

Abstract
In the present work the adsorption of doxorubicin (DOX) on the surface of single-walled carbon nanotube (SWCNT) as well as its encapsulation in SWCNT, and their dependence on the protonation of NH2 group of DOX, solvent, and the diameter of armchair (n,n) SWCNT were systematically investigated using theoretical methods such as PM6-DH2 and M06-2X in the scheme of OMIOM. It was found that the two loadings, adsorption on the sidewall of CNT and the encapsulation in CNT, have distinct solvent, protonation and diameter dependences. The encapsulation is much stronger than the adsorption of DOX on the sidewall of CNT, and the former also has significantly higher solvent and protonation effects than the latter. The adsorption primarily occurs through π-π stacking and just becomes slightly stronger as the diameter of CNT increases, while besides π-π stacking the additional C-H/N-H/O-H…π and C=O…π also contribute to the encapsulation of DOX in CNT. It seems that (8,8) CNT (diameter ~ 11Å) energetically is an onset for the encapsulation since the encapsulation turns from endothermic to exothermic as the diameter is larger than approximately 11 Å, and the optimal diameter for the encapsulation is 14Å corresponding to (10,10) CNT. Thus for the thick CNT the encapsulation may also play an important role in the loading and releasing for the CNT-based drug delivery system of the DOX.
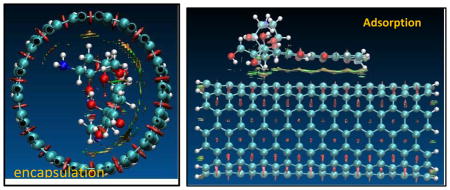
Introduction
As one of the most promising applications of carbon nanotubes (CNT), the tumor-targeted drug delivery systems on the basis of CNT have been put much effort from both experimentalists and theoreticians.1–18 The potential drug delivery systems are superior to free drug in a few aspects, such as longer circulation time, higher drug uptake and selectivity, lower dosage, and better therapeutic efficacy thereof. Doxorubicin (DOX, adriamycin, C27H29NO11) is commonly used in chemotherapy, for example, for the treatment of Kaposi’s sarcoma, ovarian carcinoma, lung, and breast cancer.19 DOX shows chemotherapeutic activity through intercalating between adjacent DNA bases pairs and preventing from replicating by causing conformational changes in the DNA molecule. However, DOX is also highly toxic especially to hearts and kidneys, limiting its therapeutic applications. Novel drug-delivery strategies for DOX are thus essential for its more effective and wide application.
For the purpose Dai et al suggested a single-walled CNT (SWCNT)-based drug delivery system for DOX,4, 6, 20–22 consisting of SWCNT, phospholipids (PL)-poly (ethylene glycol) (PEG) chain, and a cyclic tripeptide (arginine-glycine-aspartic acid, RGD). The PL-PEG functionalizes the SWCNT through noncovalent interaction between the PL chain and SWCNT, PEG pointing to aqueous solutions, while DOX can be ultra-highly loaded to the SWCNT system through its adsorption on the sidewall of SWCNT due to the high degree of π-stacking. It was estimated that the surface area of SWCNT is covered by the PL-PEG (two hydrocarbon chains of PL) and DOX in percentages of 10% and 75%, respectively. In this conjugate system (DOX-SWCNT-PL-PEG-RGD), the RGD imparts a recognition ligand for the receptors (integrin αvβ3 up-regulated in a wide range of solid tumors). The targeting moiety plays very important roles in enhancing cellular uptake of DOX, and accordingly toxicity of DOX could be reduced to normal cells as compared with free DOX.
It is very amazing that above DDS conjugates has a strong pH dependence (enhanced at high pH and reduced at low pH).4, 6 DOX remains binding to the SWCNTs at pH>7, yet readily release in acidic solution. This was mainly attributed to protonation of NH2 group of DOX at low pH values, which enhances hydrophilicity and thereof solubility of DOX. This feature has a very important implication to regulate drug loading and releasing from the SWCNT-based DDS since physiological environment is weak basic (pH=7.4) while intracellular lysosomes are acidic, enabling high DOX loading extracellularly and readily releasing intracellular of tumors. The spontaneous encapsulation of biomolecules such as single strand DNA,23–26 protein,27–29 and anticancer drug molecule 30 was well established. Although SWCNTs have a great potential to encapsulate DOX into its cavity, the relevant experimental studies 4, 6 attributed the loading to the adsorption on the surface of SWCNT through π-π stacking. The encapsulation may be achieved through cooperative π-π stacking and X-H···π (X=N and C), and may be even stronger than the surface adsorption, implying that DOX releases slowly and it actually may be important for the DDS since slow release is critically significant in drug delivery for minimizing drug lost before reaching targets and increasing circulation time.
In the present work, the adsorption on the outer surface of SWCNT (refer to the adsorption on the sidewall of CNT hereafter, DOX-CNT) as well as the encapsulation of DOX in CNT (DOX@CNT), and their dependences on the protonation of the NH2 group in DOX and diameter of armchair (n,n) SWCNT were systematically investigated with a series of theoretical methods such as PM6-DH2, and M06-2X in the scheme of OMIOM. It was found that the two loadings, on the sidewall and the encapsulation, have distinct solvent, protonation and diameter effects. (8,8) CNT may be an onset for the encapsulation since the encapsulation changes from endothermic to exothermic as the diameter is larger than approximately 11 Å, and the optimal diameter for the encapsulation is 14Å corresponding to (10,10) CNT. The encapsulation may play an important role in the loading and releasing for the CNT-based drug delivery system of the DOX.
Model and Theoretical Methods
It is well known that, in spite of great improvement over LDA, a typical GGA type of density functional theory (DFT) like the PW91 and PBE methods still greatly underestimate the binding for the systems mainly arising from weak noncovalent interaction. Hybrid meta-GGA functionals, M05-2X and M06-2x, developed by Truhlar et al 31 have been well applied to the small typical π···π stacking systems, e.g., benzene and adenine dimers, 32 and the π···π stacking systems of DNA bases and carbon nanomaterials.33–36 Recently DFT-D methods were widely used to deal with the weak systems by directly supplementing the DFT methods with van der Waals term.37, 38 However, the M06-2X as well as other dispersion corrected DFT (DFT-D) are hard to directly apply to the entire system for the SWCNT-DOX because of huge computational demanding.
