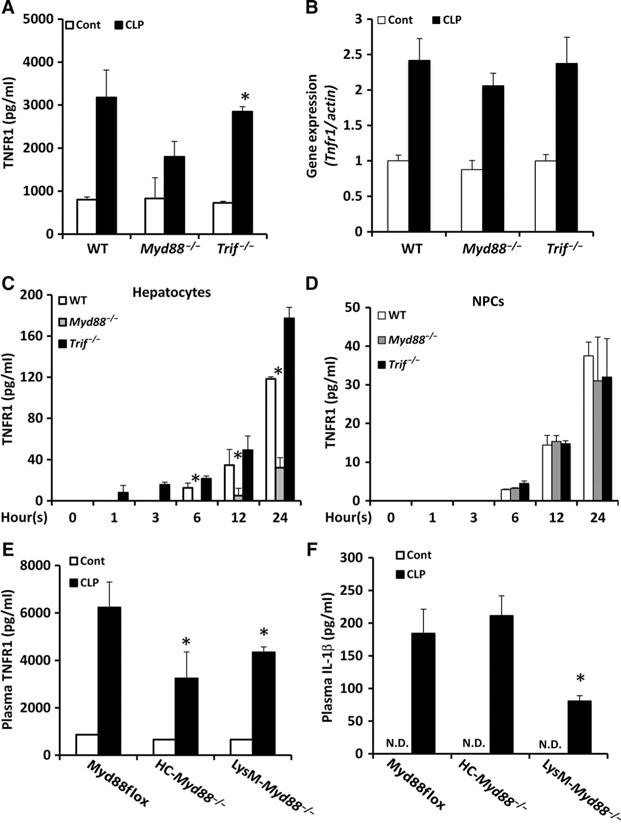
(A and B) WT, Myd88−/−, and Trif−/− mice were subjected to CLP. Eight hours later, (A) plasma sTNFR1 concentrations were determined by ELISA, and (B) Tnfr1 mRNA abundance in the liver was determined by real-time reverse transcription polymerase chain reaction (RT-PCR) analysis. Data are means ± SD from 8 to 10 mice per group from two experiments. *P < 0.05 versus control by two-tailed unpaired t test. (C and D) Hepatocytes (C) and NPCs (D) from the indicated mice were left untreated (zero-hour time point) or were treated with LPS for the indicated times. At each time point, the concentration of TNFR1 in the culture media was determined by ELISA. Data are means ± SD from three experiments. *P < 0.05 by one-way analysis of variance (ANOVA). (E and F) Myd88flox, HC-Myd88−/−, and LysM-Myd88−/− mice were subjected to CLP. Eight hours later, plasma concentrations of (E) TNFR1 and (F) IL-1β were determined by ELISA. Data are means ± SD from 8 to 10 mice per group from two experiments. *P < 0.05 versus Myd88flox by two-tailed unpaired t test. N.D., not detectable.
We and others previously reported that hepatocytes (10, 15) and immune cells (8, 16) shed TNFR1. To investigate the role of MyD88 and TRIF in TNFR1 shedding in different liver cell populations, we assessed the LPS-stimulated shedding of TNFR1 from hepatocytes and nonparenchymal cells (NPCs), including endothelial cells, Kupffer cells, and stellate cells isolated from wild-type, Myd88−/−, or Trif−/− mice. The amounts of sTNFR1 in the culture media of hepatocytes (Fig. 2C) and NPCs (Fig. 2D) from all strains of mice increased in a time-dependent manner. TNFR1 abundance in the media of hepatocytes was three- to fourfold greater than that in the media of NPCs for both wild-type and Trif−/− mice (Fig. 2, C and D), suggesting that hepatocytes are a major source of sTNFR1. Furthermore, the concentration of sTNFR1 in the culture media of Myd88−/− hepatocytes was substantially less than that in the media of wild-type and Trif−/− cells (Fig. 2C), suggesting that HC-TNFR1 shedding is MyD88-dependent. There was no statistically significant difference in the concentration of sTNFR1 in the culture media of wild-type, Myd88−/−, or Trif−/− in NPCs (Fig. 2D), suggesting that NPC-TNFR1 shedding is not dependent on either MyD88 or TRIF alone.
To further investigate the cell-specific role of MyD88 in TNFR1 shedding in vivo during sepsis, we used the Cre-loxP system to generate mouse strains with hepatocyte-specific (HC-Myd88−/−) or myeloid cell– specific (LysM-Myd88−/−) loss of MyD88. To test the effect of the Cre recombinase on cellular toxicity and the response to LPS in mice, we isolated hepatocytes and macrophages from Alb-Cre mice (overexpression of Cre specifically in hepatocytes) and Lyz-Cre mice (overexpression of Cre specifically in myeloid cells), respectively. We did not observe any alterations in cellular toxicity or the responses to LPS of hepatocytes from Alb-Cre mice or macrophages from Lyz-Cre mice, including TNFR1 shedding and IL-6 production (figs. S3 and S4). These data suggest that overexpression of the Cre recombinase specifically in these cell types did not alter their responses to a septic stimulus.
We then subjected HC-Myd88−/−, LysM-Myd88−/−, and Myd88flox mice (cell-specific knockout controls) to CLP. The plasma concentrations of sTNFR1 in HC-Myd88−/− mice and LysM-Myd88−/− mice were substantially lower than that in Myd88flox mice at 8 hours after CLP (Fig. 2E). The circulating amounts of IL-1β were similar in Myd88flox and HC-Myd88−/− mice (Fig. 2F), supporting our hypothesis that HC-TNFR1 shedding in vivo during sepsis is MyD88-dependent. However, the circulating concentration of IL-1β in LysM-Myd88−/− after CLP was markedly less than that in Myd88flox mice (Fig. 2F). IL-1β is a strong stimulator of HC-TNFR1 shedding (Fig. 2, C and D), which suggests that MyD88 in myeloid cells might indirectly regulate HC-TNFR1 shedding by stimulating the production of IL-1β. Together, these data suggest that MyD88 plays a dominant role in TNFR1 shedding during sepsis.
HC-TNFR1 shedding during sepsis is mediated through the MyD88-dependent production of iNOS
We previously showed that HC-TNFR1 shedding is mediated by iNOS (10). Therefore, we postulated that iNOS might be downstream of MyD88 in the regulation of TNFR1 shedding during sepsis. To test this hypothesis, we assessed the expression of Nos2 (the gene that encodes iNOS) in the liver. The abundance of Nos2 mRNA was increased in the liver of wild-type mice, but not Myd88−/− mice, after CLP (Fig. 3A). Furthermore, Nos2 expression in the liver of HC-Myd88−/− mice was substantially less than that in MyD88-Flox mice after CLP (Fig. 3A). The immunofluorescence signal based on antibody-dependent visualization of iNOS protein increased in the liver of wild-type mice after CLP, but no obvious increase in signal was observed in the liver of Myd88−/− mice after CLP (Fig. 3B). In vitro, the abundance of iNOS protein in LPS-treated hepatocytes from wild-type mice, but not Myd88−/− mice, increased in a time-dependent manner (Fig. 3C). Hepatocyte iNOS abundance (Fig. 3C) correlated with sTNFR1 concentration in culture media (Fig. 2C), whereas HC-TNFR1 abundance (Fig. 3C) inversely correlated with the concentrations of sTNFR1 in the culture media (Fig. 2C). These data suggest that TNFR1 shedding during sepsis requires MyD88-dependent increases in iNOS abundance in hepatocytes.
Fig. 3. Shedding of HC-TNFR1 during polymicrobial sepsis is mediated through a MyD88- and iNOS-dependent pathway.
