

Abstract
Background
Despite the worldwide circulation of human coronavirus OC43 (HCoV-OC43) and HKU1 (HCoV-HKU1), data on their molecular epidemiology and evolutionary dynamics in the tropical Southeast Asia region is lacking.
Methods
The study aimed to investigate the genetic diversity, temporal distribution, population history and clinical symptoms of betacoronavirus infections in Kuala Lumpur, Malaysia between 2012 and 2013. A total of 2,060 adults presented with acute respiratory symptoms were screened for the presence of betacoronaviruses using multiplex PCR. The spike glycoprotein, nucleocapsid and 1a genes were sequenced for phylogenetic reconstruction and Bayesian coalescent inference.
Results
A total of 48/2060 (2.4 %) specimens were tested positive for HCoV-OC43 (1.3 %) and HCoV-HKU1 (1.1 %). Both HCoV-OC43 and HCoV-HKU1 were co-circulating throughout the year, with the lowest detection rates reported in the October-January period. Phylogenetic analysis of the spike gene showed that the majority of HCoV-OC43 isolates were grouped into two previously undefined genotypes, provisionally assigned as novel lineage 1 and novel lineage 2. Sign of natural recombination was observed in these potentially novel lineages. Location mapping showed that the novel lineage 1 is currently circulating in Malaysia, Thailand, Japan and China, while novel lineage 2 can be found in Malaysia and China. Molecular dating showed the origin of HCoV-OC43 around late 1950s, before it diverged into genotypes A (1960s), B (1990s), and other genotypes (2000s). Phylogenetic analysis revealed that 27.3 % of the HCoV-HKU1 strains belong to genotype A while 72.7 % belongs to genotype B. The tree root of HCoV-HKU1 was similar to that of HCoV-OC43, with the tMRCA of genotypes A and B estimated around the 1990s and 2000s, respectively. Correlation of HCoV-OC43 and HCoV-HKU1 with the severity of respiratory symptoms was not observed.
Conclusions
The present study reported the molecular complexity and evolutionary dynamics of human betacoronaviruses among adults with acute respiratory symptoms in a tropical country. Two novel HCoV-OC43 genetic lineages were identified, warranting further investigation on their genotypic and phenotypic characteristics.
Electronic supplementary material
The online version of this article (doi:10.1186/s12985-016-0488-4) contains supplementary material, which is available to authorized users.
Keywords: Coronaviruses, Molecular epidemiology, Phylogenetics, Upper respiratory infection, Virus evolution
Background
Human coronaviruses are common cold viruses that are frequently found to be associated with acute upper respiratory tract infections (URTIs) [1]. According to the International Committee for Taxonomy of Viruses (ICTV), human coronavirus OC43 (HCoV-OC43) and HKU1 (HCoV-HKU1) belong to the betacoronavirus genus, a member of the Coronaviridae family. Coronaviruses contain the largest RNA genomes and have been established as one of the rapidly evolving viruses [2]. In addition to the high nucleotide substitution rates across the genome [3], the coronavirus genome is subjected to homologous recombination during viral replication, which is caused by RNA template switching mediated by the copy-choice mechanism [4, 5]. The genetic recombination of coronaviruses had possibly led to the emergence of lethal pathogens such as severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS-CoV), which caused up to 50 % mortality in infected individuals [6–9]. Recombination events in the spike (S), nucleocapsid (N) and the RNA dependent RNA polymerase (RdRp) within the 1a gene of HCoV-OC43 and HCoV-HKU1 leading to the emergence of unique recombinant genotypes have been reported [10, 11].
Studies have shown that HCoV-OC43 is often associated with approximately 5 % of acute respiratory infections while the more recent HCoV-HKU1 is less prevalent [12, 13]. In humans, acute upper respiratory symptoms such as nasal congestion and rhinorrhea are relatively common in HCoV infections while sore throat and hoarseness of voice are less common, with cough usually associated with HCoV-OC43 infection [14]. In tropical countries, annual shift in the predominant genotype has been documented, with more cases of HCoV-OC43 and HCoV-HKU1 infections reported during the early months of the year [15]. Despite the clinical importance and socioeconomic impact of HCoV infections [16, 17], the prevalence, seasonality, clinical and phylogenetic characteristics of HCoVs remain largely unreported in the tropical region of Southeast Asia. Based on the S, N and 1a genes of HCoV-OC43 and HCoV-HKU1 isolated from Malaysia and also globally, we attempted to delineate the genetic history and the phylodynamic profiles of human betacoronaviruses HCoV-OC43 and HCoV-HKU1 using a suite of Bayesian phylogenetic tools. We also reported the emergence of two novel HCoV-OC43 lineages, in a cross-sectional study of patients presented with acute URTI in Malaysia.
Methods
Clinical specimens
A total of 2,060 consenting outpatient adults presented with symptoms of acute URTI were recruited at the Primary Care Clinics of University Malaya Medical Centre in Kuala Lumpur, Malaysia between March 2012 and February 2013. Prior to collection of nasopharyngeal swabs, demographic data such as age, gender and ethnicity were obtained. In addition, the severities of symptoms (sneezing, nasal discharge, nasal congestion, headache, sore throat, voice hoarseness, muscle ache and cough) were graded based on previously reported criteria [18–21]. The scoring scheme used had been validated earlier on the adult populations with common cold [19]. The nasopharyngeal swabs were transferred to the laboratory in universal transport media and stored in −80 °C.
Molecular detection of HCoV-OC43 and HCoV-HKU1
Total nucleic acids were extracted from nasopharyngeal swabs using the magnetic beads-based protocols implemented in the NucliSENS easyMAG automated nucleic acid extraction system (BioMérieux, USA) [22, 23]. Specimens were screened for the presence of respiratory viruses using the xTAG Respiratory Virus Panel FAST multiplex RT-PCR assay (Luminex Molecular Diagnostics, USA) which can detect HCoV-OC43, HCoV-HKU1 and other respiratory viruses and subtypes [24].
Genetic analysis of HCoV-OC43 and HCoV-HKU1
RNA from nasopharyngeal swabs positive for HCoV-OC43 and HCoV-HKU1 was reverse transcribed into cDNA using SuperScript III kit (Invitrogen, USA) with random hexamers (Applied Biosystems, USA). The partial S gene (S1 domain) [HCoV-OC43; 848 bp (24,030-24,865) and HCoV-HKU1; 897 bp (23,300-24,196)], complete N gene [HCoV-OC43; 1,482 bp (28,997-30, 478) and HCoV-HKU1; 1,458 bp (28,241-29,688)] and partial 1a (nsp3) gene [HCoV-OC43; 1,161 bp (6,168- 7,328) and HCoV-HKU1; 1,115 bp (6,472-7,586)] were amplified either by single or nested PCR, using 10 μM of the newly designed or previously described primers listed in Table 1. The PCR mixture (25 μl) contained cDNA, PCR buffer (10 mM Tris–HCl, 50 mM KCl, 3 mM MgCl, 0.01 % gelatin), 100 μM (each) deoxynucleoside triphosphates, Hi-Spec Additive and 4u/μl BIO-X-ACT Short DNA polymerase (BioLine, USA). The cycling conditions were as follows: initial denaturation at 95 °C for 5 min followed by 40 cycles of 94 °C for 1 min, 54.5 °C for 1 min, 72 °C for 1 min and a final extension at 72 °C for 10 min, performed in a C1000 Touch automated thermal cycler (Bio-Rad, USA). Nested/semi-nested PCR was conducted for each genetic region if necessary, under the same cycling conditions at 30 cycles. Purified PCR products were sequenced using the ABI PRISM 3730XL DNA Analyzer (Applied Biosystems, USA). The nucleotide sequences were codon-aligned with previously described complete and partial HCoV-OC43 and HCoV-HKU1 reference sequences retrieved from GenBank [11, 25–32].
