

Abstract
Bacterial chemoreceptors associate with the histidine kinase CheA and coupling protein CheW to form extended membrane arrays that receive and transduce environmental signals. A receptor trimers-of-dimers resides at each vertex of the hexagonal protein lattice. CheA is fully activated and regulated when it is integrated into the receptor assembly. To mimic these states in solution, we have engineered chemoreceptor cytoplasmic kinase-control modules (KCMs) based on the Escherichia coli aspartate receptor Tar that are covalently fused and trimerized by a foldon domain (TarFO). Small-angle X-ray scattering, multi-angle light scattering, and pulsed-dipolar electron spin resonance spectroscopy of spin-labeled proteins indicate that the TarFO modules assemble into homogeneous trimers wherein the protein interaction regions closely associate at the end opposite to the foldon domains. The TarFO variants greatly increase the saturation levels of phosphorylated CheA (CheA-P), indicating that the association with a trimer of receptor dimers changes the fraction of active kinase. However, the rate constants for CheA-P formation with the Tar variants are low compared to those for autophosphorylation by free CheA, and net phosphotransfer from CheA to CheY does not increase commensurately with CheA autophosphorylation. Thus, the Tar variants facilitate slow conversion to an active form of CheA that then undergoes stable autophosphorylation and is capable of subsequent phosphotransfer to CheY. Free CheA is largely incapable of phosphorylation but contains a small active fraction. Addition of TarFO to CheA promotes a planar conformation of the regulatory domains consistent with array models for the assembly state of the ternary complex and different from that observed with a single inhibitory receptor. Introduction of TarFO into E. coli cells activates endogenous CheA to produce increased clockwise flagellar rotation, with the effects increasing in the presence of the chemotaxis methylation system (CheB/CheR). Overall, the TarFO modules demonstrate that trimerized signaling tips self-associate, bind CheA and CheW, and facilitate conversion of CheA to an active conformation.
The molecular signal transduction pathway intrinsic to bacterial chemotaxis is known for high sensitivity, dynamic range, and signal gain.1−4 The chemotaxis detection system is comprised of ordered membrane arrays of chemoreceptors [methyl-accepting chemotaxis proteins (MCPs)], the histidine kinase CheA, and the coupling protein CheW (Figure 1A).5−8 Binding of ligand to the periplasmic domains of the MCPs regulates the kinase activity of CheA, which interacts along with CheW at the receptor cytoplasmic tips. CheA activity in turn determines the level of the phosphorylated response regulator CheY that binds directly to the flagellar rotor. In Escherichia coli, repellent binding (or attractant release) to (or from) MCPs produces a kinase-on state and clockwise (CW) flagellar rotation, whereas attractant binding produces a kinase-off state and counterclockwise (CCW) flagellar rotation.
Figure 1.

(A) Cartoon depiction of the chemoreceptor array based on models from electron microscopy and crystallography.6,23 (B) Cartoon depiction of the MCP dimer with domains labeled, and the kinase control module (KCM) is colored gray. (C) Target complexes of CheA, CheW, and Tar variants, and how they may mimic the minimal activating unit composed of six MCP KCMs with a 1:2 dimeric CheA:CheW ratio. Trimerized MCP dimers are produced with and without the adaptation region. (D) Trimeric foldon motif from bacteriophage T4 fibrin that forms a β-propeller from three β-hairpins. Dotted lines represent backbone hydrogen bonds.
MCPs in general have six structural modules: the periplasmic domain for ligand binding, the transmembrane domain, a signal conversion module called the HAMP domain (named after proteins in which it is found, histidine kinases, adenylyl cyclases, methyl-accepting chemotaxis proteins, and phosphatases), the adaptation region, the glycine-rich flexible bundle, and the protein interaction region (PIR) at the cytoplasmic tip (Figure 1B).1 The last three regions comprise the kinase control module (KCM). Within the adaptation region, conserved glutamate residues undergo reversible methylation through the action of the methyltransferase CheR and the methylesterase CheB. For E. coli MCPs, methylation counters attractant binding by reactivating CheA and reducing sensitivity to ligands.9 Substitution of the glutamate residues with glutamine mimics MCP methylation.10−14 CheB and CheR are part of the feedback mechanism to reset the receptor response and allow sensitive detection over a wide concentration gradient.