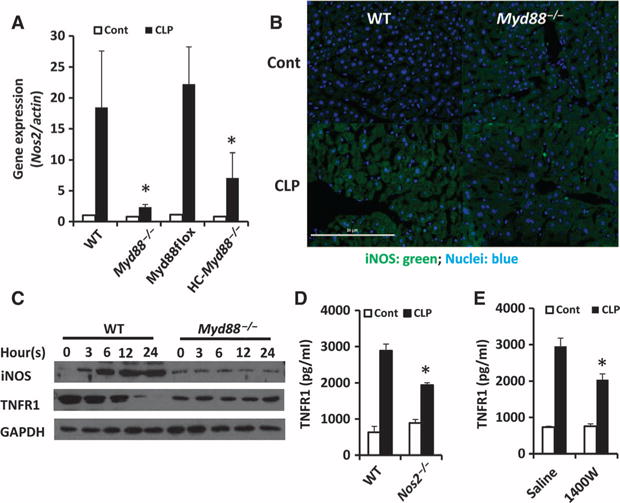
(A and B) Mice of the indicated genotypes were subjected to CLP for 8 hours. (A) The relative expression of Nos2 was determined by real-time RT-PCR analysis. Data are means ± SD from six to eight mice of each genotype from two experiments. *P < 0.05 by one-way ANOVA. (B) The production of iNOS in the liver of the indicated mice was analyzed by immunofluorescence microscopy. Nuclear staining is in blue, whereas iNOS is shown in green. Scale bar, 50 μm. (C) Hepatocytes isolated from the indicated mice were left untreated (zero time point) or were treated with LPS (100 ng/ml) for the indicated times. Whole-cell lysates were then prepared and analyzed by Western blotting with antibodies specific for iNOS and TNFR1. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) abundance was analyzed to control for loading. Western blots are representative of three independent experiments. (D) WT and Nos2−/− mice were subjected to CLP. Eight hours later, plasma concentrations of TNFR1 were determined by ELISA. Data are means ± SD from six mice per group from two experiments. *P < 0.05 by one-way ANOVA. (E) WT mice were subjected to CLP and then were left untreated or were treated with 1400W (5 mg/kg, administered subcutaneously). Eight hours after CLP, plasma concentrations of TNFR1 were determined by ELISA. Data are means ± SD from six mice per group from two experiments. *P < 0.05 by one-way ANOVA.
To further confirm the hypothesis that TNFR1 shedding in sepsis was iNOS-dependent, we treated wild-type mice with 1400W (an iNOS inhibitor) and subjected them and Nos2−/− mice to CLP. As expected, the concentration of sTNFR1 in the circulation was substantially less in Nos2−/− mice and 1400W-treated mice than in control mice (Fig. 3, D and E). Together, these data suggest that TNFR1 shedding during polymicrobial sepsis is partly mediated through the MyD88-iNOS pathway.
Activation of TACE by cGMP induces hepatic TNFR1 shedding during sepsis
Our previous study showed that cGMP was required for iNOS-induced TACE activation and TNFR1 shedding in hepatocytes (10). Therefore, we hypothesized that iNOS-induced cGMP production might activate TACE to cause TNFR1 shedding from hepatocytes during sepsis. To test this hypothesis, we determined the amounts of cGMP in liver homogenates from mice treated with saline or the iNOS inhibitor 1400W. Hepatic cGMP concentrations in wild-type mice increased substantially at 8 hours after CLP compared to those in the saline-treated control mice, whereas cGMP concentrations remained at baseline after CLP in the 1400W-treated mice (Fig. 4A).
Fig. 4. iNOS-stimulated HC-TNFR1 shedding during polymicrobial sepsis is mediated by cGMP and TACE.
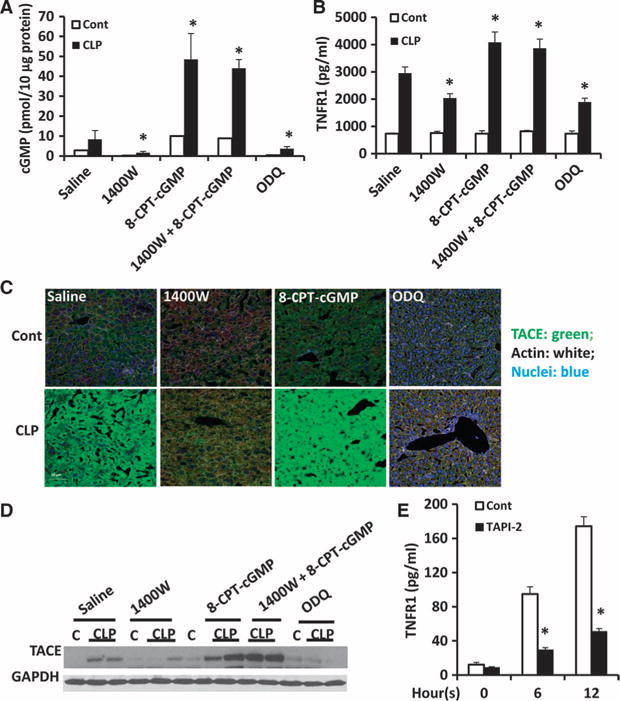
(A to D) WT mice were subjected to CLP and then were immediately treated subcutaneously with saline, 1400W (5 mg/kg), 8-CPT-cGMP (5 mg/kg), both 1400W and 8-CPT-cGMP, or ODQ (20 mg/kg). Eight hours after CLP, the concentrations of (A) cGMP in liver homogenates and (B) TNFR1 in plasma were determined. Data are means ± SD from six to eight mice per group from two experiments. *P < 0.05 by one-way ANOVA. (C) Some of the same mice were analyzed by immunofluorescence microscopy to detect active TACE in the liver. TACE is shown in green, nuclei are in blue, and actin is shown in white. Scale bar, 50 μm. (D) Liver homogenates from the same mice were analyzed by Western blotting to detect TACE. GAPDH was used as a loading control. Western blots are representative of two independent experiments. (E) Hepatocytes isolated from WT mice were left untreated (zero-hour time point) or were treated in vitro with LPS (100 ng/ml) in the absence or presence of 400 nM TAPI-2 for the indicated times. The concentration of TNFR1 in the culture media was determined by ELISA. Data are means ± SD from three independent experiments. *P < 0.05 by one-way ANOVA.
Soluble guanylyl cyclase (sGC) is an enzyme that is activated by NO to produce cGMP from GTP (guanosine triphosphate). We next used the cGMP analog 8-(4-chlorophenylthio)-cGMP (8-CPT-cGMP) and the sGC inhibitor 1H-[1,2,4]oxadiazolo[4,3,-a]quinoxalin-1-one (ODQ) to modify hepatic cGMP amounts after CLP. The abundance of cGMP in the liver of 8-CPT-cGMP–treated mice increased fivefold after CLP compared with that in the untreated control mice (Fig. 4A). In addition, 8-CPT-cGMP restored the hepatic cGMP abundance in mice that were simultaneously treated with the iNOS inhibitor 1400W (Fig. 4A). The CLP-dependent increase in hepatic cGMP abundance was substantially suppressed by ODQ compared with that in saline-treated mice (Fig. 4A). These results suggest that treatment with exogenous cGMP or an inhibitor of sGC efficiently increased or decreased, respectively, the abundance of hepatic cGMP in vivo. Furthermore, circulating sTNFR1 concentrations correlated with hepatic cGMP abundance, which suggests that TNFR1 shedding may be dependent on hepatic cGMP (Fig. 4B). Furthermore, the immunofluorescence signal for active TACE increased in the liver parenchyma of mice after CLP compared with that in control mice (Fig. 4C). Active TACE immunofluorescence increased further in the livers of mice treated with 8-CPT-cGMP and then subjected to CLP than in the livers of mice treated with saline after CLP, whereas there was no obvious CLP-dependent increase in the TACE signal in livers from mice treated with 1400W or ODQ after CLP (Fig. 4C).
Western blotting analysis of TACE abundance in liver homogenates confirmed that the abundance of TACE correlated with hepatic cGMP amounts during sepsis (Fig. 4D). We next treated hepatocytes in vitro with or without the TACE inhibitor TAPI-2 before stimulating them with LPS for various times. We found that the concentration of sTNFR1 in the media of cells not exposed to the inhibitor increased in a time-dependent manner in response to LPS; however, this increase in sTNFR1 abundance was suppressed by TAPI-2 (Fig. 4E). These results suggest that HC-TNFR1 shedding during sepsis may be mediated through an iNOS-cGMP-TACE pathway.