It was reported that the dispersion as well as hydrogen bonds corrected PM6 (referred to as PM6-DH2)39, 40 is able to yield the most accurate results for non-covalent interactions of all the semiempirical quantum mechanical methods.41 The PM6-DH2 predicted binding energies for the non-covalent interaction between DNA bases and C60/Li@C60 agree well within 0.5 kcal/mol with those from the DFT-D method.35, 36, 42 Because of significant low computational demand, PM6-DH2 may be applied to macromolecular systems for describing H-bond and non-covalent interactions; however, it needs to be carefully validated before extensive applications. Thus, in the present work the PM6-DH2 implemented in MOPAC2009 package 43 was firstly validated for the adsorptions of the DOX on a few SWCNTs by comparing with higher level methods such as the M06-2X and DFT-D in the scheme of ONIOM44. To include solvent effect in the PM6-DH2 method, conductor-like screening model (COSMO) approach is applied.45
To validate the PM6-DH2 method for the present systems, in the present work the two layer ONIOM (M06-2X/6-31G*:UFF) was performed for the binding of the DOX on the sidewall of three CNTs (7, 7), (10, 10), and (13, 13). In the ONIOM scheme the higher level model includes DOX/DOXH+ and twenty six-member rings of the CNT just below the DOX, and the rest was treated with the lower level. To get the preliminary interaction configurations between DOX and CNT, the docking with the PATCHDOCK program 46 was employed through calculating the non-bonded terms of the molecular mechanic (MM) force field. However, for the investigated CNTs the procedure only resulted in those for the adsorption of DOX on the sidewall of CNT. Thus, the initial configurations for the encapsulation was generated through manually inserting DOX into the cavity of CNT with GaussView 5, followed by the clean to roughly adjust the distance between DOX and CNT.
Results and Discussions
A conspicuous feature of doxorubicin
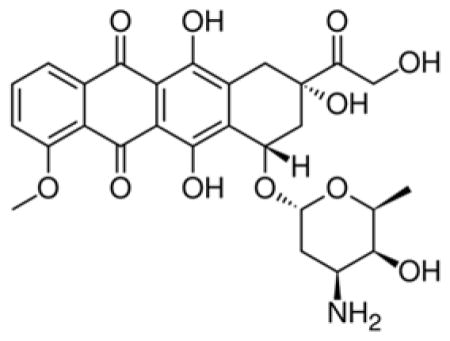
has three aromatic hydroxyanthraquinonic rings, and a grand π orbital thus delocalizes over the planar aromatic rings. A few intramolecular hydrogen bonds through O···H-O stabilize its conformation. The tetrahydropyran ring takes a chair configuration, where the hydroxyl and NH2 groups are in staggered positions. Figure 1 displays the frontier molecular orbitals of DOX and DOXH+ (protonated doxorubicin). The highest occupied molecular orbital (HOMO) of DOX delocalizes with prominent contributions from the three aromatic rings as well as the NH2 group, while for the HOMO of DOXH+ the contribution mainly comes from the three aromatic rings and the protonated NH2 group does not have clear contribution. However, the LUMO of the DOX and DOXH+ are rather similar, mainly centering over the aromatic rings.
Fig. 1.

Structures, HOMO (2nd row), and LUMO (3rd row) of doxorubicin (DOX), the protonated DOX (DOXH+), and (10,10) CNT at M06-2X/6-31G* level (the isosurface= 0.01au).
The armchair SWCNTs (n,n) (n=7–14) with a length of 24.45Å and diameter in a range of 9–20Å were used in this investigation. Both ends of CNT are saturated with hydrogen atoms to avoid the suspending carbon bonds. Because of the sp2-hybridized carbon atoms constituting CNT, a grand delocalized π frontier molecular orbital spreads over the inside and outside wall surfaces of the CNT. Comparing with the frontier orbitals of the DOX, HOMO of the CNT matches well with the LUMO of DOX when the plane of the three aromatic rings of DOX approaches the surface of CNT in parallel along with the central axis of CNT, while the HOMO of DOX does not match the LUMO of CNT. The former gap (2.50eV) for DOX-(10,10) CNT is also significantly smaller than the latter one (3.95eV). This implies that they may weakly interact with each other through the HOMO of CNT and the LUMO of DOX when both planes approach mutually. Table 1 lists the energy levels of the frontier molecular orbitals of DOX, DOXH+, CNT (n,n) (n=7, 9, 10, and 12), and the complexes for DOX, DOXH+ with (10,10) CNT.
Table 1.
The HOMO (au), LUMO (au) and gap(eV) for DOX, DOXH+ and CNTs at M06-2X/6-31G(d)
| HOMO | LUMO | gap | |
|---|---|---|---|
| DOX | −0.2658 | −0.0794 | 5.07 |
| DOXH+ | −0.3749 | −0.1896 | 5.04 |
| (7,7) | −0.1772 | −0.1124 | 1.76 |
| (9,9) | −0.1740 | −0.1176 | 1.54 |
| (10,10) | −0.1716 | −0.1205 | 1.39 |
| (12,12) | −0.1695 | −0.1230 | 1.26 |
| (13,13) | −0.1687 | −0.1240 | 1.22 |
| DOX-(10,10)* | −0.1721 | −0.1208 | 1.39 |
| DOX @(10,10)** | −0.1716 | −0.1200 | 1.40 |
| DOXH+-(10,10)* | −0.2074 | −0.1565 | 1.38 |
adsorption on the surface;
encapsulation
Figure 2 displays the orbital energy changes of both LUMOs and HOMOs with respect to the diameters of (n,n) (n=7–14) CNT at the M06-02X/6-31G* level. It can be seen that the energies of HOMOs of CNT are in the range of −4.82 – −4.59 eV and that of LUMOs of CNT in the range of −3.06 – −3.38eV. The former goes up slightly with increasing the diameter and the latter gently goes down. In spite of lower variation the HOMO and LUMO from PM6-DH2 for the CNTs support this trend.
