

Abstract
The phylogenetic and taxonomic relationships among the Old World leaf-nosed bats (Hipposideridae) and the closely related horseshoe bats (Rhinolophidae) remain unresolved. In this study, we generated a novel approximately 10-kb molecular data set of 19 nuclear exon and intron gene fragments for 40 bat species to elucidate the phylogenetic relationships within the families Rhinolophidae and Hipposideridae. We estimated divergence times and explored potential reasons for any incongruent phylogenetic signal. We demonstrated the effects of outlier taxa and genes on phylogenetic reconstructions and compared the relative performance of intron and exon data to resolve phylogenetic relationships. Phylogenetic analyses produced a well-resolved phylogeny, supporting the familial status of Hipposideridae and demonstrated the paraphyly of the largest genus, Hipposideros. A fossil-calibrated timetree and biogeographical analyses estimated that Rhinolophidae and Hipposideridae diverged in Africa during the Eocene approximately 42 Ma. The phylogram, the timetree, and a unique retrotransposon insertion supported the elevation of the subtribe Rhinonycterina to family level and which is diagnosed herein. Comparative analysis of diversification rates showed that the speciose genera Rhinolophus and Hipposideros underwent diversification during the Mid-Miocene Climatic Optimum. The intron versus exon analyses demonstrated the improved nodal support provided by introns for our optimal tree, an important finding for large-scale phylogenomic studies, which typically rely on exon data alone. With the recent outbreak of Middle East respiratory syndrome, caused by a novel coronavirus, the study of these species is urgent as they are considered the natural reservoir for emergent severe acute respiratory syndrome (SARS)-like coronaviruses. It has been shown that host phylogeny is the primary factor that determines a virus’s persistence, replicative ability, and can act as a predictor of new emerging disease. Therefore, this newly resolved phylogeny can be used to direct future assessments of viral diversity and to elucidate the origin and development of SARS-like coronaviruses in mammals.
Keywords: phylogenetics, mammals, virus, exon versus intron, biogeography, Rhinonycteridae
Introduction
The Hipposideridae, commonly known as the Old World leaf-nosed bats, are widespread and are distributed in tropical and subtropical areas of the Old World extending from western Africa, throughout Australasia, and marginally into the Palearctic (Koopman 1994; Bogdanowicz and Owen 1998). They consist of nine extant genera (see table 1 for classification) and at least 82 species (Simmons 2005). The Rhinolophidae, commonly known as horseshoe bats, comprise 77 species encompassed in a single genus, Rhinolophus (Simmons 2005). Rhinolophidae are found in diverse habitats throughout the temperate and tropical regions of the Old World (Nowak and Paradiso 1999). Hipposideridae and their sister taxon, the Rhinolophidae, are of exceptional scientific interest, having arguably the most sophisticated echolocation system (Jones and Teeling 2006) and are considered as the reservoir host species for the emergent severe acute respiratory syndrome (SARS) -like coronaviruses (Li et al. 2005; Drexler et al. 2010; Ar Gouilh et al. 2011; Anthony et al. 2013).
Table 1.
List of Taxa and Taxonomic Levels Used in This Analysis—Following Simmons (2005) Unless Otherwise Stated.
| Suborder | Superfamily | Family | Subtribe | Genus | Species |
|---|---|---|---|---|---|
| Yangochiropteraa | Vespertilionidae | Myotis | myotis | ||
| Mormoopidae | Pteronotus | parnelli | |||
| Yinpterochiropteraa | Pteropodidae | Cynopterus | brachyotis | ||
| Nyctimene | albiventer | ||||
| Rousettus | lanosus | ||||
| Rhinolophoidea | Rhinopomatidae | Rhinopomab | hardwickii | ||
| microphyllum | |||||
| Craseonycteridae | Craseonycteris | thonglongyai | |||
| Megadermatidae | Megaderma | spasma | |||
| lyra | |||||
| Rhinolophidae | Rhinolophus | hipposideros | |||
| shameli | |||||
| pusillus | |||||
| creaghi | |||||
| euryale | |||||
| trifoliatus | |||||
| ferrumequinum | |||||
| sinicus | |||||
| luctus | |||||
| pearsoni | |||||
| Hipposideridae | Anthops | ornatus | |||
| Asellia | tridens | ||||
| Aselliscus | stoliczkanus | ||||
| Coelops | frithii | ||||
| Hipposideros | armiger | ||||
| larvatus | |||||
| commersonic | |||||
| vittatus | |||||
| jonesi | |||||
| galeritus | |||||
| caffer | |||||
| pomona | |||||
| halophyllus | |||||
| abae | |||||
| Rhinonycterinad | Cloeotis | percivali | |||
| Rhinonicteris | aurantia | ||||
| Paratriaenopse | furculus | ||||
| auritus | |||||
| Triaenops | persicus | ||||
| menamena | |||||
aSubordinal and superfamilial revisions made by Springer et al. (2001).
bThe recent revision of Rhinopoma spp. (Hulva et al. 2007), which has seen this species as defined in Simmons (2005) split into two species Rhinopoma hardwickii and R. cystops, as such this study uses the classification defined by this revision.
cSensu lato referring to Hipposideros commersoni specimens identified from mainland Africa, which may include species from the commersoni species group; see Simmons (2005) and Tate (1941b).
dThe Subtribe Rhinonycterina first described by Gray (1866) and later expanded by Hill (1982) to include Triaenops and Cloeotis.
eRecent generic revisions made by Benda and Vallo (2009), which spilt the genus Triaenops with displaced taxa positioned in Paratriaenops.
Taxonomic and Phylogenetic Conflict
The evolutionary history of the Hipposideridae remains a source of phylogenetic controversy stemming from conflict between morphological and molecular data (fig. 1). The principal quantitative morphological phylogenies were conducted by Bogdanowicz and Owen (1998) and Hand and Kirsch (1998), but the resulting trees are incongruent (fig. 1a and b, respectively). Recently, a number of molecular phylogenies have emerged which typically sample only two or three Hipposideridae genera (Wang et al. 2003; Li et al. 2007; Gu et al. 2008; Benda and Vallo 2009). Although subsequent studies have increased generic sampling, they are still too underrepresented taxonomically to significantly advance our knowledge of higher level relationships among hipposiderid bats (Eick et al. 2005; Murray et al. 2012) (fig. 1c and d, respectively). The familial status of the Hipposideridae, the monophyly of its most speciose genus Hipposideros, and the biogeographical origin of the putative family and its closest relative, the Rhinolophidae (Simmons 2005), represent several areas of outstanding phylogenetic controversy. The distinctiveness of hipposiderid and rhinolophid bats was first recognized with the establishment of the subtribe Rhinonycterina (Gray 1866), later elevated to subfamily by McKenna and Bell (1997), and the subfamily Hipposiderinae (Flower and Lydekker 1891). Classification of the Hipposideridae at the family level still remains unresolved with many authors preferring a subfamilial status within Rhinolophidae (Koopman 1993, 1994; McKenna and Bell 1997; Simmons 1998; Simmons and Geisler 1998; Teeling et al. 2002), whereas others support a full familial classification (Pierson 1986; Bogdanowicz and Owen 1998; Hand and Kirsch 1998; Simmons 2005; Murray et al. 2012). A recent revision of the hipposiderid genus Triaenops resulted in a separation between Triaenops and a new genus Paratriaenops (Benda and Vallo 2009). Another recent revision rendered the genus Paracoelops invalid because a re-examination of the holotype showed that it was misidentified originally and actually best assigned to Hipposideros (Thong, Dietz, et al. 2012) (see table 1 for full classification).
Fig. 1.

(a) Tree derived from Parsimony analysis of morphological discrete state data using Nelson-like consensus cladogram from Bogdanowicz and Owen (1998). (b) Consensus tree from Parsimony analysis of unordered morphological characters on 30 taxa common to this study described in (a) from Hand and Kirsch (1998). (c) Single ML tree derived from PAUP* analysis of intron supermatrix from Eick et al. (2005). (d) ML tree derived from PAUP* analysis of ND2 and RAG1 from Murray et al. (2012).
Hipposideros is the most speciose hipposiderid genus, accounting for 67 of the 82 recognized species (Simmons 2005), a number that is increasing due to the description of new cryptic species (e.g., Thong, Dietz, et al. 2012; Thong, Puechmaille, Denzinger, Bates, et al. 2012). However, the monophyly of this genus is questioned. Morphological studies, which include up to eight of the nine Hipposideridae genera, have suggested that Hipposideros is paraphyletic (Sigé 1968; Legendre 1982; Bogdanowicz and Owen 1998). However, a recent molecular phylogenetic study, which included four of the nine Hipposideridae genera, based on a single mitochondrial and nuclear genes supported the monophyly of the genus (Murray et al. 2012) (fig. 1). Rhinolophus is the sole genus of the family Rhinolophidae and is composed of 77 recognized species falling into 12 species groups (Simmons 2005). The clade that is most basal within extant Rhinolophidae is still controversial. Previous phylogenetic reconstructions of the Rhinolophidae are characterized by poor resolution at deeper nodes suggesting a rapid radiation in this family (Guillen-Servent et al. 2003). Based on a Cyt b tree, Guillen-Servent et al. (2003) recovered a basal clade containing the Rhinolophus trifoliatus and the R. hipposideros species groups. However, based on a combined data set of Cyt b and nuclear introns, Stoffberg et al. (2010) reported a single species, R. pearsoni, as the basal lineage with all other species (including R. hipposideros) forming two geographic clades, which are predominantly Oriental or Afrotropical. Nevertheless, Stoffberg et al. (2010) did not include members of the R. trifoliatus clade.
As discussed above, it is still uncertain whether both the sister groups Rhinolophidae and Hipposideridae warrant independent familial status. Previous estimates of the time of divergence of these two groups indicate that they diverged about 39–45 Ma (Eick et al. 2005; Teeling et al. 2005; Miller-Butterworth et al. 2007), which is comparable to divergence time estimates obtained for other bat families (Teeling 2009). Their unstable family-level classification has also led to uncertainty concerning the biogeographical origin of these two groups. The early suggestions regarding the origin of the Hipposideridae, based on morphological data, favored either an African or an Asian origin (Koopman 1970; Sigé 1991). Neontological data, which supported family status for both groups, suggested that an Asian origin was most likely (Bogdanowicz and Owen 1998). Koopman (1970) and Teeling et al. (2005) who regarded the Hipposideridae as a subfamily of the Rhinolophidae suggest an Asian origin for Rhinolophidae and by extension for the Hipposideridae. A re-evaluation of divergence time estimates using a resolved phylogeny for these groups based on broad taxonomic sampling is required to elucidate the familial or nonfamilial status of the Hipposideridae and will also enable better biogeographic inferences.
The biogeographic origin of the Rhinolophidae has been a controversial topic in recent years with several competing hypotheses emerging from diverse data types. Molecular data, based on Cyt b, have placed the Rhinolophidae center of origin in Europe and suggest that from there, they expanded their geographical range into Asia and Africa (Guillen-Servent et al. 2003). The exon and 3’-UTR-derived tree of Teeling et al. (2005) and the nuclear intron and Cyt b-derived tree of Stoffberg et al. (2010) supported an Asian origin, which conflicts with the African origin proposed by Eick et al. (2005) based on nuclear introns. To provide clarity in interpreting the biogeographic origin of the Hipposideridae and Rhinolophidae, it is essential to test these hypotheses using a resolved taxonomy at the family level and building phylogenies using diverse data types.
The genera Rhinolophus and Hipposideros are notable for their high species number and cryptic diversity (Kingston et al. 2001; Thabah et al. 2006; Soisook et al. 2008; Sun et al. 2008; Thong, Puechmaille, Denzinger, Bates, et al. 2012; Thong, Puechmaille, Denzinger, Dietz, et al. 2012). Rapid diversification in many groups of organisms are associated with macroevolutionary events typified by periods of global change over the course of geological time, such as extinctions, and also periods of global warming and cooling (Hedges et al. 1996; McElwain and Punyasena 2007; Vieites et al. 2007; Meredith et al. 2011). In part, associated with their broad geographical distributions and diversity, Rhinolophus and Hipposideros represent good candidate taxa to investigate whether there is a common macroevolutionary factor underlying the high species numbers observed in these genera. Comparisons of the rate of diversification between these genera in combination with divergence time estimates can enrich our understanding of the evolution of these speciose genera.
Coevolution of SARS-Like Coronavirus
Since the outbreak of SARS in 2003 in China, there has been a surge of interest in understanding the evolution and the emergence of this deadly coronavirus (Balboni et al. 2012). Research arising from the outbreak has identified bats as a likely natural reservoir for several zoonotic viruses, including SARS-like coronavirus (Li et al. 2005; Calisher et al. 2006; Anthony et al. 2013; Chan et al. 2013; Luis et al. 2013). Among bats, the Rhinolophidae are of particular interest because R. sinicus, R. pusillus, R. macrotis, and R. ferrumequinum were suspected to be directly implicated in the outbreak of SARS in China (Lau et al. 2005; Li et al. 2005; Anthony et al. 2013), and since then, a wider diversity of coronaviruses has been described from the Rhinolophidae (Li et al. 2005; Ren et al. 2006; Woo et al. 2006; Cui et al. 2007; Drexler et al. 2010; Rihtaric et al. 2010; Yuan et al. 2010). Recent research has provided evidence to support that Rhinolophidae are a likely natural reservoir of the SARS-CoV (Ge et al. 2013). Betacoronaviruses-b, from the SARS group, have also been found in the close relatives of the Rhinolophidae, Hipposideros larvatus (Ar Gouilh et al. 2011). Hipposideridae are considered to be understudied in estimations of novel coronaviruses in wild animal populations (Ar Gouilh et al. 2011). The emergence and subsequent deaths caused by coronaviruses-like SARS-CoV and more recently Middle East respiratory syndrome coronavirus (MERS-CoV) have brought into sharp focus the potential health risks arising from the emergence of novel coronaviruses and further highlight the importance of their efficient detection and monitoring.