Table 1.
PCR primers of HCoV-OC43 and HCoV-HKU1
| Target gene | HCoV | Primer | Locationa | Sequence (5'-3') | Reference |
|---|---|---|---|---|---|
| Spike (S) | OC43 | LPW 1261 | 24010-24029 | Forward: CTRCTATARYTATAGGTAGT | [11] |
| LPW 2094 | 24866-24887 | Reverse: GCCCAAATTACCCAATTGTAGG | [11] | ||
| HKU1 | LPW 1832 | 23275-23299 | Forward: TATGTTAATAAWACTTTGTATAGTG | [40] | |
| LPW 1866 | 24197-24218 | Reverse: TACAATTGACAAGAACTAGAAG | [40] | ||
| Nucleocapsid (N) | OC43 & HKU1 | βN-F | OC43: 28974-28996 | Forward: GCTGTTTWTGTTAAGTCYAAAGT | this study |
| HKU1: 28218-28240 | |||||
| βN-R | OC43: 30479-30501 | Reverse: CATTCTGATAGAGAGTGCYTATY | this study | ||
| HKU1: 29699-29721 | |||||
| βN-Fn | OC43: 29046-29069 | Forward (nested): GCMTTGTTRAGARMTWAWATCTAA | this study | ||
| HKU1: 28287-28310 | |||||
| βN-Rn | OC43: 30447-30466 | Reverse (nested): GCGAGGGGTTACCACCWRRT | this study | ||
| HKU1: 29671-29690 | |||||
| 1a | OC43 | OC43-1aF | 6145-6167 | Forward: CTTTTGGTAAACCTGTTATATGG | this study |
| OC43-1aR | 7329-7351 | Reverse: AGCTTAATAAAAGAGGCAATAAT | this study | ||
| OC43-1aFn | 6183-6199 | Forward (semi-nested): GCTTCYCTCAATTCTTTAACAT | this study | ||
| HKU1 | HKU1-1aF | 6448-6471 | Forward: TTCTCTTACTTATTTTAATAAACC | this study | |
| HKU1-1aR | 7587-7610 | Reverse: CTTTATACATAGCAGTAACAACTA | this study |
aNucleotide location was determined based on the HCoV-OC43 ATCC VR-759 (AY585228) and HCoV-HKU1 (NC_06577) reference sequences
Maximum clade credibility (MCC) trees for the partial S (S1 domain), complete N and partial 1a (nsp3) genes were reconstructed in BEAST (version 1.7) [27, 33, 34]. MCC trees were generated using a relaxed molecular clock, assuming uncorrelated lognormal distribution under the general time-reversible nucleotide substitution model with a proportion of invariant sites (GTR + I) and a constant coalescent tree model. The Markov chain Monte Carlo (MCMC) run was set at 3 × 106 steps long sampled every 10,000 state. The trees were annotated using Tree Annotator program included in the BEAST package, after a 10 % burn-in, and visualized in FigureTree (http://tree.bio.ed.ac.uk/software/Figuretree/). Neighbor joining (NJ) trees for the partial S (S1 domain), complete N and partial 1a (nsp3) genes were also reconstructed, using Kimura 2-parameter model in MEGA 5.1 [35]. The reliability of the branching order was evaluated by bootstrap analysis of 1000 replicates. In addition, to explore the genetic relatedness between HCoV-OC43 and HCoV-HKU1 genotypes, the pairwise genetic distances among sequences of the S gene were estimated. Inter- and intra-genotype nucleotide distances were estimated by the bootstrap analysis with 1000 replicates using MEGA 5.1. Such analysis has not been done for the N and the 1a genes because those regions were highly conserved across genotypes [10, 11, 32]. To test for the presence of recombination in HCoV-OC43, the S gene was subjected to pairwise distance-based bootscanning analysis using SimPlot version 3.5 [10, 36]. Established reference genomes for HCoV-OC43 genotype A (ATCC VR-759), B (87309 Belgium 2003), and C (HK04-01) were used as putative parental lineages, with a sliding window and step size of 160 bp and 20 bp, respectively. In addition, MaxChi recombination test [37] was performed in the Recombination Detection Program (RDP) version 4.0 [38]. In RDP the highest acceptable p value (the probability that sequences could share high identities in potentially recombinant regions by chance alone) was set at 0.05, with the standard multiple comparisons corrected using the sequential Bonferroni method with 1,000 permutations [39].
Estimation of divergence time
The origin and divergence time (in calendar year) of HCoV-OC43 and HCoV-HKU1 genotypes were estimated using the MCMC approach as implemented in BEAST. Analyses were performed under the relaxed molecular clock with GTR + I nucleotide substitution models and constant-size and exponential demographic models. The MCMC analysis was computed at 3 × 106 states sampled every 10,000 steps. The mean divergence time and the 95 % highest posterior density (HPD) regions were estimated, with the best-fitting models were selected by Bayes factor inference using marginal likelihood analysis implemented in Tracer (version 1.5) [33]. The evolutionary rate for S gene of betacoronaviruses (6.1 × 10−4 substitutions/site/year) reported previously was used for analysis [36].
Statistical analysis
The association of HCoV-OC43 and HCoV-HKU1 infections with specific acute URTI symptoms and its severity (none, moderate and severe) as well as demographic data were evaluated using the Fisher’s exact test/Chi-square test carried out in the statistical package for the social sciences (SPSS, version 16; IBM Corp).
Results
Detection of HCoV-OC43 and HCoV-HKU1 in nasopharyngeal swabs
During the 12-month study period (March 2012 to February 2013), all nasopharyngeal swab specimens from 2,060 patients collected from Kuala Lumpur, Malaysia were screened for the presence of HCoV-OC43 and HCoV-HKU1 using multiplex RT-PCR method, in which a total of 48 (2.4 %) subjects were found positive for betacoronavirus. HCoV-OC43 and HCoV-HKU1 was detected in 26/2060 (1.3 %) and 22/2060 (1.1 %) patients, respectively, while no HCoV-OC43/HCoV-HKU1 co-infection was observed. Age, gender and ethnicity of the patients were summarized in Table 2. The median age of subjects infected with HCoV-OC43 and HCoV-HKU1 was 53.0 and 48.5, respectively. Both HCoV-OC43 and HCoV-HKU1 were co-circulating throughout the year, although lower numbers of HCoV-OC43 were detected between October 2012 and January 2013 while no HCoV-HKU1 was detected during these months (Fig. 1).
Table 2.
Demographic data on 48 outpatients infected with human betacoronavirus in Kuala-Lumpur, Malaysia, 2012-2013
| HCoV-OC43 (n = 26) | HCoV-HKU1 (n = 22) | P-Value | |
|---|---|---|---|
| Gender | |||
| Male | 11(42.3 %) | 8(36.4 %) | 0.77 |
| Female | 15(57.7 %) | 14(63.6 %) | |
| Age | |||
| <40 | 9(34.6 %) | 10(45.4 %) | 0.33 |
| 40–60 | 10(38.5 %) | 4(18.2 %) | |
| >60 | 7(26.9 %) | 8(36.4 %) | |
| Symptoms | |||
| Sneezing | 21(80.8 %) | 14(63.6 %) | 0.99 |
| Nasal discharge | 20(76.9 %) | 19(86.4 %) | |
| Nasal congestion | 19(73.1 %) | 14(63.6 %) | |
| Headache | 18(69.2 %) | 16(72.7 %) | |
| Sore throat | 16(61.5 %) | 14(63.6 %) | |
| Hoarseness of voice | 20(76.9 %) | 18(81.8 %) | |
| Muscle ache | 17(65.4 %) | 14(63.6 %) | |
| Cough | 23(88.5 %) | 19(86.4 %) | |
| Ethnicity | |||
| Malay | 10(38.5 %) | 10(45.4 %) | 0.19 |
| Chinese | 3(11.5 %) | 6(27.3 %) | |
| Indian | 13(50.0 %) | 6(27.3 %) | |
| Others | 0(0.0 %) | 0(0.0 %) | |
Fig. 1.