Fig. 2.
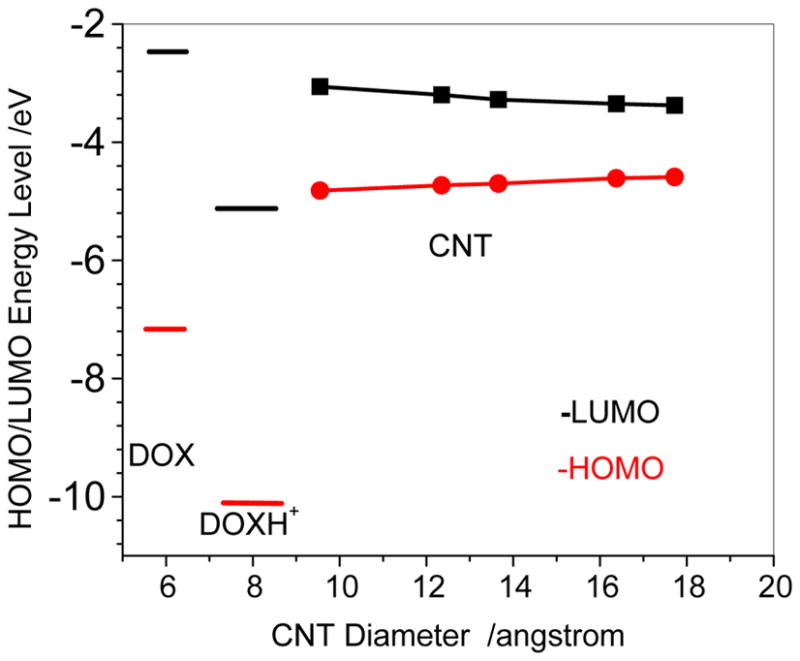
LUMOs and HOMOs of doxorubicin (DOX), protonated DOX (DOXH+), and carbon nanotubes (CNT) at M06-2X/6-31G* level.
Two types of DOX loading to CNT, adsorption on the outer sidewall and encapsulation in the CNT, were thoroughly located. To ensure the minimized structures for the current large systems, a few configurations were explored by translating the doxorubicin along the axis of the CNT from left to right for the encapsulation, and wrapping the doxorubicin in a few places for the adsorption on the surface of CNT. The binding energy was calculated for the most stable structure for the given configuration. According to Figure 3 in spite of rather low isosurface (0.01au) both the HOMO and the LUMO for the complexes through the adsorption on the sidewall dominantly come from the CNT. Their energy levels in Table 1 are only slightly different than those of the CNT. Although the interaction between the DOX and CNT is hard to be reflected by the HOMO and LUMO, the π-π stacking can be obviously visualized via the isosurface of sign(λ2)×ρ defined by Yang et al, 47 which will be discussed below. Similar to the other complexes via π-π stacking, in the complex of Figure 3 the three conjugated six-member rings are parallel to CNT, and they sit above the CNT 6-member ring in displaced configuration in a vertical distance of approximately 3.3Å.
Fig. 3.

The HOMO (left, isosurface=0.01au) and LUMO (right) for the adsorption between DOX and (10,10) CNT at M06-2X/6-31G* level.
Figure 4 shows the binding energy of DOX-loaded on the sidewall of CNT at the PM6-DH2 level for the most stable loading of the given CNT. In the gas phase, with the diameter increasing from CNT (7,7) to CNT(14,14) the binding energy changes from -33.2 kcal/mol to −40.0 kcal/mol, which qualitatively agrees with the trend of π-stacking between aromatic bio-molecules and SWCNT.33, 34 The weaker adsorption on the thinner CNT may be mainly attributed to the bigger curvature of the nanotube, which leads to less contact of the outer surface of CNT with the three aromatic rings of DOX. With the diameter increasing higher degree of contact between CNT and DOX strengthens the adsorption. The similar trend also appears on the adsorption in aqueous solution. In the aqueous solution the adsorption energies change from −27.9 to −32.6 kcal/mol with increasing the diameter of CNT. Due to the solvation of DOX, the binding strength of DOX on CNTs is weakened by 5.3–7.4 kcal/mol as compared with that in the gas phase. However, it can be found that the protonation effect on the binding is predicted to be much less than the solvent effect. In Figure 4, in the aqueous solution the bindings of DOXH+ on the CNT are only slightly weaker by approximately 1.0 kcal/mol than those of DOX on the CNT until (10,10), and the bindings of the DOX and DOXH+ on the larger diameter CNT are then rather close. The result implies that it is hard to attribute the strong pH dependence for the releasing of DOX from the CNT to the loading on the surface of CNT.
Fig. 4.
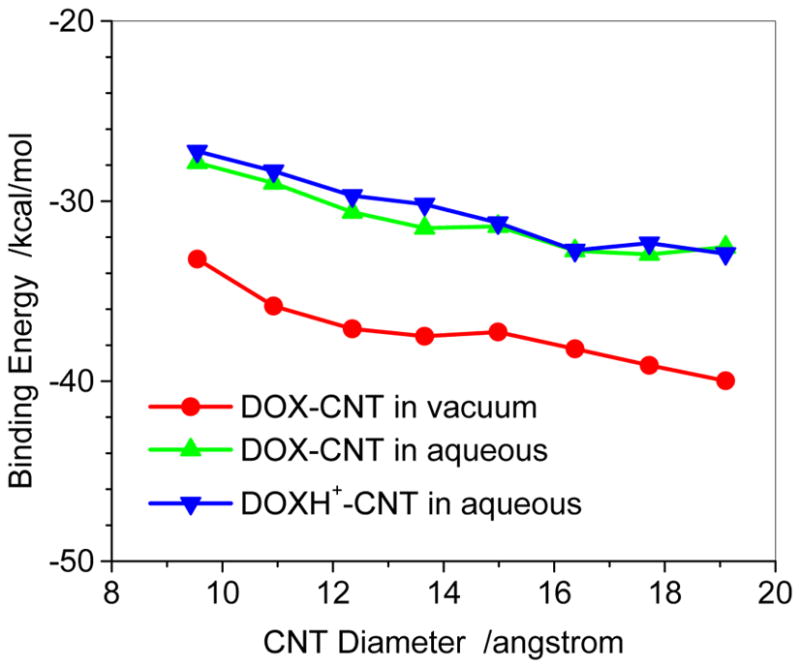
Binding energies of DOX-loaded on outer surface of CNT with respect to CNT diameter with PM6-DH2method.
