

Abstract
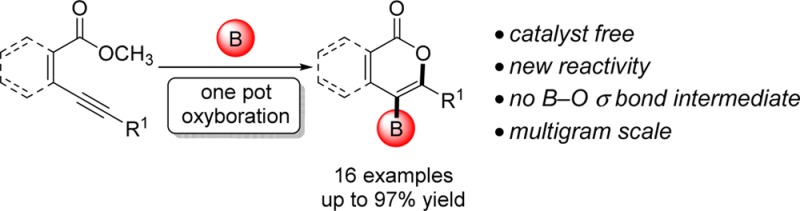
A catalyst-free oxyboration reaction of alkynes is developed. The resulting borylated isocoumarins and 2-pyrones are isolated as boronic acids, pinacolboronate esters, or potassium organotrifluoroborate salts, providing a variety of bench-stable organoboron building blocks for downstream functionalization. This method has functional group compatibility, is scalable, and proceeds with readily available materials: B-chlorocatecholborane and methyl esters. Mechanistic studies indicate that the B-chlorocatecholborane acts as a carbophilic Lewis acid toward the alkyne, providing a mechanistically distinct pathway for oxyboration that avoids B–O σ bond formation and enables this catalyst-free route.
Addition reactions of boron reagents to C–C π systems have provided powerful routes to organoboron compounds for over 65 years.1 The first oxyboration reaction of C–C π systems was only recently reported in 2014,7 possibly due to the high strength of B–O σ bonds (∼136 kcal/mol).9 This reaction proceeded through a B–O σ bond intermediate and required a gold catalyst. We report a boron reagent that promotes oxyboration of alkynes in the absence of a catalyst. This reaction does not proceed via a B–O σ bond intermediate, instead accessing an electrophilic oxycyclization pathway. The fact that boron is able to access an oxycyclization pathway—previously known only for other elements10—provides the first example of an important class of mechanistically distinct oxyboration reactions, which yield borylated heterocycles without the use of strongly basic reagents17 or transition-metal catalysts (Figure 1).18 The absence of previously reported oxyboration/cyclization reactions with electrophilic boron may be due to competitive formation of B–O bonds, formation of which are here shown surprisingly to inhibit oxyboration rather than promote it. We apply this method to the synthesis of borylated isocoumarins and 2-pyrones, classes of compounds with important biological activity20 but with few prior reports of their borylated analogues.22 Demonstration of this mechanistically distinct pathway for oxyboration will open up new pathways for the practical synthesis of borylated heterocyclic building blocks for drug discovery and materials synthesis.
Figure 1.
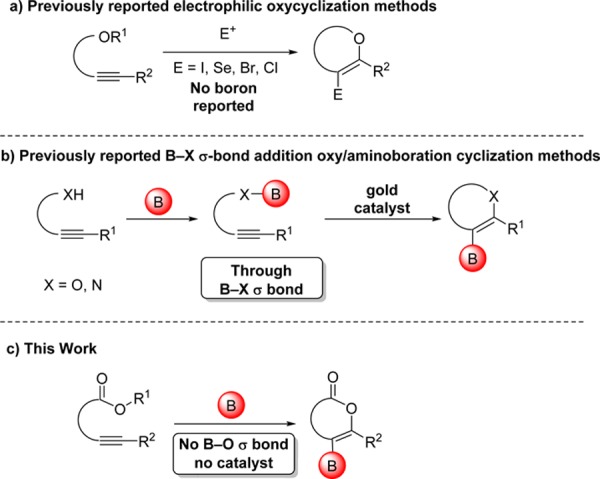
(a) Previously reported electrophilic cyclization/dealkylation methods to generate O-heterocycles from ethers or esters. (b) Previously reported heterocycle-forming B–X σ bond addition. (c) This work demonstrating mechanistically distinct oxyboration.
Primary competing strategies to synthesize borylated heterocycles include lithiation/electrophilic trapping17 and transition-metal-catalyzed borylation.18,25 Prior reports of borylated lactones employed Pd-catalyzed cross-coupling26 and lithiation/borylation.27 The oxyboration strategy demonstrated here provides complementary functional group tolerance to these leading alternative borylation strategies.