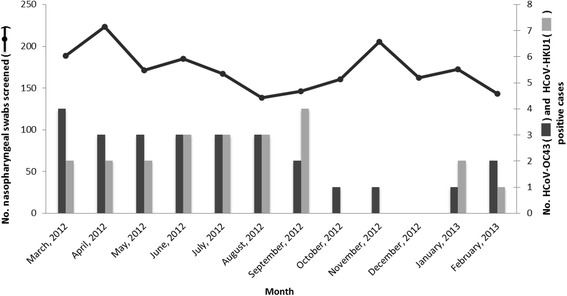
Annual distribution of HCoV-OC43 and HCoV-HKU1 among adults with acute in Malaysia. The monthly detection of HCoV-OC43 and HCoV-HKU1 (right axis, in bars) and the total number of nasopharyngeal swabs screened (left axis, in solid line) between March 2012 and February 2013 were shown
Phylogenetic analysis of the S, N and 1a genes
The partial S (S1 domain), complete N and partial 1a (nsp3) genes of 23 HCoV-OC43 isolates were successfully sequenced, while another three xTAG-positive HCoV-OC43 isolates could not be amplified, probably due to low viral copy number in these specimens. Based on the phylogenetic analysis of the S gene, one subject (1/23, 4.3 %) was grouped with HCoV-OC43 genotype B reference sequences while another subject (1/23, 4.3 %) was grouped with HCoV-OC43 genotype D sequences. The remaining 21 isolates formed two phylogenetically discrete clades that were distinct from other previously established genotypes A, B, C, D (genotype D is a recombinant lineage that is not readily distinguished from genotype C in the S and N phylogenetic trees) and E [11, 32] (Fig. 2 and Additional file 1: Figure S1). Of the 21 isolates, ten isolates have formed a cluster with other recently reported isolates from Japan, Thailand and China [31, 32] supported by the posterior probability value of 1.0 and bootstrap value of 36 % at the internal tree node of the MCC and NJ trees, respectively with intra-group pairwise genetic distance of 0.003 ± 0.001. These isolates were provisionally designated as novel lineage 1. Spatial structure was observed within novel lineage 1, with an isolate from China sampled in year 2008 located at the base of the phylogeny. Moreover, another eleven HCoV-OC43 isolates have formed a second distinct cluster supported by significant posterior probability and bootstrap values at the internal tree node (1.0 and 98 %, respectively) and intra-group pairwise genetic distance of 0.004 ± 0.001. The cluster contained Malaysian and Chinese isolates [32] only, and was denoted as novel lineage 2. Based on the phylogenetic inference of the conserved N gene, only one subject was grouped with the genotype B reference in concordance with the S gene (Additional file 2: Figure S2). Unlike the phylogenetic inference of the S gene, the remaining 22 isolates were seen intermingled with each other forming a single cluster together with isolates indicated as novel lineages 1 and 2 in the S gene, in addition to one genotype D strain. It is however important to note that the tree resolution was poor, due primarily to the lack of the N gene reference sequences in the public database. On the other hand, phylogenetic analysis of the 1a (nsp3) gene (Additional file 3: Figure S3) revealed that all except genotype A could not be differentiated clearly within this region, due mainly to the low genetic diversity between genotypes. The limited number of 1a reference sequences available in the public database could have also resulted in a poor 1a tree topology. In addition, phylogenetic trees of previously described complete and partial S gene sequences as well as partial 1a (nsp3) and complete RdRp gene sequences were reconstructed to further confirm the reliability of the partial S1 and nsp3 for identification of HCoV-OC43 genotypes (Additional file 4: Figure S4 and Additional file 5: Figure S5).
Fig. 2.
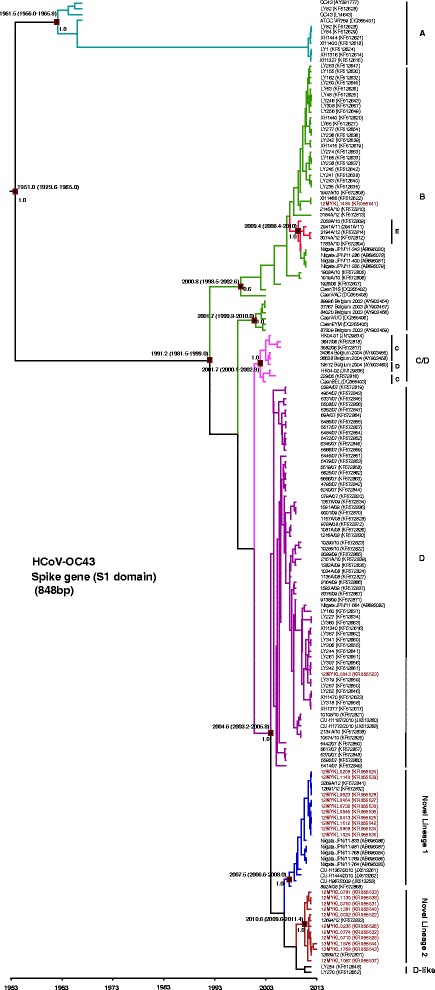
Maximum clade credibility (MCC) tree of HCoV-OC43 genotypes. Estimation of the time of the most recent common ancestors (tMRCA) with 95 % highest posterior density (95 % HPD) of HCoV-OC43 genotypes based on the spike gene (S1 domain) (848 bp). Data were analyzed under relaxed molecular clock with GTR + I substitution model and a constant size coalescent model implemented in BEAST. The Malaysian HCoV-OC43 isolates obtained in this study were color-coded and the HCoV-OC43 genotypes (a) to (e) as well as novel lineages 1 and 2 were indicated. The MCC posterior probability values were indicated on the nodes of each genotype
To assess the diversity between HCoV-OC43 genotypes, inter-genotype pairwise genetic distance was estimated for the S gene, listed in Table 3. Using the oldest genotype as reference i.e. genotype A, genetic variation between genotype A and genotypes B to E was 2.2–2.7 %. Genetic distance between novel lineages 1 and 2 compared to genotype A was 3.2 % and 3.1 %, respectively, higher than that of other established genotypes. Taken together, the distinct inter-genotype genetic variations of the two novel lineages 1 and 2 against other previously established genotypes corroborated with the MCC inference (Fig. 2) in which both lineages formed distinct phylogenetic topologies.
Table 3.
