

Abstract
Exchange proteins directly activated by cAMP (EPAC) as guanine nucleotide exchange factors mediate the effects of the pivotal second messenger cAMP, thereby regulating a wide variety of intracellular physiological and pathophysiological processes. A series of novel 2-(isoxazol-3-yl)-2-oxo-N′-phenyl-acetohydrazonoyl cyanide EPAC antagonists was synthesized and evaluated in an effort to optimize properties of the previously identified high-throughput (HTS) hit 1 (ESI-09). Structure–activity relationship (SAR) analysis led to the discovery of several more active EPAC antagonists (e.g., 22 (HJC0726), 35 (NY0123), and 47 (NY0173)) with low micromolar inhibitory activity. These inhibitors may serve as valuable pharmacological probes to facilitate our efforts in elucidating the biological functions of EPAC and developing potential novel therapeutics against human diseases. Our SAR results have also revealed that further modification at the 3-, 4-, and 5-positions of the phenyl ring as well as the 5-position of the isoxazole moiety may allow for the development of more potent EPAC antagonists.
Graphical abstract
Introduction
The pivotal second messenger cAMP can regulate a wide range of physiological and pathological processes via signaling pathways that are involved in many disease states including cancer, diabetes, inflammation, heart failure, and neurological disorders.1 The physiological effects of cAMP are mainly transduced by three different downstream effectors, including protein kinase A (PKA),2 cyclic nucleotide-activated ion channels,3,4 and the most recently discovered exchange proteins directly activated by cAMP (EPAC).5–10 After being activated by the elevation in the intracellular concentration of cAMP, EPAC proteins act as guanine nucleotide exchange factors (GEFs) to catalyze the exchange of GDP for GTP on the Raslike small GTPases, Rap1 and Rap2,5,6,11 thereby controlling Rap-mediated biological processes. Two isoforms of EPAC proteins have been identified, EPAC1 and EPAC2,5,6,11 which are coded by two independent genes in both mature and developing tissues of mammals and have distinct expression patterns. EPAC1, also known as cAMP-GEF-I (RAPGEF3 in humans), is nearly ubiquitously expressed, whereas the expression of EPAC2, also known as cAMP-GEF-II (RAPGEF4 in humans), is relatively restricted. At least three splice variants of human EPAC2 have been found to exhibit a confined expression pattern.12 Epac2A is the most broadly expressed and is largely found in brain, pituitary, and pancreatic islets.6,13,14 By contrast, Epac2B has so far been detected only in the adrenal and testis,13,15 whereas Epac2C expression is restricted to the liver.16
Since their discovery in 1998, EPAC proteins have evoked significant interest toward elucidating their roles in physiological processes and pathogenesis as well as the molecular mechanisms of EPAC activation. Accumulating evidence has demonstrated that EPAC proteins play a critical role in insulin secretion, cardiac contraction, osteoclast differentiation, vascular permeability, neurotransmitter release, Treg-mediated immune suppression, integrin-mediated cell adhesion, cell migration and proliferation, cell exocytosis, and apoptosis as well as gene transcription and chromosomal integrity.17–23 Meanwhile, a substantial number of small-molecule EPAC modulators including various cAMP analogues and newly discovered EPAC-specific antagonists has been discovered and exploited for uncovering the biological functions of EPAC.10 Various cAMP derivatives have been reported,24,25 such as 8-pCPT-2′-O-Me-cAMP (007),26–28 8-pCPT-2′-O-Me-cAMP-AM (007-AM),29 and Sp-8-pCPT-2′-O-Me-cAMPS,29–31 which have been used as powerful pharmacological tools in EPAC-related research. However, these cAMP analogues as EPAC ligands are limited in their use due to a lack of selectivity among the isoforms of EPAC and potential off-target effects. Recently, substantial effort has been made to focus on nonnucleotide small molecules as EPAC-selective or isoform-specific antagonists, such as 4-cyclopentyl-2-((2,5-dimethylbenzyl)thio)-6-oxo-1,6-dihydropyrimidine-5-carbonitrile (HJC0197) and 4-cyclopropyl-2-((2,5-dimethylbenzyl)-thio)-6-oxo-1,6-dihydropyrimidine-5-carbonitrile (HJC0198) as EPAC antagonists,32 5,7-dibromo-6-fluoro-3,4-dihydro-2-meth-yl-1(2H)-quinolinecarboxaldehyde (CE3F4) as an EPAC1-selective inhibitor,33,34 and 1,3,5-trimethyl-2-tosylbenzene (ESI-05) and 1-(mesitylsulfonyl)-2,4-dimethyl-1H-pyrrole (HJC0350) as specific EPAC2 antagonists.35
To identify new pharmacological probes with a high capability of specifically inhibiting EPAC activity, our team developed an automated, robust, and sensitive fluorescence-based high-throughput screening (HTS) assay, which is based on detecting the dose-dependent change in fluorescence intensity of a fluorescent cyclic nucleotide analogue, 8-NBD-cAMP, with purified full-length EPAC2 protein.36,37 After analyzing the EPAC2 8-NBD-cAMP competition activity, selected compounds were evaluated for their ability to inhibit cAMP-mediated EPAC1 and EPAC2 GEF activity using purified recombinant full-length EPAC1 and EPAC2 proteins. A Maybridge Hitfinder compound library of 14 400 structurally diverse small molecules has been screened.32,35,38 Among these, 2-(5-tert-butylisoxazol-3-yl)-N′-(3-chlorophenyl)-2-oxoacetohy-drazonoyl cyanide (ESI-09, 1) has been identified and characterized as a novel noncyclic nucleotide EPAC-specific antagonist with no effect toward PKA (Figure 1).38 NMR spectroscopy experiments39 have also verified that 1 specifically interacts with the cAMP binding domain (CBD) of human EPAC1 (aa. 148–318).40–49 1 dose-dependently competes with 8-NBD-cAMP in binding to EPAC2, with an IC50 value of 9.0 μM. In the presence of 20 μM cAMP, 1 also dose-dependently displays cAMP-mediated GEF inhibitory activity of full-length EPAC2 and EPAC1, with apparent IC50 values of 4.4 and 10.8 μM, respectively.39
Figure 1.

Structures of cAMP and HTS hit 1.
1, as a specific EPAC inhibitor, has been a useful tool compound in revealing the roles of cAMP in regulating Plasmodium falciparum merozoite invasion of human erythrocytes,50 restoring the endothelial barrier after thrombin-induced hyperpermeability,51 and uncovering the functions of EPAC in osteoclast formation23 and cAMP-dependent regulation of Schwann cell proliferation and differentiation,52 in addition to other fields.53–55 Further biological functional investigations have also demonstrated that 1 can specifically block intracellular Akt phosphorylation, EPAC-mediated Rap1 activation, and insulin secretion in pancreatic β cells. Treatment of pancreatic ductal adenocarcinoma (PDA) cells with 1 has no obvious effect on cell proliferation and viability, but it can give rise to a significant decrease in cell migration and invasion.38 Furthermore, by employing an orthotopic metastatic PDA mouse model, 1 was found to reduce local and distant spread of MIA PaCa-2 cells and significantly decrease metastasis to the liver at a dose of 10 mg/kg via i.p. injection for 3 weeks.20 Compound 1 also enhances leptin signaling in an organotypic hypothalamic slice culture system. Administration of 1 in wildtype mice at a dose of 50 mg/kg by oral gavage for 3 weeks significantly reduces plasma leptin.56 Moreover, treatment with 1 at a nontoxic concentration can attenuate the formation of cytopathic effects, significantly reduce viral yields, and effectively protect permissive cells against Middle East respiratory syndrome coronavirus (MERS-Cov) infection by inhibiting viral RNA replication and protein expression of MERS-CoV without affecting the expression and localization of EPAC protein.57 It was also shown that hit 1 can completely recapitulate the EPAC1 knockout phenotype in vivo via pharmacological inhibition of EPAC1 and can significantly block the early stage of rickettsial attachment and invasion into nonphagocytic host cells.58 Treatment with 1 at a dose of 10 mg/kg via i.p. injection for 12 days significantly protects wild-type mice against rickettsial infection, resulting in much milder disease manifestations and dramatically improved survival.58 Taken together, these findings support compound 1 as being a selective pharmacological probe in unraveling the in vivo functions of EPAC and may provide potential novel therapeutics for the prevention and treatment of various human diseases including pancreatic cancer, diabetes, obesity, Middle East respiratory syndrome coronavirus infections, and fatal rickettsioses.
Compound 1 displays excellent bioavailability,58 low toxicity to animals,58 good membrane permeability, no significant inhibitory effects on PDEs,10 and very weak inhibitory activities toward hERG and CYP450 enzymes.10 All of these combined observations support the notion that non-nucleotide small molecule 1 may have superior advantages in terms of off-target effects, selectivity, and toxicities over those of traditional cAMP analogues. Despite a potential concern associated with its protein denaturing properties at high concentrations,59 our extensive biochemical and pharmacological study39 has defined its therapeutic window and validated that 1 indeed acts as an EPAC-specific antagonist. Therefore, it is imperative to further optimize 1 through rational drug design approaches to develop advanced leads with enhanced activity and specificity as well as better druglike properties.
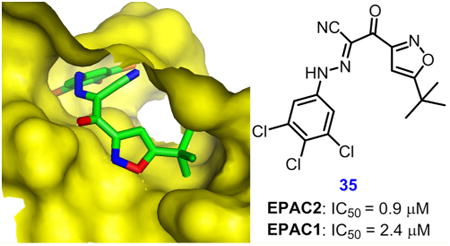
Herein, we report our chemical optimization efforts using HTS hit 1 as the chemical lead as well as detailed structure–activity relationship (SAR) studies on a series of substituted 2-(isoxazol-3-yl)-2-oxo-N′-phenyl-acetohydrazonoyl cyanide analogues. Several new molecules, such as 2-(5-(tert-butyl)isoxazol-3-yl)-N-(3,5-dichlorophenyl)-2-oxoacetohydrazonoyl cyanide (22, HJC0726),60 2-(5-(tert-butyl)isoxazol-3-yl)-2-oxo-N-(3,4,5-trichlorophenyl)acetohydrazonoyl cyanide (35, NY0123),60 and 2-(5-cyclohexylisoxazol-3-yl)-2-oxo-N-(3,4,5-trichlorophenyl)acetohydrazonoyl cyanide (47, NY0173,)60 have been identified as potent EPAC antagonists with low micromolar inhibitory activity.
Results and Discussion
Design
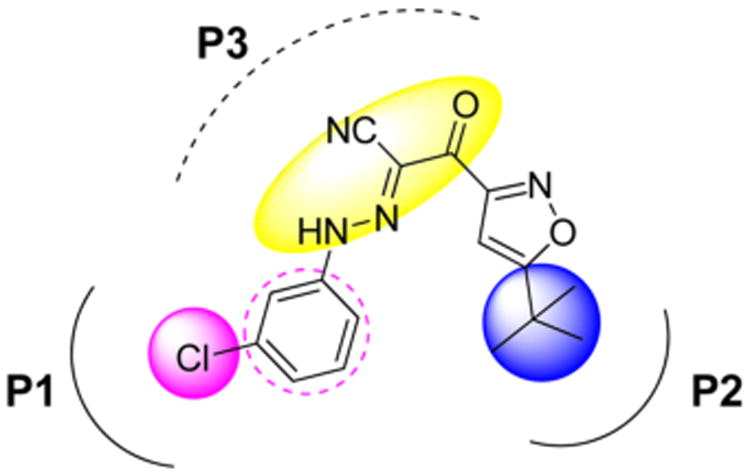
Given the well-characterized X-ray crystal structures of inactive61 and active62 forms of EPAC2 proteins, we examined the predicted the binding mode via molecular docking studies of 1 into the cAMP binding domain B of active EPAC2 protein using the AutoDock Vina algorithm,10 thereby understanding an effective interaction mode of the 2-(isoxazol-3-yl)-2-oxo-N′-phenyl-acetohydrazonoyl cyanide-based chemotype. Results suggest that the binding of the inhibitor to the EPAC2 protein may primarily occur through two terminal hydrophobic interactions and a unique linker. In the present work, we focus our new molecular design on the optimization of the aforementioned two main terminal hydrophobic pharmacophores, the 3-chlorophenyl moiety (P1) and the 5-tert-butyl group on the isoxazole (P2), as well as the exploration of the linker (P3) for systematic chemical modifications, as depicted in Figure 2.
Figure 2.
Proposed structural modifications on 1.
Chemistry
As outlined in Scheme 1, key intermediates 4a–e were generated by condensation of commercially available pinacolone 2a–e with oxalic acid diethyl ester 3 in the presence of NaH (60%) followed by oximation with hydroxylamine hydrochloride and subsequent cyclization by heating in a one-pot procedure in 30–87% yields.63 Ethyl esters 4a–e were then converted into the corresponding ketonitriles 5a–e by treatment with MeLi as the base and CH3CN as the cyanide source under our modified Kowalsko's protocol.63 Requisite aromatic amines 6 were treated with sodium nitrite and 2 N hydrochloric acid to give the corresponding aryldiazonium chlorides 7. Direct coupling of the aryldiazonium salts 7 with the crude cyanomethyl ketones 5a–e in the presence of NaOAc as the catalyst at 0 °C afforded new substituted 2-(isoxazol-3-yl)-2-oxo-N ′-phenyl-acetohydrazonoyl cyanide derivatives 8–48 in 24–76% yields in two steps from 4a–e (Scheme 1).
Scheme 1. Synthesis of Substituted 2-(Isoxazol-3-yl)-2-oxo-N′-phenyl-acetohydrazonoyl Cyanide Analogues with Modification on the Phenyl and Isoxazol Ringsa.
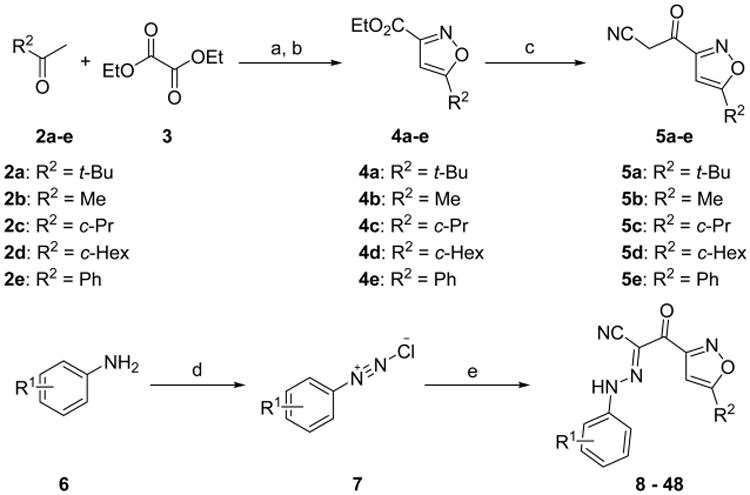
aReagents and conditions: (a) NaH, THF, 0 °C to rt; (b) NH2OH·HCl, EtOH/THF, rt to reflux, 30–87% for two steps; (c) CH3CN, MeLi, THF, −78 °C; (d) 2 N HCl, NaNO2, H2O, 0 °C; (e) 5a–e, NaOAc, EtOH, 24–76% for three steps.
As depicted in Schemes 2 and 3, amides 51–53 and 55, urea 54, carbohydrazide 56, and pyrazole 57 were prepared as novel substituted 2-(isoxazol-3-yl)-2-oxo-N′-phenyl-acetohydrazonoyl cyanide analogues bearing different linkers between the isoxazole and chlorobenzene of 1. Condensation of commercially available isoxazol-3-ylamines 49a,b with 2-cyanoacetic acid in the presence of EDCI and DIPEA yielded the corresponding intermediates 50a,b, which were then converted to 51 and 52 by coupling with 3-chlorophenyldiazonium salt upon treatment with NaOAc in 87 and 53% yields over two steps, respectively. Amide 53 and urea 54 were prepared by condensation of 49a with 3-chlorobenzoic acid and 1-isocyanato-4-chlorobenzene in 33 and 17% yields, respectively. Compound 55, the reverse amide of 53, was prepared first by removing the protection group of ester 4a, followed by condensation with 3-chloroaniline in the presence of HBTU and DIPEA, resulting in a 48% yield for two steps. Hydrazinolysis of ester 4a and then condensation with 3-chlorobenzaldehyde provided carbohydrazide 56 with a unique linker in 61% yield over two steps.64 To mask the ketone group, pyrazole 57 was prepared by cyclization of the linker of 1 with hydrazine hydrate in refluxing ethanol in 64% yield.65
Scheme 2. Synthesis of Substituted 2-(Isoxazol-3-yl)-2-oxo-N′-phenyl-acetohydrazonoyl Cyanide Analogues with Modification on the Linkera.