MCPs form extended ordered arrays composed of mixtures of thousands of receptors at the poles of cells.6,7,15−22 The arrays have a hexagonal honeycomb architecture with three MCP dimers (trimers-of-dimers) residing at the vertices of the hexagons (Figure 1A). The MCP PIR binds to CheA and CheW, provides trimerization contacts among MCP dimers, and has the highest degree of sequence conservation among receptors across bacterial species.7,23,24 The dimeric CheA kinase has five domains per subunit. P1 contains the phosphorylatable His residue. P2 docks CheY and CheB for phosphotransfer from P1. P3 dimerizes the kinase. P4 binds ATP and transfers the γ-phosphate to P1. P5 binds to CheW and the receptor tip.25,26 In the membrane arrays, P5 and its homologue, CheW, form rings with each alternating module binding one receptor tip from an adjacent trimer.6,18,23
The trimer-of-dimers arrangement of MCPs was first observed in the crystal structure of the serine-sensing receptor Tsr in E. coli.27 It has subsequently become clear that a highly ordered molecular arrangement based on trimeric receptor dimers underlies clustering.1,28,29 Substitution of the trimerization contact residues produces defective chemotaxis,30 and in vivo cross-linking experiments by a trivalent reagent support the MCP trimer-of-dimers assembly.30,31 Electron microscopy (EM) and electron cryo-tomography (ECT) revealed the trimeric architecture of receptor dimers as a conserved feature of native receptors in cells (Figure 1A).6,7,16−18 ECT studies show that the trimers-of-dimers are configured in an edge-on-edge arrangement to produce a honeycomb lattice of receptors, CheA, and CheW.6,18
Trimerization of MCPs plays a pivotal role in CheA kinase activation. MCPs associated into nanodiscs activate CheA only when there are at least three parallel receptor dimers in the same disc.32 Furthermore, the minimal stoichiometry for kinase activation involves two such nanodiscs (i.e., two trimers of dimers) per CheA dimer.33,34 This assembly state (Figure 1C) is consistent with that of the extended lattice model proposed from a combination of crystallographic and ECT data.18 In the honeycomb arrangement of the lattice model, a receptor dimer contacts either CheW or CheA through the same interface (Figure 1A).23,35
Certain MCPs do not contain transmembrane regions, yet they also appear to assemble into soluble honeycomb lattices.36,37 Nonetheless, recombinant MCP cytoplasmic KCMs generally do not produce trimers in solution. In some cases, individual MCP KCMs inhibit CheA activity, whereas in other cases, they are activating.36,38 However, if the KCMs are templated to lipid vesicles39−41 or treated with osmolytes,42 trimers capable of activating CheA form. Addition of leucine zipper domains43−46 and surrogate HAMP modules fused N-terminal of the KCMs47,48 have also been proven to be effective at activating CheA in vitro and in vivo. However, in these and other cases, the oligomeric and activity states of the receptor–kinase complexes are heterogeneous,37,49 and it is thus difficult to attribute particular activities to specific kinase conformations.
Herein, we have engineered chemoreceptor mimetics that preform the trimer-of-dimers module in solution with the goal of producing soluble active ternary complexes with CheA and CheW. Receptor KCMs were fused to known trimerization motifs, such as Leu zipper motifs that form trimers and the foldon from bacteriophage T4 fibritin.50−52 Despite its small size, the foldon forms a stable β-propeller trimer with each of three intertwined β-hairpins supplied from each subunit (Figure 1D).50−52 Foldon subunits trimerize rapidly and have been previously used to oligomerize fused proteins.50 Single-chain variants (fused dimers) of chemoreceptor KCMs with C-terminal foldon tags assemble into soluble trimers (TarFO) with interacting tips that are capable of increasing the level of CheA autophosphorylation to a degree similar to that observed in membrane arrays. Furthermore, pulsed-dipolar ESR spectroscopy (PDS) measurements of spin-labeled proteins reveal that CheA bound to a trimer-of-dimers mimetic assumes a conformation that matches expectations from the modeled cellular receptor arrays.