Genetic distance among HCoV-OC43 and HCoV-HKU1 genotypes in the spike gene
| HCoV | Genetic distance | |||||||
|---|---|---|---|---|---|---|---|---|
| OC43 | genotype A | genotype B | genotype C | genotype D | genotype E | Novel lineage 1 | Novel lineage 2 | |
| genotype A | - | |||||||
| genotype B | 2.7 | - | ||||||
| genotype C | 2.2 | 1.5 | - | |||||
| genotype D | 2.7 | 1.8 | 0.8 | - | ||||
| genotype E | 2.5 | 0.9 | 1.2 | 1.6 | - | |||
| Novel lineage 1 | 3.2 | 2.0 | 1.3 | 0.7 | 1.9 | - | ||
| Novel lineage 2 | 3.1 | 2.9 | 1.8 | 1.4 | 2.6 | 1.7 | - | |
| HKU1 | genotype A | genotype B | genotype C | |||||
| genotype A | - | |||||||
| genotype B | 15.7 | - | ||||||
| genotype C | 15.2 | 1.3 | - | |||||
Pairwise genetic distances are expressed in percentage (%) difference
On the other hand, phylogenetic analysis of 22 HCoV-HKU1 S and N genes indicated the predominance of HCoV-HKU1 genotype B (72.7 %, 16/22), followed by HCoV-HKU1 genotype A (27.3 %, 6/22) (Fig. 3, Additional file 6: Figure S6 and Additional file 7: Figure S7). Interestingly, the S and N genes of HCoV-HKU1 were equally informative for genotype assignment, while genotypes A, B and C were less distinctive based on the 1a gene phylogenetic analysis due to the high genetic conservation within this region (Additional file 8: Figure S8). Inter-genotype genetic diversity among HCoV-HKU1 genotypes showed that genotype A was more genetically diverse than genotypes B and C based on the genetic data of the S gene (Table 3). The difference in genetic distance between genotype A and genotypes B and C was 15.2–15.7 %, while the difference in genetic distance between genotypes B and C was 1.3 %.
Fig. 3.
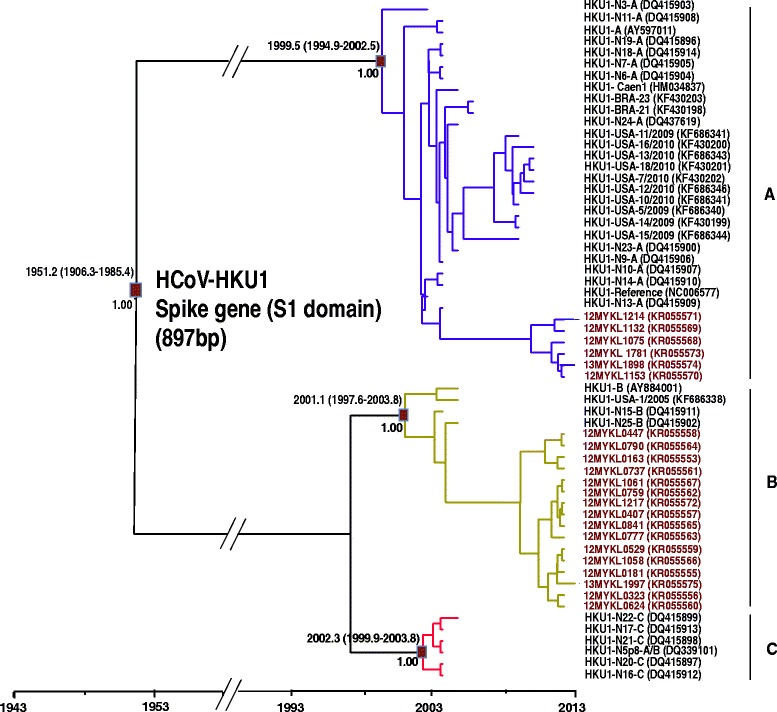
Maximum clade credibility (MCC) tree of HCoV-HKU1 genotypes. Estimation of the time of the most recent common ancestors (tMRCA) with 95 % highest posterior density (95 % HPD) of HCoV-HKU1 genotypes based on the spike gene (S1 domain) (897 bp). Data were analyzed under relaxed molecular clock with GTR + I substitution model and a constant size coalescent model implemented in BEAST. The Malaysian HCoV-HKU1isolates obtained in this study were color-coded and the HCoV-HKU1 genotypes (a) to (c) were indicated. The MCC posterior probability values were indicated on the nodes of each genotype
Evidence of possible recombination was observed in the S gene of novel lineage 1, involving genotypes B and C (Fig. 4). All isolates within novel lineage 1 showed similar recombination structures (representative isolates from Malaysia (12MYKL0208), Japan (Niigata.JPN/11-764), Thailand (CU-H967_2009) and China (892A/08) were shown). Similarly, sign of possible recombination was noticed within novel lineage 2 (Fig. 4). All Malaysian and Chinese isolates showed similar recombination structures in the S gene involving genotypes A and B (12MYKL0002, 12MYKL0760 and 12689/12 representative sequences were shown). Moreover, using the aforementioned putative parental and representative strains, MaxChi analysis of the novel lineages 1 and 2 isolates supported the hypothesis of recombination in the S gene (p < 0.05) (Additional file 9: Figure S9). Taken together, the emergence of novel lineage 1 and novel lineage 2 in these Asian countries was likely to be driven by natural recombination events.
Fig. 4.
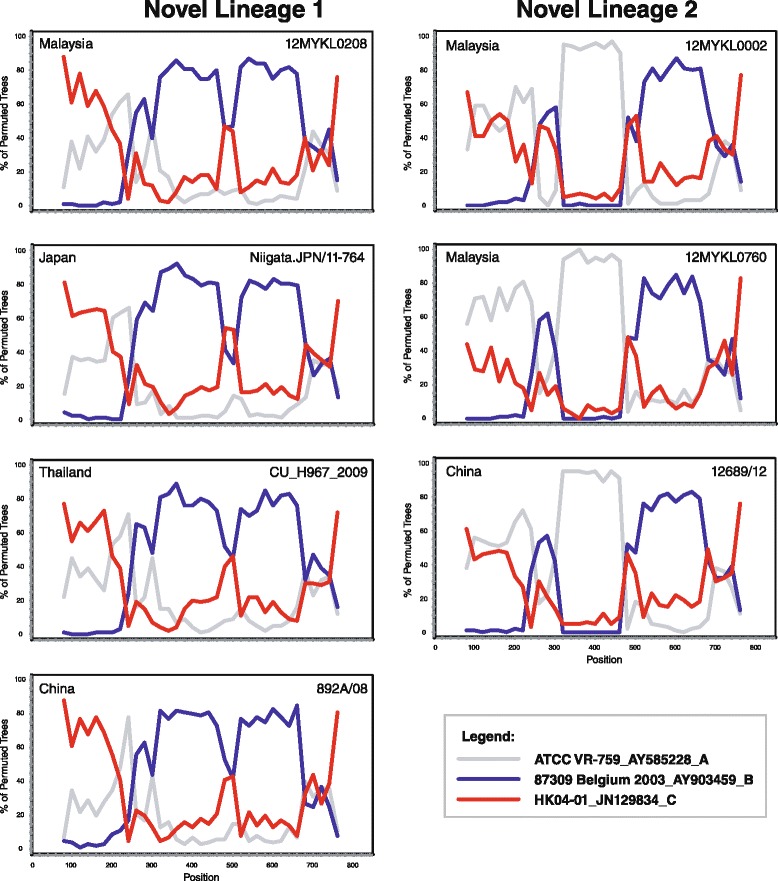
Recombination analyses of HCoV-OC43 novel lineages 1 and 2. Reference strains of HCoV-OC43 genotype A (ATCC VR-759), B (87309 Belgium 2003), and C (HK04-01) were used as the putative parental strains. The bootstrap values were plotted for a window of 160 bp moving in increments of 20 bp along the alignment. Samples 12MYKL0208, Niigata.JPN/11-764, CU-H967_2009, 892A/08 were used as representative sequences for novel lineage 1 in addition to 12MYKL0002, 12MYKL0760 and 12689/12 isolates as representatives for novel lineage 2
Estimation of divergence times
The divergence times of HCoV-OC43 and HCoV-HKU1 were estimated using the coalescent-based Bayesian relaxed molecular clock under the constant and exponential tree models (Fig. 2 and Fig. 3; Table 4). The newly estimated mean evolutionary rate for the S gene of HCoV-OC43 was 7.2 (5.0 – 9.3) × 10−4 substitutions/site/year. On the other hand, the evolutionary rate for the S gene of HCoV-HKU1 was newly estimated at 6.2 (4.2–7.8) × 10−4 substitutions/site/year. These estimates were comparable to previous findings of 6.1–6.7 × 10−4 substitutions/site/year for the S gene reported elsewhere [11].