aReagents and conditions: (a) 2-cyanoacetic acid, EDCI, DIPEA, DCM, rt; (b) 7, NaOAc, EtOH, 0 °C to rt, 53-87% yields for two steps; (c) 3-chlorobenzoic acid, (COCl)2, Et3N, DCM, 0 °C to rt, 33%; (d) 1-isocyanato-3-chlorobenzene, DCM, rt, 17%; (e) LiOH, THF/H2O, rt; (f) 3-chloroaniline, HBTU, DIPEA, rt, 48% for two steps; (g) NH2NH2, EtOH, reflux; (h) 3-chlorobenzaldehyde, EtOH, rt, 61% for two steps.
Scheme 3. Synthesis of Substituted 2-(Isoxazol-3-yl)-2-oxo-N′-phenyl-acetohydrazonoyl Cyanide Analogue 57 with Cyclization of the Linker.

In Vitro Evaluation of EPAC2 Binding
All newly synthesized compounds have been evaluated for their ability to compete with 8-NBD-cAMP binding to recombinant EPAC2 proteins to determine IC50 values.36 Previous hit 1 and cAMP were used as the reference compounds, and our data were almost identical to those previously reported,10,39 with IC50 values of 8.9 and 32.0 μM in inhibiting EPAC2, respectively.
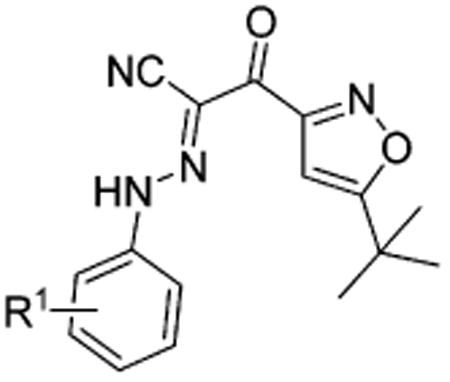
As shown in Table 1, we initially focused on investigating the effect of various substitutions on the phenyl ring of our previous hit 1. To evaluate the importance of substitutions, we removed 3-chloro on the phenyl ring of 1, leading to compound 8, which has about a 7-fold loss of binding affinity with an IC50 of 59.8 μM. Adjustment of 3-chloro to either the 2- or 4-position (as in compound 9 or 10) resulted in a 3-fold decreased inhibitory activity in comparison with that of 1, suggesting that a proper substitution at the 3-position is beneficial to its activity. We next explored the electronic effect of substitutions at the 3-position of the phenyl ring in detail. Replacement of 3-chloro with its congeners bromo and fluoro (as in compounds 11 and 12) led to a slight loss of activity compared to that of 1, whereas it increased activity by about 3–4-fold compared to that of nonsubstituted analogue 8, with IC50 values of 14.0 and 22.4 μM, respectively. Compounds 13 and 14, with other electron-withdrawing groups such as trifluoromethyl and nitro, led to approximately 5- and 2-fold increases in potency when compared to that of 8, with IC50 values of 11.9 and 31.3 μM, respectively. Compounds 15, with ethyl carboxyl, and 16, with a cyano group, both retained a similar inhibitory activity to that of 8. However, compound 17, with an acetyl group, displayed a dramatic loss of activity. Introduction of electron-donating groups, such as methoxy and its isomer, hydroxymethyl, afforded compounds 18 and 19, which also resulted in a significant loss of activity. Interestingly, compound 20 with a methyl group was about 2-fold more potent than that of nonsubstituted analogue 8, with an IC50 of 25.1 μM. These results suggest that the 3-position of the phenyl ring of 1 can tolerate chemical modifications with either electron-withdrawing or -donating groups.
Table 1. Apparent IC50 Values of Substituted 2-(Isoxazol-3-yl)-2-oxo-N′-phenyl-acetohydrazonoyl Cyanide Scaffolds with Modifications on the Phenyl Ring for Competing with 8-NBD-cAMP in Binding to EPAC2.
| |||
|---|---|---|---|
|
| |||
| Compd | R1 | IC50 (μM)a | relative affinity (RA)b |
| cAMP | 32.0 ± 5.9 | 1.0 | |
| 1 | 3-Cl | 8.9 ± 1.2 | 3.6 |
| 8 | H | 59.8 ± 9.4 | 0.5 |
| 9 | 2-Cl | 28.3 ± 4.0 | 1.1 |
| 10 | 4-Cl | 26.5 ± 4.1 | 1.2 |
| 11 | 3-Br | 14.0 ± 2.4 | 2.3 |
| 12 | 3-F | 22.4 ± 2.8 | 1.4 |
| 13 | 3-CF3 | 11.9 ± 1.7 | 2.7 |
| 14 | 3-NO2 | 31.3 ± 5.5 | 1.0 |
| 15 | 3-COOEt | 71.4 ± 23.8 | 0.4 |
| 16 | 3-CN | 67.8 ± 23.5 | 0.5 |
| 17 | 3-COCH3 | NEc | |
| 18 | 3-OMe | NE | |
| 19 | 3-CH2OH | NE | |
| 20 | 3-Me | 25.1 ± 4.8 | 1.3 |
| 21 | 2,5-di-Cl | 7.3 ± 1.2 | 4.4 |
| 22 | 3,5-di-Cl | 1.9 ± 0.339 | 16.8 |
| 23 | 3,4-di-Cl | 3.0 ± 0.6 | 10.7 |
| 24 | 2,3-di-Cl | 35.6 ± 7.9 | 0.9 |
| 25 | 3,5-di-CF3 | 6.4 ± 0.9 | 5.0 |
| 26 | 3-F, 5-CF3 | 5.8 ± 0.9 | 5.5 |
| 27 | 3,5-di-F | 5.3 ± 0.8 | 6.0 |
| 28 | 3-Cl, 5-CF3 | 1.6 ± 0.3 | 20.0 |
| 29 | 3-F, 5-Cl | 2.5 ± 0.4 | 12.8 |
| 30 | 3-F, 4-Cl | 4.3 ± 0.5 | 7.4 |
| 31 | 3-Cl, 4-F | 2.7 ± 0.3 | 11.9 |
| 32 | 3-CF3, 4-Cl | 2.9 ± 0.5 | 11.0 |
| 33 | 3,4-di-F | 6.6 ± 0.8 | 4.8 |
| 34 | 3-Cl, 4-CF3 | 3.6 ± 0.6 | 8.9 |
| 35 | 3,4,5-tri-Cl | 0.9 ± 0.2 | 35.6 |
| 36 | 3,4,5-tri-F | 1.1 ± 0.2 | 29.1 |
| 37 | 2,4,6-tri-Cl | 19.0 ± 1.7 | 1.7 |
The values are the mean ± SE of at least three independent experiments.
RA = IC50,cAMP/IC50,compound.
NE: no effect, indicating that a specific IC50 cannot be calculated from the data points collected.
We then explored the effect of disubstituted phenyl rings. By keeping 3-chloro and introducing another chloro group on the 2-, 4-, 5-, or 6-position (as in compounds 21–24), 3,4- and 3,5-dichloro analogues 22 and 23 bearing the best binding affinity were obtained and were about 5- and 3-fold more potent than that of parent compound 1, with IC50 values of 1.9 and 3.0 μM, respectively. We then explored fluorinated compounds given their notable successes in drug discovery, with an increasing number of fluorine-containing drugs on the market. Due to the unique nature of fluorine, fluorine substitution is commonly used in contemporary medicinal chemistry and drug design to alter physicochemical and conformational properties as well as to generate selective reactivity of developing candidates, thereby improving metabolic stability, solubility, bioavailability, and protein-ligand interactions.66,67 Moreover, 3-fluoro and 3-trifluoromethyl analogues 12 and 13 from the above monosubstituted SAR studies also exhibited comparable potency to that of 3-chloro (as in compound 1). Therefore, a series of compounds with fluoro, trifluoromethyl, and chloro groups in 3,4- or 3,5-disubstituted style was designed and synthesized (as in compounds 25–34). All of these compounds were approximately 2–6-fold more potent than hit 1. Compounds 28, 29, 31, and 32 displayed comparable potency to that of compounds 22 and 23, with IC50 values of 1.6–3.0 μM (Figure 3). Inspired by the results from the 3,4- and 3,5-disubstituted designs, we attempted to combine their substituted effects and study 3,4,5-trisubstituted analogues. Intriguingly, 3,4,5-trichloro-substituted analogue 35 and 3,4,5-trifluoro-substituted analogue 36 demonstrated enhanced potency, with IC50 values of 0.9 and 1.1 μM, respectively (Figure 3), whereas 2,4,6-trichloro-substituted analogue 37 resulted in a dramatic loss of activity, with an IC50 of 19.0 μM. These findings suggest that further optimization on the substitutions at the 3-, 4-, and/or 5-positions of the phenyl ring may offer the potential to develop new and more potent EPAC2 inhibitors.
Figure 3.
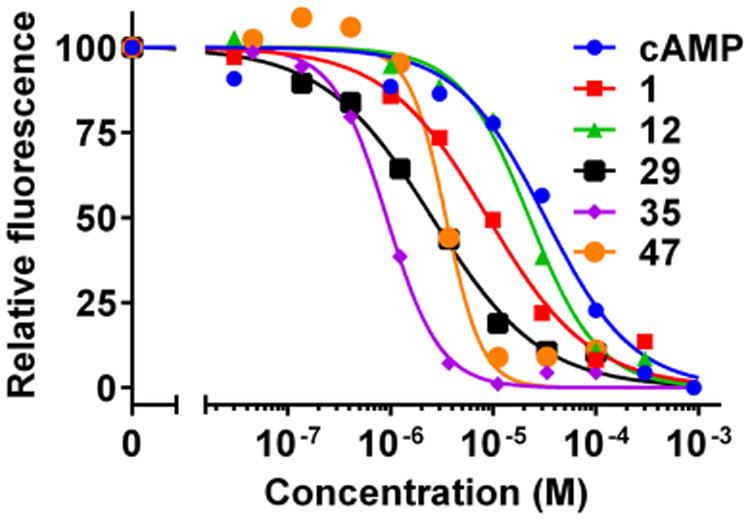
Relative binding affinity of EPAC2 antagonists. Dose-dependent competition of EPAC2 antagonists with 8-NBD-cAMP in binding to EPAC2.
Having identified an optimal substitution for the phenyl ring that maintains good potency, we then directed our effort on tuning the isoxazole scaffold. Compounds 1, 22, and 35 were chosen as reference compounds. By keeping 3-Cl, 3,5-di-Cl, and 3,4,5-tri-Cl as constant substitutions on the phenyl ring, we embarked on exploring the steric hindrance effects of substitutions on the 5-position of the isoxazole ring via replacing t-butyl group of the reference compound with a methyl, c-propyl, c-hexyl, or phenyl group. As shown in Table 2, compounds 38–41, 42–45, and 46–48 were three small parallel series, which mainly displayed good EPAC2 inhibitory effects in the order t-Bu > c-Hex ≥ c-Pr > Me > Ph. Among these, compounds 43, 44, 46, and 47 were more potent than hit 1, with IC50 values of 3.45–8.8 μM (Figure 3). Notably, compounds 41 and 48 with phenyl substitutions were totally inactive, indicating that more conformationally constricted biaryl moieties may be unsuitable for fitting into the shallow hydrophobic pockets of EPAC proteins. These results suggest that appropriate bulky alkyl substitutions offering sufficient lipophilicity are more favorable for the ligand to interact with EPAC2 proteins.
Table 2. Apparent IC50 Values of Substituted 2-(Isoxazol-3-yl)-2-oxo-N′-phenyl-acetohydrazonoyl Cyanide Scaffolds with Modifications on the Isoxazole Ring for Competing with 8-NBD-cAMP in Binding to EPAC2.
| ||||
|---|---|---|---|---|
|
| ||||
| compd | R1 | R2 | IC50 (μM)a | relative affinity (RA)b |
| cAMP | 32.0 ± 5.9 | 1.0 | ||
| 1 | 3-Cl | t-Bu | 8.9 ± 1.2 | 3.6 |
| 38 | 3-Cl | Me | NEc | |
| 39 | 3-Cl | c-Pr | 60.0 ± 20.9 | 0.5 |
| 40 | 3-Cl | c-Hex | 11.3 ± 2.1 | 2.8 |
| 41 | 3-Cl | Ph | NE | |
| 22 | 3,5-di-Cl | t-Bu | 1.9 ± 0.339 | 16.8 |
| 42 | 3,5-di-Cl | Me | 22.3 ± 0.4 | 1.4 |
| 43 | 3,5-di-Cl | c-Pr | 5.5 ± 1.5 | 5.8 |
| 44 | 3,5-di-Cl | c-Hex | 4.8 ± 1.2 | 6.7 |
| 45 | 3,5-di-Cl | Ph | 28.0 ± 4.0 | 1.1 |
| 35 | 3,4,5-tri-Cl | t-Bu | 0.9 ± 0.2 | 35.6 |
| 46 | 3,4,5-tri-Cl | c-Pr | 8.8 ± 2.3 | 3.6 |
| 47 | 3,4,5-tri-Cl | c-Hex | 3.5 ± 0.8 | 9.1 |
| 48 | 3,4,5-tri-Cl | Ph | NE | |
The values are the mean ± SE of at least three independent experiments.
RA = IC50,cAMP/IC50,compound.
NE: no effect, indicating that a specific IC50 cannot be calculated from the data points collected.
Amides 51–53 and 55, urea 54, carbohydrazide 56, and pyrazole 57 represented a subseries of novel substituted 2-(isoxazol-3-yl)-2-oxo-N′-phenyl-acetohydrazonoyl cyanide analogues bearing different linkages between the isoxazole and chlorobenzene by modifications of the linker length and type. Interestingly, compounds 51 and 52 with an additional NH fragment inserted into the linker of 1 and 38 were completely inactive in binding to EPAC2 (Table 3). Other linkages, including amide for 53 and 55, urea for 54, carbohydrazide for 56, and pyrazole for 57, also led to an entire loss of potency toward EPAC2 proteins. These results indicate that the length, shape, and type of the linkages are important for the ligand to interact with EPAC2 proteins.
Table 3. Apparent IC50 Values of Substituted 2-(Isoxazol-3-yl)-2-oxo-N′-phenyl-acetohydrazonoyl Cyanide Scaffolds with Modifications on the Linker for Competing with 8-NBD-cAMP in Binding to EPAC2.
| |||
|---|---|---|---|
|
| |||
| Compd | Linker | R3 | IC50 (μM)a |
| 1 |
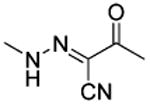
|
t-Bu | 8.9 |
| 51 |
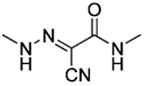
|
Me | NEb |
| 52 |
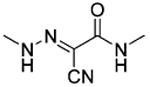
|
t-Bu | NE |
| 53 |
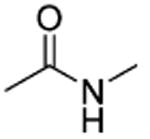
|
t-Bu | NE |
| 54 |
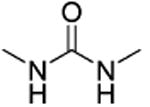
|
t-Bu | NE |
| 55 |
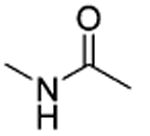
|
t-Bu | NE |
| 56 |
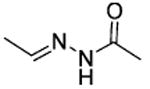
|
t-Bu | NE |
| 57 |
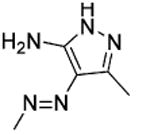
|
t-Bu | NE |
The values are the mean ± SE of at least three independent experiments.
NE: no effect, indicating that a specific IC50 cannot be calculated from the data points collected.
In Vitro Evaluation of EPAC1 Inhibition
From the biological results discussed above, compounds 22, 25, 28, 29, 31, 32, 34–36, 44, 46, and 47 were identified as potent EPAC2 binders with IC50 values lower than 10 μM. Therefore, these selected compounds together with hit 1 were further evaluated for their ability to inhibit EPAC1-mediated Rap1b-bGDP exchange activity. As shown in Table 4, previous hit 1 is slightly less potent toward EPAC1 inhibition than that on EPAC2, with an IC50 value of 10.8 μM. Interestingly, almost all of our selected, newly synthesized analogues exhibited significantly enhanced binding affinity compared to that of 1. Among these, compounds 22 and 35 shared the best inhibitory activity as EPAC1 inhibitors, with IC50 values of 2.4 μM (Figure 4). Compounds such as 22 and 35 with IC50 values lower than 5 μM for both EPAC1 and EPAC2 may serve as valuable pharmacological tools to probe the functions of EPAC in disease or as potential drug candidates for further preclinical development.