Given that boron halides are known to dealkylate esters to generate B–O σ bonds,28 we anticipated that boron trihalides should promote oxyboration of 1a due to previously reported carboboration and haloboration reactivity with alkynes.30 Both trihalogenated boron sources BBr3 and BCl3 (Table 1, entries 1 and 2, respectively) failed to yield any desired borylated isocoumarin 3aa. B-Chlorocatecholborane (ClBcat), which to our knowledge has not been previously used for alkyne activation, provided the borylated isocoumarin in yields of 25 and 75% at 45 and 100 °C, respectively (entries 3 and 4). The use of B-bromocatecholborane, which is known to demethylate methyl esters more quickly than ClBcat (and thus would be expected to yield 2 more quickly or at the same rate),31 provided a lower isolated yield of the desired oxyboration product (entry 5). These results provided an early indication that the operative oxyboration pathway proceeded without initial dealkylation/B–O σ bond formation and may be mechanistically distinct from prior reports that proceeded through the B–O σ bond.
Table 1. Boron Reagent Variation.

| entry | boron electrophile [B] | temp (°C) | yield (%)a of 3aa |
|---|---|---|---|
| 1 | BBr3b | 45 | 0 |
| 2 | BCl3b | 45 | 0 |
| 3 | B-chlorocatecholborane | 45 | 25 |
| 4 | B-chlorocatecholborane | 100 | 75 |
| 5 | B-bromocatecholborane | 100 | 48 |
Isolated yield.
1.0 M solution in DCM.
Commercially available ClBcat (1.4 equiv) was identified as the electrophile that provided the best yield, and 100 °C was identified as the optimal temperature at 1.0 M concentration, with the mass balance at lower temperatures being starting material (see SI for optimization data).
Product isolation scope and substrate scope were next investigated (Table 2). For synthetic variety, the products can be isolated three different ways: as the pinacolboronic ester (3aa), the boronic acid (3ab), or the potassium organotrifluoroborate salt (3ac). Each method provides complementary advantages. Pinacolboronic esters are stable toward silica gel chromatography, provided the best isolated yield for the test compound, and can be easily cross-coupled under basic conditions; it was therefore chosen as the preferred isolation method.32 Boronic acids, although not as bench-stable as the other options, are a preferred transition-metal-catalyzed cross-coupling partner and provide the best atom economy.33 Potassium organotrifluoroborates, although slightly lower yielding, provide a column-free workup procedure after oxyboration, making them a practical target for large-scale synthesis.34 The use of B-chloropinacolborane rather than ClBcat, which would provide a direct route to analogous isolable products, was avoided due to its lack of commercial availability and poor thermal stability (decomposition above −70 °C),36 precluding oxyboration reactions above this temperature.
Table 2. Synthesis of Borylated Isocoumarins and 2-Pyrones via the Oxyboration Reactiona,b.

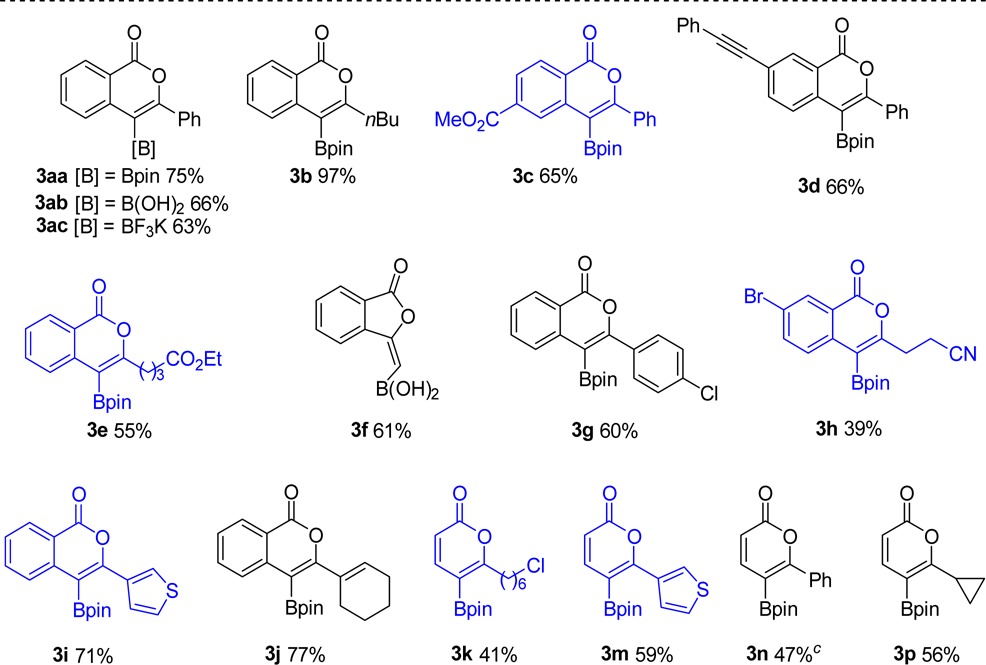
Isolated yield.