Table 4.
Evolutionary characteristics of HCoV-OC43 and HCoV-HKU1 genotypes
| Subtype-gene evolutionary ratea | Genotype | tMRCAb |
|---|---|---|
| OC43-Spike 7.2 (5.2–9.4) | ||
| all genotypes | 1952.2 (1931.0–1965.2) | |
| genotype A | 1961.8 (1955.1–1966.0) | |
| genotype B | 1991.0 (1981.4–1999.0) | |
| genotype C/D | 2001.7 (2000.1–2002.9) | |
| genotype D | 2004.5 (2003.3–2005.8) | |
| genotype E | 2009.3 (2008.3–2010.0) | |
| novel lineage 1 | 2007.5 (2006.6–2008.0) | |
| novel lineage 2 | 2010.5 (2009.5–2011.4) | |
| HKU1-Spike 6.2 (4.5–8.0) | ||
| all genotypes | 1957.2 (1920.3–1987.5) | |
| genotype A | 1999.4 (1994.8–2002.5) | |
| genotype B | 2001.2 (1997.6–2003.6) | |
| genotype C | 2002.3 (1999.8–2003.8) | |
| HKU1-Nucleocapsid 4.3 (2.8–5.8) | ||
| all genotypes | 1962.0 (1915.1–1994.8) | |
| genotype A | 1986.8 (1970.8–1999.0) | |
| genotype B | 2002.2 (1999.4–2002.2) | |
| genotype C | 2002.3 (2000.1–2003.8) |
aEstimated mean rates of evolution expressed as 10−4 nucleotide substitutions/site/year under a relaxed molecular clock with GTR + I substitution model and an Exponential tree model. The 95 % highest posterior density (HPD) confidence intervals are included in parentheses
bMean time of the most common ancestor (tMRCA, in calendar year). The 95 % highest posterior density (HPD) confidence intervals are indicated
Based on these evolutionary estimates of the S gene, the common ancestor of HCoV-OC43 was dated back to the 1950s. Divergence time of genotype A was dated back to early 1960s, followed by genotype B around 1990s. Interestingly, genotypes C, D, E, and novel lineages 1 and 2 were all traced back to the 2000s (Fig. 2). Moreover, the common ancestor of HCoV-HKU1 was traced back to early 1950s, as estimated from the S gene. Subsequently, HCoV-HKU1 continued to diverge further into distinctive genotypes (A-C). Genotype A was dated to the late 1990 and genotypes B and C were both traced back to early 2000s (Fig. 3). Bayes factor analysis showed insignificant differences (Bayes factor <3.0) between the constant and exponential coalescent models of demographic analysis. Divergence times generated using the exponential tree model were slightly (but not significantly) different from those estimated using the constant coalescent model (Table 4). Of note, HCoV-OC43 and HCoV-HKU1 genotype assignments were less distinctive within the N and 1a genes (as compared to the S gene); these regions were therefore deemed unsuitable for divergence time estimations in this study.
Clinical symptoms assessment
The type of URTI symptoms (sneezing, nasal discharge, nasal congestion, headache, sore throat, hoarseness of voice, muscle ache and cough) and their severities during HCoV-OC43 and HCoV-HKU1 infections were analyzed. Fisher’s exact test analysis suggested that the severity of symptoms was not significantly associated with HCoV-OC43 and HCoV-HKU1 infections (p values > 0.05), this is due to the fact that the majority (61 % and 55 %) of the patients infected with HCoV-OC43 and HCoV-HKU1 respectively were presented with at least one respiratory symptom at moderate level of symptom severity. In addition, no significant association between HCoV-OC43 and HCoV-HKU1 genotypes with disease severity was observed.
Discussion
In the present cohort, over 2000 patients with URTI symptoms were recruited and screened, of whom 1.3 % (26/2060) and 1.1 % (22/2060) of the subjects were infected with HCoV-OC43 and HCoV-HKU1, respectively. These estimates corroborate with the previously reported average incidence of HCoV-OC43 and HCoV-HKU1 at 0.2–4.3 % and 0.3–4.4 %, respectively [12, 15, 40–45]. Although HCoV-OC43 and HCoV-HKU1 are not as common as other respiratory viruses, several studies have reported an elevated incidence of HCoV-OC43 (up to 67 %) due to sporadic outbreaks with fatality rate up to 8 % [46, 47]. This 12-month study showed that HCoV-OC43 and HCoV-HKU1 infections were frequently detected during March 2012 to September 2012 and decreased thereafter, in line with findings reported from other tropical Southeast Asian country [15]. However, such patterns differ from that in temperate areas where the prevalence peaks during winter seasons, but few or no detections in the summer [43]. It is also important to note that the study was performed in a relatively short duration, therefore limiting the epidemiological and disease trend comparison with reports from other countries.
Phylogenetic inference based on the S gene of HCoV-OC43 suggested the emergence of two potentially novel genotypes (designated as novel lineage 1 and novel lineage 2), supported by phylogenetic evidence and shared recombination structures. The relatively low mean intra-cluster genetic variation reflects the high intra-genotype genetic homogeneity of each novel lineage. Inter-genotype genetic distances between HCoV-OC43 genotypes further supported that the novel lineages 1 and 2 are distinct from the previously described genotypes [11, 17, 32] in which the genetic distances between each of these two genotypes and the others were notably high (up to 3.2 %) (Table 3). Phylogenetic analysis also revealed that novel lineage 1 includes isolates from Malaysia, Thailand, China and Japan while novel lineage 2 isolates are all from Malaysia and China. Spatiotemporal characteristic observed within the novel lineage 1 phylogeny (Fig. 2) may suggest the origin of this lineage in China, before it spread to other regions in the East and Southeast Asia. In order to clearly define the genetic characteristic of the putative novel lineages 1 and 2 (and also any other isolates with discordant phylogenetic patterns), complete genome sequencing and phylogenetic analysis need to be carried out.
Based on the newly estimated substitution rates, the divergence times for HCoV-OC43 and HCoV-HKU1 were phylogenetically inferred. Interestingly, although HCoV-OC43 was the first human coronavirus discovered in 1965 [48, 49], and the HCoV-HKU1 was first described much later in 2005 [50], the S gene analysis of HCoV-OC43 and HCoV-HKU1 revealed that the respective common ancestors of both viruses have emerged since 1950s. Furthermore, the divergence times of HCoV-OC43 genotypes predicted in this study are comparable to those described in previous studies [11, 27]. Phylogenetic, recombination and molecular clock analysis suggest the emergence of novel lineages 1 and 2 around the mid-2000s and late 2000s, respectively, probably by natural recombination events involving genotypes B and C (for lineage 1) and genotypes A and B (for lineage 2).
