

Abstract
Complexins are synaptic SNARE complex‐binding proteins that cooperate with synaptotagmins in activating Ca2+‐stimulated, synaptotagmin‐dependent synaptic vesicle exocytosis and in clamping spontaneous, synaptotagmin‐independent synaptic vesicle exocytosis. Here, we show that complexin sequences are conserved in some non‐metazoan unicellular organisms and in all metazoans, suggesting that complexins are a universal feature of metazoans that predate metazoan evolution. We show that complexin from Nematostella vectensis, a cnidarian sea anemone far separated from mammals in metazoan evolution, functionally replaces mouse complexins in activating Ca2+‐triggered exocytosis, but is unable to clamp spontaneous exocytosis. Thus, the activating function of complexins is likely conserved throughout metazoan evolution.
Keywords: evolution, membrane fusion, SNARE proteins, synapse, synaptotagmin
Subject Categories: Evolution, Membrane & Intracellular Transport
Introduction
Neurotransmitter release is mediated by synaptic vesicle fusion that is triggered by Ca2+ binding to synaptotagmins. SNARE and SM proteins catalyze fusion by forming a tight complex that forces the membranes into close proximity. Synaptotagmins promote fusion via a Ca2+‐dependent interaction with both phospholipid membranes and SNARE/SM protein complexes 1. However, synaptotagmins do not act alone but require complexins as cofactors. Complexins are small (~130 residues) SNARE‐binding proteins. In vertebrates, complexins primarily perform an activating function in release by rendering the SNARE complex competent for Ca2+ triggering of fusion and boosting the priming of synaptic vesicles. In addition, vertebrate complexins clamp spontaneous release in most synapses. In invertebrates, however, complexins appear to be more prominently involved in clamping release, although an activating function has also been observed 2.
These findings led to the hypothesis that evolutionarily, complexins may have originated as a clamp in invertebrates and subsequently acquired activator functions in vertebrates 3. Other studies, however, suggested that invertebrate and vertebrate complexins similarly act as both clamps and activators in release 4, 5, 6, 7, 8, and in vitro reconstitutions with mammalian complexins reproduce both activities 9, 10, 11, 12. Moreover, no complexins from organisms that are evolutionarily more ancient than D. melanogaster and C. elegans have been examined.
Structurally, complexins contain an N‐terminal unstructured region followed by an accessory α‐helix, a SNARE‐binding central α‐helix, and a longer unstructured C‐terminal region. The three functions of vertebrate complexins (priming, Ca2+ triggering, and clamping) exhibit distinct sequence requirements, such that the N‐terminal region is selectively necessary for Ca2+ triggering of exocytosis, the accessory α‐helix for clamping, and the C‐terminal unstructured region is involved in both clamping and priming but not Ca2+ triggering, while the central α‐helix is required for all complexin functions 5, 8, 13, 14, 15. The differential sequence dependence of complexin functions strongly suggests that these functions are mechanistically distinct.
Here, we show that complexin sequences are not only encoded by all metazoan genomes, but are also present in the genomes of a subset of unicellular organisms that are evolutionarily older than metazoans, such as choanoflagellates. We found that the genomes of primitive metazoans, such as that of the sea anemone Nematostella vectensis, encode one or two complexin genes. Nematostella is a cnidarian that belongs to the simplest eumetazoans and develops primitive neuron‐like cells, but lacks a central nervous system 16. Furthermore, we demonstrate that re‐introduction of Nematostella complexin‐1 into complexin‐deficient mouse neurons fully rescued the inactivation of evoked neurotransmitter release, but did not reverse the unclamping of spontaneous mini release. Thus, Nematostella complexin‐1—similar to mouse complexin‐1—functions as an activator of release, but may lack clamping activity. Moreover, we demonstrate that Nematostella complexin‐1 exhibits a similar structure/function dependence as vertebrate complexins. Our data suggest that complexins activate exocytosis by mechanisms that are conserved throughout metazoan evolution and likely originate in evolution prior to the emergence of metazoans.
Results and Discussion
Complexin evolution
We performed sequence similarity searches for complexins by PSI‐BLAST. Complexin sequences were found in all major groups of metazoans (four basal metazoan groups: Porifera, Ctenophora, Cnidaria, and Placozoa and three Bilateria groups: Ecdysozoa, Lophotrochozoa, and Deuterostomia). Vertebrate organisms have four or more complexin sequences, possibly contributed by whole genome duplications. Invertebrate organisms, on the other hand, often have only one complexin gene. A few non‐vertebrate metazoans have two complexins, such as Caenorhabditis elegans, sea urchin (Strongylocentrotus purpuratus), and Nematostella vectensis, which may be attributed to lineage‐specific gene duplication events (Fig 1).
Figure 1. Sequence alignments reveal that complexins are conserved from choanoflagellates to mammals.