TNFR1 shedding through the iNOS-cGMP pathway attenuates the inflammatory response and liver injury during sepsis
We and others showed that TNF signaling through TNFR1 stimulates cytokine production in immune cells and apoptosis in parenchymal cells, such as hepatocytes (9). Therefore, we postulated that regulation of HC-TNFR1 shedding through the iNOS-cGMP pathway might modulate the systemic inflammatory response and organ damage during sepsis. We collected plasma from mice after they were subjected to CLP and then treated with either the cGMP analog 8-CPT-cGMP (to increase cGMP abundance) or the sGC inhibitor ODQ (to decrease cGMP abundance). The amounts of secreted cytokines were determined by ELISA to assess the regulation of the systemic inflammatory response. Plasma IL-6 was not detectable in the control mice, but it was readily detectable in mice after CLP (Fig. 5A). In mice treated with 1400W to inhibit iNOS, plasma IL-6 concentrations were increased compared to those in mice treated with saline (Fig. 5A). Administration of exogenous cGMP (8-CPT-cGMP) substantially reduced circulating amounts of IL-6 after CLP (Fig. 5A). Administration of 8-CPT-cGMP also reduced the amount of IL-6 in the plasma of mice treated with 1400W after CLP. Furthermore, the amounts of plasma IL-6 in mice treated with ODQ (which inhibits cGMP production) were increased substantially compared to those in saline-treated mice (Fig. 5A). The plasma concentrations of early inflammatory factors, including TNF, KC, and MIP-1α, as well as of late inflammatory factors, such as HMGB1, showed the same trend as that of IL-6 (Fig. 5, B to E). Increasing the bioavailability of cGMP in the liver by treatment with 8-CPT-cGMP prevented liver injury, as determined by measurement of alanine aminotransferase (ALT) concentrations, whereas inhibition of cGMP production with ODQ exaggerated liver injury compared with that in saline-treated mice (Fig. 5F). Thus, the inflammatory response and organ injury correlated with the amount of hepatic cGMP (Fig. 4A) and were inversely proportional to the plasma sTNFR1 concentration (Fig. 4B). This suggests that the regulation of HC-TNFR1 shedding by cGMP can modulate the inflammatory response and liver injury during sepsis in mice.
Fig. 5. Regulation of hepatic TNFR1 shedding through the iNOS-cGMP pathway modulates inflammatory responses and liver injury during sepsis.
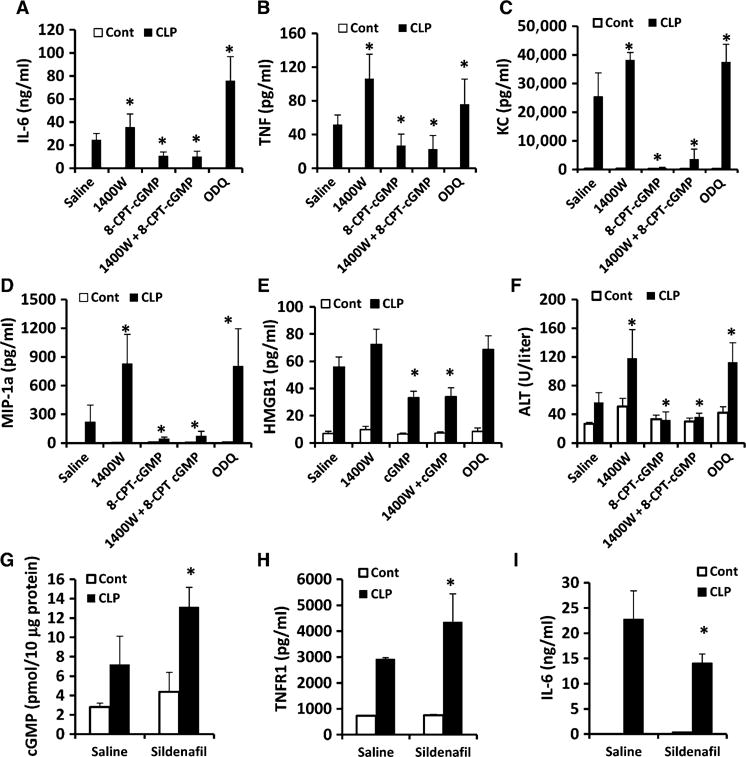
(A to F) WT mice were subjected to CLP and then were immediately treated subcutaneously with saline, 1400W (5 mg/kg), 8-CPT-cGMP (5 mg/kg), both 1400W and 8-CPT-cGMP, or ODQ (20 mg/kg). Eight hours after CLP, plasma concentrations of (A) IL-6, (B) TNF, (C) KC, (D) MIP-1α, (E) HMGB1, and (F) ALT were measured by ELISA. Data are means ± SD from 8 to 10 mice per group from two experiments. *P < 0.05 by one-way ANOVA. (G to I) WT mice were subjected to CLP and then were immediately treated subcutaneously with saline or sildenafil (50 mg/kg). Eight hours after CLP, the concentrations of (G) cGMP in liver homogenates and of (H) TNFR1 and (I) IL-6 in the plasma were determined. Data are means ± SD from six mice per group. *P < 0.05 by one-way ANOVA.
We next tested whether sildenafil, an inhibitor of phosphodiesterase 5 (PDE5) that is used in the clinic, could regulate TNFR1 shedding during sepsis. Sildenafil blocks the degradation of cGMP (17). Mice were subjected to CLP and then were treated with either sildenafil (50 mg/kg) or saline, and plasma was collected after 8 hours. Hepatic cGMP amounts increased slightly after exposure to sildenafil (4.36 ± 2.0 pmol/10 μg protein) compared with those in saline-treated control mice (2.8 ± 0.4 pmol/10 μg protein). However, after CLP was performed, hepatic cGMP amounts were substantially greater in the sildenafil-treated mice (13.1 ± 2.0 pmol/10 μg protein) than in the saline-treated mice (7.23 ± 2.9 pmol/10 μg protein) (Fig. 5G). As expected, plasma sTNFR1 amounts correlated with the abundance of hepatic cGMP (Fig. 5H). Furthermore, circulating IL-6 amounts in mice that received sildenafil after CLP were substantially lower than those in mice that were treated with saline after CLP (Fig. 5I). Together, these data suggest that TNFR1 shedding during sepsis is mediated through the iNOS-cGMP pathway. Increases in TNFR1 shedding through this pathway may reduce the inflammatory response and improve organ damage during sepsis.
Human HC-TNFR1 shedding stimulated by LPS and IL-1β is dependent on the iNOS-cGMP-TACE pathway
Our earlier results suggest that HC-TNFR1 shedding is mediated through the MyD88-iNOS-cGMP-TACE pathway in mice. To test whether TNFR1 shedding in human hepatocytes was also mediated through this pathway, we isolated human hepatocytes and treated them with LPS or IL-1β. The amounts of TNFR1 in the culture media increased in a time-dependent manner after treatment with LPS or IL-1β (Fig. 6A). The immunofluorescence signals for iNOS (red) and TACE (green) increased markedly at 6 hours after treatment with LPS compared with those in untreated control cells (Fig. 6B). Furthermore, TACE colocalized with iNOS and translocated from the perinuclear area to the perimembrane area in the cytoplasm (Fig. 6B).
Fig. 6. TNFR1 shedding by human hepatocytes in vitro is mediated through an iNOS-cGMP-TACE pathway.