There is a growing body of support to show that bats and viruses form host/virus associations (Daszak 2010; Drexler et al. 2010; Streicker et al. 2010; Ar Gouilh et al. 2011; Drexler et al. 2012; Anthony et al. 2013). It has been demonstrated that host phylogeny is the primary factor that determines virus persistence and replicative ability in new hosts and perhaps most importantly that knowledge of host phylogeny can predict the source of new emerging diseases (Longdon et al. 2011). Extrapolating from these phylogenetic associations represents a novel way in which coronavirus diversity can be more efficiently surveyed and assessed. Systematic surveillance for emerging coronaviruses should be orientated based on resolved phylogenies highlighting additional potential reservoir species. The successful detection of novel coronaviruses based on the limited studies conducted on Hipposideridae bats indicate that this family contains candidates in which to search for undescribed coronaviruses (Pfefferle et al. 2009; Quan et al. 2010; Ar Gouilh et al. 2011). However, one complicating factor is that the phylogenetic history and the taxonomy of the Hipposideridae remain unresolved. Therefore, deciphering these phylogenetic issues, as proposed, will be instrumental for detecting novel coronaviruses and for “systematically” guiding the surveillance of reservoir hosts to predict the emergence of novel pathogens.
Choice of Molecular Markers
Previous molecular phylogenies have mainly focused on the relationship between a limited number of Hipposideridae genera and have predominantly used mitochondrial data (Wang et al. 2003; Li et al. 2007; Gu et al. 2008; Murray et al. 2012). The phylogeny of Eick et al. (2005) focused on interfamilial relationships among bats and was constructed using slower evolving nuclear introns. Herein, we generated a data set comprising differentially evolving nuclear exons and introns to ascertain the familial status of the Hipposideridae and to elucidate the intergeneric phylogenetic relationships of the speciose and presumed rapidly diversifying genera Rhinolophus and Hipposideros. We hypothesize that slower evolving exons should provide resolution of deeper nodes, following their successful use in resolving phylogenies focusing on deep time scales at interordinal level in mammals (Meredith et al. 2011) and interfamilial levels in bats (Teeling et al. 2002, 2003, 2005; Miller-Butterworth et al. 2007; Lack et al. 2010) and that faster evolving introns should provide resolution of more recent nodes in a tree, following their widespread use in resolving intergeneric and species level relationships (Lim et al. 2008; Stoffberg et al. 2010; Puechmaille et al. 2011; Salicini et al. 2011). In this study, we test the validity of these assumptions and the resolving power of exons and introns.
Objectives
A combination of state-of-the-art phylogenetic methods, divergence time estimates, and biogeographical analyses were employed to elucidate the evolutionary history of the Hipposideridae and Rhinolophidae and to explore the reasons for any phylogenetic incongruence. A comparative diversification analysis focusing on Rhinolophus and Hipposideros was used to elucidate if the diversification patterns of these speciose lineages are similar through time. In particular, we explore potential impediments to phylogenetic resolution in our data by examining the effects of removing outlying data and compare the resolving abilities of exons and introns. Our phylogeny also addresses the following outstanding phylogenetic questions: 1) Do hipposiderid bats represent an independent family or a subfamily of the Rhinolophidae? 2) Is Hipposideros a monophyletic genus? 3) When did hipposiderid and rhinolophid bats diverge and where did they originate? 4) Which species or clade is the most basal lineage of the Rhinolophidae? Finally, we explore the use of our phylogeny for future systematic surveying for novel emergent coronaviruses and for the on-going conservation of these species.
Results
Alignments and Outlier Detection
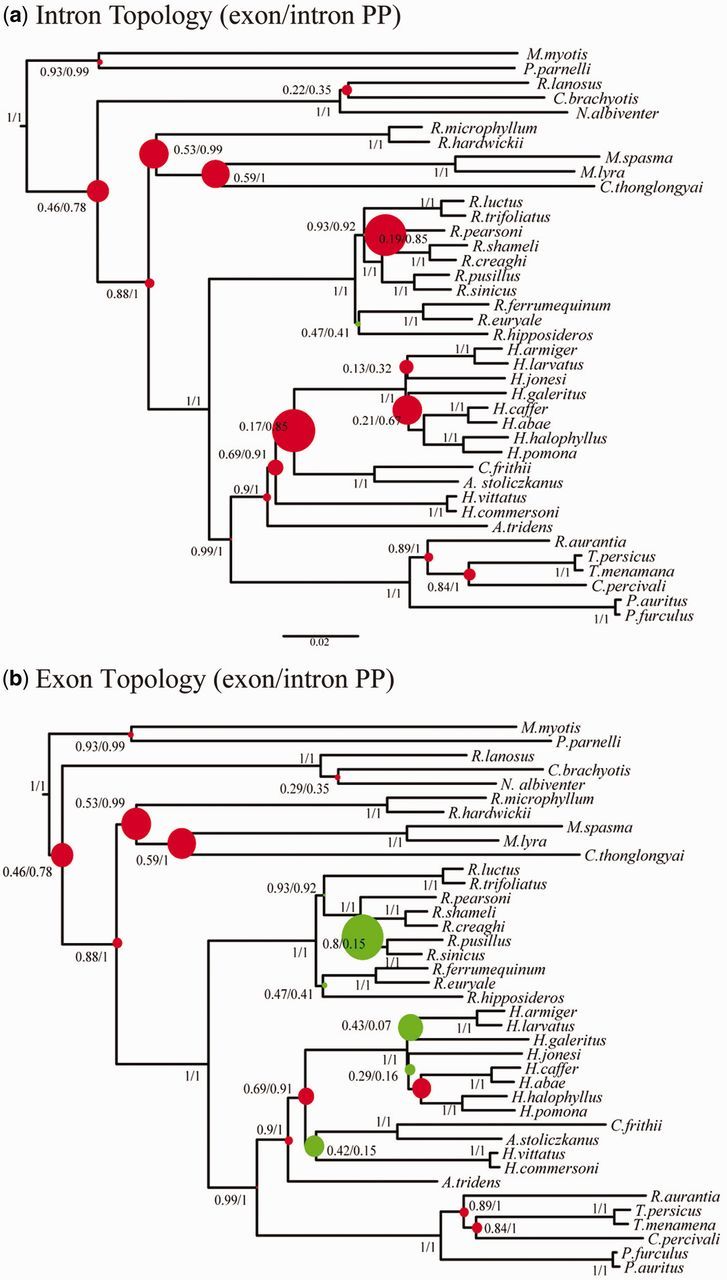
Concatenation of 12 exons and 3’-UTR gene fragments resulted in 7,888 aligned positions for 39 taxa. Concatenation of seven intron gene fragments resulted in 2,532 aligned positions. These two data sets were then further combined resulting in an alignment of 10,420 positions. Of note, analysis of the alignment with Repeatmasker identified a 128-bp insertion in the intron THY as a class 1 retrotransposon, which was common to the genera Triaenops, Cloeotis, Paratrianeops, and Rhinonicteris. jModeltest indicated the most appropriate model of sequence evolution for each individual gene fragment using the Akaike Information Criterion (AIC; see supplementary table S1, Supplementary Material online). Each data set, exons, introns, and exons+introns were analyzed with and without the outliers identified by the Phylo-MCOA analysis. Topologies of trees resulting from the exon+intron and exon+intron-outliers removed data sets were identical (fig. 2). Following the removal of outlying data, minimal changes in posteriors were observed at seven nodes with a resulting average minimal posterior change of −0.00167. Furthermore, maximum observed changes across these seven nodes were also minor, with a maximum increase in posterior values of 0.02 and a maximum decrease of 0.03.
Fig. 2.

Phylogram inferred from Bayesian Analysis in BEAST on the exon+intron-outliers removed data set, 10,420 bp comprising 12 nuclear exons and 7 nuclear introns, under a fully partitioned model. Nodal support for the exon+intron-outliers removed data set is summarized on the tree for all four analyses—RaxML, BEAST, MrBayes, and PhyloBayes. All numeric support values are shown as percentages and refer to each analysis in the order listed above. Black squares denote highly supported nodes all of which received support >99 BSS or 0.99 PP across all four analyses. A “-” indicates that this relationship was not supported by the analysis. See Systematic Summary for full description of the newly elevated family Rhinonycteridae. Frontal views of nose leaves of representatives of the major clades are shown as follows: Rhinolophidae—Rhinolophus pearsoni and Hipposideridae—Hipposideros spp. (photo credit—Sébastien J. Puechmaille) and Rhinonycteridae—Triaenops (photo credit—Paul Webala).
Phylogenetic Analysis
Each of the six data sets for phylogenetic analysis produced largely congruent topologies across all analyses. Differences between intron topology and both exon and exon+intron topologies are shown in supplementary figure S1, Supplementary Material online. Supplementary table S2, Supplementary Material online, shows a detailed breakdown of bootstrap, and posterior probability supports for major clades in the tree for each of the six data sets across all four analyses (RAxML, BEAST, MrBayes, and PhyloBayes). These values arising from analysis of the exon+intron-outliers removed data set are summarized in figure 2.
Higher Level Relationships
Higher level relationships were well resolved (see supplementary table S2, Supplementary Material online, and fig. 2). Results of phylogenetic analysis are shortened throughout as Bayesian analyses (BA); posterior probability (PP); and bootstrap support (BSS). At the subordinal level among the Yinpterochiroptera, the association between Pteropodidae and Rhinolophoidea bats was recovered with full support by all analyses except BEAST, which reported much lower support, 52–71 PP (fig. 2). The superfamily grouping of Rhinolophoidea received full support for all analyses. Each analyses also found strong support, >90/0.9–100/1 BSS/PP, for the associations between Craseonycteridae, Megadermatidae, and Rhinopomatidae. The sister taxa relationship between the Hipposideridae and Rhinolophidae was fully supported in all analyses.
Hipposideridae and Rhinonycterina
Within the traditional grouping of Hipposideridae, there is a basal division of two strongly supported monophyletic groups. The subtribe Rhinonycterina (sensu Gray 1866: 81) is supported by a long branch indicative of significant phylogenetic distance and received full support from all analyses. The other monophyletic clade is well supported (>98 BSS/0.99PP) and contains all other Hipposideridae with Asellia basal in all analyses. The genus Hipposideros is paraphyletic in all analyses. Hipposideros commersoni and H. vittatus are distinct from the main species group Hipposideros and form a strongly supported clade with Aselliscus and Coelops (fig. 2) based on analysis of the exon and exon+intron data sets; BA 0.98–1.0 PP, maximum likelihood (ML) 79–93 BSS (supplementary table S2, Supplementary Material online). In contrast to this topology, analysis of the intron data set consistently results in a poorly supported alternative topology in which the sister taxa H. vittatus and H. commersoni fall outside a clade containing the sister taxa Aselliscus and Coelops, as well as all other Hipposideros spp. approximately 70 BSS/0.7 PP. The remaining Hipposideros spp. form a monophyletic group with full support from all analyses.
The monophyletic grouping of the Rhinonycterina is further supported by a unique shared indel in the THY gene fragment. Relationships within this group are well supported. Cloeotis and Triaenops are sister taxa, ML 97–100 BSS, BA 1.0 PP. The association between the geographically disparate Rhinonicteris, Cloeotis, and Triaenops also receives strong support from all analyses, ML 98–100 BSS, BA 1.0 PP. Given this group’s distinctiveness, we propose to elevate this subtribe to full familial status, see systematic survey below.
Systematic Summary
Order Chiroptera Blumenbach (1799:58,74).
Suborder Yinpterochiroptera Springer et al. (2001:6243).
Superfamily Rhinolophoidea J.E. Gray (1825:338).
Family Rhinonycteridae J.E. Gray, 1866. Proc. Zool. Soc. Lond. 1866:81, new rank. Old World leaf-nosed bats.
(=Rhinonycterina J.E. Gray 1866:81; including the Subfamily Rhinonycterinae J.E. Gray 1866:81; Tribe Rhinonycterini J.E. Gray 1866:81; Tribe Triaenopini Benda and Vallo 2009:33; Subtribe Rhinonycterina J.E. Gray 1866:81 (including [fossil taxa with †] Rhinonicteris J.E. Gray, 1847:16; Cloeotis Thomas 1901:28; Triaenops Dobson 1871:455; †Brachipposideros Sigé, 1968:83; †Brevipalatus Hand and Archer 2005:372; Paratriaenops Benda and Vallo 2009:31.)
Type Genus—Rhinonicteris J.E. Gray, 1847
Type genus is Rhinonicteris J.E. Gray, 1847:16, which was included in J.E. Gray's (1866:81) supra-generic grouping, the Rhinonycterina, which he called “leaf-nosed bats”. Correct generic spelling was discussed by Simmons (2005:378) and resolved by Armstrong (2006), see also Mahoney and Walton (1988:127) (see derivato nominis section of the supplementary information for further discussion).