The binding energy of the DOX with the sidewall of three CNTs (7, 7), (10, 10), and (13, 13) predicted with the ONIOM (M06-2X/6-31G*:UFF) method was plotted in Figure 5. In the ONIOM scheme the higher level model includes DOX/DOXH+ and twenty six-member rings of the CNT. Figure 5 also shows that the binding decreases with increasing the diameter. This is the same trend as predicted with the PM6-DH2 level although the absolute binding energies for the DOX in the gas and the aqueous solution are approximately 8 and 4 kcal/mol, respectively, less than the corresponding values at PM6-DH2 level. At M06-2X/6-31G* level the binding energies of the DOXH+ on CNTs (7,7), (10,10) and (13,13) in the aqueous solution are predicted to be −20.4, −24.8, and −25.6 kcal/mol, respectively, which reasonably agrees with those predicted by the PM6-DH2, slightly weaker than those of DOX-CNT in aqueous solutions by 3.3, 1.5, and 3.0 kcal/mol, respectively, implying that the protonation of DOX can enhance somewhat the release of DOX from the adsorption situation. The PM6-DH2 method somewhat underestimates the inductive action of –NH3+ on the three aromatic rings. However, because of the binding trend consistency predicted by the two theoretic methods, the semi-empirical PM6-DH2 may provide reasonable geometry and binding energy especially for such large system as DOX-CNT after the dispersion force was explicitly introduced.
Fig. 5.
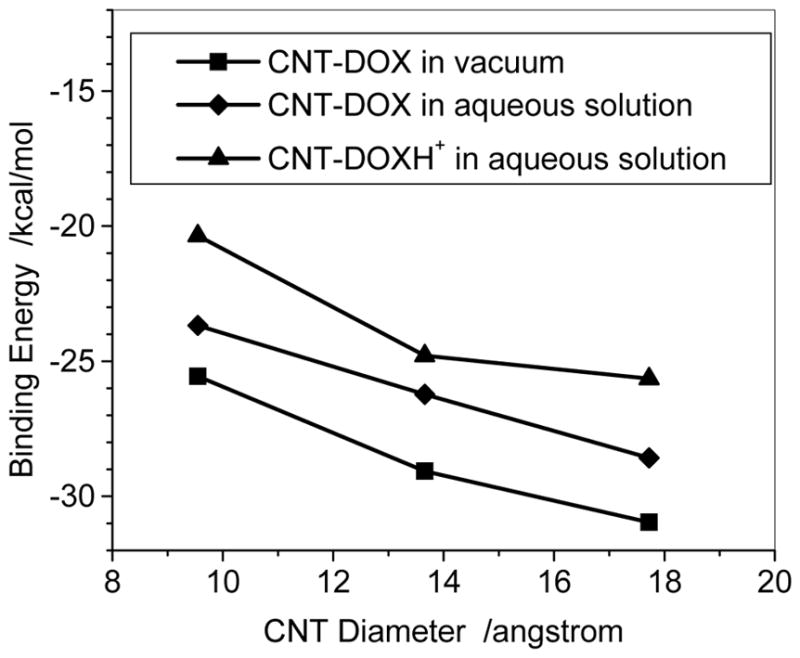
Binding energies of DOX-loaded on outer surface of CNT with respect to CNT diameters, calculated at the ONIOM(M06-2X/6-31G*:UFF) level.
Figure 6 shows the encapsulation of the DOX in the (n,n) SWCNTs (n=7, 8, 9 and 11). The encapsulation of DOX in (7,7) leads to a significant deformation of the CNT (Figure 6a), accompanied by a rather high deformation energy of approximately 120kcal/mol (Figure 7) and positive binding energies (46.5 and 76.2 kcal/mol in the respective gas phase and aqueous solution) for the DOX @(7,7) CNT. The encapsulation of DOX in (7,7) CNT is endothermic because of the approximate volume of DOX 14.9×10.8×6.4 Å3 in which the width of 10.8Å is even larger than the diameter of 9.5 Å of (7,7) CNT. This spatial effect is weakened rapidly with an increase of CNT diameter. For the (8,8) CNT its binding becomes exothermic due to a considerable decrease of the deformation energy upon the encapsulation, as shown below in Figure 8. It is apparent that the spatial effect disappears for the (9,9) CNT (diameter, 12.4 Å) and thicker CNTs with only a few kcal/mol of the deformation energy that is mainly resulted from the noncovalent interaction between the DOX and the thicker CNT. Thus, the (8,8) CNT with a diameter of 10.9Å can be considered as an onset for the encapsulation loading of the DOX with respect to the exothermic binding.
Fig. 6.

The encapsulation of DOX@(n,n) CNT (n=7,8,9 and 11) predicted by PM6-DH2.
Fig. 7.