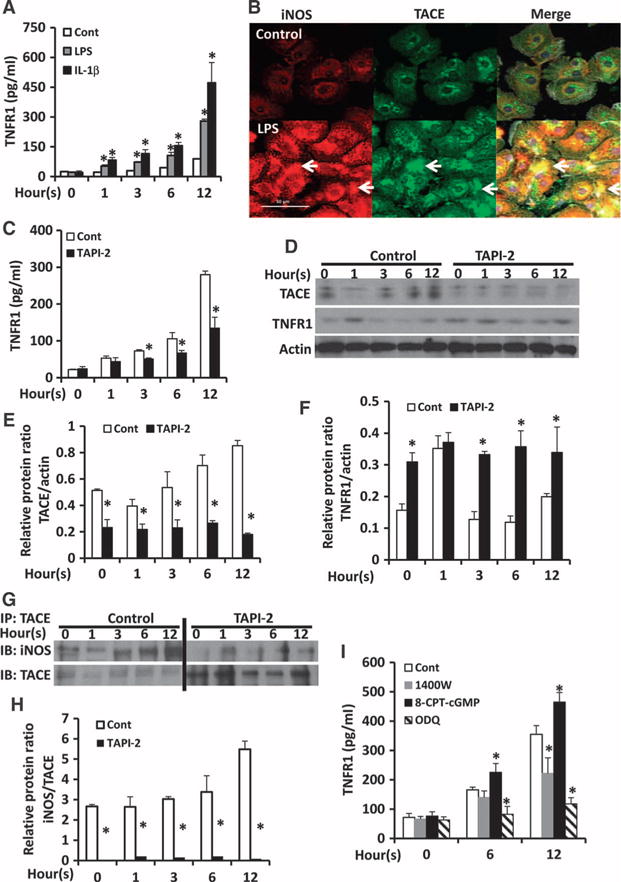
(A) Human hepatocytes were left untreated (Control) or treated with LPS (100 ng/ml) or 400 nM IL-1β for the indicated times. The concentration of TNFR1 in the culture media was determined by ELISA. Data are means ± SD from three independent experiments. *P < 0.05 by one-way ANOVA. (B) Human hepatocytes were left untreated (Control) or were treated with LPS (100 ng/ml). Cells were then analyzed by immunofluorescence microscopy to detect iNOS (red) and TACE (green). Arrows indicate the colocalization of iNOS and TACE. Scale bar, 50 mm. Images are representative of two independent experiments. (C to F) Human hepatocytes were left untreated (zero-hour time point) or were treated with LPS (100 ng/ml) in the absence (Cont) or presence of 400 nM TAPI-2 for the indicated times. (C) The concentration of TNFR1 in the culture media was determined by ELISA. Data are means ± SD from three independent experiments. *P < 0.05 by one-way ANOVA. (D) The hepatocytes were analyzed by Western blotting with antibodies specific for TACE and TNFR1. Densitometric analysis was performed to determine the relative abundances of (E) TACE and (F) TNFR1 proteins normalized to the abundance of actin. Data are means ± SD from three independent experiments. *P < 0.05 by two-tailed unpaired t test. (G and H) Human hepatocytes were left untreated (zero-hour time point) or were treated with LPS (100 mg/ml) in the absence (Cont) or presence of 400 nM TAPI-2 for the indicated times. (G) Samples were then subjected to immunoprecipitation (IP) with an anti-TACE antibody and were analyzed by Western blotting (IB) with antibodies specific for TACE and iNOS. Bands corresponding to iNOS in the TAPI-2–treated samples were only detectable when Western blots were separately developed with SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific). (H) Densitometric analysis was performed to determine the relative abundance of iNOS protein normalized to that of TACE. Data are means ± SD from three independent experiments. *P < 0.05 by two-tailed unpaired t test. (I) Human hepatocytes were treated with vehicle (0.01% DMSO), 20 μM 1400W, 100 μM 8-CPT-cGMP, or 20 μM ODQ, and then were stimulated with LPS (100 ng/ml) for the indicated times. The concentration of TNFR1 in the culture media was determined by ELISA. Data are means ± SD from three experiments. *P < 0.05 by one-way ANOVA.
The TACE inhibitor TAPI-2 reduced the sTNFR1 abundance in the culture media 6 and 12 hours after treatment with LPS (Fig. 6C). In response to stimulation with LPS, TACE abundance increased in human hepatocytes in a time-dependent manner (Fig. 6, D and E), whereas TNFR1 amounts in cell lysates decreased (Fig. 6, D and F). TAPI-2 inhibited the increase in the abundance of TACE in LPS-treated cells (Fig. 6, D and E). Furthermore, TNFR1 amounts in cells did not decrease in the TAPI-2–treated human hepatocytes after stimulation with LPS (Fig. 6, D and F). To confirm whether HC-TNFR1 shedding was mediated through an iNOS-TACE interaction, we immunoprecipitated TACE from whole-cell lysates and then analyzed the samples by Western blotting with antibodies specific for TACE and iNOS. The interaction between iNOS and TACE increased in a time-dependent manner in LPS-stimulated cells (Fig. 6, G and H). Thus, human HC-TNFR1 shedding was dependent on the iNOS-TACE pathway, which may involve a direct interaction between iNOS and TACE.
Next, we tested whether TNFR1 shedding by human hepatocytes was dependent on iNOS and cGMP. Isolated human hepatocytes were treated with vehicle [0.01% dimethyl sulfoxide (DMSO)], the iNOS inhibitor 1400W, the cGMP analog 8-CPT-cGMP, or the sGC inhibitor ODQ, and then were stimulated with LPS for 6 or 12 hours. The amounts of sTNFR1 in the culture media increased in a time-dependent manner after LPS stimulation (Fig. 6I). Inhibition of iNOS with 1400W substantially decreased sTNFR1 abundance in the culture media after 12 hours of stimulation with LPS. The amount of sTNFR1 in the culture media was substantially increased in cells treated with 8-CPT-cGMP (Fig. 6I). Inhibition of cGMP production with ODQ resulted in a substantial reduction in sTNFR1 abundance in the culture media after stimulation with LPS (Fig. 6I). These results suggest that, similar to mouse hepatocytes, TNFR1 shedding from human hepatocytes is also mediated through the iNOS-cGMP-TACE pathway in vitro.
DISCUSSION
TNFR1 shedding is an important protective mechanism to limit the bioactivity of TNF in inflammatory processes. We previously showed that TNFR1 shedding is mediated through the iNOS-cGMP-TACE signaling pathway in hepatocytes (10). Therefore, we extended our studies to determine the mechanism of HC-TNFR1 shedding during sepsis in a clinically relevant model of peritonitis. Our data indicate that circulating sTNFR1 amounts correlate with the severity of sepsis. Multiple TLR ligands and cytokines stimulated HC-TNFR1 shedding in vitro, whereas one of the major pathways that stimulated TNFR1 shedding during sepsis was regulated by the MyD88-dependent production of iNOS and the cGMP-mediated activation of TACE. Furthermore, we showed that regulation of HC-TNFR1 shedding through manipulation of the iNOS-cGMP pathway modulated systemic inflammatory responses and liver injury during sepsis. We also showed that human HC-TNFR1 shedding was similarly dependent on iNOS- and cGMP-mediated TACE activation and its translocation to the cell surface. Together with our previous finding (10), these data suggest that HC-TNFR1 shedding mediated through the MyD88-iNOS-cGMP-TACE signaling pathway limits inflammatory responses and organ damage during sepsis. We also showed that this pathway can be manipulated to enhance the amount of plasma sTNFR1 in vivo during sepsis.
TNF is a potent cytokine that exerts proinflammatory activities during host defense in infectious disease; however, excessive TNF activity leads to tissue toxicity. TNFR1 shedding is a self-protective mechanism to buffer the effects of excessive TNF amounts and block intracellular signaling by TNF (18, 19). TNFR1 shedding is thought to be an important host defense mechanism during infections with Staphylococcus aureus (20) and L. monocytogenes (19); however, these studies are based on models of infection with single pathogen. The CLP model is thought to be the model that is most representative of the sepsis commonly encountered by patients with polymicrobial intraperitoneal infection associated with fecal contamination (21). Therefore, we performed our study in a CLP model with antibiotics to investigate the influence of TNFR1 shedding on the inflammatory response in sepsis. We showed that the concentration of circulating sTNFR1 correlated with the severity of sepsis. Circulating amounts of sTNFR1 inversely correlated with the amounts of circulating inflammatory cytokines, suggesting that TNFR1 shedding is regulated to modulate inflammatory response during sepsis. However, circulating amounts of sTNFR1 increased most substantially after the early increase in TNF concentration, which suggests that the mechanisms that control TNFR1 shedding may be inadequate to counteract an early excess of TNF encountered during severe sepsis.