Table 4. Apparent IC50 Values of Substituted 2-(Isoxazol-3-yl)-2-oxo-N′-phenyl-acetohydrazonoyl Cyanide Scaffolds for Inhibiting EPAC1 GEF Activity.
| compd | EPAC1 IC50 (μM) |
|---|---|
| 1 | 10.8 ± 1.6 |
| 22 | 2.4 ± 0.2 |
| 25 | 5.7 ± 1.7 |
| 28 | 5.7 ± 1.5 |
| 29 | 11.6 ± 3.1 |
| 31 | 7.8 ± 2.0 |
| 32 | 9.7 ± 2.3 |
| 34 | 4.3 ± 0.9 |
| 35 | 2.4 ± 0.3 |
| 36 | 8.9 ± 2.2 |
| 44 | 2.6 ± 0.6 |
| 46 | 4.1 ± 0.7 |
| 47 | 4.0 ± 0.9 |
Figure 4.
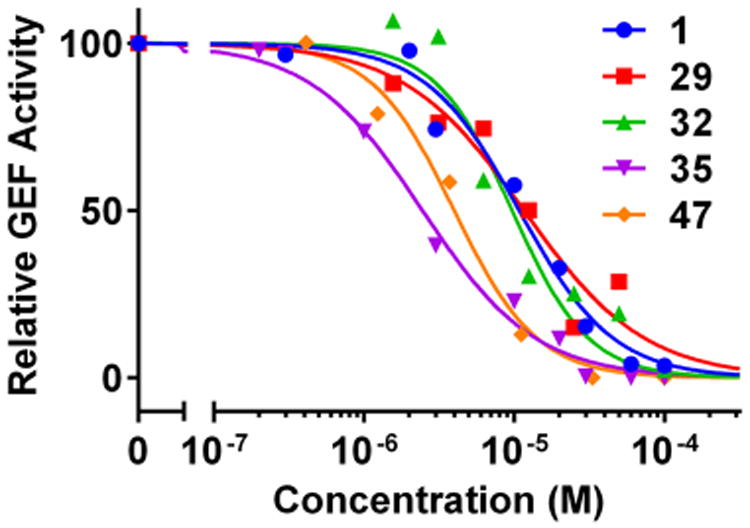
Relative inhibitory activity for EPAC1-mediated Rap1b-bGDP exchange.
Solubility of Selected Ligands in Water and Ethanol
To examine whether the synthesized substituted 2-(isoxazol-3-yl)-2-oxo-N′-phenyl-acetohydrazonoyl cyanide analogues have better solubility than 1 and to define their therapeutic window, a previously reported HPLC analysis method68,69 was employed to measure the solubility of several selected analogues, such as 25, 28, 31, 35, 36, and 47 (Table 5). One-point calibration was performed against standards with known concentrations of the sample compounds to determine concentrations of the indicated compounds in samples. 1 itself has limited aqueous solubility, with a maximum concentration around 5.95 μg/mL, whereas it possesses excellent solubility in ethanol (14.98 mg/mL). The addition of another or two chloro atoms onto the phenyl ring of 1 not only enhanced their binding affinity on EPAC1 and EPAC2 but also improved their aqueous solubility. For instance, aqueous solubility of analogue 22, with 3,5-di-Cl on the phenyl ring, is enhanced with a saturated concentration of 45.4 μg/mL, and those of 3,4,5-tri-Cl derivatives 35 and 47 are 12.7 and 10.1 μg/mL, respectively. Meanwhile, fluorinated compounds 25, 28, 31, and 36 also exhibit improved both potency and aqueous solubility. In particular, compound 25, with 3,5-di-CF3 on the phenyl ring of 1, displays a significantly improved solubility both in water and ethanol, with saturated concentrations of 418.5 μg/mL and more than 150 mg/mL, indicating approximately 70- and 10-fold improvements in comparison with that of 1, respectively (Table 5).
Table 5. Solubility of Selected Compounds in Water and Ethanol.
| compd | MW | solubility in watera | solubility in ethanola |
|---|---|---|---|
| 139 | 330.769 | 5.95 ± 0.83 μg/mL | 14.98 ± 0.39 mg/mL |
| (18.0 ± 2.5 μM) | (45.3 ± 1.2 mM) | ||
| 2239 | 365.214 | 45.4 ± 1.4 μg/mL | 6.8 ± 0.86 mg/mL |
| (124.3 ± 3.8 μM) | (18.6 ± 2.3 mM) | ||
| 25 | 432.320 | 418.5 ± 17.3 μg/mL | >150 mg/mL |
| (968.0 ± 40.0 μM) | (>347.0 mM) | ||
| 28 | 398.770 | 20.1 ± 0.8 μg/mL | 14.6 ± 0.9 mg/mL |
| (50.4 ± 2.0 μM) | (36.6 ± 2.3 mM) | ||
| 31 | 348.762 | 82.6 ± 3.6 μg/mL | 8.21 ± 0.9 mg/mL |
| (236.8 ± 10.3 μM) | (23.5 ± 2.6 mM) | ||
| 35 | 399.659 | 12.7 ± 1.0 μg/mL | 4.37 ± 0.52 mg/mL |
| (31.8 ± 2.5 μM) | (10.9 ± 1.3 mM) | ||
| 36 | 350.301 | 75.7 ± 7.1 μg/mL | 28.6 ± 1.4 mg/mL |
| (216.0 ± 20.3 μM) | (81.6 ± 4.0 mM) | ||
| 47 | 425.694 | 10.1 ± 0.79 μg/mL | 3.63 ± 0.46 mg/mL |
| (23.7 ± 1.9 μM) | (8.5 ± 1.1 mM) |
The values are the mean ± SE of at least three independent experiments.
Predicted Docking Mode of Compound 47 with the cAMP Binding Domain (CBD) of EPAC2 Protein
Since our robust HTS assay is particularly sensitive for identifying compounds that directly compete with 8-NBD-cAMP in binding to EPAC2, we predicted that our compounds may bind to the cAMP binding domain (CBD) of EPAC2. Molecular docking studies were performed to help us better understand the structure–activity relationships of these new compounds toward EPAC2. Using the AutoDock 4.0 method, the docking results revealed that our new compounds fit nicely into the functional cAMP binding pocket of active EPAC2 (PDB code: 3CF6).32,62 To further characterize the binding pose, we selected analogue 47 as a case study for our theoretical investigation. As shown in Figure 5A, the hydrophobic cyclohexylisoxazolyl moiety interacts with the hydrophobic residues of Phe 367, Leu406, Ala407, and Ala415, whereas the 3,4,5-trichlorophenyl fragment forms hydrophobic interactions with residues Val386 and Leu397. Moreover, this binding mode is further stabilized by the occurrence of hydrogen bonds between the NH hydrogen atom of the linker and the oxygen atom of residue Asp402. These results are in full agreement with our established SAR data that hydrophobic groups at the isoxazolyl moiety and phenyl scaffold as well as the types of the linkers are crucial to target EPAC2. The molecular docking studies of 47 also comply with our previous results with hit 110 through the overlay analysis of the two ligands, as depicted in Figure 5B. Given the critical role of the linker, these docking findings may also help us to understand why amides 53 and 55, urea 54, carbohydrazide 56, and pyrazole 57 display no significant activity. Taken together, these studies suggest that optimization of the substitutions at the isoxazolyl moiety and phenyl scaffold while keeping the NH groups on the linker may provide further opportunities to improve the binding affinity and specificity of the substituted 2-(isoxazol-3-yl)-2-oxo-N′-phenyl-acetohydrazonoyl cyanide analogues toward EPAC proteins.
Figure 5.
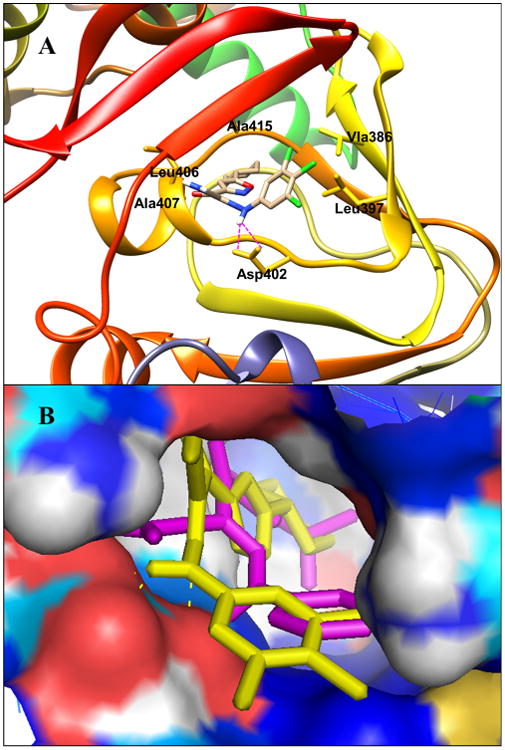
(A) Predicted binding mode and molecular docking of 47 into the cAMP binding domain B (CBD) of EPAC2 protein. Important residues are drawn in sticks. Hydrogen bonds are shown as dashed purple lines. (B) Overlay analysis of ligands 1 and 47. 1 is shown in purple, and 47 is depicted in yellow.
Conclusions
In summary, a series of substituted 2-(isoxazol-3-yl)-2-oxo-N′-phenyl-acetohydrazonoyl cyanide derivatives has been designed, synthesized, and biologically evaluated with respect to their inhibitory capability toward EPAC proteins. The SAR results based on EPAC2 activity were consistent with our docking studies, indicating that further modifications at the 3-, 4-, and 5-positions of the phenyl scaffold and the 5-position of the isoxazole moiety may allow us to tune the original hit 1 to achieve more potent EPAC antagonists. Interestingly, various linkers explored led to a significant loss in activity, indicating that the linker in the parent compound plays a critical role in the binding activity toward EPAC. Furthermore, several new molecules, such as compounds 22, 35, and 47, have been identified as potent EPAC1 and EPAC2 inhibitors with IC50 values in the low micromolar to submicromolar range. These compounds may hold promise as potential drug candidates for the preclinical development to IND-enabling studies toward novel therapeutics against human diseases. Additionally, these small-molecule inhibitors may serve as valuable pharmacological probes to facilitate our efforts in elucidating the physiological functions of EPAC in disease and associated signaling pathways. Further studies of selected compounds in various human disease models and a systematic optimization based upon identified advanced chemical leads toward EPAC subtype selectivity are under way and will be reported in due course.
Experimental Section
General Chemistry Information
All commercially available starting materials and solvents were reagent grade and used without further purification. Reactions were performed under a nitrogen atmosphere in dry glassware with magnetic stirring. Preparative column chromatography was performed using silica gel 60, particle size 0.063–0.200 mm (70–230 mesh, flash). Analytical TLC was carried out employing silica gel 60 F254 plates (Merck, Darmstadt). Visualization of the developed chromatograms was performed with UV detection (254 nm). NMR spectra were recorded on a Bruker-600 or Bruker-300 (1H, 600 and 300 MHz; 13C, 150 and 75 MHz) spectrometer. 1H and 13C NMR spectra were recorded with TMS as an internal reference. Chemical shifts are expressed in ppm, and J values are given in Hz. High-resolution mass spectra (HRMS) were obtained from Thermo Fisher LTQ Orbitrap Elite mass spectrometer. Parameters include the following: nano ESI spray voltage was 1.8 kV, capillary temperature was 275 °C, resolution was 60 000, and ionization was achieved in positive mode. Melting points were measured on a Thermo Scientific electrothermal digital melting point apparatus and are uncorrected. Purities of final compounds were established by analytical HPLC, which was carried out on a Shimadzu HPLC system (model no. CBM-20A LC-20AD SPD-20A UV/vis). HPLC analysis conditions were as follows: Waters μBondapak C18 (300 × 3.9 mm), flow rate of 0.5 mL/min, UV detection at 270 and 254 nm, and linear gradient from 10% acetonitrile in water to 100% acetonitrile in water in 20 min followed by 30 min of the last-named solvent (0.1% TFA was added into both acetonitrile and water). All biologically evaluated compounds are >95% pure.
Ethyl 5-tert-Butylisoxazole-3-carboxylate (4a)
To a solution of pinacolone (500 mg, 5.0 mmol) in THF (10 mL) was added NaH (60%) (220 mg, 5.5 mmol) at 0 °C. The reaction mixture was stirred at rt for about 30 min. Diethyl oxalate (730 mg, 5.0 mmol) was added at 0 °C, and the mixture was then stirred at rt overnight. To the resulting solution was added hydroxylamine hydrochloride (380 mg, 5.5 mmol) in ethanol (10 mL) dropwise. The mixture was heated at reflux for 16 h. After this time, the sodium chloride was removed by filtration, and the filtrate was concentrated under reduced pressure. The residue was purified by short column chromatography on silica gel, eluting with hexane/ethyl acetate (4:1) to provide 4a as a colorless oil (855 mg, 87%). 1H NMR (600 MHz, CDCl3) δ 6.37 (s, 1H), 4.43 (q, J = 7.2 Hz, 2H), 1.41 (t, J = 7.2 Hz, 3H), 1.37 (s, 9H).
Ethyl 5-Methylisoxazole-3-carboxylate (4b)
Compound 4b was prepared in 55% yield (two steps from acetone) by a procedure similar to that used to prepare compound 4a. 1H NMR (300 MHz, CDCl3) δ 6.36 (s, 1H), 4.38 (q, J = 7.2 Hz, 2H), 2.48 (s, 3H), 1.34 (t, J = 7.2 Hz, 3H).
Ethyl 5-Cyclopropylisoxazole-3-carboxylate (4c)
Compound 4c was prepared in 74% yield (two steps from 1-cyclopropylethanone) by a procedure similar to that used to prepare compound 4a. 1H NMR (300 MHz, CDCl3) δ 6.32 (s, 1H), 4.44 (q, J = 7.1 Hz, 2H), 2.07 (m, 1H), 1.42 (t, J = 7.1 Hz, 3H), 1.18–1.10 (m, 2H), 1.06–0.98 (m, 2H).
Ethyl 5-Cyclohexylisoxazole-3-carboxylate (4d)
Compound 4d was prepared in 30% yield (two steps from 1-cyclohexylethanone) by a procedure similar to that used to prepare compound 4a. 1H NMR (300 MHz, CDCl3) δ 6.37 (s, 1H), 4.42 (q, J = 7.2 Hz, 2H), 2.91–2.78 (m, 1H), 2.12–2.04 (m, 2H), 1.91–1.62 (m, 3H), 1.49–1.25 (m, 8H).
Ethyl 5-Phenylisoxazole-3-carboxylate (4e)
Compound 4e was prepared in 73% yield (two steps from acetophenone) by a procedure similar to that used to prepare compound 4a. 1H NMR (300 MHz, CDCl3) δ 7.78 (dd, J = 6.5, 3.2 Hz, 2H), 7.45 (dd, J = 5.0, 1.8 Hz, 3H), 6.91 (s, 1H), 4.45 (q, J = 7.1 Hz, 2H), 1.42 (t, J = 7.1 Hz, 3H).
2-(5-tert-Butylisoxazol-3-yl)-N′-phenyl-2-oxoacetohydrazonoyl Cyanide (8)
To a solution of CH3CN (410 mg, 10.0 mmol) in anhydrous THF (5 mL) was added 1.6 M methyl lithium in diethyl ether (3.1 mL, 5.0 mmol) at −78 °C under nitrogen. The mixture was stirred at −78 °C for 0.5 h, and 4a (500 g, 2.5 mmol) in THF (5 mL) was then added dropwise. The solution was stirred at −78 °C for 1 h and then quenched with acetic acid (0.3 mL, 5.0 mmol). The mixture was warmed to 0 °C, poured onto ice/water (10 mL), and extracted with ethyl acetate (50 mL). The organic lay was dried over Na2SO4, filtered, and concentrated under reduced pressure. Crude residue 5a (490 mg) was obtained as a yellow oil and directly used in the next step without further purification.