Human coronaviruses are progressively recognized as respiratory pathogens associated with an increasing range of clinical outcomes. Our results indicated that most patients infected with HCoV-OC43 and HCoV-HKU1 were presented with moderate respiratory symptoms (data not shown) in accordance with previously reported clinical results [16, 51–53] where they were recognized as common cold viruses associated with URTI symptoms.
Conclusions
In conclusion, epidemiological and evolutionary dynamics investigation revealed the genetic complexity of human betacoronaviruses HCoV-OC43 and HCoV-HKU1 infections in Malaysia, identifying two potentially novel HCoV-OC43 lineages among adults with acute respiratory tract infections. The reported findings warrant continuous molecular surveillance in the region, and detailed genotypic and phenotypic characterization of the novel betacoronavirus lineages.
Declarations
Ethics statement
The study was approved by the University of Malaya Medical Ethics Committee (MEC890.1). Standard, multilingual consent forms allowed by the Medical Ethics Committee were used. Written consents were obtained from all study participants.
Consent for publication
Not applicable.
Availability of data and materials
HCoV-OC43 and HCoV-HKU1 nucleotide sequences generated in the study are available in GenBank under the accession numbers KR055512-KR055644.
Acknowledgements
We would like to thank Nyoke Pin Wong, Nur Ezreen Syafina, Farhat A. Avin, Chor Yau Ooi, Sujarita Ramanujah, Nirmala K. Sambandam, Nagammai Thiagarajan and See Wie Teoh for assistance and support.
Funding
This work was supported by grants from the Ministry of Education, Malaysia: High Impact Research High Impact Research UM.C/625/1/HIR/MOE/CHAN/02/02 to KKT. The funders were not involved in study design or data collection and analysis.
Abbreviations
- GTR + I
general time-reversible nucleotide substitution model with invariant sites
- HCoV-HKU1
human coronavirus HKU1
- HCoV-OC43
human coronavirus OC43
- HPD
highest posterior density
- ICTV
International Committee for Taxonomy of Viruses
- MCC
maximum clade credibility
- MCMC
Markov chain Monte Carlo
- MERS-CoV
Middle East respiratory syndrome coronavirus
- NJ
neighbor joining
- RdRp
RNA dependent RNA polymerase
- SARS-CoV
severe acute respiratory syndrome coronavirus
- tMRCA
time of the most recent common ancestors
- URTI
upper respiratory tract infection
Additional files
Phylogenetic analysis of the HCoV-OC43 spike gene (S1 domain). Trees were reconstructed using neighbor-joining method. Bootstrap values were calculated from 1,000 trees. The scale bar of individual tree was indicated in substitutions per site, using Kimura 2-parameter model in MEGA (version 5.1) to estimate pair-wise evolutionary distance. The Malaysian isolates obtained in this study were color-coded and the HCoV-OC43 genotypes A to E as well as novel lineages 1 and 2 were indicated. (PDF 242 kb)
Phylogenetic analysis of the HCoV-OC43 nucleocapsid gene. Trees were reconstructed using neighbor-joining method. Bootstrap values were calculated from 1,000 trees. Bootstrap values of greater than 70 % were indicated on the branch nodes. The scale bar of individual tree was indicated in substitutions per site, using Kimura 2-parameter model in MEGA (version 5.1) to estimate pair-wise evolutionary distance. The Malaysian isolates obtained in this study were color-coded and the HCoV-OC43 genotypes A to E as well as novel lineages 1 and 2 were indicated. Each HCoV-OC43 sequence was assigned to its proper genotype based on the S1 phylogenetic analysis. NL1= novel lineage 1. (PDF 253 kb)
Phylogenetic analysis of the HCoV-OC43 1a gene (nsp3). Tree was reconstructed using neighbor-joining method. Bootstrap values were calculated from 1,000 trees. Bootstrap values of greater than 70% were indicated on the branch nodes. The scale bar of individual tree was indicated in substitutions per site, using Kimura 2-parameter model in MEGA (version 5.1) to estimate pair-wise evolutionary distance. The Malaysian isolates obtained in this study were color-coded. Each HCoV-OC43 sequence was assigned to its proper genotype based on the S1 phylogenetic analysis. (PDF 284 kb)
Phylogenetic analysis of the HCoV-OC43 complete and partial S gene. Trees were reconstructed using neighbor-joining method. Bootstrap values were calculated from 1,000 trees. Bootstrap values of greater than 70 % were indicated on the branch nodes. The scale bar of individual tree was indicated in substitutions per site, using Kimura 2-parameter model in MEGA (version 5.1) to estimate pair-wise evolutionary distance. NL1= novel lineage 1, NL2= novel lineage 2. (PDF 208 kb)
Phylogenetic analysis of the HCoV-OC43 1a (nsp3) and RdRp gene. Trees were reconstructed using neighbor-joining method. Bootstrap values were calculated from 1,000 trees. Bootstrap values of greater than 70 % were indicated on the branch nodes. The scale bar of individual tree was indicated in substitutions per site, using Kimura 2-parameter model in MEGA (version 5.1) to estimate pair-wise evolutionary distance. (PDF 119 kb)
Phylogenetic analysis of the HCoV-HKU1 spike gene (S1 domain). Tree was reconstructed using neighbor-joining method. Bootstrap values were calculated from 1,000 trees. Bootstrap values of greater than 70% were indicated on the branch nodes. The scale bar of individual tree was indicated in substitutions per site, using Kimura 2-parameter model in MEGA (version 5.1) to estimate pair-wise evolutionary distance. The Malaysian isolates obtained in this study were color-coded. (PDF 360 kb)
Phylogenetic analysis of the HCoV-HKU1 nucleocapsid gene. Tree was reconstructed using neighbor-joining method. Bootstrap values were calculated from 1,000 trees. Bootstrap values of greater than 70 % were indicated on the branch nodes. The scale bar of individual tree was indicated in substitutions per site, using Kimura 2-parameter model in MEGA (version 5.1) to estimate pair-wise evolutionary distance. The Malaysian isolates obtained in this study were color-coded. (PDF 347 kb)
Phylogenetic analysis of the HCoV-HKU1 1a gene (nsp3). Trees was reconstructed using neighbor-joining method. Bootstrap values were calculated from 1,000 trees. Bootstrap values of greater than 70% were indicated on the branch nodes. The scale bar of individual tree was indicated in substitutions per site, using Kimura 2-parameter model in MEGA (version 5.1) to estimate pair-wise evolutionary distance. The Malaysian isolates obtained in this study were color-coded. (PDF 133 kb)
Recombination analysis in HCoV-OC43 novel lineages 1 and 2. Analysis of the partial S gene was carried out using the MaxChi method in RDP. The x-axis gives the nucleotide positions of the alignment, whereas the y-axis presents the particular test statistics. Peaks in the log P of χ2 values in the MaxChi test marks potential points of recombination. Dashed lines represent p value cut-offs: uncorrected (lower line) and corrected for multiple comparisons (upper line) at the 0.05 level. (PDF 330 kb)
Footnotes
Competing interests
Co-author Kok Keng Tee is an Associate Editor for Virology Journal. This does not alter the authors’ adherence to all the Virology Journal policies on sharing data and materials.
Other authors do not have any competing interests in the manuscript.
Authors' contributions
Conceived and designed the experiments: MNA and KKT. Performed the experiments: MNA, KTN, XYO, and KKT. Analyzed the data: MNA, KTN, XYO, and KKT. Contributed reagents/materials/analysis tools: MNA, KTN, XYO, YKP, YT, JBC, NSH, AK, and KKT. Wrote the paper: MNA, KTN, and KKT. All authors read and approved the final version of the manuscript.