TNFR1 shedding in a complex and systemic process such as sepsis is likely to involve several mechanisms in multiple cell types. Many studies reported that during inflammation, TNFR1 is shed from mesenchymal cells, such as epithelial cells (22–24), lung endothelial cells (25), macrophages (8, 20, 26), and neutrophils (27). We and others showed that parenchymal cells, such as hepatocytes (10, 15), also shed TNFR1. Shedding mechanisms in these cells depend on TACE. TACE is an important enzyme during inflammation and tissue regeneration through its activation and cleavage (and thus release) of multiple proteins, including cytokines (TNF and transforming growth factor–β) and receptors (TNFR1, TNFR2, and IL-6R), as well as activation of ligands that activate the epithelial growth factor receptor (28, 29). However, the exact mechanisms of TACE activation are unclear and may be cell type–, receptor type–, or tissue-specific. McMahan et al. (30) showed that TACE was important for TNFR1 shedding from hepatocytes and myeloid cells in an endotoxemia model. For other cell types, TACE-dependent TNFR1 shedding requires the extracellular signal–regulated kinase (ERK) signaling pathway (20, 24) or cGMP-independent Ca2+ signaling (25). Okuyama et al. (31) showed that NO increased the abundance and shedding of TNFR1 in a human umbilical vein cell line through metalloproteinases independently of cGMP.
Our previous (10) and current studies demonstrated that HC-TNFR1 shedding is mediated, in part, through a MyD88-iNOS-cGMP-TACE pathway in vitro and in vivo, and that this pathway is dependent on a direct interaction between iNOS and TACE. Whether ERK or Ca2+ signaling is upstream of this pathway in hepatocytes was not addressed. The nitrosylation of TACE is involved in TNF maturation in macrophages (32); therefore, it is reasonable to speculate that iNOS contributes to TACE activation not only through cGMP but also through a direct interaction with TACE through targeted nitrosylation. That the extent of TNFR1 shedding is only partially suppressed by inhibition of the iNOS-cGMP-TACE pathway in hepatocytes during CLP suggests that other mechanisms for TNFR1 shedding operate during sepsis.
In polymicrobial sepsis, pathogen-associated molecular patterns (PAMPs), damage-associated molecular patterns (DAMPs), and cytokines are involved in the induction of inflammatory responses (33). TNFR1 shedding can be induced by PAMPs, such as LPS (10), CpG (8), and double-stranded RNA (dsRNA) (16), as well as by IL-1β (23). Our in vitro study showed that HC-TNFR1 shedding is induced by multiple TLR ligands and IL-1β, suggesting that multiple stimuli trigger HC-TNFR1 shedding in a polymicrobial sepsis model. Our in vivo study showed that prevention of IL-1β maturation in Casp-1−/− and Nalp3−/− mice or of IL-1β signaling in Il1r−/− mice reduced circulating sTNFR1 concentrations after CLP. Note that inflammasomes activate iNOS in either an IL-1β–dependent (34) or an IL-1β–independent manner (35). Whether inflammasomes can increase iNOS generation independently of IL-1β production to induce HC-TNFR1 shedding needs to be further investigated.
MyD88 and TRIF are receptor-proximal signaling adaptor molecules that mediate signaling activated by various pattern recognition receptors. Yu et al. (16) showed that virus-derived dsRNA induced TNFR1 shedding from human airway endothelial cells through a TLR3-TRIF-RIP pathway; however, Myddosome formation is the proximal event in the signaling of all TLRs (with the exception of TLR3) and IL-1R (36). TLR ligands, such as LPS (10), and IL-1β (14) are potent inducers of iNOS production in hepatocytes. Our in vitro and in vivo results suggest that MyD88 is required for increased iNOS production in hepatocytes, and they support a model in which hepatocytes respond directly to TLR activators or to IL-1β generated from other cells (such as myeloid cells) to enhance MyD88-dependent signaling. Activation of the Myddosome increases iNOS production in hepatocytes, which leads to TACE activation and TNFR1 shedding during sepsis.
The amounts of cGMP in the circulation increase when NO activates sGC in the cytoplasm (37). Therefore, NO generated by iNOS is likely to be one of the mechanisms responsible for the increase in cGMP concentration observed in the circulation during sepsis (38, 39). Our previous study showed that iNOS- and NO-mediated increases in cGMP concentration result in the activation of TACE and iRhom2 (a serine protease of the rhomboid family) and the trafficking of the TACE-iRhom2 complex to the cell surface in hepatocytes and the liver (10). We suggest that cGMP activates PKG, which then participates in the phosphorylation of both TACE and iRhom2. Here, our data from experiments with both mouse and human hepatocytes suggest that cGMP stimulates TACE activity and TNFR1 release in hepatocytes and in the liver during CLP sepsis. Thus, the bioavailability of cGMP appears to be an important determinant of both the cell surface abundance of TNFR1 and the circulating concentration of sTNFR1. Indeed, treatments that modulate cGMP abundance have therapeutic benefit in sepsis models, although the mechanisms involved have not always been clear. Yu et al. (38) showed that restoration of the NO-cGMP pathway with propofol improved endothelial dysfunction after CLP. Fernandes et al. (39) reported that cGMP was important for survival in the early phase of sepsis. Buys et al. (40) showed that cGMP generated by sGCα1β1 protected against cardiac dysfunction and mortality during endotoxin- and TNF-induced shock.
Our data suggest that one mechanism by which cGMP mediates protection from TNF toxicity in sepsis could be through the enhanced shedding of TNFR1. We also showed that sildenafil, which is currently approved for use in patients and has acceptable safety profiles, is an effective agent to increase cGMP availability in experimental sepsis. Sildenafil is a PDE5 inhibitor, which prevents cGMP degradation (17). Cadirci et al. (41) showed that sildenafil is protective against lung and kidney damage caused by CLP-induced sepsis by preventing oxidant-induced injury. Therefore, maintaining or enhancing cGMP abundance during sepsis could have beneficial effects through multiple interrelated mechanisms.
In summary, the integrated response to sepsis includes the increased iNOS production through MyD88-dependent pathways within hepatocytes, as well as through the MyD88-dependent production of IL-1β in myeloid cells. Within hepatocytes, increased iNOS production promotes the cGMP-dependent activation of TACE and TNFR1 shedding, which is associated with the suppression of inflammation and organ damage. Pharmacologic manipulation of the NO-cGMP-TACE-TNFR1 pathway could be of benefit in the early clinical course of severe sepsis.
MATERIALS AND METHODS
Reagents
Ultrapure LPS (from Escherichia coli 0111:B4) was from List Biological Laboratories Inc. The mouse TLR1 to TLR9 agonist kit was from InvivoGen. Sildenafil, ODQ, 1400W, and 8-CPT-cGMP were obtained from Sigma.
Mice
Experimental protocols were approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh. Male wild-type C57BL/6 mice, Il1r−/− (Il1r1tm1Imx) mice, Albumin-Cre (Alb-Cre) mice [Tg(Alb-Cre) 21MgN)], Lyz-Cre mice [Lyz2tm1(cre)Ifo], and MyD88-Flox (Myd88tm1Defr) mice were purchased from The Jackson Laboratory. Myd88−/− (Myd88tm1Aki) mice on a C57BL/6 background were a gift from R. Medzhitov [Howard Hughes Medical Institute (HHMI), Yale University, New Haven, CT]. Casp1−/− mice (Casptm1Flv) on a C57BL/6 background were a gift from R. Flavell (HHMI, Yale University). Nalp3−/− (Nlrp3tm1Flv) mice on a C56BL/6 background were a gift from R. Flavell through a material transfer agreement with Millennium Pharmaceuticals. Trif−/− mice (Ticam1Lps2) on a C57BL/6 background were a gift from B. Beutler (Scripps Research Institute). Myd88−/− mice, Casp1−/− mice, Nalp3−/− mice, and Trif−/− mice were bred in our animal facility. Tlr4−/− (Tlr4tm1Djh) and Nos2−/− (Nos2tm1Mrl) mice were generated in our laboratory on a C57BL/6 background and backcrossed at least six times. Hepatocyte-specific Myd88−/− mice (HC-Myd88−/−) and myeloid cell–specific Myd88−/− mice (LysM-Myd88−/−) were generated with the Cre-loxP technique and were identified by PCR-based genotyping with multiple primer pairs, as described previously (42). These animals were bred in our facility.