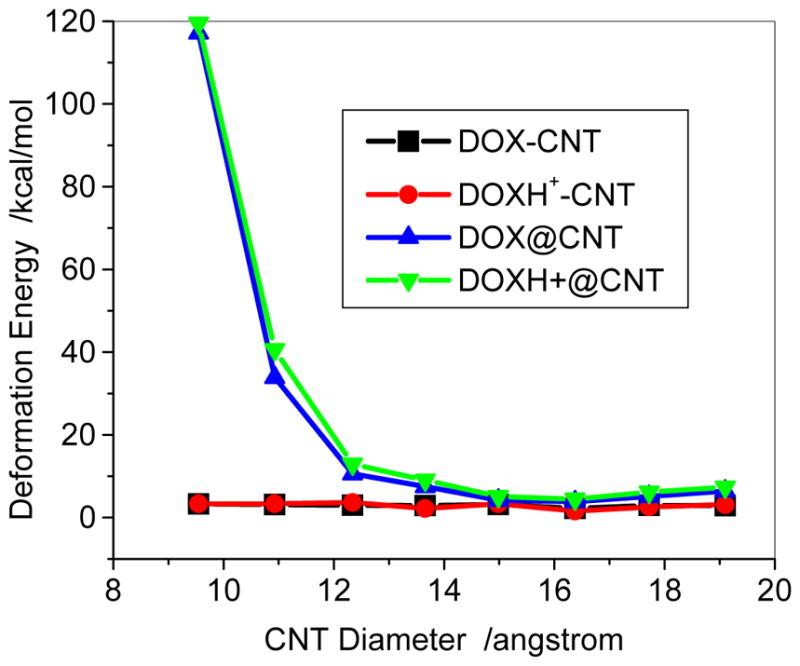
Deformation energy of DOX-CNT and DOX@CNT complexes with respect to diameters predicted with PH6-DH2. The deformation energy is the difference of the heat of formation between DOX/CNT of DOX@CNT and the individually optimized DOX/CNT, =ΔHf (DOX in the complex, DOX-CNT or DOX@CNT)+ΔHf (CNT in the complex)−ΔHf(DOX)−ΔHf(CNT).
Fig. 8.
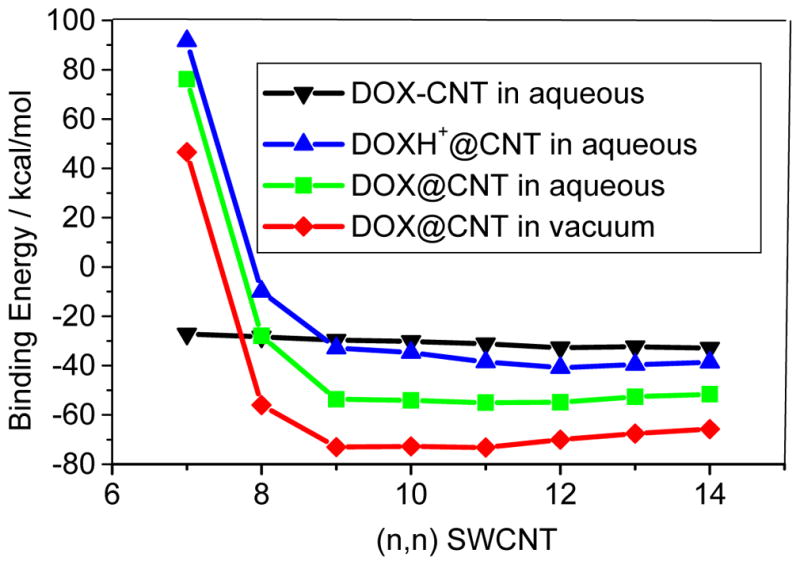
Binding energies of DOX-loaded on inner surface of CNT (encapsulation, DOX@CNT and DOXH+@CNT) with respect to CNT diameters with PM6-DH2.
The deformation energies of the SWCNT were sown in Figure 7 for both DOX-CNT and DOXH+-CNT in aqueous solutions. It can be seen that for the sidewall adsorption the deformation energies of both DOX-CNT and DOXH+-CNT are only approximately 3kcal/mol. The sidewall adsorption only slightly changes the geometric conformation because of the absence of spatial limitation. However, for the encapsulation there are quite high deformation energies for both DOX and DOXH+ loaded on the cavity of CNTs (7, 7) and (8,8) due to the considerably spatial limitation. In the cases of encapsulation, not only DOX is deformed considerably (deformation energy: 87 and 26 kcal/mol for DOX@(7,7) and DOX@(8,8)) but also the CNT (30 and 7.2 kcal/mol) is changed seriously from circle into ellipse. However, with increasing the diameter of CNT the deformation of CNT decreases gradually and the deformation energies for DOX and DOXH+@CNT (12,12) are 3.8 and 4.5 kcal/mol, respectively, which are slightly greater than that of the corresponding adsorption.
Figure 8 displays the binding energy of DOX-loaded in the cavity of the CNT, i.e., encapsulation at the PM6-DH2 level. For the smaller diameter CNTs than the (8,8) CNT the drug loading dominantly occur through the sidewall adsorption because of their endothermic encapsulation. It is plausible for the encapsulation of the DOX to the (9,9) and thicker CNTs. From Figure 8 the binding energies for the DOX encapsulation in (n,n) CNTs (n=9–14) are −65.8 ~ −73.0 kcal/mol in vacuum phases, and drop to −51.6 ~ −53.7 kcal/mol in aqueous solutions. The solvent effects (~15–20 kcal/mol) for DOX@CNT are higher than those of DOX-CNT, where solvent effects are only a few kcal/mol as shown in Figure 4. For (8,8) CNT, the binding strengths of the encapsulation and sidewall adsorption in aqueous solution are rather close, −28kcal/mol, while for the thicker CNTs the former becomes much stronger than the latter by approximately 20 kcal/mol. The encapsulation with the strong binding to the CNT may prolong the circulation time of the drug, which is actually desired for designing novel drug delivery system. In addition, the encapsulation of the DOX varies differently with the diameter of the CNT than the adsorption on the sidewall of the CNT. The encapsulation becomes stronger until (10,10) CNT in the vacuum and then tends to be slightly weaker for the thicker CNT. This is due to the further contact of the DOX with the CNTs and van der Waals consequently becomes less for the thicker CNTs, which will be discussed in detail below. The results indicate that the encapsulation of DOX in CNT becomes exothermic as the diameter is larger than approximately 11Å, and the optimal diameter for the encapsulation is 14Å.