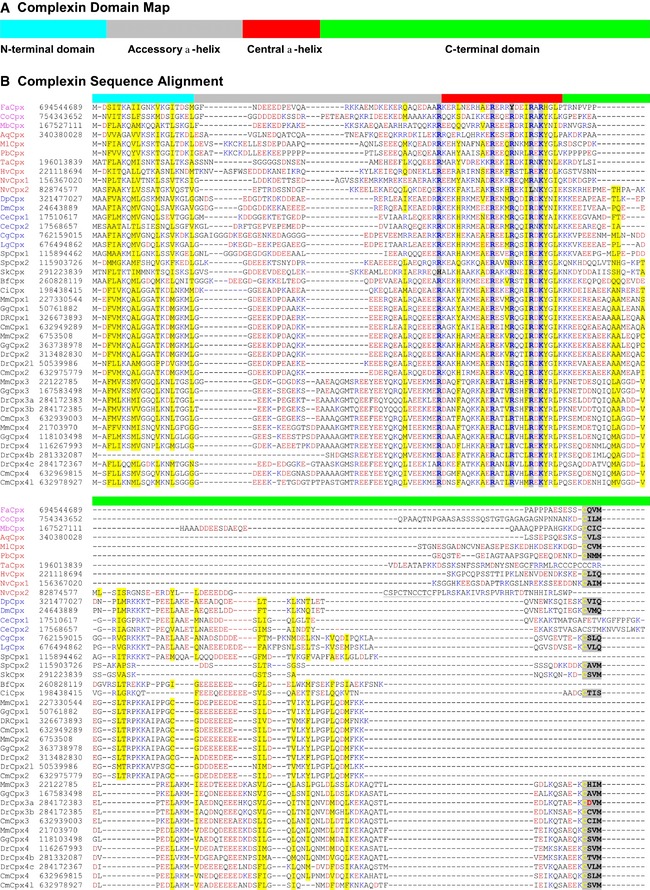
-
A, BComplexin sequences from the indicated organisms (represented as organism name abbreviations followed by Cpx and optional numbers) are aligned in single‐letter code (B). NCBI gene identification numbers follow the sequence names, except for two Ctenophora sequences (MlCpx and PbCpx) that are derived from transcriptome assembly data. Color codes of shaded positions: yellow, mainly hydrophobic residues; blue, mainly positively charged residues; and gray, C‐terminal CAAX boxes. Color codes of residues: blue, R and K; red, D and E. The four domains of complexins are indicated by the colored bar on top (A): cyan, N‐terminal domain; gray, accessory α‐helix; red, central α‐helix; and green, C‐terminal extended sequence. Sequence names are colored as follows: non‐metazoans: magenta; basal metazoans: red; protostomes: blue; and deuterostomes: black. Organism name abbreviations are as follows: Aq, Amphimedon queenslandica; Bf, Branchiostoma floridae; Ce, Caenorhabditis elegans; Cm, Callorhinchus milii; Co, Capsaspora owczarzaki; Ci, Ciona intestinalis; Cg, Crassostrea gigas; Dr, Danio rerio; Dp, Daphnia pulex; Dm, Drosophila melanogaster; Fa, Fonticula alba; Gg, Gallus gallus; Hv, Hydra vulgaris; Lg, Lottia gigantea; Mb, Monosiga brevicollis; Mm, Mus musculus; Nv, Nematostella vectensis; Sk, Saccoglossus kowalevskii; Sp, Strongylocentrotus purpuratus; and Ta, Trichoplax adhaerens.
Complexin sequences encode four domains: a short N‐terminal domain with a conserved pattern of hydrophobic and positively charged residues, a charged accessory α‐helix, a central α‐helix that binds to SNARE complexes, and a C‐terminal region that often accounts for most of the complexin sequence (Fig 1A). The N‐terminal domain and central α‐helix exhibit the most conservation across species, whereas the C‐terminal region is the most variable (Fig 1B). Negatively charged residue patches are often present in the accessory α‐helix (all complexins) and the C‐terminal domain (mostly complexins from vertebrates). Many but not all complexin sequences have a C‐terminal CAAX motif and are likely isoprenylated. All four domains were detected in all complexin sequences (except zebrafish Complexin 4b that lacks the N‐terminal domain), suggesting that they are obligatory complexin components (Fig 1B).
Surprisingly, we also observed that a few non‐metazoan single‐cell organisms belonging to groups such as Choanoflagellatea, Filasterea, and Nuclearia, but not in Fungi, Amoebozoa, and Unikonta, contain single complexin sequences with an apparently similar domain organization (Fig 1). Specifically, we detected complexin sequences in the choanoflagellate Monosiga brevicollis, the cellular slime mold Fonticula alba, and the filasterean Capsaspora owczarzaki, as previously noted in the Supplementary Materials of Burckhard et al 17. The unicellular complexins include all of the typical complexin features, including the N‐terminal sequence containing the typical amphipathic pattern of hydrophobic and positively charged residues, the classical central α‐helix that is highly similar to that of other complexins, and a C‐terminal region with a canonical isoprenylation sequence (CAAX box). The major difference between the non‐metazoan and metazoan complexins is that the unstructured C‐terminal half of the molecule that is involved in clamping and priming in mammalian complexins 5, 8, 18 is shorter and more variable. However, this part is the least conserved sequence in all complexins and probably exerts a primarily modulatory function 18.
We next sought to investigate the relation of the various complexin sequences we identified by constructing a phylogenetic tree. The small size and limited positions in complexins coupled with sequence divergence make phylogenetic signals weak, and the phylogenetic construction by MOLPHY did not yield significant supports in most groups (Fig 2). Nevertheless, the analyses clearly show that there is no clear‐cut evolutionary division of complexins into evolutionarily distinct classes. The most robust separation of complexins into classes was observed for the vertebrate sequences which formed two major groups, one including mouse complexin‐1 and ‐2 (without a CAAX motif), and the other including mouse complexin‐3 and ‐4 (with a CAAX motif).
Figure 2. A phylogenetic tree of complexins.
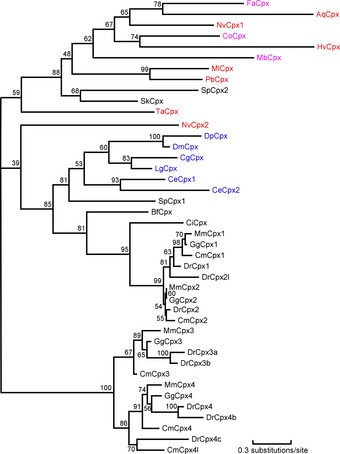
This tree was obtained by the MOLPHY package based on the alignment shown in Fig 1 (positions with 50% or more gaps were discarded before tree reconstruction). The coloring of sequence names is as follows: non‐metazoans: magenta; basal metazoans: red; protostomes: blue; and deuterostomes: black.