CLP procedure
Sepsis was induced by CLP. Mice that were 25 to 30 g in weight were used. The skin was disinfected with a 2% iodine tincture. Laparotomy was performed under 2% isoflurane (Piramal Critical Care) with oxygen. For the sublethal model, 50% of the cecum was ligated and punctured twice with a 22-gauge needle. For the lethal model, 75% of the cecum was ligated and punctured twice with an 18-gauge needle. Saline (1 ml) was given subcutaneously for resuscitation immediately after operation. Primaxin (25 mg/kg, Merck) was dissolved in 5% dextrose (Baxter) and injected into mice subcutaneously every 12 hours starting 2 hours after CLP was performed. In the control mice, laparotomy and cecal manipulation were performed without ligation and puncture.
Isolation and culture of hepatocytes
Cells were isolated from mice through an in situ collagenase (type VI, Sigma) perfusion technique, modified as described previously (13). Heptocyte purity exceeded 99% by flow cytometric assay. Cell viability was typically >90% by trypan blue exclusion. Hepatocytes (150,000 cells/ml) were plated on gelatin-coated culture plates or coverslips coated with collagen I (BD Pharmingen) in William’s medium E with 10% calf serum, 15 mM Hepes, 1 μM insulin, 2 mM L-glutamine, penicillin (100 U/ml), and streptomycin (100 U/ml). Hepatocytes were allowed to attach to plates overnight, and the culture medium was replaced with fresh medium before the cells were treated for experiments. After treatment, 1 ml of culture medium was collected and centrifuged at 400g for 5 min. The supernatant was removed to a new tube and stored at −80°C for further analysis.
Crystal violet staining
Hepatocytes were plated in 12-well plates. After treatment, hepatocytes were stained with 0.1% crystal violet (Sigma) for 15 min and then washed with sterile distilled water. The rinsed plates were left to air-dry overnight. The bound dye was released by the addition of 1% SDS. The elution liquid of crystal violet was removed to a new microplate, and the absorbance (A) was measured at a wavelength of 575 nm. The percentage of viable cells (% viability) was calculated by dividing the A575 of the treated sample by the A575 of the control sample and multiplying by 100.
Assessment of TNFR1 and cytokine abundance
Plasma and cell culture media samples were analyzed with ELISA kits specific for IL-6, TNF, KC, MIP-1α, and TNFR1 (R&D Systems Inc.) to assess their concentrations.
cGMP assay
The abundance of cGMP in liver tissue was analyzed with a cGMP EIA kit (Cayman Chemical). Briefly, frozen liver tissue (50 mg) was homogenized with 5% trichloroacetic acid (TCA) on ice. After centrifugation, the TCA was extracted from the supernatant with water-saturated ether. After TCA extraction, the residual ether was removed by heating the sample at 70°C for 5 min. Samples (50 μl) were incubated with cGMP AchE Tracer at 4°C overnight and developed with Ellman’s reagent. The amount of cGMP in each sample was assessed by measuring the absorbance of the samples at 405 nm.
Isolation of human hepatocytes
Human hepatocytes were isolated from histologically normal liver and were provided by D. Geller (Department of Surgery, University of Pittsburgh, Pittsburgh, PA) according to a protocol approved by the Institutional Review Board. Human hepatocytes were prepared by a three-step collagenase perfusion technique, modified as described previously (43). Isolated human hepatocytes were cultured in William’s medium E supplemented with 5% calf serum (GE Healthcare Life Sciences), penicillin (100 U/ml), streptomycin (100 U/ml), 2 mM L-glutamine, and 15 mM Hepes.
Cell lysis and Western blotting analysis
For in vitro experiments, hepatocytes were washed with cold phosphate-buffered saline (PBS), collected in lysis buffer (Cell Signaling Technology), sonicated, and centrifuged at 16,000g for 15 min, after which the supernatant was collected. For in vivo experiments, snap-frozen liver (median lobe) was homogenized in lysis buffer and centrifuged at 16,000g for 15 min, after which the supernatant was collected. Protein concentrations were determined with the BCA (bicinchoninic acid) protein assay kit (Thermo Fisher Scientific). Loading buffer was added to the samples, which were then resolved by 10 or 15% SDS–polyacrylamide gel electrophoresis. Samples were then transferred onto a polyvinylidene difluoride membrane at 250 mA for 2 hours. The membrane was blocked in 5% milk for 1 hour and then incubated overnight with primary antibody in 1% milk. Membranes were washed in tris-buffered saline containing Tween (TBS-T) for 10 min, incubated with horseradish peroxidase– conjugated secondary antibody for 1 hour, and then washed for 1 hour in TBS-T, before being developed for chemiluminescence (Thermo Fisher Scientific). The primary antibodies used were anti-iNOS (1:1000, R&D Systems Inc.), anti-TNFR1, anti-TACE, and anti-GAPDH (1:1000 for all antibodies, Abcam). Protein band intensities were quantified with ImageJ software (National Institutes of Health).
Comparative PCR analysis
The median lobe of the liver was frozen in liquid nitrogen and stored at −80°C for comparative PCR analysis. Total RNA was extracted with the RNeasy Mini extraction kit (Qiagen) according to the manufacturer’s instructions. From 1 μg of RNA and with oligo-dT primers (Qiagen) and Omniscript reverse transcriptase (Qiagen), complementary DNA was generated and used for real-time PCR analysis. SYBR Green PCR Master Mix (PE Applied Bio-systems) was used to prepare the PCR reaction mixes. Two-step real-time RT-PCR was performed with prevalidated forward and reverse primer pairs that were specific for Tnfr1 and Nos2 (Qiagen). All samples were assayed in triplicate, and data were normalized to actin mRNA abundance.
Immunofluorescence microscopy
The left lateral lobe of the liver was perfused with PBS and fixed in 2% paraformaldehyde according to a standardized protocol for cryopreservation (44). Livers were sectioned in a cryostat and stained as follows. Liver sections (5 mm) were incubated with 2% bovine serum albumin (BSA) in PBS for 1 hour, followed by five washes with PBS containing 0.5% BSA (PBB). The samples were then incubated overnight with primary antibodies specific for iNOS (1:500, R&D Systems Inc.) and TACE (1:300, Abcam) in PBB, 0.1% Triton X-100. Samples were washed five times with PBB and then were incubated with the appropriate Alexa Fluor 488–labeled (1:500, Invitrogen) and Cy3-labeled (1:1000, Jackson ImmunoResearch Laboratories) secondary antibodies diluted in PBB. Samples were washed three times with PBB, followed by a single wash with PBS, before being incubated for 30 s with Hoechst nuclear stain. The nuclear stain was removed, and samples were washed with PBS before being placed on a coverslip using Gelvatol (EMD Millipore). Positively stained cells in six random fields were imaged with a FluoView 1000 confocal scanning microscope (Olympus). Imaging conditions were maintained at identical settings within each antibody labeling experiment, with original gating performed with the negative control.
Statistical analysis
The data are presented as means ± SD. The experimental results were analyzed for statistical significance with GraphPad Prism (GraphPad Software). Normal distribution of samples was tested with a two-tailed Student’s t test or by one-way ANOVA for multiple comparisons as indicated in the figure legends. Statistical significance was established at the 95% confidence level (P < 0.05).
Supplementary Material
Fig. S1. The kinetics of TNFR1 and TNF release after CLP.
Fig. S2. Analysis of plasma IL-1β concentrations at 8 hours after CLP.
Fig. S3. The responses to LPS of hepatocytes from Alb-Cre mice are similar to those of hepatocytes from wild-type mice.
Fig. S4. The responses to LPS of cells from Lyz-Cre mice are similar to those of cells from wild-type mice.
Acknowledgments
We thank H. Liao, M. Bo, A. Frank, and N. Martik-Hays for their excellent technical support.
Funding: This work was supported by NIH grants R37-GM-044100 and R01-GM-050441.
Footnotes
Author contributions: M.D. performed most of the experiments and contributed to the experimental design and writing of the manuscript; P.A.L. performed all of the immunofluorescence experiments; L.Z. performed all of the immunoprecipitation experiments; M.J.S. contributed to the experimental design and edited the manuscript; and T.R.B. designed most of the research project, provided oversight of experiments, and edited the manuscript.