According to Figure 8, the binding energies for the encapsulations of the protonated DOX (DOXH+) in (n,n) CNT (n=9–14) in aqueous solutions (the blue line) are predicted to be in a range of −32.9 – −38.7 kcal/mol, 13–20 kcal/mol weaker than those of the DOX (the green line). This distinct binding between the DOX and DOXH+ has very important implication to the cancer-targeted drug delivery system, suggesting that the DOX loading in the CNT conjugates may have a strong pH dependence, enhanced at high pH and reduced at low pH. The DOX remains strong binding to the SWCNTs at pH>7, yet the protonated DOXH+ readily releases in acidic solution. This was mainly attributed to the protonation of NH2 group of DOX at low pH values, which enhances hydrophilicity and thereof solubility of the DOX. Due to more significant decrease of the binding energy, the protonated-DOX-CNT complex is more readily to dissociate in acidic circumstance. This feature has a very important implication to regulate drug loading and releasing from the SWCNT-based DDS since physiological environment is weak basic (pH=7.4) while intracellular lysoomes are acidic, enabling high DOX loading extracellularly and readily releasing intracellular of tumors.
Compared with the adsorption on the sidewall of the CNT, the encapsulation of the DOX is much more sensitive to the protonation that decreases its binding with the CNT by 13–20 kcal/mol. However, for the former the protonation only slightly weaken its binding with the CNT as shown in Figure 4. Thus, the encapsulation of the DOX may also play an important role in the loading and releasing of the DDS. Even for the strong encapsulation, the frontier molecular orbitals (HOMO and LUMO, not shown) still dominantly come from the CNT.
To understand the binding difference for the adsorption and the encapsulation, the noncovalent interactions for the encapsulation in and the adsorption on the surface of (9,9) CNT were visualized via the isosurface of sign(λ2)×ρ defined by Yang et al.47 According to the color code that green/yellow shows van der Waals (vdw) interaction and red strong nonbonded overlap, vdw interaction for the encapsulation is very obvious via cooperative π···π, N-H···π as well as C-H···π, while for the adsorption on sidewall the vdw comes mainly from the π···π stacking. Inside the CNT the NH2 and a few CH2 groups are approximately 3.0–3.5 Å from the inner wall carbon of the CNT, and the vdw between the NH2/CH2/C=O groups and the SWCNT enhances their binding with N-H···π, C-H···π, OH···π and C=O···π in a few areas.
The vdw contribution (EVDW) was also quantitatively estimated for the DOX-CNT complexes in the gas phase through the binding energy difference, EVDW=ΔE(PM6−DH2)−ΔE(PM6), which was shown in Figure 10. For the adsorption of DOX on the sidewall of the CNTs the van der Waals contribution (EVDW) is basically in a short range of −27.8 – −33.6 kcal/mol; while for the encapsulation EVDW becomes rather high to −106 kcal/mol for the small diameter (7,7) and −50 kcal/mol for the large diameter (14,14), showing that the small inner space of CNT has the higher van der Waals interaction between DOX and the CNT because of the closer contact. The van der Waals contribution approximately accounts for 80% of the total binding energy for the adsorptions, which can be primarily attributed to the π-π stacking between DOX and CNT. The high EVDW (−106 kcal/mol) for the encapsulations in (7,7) SWCNT is not able to compensate the large deformation due to the encapsulation. The EVDW (−85 kcal/mol) is dominantly responsible for the encapsulations in (8,8) SWCNT, and leads to an exothermic binding energy of −56 kcal/mol. The EVDW for the encapsulation tends to be weaker with increasing the diameter of CNT, and it is dominantly resulted from the π-π stacking, CH-π, NH-π, as well as C=O-π interactions.
Fig. 10.

Van der Waals contribution to the binding energy for DOX-CNT and DOX@CNT complexes with respect to CNT diameters. The EVDW is the binding energy difference predicted by the PM6-DH2 and PM6.
Table 2 lists the amounts of charges carried by the DOX/DOXH+ upon their binding with CNT. For the adsorptions, the charge slightly decreases with increasing the diameter of the CNT and the transferred charge from the CNT to DOX is similar to that from CNT to DOXH+. Opposite to the sidewall adsorption, for the encapsulation only little amount of approximately 0.01e charge is transferred from the CNT to DOX and DOXH+, and again charges carried by the DOX and DOXH+ are very close. The very little transferred electron also highlights the non-covalent interaction between DOX and CNT in both adsorption and encapsulation.
Table 2.
Amounts of charges (e) carried by the DOX and DOXH+ in adsorption complexes at PM6-DH2.
| CNT (n,n) | Encapsulation | Sidewall Adsorption | ||
|---|---|---|---|---|
| DOX | DOXH+ | DOX | DOXH+ | |
| (7,7) | −0.0066 | −0.0067 | −0.1332 | −0.1601 |
| (8,8) | −0.0070 | −0.0072 | −0.0597 | −0.0730 |
| (9,9) | −0.0084 | −0.0099 | −0.0386 | −0.0512 |
| (10,10) | −0.0100 | −0.0104 | −0.0275 | −0.0366 |
| (11,11) | −0.0109 | −0.0091 | −0.0252 | −0.0316 |
| (12,12) | −0.0108 | −0.0102 | −0.0246 | −0.0278 |
| (13,13) | −0.0096 | −0.0086 | −0.0235 | −0.0273 |
| (14,14) | −0.0094 | −0.0101 | −0.0224 | −0.0251 |
Because of ultrahigh surface areas up to ~2600m2/g, Dai et al estimated that for the laser-ablation-grown SWCNT (mean diameter ~1.9nm) ~50 DOX can bound to 10nm long SWCNT.4 Due to the specificity of the frontier orbitals of SWCNT as discussed above, the adsorbates could somewhat influence each other on the adsorption strength. Thus, we took into account the DOXH+-CNT (13,13) (diameter ~1.7nm) system in the aqueous solution as an example to illustrate the effect. Figure 11 shows that for the multi-adsorption of DOXH+ on the sidewall of SWCNT the average binding energy becomes slightly less with increasing the number of the DOXH+. The positive slope is approximately 0.4 kcal/mol/DOXH+, suggesting that one more adsorbed DOXH+ can approximately decrease the average adsorption energy by 0.4 kcal/mol. Assuming that it would be applied to multi adsorbates of 40 DOXH+, the average binding energy would change to ~ −16 kcal/mol that approaches to the experimentally estimated −14.1 kcal/mol for the DOX on the laser-ablation SWCNT.4 As for the encapsulation, Figure 11 displays the binding energy comparison of DOX, DOXH+, and two DOXH+ because the inner of CNT can accommodate only a few of DOXs in the confined space. It can be seen that the average binding energies for the two DOXH+ and one DOXH+ are almost the same (−39.7 ~ −39.6 kcal/mol) although there is a significantly binding decrease in comparison with the DOX@CNT complex (~−39.7 vs −52.7 kcal/mol).