Description and Diagnosis of the Family Rhinonycteridae
The soft part characters of the rhinarium (noseleaf) outlined here are derived largely from Gray (1845) and Hill (1982), with verification of specimens in The Natural History Museum, London (BMNH). The Family Rhinonycteridae, Old World leaf-nosed bats, as diagnosed here, possess the following combination of five principal features of the rhinarium observable in extant species (reference specimens include Cleotis percivali [BMNH 56.550], Paratriaenops furculus [BMNH 78.185], Rhinonicteris aurantia [BMNH 57.10.24.10], and Triaenops persicus [BMNH 72.4372]): 1) having a sella (strap-like projection) extending forward from the internarial region of the anterior portion of the rhinarium, which distinguishes them from their closest relatives in the Hipposideridae Lydekker, in Flower and Lydekker,1891:657; 2) anterior rhinarium is deeply emarginate medianly, more so than in the Rhinolophidae (Gray, 1825:242) (see illustrations in Hill [1982] and Benda and Vallo [2009]; cf. Csorba et al. 2003); 3) strongly cellularized (more so than members of the Hipposideridae) and multipocketed posterior rhinarium; 4) either with (Cleotis, Paratriaenops, and Triaenops) or without (Rhinonicteris) a trident-like projection oriented dorsally and originating from the caudal margin (these are structurally different from the three reduced projections in the genus Asellia); and 5) a compressed longitudinal process originating from the intermediate rhinarium between the nares and central cellular pocket. For further descriptions and illustrations, see the following: Gray (1866); Dobson (1878); Hill (1982); and Benda and Vallo (2009). The Rhinonycteridae are further distinguished from Hipposideridae by a 128-bp retrotransposon insertion in the THY gene fragment.
The Rhinonycteridae differ from the Nycteridae, Van der Hoeven, 1855:1028 and Megadermatidae, H. Allen, 1864:1 based on noseleaf structure (as described above; see descriptions of the latter in Tate [1941a, 1941b] and Koopman [1994]) and by having ears that are separate, not enlarged and lacking a tragus. Like the Hipposideridae, members of the Rhinonycteridae differ from the Rhinolophidae, by having two pedal phalanges rather than three, and they lack a P3. Craniodental features of extinct and extant Rhinonycteridae show considerable variation, and the examination of relevant specimens and literature does not reveal characters that diagnose members of the Rhinonycteridae from all other rhinolophoids or from the members of the Hipposideridae; see Sigé et al. (1982) and Hand and Archer (2005) for combinations of features that distinguish fossil members of the Rhinonycteridae from members of the Hipposideridae; these differences are not necessarily unique to the Rhinonycteridae. Both the Rhinonycteridae and Hipposideridae differ in terms of their echolocation call structure from the Rhinolophidae, emitting typical pulse durations of around 15 ms or less in “search mode”, compared with >30 ms search mode calls produced by members of the Rhinolophidae.
Rhinolophidae
The Rhinolophidae form a fully supported monophyletic group but intrafamilial relationships are less well resolved than for the Hipposideridae (see supplementary table S2, Supplementary Material online). Generally, the analyses produced a topology that provided either no structure at the base of the crown group Rhinolophidae or a strict division between the African/European clade, comprising R. hipposideros, R. ferrumequinum, and R. euryale versus the Asian clade, comprising all other Rhinolophus spp. However, the African/European clade, when recovered, grouped together with relatively low support approximately 60 BSS/0.6 PP. For all analyses using the CAT model in PhyloBayes, a topology with no structure at the base of the crown group Rhinolophidae was supported. Uniquely, the intron data set with outliers included under a BEAST analysis provided an alternative topology in which R. hipposideros was the basal clade; however, this was poorly supported, 0.38 PP. The association of Asian taxa was supported across all BA 0.88–1.0 PP, whereas ML analyses provided variable support depending on the data set. The exon and exon+introns data sets strongly support the association of the Asian taxa, 98–100 BSS; however, less support for this association is found in the introns analyses 63–65 BSS.
Anthops Subtree
The three intron-based subtree shows that Anthops falls within the monophyletic grouping of Hipposideros with full support from the BA (fig. 3). However, the exact position of Anthops within Hipposideros is less certain with the grouping of Anthops, H. jonesi, H. armiger, and H. larvatus being poorly supported, 0.41 PP.
Fig. 3.

Bayesian Tree derived from BEAST analysis of 1,223 bp comprising three nuclear introns—STAT5A, PRKC1, and THY under GTR+G substitution model, highlighting the position of Anthops ornatus.
Alternative Topology Tests
Alternative topology tests, Kishino–Hasegawa (KH), Shimodaira–Hasegawa (SH), approximately unbiased (AU), were carried out to test a number of competing hypotheses and alternative topologies arising from our data, results are summarized in table 2. The statistical tests were unable to differentiate between the phylogenetic hypothesis in which H. vittatus and H. commersoni fall outside a clade containing the sister taxa Aselliscus and Coelops and all other Hipposideros spp. (KH P = 0.104, SH P = 0.104, and AU P = 0.89) and the hypothesis in which H. commersoni and H. vittatus are sister taxa to Aselliscus stoliczkanus and Coelops frithii (KH P = 0.896, SH P = 0.896, and AU P = 0.911). The paraphyly of Hipposideros is strongly supported by our phylogenetic analyses; statistical tests also rejected the alternative hypothesis in which Hipposideros is monophyletic (KH P = 0.037, SH P = 0.037, and AU P = 0.018). Four alternative topologies of Rhinolophidae arising from our data were tested. Topology tests could not reject the hypothesis in which R. hipposideros was basal (KH P = 0.363, SH P = 0.784, and AU P = 0.37). The topology in which the R. trifoliatus and R. luctus was the basal clade was rejected by the AU (P = 0.022) and KH (P = 0.047) tests, but the SH test (P = 0.268) was unable to reject this hypothesis. The topology in which R. pearsoni is basal was also rejected (KH P = 0.000, SH P = 0.000, and AU P = 0.00). The topology arising from the consensus tree, which supports a basal division between European/African versus Asian clades, could not be rejected in favor of any other topology tested (KH P = 0.637, SH P = 0.911, and AU P = 0.765).
Table 2.
P Values Resulting from Statistical Comparison of Alternative Phylogenetic Hypotheses Using Topology Tests in Tree Puzzle and Consel.
| Tree | Tree puzzle |
Consel |
||||
|---|---|---|---|---|---|---|
| Log likelihood | AU | KH | SH | WKH | WSH | |
| Exon vs. intron topology—position of Hipposideros commersoni, H. vittatus | ||||||
| Exon topology (H. commersoni, H. vittatus) (Aselliscus stoliczkanus, Coelops frithii) | −60,264.89 | 0.911 | 0.896 | 0.896 | 0.896 | 0.896 |
| Intron Topology (H. commersoni, H. vittatus) (As. stoliczkanus, C. frithii) | −602,745.00 | 0.89 | 0.104 | 0.104 | 0.104 | 0.104 |
| Hipposideros—monophyletic vs. paraphyletic | ||||||
| Paraphyletic | −60,264.89 | 0.982 | 0.963 | 0.963 | 0.963 | 0.963 |
| Monophyletic | −60,277.81 | 0.018* | 0.037* | 0.037* | 0.037* | 0.037* |
| Rhinolophidae—basal clade | ||||||
| Rhinolophus hipposideros basal (arising from BEAST analysis of introns—cf. table S1) | −60,265.26 | 0.37 | 0.363 | 0.784 | 0.363 | 0.714 |
| R. trifoliatus and R. luctus basal (Guillen-Servent et al. 2003) | −60,282.96 | 0.022* | 0.047* | 0.268 | 0.047* | 0.083 |
| R. pearsoni basal (Stoffberg et al. 2010) | −60,381.35 | <0.001* | <0.001* | <0.001* | <0.001* | <0.001* |
| Basal division of European/African vs. Asian clades (consensus tree—fig. 2) | −60,264.89 | 0.765 | 0.637 | 0.911 | 0.637 | 0.939 |
*Significant.
Divergence Time Estimates and Biogeography
Results from the divergence time estimates indicate that Rhinolophoidea and Pteropodidae diverged approximately 59 Ma (fig. 4). The Rhinolophidae and Hipposideridae diverged in Africa from their common ancestor approximately 42 Ma during the Eocene. The family Rhinonycteridae separated from the rest of the Hipposideridae approximately 39 Ma, also in Africa. The monophyletic Hipposideros clade diverged from other Hipposideridae approximately 31 Ma during the Oligocene in Africa. The monophyletic crown groups Hipposideros and Rhinolophus diversified during the Miocene at approximately 15 Ma and approximately 17 Ma, respectively. There was no difference in dating estimates when using the different stratigraphic dating sources. The biogeographic analysis reveals distinct geographical origins for two of the three of the Rhinolophidae clades; the clade comprising R. trifoliatus and R. luctus diverged in Eastern/South Eastern Asia, and the clade formed by R. pearsoni, R. sinicus, R. creaghi, R. shameli, and R. pusillus diverged in East Asia. The geographical origin of the clade containing R. hipposideros, R. ferrumequinum, and R. euryale is less clear with the analysis returning a combination of widespread geographical areas including Europe, East Asia, Middle East, and Africa. The analysis was repeated removing South America to investigate whether this disparate area represented an outlier in our data. No significant changes were observed in the clades of interest, with the reconstruction after the removal of South America shown on the right; namely: The divergence of Rhinolophidae and Hipposideridae (H, H), the divergence of Hipposideridae and Rhinonycteridae (H, H), the crown group Rhinolophidae (ACEH, AFEH), the crown group Hipposideridae (H, H), and the crown group Rhinonycteridae (H, DH).
Fig. 4.

Molecular time scale resulting from MCMCTREE analysis in PAML using the BEAST topology shown in figure 2, four fossil calibrations (as described in Materials and Methods using stratigraphic bounding), and a root prior of 64–65 Ma. Numbers at nodes are divergence time estimates in millions of years and the 95% confidence interval for each estimate is denoted by a blue shaded bar. Biogeographic reconstructions resulting from ML analysis in Lagrange under the same topology are shown as letters at each node. Areas are coded as follows: A—Europe, B—South America, C—South East Asia, D—India, E—Middle East, F—East Asia, G—Australia, and H—Africa.
Diversification Analysis
Concatenation of the four data sets for diversification analysis resulted in the following alignments: Hipposideros Cyt b—980 bp comprising 87 sequences; Rhinolophus Cyt b—1,140 bp comprising 58 sequences; Hipposideros Cox1—658 bp comprising 50 sequences; and Rhinolophus Cox1—658 bp comprising 51 sequences, where all sequences are representative of putative species. The resulting BEAST trees were used to generate and compare lineage-through-time (LTT) plots (fig. 5). Comparison of the rates of diversification for each gene using the Kolmogorov–Smirnov (K-S) test revealed no significant differences between Rhinolophus spp. and Hipposideros spp. (Cyt b, D = 0.1159, P = 0.733; Cox1, D = 0.919, P = 0.985). Rates of diversification obtained from Cyt b and Cox1 were not significantly different (K-S test; D = 0.101, P = 0.908 for Hipposideros; D = 0.114, P = 0.869 for Rhinolophus).
Fig. 5.

LTT plot showing the diversification rate of the genera Rhinolophus and Hipposideros for Cyt b and Cox1, where time is represented by arbitrary values with 0.0 representing the present.
Performance Evaluation: Differential Resolving Power of Intron and Exon Data?
For the ten jack-knifed exons subsets (intron or exon topology), node height was a significant predictor of posterior (GLM, all P < 0.05) with higher nodes (i.e., older nodes) being associated with lower posteriors (fig. 6). In contrast, for the intron data set for either the intron or the exon topology, node height was not a significant predictor of posterior (GLM, P = 0.42 and 0.72, respectively). Average posteriors of the ten exon subsets with jack-knifed sites were significantly lower than intron posteriors for the 33 nodes that were congruent between intron and exons topologies (exon PP = 0.90, intron PP = 0.97; asymptotic Wilcoxon Mann–Whitney rank sum test, P = 0.036). Results were similar when gene fragments were jack-knifed (see supplementary fig S2, Supplementary Material online) (exon PP = 0.86, intron PP = 0.95; asymptotic Wilcoxon Mann–Whitney rank sum test, P = 0.028).
Fig. 6.
Showing the relative difference in nodal support between intron and jack-knifed exon data sets of equal size, where colored dots indicate the proportion by which nodes are better resolved by either intron (red) or exon (green) data for (a) intron topology and (b) the exon topology. Species names are as in figure 2.
Discussion
Performance Evaluation: Removal of Outliers and Differential Resolving Power of Introns and Exons?
Our results show that removal of outlying data had no effect on the overall tree topology and had a very minimal effect on posterior probabilities with a minor average posterior change of − 0.00167. Minor decreases (maximum decrease 0.03) in PP occurred at three nodes. It would be expected that phylogenetic trees that contain both old and young divergences would be best resolved by a combination of exon and intron data. In theory, exon data, which are slower evolving, should be best suited to resolve deep nodes in a tree and faster evolving intron data should better resolve young nodes. In contrast to these expectations, our analyses show there is no significant difference in the relationship between node height and posteriors for intron data (fig. 6), yet exons showed poorer support for most of the oldest nodes in our phylogeny.