Nematostella complexin‐1 is a functional activator of neurotransmitter release in mouse neurons
Given the small size of complexins, their surprising conservation in the most primitive metazoans and even in some unicellular organisms raises the question whether the complexins observed in these species actually function as complexins. To address this question, we focused on two complexin sequences that we had identified in the Nematostella vectensis genome 19 (Figs 1 and EV1). One of the two Nematostella complexins (complexin‐1 [referred to as “nvCpx1”]) contains a C‐terminal CAAX box, whereas the other Nematostella complexin (nvCpx2) does not.
Figure EV1. Alignment of mouse and Nematostella complexin sequences.

Alignment of complexin sequences from mouse (mCpx1–4) and Nematostella vectoriensis (nvCpx1 and 2). Sequences are shown in single‐letter amino acid code; residues shared by 6, 5, or 4 of the 6 sequences are highlighted in yellow, green, and blue, respectively. Conserved hydrophobic sequences are highlighted in red, and C‐terminal cysteine residues that are presumably isoprenylated are displayed on a black background.
To test whether Nematostella complexins are functional and whether the function of complexin has been evolutionarily conserved, we constructed expression vectors encoding nvCpx1 and nvCpx2. We then examined whether Nematostella complexins could rescue the synaptic phenotype of complexin‐deficient mouse neurons. Toward this goal, we suppressed endogenous complexin‐1 and ‐2 expression by virally delivered shRNAs (referred to as the double knockdown [DKD]) 5, 8 and examined rescue of the synaptic phenotype of complexin‐deficient mouse neurons by Nematostella complexins. This approach was chosen because no tests of complexin function in Nematostella itself are readily available—these metazoans do not have synapses that could be recorded from, and no functional assays of their morphologically identified neurosecretory cells have been reported—and because rescue of a loss‐of‐function over such a large evolutionary distance as that separating Cnidaria from rodents could be considered the most compelling evidence for functional similarity.
We first compared the nvCpx1 and nvCpx2 in parallel. To assess their potential clamping functions, we measured miniature excitatory postsynaptic currents (mEPSCs) in control mouse neurons and mouse neurons subjected to the complexin DKD without or with expression of either nvCpx1 or nvCpx2 (Fig 3). Consistent with previous data 5, we found that the complexin DKD significantly increased the mEPSC frequency, that is, unclamped mini release. This phenotype was ameliorated but not completely reversed by nvCpx1 and was even exacerbated by nvCpx2 (Fig 3A).
Figure 3. Testing rescue of complexin‐deficient neurons with Nematostella complexin‐1 (nvCpx1) or ‐2 (nvCpx2).
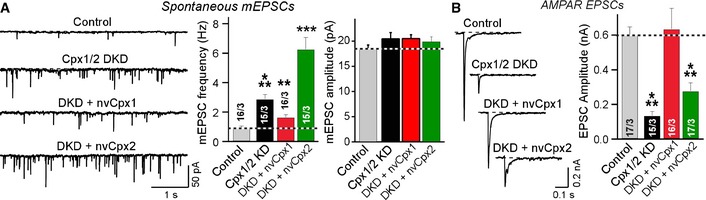
- Rescue effects of Nematostella nvCpx1 and nvCpx2 on clamping spontaneous release in complexin‐deficient mouse neurons. Panels show sample traces (left) and summary graphs of the frequency (center) and amplitude (right) of mEPSCs recorded in WT cortical neurons that were infected with a control lentivirus (Control) or a lentivirus expressing complexin shRNAs (Cpx1/2 DKD), without or with co‐expression of Nematostella complexin‐1 (nvCpx1) or ‐2 (nvCpx2).
- Rescue of impaired evoked release in complexin‐deficient mouse neurons by Nematostella nvCpx1 and nvCpx2. Panels show sample traces (left) and summary graphs of the amplitude of evoked AMPAR‐mediated EPSCs that were induced by isolated action potentials.
We then assessed the potential activator function of nvCpx1 and nvCpx2 by measuring action potential‐evoked release (Fig 3B). We found that nvCpx1 fully rescued the impairment of evoked synaptic transmission in mouse neurons lacking complexins, whereas nvCpx2 had a much smaller rescue effect. Viewed together, these experiments suggest that nvCpx1 surprisingly may be a functional activator of release in mammalian synapses, whereas nvCpx2 is only weakly active, possibly because of expression impairments or evolutionary constraints. Thus, for further analyses, we focused on nvCpx1.
Nematostella complexin‐1 activates neurotransmitter release similar to mammalian complexins
In the next set of experiments, we asked how the function of NvCpx1 in neurotransmitter release quantitatively compared to that of a mammalian complexin. We examined the relative effects of rat Cpx1 (which is identical in amino acid sequence to mouse Cpx 1 20) and nvCpx1 on spontaneous mEPSCs in mouse cortical neurons (Fig 4A). As expected, nvCpx1 neither aggravated nor rescued the unclamping of mini release in complexin DKD neurons, whereas rat Cpx1 fully rescued. We then examined the activating function of complexin. Complexins activate Ca2+‐triggered exocytosis by two sequential mechanisms: enhancement of synaptic vesicle priming and enabling of Ca2+ triggering by synaptotagmins. Only the former mechanism requires the C‐terminal region of complexins 18. To assess the activating function of nvCpx1, we measured action potential‐evoked (i.e., Ca2+‐triggered) EPSCs, which are largely mediated by AMPA‐type glutamate receptors (AMPARs). We found that nvCpx1 fully rescued the ~3‐fold decrease in evoked EPSC amplitude induced by the complexin DKD (Fig 4B). Moreover, we measured the size of the readily releasable pool (RRP) of vesicles, monitored as the synaptic charge transfer that occurs during an EPSC induced by hypertonic sucrose 21. Again, we found that nvCpx1 rescued the ~2‐fold decrease in the RRP induced by the complexin DKD (Fig 4C). These data indicate that nvCpx1 can substitute for mammalian complexin in activating both the Ca2+ triggering and the priming of vesicles for release, an unexpected finding given the evolutionary distance between Nematostella and mammals.