Blue molecules contain functional groups not compatible with other leading borylation strategies.
From ethyl ester.
We attempted an alternative oxyboration through the corresponding carboxylic acid rather than methyl ester. An intractable product mixture was produced. The route from the methyl ester is fortunately much cleaner. The methyl esters are also bench-stable and a more practical synthetic precursor than the o-alkynylbenzoic acids, which decompose via tautomerization/cyclization.
Functional groups that can be tolerated with this oxyboration strategy include esters, cyanides, aryl bromides and chlorides, and thiophenes, which are incompatible with competing lithiation/borylation routes and/or Pd-catalyzed oxidative addition routes. The tolerance toward 3c was particularly noteworthy because these boron reagents dealkylate esters; this tolerance was examined further in mechanistic studies. Similarly, tolerance of alkynes distal to 3d implies that independent reactivity of the alkyne (e.g., haloboration5,6) is not part of the operative pathway. An aromatic backbone was not a requirement for the oxyboration reaction. Alkenyl esters also underwent the oxyboration reaction to produce 2-pyrones 3k–3p in lower yields. Because of ClBcat’s reactivity, ethers, an O-TBDPS protecting group, furans, and a ketone with α protons were not tolerated by the oxyboration reaction.
The oxyboration reaction could theoretically produce either the regioisomer from 5-exo-dig or 6-endo-dig cyclization.37 X-ray crystallographic analysis of 3aa confirmed it was the product of 6-endo-dig cyclization (Figure 2). No other regioisomer was observed in the crude 1H NMR spectrum. 3f is the only product formed from 5-exo-dig cyclization (see SI for characterization data). Consistent with the mechanistic proposal, formation of the unobserved 6-endo-dig product required disfavored buildup of primary cationic character on the terminal carbon of the unsubstituted alkyne.
Figure 2.
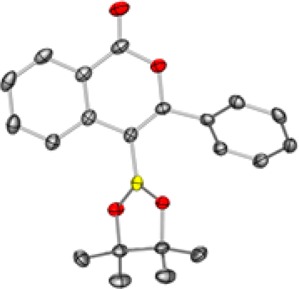
X-ray crystallographic structure of 3aa, confirming six-membered ring formation, with the thermal ellipsoids shown at 50% probability (B, yellow; C, gray; O, red).
Two mechanistic pathways were considered for this oxyboration reaction (Scheme 1). In the top pathway, dealkylation occurs first to produce intermediate 4, followed by the oxyboration/cyclization with the alkyne. In the bottom pathway, boron-induced electrophilic cyclization, possibly through a formal vinylic cation, 5, or alternatively directly from 1 to 6, as has been proposed for alkyne activation by BCl3,38 precedes dealkylation. Cyclized oxocarbenium ion 6 was then primed for rapid dealkylation due to the increased positive charge on the oxygen. Oxygen in 4 would be less nucleophilic toward cyclization than oxygen in 5 due to donation of the electron density of the carboxy group into the empty p orbital on boron. This decrease in nucleophilicity may rationalize why direct dealkylation of 1 via the top pathway inhibits the oxyboration reaction rather than promotes it.
Scheme 1. Two Potential Oxyboration Pathways.