Competing interests: The authors declare that they have no competing interests.
SUPPLEMENTARY MATERIALS
REFERENCES AND NOTES
- 1.Cauwels A, Brouckaert P. Survival of TNF toxicity: Dependence on caspases and NO. Arch Biochem Biophys. 2007;462:132–139. doi: 10.1016/j.abb.2007.01.021. [DOI] [PubMed] [Google Scholar]
- 2.Wang Y, Kim PK, Peng X, Loughran P, Vodovotz Y, Zhang B, Billiar TR. Cyclic AMP and cyclic GMP suppress TNFα-induced hepatocyte apoptosis by inhibiting FADD up-regulation via a protein kinase A-dependent pathway. Apoptosis. 2006;11:441–451. doi: 10.1007/s10495-005-4293-6. [DOI] [PubMed] [Google Scholar]
- 3.Eum HA, Billiar TR. TNF/TNF receptor 1-mediated apoptosis in hepatocytes. Adv Exp Med Biol. 2011;691:617–624. doi: 10.1007/978-1-4419-6612-4_65. [DOI] [PubMed] [Google Scholar]
- 4.Kim YM, de Vera ME, Watkins SC, Billiar TR. Nitric oxide protects cultured rat hepatocytes from tumor necrosis factor-α-induced apoptosis by inducing heat shock protein 70 expression. J Biol Chem. 1997;272:1402–1411. doi: 10.1074/jbc.272.2.1402. [DOI] [PubMed] [Google Scholar]
- 5.Damas P, Reuter A, Gysen P, Demonty J, Lamy M, Franchimont P. Tumor necrosis factor and interleukin-1 serum levels during severe sepsis in humans. Crit Care Med. 1989;17:975–978. doi: 10.1097/00003246-198910000-00001. [DOI] [PubMed] [Google Scholar]
- 6.Barochia AV, Cui X, Vitberg D, Suffredini AF, O’Grady NP, Banks SM, Minneci P, Kern SJ, Danner RL, Natanson C, Eichacker PQ. Bundled care for septic shock: An analysis of clinical trials. Crit Care Med. 2010;38:668–678. doi: 10.1097/CCM.0b013e3181cb0ddf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morris PE, Zeno B, Bernard AC, Huang X, Das S, Edeki T, Simonson SG, Bernard GR. A placebo-controlled, double-blind, dose-escalation study to assess the safety, tolerability and pharmacokinetics/pharmacodynamics of single and multiple intravenous infusions of AZD9773 in patients with severe sepsis and septic shock. Crit Care. 2012;16:R31. doi: 10.1186/cc11203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jin L, Raymond DP, Crabtree TD, Pelletier SJ, Houlgrave CW, Pruett TL, Sawyer RG. Enhanced murine macrophage TNF receptor shedding by cytosine-guanine sequences in oligodeoxynucleotides. J Immunol. 2000;165:5153–5160. doi: 10.4049/jimmunol.165.9.5153. [DOI] [PubMed] [Google Scholar]
- 9.Kim YM, Kim TH, Chung HT, Talanian RV, Yin XM, Billiar TR. Nitric oxide prevents tumor necrosis factor a-induced rat hepatocyte apoptosis by the interruption of mitochondrial apoptotic signaling through S-nitrosylation of caspase-8. Hepatology. 2000;32:770–778. doi: 10.1053/jhep.2000.18291. [DOI] [PubMed] [Google Scholar]
- 10.Chanthaphavong RS, Loughran PA, Lee TY, Scott MJ, Billiar TR. A role for cGMP in inducible nitric-oxide synthase (iNOS)-induced tumor necrosis factor (TNF) a-converting enzyme (TACE/ADAM17) activation, translocation, and TNF receptor 1 (TNFR1) shedding in hepatocytes. J Biol Chem. 2012;287:35887–35898. doi: 10.1074/jbc.M112.365171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lucas R, Lou J, Morel DR, Ricou B, Suter PM, Grau GE. TNF receptors in the microvascular pathology of acute respiratory distress syndrome and cerebral malaria. J Leukoc Biol. 1997;61:551–558. doi: 10.1002/jlb.61.5.551. [DOI] [PubMed] [Google Scholar]
- 12.Knotek M, Rogachev B, Wang W, Ecder T, Melnikov V, Gengaro PE, Esson M, Edelstein CL, Dinarello CA, Schrier RW. Endotoxemic renal failure in mice: Role of tumor necrosis factor independent of inducible nitric oxide synthase. Kidney Int. 2001;59:2243–2249. doi: 10.1046/j.1523-1755.2001.00740.x. [DOI] [PubMed] [Google Scholar]
- 13.Garcia I, Miyazaki Y, Araki K, Araki M, Lucas R, Grau GE, Milon G, Belkaid Y, Montixi C, Lesslauer W, Vassalli P. Transgenic mice expressing high levels of soluble TNF-R1 fusion protein are protected from lethal septic shock and cerebral malaria, and are highly sensitive to Listeria monocytogenes and Leishmania major infections. Eur J Immunol. 1995;25:2401–2407. doi: 10.1002/eji.1830250841. [DOI] [PubMed] [Google Scholar]
- 14.Geller DA, de Vera ME, Russell DA, Shapiro RA, Nussler AK, Simmons RL, Billiar TR. A central role for IL-1 beta in the in vitro and in vivo regulation of hepatic inducible nitric oxide synthase. IL-1 beta induces hepatic nitric oxide synthesis. J Immunol. 1995;155:4890–4898. [PubMed] [Google Scholar]
- 15.Xia M, Xue SB, Xu CS. Shedding of TNFR1 in regenerative liver can be induced with TNFα and PMA. World J Gastroenterol. 2002;8:1129–1133. doi: 10.3748/wjg.v8.i6.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu M, Lam J, Rada B, Leto TL, Levine SJ. Double-stranded RNA induces shedding of the 34-kDa soluble TNFR1 from human airway epithelial cells via TLR3–TRIF–RIP1-dependent signaling: Roles for dual oxidase 2- and caspase-dependent pathways. J Immunol. 2011;186:1180–1188. doi: 10.4049/jimmunol.1001499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ghofrani HA, Osterloh IH, Grimminger F. Sildenafil: From angina to erectile dysfunction to pulmonary hypertension and beyond. Nat Rev Drug Discov. 2006;5:689–702. doi: 10.1038/nrd2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aderka D, Sorkine P, Abu-Abid S, Lev D, Setton A, Cope AP, Wallach D, Klausner J. Shedding kinetics of soluble tumor necrosis factor (TNF) receptors after systemic TNF leaking during isolated limb perfusion. Relevance to the pathophysiology of septic shock. J Clin Invest. 1998;101:650–659. doi: 10.1172/JCI694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xanthoulea S, Pasparakis M, Kousteni S, Brakebusch C, Wallach D, Bauer J, Lassmann H, Kollias G. Tumor necrosis factor (TNF) receptor shedding controls thresholds of innate immune activation that balance opposing TNF functions in infectious and inflammatory diseases. J Exp Med. 2004;200:367–376. doi: 10.1084/jem.20040435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Giai C, Gonzalez C, Ledo C, Garofalo A, Di Genaro MS, Sordelli DO, Gomez MI. Shedding of tumor necrosis factor receptor 1 induced by protein A decreases tumor necrosis factor alpha availability and inflammation during systemic Staphylococcus aureus infection. Infect Immun. 2013;81:4200–4207. doi: 10.1128/IAI.00593-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rittirsch D, Huber-Lang MS, Flierl MA, Ward PA. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat Protoc. 2009;4:31–36. doi: 10.1038/nprot.2008.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sakimoto S, Tsukamoto Y, Saito Y. Removal of silicone oil droplet adhering to a silicone intraocular lens using 25-gauge instrumentation. J Cataract Refract Surg. 2009;35:383–385. doi: 10.1016/j.jcrs.2008.07.041. [DOI] [PubMed] [Google Scholar]
- 23.Levine SJ, Logun C, Chopra DP, Rhim JS, Shelhamer JH. Protein kinase C, interleukin-1 beta, and corticosteroids regulate shedding of the type I, 55 kDa TNF receptor from human airway epithelial cells. Am J Respir Cell Mol Biol. 1996;14:254–261. doi: 10.1165/ajrcmb.14.3.8845176. [DOI] [PubMed] [Google Scholar]