Experimental Procedures
Construction and Cloning of the Trimer-of-Dimers Mimetics
The KCM of the aspartate receptor Tar plus a sequence that encodes a seven-amino acid peptide, GASGGTG, at the 3′ end was cloned into pET28a between 5′ NdeI and 3′ BamHI restriction sites. A second Tar fragment was then cloned in frame into the same vector between 5′ BamHI and 3′ HindIII restriction sites. The NdeI-TarC-BamHI-TarC′-stop-HindIII construct encodes a Tar covalent single-chain “dimer” (TarSC). The foldon trimerization motif was introduced through polymerase chain reaction (PCR) cloning from the GP67 vector template (a gift from G. Whittaker, Cornell University) with introduced restriction sites for fusion onto the C-terminus of TarSC (5′ HindIII and 3′ XhoI sites), or for fusion onto the N-terminus (5′ NheI and 3′ NdeI sites). Constructs of the general formulation NcoI-foldon-NdeI-Tarc-BamHI-Tarc′-HindIII or NdeI-Tarc-BamHI-Tarc′-HindIII-foldon-stop-XhoI produced variants of the Tar foldon (TarFO) with different Tar subunit lengths and foldon linkages (Figure 1 of the Supporting Information). The TarFO, short construct was produced as NdeI-Tar353–424-BamHI-SpeI-Tar353–424-ScaI-foldon-stop-NotI-XhoI. For N-terminally fused foldons, an N-terminal His6 tag and thrombin cleavage site were retained on the foldon fragment for purification; for C-terminally fused foldons, the His6 tag and thrombin site were removed prior to Tar fusion by PCR cloning the foldon unit alone into vector pET28a and then ligating the His6 tag free foldon into the Tar-containing vector (Figure 1 of the Supporting Information). TarFO was grown in Luria-Bertani broth with kanamycin (50 mg/L). Cells were grown until the optical density reached an A600 of 0.6, and the temperature was then lowered to 17 °C before cells were induced with IPTG (35 mg/L) and left overnight before being pelleted. During purification, all buffers included 10% glycerol to improve stability. Prior to sonication, 0.1 mM PMSF was added to lysis buffer [50 mM Tris (pH 7.5), 150 mM NaCl, 5 mM imidazole, and 10% glycerol] to limit proteolysis. After sonication, the lysate was centrifuged and applied to Ni2+-NTA affinity resin. Nonspecific binding was removed by washing with 50 mM Tris (pH 7.5), 150 mM NaCl, 20 mM imidazole, and 10% glycerol, and the TarFO was then eluted with 50 mM Tris (pH 7.5), 150 mM NaCl, 200 mM imidazole, and 10% glycerol. Eluted protein was incubated overnight with thrombin to cleave the His6 tag. The protein was then further purified on a Sephadex 200 SEC column with buffer consisting of 50 mM Tris (pH 7.5), 150 mM NaCl, and 10% glycerol before further concentration with a 50 kDa molecular weight concentrator (Amicon).
Cloning, Mutagenesis, and Spin-Labeling of Proteins
Two Glu residues in the adaptation region of Tar (E302 and E491) were mutated to Gln via QuikChange mutagenesis (Agilent Technologies). For site-specific spin-labeling, Glu389 on TarFO was mutated to Cys (Quikchange) and spin-labeled as previously described.53 TarFO is otherwise Cys free. Within the P3P4P5 portion of Thermotoga maritima CheA (Δ289), Gln545 was changed to Cys (Quikchange) and spin-labeled as previously described.53 CheA P3P4P5 is otherwise Cys free.