Figure 4. Nematostella complexin‐1 functionally substitutes for mammalian complexins in enabling neurotransmitter release.
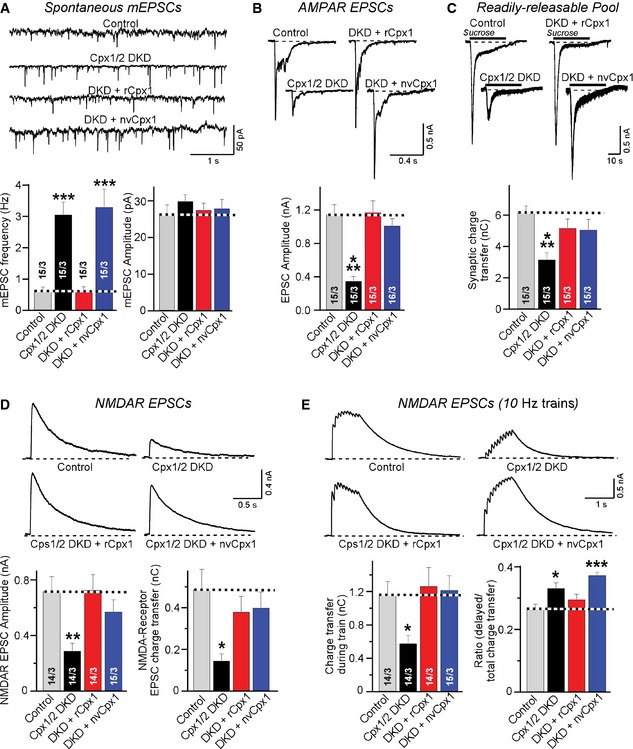
- Sample traces (top) and summary graphs (bottom) of mEPSCs recorded in WT cortical neurons that were infected with a control lentivirus (Control) or a lentivirus expressing complexin shRNAs (Cpx1/2 DKD), without or with co‐expression of rat (rCpx1) or Nematostella complexin‐1 (nvCpx1).
- Sample traces (top) and summary graphs (bottom) of evoked AMPAR‐mediated EPSCs that were induced by isolated action potentials. EPSCs were monitored as described for (A).
- Sample traces (top) and summary graphs (bottom) of EPSCs evoked by 0.5 M sucrose, recorded from neurons as described for (A).
- Sample traces (top) and summary graphs (bottom) of NMDAR‐mediated EPSCs evoked by isolated action potentials recorded from neurons as described for (A).
- Sample traces (top) and summary graphs of NMDAR‐mediated EPSCs induced by a 10‐Hz, 1‐s stimulus train (bottom). Recordings were performed as in (A).
To corroborate this conclusion, we inquired whether nvCpx1 could also support neurotransmitter release during high‐frequency stimulus trains. In high‐density cultures of neurons, EPSCs induced by stimulus trains can only be monitored by measuring slower EPSCs mediated by NMDA‐type glutamate receptors (NMDARs) with simultaneous inhibition of AMPARs that drive network activity 8. We first confirmed that the rescue of evoked neurotransmitter release with nvCpx1 could also be observed in NMDAR‐mediated EPSCs (Fig 4D). We then measured synaptic responses to a 10‐Hz stimulus train monitored by NMDAR‐mediated EPSCs. We observed that nvCpx1 was as competent as rat complexin‐1 in rescuing the impairment of synaptic release induced by stimulus trains in complexin‐deficient neurons (Fig 4E). We examined the release kinetics during the stimulus trains by plotting the cumulative charge transfer as a function of stimulus number, and detected no major differences in the kinetics of release during the stimulus train between neurons expressing rat or Nematostella complexin‐1 (Fig EV2), suggesting that nvCpx1 fully supports release during the train. However, nvCpx1 did not reverse the slight increase in delayed release that is observed in complexin‐deficient neurons (Fig 4E) 8. Delayed release represents the release that continues after a stimulus train has ended 22. The inability of nvCpx1 to reverse the increase in delayed release in complexin‐deficient neurons is consistent with its lack of a clamping function (Fig 4A), since the enhanced delayed release likely reflects, at least in part, an enhanced Ca2+‐dependent rate of spontaneous release 8.
Figure EV2. Analysis of the kinetics of evoked NMDA receptor‐dependent EPSCs (related to Fig 4E).
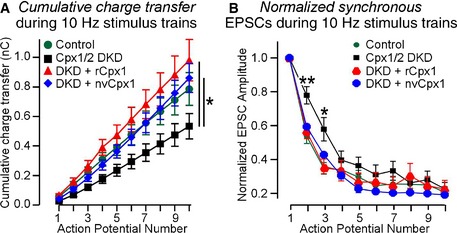
- The plot of cumulative charge transfer during the 10‐Hz stimulus train as a function of the action potential number is recorded in WT cortical neurons that were infected with a control lentivirus (Control) or a lentivirus expressing complexin shRNAs (Cpx1/2 DKD) without or with co‐expression of rCpx1 or nvCpx1.