Contributor Information
Maryam Nabiel Al-Khannaq, Email: maryasala@gmail.com.
Kim Tien Ng, Email: kimtien@siswa.um.edu.my.
Xiang Yong Oong, Email: oxy123@siswa.um.edu.my.
Yong Kek Pang, Email: ykpang@ummc.edu.my.
Yutaka Takebe, Email: takebe@niid.go.jp.
Jack Bee Chook, Email: jackbee2002@um.edu.my.
Nik Sherina Hanafi, Email: sherina@ummc.edu.my.
Adeeba Kamarulzaman, Email: adeeba@ummc.edu.my.
Kok Keng Tee, Phone: +60-3-7967-7833, Email: k2tee@um.edu.my.
References
- 1.Arden KE, McErlean P, Nissen MD, Sloots TP, Mackay IM. Frequent detection of human rhinoviruses, paramyxoviruses, coronaviruses, and bocavirus during acute respiratory tract infections. J Med Virol. 2006;78(9):1232–40. doi: 10.1002/jmv.20689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lai MM, Cavanagh D. The molecular biology of coronaviruses. Adv Virus Res. 1997;48:01–00. doi: 10.1016/S0065-3527(08)60286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vijgen L, Keyaerts E, Moes E, Maes P, Duson G, Van Ranst M. Development of one-step, real-time, quantitative reverse transcriptase PCR assays for absolute quantitation of human coronaviruses OC43 and 229E. J Clin Microbiol. 2005;43(11):5452–6. doi: 10.1128/JCM.43.11.5452-5456.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Makino S, Keck JG, Stohlman SA, Lai M. High-frequency RNA recombination of murine coronaviruses. J Virol. 1986;57(3):729–37. doi: 10.1128/jvi.57.3.729-737.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brian DA, Spaan WJ. Recombination and coronavirus defective interfering RNAs. Seminars in Virology. Academic Press. 1997;8(2):101–11. doi: 10.1006/smvy.1997.0109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhong N. Management and prevention of SARS in China. Philos Trans Phys Sci Eng. 2004;359(1447):1115–6. doi: 10.1098/rstb.2004.1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peiris J, Guan Y, Yuen K. Severe acute respiratory syndrome. Nat Med. 2004;10:88–97. doi: 10.1038/nm1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Graham RL, Donaldson EF, Baric RS. A decade after SARS: strategies for controlling emerging coronaviruses. Nat Rev Microbiol. 2013;11(12):836–48. doi: 10.1038/nrmicro3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stavrinides J, Guttman DS. Mosaic evolution of the severe acute respiratory syndrome coronavirus. J Virol. 2004;78(1):76–82. doi: 10.1128/JVI.78.1.76-82.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Woo PC, Lau SK, Yip CC, Huang Y, Tsoi HW, Chan KH, et al. Comparative analysis of 22 coronavirus HKU1 genomes reveals a novel genotype and evidence of natural recombination in coronavirus HKU1. J Virol. 2006;80(14):7136–45. doi: 10.1128/JVI.00509-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lau SK, Lee P, Tsang AK, Yip CC, Tse H, Lee RA, et al. Molecular epidemiology of human coronavirus OC43 reveals evolution of different genotypes over time and recent emergence of a novel genotype due to natural recombination. J Virol. 2011;85(21):11325–37. doi: 10.1128/JVI.05512-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Esper F, Weibel C, Ferguson D, Landry ML, Kahn JS. Coronavirus HKU1 infection in the United States. Emerg Infect Dis. 2006;12(5):775–9. doi: 10.3201/eid1205.051316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Woo PC, Lau SK, Huang Y, Tsoi HW, Chan KH, Yuen KY. Phylogenetic and recombination analysis of coronavirus HKU1, a novel coronavirus from patients with pneumonia. Arch Virol. 2005;150(11):2299–11. doi: 10.1007/s00705-005-0573-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walsh EE, Shin JH, Falsey AR. Clinical impact of human coronaviruses 229E and OC43 infection in diverse adult populations. J Infect Dis. 2013;208(10):1634–42. doi: 10.1093/infdis/jit393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dare RK, Fry AM, Chittaganpitch M, Sawanpanyalert P, Olsen SJ, Erdman DD. Human coronavirus infections in rural Thailand: a comprehensive study using real-time reverse-transcription polymerase chain reaction assays. J Infect Dis. 2007;196(9):1321–8. doi: 10.1086/521308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pyrc K, Berkhout B, van der Hoek L. The novel human coronaviruses NL63 and HKU1. J Virol. 2007;81(7):3051–7. doi: 10.1128/JVI.01466-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu Q, Lu R, Peng K, Duan X, Wang Y, Zhao Y, et al. Prevalence and genetic diversity analysis of human coronavirus OC43 among adult patients with acute respiratory infections in Beijing, 2012. PLoS ONE. 2014;9(7):e100781. doi: 10.1371/journal.pone.0100781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jackson GG, Dowling HF, Spiesman IG, Boand AV. Transmission of the common cold to volunteers under controlled conditions. I. The common cold as a clinical entity. AMA Arch Intern Med. 1958;101(2):267–78. doi: 10.1001/archinte.1958.00260140099015. [DOI] [PubMed] [Google Scholar]
- 19.Turner RB, Wecker MT, Pohl G, Witek TJ, McNally E, St George R, et al. Efficacy of tremacamra, a soluble intercellular adhesion molecule 1, for experimental rhinovirus infection: a randomized clinical trial. JAMA. 1999;281(19):1797–04. doi: 10.1001/jama.281.19.1797. [DOI] [PubMed] [Google Scholar]
- 20.Yale SH, Liu K. Echinacea purpurea therapy for the treatment of the common cold: a randomized, double-blind, placebo-controlled clinical trial. Arch Intern Med. 2004;164(11):1237–41. doi: 10.1001/archinte.164.11.1237. [DOI] [PubMed] [Google Scholar]
- 21.Zitter JN, Mazonson PD, Miller DP, Hulley SB, Balmes JR. Aircraft cabin air recirculation and symptoms of the common cold. JAMA. 2002;288(4):483–6. doi: 10.1001/jama.288.4.483. [DOI] [PubMed] [Google Scholar]
- 22.Boom R, Sol C, Salimans M, Jansen C, Wertheim-van Dillen P, Van der Noordaa J. Rapid and simple method for purification of nucleic acids. J Clin Microbiol. 1990;28(3):495–03. doi: 10.1128/jcm.28.3.495-503.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chan KH, Yam WC, Pang CM, Chan KM, Lam SY, Lo KF, et al. Comparison of the NucliSens easyMAG and Qiagen BioRobot 9604 nucleic acid extraction systems for detection of RNA and DNA respiratory viruses in nasopharyngeal aspirate samples. J Clin Microbiol. 2008;46(7):2195–9. doi: 10.1128/JCM.00315-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jokela P, Piiparinen H, Mannonen L, Auvinen E, Lappalainen M. Performance of the Luminex xTAG Respiratory Viral Panel Fast in a clinical laboratory setting. J Virol Methods. 2012;182(2):82–6. doi: 10.1016/j.jviromet.2012.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.St-Jean JR, Jacomy H, Desforges M, Vabret A, Freymuth F, Talbot PJ. Human respiratory coronavirus OC43: genetic stability and neuroinvasion. J Virol. 2004;78(16):8824–34. doi: 10.1128/JVI.78.16.8824-8834.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vabret A, Dina J, Mourez T, Gouarin S, Petitjean J, van der Werf S, et al. Inter- and intra-variant genetic heterogeneity of human coronavirus OC43 strains in France. J Gen Virol. 2006;87(11):3349–53. doi: 10.1099/vir.0.82065-0. [DOI] [PubMed] [Google Scholar]