To a solution of aniline (22 mg, 0.24 mmol) in H2O (1 mL) cooled to −5 °C was added 0.24 mL of 1 N HCl (aq.). To the resulting acidic aniline solution was added 1 mL of a solution of sodium nitrite (16 mg, 0.24 mmol) in H2O dropwise to generate the aryldiazonium salt solution. To the aryldiazonium salt solution was added sodium acetate (33 mg, 0.4 mmol), followed by 1 mL of a solution of crude 5a (38 mg, 0.2 mmol) in ethanol. The reaction mixture was stirred at 0 °C for 5 min, poured onto H2O (2 mL), and extracted with ethyl acetate (20 mL). The organic layer was dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by short column chromatography on silica gel, eluting with hexane/ethyl acetate (2:1) to provide desired product 8 (45 mg, 76%, two steps from 4a) as a yellow solid (mp 118–119 °C). HPLC purity 99.4% (tR = 20.50 min). 1H NMR (600 MHz, DMSO-d6) δ 12.60 (bs, 1H), 7.49 (d, 2H, J = 7.8 Hz), 7.42 (d, 2H, J = 7.8 Hz), 7.20–7.23 (m, 1H), 6.66 (s, 1H), 1.37 (s, 9H). 13C NMR (150 MHz, DMSO-d6) δ 180.8, 179.2, 160.0, 142.1, 129.3, 125.6, 116.8, 112.2, 110.7, 100.2, 32.4, 28.3. HRMS (ESI) calcd for C16H17N4O2 (M + H)+, 297.1346; found, 297.1355.
2-(5-tert-Butylisoxazol-3-yl)-N′-(2-chlorophenyl)-2-oxoacetohydrazonoyl Cyanide (9)
Compound 9 was prepared in 76% yield (two steps from 4a) by a procedure similar to that used to prepare compound 8. The title compound was obtained as a yellow solid (mp 143–144 °C). HPLC purity 96.6% (tR = 22.77 min). 1H NMR (600 MHz, DMSO-d6) δ 7.53–7.60 (m, 1H), 7.47–7.52 (m, 1H), 7.37–7.46 (m, 1H), 7.22–7.36 (m, 1H), 6.66 (s, 1H), 1.34 (s, 9H). 13C NMR (150 MHz, DMSO-d6) δ 180.8, 178.6, 159.9, 139.3, 129.9, 128.1, 126.9, 123.5, 119.6, 114.4, 110.7, 99.9, 32.3, 28.2. HRMS (ESI) calcd for C16H16ClN4O2 (M + H)+, 331.0956; found, 331.0969.
2-(5-tert-Butylisoxazol-3-yl)-N′-(4-chlorophenyl)-2-oxoacetohydrazonoyl Cyanide (10)
Compound 10 was prepared in 75% yield (two steps from 4a) by a procedure similar to that used to prepare compound 8. The title compound was obtained as a yellow solid (mp 142–143 °C). HPLC purity 98.1% (tR = 21.74 min). 1H NMR (600 MHz, DMSO-d6) δ 7.46–7.60 (m, 4H), 6.64 (s, 1H), 1.36 (s, 9H). 13C NMR (150 MHz, DMSO-d6) δ 181.5, 179.6, 160.8, 142.5, 130.1, 129.9, 119.2, 113.3, 111.6, 100.8, 33.0, 29.0. HRMS (ESI) calcd for C16H16ClN4O2 (M + H)+, 331.0956; found, 331.0963.
2-(5-tert-Butylisoxazol-3-yl)-N′-(3-bromophenyl)-2-oxoacetohydrazonoyl Cyanide (11)
Compound 11 was prepared in 75% yield (two steps from 4a) by a procedure similar to that used to prepare compound 8. The title compound was obtained as a yellow solid (mp 162–163 °C). HPLC purity 98.3% (tR = 21.93 min). 1H NMR (600 MHz, DMSO-d6) δ 12.84 (s, 1H), 7.60 (s, 1H), 7.48–7.52 (m, 1H), 7.36–7.40 (m, 2H), 6.69 (s, 1H), 1.38 (s, 9H). 13C NMR (150 MHz, DMSO-d6) δ 181.0, 179.3, 160.0, 143.8, 131.4, 127.9, 122.3, 119.0, 116.2, 113.3, 110.5, 100.3, 32.5, 28.4. HRMS (ESI) calcd for C16H16BrN4O2 (M + H)+, 375.0451; found, 375.0456.
2-(5-tert-Butylisoxazol-3-yl)-N′-(3-fluorophenyl)-2-oxoacetohydrazonoyl Cyanide (12)
Compound 12 was prepared in 62% yield (two steps from 4a) by a procedure similar to that used to prepare compound 8. The title compound was obtained as a yellow solid (mp 140–141 °C). HPLC purity 99.4% (tR = 21.73 min). 1H NMR (600 MHz, DMSO-d6) δ 12.88 (s, 1H), 7.46 (td, J = 8.2, 6.5 Hz, 1H), 7.38–7.31 (m, 1H), 7.22 (d, J = 10.8 Hz, 1H), 7.03 (td, J = 8.4, 2.2 Hz, 1H), 6.67 (s, 1H), 1.36 (s, 9H). 13C NMR (150 MHz, DMSO-d6) δ 180.9, 179.2, 162.6 (d, J = 242.3 Hz), 160.2, 131.2 (d, J = 8.85 Hz), 113.3, 112.9, 111.9 (d, J = 21.2 Hz), 110.8, 103.7, 103.6, 100.3, 32.4, 28.4. HRMS (ESI) calcd for C16H16FN4O2 (M + H)+, 315.1252; found, 315.1248.
2-(5-tert-Butylisoxazol-3-yl)-N′-(3-trifluoromethylphenyl)-2-oxoacetohydrazonoyl Cyanide (13)
Compound 13 was prepared in 33% yield (two steps from 4a) by a procedure similar to that used to prepare compound 8. The title compound was obtained as a yellow solid (mp 147–148 °C). HPLC purity 96.0% (tR = 21.80 min). 1H NMR (600 MHz, DMSO-d6) δ 7.74 (d, J = 8.0 Hz, 1H), 7.70 (s, 1H), 7.64 (t, J = 7.9 Hz, 1H), 7.51 (d, J = 7.6 Hz, 1H), 6.63 (s, 1H), 1.36 (s, 9H). 13C NMR (150 MHz, DMSO-d6) δ 181.2, 179.7, 161.1, 131.1, 130.5 (q, J = 31.5 Hz), 125.5, 123.5, 122.4, 121.8, 113.7, 112.2, 100.6, 32.9, 28.9. HRMS (ESI) calcd for C17H16F3N4O2 (M + H)+, 365.1220; found, 365.1230.
2-(5-tert-Butylisoxazol-3-yl)-N′-(3-nitrophenyl)-2-oxoacetohydrazonoyl Cyanide (13)
Compound 14 was prepared in 29% yield (two steps from 4a) by a procedure similar to that used to prepare compound 8. The title compound was obtained as a yellow solid (mp 151–152 °C). HPLC purity 96.4% (tR = 20.33 min). 1H NMR (600 MHz, DMSO-d6) δ 12.03 (s, 1H), 8.19 (s, 1H), 7.97 (d, 1H, J = 8.4 Hz), 7.85 (d, 1H, J = 7.8 Hz), 7.67 (t, 1H, J = 7.8 Hz), 6.62 (s, 1H), 1.37 (s, 9H). 13C NMR (150 MHz, DMSO-d6) δ 180.5, 179.0, 171.9, 160.8, 148.5, 130.7, 124.6, 119.1, 113.4, 112.1, 111.2, 100.1, 32.4, 28.4. HRMS (ESI) calcd for C16H16N5O4 (M + H)+, 342.1197; found, 342.1207.
Ethyl 3-(2-(2-(5-tert-Butylisoxazol-3-yl)-1-cyano-2-oxoethylidene)hydrazinyl)benzoate (15)
Compound 15 was prepared in 74% yield (two steps from 4a) by a procedure similar to that used to prepare compound 8. The title compound was obtained as a yellow solid (mp 155–156 °C). HPLC purity 98.8% (tR = 21.53 min). 1H NMR (600 MHz, DMSO-d6) δ 12.92 (s, 1H), 8.16 (s, 1H), 7.76 (d, 1H, J = 7.8 Hz), 7.74 (d, 1H, J = 8.4 Hz), 7.57 (t, 1H, J = 8.4 Hz), 6.67 (s, 1H), 4.32 (q, 2H, J = 7.2 Hz), 1.37 (s, 9H), 1.34 (t, 3H, J = 7.2 Hz). 13C NMR (150 MHz, DMSO-d6) δ 181.1, 179.2, 165.1, 159.9, 142.4, 131.2, 129.8, 125.9, 121.0, 117.3, 113.2, 110.5, 100.1, 61.0, 32.5, 28.4, 14.1. HRMS (ESI) calcd for C19H21N4O4 (M + H)+, 369.1557; found, 369.1558.
2-(5-tert-Butylisoxazol-3-yl)-N′-(3-cyanophenyl)-2-oxoacetohydrazonoyl Cyanide (16)
Compound 16 was prepared in 58% yield (two steps from 4a) by a procedure similar to that used to prepare compound 8. The title compound was obtained as a yellow solid (mp 181–182 °C). HPLC purity 99.3% (tR = 19.87 min). 1H NMR (600 MHz, DMSO-d6) δ 7.75–7.77 (m, 1H), 7.69 (s, 1H), 7.61–7.63 (m, 2H), 6.69 (s, 1H), 1.36 (s, 9H). 13C NMR (150 MHz, DMSO-d6) δ 181.3, 179.5, 160.3, 143.8, 131.0, 128.7, 122.0, 119.8, 118.4, 113.9, 112.4, 110.8, 100.5, 32.7, 28.6. HRMS (ESI) calcd for C17H16N5O2 (M + H)+, 322.1299; found, 322.1303.
N′-(3-Acetylphenyl)-2-(5-tert-butylisoxazol-3-yl)-2-oxoacetohydrazonoyl Cyanide (17)
Compound 17 was prepared in 68% yield (two steps from 4a) by a procedure similar to that used to prepare compound 8. The title compound was obtained as a yellow solid (mp 152–153 °C). HPLC purity 98.2% (tR = 19.80 min). 1H NMR (600 MHz, DMSO-d6) δ 8.05 (s, 1H), 7.79 (d, 1H, J = 7.8 Hz), 7.72 (d, 1H, J = 8.4 Hz), 7.57 (t, 1H, J = 7.8 Hz), 6.66 (s, 1H), 2.57 (s, 3H), 1.37 (s, 9H). 13C NMR (150 MHz, DMSO-d6) δ 197.4, 181.4, 179.6, 160.2, 142.7, 138.0, 130.1, 125.6, 121.3, 116.0, 113.3, 100.7, 100.3, 32.7, 28.6, 26.8. HRMS (ESI) calcd for C18H19N4O3 (M + H)+, 339.1452; found, 339.1459.
2-(5-tert-Butylisoxazol-3-yl)-N′-(3-methoxyphenyl)-2-oxoacetohydrazonoyl Cyanide (18)
Compound 18 was prepared in 61% yield (two steps from 4a) by a procedure similar to that used to prepare compound 8. The title compound was obtained as a yellow solid (mp 125–126 °C). HPLC purity 95% (tR = 21.73 min). 1H NMR (600 MHz, DMSO-d6) δ 7.32 (t, J = 8.1 Hz, 1H), 7.11 (m, 2H), 6.78 (dt, J = 9.0, 4.5 Hz, 1H), 6.65 (s, 1H), 3.77 (s, 3H), 1.36 (s, 9H). 13C NMR (150 MHz, DMSO-d6) δ 181.0, 179.4, 160.1, 130.3, 112.3, 111.4, 110.7, 109.3, 102.5, 100.1, 55.1, 32.4, 28.4. HRMS (ESI) calcd for C17H19N4O3 (M + H)+, 327.1452; found, 327.1450.
2-(5-tert-Butylisoxazol-3-yl)-N′-(3-(hydroxymethyl)phenyl)-2-oxoacetohydrazonoyl Cyanide (19)
Compound 19 was prepared in 63% yield (two steps from 4a) by a procedure similar to that used to prepare compound 7. The title compound was obtained as a yellow solid (mp 147–148 °C). HPLC purity 99.6% (tR = 17.86 min). 1H NMR (600 MHz, DMSO-d6) δ 12.81 (s, 1H), 8.28 (s, 1H), 7.46 (s, 1H), 7.35–7.40 (m, 1H), 7.13–7.18 (m, 1H), 6.65 (s, 1H), 4.49 (s, 2H), 1.36 (s, 9H). 13C NMR (150 MHz, DMSO-d6) δ 180.9, 179.4, 160.0, 144.2, 142.0, 129.1, 123.6, 115.4, 114.5, 112.4, 110.7, 100.2, 62.5, 32.4, 28.4. HRMS (ESI) calcd for C17H19N4O3 (M + H)+, 327.1452; found, 327.1457.
2-(5-tert-Butylisoxazol-3-yl)-2-oxo-N′-m-tolylacetohydrazonoyl Cyanide (20)
Compound 20 was prepared in 50% yield (two steps from 4a) by a procedure similar to that used to prepare compound 8. The title compound was obtained as a yellow solid (mp 131–132 °C). HPLC purity 99.0% (tR = 21.29 min). 1H NMR (600 MHz, DMSO-d6) δ 12.73 (s, 1H), 7.31 (m, 2H), 7.28 (s, 1H), 7.04 (d, J = 6.0 Hz, 1H), 6.68 (s, 1H), 2.30 (s, 3H), 1.37 (s, 9H). 13C NMR (150 MHz, DMSO-d6) δ 180.9, 179.4, 160.0, 142.0, 138.9, 129.3, 126.5, 117.0, 114.4, 112.3, 110.7, 100.3, 32.4, 28.4, 21.1. HRMS (ESI) calcd for C17H19N4O2 (M + H)+, 311.1503; found, 311.1514.
2-(5-tert-Butylisoxazol-3-yl)-N′-(2,5-dichlorophenyl)-2-oxoacetohydrazonoyl Cyanide (21)
Compound 21 was prepared in 62% yield (two steps from 4a) by a procedure similar to that used to prepare compound 8. The title compound was obtained as a yellow solid (mp 193–194 °C). HPLC purity 97.1% (tR = 23.69 min). 1H NMR (600 MHz, DMSO-d6) δ 7.45–7.54 (m, 1H), 7.34 (s, 1H), 7.13–7.22 (m, 1H), 6.56 (s, 1H), 1.37 (s, 9H). 13C NMR (150 MHz, DMSO-d6) δ 180.4, 179.6, 162.1, 147.3, 132.8, 132.0, 125.7, 125.4, 118.7, 114.4, 113.7, 100.8, 31.2, 29.0. HRMS (ESI) calcd for C16H15Cl2N4O2 (M + H)+, 365.0567; found, 365.0576.
2-(5-tert-Butylisoxazol-3-yl)-N′-(2,5-dichlorophenyl)-2-oxoacetohydrazonoyl Cyanide (21)
Compound 21 was prepared in 62% yield (two steps from 4a) by a procedure similar to that used to prepare compound 8. The title compound was obtained as a yellow solid (mp 193–194 °C). HPLC purity 97.1% (tR = 23.69 min). 1H NMR (600 MHz, DMSO-d6) δ 7.45–7.54 (m, 1H), 7.34 (s, 1H), 7.13–7.22 (m, 1H), 6.56 (s, 1H), 1.37 (s, 9H). 13C NMR (150 MHz, DMSO-d6) δ 180.4, 179.6, 162.1, 147.3, 132.8, 132.0, 125.7, 125.4, 118.7, 114.4, 113.7, 100.8, 31.2, 29.0. HRMS (ESI) calcd for C16H15Cl2N4O2 (M + H)+, 365.0567; found, 365.0576.
2-(5-tert-Butylisoxazol-3-yl)-N′-(3,5-dichlorophenyl)-2-oxoacetohydrazonoyl Cyanide (22)
Compound 22 was prepared in 41% yield (two steps from 4a) by a procedure similar to that used to prepare compound 8. The title compound was obtained as a yellow solid (mp 191–192 °C). HPLC purity 99.0% (tR = 23.20 min). 1H NMR (300 MHz, DMSO-d6) δ 7.42 (s, 2H), 7.39 (s, 1H), 6.70 (s, 1H), 1.38 (s, 9H). 13C NMR (75 MHz, DMSO-d6) δ 181.6, 179.7, 160.5, 145.4, 135.3, 124.7, 115.9, 114.7, 110.9, 100.9, 33.0, 29.0. HRMS (ESI) calcd for C16H15Cl2N4O2 (M + H)+, 365.0567; found, 365.0563.