- The degree of synaptic depression as a function of the action potential number is plotted as described for (A).
Data information: Data shown are means ± SEM; statistical assessments were performed by two‐way ANOVA (A) or Student's t‐test (B) comparing each condition to control (*P < 0.05; **P < 0.01). The data from the experiments shown in Fig 4E, and the number of neurons/cultures analyzed correspond to those listed in the legend to Fig 4E.
Nematostella and mammalian complexin‐1 exhibit similar functional domain architectures
To determine whether nvCpx1 activates priming and Ca2+ triggering of release by mechanisms similar to those of mammalian complexin‐1, we tested whether the N‐ and C‐terminal regions of nvCpx1 were essential for Ca2+ triggering and priming of release, similar to the corresponding regions of mammalian complexin‐1 5, 8, 15, 18.
When we examined spontaneous mEPSCs, we found that neither the N‐terminal nor the C‐terminal truncation of nvCpx1 endowed it with a clamping activity (Fig 5A), consistent with the initial results (Figs 3 and 4). We then measured Ca2+‐triggered release monitored by AMPAR‐mediated EPSCs induced by single action potentials. We observed that the N‐ but not the C‐terminal truncation blocked the Ca2+‐triggering function of nvCpx1 (Fig 5B), similar to mammalian complexin‐1 5, 15, 23. However, the impairment in complexin‐deficient neurons of sucrose‐induced release, used to monitor synaptic vesicle priming, was not rescued by either C‐ or N‐terminally truncated nvCpx1 (Fig 5C). Moreover, when we examined Ca2+‐triggered release in complexin‐deficient neurons by monitoring NMDAR‐mediated EPSCs, we also observed full rescue by C‐ but not by N‐terminally truncated nvCpx1 (Fig 5D). Finally, release induced by high‐frequency stimulus trains was also only rescued by C‐terminally truncated nvCpx1, although as before nvCpx1 was unable to reverse the increase in delayed release induced by complexin DKD (Figs 5E and EV3). Thus, the activating and not the clamping function of complexin is evolutionarily conserved, demonstrating that there is no evolutionary switch in complexins from a primarily clamping to a primarily activating function, but that the activating functions of complexins are central to their role in all metazoans.
Figure 5. Functional domain organization of Nematostella complexin‐1 is similar to that of mammalian complexins.
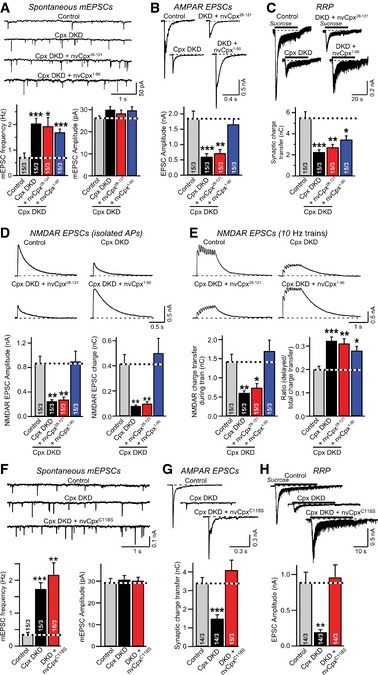
- Sample traces (top) and summary graphs (bottom) of mEPSCs, recorded in cortical neurons that were infected with a control lentivirus (Control) or lentiviruses expressing complexin shRNAs (Cpx DKD) without or with co‐expression of N‐ (nvCpx28‐121) or C‐terminally truncated Nematostella complexin‐1 (nvCpx1‐90).
- Sample traces (top) and summary graphs (bottom) of action potential‐evoked AMPAR‐mediated EPSCs monitored in neurons as described for (A).
- Sample traces (top) and summary graphs of AMPAR‐mediated EPSCs evoked by 0.5 M sucrose (bottom), recorded as described for (A).
- Sample traces (top) and summary graphs (bottom) of NMDAR‐mediated EPSCs evoked by isolated action potentials recorded as described for (A).
- Sample traces (top) and summary graphs (bottom) of NMDAR‐mediated EPSCs evoked by action potential trains (10 Hz for 1 s) recorded as described for (A).
- Sample traces (top) and summary graphs (bottom) of mEPSCs, recorded in WT cortical neurons that were infected with a control lentivirus (Control) or a lentivirus expressing complexin shRNAs (Cpx DKD) without or with rescue with mutant Nematostella complexin‐1 in which the C‐terminal cysteine that is presumably isoprenylated (Fig 1A) was converted to a serine (nvCpxC118S).
- Sample traces (top) and summary graphs (bottom) of action potential‐evoked AMPAR‐mediated EPSCs monitored as described for (A).
- Sample traces (top) and summary graphs of EPSCs evoked by 0.5 M sucrose (bottom), recorded as described for (A).
Figure EV3. Analysis of the kinetics of evoked NMDA receptor‐dependent EPSCs (related to Fig 5E).
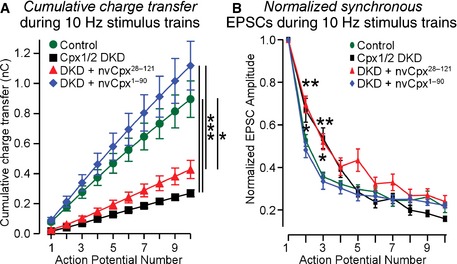
- The plot of cumulative charge transfer during the 10‐Hz stimulus train as a function of the action potential number is recorded in WT cortical neurons that were infected with a control lentivirus (Control) or a lentivirus expressing complexin shRNAs (Cpx1/2 DKD) without or with co‐expression of nvCpx28‐121 or nvCpx1‐90.