- 24.Cook EB, Stahl JL, Graziano FM, Barney NP. Regulation of the receptor for TNFα, TNFR1, in human conjunctival epithelial cells. Invest Ophthalmol Vis Sci. 2008;49:3992–3998. doi: 10.1167/iovs.08-1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rowlands DJ, Islam MN, Das SR, Huertas A, Quadri SK, Horiuchi K, Inamdar N, Emin MT, Lindert J, Ten VS, Bhattacharya S, Bhattacharya J. Activation of TNFR1 ectodomain shedding by mitochondrial Ca2+ determines the severity of inflammation in mouse lung microvessels. J Clin Invest. 2011;121:1986–1999. doi: 10.1172/JCI43839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rodrigues MF, Alves CC, Figueiredo BB, Rezende AB, Wohlres-Viana S, Silva VL, Machado MA, Teixeira HC. Tumour necrosis factor receptors and apoptosis of alveolar macrophages during early infection with attenuated and virulent Mycobacterium bovis. Immunology. 2013;139:503–512. doi: 10.1111/imm.12097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garton KJ, Gough PJ, Raines EW. Emerging roles for ectodomain shedding in the regulation of inflammatory responses. J Leukoc Biol. 2006;79:1105–1116. doi: 10.1189/jlb.0106038. [DOI] [PubMed] [Google Scholar]
- 28.Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, Castner BJ, Stocking KL, Reddy P, Srinivasan S, Nelson N, Boiani N, Schooley KA, Gerhart M, Davis R, Fitzner JN, Johnson RS, Paxton RJ, March CJ, Cerretti DP. A metalloproteinase disintegrin that releases tumour-necrosis factor-α from cells. Nature. 1997;385:729–733. doi: 10.1038/385729a0. [DOI] [PubMed] [Google Scholar]
- 29.Moss ML, Jin SL, Becherer JD, Bickett DM, Burkhart W, Chen WJ, Hassler D, Leesnitzer MT, McGeehan G, Milla M, Moyer M, Rocque W, Seaton T, Schoenen F, Warner J, Willard D. Structural features and biochemical properties of TNF-α converting enzyme (TACE) J Neuroimmunol. 1997;72:127–129. doi: 10.1016/s0165-5728(96)00180-4. [DOI] [PubMed] [Google Scholar]
- 30.McMahan RS, Riehle KJ, Fausto N, Campbell JS. A disintegrin and metalloproteinase 17 regulates TNF and TNFR1 levels in inflammation and liver regeneration in mice. Am J Physiol Gastrointest Liver Physiol. 2013;305:G25–G34. doi: 10.1152/ajpgi.00326.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Okuyama M, Yamaguchi S, Yamaoka M, Nitobe J, Fujii S, Yoshimura T, Tomoike H. Nitric oxide enhances expression and shedding of tumor necrosis factor receptor I (p55) in endothelial cells. Arterioscler Thromb Vasc Biol. 2000;20:1506–1511. doi: 10.1161/01.atv.20.6.1506. [DOI] [PubMed] [Google Scholar]
- 32.Zhang Z, Kolls JK, Oliver P, Good D, Schwarzenberger PO, Joshi MS, Ponthier JL, Lancaster JR., Jr Activation of tumor necrosis factor-α-converting enzyme-mediated ectodomain shedding by nitric oxide. J Biol Chem. 2000;275:15839–15844. doi: 10.1074/jbc.M000604200. [DOI] [PubMed] [Google Scholar]
- 33.Gentile LF, Moldawer LL. DAMPs, PAMPs, and the origins of SIRS in bacterial sepsis. Shock. 2013;39:113–114. doi: 10.1097/SHK.0b013e318277109c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jüttler E, Bonmann E, Spranger M, Kolb-Bachofen V, Suschek CV. A novel role of interleukin-1-converting enzyme in cytokine-mediated inducible nitric oxide synthase gene expression: Implications for neuroinflammatory diseases. Mol Cell Neurosci. 2007;34:612–620. doi: 10.1016/j.mcn.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 35.Buzzo CL, Campopiano JC, Massis LM, Lage SL, Cassado AA, Leme-Souza R, Cunha LD, Russo M, Zamboni DS, Amarante-Mendes GP, Bortoluci KR. A novel pathway for inducible nitric-oxide synthase activation through inflammasomes. J Biol Chem. 2010;285:32087–32095. doi: 10.1074/jbc.M110.124297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gay NJ, Gangloff M, O’Neill LA. What the Myddosome structure tells us about the initiation of innate immunity. Trends Immunol. 2011;32:104–109. doi: 10.1016/j.it.2010.12.005. [DOI] [PubMed] [Google Scholar]
- 37.Martínez-Ruiz A, Cadenas S, Lamas S. Nitric oxide signaling: Classical, less classical, and nonclassical mechanisms. Free Radic Biol Med. 2011;51:17–29. doi: 10.1016/j.freeradbiomed.2011.04.010. [DOI] [PubMed] [Google Scholar]
- 38.Yu HP, Lui PW, Hwang TL, Yen CH, Lau YT. Propofol improves endothelial dysfunction and attenuates vascular superoxide production in septic rats. Crit Care Med. 2006;34:453–460. doi: 10.1097/01.ccm.0000198530.68343.21. [DOI] [PubMed] [Google Scholar]
- 39.Fernandes MA, Marques RJ, Vicente JA, Santos MS, Monteiro P, Moreno AJ, Custódio JB. Sildenafil citrate concentrations not affecting oxidative phosphorylation depress H2O2 generation by rat heart mitochondria. Mol Cell Biochem. 2008;309:77–85. doi: 10.1007/s11010-007-9645-9. [DOI] [PubMed] [Google Scholar]
- 40.Buys ES, Cauwels A, Raher MJ, Passeri JJ, Hobai I, Cawley SM, Rauwerdink KM, Thibault H, Sips PY, Thoonen R, Scherrer-Crosbie M, Ichinose F, Brouckaert P, Bloch KD. sGCα1β1 attenuates cardiac dysfunction and mortality in murine inflammatory shock models. Am J Physiol Heart Circ Physiol. 2009;297:H654–H663. doi: 10.1152/ajpheart.00367.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cadirci E, Halici Z, Odabasoglu F, Albayrak A, Karakus E, Unal D, Atalay F, Ferah I, Unal B. Sildenafil treatment attenuates lung and kidney injury due to overproduction of oxidant activity in a rat model of sepsis: A biochemical and histopathological study. Clin Exp Immunol. 2011;166:374–384. doi: 10.1111/j.1365-2249.2011.04483.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sodhi CP, Neal MD, Siggers R, Sho S, Ma C, Branca MF, Prindle T, Jr, Russo AM, Afrazi A, Good M, Brower-Sinning R, Firek B, Morowitz MJ, Ozolek JA, Gittes GK, Billiar TR, Hackam DJ. Intestinal epithelial toll-like receptor 4 regulates goblet cell development and is required for necrotizing enterocolitis in mice. Gastroenterology. 2012;143:708–718.e5. doi: 10.1053/j.gastro.2012.05.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Park KS, Guo Z, Shao L, Du Q, Geller DA. A far-upstream Oct-1 motif regulates cytokine-induced transcription of the human inducible nitric oxide synthase gene. J Mol Biol. 2009;390:595–603. doi: 10.1016/j.jmb.2009.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eum HA, Vallabhaneni R, Wang Y, Loughran PA, Stolz DB, Billiar TR. Characterization of DISC formation and TNFR1 translocation to mitochondria in TNF-α– treated hepatocytes. Am J Pathol. 2011;179:1221–1229. doi: 10.1016/j.ajpath.2011.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. The kinetics of TNFR1 and TNF release after CLP.
Fig. S2. Analysis of plasma IL-1β concentrations at 8 hours after CLP.
Fig. S3. The responses to LPS of hepatocytes from Alb-Cre mice are similar to those of hepatocytes from wild-type mice.
Fig. S4. The responses to LPS of cells from Lyz-Cre mice are similar to those of cells from wild-type mice.