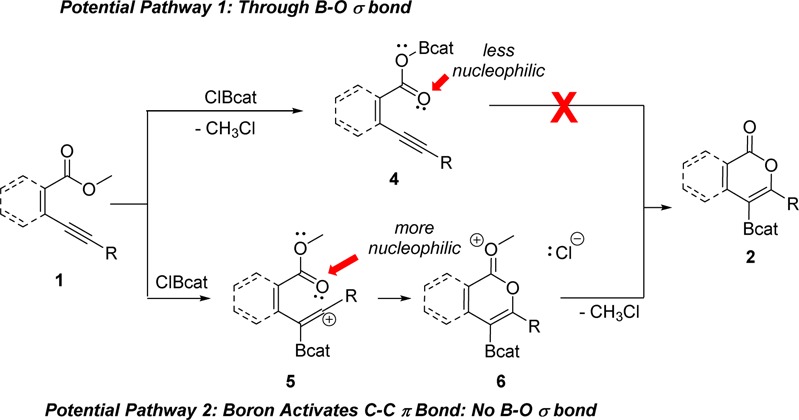
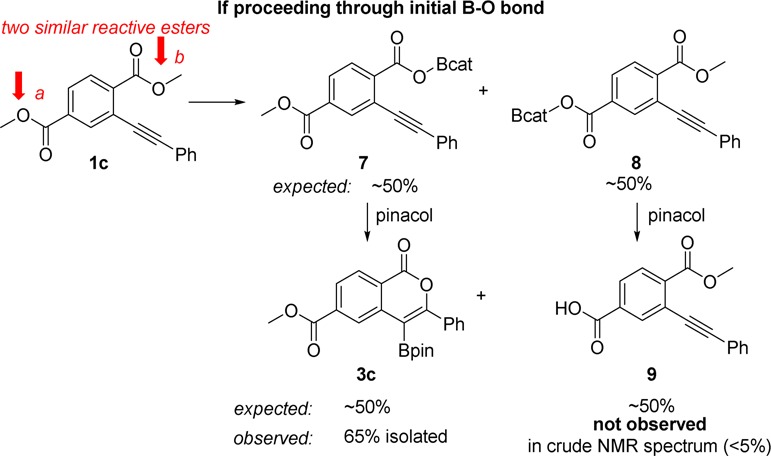
If demethylation occurs before cyclization, in the operable pathway to the oxyboration product, then similar esters (a and b) in 1c should demethylate at similar rates (Scheme 2). This demethylation would produce intermediates 7 and 8 in approximately equal quantities, resulting in formation of 3c and 9. 9 was not observed in the crude reaction mixture by 1H NMR spectroscopy. 3c was isolated in 65% yield, with the majority of the mass balance being unreacted 1c. Therefore, ester b demethylates significantly faster than ester a, consistent with cyclization preceding demethylation. The position of ester a does not permit cyclization, thus it does not have access to that pathway for demethylation. This is inconsistent with operation of the top pathway (dealkylation/cyclization) and is consistent with the bottom pathway (cyclization/dealkylation) in the overall oxyboration reaction.
Scheme 2. Intramolecular Competition Experiment.
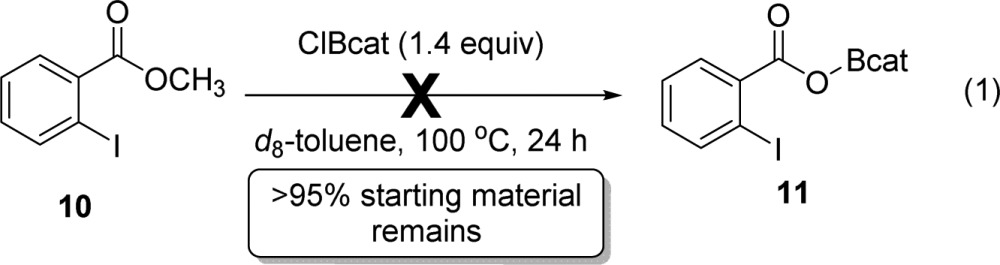
Demethylation of test compound 10 was examined. 10 has no alkyne; therefore, if demethylation occurs, it must proceed directly, rather than through a precyclization pathway. Under identical conditions that produced 3aa from 1a in 75% isolated yield, 10 led to no detectable decrease in starting methyl ester, as determined by 1H NMR spectroscopy relative to a 1,3,5-triisopropylbenzene internal standard (<5%, eq 1). No borenium species were detected via 11B NMR spectroscopy, in contrast to the arene borylation conditions reported by Ingleson.40 Thus, the rate of reactivity of methyl esters with ClBcat in the absence of tethered alkynes is insufficiently rapid to account for the observed oxyboration reactivity. These data support that cyclization precedes demethylation in the operative oxyboration reaction mechanism (Scheme 1, bottom).