2-(5-tert-Butylisoxazol-3-yl)-N′-(3,4-dichlorophenyl)-2-oxoacetohydrazonoyl Cyanide (23)
Compound 23 was prepared in 55% yield (two steps from 4a) by a procedure similar to that used to prepare compound 8. The title compound was obtained as a yellow solid (mp 189–190 °C). HPLC purity 97.5% (tR = 23.74 min). 1H NMR (300 MHz, DMSO-d6) δ 7.66 (d, J = 8.8 Hz, 1H), 7.56 (d, J = 2.2 Hz, 1H), 7.42 (dd, J = 8.7, 2.4 Hz, 1H), 6.62 (s, 1H), 1.37 (s, 9H). 13C NMR (75 MHz, DMSO-d6) δ 181.1, 179.5, 161.2, 145.6, 132.3, 131.7, 127.3, 118.9, 118.8, 113.7, 112.3, 100.8, 33.0, 29.0. HRMS (ESI) calcd for C16H15Cl2N4O2 (M + H)+, 365.0567; found, 365.0577.
2-(5-tert-Butylisoxazol-3-yl)-N′-(2,3-dichlorophenyl)-2-oxoacetohydrazonoyl Cyanide (24)
Compound 24 was prepared in 68% yield (two steps from 4a) by a procedure similar to that used to prepare compound 8. The title compound was obtained as a yellow solid (mp 187–188 °C). HPLC purity 97.5% (tR = 23.74 min). 1H NMR (300 MHz, DMSO-d6) δ 7.35–7.62 (m, 3H), 6.65 (s, 1H), 1.35 (s, 9H). 13C NMR (75 MHz, DMSO-d6) δ 181.4, 179.2, 160.8, 133.0, 129.1, 127.4, 118.6, 100.5, 33.0, 29.0. HRMS (ESI) calcd for C16H15Cl2N4O2 (M + H)+, 365.0567; found, 365.0568.
N′-(3,5-Bis(trifluoromethyl)phenyl)-2-(5-tert-butylisoxazol-3-yl)-2-oxoacetohydrazonoyl Cyanide (25)
Compound 25 was prepared in 43% yield (two steps from 4a) by a procedure similar to that used to prepare compound 8. The title compound was obtained as a yellow solid (mp 138–139 °C). HPLC purity 96.4% (tR = 22.96 min). 1H NMR (600 MHz, DMSO-d6) δ 7.91 (s, 2H), 7.76 (s, 1H), 6.58 (s, 1H), 1.33 (s, 9H). 13C NMR (150 MHz, DMSO-d6) δ 180.6, 179.3, 161.1, 149.0, 131.3 (q, J = 32.6 Hz), 123.3 (q, J = 271.5 Hz), 118.2, 117.2, 114.1, 112.5, 100.2, 32.5, 28.5. HRMS (ESI) calcd for C18H15F6N4O2 (M + H)+, 433.1094; found, 433.1098.
2-(5-tert-Butylisoxazol-3-yl)-N′-(3-fluoro-5-(trifluoromethyl)-phenyl)-2-oxoacetohydrazonoyl Cyanide (26)
Compound 26 was prepared in 47% yield (two steps from 4a) by a procedure similar to that used to prepare compound 8. The title compound was obtained as a yellow solid (mp 160–161 °C). HPLC purity 97.5% (tR = 22.36 min). 1H NMR (300 MHz, DMSO-d6) δ 7.60 (s, 1H), 7.54–7.42 (m, 2H), 6.68 (s, 1H), 1.36 (s, 9H). 13C NMR (75 MHz, DMSO-d6) δ 181.5, 179.7, 163.02 (d, J = 244.5 Hz), 160.6, 146.6, 132.2 (qd, J = 33.0 and 10.5 Hz), 125.4, 121.7, 114.7, 111.2, 110.1, 109.4, 108.5, 108.2, 100.7, 33.0, 28.8. HRMS (ESI) calcd for C17H15F4N4O2 (M + H)+, 383.1131; found, 383.1124.
2-(5-tert-Butylisoxazol-3-yl)-N′-(3,5-difluorophenyl)-2-oxoacetohydrazonoyl Cyanide (27)
Compound 27 was prepared in 50% yield (two steps from 4a) by a procedure similar to that used to prepare compound 8. The title compound was obtained as a yellow solid (mp 183–184 °C). HPLC purity 96.1% (tR = 21.56 min). 1H NMR (300 MHz, DMSO-d6) δ 7.21–6.98 (m, 3H), 6.69 (s, 1H), 1.36 (s, 9H). 13C NMR (75 MHz, DMSO-d6) δ 181.6, 179.7, 163.5 (d, J = 243.0 Hz), 163.3 (d, J = 243. Eight Hz), 160.5, 145.5 (t, J = 12.0 Hz), 114.4, 110.8, 101.1, 100.9, 100.8, 100.5, 100.4, 33.0, 28.9. HRMS (ESI) calcd for C16H15F2N4O2 (M + H)+, 333.1163; found, 333.1162.
2-(5-tert-Butylisoxazol-3-yl)-N′-(3-chloro-5-(trifluoromethyl)-phenyl)-2-oxoacetohydrazonoyl Cyanide (28)
Compound 28 was prepared in 46% yield (two steps from 4a) by a procedure similar to that used to prepare compound 8. The title compound was obtained as a yellow solid (mp 173–174 °C). HPLC purity 99.4% (tR = 23.11 min). 1H NMR (300 MHz, DMSO-d6) δ 7.71 (s, 2H), 7.64 (s, 1H), 6.71 (s, 1H), 1.36 (s, 9H). 13C NMR (75 MHz, DMSO-d6) δ 181.7, 179.8, 160.4, 145.1, 135.5, 132.2 (q, J = 32.3 Hz), 125.3, 121.6 (d, J = 3.8 Hz), 120.6, 115.1, 112.6 (d, J = 3.8 Hz), 110.9, 100.8, 33.0, 28.9. HRMS (ESI) calcd for C17H15ClF3N4O2 (M + H)+, 399.0836; found, 399.0830.
2-(5-tert-Butylisoxazol-3-yl)-N′-(3-chloro-5-fluorophenyl)-2-oxoacetohydrazonoyl Cyanide (29)
Compound 29 was prepared in 52% yield (two steps from 4a) by a procedure similar to that used to prepare compound 8. The title compound was obtained as a yellow solid (mp 175–176 °C). HPLC purity 98.3% (tR = 22.31 min). 1H NMR (300 MHz, DMSO-d6) δ 7.31 (s, 1H), 7.23 (s, 2H), 6.69 (s, 1H), 1.37 (s, 9H). 13C NMR (75 MHz, DMSO-d6) δ 186.4, 184.5, 167.8 (d, J = 245.3 Hz), 165.2, 150.4, 140.1 (d, J = 12.8 Hz), 119.3, 118.1, 117.7, 117.3, 115.7, 108.4, 108.1, 105.6, 37.7, 33.7. HRMS (ESI) calcd for C16H15ClFN4O2 (M + H)+, 349.0868; found, 349.0862.
2-(5-tert-Butylisoxazol-3-yl)-N′-(4-chloro-3-fluorophenyl)-2-oxoacetohydrazonoyl Cyanide (30)
Compound 30 was prepared in 40% yield (two steps from 4a) by a procedure similar to that used to prepare compound 8. The title compound was obtained as a yellow solid (mp 170–171 °C). HPLC purity 99.2% (tR = 21.94 min). 1H NMR (300 MHz, DMSO-d6) δ 7.65 (t, J = 8.4 Hz, 1H), 7.36 (t, J = 9.0 Hz, 2H), 6.68 (s, 1H), 1.37 (s, 9H). 13C NMR (75 MHz, DMSO-d6) δ 181.6, 179.6, 160.6, 158.0 (d, J = 244.4 Hz), 143.5, 132.0, 116.1, 115.9, 114.5, 114.1, 111.0, 105.7, 105.4, 100.8, 33.0, 28.9. HRMS (ESI) calcd for C16H15ClFN4O2 (M + H)+, 349.0868; found, 349.0860.
2-(5-tert-Butylisoxazol-3-yl)-N′-(3-chloro-4-fluorophenyl)-2-oxoacetohydrazonoyl Cyanide (31)
Compound 31 was prepared in 40% yield (two steps from 4a) by a procedure similar to that used to prepare compound 8. The title compound was obtained as a yellow solid (mp 173–174 °C). HPLC purity 97.4% (tR = 21.70 min). 1H NMR (300 MHz, DMSO-d6) δ 7.55 (d, J = 5.9 Hz, 1H), 7.52–7.45 (m, 2H), 6.69 (s, 1H), 1.37 (s, 9H). 13C NMR (75 MHz, DMSO-d6) δ 181.6, 179.7, 160.6, 155.2 (d, J = 243.7 Hz), 140.3, 121.1, 120.8, 118.7, 118.4, 118.1, 117.9, 113.6, 111.1, 100.8, 33.0, 29.0. HRMS (ESI) calcd for C16H15ClFN4O2 (M + H)+, 349.0868; found, 349.0859.
2-(5-tert-Butylisoxazol-3-yl)-N′-(4-chloro-3-(trifluoromethyl)-phenyl)-2-oxoacetohydrazonoyl Cyanide (32)
Compound 32 was prepared in 42% yield (two steps from 4a) by a procedure similar to that used to prepare compound 8. The title compound was obtained as a yellow solid (mp 175–176 °C). HPLC purity 99.8% (tR = 22.58 min). 1H NMR (300 MHz, DMSO-d6) δ 7.91 (s, 1H), 7.79 (d, J = 8.6 Hz, 1H), 7.72 (d, J = 8.7 Hz, 1H), 6.67 (s, 1H), 1.35 (s, 9H). 13C NMR (75 MHz, DMSO-d6) δ 181.7, 179.7, 160.5, 142.4, 133.3, 128.0 (q, J = 30.8 Hz), 126.7, 124.8, 122.3, 121.2, 116.3, 116.2, 114.5, 111.1, 100.6, 33.0, 28.9. HRMS (ESI) calcd for C17H15ClF3N4O2 (M + H)+, 399.0836; found, 399.0829.
2-(5-tert-Butylisoxazol-3-yl)-N′-(3,4-difluorophenyl)-2-oxoacetohydrazonoyl Cyanide (33)
Compound 33 was prepared in 45% yield (two steps from 4a) by a procedure similar to that used to prepare compound 8. The title compound was obtained as a yellow solid (mp 185–186 °C). HPLC purity 98.2% (tR = 21.12 min). 1H NMR (300 MHz, DMSO-d6) δ 7.53 (dd, J = 18.7, 9.3 Hz, 1H), 7.45–7.29 (m, 2H), 6.68 (s, 1H), 1.37 (s, 9H). 13C NMR (75 MHz, DMSO-d6) δ 181.6, 179.7, 160.6, 150.2 (dd, J = 13.8 and 244.3 Hz), 148.0 (dd, J = 12.7 and 167.7 Hz), 140.0 (d, J = 7.4 Hz), 118.9 (d, J = 18.5 Hz), 113.9, 113.5, 111.1, 106.4 (d, J = 21.9 Hz), 100.8, 33.0, 28.9. HRMS (ESI) calcd for C16H15F2N4O2 (M + H)+, 333.1163; found, 333.1154.
2-(5-tert-Butylisoxazol-3-yl)-N′-(3-chloro-4-(trifluoromethyl)-phenyl)-2-oxoacetohydrazonoyl Cyanide (34)
Compound 34 was prepared in 35% yield (two steps from 4a) by a procedure similar to that used to prepare compound 8. The title compound was obtained as a yellow solid (mp 176–177 °C). HPLC purity 98.9% (tR = 22.61 min). 1H NMR (300 MHz, DMSO-d6) δ 7.90 (d, J = 8.7 Hz, 1H), 7.63 (s, 1H), 7.56 (d, J = 8.8 Hz, 1H), 6.70 (s, 1H), 1.38 (s, 9H). 13C NMR (75 MHz, DMSO-d6) δ 181.7, 179.7, 160.5, 147.7, 132.4, 129.8, 122.6 (q, J = 31.2 Hz), 119.2, 116.2, 115.4, 111.0, 100.9, 33.0, 29.0. HRMS (ESI) calcd for C17H15ClF3N4O2 (M + H)+, 399.0836; found, 399.0828.
2-(5-tert-Butylisoxazol-3-yl)-2-oxo-N′-(3,4,5-trichlorophenyl)acetohydrazonoyl Cyanide (35)
Compound 35 was prepared in 52% yield (two steps from 4a) by a procedure similar to that used to prepare compound 8. The title compound was obtained as a yellow solid (mp 208–209 °C). HPLC purity 98.6% (tR = 13.22 min). 1H NMR (300 MHz, DMSO-d6) δ 7.50 (s, 2H), 6.55 (s, 1H), 1.37 (s, 9H). 13C NMR (75 MHz, DMSO-d6) δ 180.6, 179.4, 161.8, 133.8, 124.9, 119.1, 113.6, 100.8, 32.9, 29.0. HRMS (ESI) calcd for C16H14Cl3N4O2 (M + H)+, 399.0177; found, 399.0171.
2-(5-tert-Butylisoxazol-3-yl)-2-oxo-N′-(3,4,5-trifluorophenyl)acetohydrazonoyl Cyanide (36)
Compound 36 was prepared in 24% yield (two steps from 4a) by a procedure similar to that used to prepare compound 8. The title compound was obtained as a yellow solid (mp 190–191 °C). HPLC purity 97.7% (tR = 21.62 min). 1H NMR (300 MHz, DMSO-d6) δ 7.28 (d, J = 6.3 Hz, 1H), 7.26 (d, J = 6.3 Hz, 1H), 6.69 (s, 1H), 1.37 (s, 9H). 13C NMR (75 MHz, DMSO-d6) δ 181.6, 179.5, 160.6, 152.8 (dd, J = 10.5, 5.2 Hz), 149.6 (dd, J = 10.4, 4.9 Hz), 139.7, 114.2, 111.1, 102.2, 101.8, 100.8, 33.0, 28.9. HRMS (ESI) calcd for C17H15ClF3N4O2 (M + H)+, 399.0836; found, 399.0828.
2-(5-tert-Butylisoxazol-3-yl)-2-oxo-N′-(2,4,6-trichlorophenyl)acetohydrazonoyl Cyanide (37)
Compound 37 was prepared in 37% yield (two steps from 4a) by a procedure similar to that used to prepare compound 8. The title compound was obtained as a yellow solid (mp 185–186 °C). HPLC purity 98.9% (tR = 12.77 min). 1H NMR (600 MHz, DMSO-d6) δ 7.55 (s, 2H), 6.39 (s, 1H), 1.27 (s, 9H). 13C NMR (150 MHz, DMSO-d6) δ 179.3, 179.0, 162.7, 148.2, 128.9, 128.5, 128.2, 116.2, 113.5, 100.5, 32.7, 29.0. HRMS (ESI) calcd for C16H14Cl3N4O2 (M + H)+, 399.0177; found, 399.0169.
N′-(3-Chlorophenyl)-2-(5-methylisoxazol-3-yl)-2-oxoacetohydrazonoyl Cyanide (38)
Compound 38 was prepared in 42% yield (two steps from 4b) by a procedure similar to that used to prepare compound 8. The title compound was obtained as a yellow solid (mp 143–144 °C). HPLC purity 98.4% (tR = 19.18 min). 1H NMR (600 MHz, DMSO-d6) δ 7.51 (s, 1H), 7.41–7.45 (m, 2H), 7.20–7.25 (m, 1H), 6.63 (s, 1H), 2.51 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 179.3, 170.3, 160.6, 144.4, 134.1, 131.3, 125.2, 116.8, 115.9, 113.3, 111.0, 103.0, 11.8. HRMS (ESI) calcd for C13H10ClN4O2 (M + H)+, 289.0487; found, 289.0492.
N′-(3-Chlorophenyl)-2-(5-cyclopropylisoxazol-3-yl)-2-oxoacetohydrazonoyl Cyanide (39)
Compound 39 was prepared in 66% yield (two steps from 4c) by a procedure similar to that used to prepare compound 8. The title compound was obtained as a yellow solid (mp 169–170 °C). HPLC purity 99.6% (tR = 10.02 min). 1H NMR (300 MHz, DMSO-d6) δ 7.51 (s, 1H), 7.45 (d, J = 4.6 Hz, 2H), 7.25 (s, 1H), 6.62 (s, 1H), 2.25 (m, 1H), 1.14 (m, 2H), 0.98 (m, 2H). 13C NMR (75 MHz, DMSO-d6) δ 179.7, 176.1, 160.7, 144.0, 134.4, 131.7, 125.6, 116.9, 116.1, 113.8, 110.9, 100.2, 9.1, 8.0. HRMS (ESI) calcd for C15H12ClN4O2 (M + H)+, 315.0643; found, 315.0625.