Multi-angle Light Scattering (MALS)
A 5.0 mg/mL solution of bovine serum albumin (BSA, Sigma) was injected onto a Phenomenex Bio Sep-SEC-s 300 column that had been equilibrated in GF buffer containing 50 mM Tris (pH 7.5) and 150 mM NaCl to normalize the light scattering detectors and act as a calibration control for both peak alignment and molecular weight determinations. Purified protein samples (1–10 mg/mL) were then injected onto the same column. For ternary complexes, proteins were mixed with 1:1:6 CheA:CheW:TarFO (or TarSC) subunit ratios. The SEC is coupled to a static 18-angle light scattering detector (DAWN HELEOS-II), a refractive index detector (Optilab T-rEX, Wyatt Technology), and a dynamic light-scattering device (WyattQELS). Data were collected every second for 30 min at a flow rate of 1 mL/min and 25 °C. ASTRA V software was used to extract the molar weight distribution, root-mean-square (rms) radius, radius of hydration, and polydispersity of each resolved peak, which were taken as averages across the elution peaks. Concentrations were determined by the refractive index indicator and further verified by the absorbance at 280 nm with molar extinction coefficients (ε280) calculated from the protein sequence.
Small-Angle X-ray Scattering (SAXS)
Protein buffer was exchanged with 50 mM HEPES (pH 7.5), 150 mM NaCl, 2 mM DTT, and 5% glycerol. Three concentrations of 4, 3, and 2 μM were used for TarFO 4Q and TarSC 4Q. For the TarFO short, three concentrations were used, 31, 15, and 7 μM. The samples were centrifuged at 13000 rpm for 20 min and kept in a 4 °C chilling tray prior to data collection. Data were collected at G1 of CHESS with a Pilatus 100K detector.54 Samples were exposed to X-rays for 2 s per frame for 10 frames. During X-ray exposures, 30 μL of the sample is continuously oscillated through the illuminated volume (0.125 μL) at a rate of 2–4 μL s–1, thereby reducing the absorbed dose by 2 orders of magnitude.55 RAW56 and Primus57 were used to generate Guinier and Kratky plots. Molecular weight prediction was based upon a standard of 3 mg/mL glucose isomerase (173 kDa). Envelope reconstructions were calculated using ATSAS58−60 programs. A total 10 independent models were generated and averaged using Damaver and then combined into one envelope.
Protein Interactions Assessed by Pull-Down Assays
Binding affinities of untagged TarFO 4Q, Tm14, and T. maritima CheW for His6-tagged T. maritima CheA Δ289 (P3P4P5) were tested with pull-down assays. Proteins were incubated together with 30 μL of Ni2+-NTA affinity resin [equilibrated with 50 mM HEPES (pH 7.5), 150 mM NaCl, 5 mM imidazole, and 10% glycerol] and mixed on a rocker for 1 h at room temperature. Samples were microcentrifuged and resuspended several times in wash buffer [50 mM HEPES (pH 7.5), 150 mM NaCl, 50 mM imidazole, and 10% glycerol] to remove nonspecifically bound proteins. SDS Coomassie loading dye (30 μL) was added to the sample, which was then heated at 90 °C for 5 min prior to being run on a denaturing Nu PAGE gel (Invitrogen). Gel bands were quantified by densitometry and analyzed with ImageJ. Nonspecific binding of target proteins to Ni2+-NTA resin was determined and subtracted from values obtained with the His6-tagged CheA bait, and peak intensity was weighted by MW to obtain relative molar amounts (designated as “extent pulled down”).
CheA Autophosphorylation Assays
CheA monomer (1–2.5 μM), CheW (1–2.5 μM), and TarFO 4Q or short (1–2.5 μM) or TarSC 4Q (3–6 μM) were mixed and left to form a complex for 1 h at room temperature in TKEDM buffer [50 mM KCl, 10 mM MgCl2, 0.5 mM DTT, 0.5 mM EDTA, and 5 mM Tris (pH 7.5)]. Stoichiometries for CheW and MCP variants were optimized to a 1:1:3 CheA monomer:CheW:MCP dimer ratio. After incubation, 2 μL of 2.3 mM cold ATP and 3–8 μL of a [γ-32P]ATP (3000 Ci/mmol, 10 mCi/mL, PerkinElmer) solution were added to the sample to produce a total volume of 25 μL. After [γ-32P]ATP exposure times from 10 s to 12 min (up to 40 min for TarFO short), the sample was quenched with 25 μL of 3× SDS with 50 mM EDTA (pH 8.0) and then subjected to gel electrophoresis on a 4–20% gradient Tris-glycine gel. The gel was stained with Coomassie blue (10 min), destained with water, and then dried with a GelAir dryer (Bio-Rad). The dry gel was placed in an imaging cassette for at least 24 h and then imaged with a Storm phosphorimager (GE Healthcare). The resulting band intensities were analyzed with ImageJ. The kinetic data were fit to the first-order expression Pt = A0(1 – e–k1t), where Pt represents CheA-P at time t, A0 represents the saturation level of CheA-P, and k1 is the first-order rate constant.