Figure 11.

Average binding energy of DOX, (DOXH+) – (13, 13) CNT complex with respect to charge with PH6-DH2.
In addition, the mutual influence of the adsorption and encapsulation DOX-CNT(13,13) was investigated (Figure 12). As the two DOX molecules are rather far from each other as in case a, for a few located configurations the total binding energies are slightly lower than the sum of the individual DOX (−83.6 vs −85.5 kcal/mol). However, if the encapsulation just beneath the surface adsorption as in case b the total binding energy is generally stronger than the sum of the individual DOX binding energy (−86.6 vs −85.5 kcal/mol), suggesting a weak π···π stacking cooperativity.48
Figure 12.

Mutual influence of adsorption and encapsulation of DOXH+-CNT(13,13) in the case of rather separated (a), and in the case of neighbor (b).
Conclusion
The dispersion and hydrogen bonding corrected semi-empirical quantum method, PM6-DH2 and the meta-hybrid density functional theory M06-2X in the scheme of OMIOM were employed to systematically investigate the loading of doxorubicin (DOX) on the single-walled carbon nanotube (SWCNT), including the adsorption on the sidewall of SWCNT, encapsulation in CNT, and their dependences on the protonation of the NH2 group of DOX and the diameter of armchair (n,n) SWCNT. The adsorption primarily occurs through π-π stacking and just becomes slightly stronger as the diameter of CNT is increased. The encapsulation is much stronger than the adsorption because of the C-H/N/O-H…π and C=O…π interaction besides the π-π stacking as in the adsorption. (8,8) CNT (diameter ~ 11Å) energetically is an onset for the encapsulation since the encapsulation turns from endothermic to exothermic as the diameter is larger than approximately 11 Å, and the optimal diameter for the encapsulation is 14Å corresponding to (10,10) CNT. Thus for the thick CNT the encapsulation may also play an important role in the loading and releasing for the CNT-based drug delivery system of the DOX.
Fig. 9.

Reduced density gradient isosurface (0.5 au) for the adsorption of the DOX on (10,10) CNT (right) and its encapsulation DOX@CNT (left). Green and yellow indicate van der Waals interaction and red strong nonbonded overlap.
Acknowledgments
This work was supported by the National Institute of General Medical Science of the National Institute of Health (SC3GM105576 and SC3GM082324).
References
- 1.Singh R, Pantarotto D, McCarthy D, Chaloin O, Hoebeke J, Partidos CD, Briand JP, Prato M, Bianco A, Kostarelos K. J Am Chem Soc. 2005;127:4388–4396. doi: 10.1021/ja0441561. [DOI] [PubMed] [Google Scholar]
- 2.Kam NWS, Dai HJ. J Am Chem Soc. 2005;127:6021–6026. [Google Scholar]
- 3.Kam NWS, O’Connell M, Wisdom JA, Dai HJ. Proc Natl Acad Sci USA. 2005;102:11600–11605. doi: 10.1073/pnas.0502680102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu Z, Sun X, Nakayama-Ratchford N, Dai H. ACS Nano. 2007;1:50–56. doi: 10.1021/nn700040t. [DOI] [PubMed] [Google Scholar]
- 5.Liu Z, Tabakman S, Welsher K, Dai H. Nano Res. 2009;2:85–120. doi: 10.1007/s12274-009-9009-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu Z, Fan AC, Rakhra K, Sherlock S, Goodwin A, Chen X, Yang Q, Felsher DW, Dai H. Angew Chem, Int Ed. 2009;48:7668–7672. doi: 10.1002/anie.200902612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prencipe G, Tabakman SM, Welsher K, Liu Z, Goodwin AP, Zhang L, Henry J, Dai H. J Am Chem Soc. 2009;131:4783–4787. doi: 10.1021/ja809086q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu F, Gu L, Meziani MJ, Wang X, Luo PG, Veca LM, Cao L, Sun YP. Adv Mater. 2009;21:139–152. [Google Scholar]
- 9.Liu Z, Robinson JT, Tabakman SM, Yang K, Dai H. Materials Today. 2011;14:316–323. [Google Scholar]
- 10.Fabbro C, Ali-Boucetta H, Da Ros T, Kostarelos K, Bianco A, Prato M. Chem Commun (Camb) 2012;48:3911–3926. doi: 10.1039/c2cc17995d. [DOI] [PubMed] [Google Scholar]
- 11.Heister E, Neves V, Lamprecht C, Silva SRP, Coley HM, McFadden J. Carbon. 2012;50:622–632. [Google Scholar]
- 12.Lay CL, Liu J, Liu Y. Expert Rev Med Dev. 2011;8:561–566. doi: 10.1586/erd.11.34. [DOI] [PubMed] [Google Scholar]
- 13.Gu YJ, Cheng J, Jin J, Cheng SH, Wong WT. Int J Nanomed. 2011;6:2889–2898. doi: 10.2147/IJN.S25162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meng L, Zhang X, Lu Q, Fei Z, Dyson PJ. Biomater. 2012;33:1689–1698. doi: 10.1016/j.biomaterials.2011.11.004. [DOI] [PubMed] [Google Scholar]