N′-(3-Chlorophenyl)-2-(5-cyclohexylisoxazol-3-yl)-2-oxoacetohydrazonoyl Cyanide (40)
Compound 40 was prepared in 58% yield (two steps from 4d) by a procedure similar to that used to prepare compound 8. The title compound was obtained as a yellow solid (mp 176–177 °C). HPLC purity 99.7% (tR = 12.20 min). 1H NMR (300 MHz, DMSO-d6) δ 7.46 (t, J = 5.1 Hz, 3H), 7.25 (dt, J = 5.1, 2.5 Hz, 1H), 6.68 (s, 1H), 3.00–2.86 (m, 1H), 2.04 (d, J = 11.5 Hz, 2H), 1.83–1.63 (m, 3H), 1.42 (dt, J = 25.3, 11.9 Hz, 4H), 1.31–1.17 (m, 1H). 13C NMR (75 MHz, DMSO-d6) δ 179.9, 178.3, 160.5, 144.0, 134.5, 131.7, 125.6, 116.7, 116.2, 113.9, 110.9, 101.4, 35.8, 31.1, 25.7, 25.5. HRMS (ESI) calcd for C18H18ClN4O2 (M + H)+, 357.1118; found, 357.1128.
N′-(3-Chlorophenyl)-2-oxo-2-(5-phenylisoxazol-3-yl)-acetohydrazonoyl Cyanide (41)
Compound 41 was prepared in 55% yield (two steps from 4e) by a procedure similar to that used to prepare compound 8. The title compound was obtained as a yellow solid (mp 199–200 °C). HPLC purity 99.8% (tR = 21.88 min). 1H NMR (300 MHz, DMSO-d6) δ 7.98 (dd, J = 7.4, 2.1 Hz, 2H), 7.57 (m, 4H), 7.47 (m, 3H), 7.26 (d, J = 7.4 Hz, 1H). 13C NMR (75 MHz, DMSO-d6) δ 179.4, 170.1, 161.5, 144.1, 134.4, 131.7, 131.4, 129.8, 126.6, 126.3, 125.7, 117.1, 116.2, 113.8, 111.0, 102.0. HRMS (ESI) calcd for C18H12ClN4O2 (M + H)+, 351.0649; found, 351.0641.
N′-(3,5-Dichlorophenyl)-2-(5-methylisoxazol-3-yl)-2-oxoacetohydrazonoyl Cyanide (42)
Compound 42 was prepared in 35% yield (two steps from 4b) by a procedure similar to that used to prepare compound 8. The title compound was obtained as a yellow solid (mp 185–186 °C). HPLC purity 98.4% (tR = 20.79 min). 1H NMR (600 MHz, DMSO-d6) δ 7.45 (s, 2H), 7.36 (s, 1H), 6.63 (s, 1H), 2.50 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 179.1, 170.2, 160.8, 146.4, 134.9, 124.1, 116.0, 113.9, 111.2, 103.0, 11.8. HRMS (ESI) calcd for C13H9Cl2N4O2 (M + H)+, 323.0097; found, 323.0103.
2-(5-Cyclopropylisoxazol-3-yl)-N′-(3,5-dichlorophenyl)-2-oxoacetohydrazonoyl Cyanide (43)
Compound 43 was prepared in 44% yield (two steps from 4c) by a procedure similar to that used to prepare compound 8. The title compound was obtained as a yellow solid (mp 209–210 °C). HPLC purity 98.7% (tR = 11.05 min). 1H NMR (300 MHz, DMSO-d6) δ 7.45 (s, 2H), 7.39 (s, 1H), 6.61 (s, 1H), 2.30–2.19 (m, 1H), 1.14 (m, 2H), 1.03–0.92 (m, 2H). 13C NMR (75 MHz, DMSO-d6) δ 179.5, 176.1, 160.8, 145.7, 135.3, 124.6, 116.1, 114.5, 111.1, 100.2, 9.2, 8.0. HRMS (ESI) calcd for C15H11Cl2N4O2 (M + H)+, 349.0259; found, 349.0257.
2-(5-Cyclohexylisoxazol-3-yl)-N′-(3,5-dichlorophenyl)-2-oxoacetohydrazonoyl Cyanide (44)
Compound 44 was prepared in 48% yield (two steps from 4d) by a procedure similar to that used to prepare compound 8. The title compound was obtained as a yellow solid (mp 215–216 °C). HPLC purity 99.7% (tR = 13.30 min). 1H NMR (300 MHz, DMSO-d6) δ 7.44 (d, J = 1.8 Hz, 2H), 7.39 (t, J = 2.0 Hz, 1H), 6.68 (s, 1H), 2.93 (t, J = 6.5 Hz, 1H), 2.04 (d, J = 11.5 Hz, 2H), 1.80–1.66 (m, 3H), 1.54–1.33 (m, 4H), 1.26 (t, J = 11.2 Hz, 1H). 13C NMR (75 MHz, DMSO-d6) δ 179.6, 178.3, 160.5, 145.7, 135.3, 124.6, 116.1, 114.7, 111.0, 101.3, 35.9, 31.2, 25.7, 25.5. HRMS (ESI) calcd for C18H17Cl2N4O2 (M + H)+, 391.0729; found, 391.0724.
N′-(3,5-Dichlorophenyl)-2-oxo-2-(5-phenylisoxazol-3-yl)-acetohydrazonoyl Cyanide (45)
Compound 45 was prepared in 40% yield (two steps from 4e) by a procedure similar to that used to prepare compound 8. The title compound was obtained as a yellow solid (mp 213–214 °C). HPLC purity 97.7% (tR = 23.17 min). 1H NMR (300 MHz, DMSO-d6) δ 7.96 (m, 2H), 7.57 (m, 3H), 7.47 (s, 3H), 7.36 (s, 1H). 13C NMR (75 MHz, DMSO-d6) δ 179.0, 169.9, 161.8, 146.9, 135.2, 131.3, 129.8, 126.7, 126.3, 124.6, 116.5, 114.3, 111.7, 102.1. HRMS (ESI) calcd for C18H11Cl2N4O2 (M + H)+, 385.0259; found, 385.0252.
2-(5-Cyclopropylisoxazol-3-yl)-2-oxo-N′-(3, 4, 5-trichlorophenyl)acetohydrazonoyl Cyanide (46)
Compound 46 was prepared in 36% yield (two steps from 4c) by a procedure similar to that used to prepare compound 8. The title compound was obtained as a yellow solid (mp 220–221 °C). HPLC purity 98.3% (tR = 12.18 min). 1H NMR (300 MHz, DMSO-d6) δ 7.53 (s, 2H), 6.49 (s, 1H), 2.21 (dt, J = 8.3, 3.8 Hz, 1H), 1.11 (dt, J = 8.6, 3.2 Hz, 2H), 0.99–0.91 (m, 2H). 13C NMR (75 MHz, DMSO-d6) δ 179.2, 175.2, 162.0, 133.8, 125.0, 119.0, 113.6, 100.1, 9.0, 8.0. HRMS (ESI) calcd for C15H10N4O2 (M + H)+, 382.9869; found, 382.9881.
2-(5-Cyclohexylisoxazol-3-yl)-2-oxo-N′-(3,4,5-trichlorophenyl)acetohydrazonoyl Cyanide (47)
Compound 47 was prepared in 35% yield (two steps from 4d) by a procedure similar to that used to prepare compound 8. The title compound was obtained as a yellow solid (mp 228–229 °C). HPLC purity 99.2% (tR = 13.85 min). 1H NMR (300 MHz, DMSO-d6) δ 7.57 (s, 2H), 6.64 (s, 1H), 2.94–2.87 (m, 1H), 2.03 (d, J = 11.2 Hz, 2H), 1.79–1.64 (m, 3H), 1.54–1.36 (m, 4H), 1.29–1.17 (m, 1H). 13C NMR (75 MHz, DMSO-d6) δ 179.5, 178.1, 160.7, 134.0, 125.7, 117.9, 114.7, 111.4, 101.4, 35.8, 31.2, 25.7, 25.5. HRMS (ESI) calcd for C18H16ClN4O2 (M + H)+, 425.0335; found, 425.0338.
N′-(3,4,5-Trichlorophenyl)-2-oxo-2-(5-phenylisoxazol-3-yl)-acetohydrazonoyl Cyanide (48)
Compound 48 was prepared in 51% yield (two steps from 4e) by a procedure similar to that used to prepare compound 8. The title compound was obtained as a yellow solid (mp 230–231 °C). HPLC purity 98.5% (tR = 24.07 min). 1H NMR (300 MHz, DMSO-d6) δ 7.98–7.93 (m, 2H), 7.65 (s, 2H), 7.57 (m, 3H), 7.48 (s, 1H). 13C NMR (75 MHz, DMSO-d6) δ 179.0, 170.0, 161.7, 144.2, 134.1, 131.4, 129.8, 126.7, 126.3, 125.8, 118.1, 114.6, 102.1. HRMS (ESI) calcd for C18H10Cl3N4O2 (M + H)+, 418.9864; found, 418.9864.
N-(5-tert-Butyl-isoxazol-3-yl)-2-[(3-chlorophenyl)-hydrazono]-2-cyanoacetamide (51)
To a solution of 5-tert-butyl-isoxazol-3-ylamine 49a (140 mg, 1.0 mmol) and cyanoacetic acid (85 mg, 1.0 mmol) in 10 mL of DCM was added DIPEA (258 mg, 2.0 mmol). EDCI (191 mg, 1.0 mmol) was added at 0 °C. The resulting mixture was stirred at rt for 16 h. The solution was diluted with DCM (50 mL) and washed with 1 N HCl (aq.) (10 mL) and brine (10 mL). The organic layer was dried over anhydrous Na2SO4 and then concentrated under reduced pressure. The residue was purified by silica gel column chromatography (hexane/EtOAc 1:1) to give desired product 50a as a white solid (163 mg, 90%). 1H NMR (600 MHz, CDCl3) δ 10.59 (s, 1H), 6.70 (s, 1H), 3.65 (s, 2H), 1.36 (s, 9H). 13C NMR (150 MHz, CDCl3) δ 182.9, 159.8, 157.6, 113.4, 93.7, 33.4, 28.7, 26.9.
To a solution of 3-chloroaniline (25 mg, 0.2 mmol) in H2O (1 mL cooled to −5 °C) was added 0.2 mL of 1 N HCl (aq.). To the resulting acidic aniline solution was added 1 mL of a solution of sodium nitrite (14 mg, 0.2 mmol) in H2O dropwise to generate the aryldiazonium salt solution. To the aryldiazonium salt solution was added sodium acetate (33 mg, 0.4 mmol), followed by 1 mL of a solution of 50a (29 mg, 0.14 mmol) in ethanol. The reaction mixture was stirred at 0 °C for 5 min, poured onto H2O (2 mL), and extracted with ethyl acetate (20 mL). The organic layer was dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by short column chromatography on silica gel, eluting with hexane/ethyl acetate (2:1) to provide desired product 51 (32 mg, 66%) as a yellow solid (mp 238–240 °C). HPLC purity 96.7% (tR = 20.97 min). 1H NMR (600 MHz, DMSO-d6) δ 12.04 (s, 1H), 11.22 (s, 1H), 7.98 (s, 1H), 7.69 (d, 1H, J = 9.0 Hz), 7.40 (t, 1H, J = 7.8 Hz), 7.17 (d, 1H, J = 7.8 Hz), 6.63 (s, 1H), 1.32 (s, 9H). 13C NMR (150 MHz, DMSO-d6) δ 180.2, 159.5, 157.2, 143.0, 133.6, 130.4, 123.8, 115.8, 115.0, 110.6, 107.9, 93.6, 32.2, 28.1. HRMS (ESI) calcd for C16H17ClN5O2 (M + H)+, 346.1065; found, 346.1074.
2-[(3-Chlorophenyl)-hydrazono]-2-cyano-N-(5-methyl-isoxazol-3-yl)acetamide (52)
Compound 52 was prepared in 53% yield (two steps) by a procedure similar to that used to prepare compound 51. The title compound was obtained as a yellow solid (mp 241–243 °C). HPLC purity 98.5% (tR = 18.55 min). 1H NMR (600 MHz, DMSO-d6) δ 12.03 (s, 1H), 11.19 (s, 1H), 7.98 (s, 1H), 7.69 (d, 1H, J = 8.4 Hz), 7.39 (t, 1H, J = 7.8 Hz), 7.18 (d, 1H, J = 6.6 Hz), 6.67 (s, 1H), 2.43 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 169.6, 159.8, 157.9, 143.4, 133.9, 130.7, 124.1, 116.1, 115.3, 110.9, 108.3, 97.0, 12.2. HRMS (ESI) calcd for C13H11ClN5O2 (M + H)+, 304.0596; found, 304.0606.
N-(5-(tert-Butyl)isoxazol-3-yl)-3-chlorobenzamide (53)
To a solution of 3-chlorobenzoic acid (35 mg, 0.25 mmol) in 5 mL of DCM was added (COCl)2 (212 μL, 2.5 mmol) at 0 °C, and the mixture was allowed to stir rt for 1 h. The solution was concentrated and then dissolved in 5 mL of DCM. 5-(tert-Butyl)isoxazol-3-amine 49a (78 mg, 0.5 mmol) and Et3N (151 mg, 1.5 mmol) were added at 0 °C; then, the mixture was stirred at rt overnight. The solution was diluted by DCM and washed with 1 N NaHSO4, saturated NaHCO3, and brine (5 mL each). The organic layer was concentrated and purified by PTLC to obtain 53 (25 mg, 33%) as a white solid (mp 145–146 °C). HPLC purity 96.5% (tR = 20.08 min). 1H NMR (300 MHz, CDCl3) δ 10.74 (s, 1H), 8.05 (t, J = 1.9 Hz, 1H), 7.96 (ddd, J = 7.7, 1.7, 1.1 Hz, 1H), 7.59 (ddd, J = 8.0, 2.1, 1.1 Hz, 1H), 7.47 (t, J = 7.8 Hz, 1H), 6.91 (s, 1H), 1.38 (s, 9H). 13C NMR (75 MHz, CDCl3) δ 181.8, 164.6, 135.2, 134.9, 132.4, 129.9, 128.2, 126.0, 94.0, 33.1, 28.7. HRMS (ESI) calcd for C14H16ClN2O2 (M + H)+, 279.0900; found, 279.0894.
1-(5-tert-Butylisoxazol-3-yl)-3-(3-chlorophenyl)urea (54)
A mixture of 5-(tert-butyl)isoxazol-3-amine 49a (30 mg, 0.21 mmol) and 1-chloro-3-isocyanatobenzene (33 mg, 0.21 mmol) in 5 mL of DCM was stirred at rt overnight. The mixture was concentrated under vacuum. The residue was purified with silica gel column (hexane/EtOAc 10:1 to 5:1) to obtain 54 (10 mg, 17%) as a white solid (mp 114–116 °C). HPLC purity 98.0% (tR = 19.99 min). 1H NMR (300 MHz, CDCl3) δ 11.08 (s, 1H), 9.24 (s, 1H), 7.75 (t, J = 2.0 Hz, 1H), 7.55 (d, J = 8.0 Hz, 1H), 7.36 (t, J = 8.0 Hz, 1H), 7.29 (s, 1H), 5.81 (s, 1H), 1.37 (s, 9H). 13C NMR (75 MHz, CDCl3) δ 181.6, 177.7, 158.4, 139.3, 134.4, 129.8, 126.9, 125.1, 123.1, 91.3, 33.0, 28.6. HRMS (ESI) calcd for C14H17ClN3O2 (M + H)+, 294.1009; found, 294.0992.
5-tert-Butyl-N-(3-chlorophenyl)isoxazole-3-carboxamide (55)
4a (1000 mg, 5.1 mmol) and LiOH (638 mg, 15.3 mmol) were dissolved in 30 mL of MeOH and 10 mL of H2O, respectively. Then, the solution was stirred at rt for 1 h. In an ice-cooled bath, 1 N Na2SO4 (10 mL) was added, and the mixture was extracted by ethyl acetate (20 mL × 3). The organic layer was separated and washed with brine (10 mL). After drying over anhydrous Na2SO4, the solution was concentrated to give 5-tert-butylisoxazole-3-carboxylic acid as a light yellow oil (818 mg, 95%). 1H NMR (300 MHz, CDCl3) δ 10.64 (s, 1H), 6.42 (s, 1H), 1.37 (s, 9H).