 |
1 |
Various O-alkyl esters were examined with the oxyboration method (Table 3). Oxyboration reaction tolerated methyl, ethyl, and isopropyl groups with iterative reductions in 1H NMR yields. The t-butyl ester did not furnish any of the desired borylated isocoumarin, despite successful dealkylation, as characterized by isobutylene formation and quantification of the benzoic acid derivative of 1a in 68% 1H NMR yield. This detection is consistent with the reported ability of ClBcat to dealkylate t-butyl esters at ambient temperature while ethyl esters remain unreacted31 and provides further evidence that cyclization precedes dealkylation in the pathway that generates the oxyboration product because when dealkylation occurs rapidly at ambient temperature, presumably generating B–O σ bonds, oxyboration does not occur even at elevated temperatures.
Table 3. Mechanistic Insight from O-Alkyl Group Variance of the Oxyboration Reaction.

| entry | R | 1H NMR yield (%)a of 3aa |
|---|---|---|
| 1 | Me | 81 |
| 2 | Et | 68 |
| 3 | iPr | 60 |
| 4 | tBu | 0 |
Yield determined relative to mesitylene internal standard.
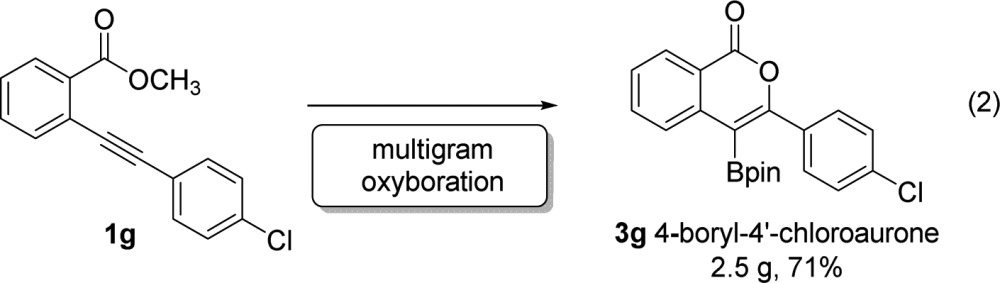
Oxyboration reaction provides scalable access to borylated building blocks of bioactive cores (eq 2). Subjecting 2.5 g of methyl benzoate ester 1g to the standard oxyboration reaction conditions yielded 2.5 g (71%) of the desired borylated isocoumarin 3g. Compound 3g is the 4-borylated analogue of the marine natural product chloroaurone, isolated from Spatoglossum variabile.41
 |
2 |
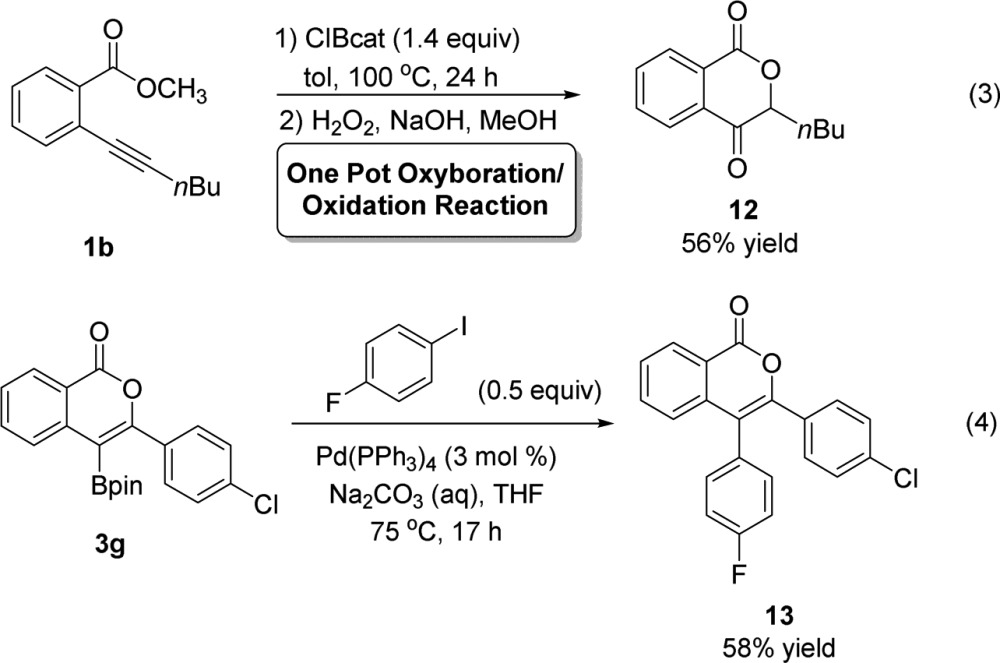
The boron functional group provides a handle for downstream functionalization of the newly formed lactone core. One example of this utility is demonstrated in the synthesis of isochroman-1,4-diones.42 Previously reported synthesis of 12 employed chromium trioxide and sulfuric acid.44 Subjecting butyl alkynyl ester 1b to the standard oxyboration conditions, followed by oxidative workup, furnished 12 in 56% yield over two steps in one pot (eq 3). The utility of these borylated isocoumarins in the construction of new C–C bonds was highlighted in a Suzuki crosss-coupling reaction of borylated lactone 3g with p-fluoroiodobenzene to generate isocoumarin 13 (eq 4).