When omitting incongruent nodes, introns systematically outperform exons in terms of nodal support. This cannot be explained by differences in fragment length as our comparisons were carried out on data sets of similar length with both jack-knifed sites and gene fragments. Rather, in line with previous studies (Chojnowski et al. 2008), we argue that for an equal fragment size and the time frame investigated (∼60 Ma), introns carry more phylogenetic signal than exons and are less prone to gene tree conflicts (Romiguier et al. 2013). Because of their faster substitution rate, introns evolve more quickly than exons and, hence, can provide more phylogenetic information. However, because they evolve faster, introns should also saturate faster but in a time frame of approximately 60 Ma, as investigated by Chojnowski et al. (2008), saturation of the phylogenetic signal was not an issue for introns. This may stem from the alignment process, whereby highly variable and divergent intronic sites are removed from analyses given the difficulty in ascertaining homology for these positions. Throughout plants, invertebrates, and vertebrates, it has been shown that introns tend to have a lower GC content than exons (Amit et al. 2012). This conserved characteristic of genome evolution has important consequences for accurate phylogenetic reconstructions as it has been shown that regions of the genome, which are GC rich have a higher amount of gene tree conflict and have greater difficulties in reconstructing well-supported consensus nodes in the placental mammal tree (Romiguier et al. 2013). This is particularly important in the context of future phylogenomic studies as to date phylogenomic tree reconstructions have typically used only coding regions (Parker et al. 2013; Seim et al. 2013; Zhang et al. 2013). A recent study, particularly focused on echolocating mammals, revealed that many coding gene sequences show signs of convergent evolution and, hence, provide phylogenetic signals that are incongruent with the true species tree (Parker et al. 2013). Although this remains to be tested, we predict that introns should be less prone to convergent evolution and should therefore provide better data to resolve species trees, especially when aligned optimally. The incongruence in tree topology observed between analysis of intron and exon data in our study highlights the importance of using a combination of markers in phylogenetic studies.
Phylogenetic Questions
For the first time, based on complete taxonomic sampling at the generic level and with diverse data types, the outstanding phylogenetic controversies surrounding the hipposiderid bats have been resolved. Although topological differences are observed in analyses of the intron data sets, these differences do not affect our interpretation of higher level relationships among these bat lineages.
Taxonomic Clarification and Revision at the Family Level
Our analyses indicate that the Hipposideridae should be recognized as a distinct family. The Hipposideridae are further divided into two well-supported monophyletic clades. On the basis of phylogenetic analysis, divergence time estimates, immunological transferrin data (Pierson 1986), previous morphological comparisons (Hill 1982), and a 128-bp endogenous retroviral insertion unique to the clade containing Paratriaenops, Triaenops, Cloeotis, and Rhinonicteris, we show that these genera are also sufficiently distinct to merit independent familial status, Rhinonycteridae (see systematic summary and supplementary information, Supplementary Material online, for taxonomic information). The phylogenetic analysis revealed strong support for the monophyly of these two families (Hipposideridae and Rhinonycteridae) across all data types and analyses performed. Our divergence time estimates show that the Rhinolophidae, Hipposideridae, and Rhinonycteridae last shared a common ancestor approximately 42 Ma, followed by the divergence of the Hipposideridae and Rhinonycteridae approximately 39 Ma. These divergence time estimates are comparable to those obtained for other established bat families in previous analyses (see Teeling 2009).
Rhinonycteridae
The original description of Rhinonycterina was recognized by later studies and was expanded to include Triaenops and Cloeotis as these taxa fitted the original description for the Rhinonycterina based primarily on noseleaf morphology (Hill 1982). The association of these genera is also supported by our molecular data, which show that Triaenops and Cloeotis are sister taxa (fig. 2) and, furthermore, form a monophyletic group with Rhinonicteris. The data also support the recent separation of Triaenops and Paratriaenops but do not support the monophyly of the recently erected Tribe Triaenopini (Benda and Vallo 2009). Strong support was found for Paratriaenops as the basal clade of the extant crown group rhinonycterids (fig. 2).
Hipposideridae
The familial delimitation of Rhinonycteridae and Hipposideridae results in Asellia being basal to all other Hipposideridae. This clade received strong bootstrap and PP support across all analyses (see supplementary table S2, Supplementary Material online). This is contrary to a previous study, which, based on morphological data, places Aselliscus basal (Hand and Kirsch 1998), despite the inclusion of Asellia in their data set. The genus Aselliscus contains two species, and its placement in the Hipposideridae phylogeny has been controversial. Pierson (1986) concluded that Aselliscus was not a member of Hipposideridae and was more closely aligned with the Rhinolophidae, based on immunological transferrin distance data. Morphological data place this genus among other Hipposideros spp. (Bogdanowicz and Owen 1998; Hand and Kirsch 1998). These morphological results are in agreement with molecular data, which found strong support for a sister taxa relationship between Aselliscus and Coelops (Li et al. 2007). Previously, Coelops has been grouped in a separate tribe Coelopini Tate (1941:11) (sensu; McKenna and Bell [1997] = Coelopinae Tate [1941:11]), which comprised Coelops and Paracoelops, the latter no longer recognized as a valid genus (Thong, Dietz, et al. 2012). Our findings provide strong support for the sister taxa relationship between Aselliscus and Coelops recovered by Li et al. (2007).
Does Hipposideros Form a Monophyletic Group?
Our data do not support the genus Hipposideros as a natural monophyletic group. Statistical tests reject the monophyly of Hipposideros (table 2). Monophyly is also rejected by morphological data (Sigé 1968; Legendre 1982; Bogdanowicz and Owen 1998). Extreme karyological conservatism is reported in the genus Hipposideros with all members investigated so far having a 2n complement of 32; however, H. commersoni sensu lato (table 1) based on a distributional context of where the material was obtained (Monadjem et al. 2010) has a 2n complement of 52 (Bogdanowicz and Owen 1998). To date, no karyological data exist for H. commersoni sensu stricto (referring specifically to specimens identified from the type locality, Madagascar). Support for the association of H. commersoni and H. vittatus as sister taxa to Aselliscus and Coelops is derived from the exons and exons+introns data set. An alternative topology, arising from the intron data sets, where H. commersoni and H. vittatus fall outside a clade containing the sister taxa Aselliscus and Coelops and all other Hipposideros spp. was poorly supported, but statistical tests were unable to reject this topology (table 2). The monophyly of the remaining Hipposideros spp. was strongly supported by all analyses. Relationships within this genus are poorly resolved and short branches illustrate the rapid diversification of the crown group.
The BEAST analysis of three nuclear introns indicated that Anthops falls within the monophyletic group of Hipposideros spp. (fig. 3). The previous study involving this genus showed that it formed a sister taxa relationship with H. caffer to which H. commersoni sensu lato was basal (Eick et al. 2005). As our study included more species of true Hipposideros, it was hoped that the position of Anthops within this group could be resolved as it has implications for the taxonomic classification of the genus. Our data show that Anthops forms a poorly supported group with H. jonesi, H. armiger, and H. larvatus. The strong support for the monophyly of this clade indicates that Anthops most likely represents an incorrectly classified member of the genus Hipposideros. However, as this is based on a small data set and only received poor support, this result is not yet conclusive. Therefore, we cannot rule out the possibility that Anthops is a distinct genus basal to Hipposideros.
Which Species or Clade Is the Most Basal in the Rhinolophidae?
To differentiate between the competing phylogenetic hypotheses, our data included taxa recovered by previous studies as basal clades in Rhinolophidae. Guillen-Servent et al. (2003) recovered a sister taxa relationship between R. trifoliatus and R. hipposideros containing clades as the basal clade based on an analysis of Cyt b. Stoffberg et al. (2010) proposed a basal position for R. pearsoni based on Cyt b and intron data, though they did not include R. trifoliatus in their data set. Phylogenetic analysis of our data provided two further alternative topologies—a basal division between Asian and African/European clades and an alternative in which R. hipposideros is basal (table 2). Statistical tests rejected the hypothesis in which R. pearsoni is basal; however, only the AU and KH tests rejected the hypothesis in which a clade containing R. trifoliatus and R. luctus was basal, and the SH test was unable to reject this hypothesis most likely due to the conservative nature of this test (Strimmer and Rambaut 2002) (table 2). The Stoffberg et al. (2010) phylogeny showed that, with the exception of the basal taxa R. pearsoni, two distinct clades are present in this family, one of African and one of Asian origin. To a certain extent, this biogeographic dichotomy is supported by our phylogeny. The Asian species grouped together with strong support to the exclusion of the European/African taxa; however, statistical tests were unable to differentiate between this hypothesis and an alternative in which R. hipposideros is basal. As a result, the structure within this group remains uncertain likely owing to the rapid diversification of the Rhinolophidae (Guillen-Servent et al. 2003) combined with a long branch leading to them. In future studies, increased taxonomic sampling and more markers will be necessary to resolve relationships within the Rhinolophidae.
How Long Ago Did Hipposideridae, Rhinonycteridae, and Rhinolophidae Diverge, and Where Did They Originate?
The divergence times estimated by our molecular dating analysis were in-line with estimates previously established for the same groups (Eick et al. 2005; Teeling et al. 2005; Miller-Butterworth et al. 2007; Teeling 2009; Stoffberg et al. 2010; Anthony et al. 2013). The Rhinolophidae are estimated to have diverged from the ancestor of the Rhinonycteridae and Hipposideridae approximately 42 Ma during the Eocene (fig. 4). Rhinonycteridae and Hipposideridae diverged 39 Ma. The Lagrange biogeographic analysis indicated that the ancestors of these three families are of African origin (fig. 4). An African origin for the Hipposideridae is in contrast to the Asian origin proposed by Teeling et al. (2005) and Bogdanowicz and Owen (1998) and the Australian origin proposed by Hand and Kirsch (1998). No previous biogeographic hypothesis exists as to the origin of the component taxa of what is recognized herein as the Rhinonycteridae, and our analysis show that these bats are of African origin. The distribution of the extant members of the Rhinonycteridae is unusual in that Rhinonicteris is endemic to Australia, whereas all other members of the family are distributed in and around Africa, including Madagascar. This unusual distribution is shared by Allodapine bees that exhibit a similar Africa/Australia split and are thought to have dispersed across the Indian Ocean by island hopping along the aerial formations of the Kergulen Plateau (Schwarz et al. 2006). However, a terrestrial continental dispersal route is better supported for Rhinonycteridae by the fossil record. The fossil taxon Brachipposideros nooraleebus, dating from the middle Miocene, is thought to be sister taxa to Rhinonicteris (Sigé 1968; Legendre 1982; Sigé et al. 1982; Hand and Kirsch 1998), based on this, Brachipposideros is thought to have dispersed into Australia 15–20 Ma, a time which correlates well with our estimated divergence time for Rhinonicteris (Sigé et al. 1982). Brachipposideros has been found in Western Europe, Asia, North Africa, and Australia (Hand and Archer 2005), which suggests a route from Africa into Europe then Asia before arriving in Australia.
Our data indicated that the family Rhinolophidae originated in Africa (fig. 4), the same conclusion as reached by Eick et al. (2005). This is in contrast to the Asian origin reported by several other phylogenetic studies (Bogdanowicz and Owen 1992; Teeling et al. 2005; Stoffberg et al. 2010) and the recent finding of fossil Rhinolophidae dating from the middle Eocene in China (Ravel et al. 2014). We did not find support for the European origin for this family as proposed by Guillen-Servent et al. (2003). However, biogeographic reconstructions were not able to pinpoint the geographic origin of the Rhinolophus crown group.
Diversification Analysis
The nuclear phylogeny (fig. 2) revealed strong similarities in tree shape and branching pattern between the Rhinolophus and Hipposideros genera despite considerable phylogenetic distance between the two groups. Molecular clock analysis (fig. 4) revealed that the divergence time estimates for these two genera are concurrent, occurring between 15 and 17 Ma. This indicated that these genera underwent a largely concurrent, rapid diversification during the Miocene, coinciding with the Mid-Miocene Climatic Optimum, which was a warm and humid period in Earth’s history, which peaked roughly 15–18 Ma (Sun and Zhang 2008). This period may have represented optimal conditions for the evolution and rapid diversification of these lineages in response to more favorable climatic conditions. The analyses showed that the rate of diversification of Rhinolophus and Hipposideros is very similar for both molecular markers (fig. 5). However, although Cox1 and Cyt b support very similar rates of diversification, they are both fragments of the mitochondrial genome, and it is yet to be verified if these diversification rates would remain consistent across a wider diversity of nuclear markers.