To a solution of 5-tert-butylisoxazole-3-carboxylic acid (70 mg, 0.41 mmol) and 3-chloroaniline (53 mg, 0.41 mmol) in 5 mL of DCM were added HBTU (395 mg, 1.23 mmol) and DIPEA (271 mg, 2.1 mmol). The mixture was stirred at rt for 18 h. The mixture was washed with 1 N Na2SO4, saturated NaHCO3, and brine (5 mL each). After drying over anhydrous Na2SO4, the solution was concentrated and purified with silica gel column (hexane/EtOAc 10:1 to 7:1) to obtain 55 (54 mg, 51%) as a colorless oil. HPLC purity 99.6% (tR = 21.11 min). 1H NMR (300 MHz, CDCl3) δ 8.58 (s, 1H), 7.81 (t, J = 2.0 Hz, H1), 7.50 (ddd, J = 8.1, 2.1, 1.0 Hz, 1H), 7.30 (d, J = 7.8 Hz, 1H), 7.16 (ddd, J = 8.0, 2.0, 1.0 Hz, 1H), 6.52 (s, 1H), 1.41 (s, 9H). 13C NMR (75 MHz, CDCl3) δ 183.8, 158.3, 157.1, 138.1, 134.8, 130.1, 124.9, 120.1, 118.0, 98.4, 33.1, 28.8. HRMS (ESI) calcd for C14H16ClN2O2 (M + H)+, 279.0900; found, 279.0895.
(E)-5-(tert-Butyl)-N′-(3-chlorobenzylidene)isoxazole-3-carbohydrazide (56)
Ethyl 5-(tert-butyl)isoxazole-3-carboxylate 4a (100 mg, 0.5 mmol) and NH2NH2 (76 mg, 1.5 mmol) were dissolved in 5 mL of EtOH and refluxed for 2 h. 3-Chlorobenzaldehyde was then added, and the solution was allowed to stir at rt overnight. Then the solution was concentrated and extracted with DCM (10 mL × 3). After drying over anhydrous Na2SO4, the solution was concentrated and purified with silica gel column (hexane/EtOAc 20:1 to 10:1) to obtain 56 (89 mg, 61% for two steps) as a white solid (mp 157–159 °C). HPLC purity 99.5% (tR = 19.74 min). 1H NMR (300 MHz, CDCl3) δ 9.88 (s, 1H), 8.23 (s, 1H), 7.84 (d, J = 1.7 Hz, 1H), 7.70–7.60 (m, 1H), 7.45–7.32 (m, 2H), 6.58 (s, 1H), 1.41 (s, 9H). 13C NMR (75 MHz, CDCl3) δ 183.6, 157.4, 155.5, 147.6, 135.1, 135.0, 130.8, 130.0, 127.5, 126.1, 98.8, 33.1, 28.8. HRMS (ESI) calcd for C15H17ClN3O2 (M + H)+, 306.1009; found, 306.1005.
3-(5-tert-Butylisoxazol-3-yl)-4-((3-chlorophenyl)diazenyl)-1H-pyrazol-5-amine (57)
To a solution of 1 (90 mg, 0.27 mmol) in EtOH (4 mL) was added hydrazine hydrate (15 μL, 0.36 mmol). The reaction mixture was refluxed for 6 h, the solvent was evaporated, and the crude product was crystallized from dichloromethane to yield 57 as a yellow solid (mp 230–232 °C) in 64% yield (60 mg). HPLC purity 99.2% (tR = 21.18 min). 1H NMR (300 MHz, DMSO-d6) δ 12.45 (s, 1H), 7.83–7.79 (m, 1H), 7.74 (d, J = 8.0 Hz, 1H), 7.51 (t, J = 7.9 Hz, 1H), 7.40 (m, 3H), 6.63 (s, 1H), 1.38 (s, 9H). 13C NMR (75 MHz, DMSO-d6) δ 180.6, 156.6, 154.8, 141.3, 140.5, 134.2, 131.2, 128.0, 121.9, 120.6, 120.6, 99.0, 32.8, 29.0. HRMS (ESI) calcd for C16H18ClN6O (M + H)+, 345.1231; found, 345.1229.
Molecular Docking Protocol
The substituted 2-(isoxazol-3-yl)-2-oxo-N′-phenyl-acetohydrazonoyl cyanide analogues were docked with EPAC2 using the X-ray structure of EPAC2 (PDB code: 3CF6) and AutoDock 4.0. Water molecules and the ligand (Sp-cAMPS) within the crystal structure were removed, and polar hydrogens were added using AutoDockTools. In the structures of the analogues, all bonds were treated as rotatable except for the aromatic, amide, cyano, and double bonds, whereas the protein was treated as rigid. Docking runs were carried out using the standard parameters of the program for interactive growing and subsequent scoring, except for the parameters for setting grid box dimensions and center. For all of the docking studies, a grid box size of 60 × 60 × 60 Å3, centered at coordinates 4.995 (x), −14.957 (y), and −60.351 (z) of the PDB structure, was used. The binding affinities of the 47 output structures ranged from −10.61 to −8.64 kcal/mol.
Determination of Solubility
Solubility in water or ethanol for 25, 28, 31, 35, 36, and 47 was determined by HPLC analysis according to a previously published protocol.68–70 First, 1 to 2 mg of 25, 28, 31, 35, 36, and 47 was weighed and added to 1 mL of water. Then, 10–15 mg of 25, 28, 31, 35, 36, and 47 was weighed and added to 0.1–0.6 mL of ethanol. The suspensions were shaken at 25 °C for 24 h and then centrifuged, and the supernatants were filtered. Aliquots (5 μL) of the supernatants were injected into the HPLC system equipped with a C18 reverse-phase column under the same conditions as those described in the Experimental Section, General Chemistry Information. One-point calibration71 was done by injecting 5 μL aliquots of the corresponding buffer solutions of 25, 28, 31, 35, 36, or 47 with known concentrations.
Biological Evaluation Methods Protein Expression and Purification
Recombinant full-length human EPAC1, mouse EPAC2, and C-terminal truncated Rap1B(1–167) were purified as previously described.72,73
8-NBD-cAMP Competition Assay
The 8-NBD-cAMP competition assay was performed in 96-well microplates from Corning Costar (Cambridge, MA, USA) as previously described.36 Reaction mix containing 50 nM EPAC2 and 60 nM 8-NBD-cAMP in 20 mM Tris-HCl, pH 7.5, buffer, with 150 mM NaCl, 1 mM EDTA, and 1 mM DDT was dispensed into a 96-well plate. Test compounds were added to the reaction mix at different doses. The fluorescence intensity signals from the 8-NBD probe before and after the addition of test compounds were measured using a SpectaMaxM2 microplate reader (Molecular Devices, Silicon Valley, CA, USA), with excitation/emission wavelengths set at 470/540 nm. Lastly, fluorescence intensity was again monitored after the addition of cAMP at a saturated final concentration of 300 μM. Data were presented by normalizing the observed fluorescence intensity (F) with the initial florescence signal (F0) before the addition of compound and the final florescence signal after cAMP addition (FcAMP) using the following equation: relative fluorescence = (F – FcAMP)/(F0 – FcAMP) × 100.
In Vitro Guanine Nucleotide Exchange Factor (GEF) Activity Assay of EPAC1
In vitro EPAC1 GEF activity was acquired as previously described.39 Briefly, the assay was performed using 500 nM Rap1b-BODIPY-GDP and 200 nM EPAC1 in buffer containing 50 mM Tris-HCl, pH 7.5, 50 mM NaCl, 5 mM MgCl2, 1 mM DTT, 50 mM GDP, and the indicated concentrations of test compounds at room temperature using half area 96-well plates (Corning Costar 3915). The exchange reaction was monitored using a Spectramax M2 plate reader (Molecular Devices), with the excitation/emission wavelengths set at 485/515 nm. The reaction rate constant (kobs) was determined by globally fitting the experimental data to a single-exponential equation. Quantification was processed by normalizing the observed kobs in the presence of inhibitor with the rate constant in the presence of 20 μM cAMP (no inhibitor) (kcAMP) and the rate constant without cAMP or inhibitor (k0) using the following equation: relative GEF activity = (kobs – k0)/(kcAMP – k0) × 100.
Supplementary Material
Acknowledgments
This work was supported by grant nos. R01 AI111464, R01 GM066170, and R01 GM106218 from the National Institutes of Health and a training fellowship from the Keck Center for Interdisciplinary Bioscience Training of the GCC (NIGMS grant T32 GM089657). We want to thank Drs. Lawrence C. Sowers and Jacob A. Theruvathu at the Department of Pharmacology as well as Dr. Tianzhi Wang at the NMR core facility of UTMB for NMR spectroscopy assistance.
Abbreviations
- EPAC
exchange proteins directly activated by cAMP
- SAR
structure–activity relationship
- cAMP
cyclic adenosine monophosphate
- 8-NBD-cAMP
8-(2-[7-nitro-4- benzofurazanyl]-aminoethylthio)adenosine-3′,5′-cyclic monophosphate
- GDP
guanosine diphosphate
- PKA
protein kinase A
- FRET
fluorescence resonance energy transfer
- GEF
guanine nucleotide exchange factor
- GTP
guanosine triphosphate
- Rap
Rasrelated protein
- HTS
high-throughput screening
- 007-AM
8-(4-chlorophenylthio)-2′-O-methyladenosine-3′,5′-cyclic monophosphate acetoxymethyl ester
- TLC
thin-layer chromatography
- UV
ultraviolet
- TMS
tetramethylsilane
- HRMS
high-resolution mass spectrometry
- HPLC
high-performance liquid chromatography
- DCM
dichloromethane
- EtOAc
ethyl acetate
- DMSO
dimethyl sulfoxide
- EDTA
ethylenediaminetetraacetic acid
- DDT
dichlorodiphenyltrichloroethane
- ADP
adenosine diphosphate
- NADH
nicotinamide adenine dinucleotide
- PEP
phospho(enol)pyruvate
- H89
N-[2-[[3-(4-bromophenyl)-2-propenyl]amino]ethyl]-5-isoquinolinesulfona-mide dihydrochloride
- CBD
cAMP binding domain
- CFP
cyan fluorescent protein
- YFP
yellow fluorescent protein
- DMEM
Dulbecco's modified Eagle's medium
- FBS
fetal bovine serum
Footnotes
Notes: The authors declare no competing financial interest.
Supporting Information: 1H and 13C NMR spectra of all new compounds. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.5b00635.
References
- 1.Beavo JA, Brunton LL. Cyclic nucleotide research – still expanding after half a century. Nat Rev Mol Cell Biol. 2002;3:710–718. doi: 10.1038/nrm911. [DOI] [PubMed] [Google Scholar]
- 2.Taylor SS, Zhang P, Steichen JM, Keshwani MM, Kornev AP. PKA: lessons learned after twenty years. Biochim Biophys Acta, Proteins Proteomics. 2013;1834:1271–1278. doi: 10.1016/j.bbapap.2013.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Biel M. Cyclic nucleotide-regulated cation channels. J Biol Chem. 2009;284:9017–9021. doi: 10.1074/jbc.R800075200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Biel M, Michalakis S. Cyclic nucleotide-gated channels. Handb Exp Pharmacol. 2009:111–136. doi: 10.1007/978-3-540-68964-5_7. [DOI] [PubMed] [Google Scholar]
- 5.de Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A, Bos JL. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature. 1998;396:474–477. doi: 10.1038/24884. [DOI] [PubMed] [Google Scholar]
- 6.Kawasaki H, Springett GM, Mochizuki N, Toki S, Nakaya M, Matsuda M, Housman DE, Graybiel AM. A family of cAMP-binding proteins that directly activate Rap1. Science. 1998;282:2275–2279. doi: 10.1126/science.282.5397.2275. [DOI] [PubMed] [Google Scholar]
- 7.Bos JL. Epac: a new cAMP target and new avenues in cAMP research. Nat Rev Mol Cell Biol. 2003;4:733–738. doi: 10.1038/nrm1197. [DOI] [PubMed] [Google Scholar]
- 8.Bos JL. Epac proteins: multi-purpose cAMP targets. Trends Biochem Sci. 2006;31:680–686. doi: 10.1016/j.tibs.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 9.Cheng X, Ji Z, Tsalkova T, Mei F. Epac and PKA: a tale of two intracellular cAMP receptors. Acta Biochim Biophys Sin. 2008;40:651–662. doi: 10.1111/j.1745-7270.2008.00438.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen H, Wild C, Zhou X, Ye N, Cheng X, Zhou J. Recent advances in the discovery of small molecules targeting exchange proteins directly activated by cAMP (EPAC) J Med Chem. 2014;57:3651–3665. doi: 10.1021/jm401425e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Rooij J, Rehmann H, van Triest M, Cool RH, Wittinghofer A, Bos JL. Mechanism of regulation of the Epac family of cAMP-dependent RapGEFs. J Biol Chem. 2000;275:20829–20836. doi: 10.1074/jbc.M001113200. [DOI] [PubMed] [Google Scholar]
- 12.Hoivik EA, Witsoe SL, Bergheim IR, Xu Y, Jakobsson I, Tengholm A, Doskeland SO, Bakke M. DNA methylation of alternative promoters directs tissue specific expression of Epac2 isoforms. PLoS One. 2013;8:e67925. doi: 10.1371/journal.pone.0067925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Niimura M, Miki T, Shibasaki T, Fujimoto W, Iwanaga T, Seino S. Critical role of the N-terminal cyclic AMP-binding domain of Epac2 in its subcellular localization and function. J Cell Physiol. 2009;219:652–658. doi: 10.1002/jcp.21709. [DOI] [PubMed] [Google Scholar]
- 14.Shibasaki T, Takahashi H, Miki T, Sunaga Y, Matsumura K, Yamanaka M, Zhang C, Tamamoto A, Satoh T, Miyazaki J, Seino S. Essential role of Epac2/Rap1 signaling in regulation of insulin granule dynamics by cAMP. Proc Natl Acad Sci U S A. 2007;104:19333–19338. doi: 10.1073/pnas.0707054104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aumo L, Rusten M, Mellgren G, Bakke M, Lewis AE. Functional roles of protein kinase A (PKA) and exchange protein directly activated by 3′,5′-cyclic adenosine 5′-monophosphate (cAMP) 2 (EPAC2) in cAMP-mediated actions in adrenocortical cells. Endocrinology. 2010;151:2151–2161. doi: 10.1210/en.2009-1139. [DOI] [PubMed] [Google Scholar]
- 16.Ueno H, Shibasaki T, Iwanaga T, Takahashi K, Yokoyama Y, Liu LM, Yokoi N, Ozaki N, Matsukura S, Yano H, Seino S. Characterization of the gene EPAC2: structure, chromosomal localization, tissue expression, and identification of the liver-specific isoform. Genomics. 2001;78:91–98. doi: 10.1006/geno.2001.6641. [DOI] [PubMed] [Google Scholar]
- 17.Grandoch M, Roscioni SS, Schmidt M. The role of Epac proteins, novel cAMP mediators, in the regulation of immune, lung and neuronal function. Br J Pharmacol. 2010;159:265–284. doi: 10.1111/j.1476-5381.2009.00458.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Breckler M, Berthouze M, Laurent AC, Crozatier B, Morel E, Lezoualc'h F. Rap-linked cAMP signaling Epac proteins: compartmentation, functioning and disease implications. Cell Signalling. 2011;23:1257–1266. doi: 10.1016/j.cellsig.2011.03.007. [DOI] [PubMed] [Google Scholar]
- 19.Gloerich M, Bos JL. Epac: defining a new mechanism for cAMP action. Annu Rev Pharmacol Toxicol. 2010;50:355–375. doi: 10.1146/annurev.pharmtox.010909.105714. [DOI] [PubMed] [Google Scholar]
- 20.Almahariq M, Chao C, Mei FC, Hellmich MR, Patrikeev I, Motamedi M, Cheng X. Pharmacological inhibition and genetic knockdown of EPAC1 reduce pancreatic cancer metastasis in vivo. Mol Pharmacol. 2014;87:142–149. doi: 10.1124/mol.114.095158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schmidt M, Dekker FJ, Maarsingh H. Exchange protein directly activated by cAMP (epac): a multidomain cAMP mediator in the regulation of diverse biological functions. Pharmacol Rev. 2013;65:670–709. doi: 10.1124/pr.110.003707. [DOI] [PubMed] [Google Scholar]