- The degree of synaptic depression as a function of the action potential number is plotted as described for (A).
Data information: Data shown are means ± SEM; statistical assessments were performed by two‐way ANOVA (A) or Student's t‐test (B) comparing each condition to control (*P < 0.05; **P < 0.01; ***P < 0.001). The data from the experiments shown in Fig 5E, and the number of neurons/cultures analyzed correspond to those listed in the legend to Fig 5E.
A possible reason for the lack of a clamping activity of nvCpx1 in our experiments may be its C‐terminal isoprenylation sequence. Mammalian complexin‐3 and ‐4 also contain such a C‐terminal sequence and lack clamping activity 18, 24, and in Drosophila and C. elegans, this sequence has been implicated in the clamping capability of complexins 13, 14, 25. To test this possibility, we abolished the isoprenylation potential of nvCpx1 by mutating the putative isoprenylated cysteine residue (C118) to a serine (nvCpx1C118S).
We then tested whether lack of isoprenylation endows nvCpx1 with a clamping ability, but observed that the mutation did not have any effect in mEPSCs (Fig 5F). Moreover, nvCpx1C118S was still capable of activating Ca2+ triggering and priming of synaptic vesicle exocytosis (Fig 5G and H), similar to the lack of an effect we observed when we converted mammalian complexin‐1 into an isoprenylated protein 26. Thus, C‐terminal isoprenylation of nvCpx1 is not functionally essential. Overall, these results indicate that nvCpx1 acts by a similar mechanism as mammalian complexins 26.
Nematostella complexin‐1 also rescues release in complexin‐1/2 double KO (DKO) neurons
The complexin DKD approach in our experiments, although well validated, may raise concerns because of potential off‐target effects 27. In a direct comparison, we previously found that the DKD and the double KO of complexin‐1 and ‐2 in mouse neurons caused identical priming and Ca2+‐triggering phenotypes, but distinct clamping phenotypes and compensatory changes in mRNA levels of complexin‐3 and ‐4 26. We thus sought to further validate our results with nvCpx1 in complexin DKD neurons using complexin DKO neurons 28.
Consistent with earlier results 26, we found a significant but small increase in spontaneous mEPSC frequency induced by the complexin‐1/‐2 DKO in cortical neurons that was rescued by the expression of rat complexin‐1 (Fig 6A). Expression of nvCpx1, however, caused a large increase in mEPSC frequency, which may be explained by earlier observations showing that overexpression of mutant complexins that are unable to clamp spontaneous release causes an unclamping of mEPSC release by a dominant‐negative mechanism 26. Strikingly, this unclamping was abolished by deletion of the C‐terminal regions from nvCpx1 (Fig 6A). Moreover, full‐length as well as C‐terminally truncated nvCpx1 was as effective as mammalian complexin‐1 in increasing evoked EPSCs in the DKO neurons, confirming that nvCpx1 is fully capable of activating release (Fig 6B). Finally, as in the DKD neurons, full‐length but not C‐terminally truncated nvCpx1 was as active as mammalian complexin‐1 in increasing the RRP size in complexin‐1/‐2 DKO neurons (Fig 6C). Together, these experiments confirm that in DKO neurons, nvCpx1 is fully competent to replace the complexin activator functions in mammalian neurons.
Figure 6. Nematostella complexin‐1 rescues evoked neurotransmitter release in cortical neurons cultured from complexin‐1/2 double KO mice.
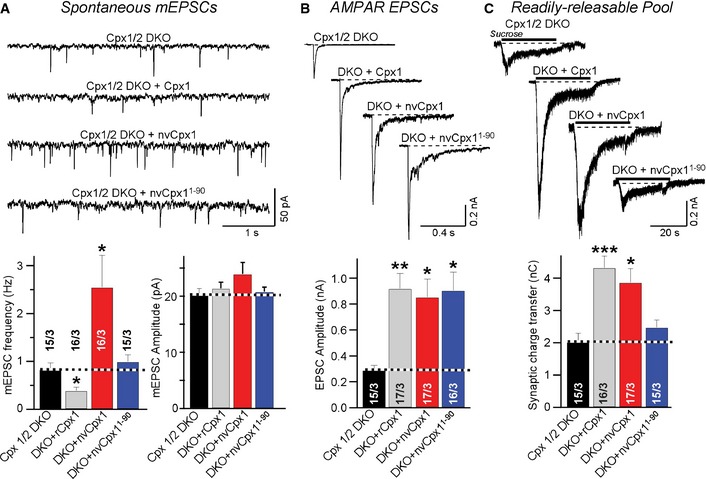
- Sample traces (top) and summary graphs (bottom) of mEPSCs, recorded in cortical neurons cultured from complexin‐1/2 double KO mice 23. Neurons were infected with a control lentivirus (Cpx1/2 DKO) or a lentivirus expressing wild‐type rat complexin‐1 (rCpx1), wild‐type Nematostella complexin‐1 (nvCpx1), or C‐terminally truncated Nematostella complexin‐1 (nvCpx1‐90).
- Sample traces (top) and summary graphs (bottom) of action potential‐evoked AMPAR‐mediated EPSCs monitored as described for (A).
- Sample traces (top) and summary graphs of EPSCs evoked by 0.5 M sucrose (bottom), recorded as described for (A).