- 27.Vijgen L, Keyaerts E, Moes E, Thoelen I, Wollants E, Lemey P, et al. Complete genomic sequence of human coronavirus OC43: molecular clock analysis suggests a relatively recent zoonotic coronavirus transmission event. J Virol. 2005;79(3):1595–04. doi: 10.1128/JVI.79.3.1595-1604.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mounir S, Talbot PJ. Molecular characterization of the S protein gene of human coronavirus OC43. J Gen Virol. 1993;74:1981–81. doi: 10.1099/0022-1317-74-9-1981. [DOI] [PubMed] [Google Scholar]
- 29.Vijgen L, Keyaerts E, Lemey P, Moes E, Li S, Vandamme AM, et al. Circulation of genetically distinct contemporary human coronavirus OC43 strains. Virol J. 2005;337(1):85–92. doi: 10.1016/j.virol.2005.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kon M, Watanabe K, Tazawa T, Watanabe K, Tamura T, Tsukagoshi H, et al. Detection of human coronavirus NL63 and OC43 in children with acute respiratory infections in Niigata, Japan, between 2010 and 2011. Jpn J Infect Dis. 2012;65(3):270–2. doi: 10.7883/yoken.65.270. [DOI] [PubMed] [Google Scholar]
- 31.Suwannakarn K, Chieochansin T, Vichiwattana P, Korkong S, Theamboonlers A, Poovorawan Y. Prevalence and genetic characterization of human coronaviruses in southern thailand from july 2009 to january 2011. Southeast Asian J Trop Med Public Health. 2014;45(2):326–36. [PubMed] [Google Scholar]
- 32.Zhang Y, Li J, Xiao Y, Zhang J, Wang Y, Chen L, et al. Genotype shift in human coronavirus OC43 and emergence of a novel genotype by natural recombination. J Infect. 2014;70:641–50. doi: 10.1016/j.jinf.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214–9. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lau SK, Li KS, Huang Y, Shek C-T, Tse H, Wang M, et al. Ecoepidemiology and complete genome comparison of different strains of severe acute respiratory syndrome-related Rhinolophus bat coronavirus in China reveal bats as a reservoir for acute, self-limiting infection that allows recombination events. J Virol. 2010;84(6):2808–19. doi: 10.1128/JVI.02219-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28(10):2731–9. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vijgen L, Keyaerts E, Lemey P, Maes P, Van Reeth K, Nauwynck H, et al. Evolutionary history of the closely related group 2 coronaviruses: Porcine hemagglutinating encephalomyelitis virus, bovine coronavirus, and human coronavirus OC43. J Virol. 2006;80(14):7270–4. doi: 10.1128/JVI.02675-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smith JM. Analyzing the mosaic structure of genes. J Mol Evol. 1992;34(2):126–9. doi: 10.1007/BF00182389. [DOI] [PubMed] [Google Scholar]
- 38.Martin DP, Murrell B, Golden M, Khoosal A, Muhire B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015;1(1):vev003. doi: 10.1093/ve/vev003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martin D, Rybicki E. RDP: detection of recombination amongst aligned sequences. J Bioinform. 2000;16(6):562–3. doi: 10.1093/bioinformatics/16.6.562. [DOI] [PubMed] [Google Scholar]
- 40.Lau SK, Woo PC, Yip CC, Tse H, Tsoi HW, Cheng VC, et al. Coronavirus HKU1 and other coronavirus infections in Hong Kong. J Clin Microbiol. 2006;44(6):2063–71. doi: 10.1128/JCM.02614-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.S-d T, Salez N, Benkouiten S, Badiaga S, Charrel R, Brouqui P. Respiratory viruses within homeless shelters in Marseille, France. BMC Res Notes. 2014;7(1):81–9. doi: 10.1186/1756-0500-7-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Garbino J, Crespo S, Aubert J-D, Rochat T, Ninet B, Deffernez C, et al. A prospective hospital-based study of the clinical impact of non–severe acute respiratory syndrome (non-SARS)–related human coronavirus infection. Clin Infect Dis. 2006;43(8):1009–15. doi: 10.1086/507898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Furuse Y, Suzuki A, Kishi M, Galang HO, Lupisan SP, Olveda RM, et al. Detection of novel respiratory viruses from influenza-like illness in the Philippines. J Med Virol. 2010;82(6):1071–4. doi: 10.1002/jmv.21763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee WJ, Chung YS, Yoon HS, Kang C, Kim K. Prevalence and molecular epidemiology of human coronavirus HKU1 in patients with acute respiratory illness. J Med Virol. 2013;85(2):309–14. doi: 10.1002/jmv.23465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dominguez SR, Shrivastava S, Berglund A, Qian Z, Goes LG, Halpin RA, et al. Isolation, propagation, genome analysis and epidemiology of HKU1 betacoronaviruses. J Gen Virol. 2014;95(4):836–48. doi: 10.1099/vir.0.059832-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vabret A, Mourez T, Gouarin S, Petitjean J, Freymuth F. An outbreak of coronavirus OC43 respiratory infection in Normandy, France. Clin Infect Dis. 2003;36(8):985–9. doi: 10.1086/374222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Patrick DM, Petric M, Skowronski DM, Guasparini R, Booth TF, Krajden M, et al. An outbreak of human coronavirus OC43 infection and serological cross-reactivity with SARS Coronavirus. Can J Infect Dis Med Microbiol. 2006;17(6):330–6. doi: 10.1155/2006/152612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tyrrell D, Bynoe M. Cultivation of viruses from a high proportion of patients with colds. Lancet. 1966;287(7428):76–7. doi: 10.1016/S0140-6736(66)92364-6. [DOI] [PubMed] [Google Scholar]
- 49.Kahn JS, McIntosh K. History and recent advances in coronavirus discovery. J Pediatric Infect Dis Soc. 2005;24(11):223–7. doi: 10.1097/01.inf.0000188166.17324.60. [DOI] [PubMed] [Google Scholar]
- 50.Woo PC, Lau SK, Chu CM, Chan KH, Tsoi HW, Huang Y, et al. Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J Virol. 2005;79(2):884–95. doi: 10.1128/JVI.79.2.884-895.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lu R, Yu X, Wang W, Duan X, Zhang L, Zhou W, et al. Characterization of human coronavirus etiology in Chinese adults with acute upper respiratory tract infection by real-time RT-PCR assays. PLoS ONE. 2012;7(6):e38638. doi: 10.1371/journal.pone.0038638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.van Elden LJ, van Alphen F, Hendriksen KA, Hoepelman AI, van Kraaij MG, Oosterheert J-J, et al. Frequent detection of human coronaviruses in clinical specimens from patients with respiratory tract infection by use of a novel real-time reverse-transcriptase polymerase chain reaction. J Infect Dis. 2004;189(4):652–7. doi: 10.1086/381207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gerna G, Percivalle E, Sarasini A, Campanini G, Piralla A, Rovida F, et al. Human respiratory coronavirus HKU1 versus other coronavirus infections in Italian hospitalised patients. J Clin Virol. 2007;38(3):244–50. doi: 10.1016/j.jcv.2006.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
HCoV-OC43 and HCoV-HKU1 nucleotide sequences generated in the study are available in GenBank under the accession numbers KR055512-KR055644.