 |
3 |
Having established the feasibility of using an external boron electrophile to generate borylated isocoumarin products, we explored the applicability of the oxyboration strategy to synthesize borylated isoxazoles (eq 5).45 Treatment of O-methyl oxime 14 with ClBcat at 100–110 °C for 72 h furnished borylated isoxazole 15 in 35% yield. This illustrates the potential for the mechanistic concept to be applied to other systems to generate value-added borylated heterocycles from simple alkylated heteroatoms.
 |
5 |
In summary, this reaction is the first report of a transition-metal-free oxyboration reaction that adds boron and oxygen to C–C π systems. It is also the first formal carboxyboration—addition of the CO2 group and boron—across alkynes. This new reactivity is enabled by dioxaborole activation of an alkyne to promote oxycyclization.47 Reactivity lessons learned converge on employing electrophilic boron reagents with the right balance of carbophilicity vs oxyphilicity and with substrates exhibiting slow competitive dealkylation prior to cyclization. These balances enable the desired reactivity by avoiding competitive formation of the strong B–O σ bond, which prevents oxyboration reactivity under these catalyst-free conditions. These balances are conveniently achieved with commercially available ClBcat and readily available methyl ester substrates. This scalable method can tolerate a variety of functional groups that are incompatible with the alternative strongly basic or oxidative addition pathways that comprise other leading borylation strategies. Additional mechanistic studies and substrate class expansions are currently ongoing in our research group. We envision that this mechanistically distinct oxyborylation strategy will serve as a springboard toward broader application of catalyst-free boron–element addition reactions to generate valuable borylated heterocyclic products.
Acknowledgments
This work was supported by a grant from the NIH (1R01GM098512-01), the University of California, Irvine, and an Allergan Foundation Graduate Fellowship to D.J.F. We thank Ms. Nicole A. Nava and Dr. Mohammad Al-Amin for synthesis of starting materials, Dr. Joseph W. Ziller and Mr. Jason R. Jones for X-ray diffraction analysis, and Dr. Phillip R. Dennison for NMR spectroscopy assistance.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.5b12989.
The authors declare the following competing financial interest(s): U.S. patent no. 9,238,661.