Summary
Overall, our experiments suggest two major conclusions. First, complexins likely predate metazoan evolution and may have a general role in membrane traffic. Their emergence prior to that of neurosecretory cells is consistent with the observation of complexins in non‐neuronal mammalian cells 20, and suggests that complexins are fundamental components of all types of regulated exocytosis. Second, the function of complexins in the core machinery of neurotransmitter release appears to be conserved throughout metazoan evolution. To the best of our knowledge, our study reports the first functional expression of a cnidarian protein in mammalian neurons, and the first demonstration that cnidarian exocytosis operates by fundamentally identical molecular mechanisms as mammals. The fact that Nematostella complexin can replace mouse complexin in activating neurotransmitter release indicates that the fundamental function of complexin consists of an activating role, a role that is essential for preparing the fusion machinery for fast regulated exocytosis.
Materials and Methods
Bioinformatics
PSI‐BLAST [ref: PubMed 9254694] was used to search for complexins against the nr database of NCBI, with the human complexin‐3 as the initial query (e‐value cutoff: 0.001). Found homologs were clustered using BLASTCLUST, and one representative sequence was selected from each cluster and used as query for further PSI‐BLAST iterations. Multiple sequence alignment of select complexins from major lineages of Metazoa and non‐metazoans was made by PROMALS3D (PMID: 18287115), followed by manual adjustment. The MOLPHY package 29 was used for phylogenetic reconstruction of these proteins based on the alignment, with positions containing 50% or more gap characters removed. The JTT amino acid substitution model 30 was used in MOLPHY. The local estimates of bootstrap percentages (shown next to branch points) were obtained by the RELL method 31 (‐R option in the ProtML program of MOLPHY).
Neuronal cultures and lentiviruses preparation
Neuronal cultures were obtained from wild‐type (WT) or complexin‐1/2 double KO mice 26, 28 as described 8. Lentiviral expression vectors and three helper plasmids (pRSV‐REV, pMDLg/pRRE, and pVSVG) were co‐transfected into HEK293 cells (ATCC, VA), and the viruses were collected 48 hr after transfection 5. All steps were performed under level II biosafety conditions. Neurons were infected with lentiviruses at DIV4 and analyzed at DIV14‐16. All mouse procedures used were approved by Stanford Institutional Review Boards.
Plasmid construction
Constructs encoding WT Nematostella complexin‐1 (nvCpx1) and mutants thereof (the N‐ (nvCpx28‐121) and C‐terminal truncations (nvCpx1‐90) and the C‐terminal cysteine substitution (nvCpxC118S)) were generated by gene synthesis and were cloned downstream of the human ubiquitin promoter in the L309 lentiviral vector 5.
Electrophysiological recordings
Electrophysiological recordings were performed in whole‐cell patch‐clamp mode. Synaptic currents were monitored with a Multiclamp 700A amplifier (Molecular Devices). The frequency, duration, and magnitude of the extracellular stimulus were controlled with a Model 2100 Isolated Pulse Stimulator (A‐M Systems) synchronized with Clampex 10 data acquisition software (Molecular Devices). AMPA receptor‐ and NMDA receptor‐mediated EPSCs (recorded at a holding potential of −70 mV and +40 mV, respectively) were isolated pharmacologically with D‐APV and picrotoxin, and with CNQX, picrotoxin, and glycine, respectively. Spontaneous mEPSCs were monitored in the presence of tetrodotoxin (TTX). Sucrose‐evoked release was triggered by a 30‐s application of 0.5 M sucrose with D‐APV, picrotoxin, and TTX, puffed by Picospritzer III (Parker).
Statistical analyses
Statistical analyses were performed with Student's t‐tests or two‐way ANOVA (for Figs EV1, EV2, EV3) comparing test to control samples analyzed in the same experiments.
Author contributions
XY, YJK‐W, and TB planned and performed the experiments, analyzed the data, and wrote the paper; JP and NVG performed bioinformatics analyses; and TCS analyzed the data and wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Review Process File
Acknowledgements
This paper was supported by grants from NINDS (K99 NS08708601 to T.B.; R01 NS077906 to T.C.S.), NIMH (P50 MH086403 to R.C.M.), NIGMS (R01 GM094575 to N.V.G.), and the National Natural Science Foundation of China (31300892 to Y.X.)