Comments on Molecular Clock and Biogeographical Analysis Methods
It is important to recognize the limitations of the methods used herein for divergence time estimation and biogeographic analysis. Caution must be taken when interpreting results provided by divergence time estimates given that errors can be introduced through the use of fossil calibrations as fossils based on limited characters can be incorrectly classified (Parham et al. 2012), and stratigraphic layers can be assigned to incorrect geological times. This is particularly applicable to bats which have a poor and limited fossil record (Teeling et al. 2005; Eiting and Gunnell 2009). Although the oldest fossil attributable to a lineage can provide a minimum age in a soft bound analysis, choosing a maximum bound is more difficult, here we use the stratigraphic bounding method of Meredith et al. (2011) to standardize the way in which maximum boundaries are chosen across the tree. Area coding is one of the key manners contributing to error in biogeographic reconstruction. Springer et al. (2011) showed that including information regarding the provenance of the oldest fossil attributable to a clade produced very different results than only coding for the extant geographical distribution of a species. Including fossil taxa introduces error in the form of taphonomy and sampling bias. In particular, with regard to the fossil record of the Hipposideridae, the inclusion of fossil data to biogeographic analysis would introduce a European/Australian bias based on the majority of species ascribed to this group to date (McKenna and Bell 1997; Hand and Kirsch 1998). To overcome the limitations inherent in the bat fossil record, we use only the distribution of extant taxa in our analysis. Furthermore, taxonomic sampling remains a problem inherent in biogeographic analyses. Our phylogeny comprises representatives from all Hipposideridae, Rhinolophidae, and Rhinonycteridae genera. Our taxonomic sampling of Rhinolophidae does not include any Sub-Saharan African representatives, and so this may hinder a definitive biogeographic analysis of this family. However, a recent study of Emballonura bats has shown that the basal clade of a group can drastically impact its biogeographical reconstruction (Ruedi et al. 2012). Although the basal clade of the Rhinolophidae remains unresolved, our reconstruction is the first to include all previously recovered basal candidates for this group as a strategy to minimize the impacts of partial taxonomic sampling of this group.
Implications of the Phylogeny
By addressing the phylogenetic controversies and taxonomic uncertainties, our phylogeny has implications for the detection and surveillance of novel coronaviruses, as well as conservation strategies for these unique lineages. The recent emergence of the novel MERS-CoV (Bermingham et al. 2013) has highlighted the importance of detecting and monitoring animal reservoirs of potentially pathological novel coronaviruses. The intermediate phylogenetic position of a SARS-like CoV between Rhinolophidae and Hipposideridae derived from Rhinonicteris (Ar Gouilh et al. 2011) is supported by the resolved phylogeny for these bats as its phylogenetic position reflects the underlying divergence of these three bat families. Although they have been underrepresented in previous studies—see table 2 in Ar Gouilh et al. (2011)—our phylogenetic results suggest that other members of the Rhinonycteridae may represent excellent survey candidates to expand knowledge of the diversity of coronaviruses, especially those related to the Betacoronavirus-b group, which includes the SARS-CoV. Given the similarity of diversification rates between Rhinolophus and Hipposideros, studies focused on detecting novel coronaviruses in Hipposideros could potentially mirror the diversity of coronaviruses detected so far in Rhinolophus. A comprehensive understanding of the diversity of coronaviruses is important for human health in that as the genetic diversity of zoonotic viruses increases so too does the possibilities of variants crossing species boundaries (Li et al. 2005).
Our data show that it is necessary to revise the conservation management plan for these groups of bats. Hipposideridae, Rhinonycteridae, and Rhinolophidae are of considerable conservation concern with a number of species being listed as endangered and near threatened (IUCN 2014). This is mostly due to the increasing levels of human disturbance, which in turn reduces the number of available roost sites, as well as direct habitat destruction (Nowak and Paradiso 1999). A resolved phylogeny is a considerable aid to conservation as it informs management plans operating under the EDGE strategy which aims to conserve not only endangered species but also conserves phylogenetic diversity (Isaac et al. 2007).
Conclusion
To elucidate possible systematic bias obscuring the phylogenetic signals, we investigated the effects of removing outlying data on phylogenetic reconstruction and compared the relative resolving power of intron and exon data for our tree. We show that removal of outlying data had no effect on tree topology and only a minimal effect on posterior probabilities. Furthermore, our analyses show that intron data provides better nodal support for our tree than exon data sets of similar size. Phylogenetic analyses resulted in a well-resolved phylogeny, which settles, for the first time, the phylogenetic controversies surrounding the Hipposideridae. A combination of phylogenetic analyses and divergence time estimates support the elevation of Hipposideridae and Rhinonycteridae to family level. Biogeographic analysis revealed that the Rhinolophidae, Hipposideridae, and Rhinonycteridae originated in Africa. The largest genus of the Hipposideridae, Hipposideros, is paraphyletic, and our study shows that Anthops ornatus may represent a species of Hipposideros. Our resolved phylogeny provides the foundation essential for “systematic surveillance" of emerging bat-based novel coronaviruses. Although they have been underrepresented in previous studies, our phylogenetic results suggest members of the Rhinonycteridae may represent excellent candidates in which to survey to expand our knowledge of the diversity of coronaviruses, especially those related to the Betacoronavirus-b group, which includes the SARS-CoV. In this way, our phylogeny provides guidance to better predict and manage these human-lethal zoonotic diseases. These analyses also highlight these three bat families as separate Evolutionary Significant Units, with their own morphological peculiarities, long phylogenetic history, and patterns of dispersal, warranting differential conservation management strategies in the future.
Materials and Methods
Taxon Sampling and Data Selection
A total of 19 nuclear gene fragments were amplified for 39 taxa. Additional data for A. ornatus were downloaded from GenBank to bring the total number of taxa analyzed to 40. These nuclear gene fragments consisted of 12 exon fragments (ADORA3, APP, ATP7A, BDNF, BUF134, PNOC, PLCB4, TTN3, TTN5, TTN6, TTN7, and RGF2) and seven intron fragment (ABHD11, ROGDI, ACOX2, BGN, THY, STAT5A, and PRKC1) (supplementary table S1, Supplementary Material online). Of the 40 taxa analyzed, ten were outgroup taxa represented by two Yangochiroptera (Teeling et al. 2005), represented by the families Vespertilionidae (n = 1) and Mormoopidae (n = 1), and four Yinpterochiroptera (Teeling et al. 2005), including the families Megadermatidae (n = 2), Rhinopomatidae (n = 2), Craseonycteridae (n = 1), and Pteropodidae (n = 3) (table 1). The remaining 30 taxa are split between the rhinolophid (n = 10) and hipposiderid bats (n = 20). The ten Rhinolophus spp. were chosen with regard to previously published phylogenies to differentiate between competing topologies.
Our 20 hipposiderid taxa represent all nine of the currently recognized genera and include Anthops (n = 1), Asellia (n = 1), Aselliscus (n = 1), Cloeotis (n = 1), Coelops (n = 1), Hipposideros (n = 10), Paratriaenops (n = 2), Rhinonicteris (n = 1), and Triaenops (n = 2). As it was not possible to obtain DNA for Anthops, only three nuclear introns (STA5A, THY, and PRKC1) could be downloaded from GenBank. Hence, a phylogenetic reconstruction was specifically carried out only with these three introns to position Anthops.
To overcome potential systematic bias resulting from cryptic diversity, where possible, all sequences were generated from the same sample for each species or from the same species collected in the same geographical area as the original sample. All sequences newly generated are deposited into GenBank accession numbers (KP175738–KP176371) and concatenated with previously published data where possible (supplementary table S3, Supplementary Material online). In total, 40 species, including A. ornatus, from eight families were analyzed.
Tissue Sampling, DNA Extraction, and Amplification
Tissues used for genetic analyses were either from wing punches from released individuals or tissue samples from voucher specimens housed in different museum collections (see Genbank Accessions for further details). DNA was isolated using a modified salt/chloroform extraction protocol (Miller et al. 1988), which included an additional chloroform/isoamyl alcohol (24/1) step after the addition of the saturated NaCl solution (Puechmaille et al. 2011). Nuclear gene fragments, comprising exon and intron sequences of varying length were amplified with primers listed in supplementary table S1, Supplementary Material online. Reactions were carried out in 25 μl solutions containing 2 μl of DNA extract, 1X polymerase chain reaction (PCR) buffer minus Mg (Invitrogen), 1.5 mM MgCl2, 0.4 μM each primer, 0.2 mM dNTPs, and 1 U Platinum Taq DNA Polymerase High Fidelity (Invitrogen). Amplifications were carried out in a DNA Engine DYAD thermocycler (MJ Research) with PCR programs: 1) Touchdown 50—initial step 10’ at 95 °C, then ten cycles of 15” at 95 °C, 30” at 60 °C (with a reduction of 2 °C every 2 cycles), 1’ at 72 °C, following by 30 cycles of 30” at 95 °C, 30” at 50 °C, and 1’ at 72 °C and a final step of 5’ at 72 °C and 2) touchdown 55—initial step 10’ at 95 °C, then ten cycles of 15” at 95 °C, 30” at 65 °C (with a reduction of 2 °C every 2 cycles), 1’ at 72 °C, following by 30 cycles of 30” at 95 °C, 30” at 55 °C, and 1’ at 72 °C and a final step of 5’ at 72 °C.
PCR products were purified using Exosap (USB) and sequenced in both directions by Macrogen (Europe) using the PCR primers listed in supplementary table S1, Supplementary Material. Complementary sequences were assembled and edited for accuracy using CodonCode Aligner 3.7.1. (CodonCode Corporation, Dedham, MA).
Alignment Optimization
Alignment optimization involves the removal of ambiguously aligned positions, which can occur partly as a result of insertion/deletion events, which have previously been shown to be phylogenetically informative (Murphy et al. 2007). To compromise between alignment optimization and the loss of potentially phylogenetically informative sites through the removal of indels, single gene fragments were preliminarily aligned with MAFFT (Katoh and Standley 2013). The program Repeatmasker Open-3.0 (Smit et al. 1996) was used to screen for and annotate indels prior to their downstream removal as part of alignment optimization.
Single gene fragments were then re-aligned using the T-Coffee web server. The analysis was carried out with default parameters using a comparison of the Mafft, Clustal W, Muscle, and T-Coffee algorithms (Notredame et al. 2000), and any ambiguous regions of the single gene alignments identified by the programs color coded system as bad or average were removed to optimize the alignment. An appropriate model of sequence evolution was chosen for each gene fragment using jModeltest (Posada 2008) as selected by the AIC (see supplementary table S1, Supplementary Material online, for selected models). Preliminary gene trees for each gene fragment were generated in Beast v.1.7.2 (Drummond and Rambaut 2007). The Markov chain Monte Carlo (MCMC) chain was run for 10 million generations sampling every 1,000 generations. Tracer v1.5 (Rambaut and Drummond 2007) was used to check for convergence. Effective sample sizes values were all more than 200 for all parameters, suggesting the MCMC run was sufficient to obtain valid estimates of the parameters (Rambaut and Drummond 2007). The resulting trees were collated using TreeAnnotator v1.7.2 (Drummond and Rambaut 2007) with 10% discarded as burn-in to obtain a single representative tree for each gene fragment.
To examine differences between different data types (exons vs. introns), single gene fragments were divided by data type and concatenated to form distinct data sets for further analysis: 1) exons, 2) introns, and 3) exons+introns. Preliminary analysis revealed topological incongruence between intron and exon data. Topological incongruence may be due to factors such as incomplete lineage sorting, hybridization, or different mutation rates, therefore it is optimal to identify these data and remove them to assess the true evolutionary signal in a data set (de Vienne et al. 2012). Given that our data comprised of introns and exons, which evolve at differential rates, we performed a search for outlying data using Phylo-MCOA (de Vienne et al. 2012) with default settings on individual gene trees to resolve the topological incongruence observed between exon and intron data in preliminary analysis. A small number of sequences (0.67%; 5/741; supplementary table S1, Supplementary Material online) were identified as outlying data and were removed to optimize each single gene alignment. jModeltest was rerun as before for each gene fragment to obtain an appropriate model of sequence evolution, there was no change from previous results (supplementary table S1, Supplementary Material online). To evaluate the effect of removing outliers, we analyzed our data with and without outliers resulting in a total of six data sets for further phylogenetic analysis: 1) exons, 2) introns, 3) exons+introns, 4) exons–outliers removed, 5) introns–outliers removed, and 6) exons+introns–outliers removed. The steps taken to optimize the alignment of each data set were then evaluated using the GUIDANCE web server (Penn et al. 2010), which reported that each position of our alignments were confidently aligned with regard to tree uncertainty. A comparison of the observed change in posteriors obtained from BEAST runs for exon+intron and exon+intron-outliers removed data sets was used to evaluate the effect of removing outliers.
Phylogenetic Analysis
Phylogenetic analysis was carried out using RAxML (Stamatakis 2006) for ML and BEAST v1.7.2. (Drummond and Rambaut 2007), MrBayes 3.2.1 (Ronquist et al. 2012), and PhyloBayes 3.3 incorporating the CAT model (Lartillot et al. 2009) for Bayesian analysis. The RAxML analysis was carried out via the RAxMLgui v.0.95 (Silvestro and Michalak 2012) using the thorough bootstrap method with 500 replicates. The analysis used a partitioned model in which each gene was allowed its own model of sequence evolution. The analysis carried out in BEAST was as described above for single gene trees. BEAST employs a single Markov chain in its analysis, whereas MrBayes uses two independent chains. To highlight any differences or avoid shortcomings in either program arising from these different tree-searching methods, we used both programs as representatives of Bayesian Analysis. The MrBayes analysis was carried out under default settings, and the resulting run was analyzed as above for single-gene trees generated using BEAST. Bayesian analysis was also carried out in PhyloBayes to implement the CAT model of site rate heterogeneity, which is not yet implemented in BEAST or MrBayes. The mixture model options in PhyloBayes for nucleotide data are restricted to variants of either a general time reversible (GTR) or Poisson process of sequence evolution. For this analysis, the more complicated GTR was chosen in combination with the CAT model and a Dirichlet process to describe rate variation across sites. Two simultaneous MCMC chains were run, sampling and checking for convergence every 100 generations. This was done by automatically discarding 20% of the trees from each independent run and comparing, every 100 generations, the resulting 50% majority rule consensus trees until the observed maximum difference in split frequencies between the two chains converged on a value of less than 0.1. To assess further the convergence of the run, the analysis was repeated three times, resulting in the same topology.