- 22.Almahariq M, Mei FC, Wang H, Cao AT, Yao S, Soong L, Sun J, Cong Y, Chen J, Cheng X. Exchange protein directly activated by cAMP modulates regulatory T-cell-mediated immunosuppression. Biochem J. 2015;465:295–303. doi: 10.1042/BJ20140952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mediero A, Perez-Aso M, Cronstein BN. Activation of EPAC1/2 is essential for osteoclast formation by modulating NFkappaB nuclear translocation and actin cytoskeleton rearrangements. FASEB J. 2014;28:4901–4913. doi: 10.1096/fj.14-255703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schwede F, Maronde E, Genieser H, Jastorff B. Cyclic nucleotide analogs as biochemical tools and prospective drugs. Pharmacol Ther. 2000;87:199–226. doi: 10.1016/s0163-7258(00)00051-6. [DOI] [PubMed] [Google Scholar]
- 25.Holz GG, Chepurny OG, Schwede F. Epac-selective cAMP analogs: new tools with which to evaluate the signal transduction properties of cAMP-regulated guanine nucleotide exchange factors. Cell Signalling. 2008;20:10–20. doi: 10.1016/j.cellsig.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stokman G, Qin Y, Genieser HG, Schwede F, de Heer E, Bos JL, Bajema IM, van de Water B, Price LS. Epac-Rap signaling reduces cellular stress and ischemia-induced kidney failure. J Am Soc Nephrol. 2011;22:859–872. doi: 10.1681/ASN.2010040423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ostroveanu A, van der Zee EA, Eisel UL, Schmidt M, Nijholt IM. Exchange protein activated by cyclic AMP 2 (Epac2) plays a specific and time-limited role in memory retrieval. Hippocampus. 2010;20:1018–1026. doi: 10.1002/hipo.20700. [DOI] [PubMed] [Google Scholar]
- 28.Christensen AE, Selheim F, de Rooij J, Dremier S, Schwede F, Dao KK, Martinez A, Maenhaut C, Bos JL, Genieser HG, Doskeland SO. cAMP analog mapping of Epac1 and cAMP kinase. Discriminating analogs demonstrate that Epac and cAMP kinase act synergistically to promote PC-12 cell neurite extension. J Biol Chem. 2003;278:35394–35402. doi: 10.1074/jbc.M302179200. [DOI] [PubMed] [Google Scholar]
- 29.Vliem MJ, Ponsioen B, Schwede F, Pannekoek WJ, Riedl J, Kooistra MR, Jalink K, Genieser HG, Bos JL, Rehmann H. 8-pCPT-2′-O-Me-cAMP-AM: an improved Epac-selective cAMP analogue. ChemBioChem. 2008;9:2052–2054. doi: 10.1002/cbic.200800216. [DOI] [PubMed] [Google Scholar]
- 30.Poppe H, Rybalkin SD, Rehmann H, Hinds TR, Tang XB, Christensen AE, Schwede F, Genieser HG, Bos JL, Doskeland SO, Beavo JA, Butt E. Cyclic nucleotide analogs as probes of signaling pathways. Nat Methods. 2008;5:277–278. doi: 10.1038/nmeth0408-277. [DOI] [PubMed] [Google Scholar]
- 31.Ouyang M, Zhang L, Zhu JJ, Schwede F, Thomas SA. Epac signaling is required for hippocampus-dependent memory retrieval. Proc Natl Acad Sci U S A. 2008;105:11993–11997. doi: 10.1073/pnas.0804172105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen H, Tsalkova T, Mei FC, Hu Y, Cheng X, Zhou J. 5-Cyano-6-oxo-1,6-dihydro-pyrimidines as potent antagonists targeting exchange proteins directly activated by cAMP. Bioorg Med Chem Lett. 2012;22:4038–4043. doi: 10.1016/j.bmcl.2012.04.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Courilleau D, Bisserier M, Jullian JC, Lucas A, Bouyssou P, Fischmeister R, Blondeau JP, Lezoualc'h F. Identification of a tetrahydroquinoline analog as a pharmacological inhibitor of the cAMP-binding protein Epac. J Biol Chem. 2012;287:44192–44202. doi: 10.1074/jbc.M112.422956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Courilleau D, Bouyssou P, Fischmeister R, Lezoualc'h F, Blondeau JP. The (R)-enantiomer of CE3F4 is a preferential inhibitor of human exchange protein directly activated by cyclic AMP isoform 1 (Epac1) Biochem Biophys Res Commun. 2013;440:443–448. doi: 10.1016/j.bbrc.2013.09.107. [DOI] [PubMed] [Google Scholar]
- 35.Chen H, Tsalkova T, Chepurny OG, Mei FC, Holz GG, Cheng X, Zhou J. Identification and characterization of small molecules as potent and specific EPAC2 antagonists. J Med Chem. 2013;56:952–962. doi: 10.1021/jm3014162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsalkova T, Mei FC, Cheng X. A fluorescence-based high-throughput assay for the discovery of exchange protein directly activated by cyclic AMP (EPAC) antagonists. PLoS One. 2012;7:e30441. doi: 10.1371/journal.pone.0030441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tsalkova T, Mei FC, Li S, Chepurny OG, Leech CA, Liu T, Holz GG, Woods VL, Jr, Cheng X. Isoform-specific antagonists of exchange proteins directly activated by cAMP. Proc Natl Acad Sci U S A. 2012;109:18613–18618. doi: 10.1073/pnas.1210209109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Almahariq M, Tsalkova T, Mei FC, Chen H, Zhou J, Sastry SK, Schwede F, Cheng X. A novel EPAC-specific inhibitor suppresses pancreatic cancer cell migration and invasion. Mol Pharmacol. 2013;83:122–128. doi: 10.1124/mol.112.080689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhu Y, Chen H, Boulton S, Mei F, Ye N, Melacini G, Zhou J, Cheng X. Biochemical and pharmacological characterizations of ESI-09 based EPAC inhibitors: defining the ESI-09 “therapeutic window”. Sci Rep. 2015;5:9344. doi: 10.1038/srep09344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mazhab-Jafari MT, Das R, Fotheringham SA, Sil Das S, Chowdhury S, Melacini G. Understanding cAMP-dependent allostery by NMR spectroscopy: comparative analysis of the EPAC1 cAMP-binding domain in its apo and cAMP-bound states. J Am Chem Soc. 2007;129:14482–14492. doi: 10.1021/ja0753703. [DOI] [PubMed] [Google Scholar]
- 41.Selvaratnam R, Chowdhury S, VanSchouwen B, Melacini G. Mapping allostery through the covariance analysis of NMR chemical shifts. Proc Natl Acad Sci U S A. 2011;108:6133–6138. doi: 10.1073/pnas.1017311108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Selvaratnam R, Mazhab-Jafari MT, Das R, Melacini G. The auto-inhibitory role of the EPAC hinge helix as mapped by NMR. PLoS One. 2012;7:e48707. doi: 10.1371/journal.pone.0048707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Das R, Chowdhury S, Mazhab-Jafari MT, Sildas S, Selvaratnam R, Melacini G. Dynamically driven ligand selectivity in cyclic nucleotide binding domains. J Biol Chem. 2009;284:23682–23696. doi: 10.1074/jbc.M109.011700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Selvaratnam R, VanSchouwen B, Fogolari F, Mazhab-Jafari MT, Das R, Melacini G. The projection analysis of NMR chemical shifts reveals extended EPAC autoinhibition determinants. Biophys J. 2012;102:630–639. doi: 10.1016/j.bpj.2011.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Das R, Mazhab-Jafari MT, Chowdhury S, Sil Das S, Selvaratnam R, Melacini G. Entropy-driven cAMP-dependent allosteric control of inhibitory interactions in exchange proteins directly activated by cAMP. J Biol Chem. 2008;283:19691–19703. doi: 10.1074/jbc.M802164200. [DOI] [PubMed] [Google Scholar]
- 46.VanSchouwen B, Selvaratnam R, Fogolari F, Melacini G. Role of dynamics in the autoinhibition and activation of the exchange protein directly activated by cyclic AMP (EPAC) J Biol Chem. 2011;286:42655–42669. doi: 10.1074/jbc.M111.277723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boulton S, Akimoto M, Selvaratnam R, Bashiri A, Melacini G. A tool set to map allosteric networks through the NMR chemical shift covariance analysis. Sci Rep. 2014;4:7306. doi: 10.1038/srep07306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boulton S, Akimoto M, VanSchouwen B, Moleschi K, Selvaratnam R, Giri R, Melacini G. Tapping the translation potential of cAMP signalling: molecular basis for selectivity in cAMP agonism and antagonism as revealed by NMR. Biochem Soc Trans. 2014;42:302–307. doi: 10.1042/BST20130282. [DOI] [PubMed] [Google Scholar]
- 49.Selvaratnam R, Akimoto M, VanSchouwen B, Melacini G. cAMP-dependent allostery and dynamics in Epac: an NMR view. Biochem Soc Trans. 2012;40:219–223. doi: 10.1042/BST20110628. [DOI] [PubMed] [Google Scholar]
- 50.Dawn A, Singh S, More KR, Siddiqui FA, Pachikara N, Ramdani G, Langsley G, Chitnis CE. The central role of cAMP in regulating Plasmodium falciparum merozoite invasion of human erythrocytes. PLoS Pathog. 2014;10:e1004520. doi: 10.1371/journal.ppat.1004520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Aslam M, Tanislav C, Troidl C, Schulz R, Hamm C, Gunduz D. cAMP controls the restoration of endothelial barrier function after thrombin-induced hyperpermeability via Rac1 activation. Physiol Rep. 2014;2:e12175. doi: 10.14814/phy2.12175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bacallao K, Monje PV. Opposing roles of PKA and EPAC in the cAMP-dependent regulation of schwann cell proliferation and differentiation [corrected] PLoS One. 2013;8:e82354. doi: 10.1371/journal.pone.0082354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ivonnet P, Salathe M, Conner GE. Hydrogen peroxide stimulation of CFTR reveals an Epac-mediated, soluble AC-dependent cAMP amplification pathway common to GPCR signalling. Br J Pharmacol. 2015;172:173–184. doi: 10.1111/bph.12934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang M, Robitaille M, Showalter AD, Huang X, Liu Y, Bhattacharjee A, Willard FS, Han J, Froese S, Wei L, Gaisano HY, Angers S, Sloop KW, Dai FF, Wheeler MB. Progesterone receptor membrane component 1 is a functional part of the glucagon-like peptide-1 (GLP-1) receptor complex in pancreatic beta cells. Mol Cell Proteomics. 2014;13:3049–3062. doi: 10.1074/mcp.M114.040196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li X, Guo Q, Gao J, Yang J, Zhang W, Liang Y, Wu D, Liu Y, Weng J, Li Q, Zhang Y. The adenylyl cyclase inhibitor MDL-12,330A potentiates insulin secretion via blockade of voltage-dependent K(+) channels in pancreatic beta cells. PLoS One. 2013;8:e77934. doi: 10.1371/journal.pone.0077934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yan J, Mei FC, Cheng H, Lao DH, Hu Y, Wei J, Patrikeev I, Hao D, Stutz SJ, Dineley KT, Motamedi M, Hommel JD, Cunningham KA, Chen J, Cheng X. Enhanced leptin sensitivity, reduced adiposity, and improved glucose homeostasis in mice lacking exchange protein directly activated by cyclic AMP isoform 1. Mol Cell Biol. 2013;33:918–926. doi: 10.1128/MCB.01227-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tao X, Mei F, Agrawal A, Peters CJ, Ksiazek TG, Cheng X, Tseng CT. Blocking of exchange proteins directly activated by cAMP leads to reduced replication of Middle East respiratory syndrome coronavirus. J Virol. 2014;88:3902–3910. doi: 10.1128/JVI.03001-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gong B, Shelite T, Mei FC, Ha T, Hu Y, Xu G, Chang Q, Wakamiya M, Ksiazek TG, Boor PJ, Bouyer DH, Popov VL, Chen J, Walker DH, Cheng X. Exchange protein directly activated by cAMP plays a critical role in bacterial invasion during fatal rickettsioses. Proc Natl Acad Sci U S A. 2013;110:19615–19620. doi: 10.1073/pnas.1314400110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rehmann H. Epac-inhibitors: facts and artefacts. Sci Rep. 2013;3:3032. doi: 10.1038/srep03032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cheng X, Zhou J, Tsalkova T, Mei F, Chen H. US 2015/0110809 and WO 2013/119931 A1 Modulators of exchange proteins directly activated by cAMP (EPACs)
- 61.Rehmann H, Das J, Knipscheer P, Wittinghofer A, Bos JL. Structure of the cyclic-AMP-responsive exchange factor Epac2 in its auto-inhibited state. Nature. 2006;439:625–628. doi: 10.1038/nature04468. [DOI] [PubMed] [Google Scholar]
- 62.Rehmann H, Arias-Palomo E, Hadders MA, Schwede F, Llorca O, Bos JL. Structure of Epac2 in complex with a cyclic AMP analogue and RAP1B. Nature. 2008;455:124–127. doi: 10.1038/nature07187. [DOI] [PubMed] [Google Scholar]
- 63.Chen H, Ding C, Wild C, Liu H, Wang T, White MA, Cheng X, Zhou J. Efficient synthesis of ESI-09, a novel non-cyclic nucleotide EPAC antagonist. Tetrahedron Lett. 2013;54:1546–1549. doi: 10.1016/j.tetlet.2013.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cherala S, Lingabathula H, Ganta R, Ampati S, Manda S. Synthesis and anti-inflammatory activity of a novel series of diphenyl-1,2,4-triazoles and related derivatives. E-J Chem. 2012;9:2510–2515. [Google Scholar]
- 65.Hassaneen HM, Hassaneen HME, Gomaa ZA. Reactivity of 3-cyanoacetylindole derivatives: synthesis of 3-hydrazonopyrazolyl and 3-thiadiazolyl indole derivatives. Int J Org Chem. 2011;1:97–104. [Google Scholar]
- 66.Purser S, Moore PR, Swallow S, Gouverneur V. Fluorine in medicinal chemistry. Chem Soc Rev. 2008;37:320–330. doi: 10.1039/b610213c. [DOI] [PubMed] [Google Scholar]
- 67.Hagmann WK. The many roles for fluorine in medicinal chemistry. J Med Chem. 2008;51:4359–4369. doi: 10.1021/jm800219f. [DOI] [PubMed] [Google Scholar]
- 68.Ding C, Zhang Y, Chen H, Yang Z, Wild C, Chu L, Liu H, Shen Q, Zhou J. Novel nitrogen-enriched oridonin analogs with thiazole-fused A-ring: protecting group-free synthesis, enhanced anticancer profile, and improved aqueous solubility. J Med Chem. 2013;56:5048–5058. doi: 10.1021/jm400367n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen H, Yang Z, Ding C, Chu L, Zhang Y, Terry K, Liu H, Shen Q, Zhou J. Discovery of O-alkylamino tethered niclosamide derivatives as potent and orally bioavailable anticancer agents. ACS Med Chem Lett. 2013;4:180–185. doi: 10.1021/ml3003082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vogel GH, editor. Drug Discovery and Evaluation: Safety and Pharmacokinetics Assay. Springer; New York: 2006. Determination of solubility by hyphenated HPLC methods; pp. 400–402. [Google Scholar]
- 71.Kiselev E, DeGuire S, Morrell A, Agama K, Dexheimer TS, Pommier Y, Cushman M. 7-azaindenoisoquinolines as topoisomerase I inhibitors and potential anticancer agents. J Med Chem. 2011;54:6106–6116. doi: 10.1021/jm200719v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mei FC, Cheng X. Interplay between exchange protein directly activated by cAMP (Epac) and microtubule cytoskeleton. Mol BioSyst. 2005;1:325–331. doi: 10.1039/b511267b. [DOI] [PubMed] [Google Scholar]
- 73.Li S, Tsalkova T, White MA, Mei FC, Liu T, Wang D, Woods VL, Jr, Cheng X. Mechanism of intracellular cAMP sensor Epac2 activation: cAMP-induced conformational changes identified by amide hydrogen/deuterium exchange mass spectrometry (DXMS) J Biol Chem. 2011;286:17889–17897. doi: 10.1074/jbc.M111.224535. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.