- 15.Saikia N, Deka RC. Comput Theor Chem. 2011;964:257–261. [Google Scholar]
- 16.Wang C, Lv P, Wei W, Tao S, Hu T, Yang J, Meng C. Nanotech. 2011;22:415101/415101–415108. doi: 10.1088/0957-4484/22/41/415101. [DOI] [PubMed] [Google Scholar]
- 17.Sornmee P, Rungrotmongkol T, Saengsawang O, Arsawang U, Remsungnen T, Hannongbua S. J Comp Theor Nanosci. 2011;8:1385–1391. doi: 10.1016/j.jmgm.2010.11.002. [DOI] [PubMed] [Google Scholar]
- 18.Kavitha T, Haider Abdi SI, Park S-Y. Phys Chem Chem Phys. 2013;15:5176–5185. doi: 10.1039/c3cp00008g. [DOI] [PubMed] [Google Scholar]
- 19.Collins Y, Lele S. J Nat Med Ass. 2005;97:1414–1416. [PMC free article] [PubMed] [Google Scholar]
- 20.Dinan NM, Atyabi F, Rouini MR, Amini M, Golabchifar AA, Dinarvand R. Mater Sci Eng C. 2014;39:47–55. doi: 10.1016/j.msec.2014.01.055. [DOI] [PubMed] [Google Scholar]
- 21.Lv S, Tang Z, Li M, Lin J, Song W, Liu H, Huang Y, Zhang Y, Chen X. Biomater. 2014;35:6118–6129. doi: 10.1016/j.biomaterials.2014.04.034. [DOI] [PubMed] [Google Scholar]
- 22.Xu H, Fan M, Elhissi AM, Zhang Z, Wan KW, Ahmed W, Phoenix DA, Sun X. Nanomed. 2015;10:1247–1262. doi: 10.2217/nnm.14.233. [DOI] [PubMed] [Google Scholar]
- 23.Kamiya K, Okada S. J Chem Phys. 2011;83:155444/155441–155446. [Google Scholar]
- 24.Xing YF, Yang CL, Mo YF, Wang MS, Ma XG. J Phys Soc Japan. 2014;83:024801/024801–024807. [Google Scholar]
- 25.Alshehri MH, Cox BJ, Hill JM. Mico & Nano Lett. 2014;9:113–118. [Google Scholar]
- 26.Cruz FJAL, de Pablo JJ, Mota JPB. RSC Advances. 2014;4:1310–1321. [Google Scholar]
- 27.Kang Y, Wang Q, Liu YC, Shen JW, Wu T. J Phys Chem B. 2010;114:2869–2875. doi: 10.1021/jp905995s. [DOI] [PubMed] [Google Scholar]
- 28.Chen Q, Wang Q, Liu YC, Wu T, Kang Y, Moore JD, Gubbins KE. J Chem Phys. 2009;131:015101/015101–015106. doi: 10.1063/1.3148025. [DOI] [PubMed] [Google Scholar]
- 29.Kang Y, Liu YC, Wang Q, Shen JW, Wu T, Guan WJ. Biomater. 2009;30:2807–2815. doi: 10.1016/j.biomaterials.2009.01.024. [DOI] [PubMed] [Google Scholar]
- 30.Mejri A, Vardanega D, Tangour B, Gharbi T, Picaud F. J Phys Chem B. 2015;119:604–611. doi: 10.1021/jp5102384. [DOI] [PubMed] [Google Scholar]
- 31.Zhao Y, Truhlar DG. Acc Chem Res. 2008;41:157. doi: 10.1021/ar700111a. [DOI] [PubMed] [Google Scholar]
- 32.Morgado CA, Jurecka P, Svozil D, Hobza P, Sponer J. Phys Chem Chem Phys. 2010;12:3522. doi: 10.1039/b924461a. [DOI] [PubMed] [Google Scholar]
- 33.Wang Y. J Phys Chem C. 2008;112:14297–14305. doi: 10.1021/jp803917t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Y, Bu Y. J Phys Chem B. 2007;111:6520–6526. doi: 10.1021/jp0700433. [DOI] [PubMed] [Google Scholar]
- 35.Sun W, Bu Y, Wang Y. J Phys Chem C. 2011;115:3220–3228. doi: 10.1021/jp108812z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sun W, Bu Y, Wang Y. J Comp Chem. 2012;33:490–501. doi: 10.1002/jcc.22881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grimme S. J Comput Chem. 2006;27:1787–1799. doi: 10.1002/jcc.20495. [DOI] [PubMed] [Google Scholar]
- 38.Schwabe T, Grimme S. Phys Chem Chem Phys. 2007;9:3397. doi: 10.1039/b704725h. [DOI] [PubMed] [Google Scholar]
- 39.Řezáč J, Fanfrlík Ji, Salahub D, Hobza P. J Chem Theory Comput. 2009;5:1749–1760. doi: 10.1021/ct9000922. [DOI] [PubMed] [Google Scholar]
- 40.Korth M, Pitoňák M, Řezáč J, Hobza P. J Chem Theory Comput. 2009;6:344–352. doi: 10.1021/ct900541n. [DOI] [PubMed] [Google Scholar]
- 41.Riley KE, Pitoňák M, Jurečka P, Hobza P. Chem Rev. 2010;110:5023–5063. doi: 10.1021/cr1000173. [DOI] [PubMed] [Google Scholar]
- 42.Xu Z, Meher BR, Eustache D, Wang Y. J Mol Graph Mod. 2014;47:8–17. doi: 10.1016/j.jmgm.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stewart JJP. MOP AC2009 James J. P. Stewart, Stewart Computational Chemistry, Version 11.052W. web: HTTP://OpenMOPAC.net.
- 44.Dapprich S, Komáromi I, Byun KS, Morokuma K, Frisch MJ. J Mol Struct (Theochem) 1999;462:1–21. [Google Scholar]
- 45.Klamt A, Schüümann G. J Chem Soc Perkin Transactions. 1993;2:799–805. [Google Scholar]
- 46.Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ. Nucleic Acids Res. 2005;33:W363–367. doi: 10.1093/nar/gki481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Johnson ER, Keinan S, Mori-Sánchez P, Contreras-García J, Cohen AJ, Yang W. J Amer Chem Soc. 2010;132:6498–6506. doi: 10.1021/ja100936w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kulkarni C, Reddy SK, George SJ, Balasubramanian S. Chem Phys Lett. 2011;515:226–230. [Google Scholar]