To determine the position of Anthops within our tree, a separate analysis was carried out on a subset of data, comprising only Hipposideridae species. The analysis was constrained to include only Hipposideridae species as it was important to maximize the number of confidently aligned positions available in this reduced intron data set to accurately determine the position of Anthops. Each gene fragment was aligned separately using T-Coffee (Notredame et al. 2000), all resulting ambiguous alignment regions were removed, and fragments were subsequently concatenated. jModeltest was used to determine the most appropriate model of sequence evolution for the concatenated alignment and AIC indicated a TVM + G model was best. Bayesian analysis was carried out in BEAST and analyzed as above. As TVM + G is not directly implemented in BEAST, our analysis was run under the next most complicated model GTR+G.
Alternative Topology Tests
The KH, SH, AU, and a number of their weighted variants were used to test the statistical significance of a number of competing phylogenetic hypotheses arising from our data and in the literature (see table 2 for description of competing hypotheses). The log likelihood of each tree was calculated in Tree Puzzle 5.2 (Schmidt et al. 2002), and KH, SH, and AU tests were implemented in Consel (Shimodaira and Hasegawa 2001) using the RELL bootstrap method with a significance limit of α = 0.05. Tests were performed using approximate parameter settings under a GTR model of sequence evolution.
Molecular Dating Analysis
The molecular dating analysis was carried out using the MCMCTREE program in PAML (Yang 2007). The analysis was carried out on the exon+intron data set-outliers removed, as this represents the most optimized combined data set. MCMCTREE requires a completely bifurcating tree, and as such, the analysis was carried out on the tree resulting from the Bayesian analysis with BEAST from the same data set, because other trees contained polytomies. The analysis was run under the HKY85 model of sequence evolution, the most comprehensive model available in PAML. Analysis was carried out using both the independent and correlated rates models. The root age was set to 64–65 Ma following consistent recovery in the following articles: Eick et al. (2005), Teeling et al. (2005), and Miller-Butterworth et al. (2007). The chain was run for 20,000 generations sampling every second generation, with 10% of the resulting trees discarded as burn-in. The analysis was repeated three times to check for convergence.
Maximum and minimum soft-bound calibrations were applied at four major nodes across the tree to constrain the analysis (described below). The oldest fossil ascribed to each lineage provided the minimum bound for the fossil calibration. Maximum bounds on fossil calibrations were calculated as two stage ages lower than the stage age containing the oldest representative fossil as described by Meredith et al. (2011). Geological stages ages were taken from Gradstein and Ogg (2009) and rounded to the nearest whole number to facilitate analysis. The analyses were also repeated with stage ages as defined by the International Commission on Stratigraphy (Cohen et al. 2013).
The base of the Rhinolophoidea was constrained following Teeling et al. (2005), which placed the maximum age of this group at the Paleocene-Eocene boundary, 56 Ma, as there are no early Eocene fossils for this group.
Hipposideros first appears in the fossil record in the Bartonian (Eiting and Gunnell 2009), which gives a minimum bound of 37 Ma. Stratigraphic bounding provided a maximum bound of 56 Ma. This constraint was applied to the split between the Rhinolophidae and Hipposideridae.
Rhinonicteris first appears in the fossil record in the Burdigalian (Eiting and Gunnell 2009), which gives a minimum bound of 16 Ma. Stratigraphic bounding provided a maximum bound of 34 Ma. This constraint was applied to the split between Rhinon. aurantia and Paratriaenops spp.
Saharaderma first appears in the fossil record in the Priabonian (Eiting and Gunnell 2009), which gives a minimum bound of 34 Ma. Saharaderma is preferred to Necromantis as its phylogenetic affinities with megadermatids are less disputed following Meredith et al. (2011). Stratigraphic bounding provided a maximum bound of 49 Ma. This constraint was applied to the split between Craseonycteridae and Megadermatidae.
Biogeographic Analysis
Biogeographic analysis was carried out using the ML method implemented in Lagrange (Ree and Smith 2008). Distribution data were gathered for all taxa in the tree from Bat Distribution Viewer (Polish Academy of Sciences, http://gis.miiz.waw.pl/webapps/thebats/iucn/, last accessed October 1, 2014) except for Rhinopoma hardwickii, which has recently been split into two species (table 1), whose distribution was recalculated in line with this revision (Hulva et al. 2007). Distributions for all species were coded as eight possible areas: A) Europe, B) South America, C) South East Asia, D) India, E) Middle East, F) East Asia, G) Australia, and H) Africa (including Madagascar). Reconstructions were performed on the tree arising from the BEAST analysis of the exon+intron-outliers removed data set. The maximum number of areas allowed in the reconstruction was set to two. Implausible geographic ranges, for example, a range where a single species is reconstructed as being present only in two disparate coding areas such as South America and India were removed from the analysis. All other parameters were run as default.
Diversification Analysis
To facilitate our comparative analysis of diversification rate in the speciose genera Rhinolophus and Hipposideros, faster evolving mitochondrial markers Cyt b and Cox1 were used to enable species level differentiation. To explore if the rate of diversification was marker specific, all available Cyt b and Cox1 sequences for Rhinolophus and Hipposideros species were downloaded from GenBank (April 30, 2013). Four data sets were prepared for the diversification analysis: 1) Rhinolophus—Cyt b; 2) Rhinolophus—Cox1; 3) Hipposideros—Cyt b, and 4) Hipposideros—Cox1. The four data sets were subsequently aligned using MAFFT v.7 alignment server (Katoh and Standley 2013). Sequences with 100% identity were identified and removed using CD-HIT server (Huang et al. 2010). Because of the high number of sequences of conspecifics, sequences with 97.5% sequence identity or above were removed from the data set in an attempt to keep only one sequence per species; 97.5% was used as the cutoff as low intraspecific mtDNA sequence divergence between 1% and 2.5% have been generally reported for bats (Ditchfield 2000). Data sets were then visually inspected to remove misidentified sequences and trimmed leaving one outgroup species. jModeltest was used to identify the most appropriate model of sequence evolution for each data set using the AIC, which resulted in the selection of GTR+I+G variants for all data sets. Gene trees for each gene fragment were generated in BEAST (Drummond and Rambaut 2007) using the same parameters as previously described. LTT plots were constructed for each data set and compared in R 2.5.2 (R Development Core Team 2012) using the APE package (Paradis et al. 2004). A nonparametric K-S statistical test was implemented in R 2.5.2 to detect significant difference between the rates of diversification inferred from the LTT plots of the Rhinolophus and Hipposideros genera.
Performance Evaluation: Differential Resolving Power of Intron and Exon Data?
To test whether exons perform better at resolving older nodes and if introns are better at resolving younger nodes, we compared the relationship between PP and height (referred to as node height and is defined as the average height of the clade across all trees in a BEAST run that support that clade) for intron and exon data sets. This analysis was carried out using the results of BEAST runs from analysis of the “exons – outliers removed" and “introns – outliers removed" data sets. It was not possible to compare directly the two outliers removed data sets (exons vs. introns) as the exon alignment was approximately three times larger (∼7,000 bp vs. ∼2,500 bp), hence it would be expected that the exons should have more resolving power simply because there are more data. To overcome this, positions from the concatenated exon alignment were jack-knifed to form ten random exon data sets of 2,532 bp using custom R scripts. For each of the ten random exon data sets, jmodeltest indicated GTR+I+G was the best model. Each data set was analyzed in BEAST as described previously. We tested whether node height was related to PP using a general linear model with a binomial function and logit link. Differences in posterior probabilities for the same node between exons (average over the ten subsets) and introns were then tested in R using an asymptotic Wilcoxon Mann–Whitney rank sum test (Hollander and Wolfe 1999). To investigate whether the results were specific to the jack-knifed data sets, we repeated the analysis using gene fragments. To obtain size comparable data sets, ten random combinations of four exon gene fragments (min: 2,499 bp, max: 2,564 bp, average: 2,532 bp) were generated for comparison to the intron data set (supplementary table S4, Supplementary Material online). jModeltest indicated GTR+I+G was the best model for these data sets.
Supplementary Material
Supplementary figures S1 and S2 and tables S1–S4 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
This project was supported in part by a Science Foundation Ireland PIYRA 06/YI3/B932 to E.C.T. and an IRCSET-Marie Curie International Mobility Fellowships in Science, Engineering and Technology to S.J.P. E.C.T. and N.M.F. are supported by the European Research Council ERC-2012- StG: 311000. For access to museum specimens, we acknowledge Paula Jenkins (BMNH). S.M.G.’s examination of specimens in the BMNH received support from the SYNTHESYS project http://www.synthesys.info/ (last accessed December 6, 2014), which is financed by European Community Research Infrastructure Action under the FP7 Integrating Activities Program. Fieldwork conducted by V.D.T was supported by Vietnam National Foundation for Science and Technology Development (NAFOSTED) under grant number 106.11-2012.02. The authors thank Nicolas Chenaval, Vanessa Lelant, and Laurent Arthur for providing samples; Meriadeg Ar Gouilh for constructive discussions in the conception of this project; John Finarelli for helpful comments on the manuscript; Paul Webala for a photograph of Triaenops; and both Ken P. Alpin and Harold G. Cogger for taxonomic discussions.