Supplementary Material
References
- a Brown H. C. Tetrahedron 1961, 12, 117. 10.1016/0040-4020(61)80107-5. [DOI] [Google Scholar]; b Miyaura N.Hydroboration, Diboration, and Stannylboration. In Catalytic Heterofunctionalization; Togni A., Grützmacher H., Eds.; Wiley-VCH: Weinheim, 2001; pp 1–46. [Google Scholar]; c Barbeyron R.; Benedetti E.; Cossy J.; Vasseur J.; Arseniyadis S.; Smietana M. Tetrahedron 2014, 70, 8431. 10.1016/j.tet.2014.06.026. [DOI] [Google Scholar]; d Sakae R.; Hirano K.; Miura M. J. Am. Chem. Soc. 2015, 137, 6460. 10.1021/jacs.5b02775. [DOI] [PubMed] [Google Scholar]; e Hara S.; Dojo H.; Takinami S.; Suzuki A. Tetrahedron Lett. 1983, 24, 731. 10.1016/S0040-4039(00)81511-7. [DOI] [Google Scholar]; f Cade I. A.; Ingleson M. J. Chem. - Eur. J. 2014, 20, 12874. 10.1002/chem.201403614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Hirner J. J.; Faizi D. J.; Blum S. A. J. Am. Chem. Soc. 2014, 136, 4740. 10.1021/ja500463p. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Hirner J. J.; Blum S. A. Tetrahedron 2015, 71, 4445. 10.1016/j.tet.2015.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson R. T.Polar Covalence; Academic Press: San Diego, CA, 1983. [Google Scholar]
- a Roy S.; Roy S.; Neuenswander B.; Hill D.; Larock R. C. J. Comb. Chem. 2009, 11, 1128. 10.1021/cc9001197. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Pal S.; Chatare V.; Pal M. Curr. Org. Chem. 2011, 15, 782. 10.2174/138527211794518970. [DOI] [Google Scholar]; c Yao T.; Larock R. C. J. Org. Chem. 2003, 68, 5936. 10.1021/jo034308v. [DOI] [PubMed] [Google Scholar]; d Oliver M. A.; Gandour R. D. J. Org. Chem. 1984, 49, 558. 10.1021/jo00177a038. [DOI] [Google Scholar]; e Yao T.; Larock R. C. Tetrahedron Lett. 2002, 43, 7401. 10.1016/S0040-4039(02)01731-8. [DOI] [Google Scholar]; f Rossi R.; Carpita A.; Bellina F.; Stabile P.; Mannina L. Tetrahedron 2003, 59, 2067. 10.1016/S0040-4020(03)00212-6. [DOI] [Google Scholar]
- g Godoi B.; Schumacher R. F.; Zeni G. Chem. Rev. 2011, 111, 2937. 10.1021/cr100214d. [DOI] [PubMed] [Google Scholar]
- Nagaki A.; Moriwaki Y.; Yoshida J. Chem. Commun. 2012, 48, 11211. 10.1039/c2cc36197c. [DOI] [PubMed] [Google Scholar]
- a Larsen M. A.; Hartwig J. F. J. Am. Chem. Soc. 2014, 136, 4287. 10.1021/ja412563e. [DOI] [PubMed] [Google Scholar]; b Ishiyama T.; Murata M.; Miyaura N. J. Org. Chem. 1995, 60, 7508. 10.1021/jo00128a024. [DOI] [Google Scholar]
- a Janecki T., Ed. Natural Lactones and Lactams: Synthesis, Occurrence and Biological Activity; Wiley-VCH: Weinheim, 2014; pp 147–192. [Google Scholar]; b Pochet L.; Frederick R.; Masereel B. Curr. Pharm. Des. 2004, 10, 3781. 10.2174/1381612043382684. [DOI] [PubMed] [Google Scholar]
- a Suzuki A. Acc. Chem. Res. 1982, 15, 178. 10.1021/ar00078a003. [DOI] [Google Scholar]; b Pelter A. Chem. Soc. Rev. 1982, 11, 191. 10.1039/cs9821100191. [DOI] [Google Scholar]; c Dimitrijević E.; Taylor M. ACS Catal. 2013, 3, 945. 10.1021/cs4000848. [DOI] [Google Scholar]
- Chong E.; Blum S. A. J. Am. Chem. Soc. 2015, 137, 10144. 10.1021/jacs.5b06678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravett E. C.; Hilton P. J.; Jones K.; Romero F. Tetrahedron Lett. 2001, 42, 9081. 10.1016/S0040-4039(01)01980-3. [DOI] [Google Scholar]
- Fletcher C. J.; Wheelhouse K. M. P.; Aggarwal V. K. Angew. Chem., Int. Ed. 2013, 52, 2503. 10.1002/anie.201208403. [DOI] [PubMed] [Google Scholar]