EMBO Reports (2015) 16: 1308–1317
References
- 1. Südhof TC (2013) Neurotransmitter release: the last millisecond in the life of a synaptic vesicle. Neuron 80: 675–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Martin JA, Hu Z, Fenz KM, Fernandez J, Dittman JS (2011) Complexin has opposite effects on two modes of synaptic vesicle fusion. Curr Biol 21: 97–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Xue M, Lin YQ, Pan H, Reim K, Deng H, Bellen HJ, Rosenmund C (2009) Tilting the balance between facilitatory and inhibitory functions of mammalian and Drosophila Complexins orchestrates synaptic vesicle exocytosis. Neuron 64: 367–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tang J, Maximov A, Shin OH, Dai H, Rizo J, Südhof TC (2006) A complexin/synaptotagmin 1 switch controls fast synaptic vesicle exocytosis. Cell 126: 1175–1187 [DOI] [PubMed] [Google Scholar]
- 5. Maximov A, Tang J, Yang X, Pang ZP, Südhof TC (2009) Complexin controls the force transfer from SNARE complexes to membranes in fusion. Science 323: 516–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cho RW, Song Y, Littleton JT (2010) Comparative analysis of Drosophila and mammalian complexins as fusion clamps and facilitators of neurotransmitter release. Mol Cell Neurosci 45: 389–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hobson RJ, Liu Q, Watanabe S, Jorgensen EM (2011) Complexin maintains vesicles in the primed state in C. elegans . Curr Biol 21: 106–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yang X, Kaeser‐Woo YJ, Pang ZP, Xu W, Südhof TC (2010) Complexin clamps asynchronous release by blocking a secondary Ca2+‐sensor via its accessory α‐helix. Neuron 68: 907–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Giraudo CG, Eng WS, Melia TJ, Rothman JE (2006) A clamping mechanism involved in SNARE‐dependent exocytosis. Science 313: 676–680 [DOI] [PubMed] [Google Scholar]
- 10. Kyoung M, Srivastava A, Zhang Y, Diao J, Vrljic M, Grob P, Nogales E, Chu S, Brunger AT (2011) In vitro system capable of differentiating fast Ca2+‐triggered content mixing from lipid exchange for mechanistic studies of neurotransmitter release. Proc Natl Acad Sci USA 108: E304–E313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Malsam J, Seiler F, Schollmeier Y, Rusu P, Krause JM, Sollner TH (2009) The carboxy‐terminal domain of complexin I stimulates liposome fusion. Proc Natl Acad Sci USA 106: 2001–2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yoon TY, Lu X, Diao J, Lee SM, Ha T, Shin YK (2008) Complexin and Ca2+ stimulate SNARE‐mediated membrane fusion. Nat Struct Mol Biol 15: 707–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Buhl LK, Jorquera RA, Akbergenova Y, Huntwork‐Rodriguez S, Volfson D, Littleton JT (2013) Differential regulation of evoked and spontaneous neurotransmitter release by C‐terminal modifications of complexin. Mol Cell Neurosci 52: 161–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Iyer J, Wahlmark CJ, Kuser‐Ahnert GA, Kawasaki F (2013) Molecular mechanisms of COMPLEXIN fusion clamp function in synaptic exocytosis revealed in a new Drosophila mutant. Mol Cell Neurosci 56: 244–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Xue M, Reim K, Chen X, Chao HT, Deng H, Rizo J, Brose N, Rosenmund C (2007) Distinct domains of complexin I differentially regulate neurotransmitter release. Nat Struct Mol Biol 14: 949–958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nakanishi N, Renfer E, Technau U, Rentzsch F (2012) Nervous systems of the sea anemone Nematostella vectensis are generated by ectoderm and endoderm and shaped by distinct mechanisms. Development 139: 347–357 [DOI] [PubMed] [Google Scholar]
- 17. Burkhardt P, Grønborg M, McDonald K, Sulur T, Wang Q, King N (2014) Evolutionary insights into premetazoan functions of the neuronal protein homer. Mol Biol Evol 31: 2342–2355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kaeser‐Woo YJ, Yang X, Südhof TC (2012) C‐terminal complexin sequence is selectively required for clamping and priming but not for Ca2+‐triggering of synaptic exocytosis. J Neurosci 32: 2877–2885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Putnam NH, Srivastava M, Hellsten U, Dirks B, Chapman J, Salamov A, Terry A, Shapiro H, Lindquist E, Kapitonov VV et al (2007) Sea anemone genome reveals ancestral eumetazoan gene repertoire and genomic organization. Science 317: 86–94 [DOI] [PubMed] [Google Scholar]
- 20. McMahon HT, Missler M, Li C, Südhof TC (1995) Complexins: cytosolic proteins that regulate SNAP receptor function. Cell 83: 111–119 [DOI] [PubMed] [Google Scholar]
- 21. Rosenmund C, Stevens CF (1996) Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron 16: 1197–1207 [DOI] [PubMed] [Google Scholar]
- 22. Maximov A, Südhof TC (2005) Autonomous function of synaptotagmin 1 in triggering synchronous release independent of asynchronous release. Neuron 48: 547–554 [DOI] [PubMed] [Google Scholar]
- 23. Xue M, Craig TK, Xu J, Chao HT, Rizo J, Rosenmund C (2010) Binding of the complexin N terminus to the SNARE complex potentiates synaptic‐vesicle fusogenicity. Nat Struct Mol Biol 17: 568–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Reim K, Wegmeyer H, Brandstätter JH, Xue M, Rosenmund C, Dresbach T, Hofmann K, Brose N (2005) Structurally and functionally unique complexins at retinal ribbon synapses. J Cell Biol 169: 669–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wragg RT, Snead D, Dong Y, Ramlall TF, Menon I, Bai J, Eliezer D, Dittman JS (2013) Synaptic vesicles position complexin to block spontaneous fusion. Neuron 77: 323–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yang X, Cao P, Südhof TC (2013) Deconstructing complexin function in activating and clamping Ca2+‐triggered exocytosis by comparing knockout and knockdown phenotypes. Proc Natl Acad Sci USA 110: 20777–20782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bacaj T, Ahmad M, Jurado S, Malenka RC, Südhof TC (2015) Synaptic function of Rab11Fip5: selective requirement for Hippocampal Long‐Term Depression. J Neurosci 35: 7460–7474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Reim K, Mansour M, Varoqueaux F, McMahon HT, Südhof TC, Brose N, Rosenmund C (2001) Complexins regulate a late step in Ca2+‐dependent neurotransmitter release. Cell 104: 71–81 [DOI] [PubMed] [Google Scholar]
- 29. Adachi J, Hasegawa M (1996) MOLPHY version 2.3, programs for molecular phylogenetics based on maximum likelihood. Computer Science Monographs 28. The Institute of Statistical Mathematics; pp. 1–150.
- 30. Jones DT, Taylor WR, Thornton JM (1992) The rapid generation of mutation data matrices from protein sequences. Comput Appl Biosci 8: 275–282 [DOI] [PubMed] [Google Scholar]
- 31. Hasegawa M, Kishino H, Saitou N (1991) On the maximum likelihood method in molecular phylogenetics. J Mol Evol 32: 443–445 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Review Process File