References
- Amit M, Donyo M, Hollander D, Goren A, Kim E, Gelfman S, Lev-Maor G, Burstein D, Schwartz S, Postolsky B. Differential GC content between exons and introns establishes distinct strategies of splice-site recognition. Cell Rep. 2012;1:543–556. doi: 10.1016/j.celrep.2012.03.013. [DOI] [PubMed] [Google Scholar]
- Anthony S, Ojeda-Flores R, Rico-Chavez O, Navarrete-Macias I, Zambrana-Torrelio C, Rostal MK, Epstein JH, Tipps T, Liang E, Sanchez-Leon M. Coronaviruses in bats from Mexico. J Gen Virol. 2013;94:1028–1038. doi: 10.1099/vir.0.049759-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ar Gouilh M, Puechmaille SJ, Gonzalez J-P, Teeling E, Kittayapong P, Manuguerra J-C. SARS-Coronavirus ancestors foot-prints in South-East Asian bat colonies and the refuge theory. Infect Genet Evol. 2011;11:1690–1702. doi: 10.1016/j.meegid.2011.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong KN. Resolving the correct nomenclature of the orange leaf-nosed bat Rhinonicteris aurantia (Gray, 1845)(Hipposideridae) Aust Mammal. 2006;28:125–130. [Google Scholar]
- Balboni A, Battilani M, Prosperi S. The SARS-like coronaviruses: the role of bats and evolutionary relationships with SARS coronavirus. New Microbiol. 2012;35:1–16. [PubMed] [Google Scholar]
- Benda P, Vallo P. Taxonomic revision of the genus Triaenops (Chiroptera: Hipposideridae) with description of a new species from southern Arabia and definitions of a new genus and tribe. Folia Zool. 2009;58:1–45. [Google Scholar]
- Bermingham A, Chand M, Brown C, Aarons E, Tong C, Langrish C, Hoschler K, Brown K, Galiano M, Myers R. Severe respiratory illness caused by a novel coronavirus, in a patient transferred to the United Kingdom from the Middle East, September 2012. Euro Surveill. 2013;17:20290. [PubMed] [Google Scholar]
- Bogdanowicz W, Owen R. In the Minotaurs labyrinth: phylogeny of the bat family Hipposideridae. In: Kunz TH, Racey PA, editors. Bat biology and conservation. Washington (DC): Smithsonian Institution Press; 1998. pp. 27–42. [Google Scholar]
- Bogdanowicz W, Owen RD. Phylogenetic analyses of the bat family Rhinolophidae. Z Zool Syst Evol. 1992;30:142–160. [Google Scholar]
- Calisher CH, Childs JE, Field HE, Holmes KV, Schountz T. Bats: important reservoir hosts of emerging viruses. Clin Microbiol Rev. 2006;19:531–545. doi: 10.1128/CMR.00017-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan JF-W, To KK-W, Tse H, Jin D-Y, Yuen K-Y. Interspecies transmission and emergence of novel viruses: lessons from bats and birds. Trends Microbiol. 2013;21:544–555. doi: 10.1016/j.tim.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chojnowski JL, Kimball RT, Braun EL. Introns outperform exons in analyses of basal avian phylogeny using clathrin heavy chain genes. Gene. 2008;410:89–96. doi: 10.1016/j.gene.2007.11.016. [DOI] [PubMed] [Google Scholar]
- Cohen KM, Finney SC, Gibbard PL, Fan JX. The ICS International Chronostratigraphic Chart. Episodes. 2013;36:199–204. [Google Scholar]
- Csorba G, Ujhelyi P, Thomas N. 2003 Horseshoe bats of the World. Shropshire UK: Alana Books. [Google Scholar]
- Cui J, Han N, Streicker D, Li G, Tang X, Shi Z, Hu Z, Zhao G, Fontanet A, Guan Y. Evolutionary relationships between bat coronaviruses and their hosts. Emerg Infect Dis. 2007;13:1526–1532. doi: 10.3201/eid1310.070448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daszak P. Bats, in black and white. Science. 2010;329:634–635. doi: 10.1126/science.1194089. [DOI] [PubMed] [Google Scholar]
- de Vienne DM, Ollier S, Aguileta G. Phylo-MCOA: a fast and efficient method to detect outlier genes and species in phylogenomics using multiple co-inertia analysis. Mol Biol Evol. 2012;29:1587–1598. doi: 10.1093/molbev/msr317. [DOI] [PubMed] [Google Scholar]
- Ditchfield A. The comparative phylogeography of Neotropical mammals: patterns of intraspecific mitochondrial DNA variation among bats contrasted to nonvolant small mammals. Mol Ecol. 2000;9:1307–1318. doi: 10.1046/j.1365-294x.2000.01013.x. [DOI] [PubMed] [Google Scholar]
- Dobson GE. Catalogue of the Chiroptera in the collection of the British Museum. 1878 London: Trustees of the British Museum. [Google Scholar]
- Drexler JF, Corman VM, Muller MA, Maganga GD, Vallo P, Binger T, Gloza-Rausch F, Rasche A, Yordanov S, Seebens A. Bats host major mammalian paramyxoviruses. Nat Commun. 2012;3:796. doi: 10.1038/ncomms1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drexler JF, Gloza-Rausch F, Glende J, Corman VM, Muth D, Goettsche M, Seebens A, Niedrig M, Pfefferle S, Yordanov S. Genomic characterization of severe acute respiratory syndrome-related coronavirus in European bats and classification of coronaviruses based on partial RNA-dependent RNA polymerase gene sequences. J Virol. 2010;84:11336–11349. doi: 10.1128/JVI.00650-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eick GN, Jacobs DS, Matthee CA. A nuclear DNA phylogenetic perspective on the evolution of echolocation and historical biogeography of extant bats (Chiroptera) Mol Biol Evol. 2005;22:1869–1886. doi: 10.1093/molbev/msi180. [DOI] [PubMed] [Google Scholar]
- Eiting TP, Gunnell GF. Global completeness of the bat fossil record. J Mamm Evol. 2009;16:151–173. [Google Scholar]
- Flower WH, Lydekker R. 1891 An introduction to the study of mammals, living and extinct. London: Adam and Charles Black. [Google Scholar]
- Ge X-Y, Li J-L, Yang X-L, Chmura AA, Zhu G, Epstein JH, Mazet JK, Hu B, Zhang W, Peng C. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature. 2013;8:503–535. doi: 10.1038/nature12711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gradstein FM, Ogg JG. The geologic time scale. In: Hedges SB, Kumar S, editors. The timetree of life. New York: Oxford University Press; 2009. pp. 26–34. [Google Scholar]
- Gray J. In: Description of some new Australian animals. Eyre E, editor. 1845. pp. 405–411. [Google Scholar]
- Gray J. A revision of the genera of Rhinolophidae, or horseshoe bats. Proc Zool Soc Lond. 1866;1866:81–83. [Google Scholar]
- Gu X-M, He S-Y, Ao L. Molecular phylogenetics among three families of bats (Chiroptera: Rhinolophidae, Hipposideridae, and Vespertilionidae) based on partial sequences of the mitochondrial 12S and 16S rRNA genes. Zool Stud. 2008;47:368–378. [Google Scholar]
- Guillen-Servent A, Francis CM, Ricklefs RE. Phylogeography and biogeography of the horseshoe bats (Chiroptera: Rhinolophidae) In: Csorba G, Ujhelyi P, Thomas N, editors. Horseshoe bats of the world. Shropshire (UK): Alana Books; 2003. pp. xii–xxiv. [Google Scholar]
- Hand S, Archer M. A new hipposiderid genus (Microchiroptera) from an early Miocene bat community in Australia. Palaeontology. 2005;48:371–383. [Google Scholar]
- Hand S, Kirsch J. A southern origin for the Hipposideridae (Microchiroptera)? Evidence from the Australian fossil record. In: Kunz TH, Racey PA, editors. Bat biology and conservation. Washington (DC): Smithsonian Institution Press; 1998. pp. 72–90. [Google Scholar]
- Hedges SB, Parker PH, Sibley CG, Kumar S. Continental breakup and the ordinal diversification of birds and mammals. Nature. 1996;381:226–229. doi: 10.1038/381226a0. [DOI] [PubMed] [Google Scholar]
- Hill J. A review of the leaf-nosed bats Rhinonycteris, Cloeotis and Triaenops (Chiroptera: Hipposideridae) Bonn Zool Beitr. 1982;33:165–186. [Google Scholar]
- Hollander M, Wolfe DA. Nonparametric statistical methods. New York: John Wiley & Sons; 1999. [Google Scholar]
- Huang Y, Niu B, Gao Y, Fu L, Li W. CD-HIT Suite: a web server for clustering and comparing biological sequences. Bioinformatics. 2010;26:680–682. doi: 10.1093/bioinformatics/btq003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulva P, Horacek I, Benda P. Molecules, morphometrics and new fossils provide an integrated view of the evolutionary history of Rhinopomatidae (Mammalia: Chiroptera) BMC Evol Biol. 2007;7:165. doi: 10.1186/1471-2148-7-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaac NJ, Turvey ST, Collen B, Waterman C, Baillie JE. Mammals on the EDGE: conservation priorities based on threat and phylogeny. PLoS One. 2007;2:e296. doi: 10.1371/journal.pone.0000296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IUCN. 2014 IUCN red list of threatened species. [cited 2014 May 4]. Available from: http://www.iucnredlist.org/ [Google Scholar]
- Jones G, Teeling EC. The evolution of echolocation in bats. Trends Ecol Evol. 2006;21:149–156. doi: 10.1016/j.tree.2006.01.001. [DOI] [PubMed] [Google Scholar]
- Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingston T, Lara MC, Jones G, Akbar Z, Kunz TH, Schneider CJ. Acoustic divergence in two cryptic Hipposideros species: a role for social selection? Proc R Soc B. 2001;268:1381–1386. doi: 10.1098/rspb.2001.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koopman K. Zoogeography of bats. In: Slaughter B, Walton D, editors. About bats: a chiropteran biology symposium. Dallas (TX): Southern Methodist University Press; 1970. pp. 29–50. [Google Scholar]
- Koopman KF. Order Chiroptera. In: Wilson D, Reeder D, editors. Mammal species of the world, a taxonomic and geographic reference. Washington (DC): Smithsonian Institution Press; 1993. pp. 137–241. [Google Scholar]
- Koopman KF. Chiroptera: Systematics. In: Andreas S-R, editor. Handbook of zoology. Berlin (Germany): Walter de Gruyter; 1994. pp. 1–217. [Google Scholar]
- Lack JB, Roehrs ZP, Stanley CE, Jr, Ruedi M, Van Den Bussche RA. Molecular phylogenetics of Myotis indicate familial-level divergence for the genus Cistugo (Chiroptera) J Mammal. 2010;91:976–992. [Google Scholar]
- Lartillot N, Lepage T, Blanquart S. PhyloBayes 3: a Bayesian software package for phylogenetic reconstruction and molecular dating. Bioinformatics. 2009;25:2286–2288. doi: 10.1093/bioinformatics/btp368. [DOI] [PubMed] [Google Scholar]
- Lau SK, Woo PC, Li KS, Huang Y, Tsoi H-W, Wong BH, Wong SS, Leung S-Y, Chan K-H, Yuen K-Y. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc Natl Acad Sci U S A. 2005;102:14040–14045. doi: 10.1073/pnas.0506735102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legendre S. Hipposideridae (Mammalia: Chiroptera) from the Mediterranean Middle and Late Neogene, and evolution of the genera Hipposideros and Asellia. J Vertebr Paleontol. 1982;2:372–385. [Google Scholar]
- Li G, Liang B, Wang Y, Zhao H, Helgen KM, Lin L, Jones G, Zhang S. Echolocation calls, diet, and phylogenetic relationships of Stoliczka's trident bat, Aselliscus stoliczkanus (Hipposideridae) J Mammal. 2007;88:736–744. [Google Scholar]
- Li W, Shi Z, Yu M, Ren W, Smith C, Epstein JH, Wang H, Crameri G, Hu Z, Zhang H. Bats are natural reservoirs of SARS-like coronaviruses. Science. 2005;310:676–679. doi: 10.1126/science.1118391. [DOI] [PubMed] [Google Scholar]
- Lim BK, Engstrom MD, Bickham JW, Patton JC. Molecular phylogeny of New World sheath-tailed bats (Emballonuridae: Diclidurini) based on loci from the four genetic transmission systems in mammals. Biol J Linn Soc. 2008;93:189–209. [Google Scholar]
- Longdon B, Hadfield JD, Webster CL, Obbard DJ, Jiggins FM. Host phylogeny determines viral persistence and replication in novel hosts. PLoS Pathog. 2011;7:e1002260. doi: 10.1371/journal.ppat.1002260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luis AD, Hayman DT, O'Shea TJ, Cryan PM, Gilbert AT, Pulliam JR, Mills JN, Timonin ME, Willis CK, Cunningham AA. A comparison of bats and rodents as reservoirs of zoonotic viruses: are bats special? Proc R Soc B. 2013;280 doi: 10.1098/rspb.2012.2753. 20122753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahoney J, Walton D. Hipposideridae. In: Bannister JL, Calaby JH, Dawson LJ, Ling JK, Mahoney JA, McKay GM, Richardson BJ, Ride WDL, Walton DW, editors. Zoological catalogue of Australia. Vol. 5. Mammalia: 1988. pp. 124–127. [Google Scholar]
- McElwain JC, Punyasena SW. Mass extinction events and the plant fossil record. Trends Ecol Evol. 2007;22:548–557. doi: 10.1016/j.tree.2007.09.003. [DOI] [PubMed] [Google Scholar]
- McKenna MC, Bell SK. Classification of mammals. New York: Columbia University Press; 1997. [Google Scholar]
- Meredith RW, Janecka JE, Gatesy J, Ryder OA, Fisher CA, Teeling EC, Goodbla A, Eizirik E, Simao TL, Stadler T. Impacts of the Cretaceous Terrestrial Revolution and KPg extinction on mammal diversification. Science. 2011;334:521–524. doi: 10.1126/science.1211028. [DOI] [PubMed] [Google Scholar]
- Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller-Butterworth CM, Murphy WJ, O'Brien SJ, Jacobs DS, Springer MS, Teeling EC. A family matter: conclusive resolution of the taxonomic position of the long-fingered bats, Miniopterus. Mol Biol Evol. 2007;24:1553–1561. doi: 10.1093/molbev/msm076. [DOI] [PubMed] [Google Scholar]
- Monadjem A, Schoeman MC, Reside A, Pio DV, Stoffberg S, Bayliss J, Cotterill F, Curran M, Kopp M, Taylor PJ. A recent inventory of the bats of Mozambique with documentation of seven new species for the country. Acta Chiropterol. 2010;12:371–391. [Google Scholar]