- a Felix A. M. J. Org. Chem. 1974, 39, 1427. 10.1021/jo00926a025. [DOI] [Google Scholar]; b Manchand P. S. J. Chem. Soc. D 1971, 667. 10.1039/c29710000667. [DOI] [PubMed] [Google Scholar]
- Warner A. J.; Lawson J. R.; Fasano V.; Ingleson M. J. Angew. Chem., Int. Ed. 2015, 54, 11245. 10.1002/anie.201505810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeckman R. K.; Potenza J. C. Tetrahedron Lett. 1985, 26, 1411. 10.1016/S0040-4039(00)99058-0. [DOI] [Google Scholar]
- Miyaura N. Chem. Rev. 1995, 95, 2457. 10.1021/cr00039a007. [DOI] [Google Scholar]
- Badone D.; Baroni M.; Cardamone R.; Ielmini A.; Guzzi U. J. Org. Chem. 1997, 62, 7170. 10.1021/jo970439i. [DOI] [PubMed] [Google Scholar]
- a Molander G. A.; Ellis N. Acc. Chem. Res. 2007, 40, 275. 10.1021/ar050199q. [DOI] [PubMed] [Google Scholar]; b Darses S.; Genet J.-P. Chem. Rev. 2008, 108, 288. 10.1021/cr0509758. [DOI] [PubMed] [Google Scholar]
- Bettinger H. F.; Filthaus M.; Bornemann H.; Oppel I. M. Angew. Chem., Int. Ed. 2008, 47, 4744. 10.1002/anie.200705936. [DOI] [PubMed] [Google Scholar]
- Marchal E.; Uriac P.; Legouin B.; Toupet L.; van de Weghe P. Tetrahedron 2007, 63, 9979. 10.1016/j.tet.2007.07.066. [DOI] [Google Scholar]
- a Lawson J. R.; Clark E. R.; Cade I. A.; Solomon S. A.; Ingleson M. J. Angew. Chem., Int. Ed. 2013, 52, 7518. 10.1002/anie.201302609. [DOI] [PMC free article] [PubMed] [Google Scholar]; b For an example of alkyne activation by boron, see:Hansmann M. M.; Melen R. L; Rudolph M.; Rominger F.; Wadepohl H.; Stephan D. W.; Hashmi A. S. K. J. Am. Chem. Soc. 2015, 137, 15469. 10.1021/jacs.5b09311. [DOI] [PubMed] [Google Scholar]
- Del Grosso A.; Singleton P. J.; Muryn C. A.; Ingleson M. J. Angew. Chem., Int. Ed. 2011, 50, 2102. 10.1002/anie.201006196. [DOI] [PubMed] [Google Scholar]
- Venkateswarlu S.; Panchagnula G.; Gottumukkala A.; Subbaraju G. Tetrahedron 2007, 63, 6909. 10.1016/j.tet.2007.04.048. [DOI] [Google Scholar]
- a Jansen R.; Kunze B.; Reichenbach H.; Hofle G. Eur. J. Org. Chem. 2002, 2002, 917.. [DOI] [Google Scholar]; b Neil A.; Gordon A.; Urquhart M. Patent Appl. WO2012/80243 A2, 2012.
- Hobson S. J.; Parkin A.; Marquez R. Org. Lett. 2008, 10, 2813. 10.1021/ol8009336. [DOI] [PubMed] [Google Scholar]
- a Liu J.; Yu L.-F.; Eaton J. B.; Caldarone B.; Cavino K.; Ruiz C.; Terry M.; Fedolak A.; Wang D.; Ghavami A.; Lowe D. A.; Brunner D.; Lukas R. J.; Kozikowski A. P. J. Med. Chem. 2011, 54, 7280. 10.1021/jm200855b. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Tu K.; Hirner J. J.; Blum S. A. Org. Lett. 2016, 18, 480. 10.1021/acs.orglett.5b03530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Electrophilic activation of C–C π systems in other contexts has been reported employing the more electrophilic boron reagents (e.g., BCl3, BBr3, BR3, and borenium species BO2L+), but not with neutral dioxyboron systems (BO2X). See refs (6 and 40) and the following:; a Hansmann M. M.; Melen R. L.; Rominger F.; Hashmi A. S. K.; Stephan D. W. Chem. Commun. 2014, 50, 7243. 10.1039/c4cc01370k. [DOI] [PubMed] [Google Scholar]; b Wilkins L. C.; Wieneke P.; Newman P. D.; Kariuki B. M.; Rominger F.; Hashmi A. S. K.; Hansmann M. M.; Melen R. L. Organometallics 2015, 34, 5298. 10.1021/acs.organomet.5b00753. [DOI] [Google Scholar]; c Yang C. H.; Zhang Y. S.; Fan W. W.; Liu G. Q.; Li Y. M. Angew. Chem., Int. Ed. 2015, 54, 12636. 10.1002/anie.201505489. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.