- Murphy WJ, Pringle TH, Crider TA, Springer MS, Miller W. Using genomic data to unravel the root of the placental mammal phylogeny. Genome Res. 2007;17:413–421. doi: 10.1101/gr.5918807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray SW, Campbell P, Kingston T, Zubaid A, Francis CM, Kunz TH. Molecular phylogeny of hipposiderid bats from Southeast Asia and evidence of cryptic diversity. Mol Phylogenet Evol. 2012;62:597–611. doi: 10.1016/j.ympev.2011.10.021. [DOI] [PubMed] [Google Scholar]
- Notredame C, Higgins DG, Heringa J. T-Coffee: a novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000;302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- Nowak RM, Paradiso JL. 1999 Walker's mammals of the world. Baltimore and London: John Hopkins University Press. [Google Scholar]
- Paradis E, Claude J, Strimmer K. APE: analyses of phylogenetics and evolution in R language. Bioinformatics. 2004;20:289–290. doi: 10.1093/bioinformatics/btg412. [DOI] [PubMed] [Google Scholar]
- Parham JF, Donoghue PC, Bell CJ, Calway TD, Head JJ, Holroyd PA, Inoue JG, Irmis RB, Joyce WG, Ksepka DT. Best practices for justifying fossil calibrations. Syst Biol. 2012;61:346–359. doi: 10.1093/sysbio/syr107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker J, Tsagkogeorga G, Cotton JA, Liu Y, Provero P, Stupka E, Rossiter SJ. Genome-wide signatures of convergent evolution in echolocating mammals. Nature. 2013;502:228–231. doi: 10.1038/nature12511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penn O, Privman E, Ashkenazy H, Landan G, Graur D, Pupko T. GUIDANCE: a web server for assessing alignment confidence scores. Nucleic Acids Res. 2010;38:W23–W28. doi: 10.1093/nar/gkq443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfefferle S, Oppong S, Drexler JF, Gloza-Rausch F, Ipsen A, Seebens A, Muller MA, Annan A, Vallo P, Adu-Sarkodie Y. Distant relatives of severe acute respiratory syndrome coronavirus and close relatives of human coronavirus 229E in bats, Ghana. Emerg Infect Dis. 2009;15:1377–1384. doi: 10.3201/eid1509.090224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierson ED. 1986 Molecular systematics of the Microchiroptera: higher taxon relationships and biogeography. PhD dissertation. University of California, Berkeley, USA. [Google Scholar]
- Posada D. jModelTest: phylogenetic model averaging. Mol Biol Evol. 2008;25:1253–1256. doi: 10.1093/molbev/msn083. [DOI] [PubMed] [Google Scholar]
- Puechmaille SJ, Ar Gouilh M, Piyapan P, Yokubol M, Mie KM, Bates PJ, Satasook C, Nwe T, Bu SSH, Mackie IJ, et al. The evolution of sensory divergence in the context of limited gene flow in the bumblebee bat. Nat Commun. 2011;2:573. doi: 10.1038/ncomms1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan P-L, Firth C, Street C, Henriquez JA, Petrosov A, Tashmukhamedova A, Hutchison SK, Egholm M, Osinubi MO, Niezgoda M. Identification of a severe acute respiratory syndrome coronavirus-like virus in a leaf-nosed bat in Nigeria. MBio. 2010;1:e00208–e00210. doi: 10.1128/mBio.00208-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team. 2012 R: a language and environment for statistical computing. Vienna (Austria): R Foundation for Statistical Computing. [cited 2014 Dec 6]. Available from: http://cran.r-project.org. [Google Scholar]
- Rambaut A, Drummond A. 2007 Tracer v1. 4. [cited 2012 Apr 14]. Available from: http://beast.bio.ed.ac.uk/Tracer. [Google Scholar]
- Ravel A, Marivaux L, Qi T, Wang Y-Q, Beard KC. New chiropterans from the middle Eocene of Shanghuang (Jiangsu Province, Coastal China): new insight into the dawn horseshoe bats (Rhinolophidae) in Asia. Zool Scr. 2014;43:1–23. [Google Scholar]
- Ree RH, Smith SA. Maximum likelihood inference of geographic range evolution by dispersal, local extinction, and cladogenesis. Syst Biol. 2008;57:4–14. doi: 10.1080/10635150701883881. [DOI] [PubMed] [Google Scholar]
- Ren W, Li W, Yu M, Hao P, Zhang Y, Zhou P, Zhang S, Zhao G, Zhong Y, Wang S. Full-length genome sequences of two SARS-like coronaviruses in horseshoe bats and genetic variation analysis. J Gen Virol. 2006;87:3355–3359. doi: 10.1099/vir.0.82220-0. [DOI] [PubMed] [Google Scholar]
- Rihtaric D, Hostnik P, Steyer A, Grom J, Toplak I. Identification of SARS-like coronaviruses in horseshoe bats (Rhinolophus hipposideros) in Slovenia. Arch Virol. 2010;155:507–514. doi: 10.1007/s00705-010-0612-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romiguier J, Ranwez V, Delsuc F, Galtier N, Douzery EJ. Less is more in mammalian phylogenomics: AT-rich genes minimize tree conflicts and unravel the root of placental mammals. Mol Biol Evol. 2013;30:2134–2144. doi: 10.1093/molbev/mst116. [DOI] [PubMed] [Google Scholar]
- Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Hoehna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61:539–542. doi: 10.1093/sysbio/sys029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruedi M, Friedli-Weyeneth N, Teeling EC, Puechmaille SJ, Goodman SM. Biogeography of Old World emballonurine bats (Chiroptera: Emballonuridae) inferred with mitochondrial and nuclear DNA. Mol Phylogenet Evol. 2012;64:204–211. doi: 10.1016/j.ympev.2012.03.019. [DOI] [PubMed] [Google Scholar]
- Salicini I, Ibanez C, Juste J. Multilocus phylogeny and species delimitation within the Natterer's bat species complex in the Western Palearctic. Mol Phylogenet Evol. 2011;61:888–898. doi: 10.1016/j.ympev.2011.08.010. [DOI] [PubMed] [Google Scholar]
- Schmidt HA, Strimmer K, Vingron M, von Haeseler A. TREE-PUZZLE: maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics. 2002;18:502–504. doi: 10.1093/bioinformatics/18.3.502. [DOI] [PubMed] [Google Scholar]
- Schwarz MP, Fuller S, Tierney SM, Cooper SJ. Molecular phylogenetics of the exoneurine allodapine bees reveal an ancient and puzzling dispersal from Africa to Australia. Syst Biol. 2006;55:31–45. doi: 10.1080/10635150500431148. [DOI] [PubMed] [Google Scholar]
- Seim I, Fang X, Xiong Z, Lobanov AV, Huang Z, Ma S, Feng Y, Turanov AA, Zhu Y, Lenz TL. Genome analysis reveals insights into physiology and longevity of the Brandts bat Myotis brandtii. Nat Commun. 2013;4:2212. doi: 10.1038/ncomms3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimodaira H, Hasegawa M. CONSEL: for assessing the confidence of phylogenetic tree selection. Bioinformatics. 2001;17:1246–1247. doi: 10.1093/bioinformatics/17.12.1246. [DOI] [PubMed] [Google Scholar]
- Sigé B. Les chiropteres du Miocene inferieur de Bouzigues. I. Etude systematique. Paleovertebrata. 1968;1:65–133. [Google Scholar]
- Sigé B. Nouveaux chiropteres de l'Oligocene moyen des phosphorites du Quercy, France. C R Acad Sci. 1991;310:1131–1137. [Google Scholar]
- Sigé B, Hand S, Archer M. An Australian Miocene Brachipposideros (Mammalia, Chiroptera) related to Miocene representatives from France. Palaeovertebrata. 1982;12:149–171. [Google Scholar]
- Silvestro D, Michalak I. raxmlGUI: a graphical front-end for RAxML. Org Divers Evol. 2012;12:335–337. [Google Scholar]
- Simmons N. A reappraisal of interfamilial relationships of bats. In: Kunz TH, Racey PA, editors. Bat biology and conservation. Washington (DC): Smithsonian Institution Press; 1998. pp. 3–26. [Google Scholar]
- Simmons NB. Order chiroptera. In: Wilson DE, Reeder DM, editors. 2005. Mammal species of the world: a taxonomic and geographic reference. Washington (DC): Smithsonian Institution Press. p. 312-529. [Google Scholar]
- Simmons NB, Geisler JH. 1998 Phylogenetic relationships of Icaronycteris, Archaeonycteris, Hassianycteris, and Palaeochiropteryx to extant bat lineages, with comments on the evolution of echolocation and foraging strategies in Microchiroptera. Bull Am Mus Nat Hist. 235:1–182. [Google Scholar]
- Smit AF, Hubley R, Green P. 1996 RepeatMasker Open-3.0. [cited 2012 Apr 14]. Available from: http://www.repeatmasker.org. [Google Scholar]
- Soisook P, Bumrungsri S, Satasook C, Thong VD, Bu SSH, Harrison DL, Bates PJ. A taxonomic review of Rhinolophus stheno and R. malayanus (Chiroptera: Rhinolophidae) from continental Southeast Asia: an evaluation of echolocation call frequency in discriminating between cryptic species. Acta Chiropterol. 2008;10:221–242. [Google Scholar]
- Springer MS, Meredith RW, Janecka JE, Murphy WJ. The historical biogeography of Mammalia. Philos Trans R Soc B. 2011;366:2478–2502. doi: 10.1098/rstb.2011.0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer MS, Teeling EC, Madsen O, Stanhope MJ, de Jong WW. Integrated fossil and molecular data reconstruct bat echolocation. Proc Natl Acad Sci U S A. 2001;98:6241–6246. doi: 10.1073/pnas.111551998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- Stoffberg S, Jacobs DS, Mackie IJ, Matthee CA. Molecular phylogenetics and historical biogeography of Rhinolophus bats. Mol Phylogenet Evol. 2010;54:1–9. doi: 10.1016/j.ympev.2009.09.021. [DOI] [PubMed] [Google Scholar]
- Streicker DG, Turmelle AS, Vonhof MJ, Kuzmin IV, McCracken GF, Rupprecht CE. Host phylogeny constrains cross-species emergence and establishment of rabies virus in bats. Science. 2010;329:676–679. doi: 10.1126/science.1188836. [DOI] [PubMed] [Google Scholar]
- Strimmer K, Rambaut A. Inferring confidence sets of possibly misspecified gene trees. Proc R Soc B. 2002;269:137–142. doi: 10.1098/rspb.2001.1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Zhang Z. Palynological evidence for the mid-Miocene climatic optimum recorded in Cenozoic sediments of the Tian Shan Range, northwestern China. Global Planet Change. 2008;64:53–68. [Google Scholar]
- Sun K-P, Feng J, Jiang T-L, Ma J, Zhang Z-Z, Jin L-R. A new cryptic species of Rhinolophus macrotis (Chiroptera: Rhinolophidae) from Jiangxi Province, China. Acta Chiropterol. 2008;10:1–10. [Google Scholar]
- Tate GHH. Results of the Archbold Expeditions No. 35. A review of the genus Hipposideros with special reference to Indo-Australian species. Bull Am Mus Nat Hist. 1941b;73:353–393. [Google Scholar]
- Tate GHH. Results of the Archbold Expeditions No. 36. Remarks on some Old World leaf-nosed Bats. Am Mus Novit. 1941a;1140:1–11. [Google Scholar]
- Teeling E. Bats (Chiroptera) In: Hedges B, Kumar S, editors. The timetree of life. New York: Oxford University Press; 2009. pp. 499–503. [Google Scholar]
- Teeling EC, Madsen O, Murphy WJ, Springer MS, O'Brien SJ. Nuclear gene sequences confirm an ancient link between New Zealand's short-tailed bat and South American noctilionoid bats. Mol Phylogenet Evol. 2003;28:308–319. doi: 10.1016/s1055-7903(03)00117-9. [DOI] [PubMed] [Google Scholar]
- Teeling EC, Madsen O, Van Den Bussche RA, de Jong WW, Stanhope MJ, Springer MS. Microbat paraphyly and the convergent evolution of a key innovation in Old World rhinolophoid microbats. Proc Natl Acad Sci U S A. 2002;99:1431–1436. doi: 10.1073/pnas.022477199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teeling EC, Springer MS, Madsen O, Bates P, O'Brien SJ, Murphy WJ. A molecular phylogeny for bats illuminates biogeography and the fossil record. Science. 2005;307:580–584. doi: 10.1126/science.1105113. [DOI] [PubMed] [Google Scholar]
- Thabah A, Rossiter SJ, Kingston T, Zhang S, Parsons S, Mya KM, Akbar Z, Jones G. Genetic divergence and echolocation call frequency in cryptic species of Hipposideros larvatus s.l. (Chiroptera: Hipposideridae) from the Indo-Malayan region. Biol J Linn Soc. 2006;88:119–130. [Google Scholar]
- Thong VD, Dietz C, Denzinger A, Bates PJ, Puechmaille SJ, Callou C, Schnitzler H-U. Resolving a mammal mystery: the identity of Paracoelops megalotis (Chiroptera: Hipposideridae) Zootaxa. 2012;3505:75–85. [Google Scholar]
- Thong VD, Puechmaille SJ, Denzinger A, Bates PJ, Dietz C, Csorba G, Soisook P, Teeling EC, Matsumura S, Furey NM. Systematics of the Hipposideros turpis complex and a description of a new subspecies from Vietnam. Mammal Rev. 2012;42:166–192. [Google Scholar]
- Thong VD, Puechmaille SJ, Denzinger A, Dietz C, Csorba G, Bates PJ, Teeling EC, Schnitzler H-U. A new species of Hipposideros (Chiroptera: Hipposideridae) from Vietnam. J Mammal. 2012;93:1–11. [Google Scholar]
- Vieites DR, Min M-S, Wake DB. Rapid diversification and dispersal during periods of global warming by plethodontid salamanders. Proc Natl Acad Sci U S A. 2007;104:19903–19907. doi: 10.1073/pnas.0705056104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Liang B, Feng J, Sheng L, Zhang S. Molecular phylogenetic of Hipposiderids (Chiroptera: Hipposideridae) and Rhinolophids (Chiroptera: Rhinolophidae) in China based on mitochondrial cytochrome b sequences. Folia Zool. 2003;52:259–268. [Google Scholar]
- Woo P, Lau S, Li K, Poon R, Wong B, Tsoi H, Yip B, Huang Y, Chan K, Yuen K. Molecular diversity of coronaviruses in bats. Virology. 2006;351:180–187. doi: 10.1016/j.virol.2006.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- Yuan J, Hon C, Li Y, Wang D, Xu G, Zhang H, Zhou P, Poon L, Lam T, Leung F. Intraspecies diversity of SARS-like coronaviruses in Rhinolophus sinicus and its implications for the origin of SARS coronaviruses in humans. J Gen Virol. 2010;91:1058–1062. doi: 10.1099/vir.0.016378-0. [DOI] [PubMed] [Google Scholar]
- Zhang G, Cowled C, Shi Z, Huang Z, Bishop-Lilly K, Fang X, Wynne J, Xiong Z, Baker M, Zhao W. Comparative analysis of bat genomes provides insight into the evolution of flight and immunity. Science. 2013;339:456–460. doi: 10.1126/science.1230835. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.