ADP/ATP Chase
CheA monomer (1 μM), CheW (1 μM), and TarFO 4Q or short (1 μM) or TarSC 4Q (3 μM) samples were prepared and exposed to [γ-32P]ATP as described previously. After exposure to 2 μL of a [γ-32P]ATP solution for 6 min, 2 μL of 2 mM nucleotide (ADP or ATP) was added and subsequently quenched after 3–30 min.
Transfer of Phosphate to CheY
CheA (subunit concentration of 1 μM), CheW (1 μM), CheY (25 μM), and TarFO 4Q or short (1 μM) or TarSC 4Q (3 μM) samples were prepared and exposed to [γ-32P]ATP as described above for 30 s or 30 min. CheY (25 μM) was then added and the reaction was quenched after various times.
Membrane Array Assays
The membrane array samples were prepared and sent by K. Piasta and J. Falke (University of Colorado, Boulder, CO).35,61 They contained CheA (5 μM), CheW (10 μM), and Tsr receptors (6.7 μM). The samples were spun down at 13000 rpm for 7 min, and the supernatant was removed and resuspended in 15 μL of 1× TKEDM buffer. Each sample contained 5 μL of the washed arrays, and 5 μL of the CheY solution (80 μM stock). The assays containing CheA (2.5 μM), CheW (5 μM), CheY (40 μM), and Tsr receptors (3.4 μM) were supplemented with 1 μL of a hot [γ-32P]ATP solution as described above and then quenched with SDS buffer after 30 s. Amounts of CheA, CheW, and CheY in the membrane assays were similar to those in assays with soluble Tar variants, as confirmed separately on sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) gels.
Pulsed-Dipolar ESR Spectroscopy
Cysteine variants were expressed in E. coli as described above. Cell lysates were applied to a Ni2+-NTA column to bind the His6-tagged target proteins, and then 5–10 mM MTSSL nitroxide spin-label [1-oxyl-2,2,5,5-tetramethylpyrroline-3-(methyl)methanethiosulfonate (Toronto research, Toronto, ON)] was added to the column and the mixture incubated at room temperature for 4 h and then overnight at 4 °C. Reaction with the free Cys thiol yields the nitroxide side chain commonly known as R1. Samples were eluted after a subsequent overnight incubation with thrombin to remove the His6 tag. Proteins were further purified on a size-exclusion column (Superdex 200, Pharmacia Biotech) and concentrated in GFB [50 mM Tris (pH 7.5) and 150 mM NaCl]. Samples contained 50–100 μL of 38–50 μM spin-labeled protein (T. maritima P3P4P5 Q545C-R1 or E. coli TarFO 4Q E389C-R1). For PDS measurements, four pulse double electron–electron resonance (DEER) experiments were conducted at 60 K on a 17.3 GHz FT EPR spectrometer, which is modified to perform PDS experiments.62−66 The baseline used for data processing was approximated by a linear polynomial. Distance distributions of spin separations within the sample were calculated by the Tikhonov method67 and refined by the maximum entropy regularization method (MEM).68
Quantification of Flagellar Rotation Patterns
TarFO and TarSC constructs were recloned into vector pKG116 with NdeI and HindIII sites to vector pKG116, which contained a salicylate inducible promoter. Plasmids were transferred into host strain (UU2612) +CheB/R or (UU2610) −CheB/R (generously provided by J. S. Parkinson). Direct measurements of flagellar rotation patterns were taken using a tethered cell assay similar to that published by Parkinson and Slocum.69 Specifically, E. coli cells harboring TarFO, TarSC, full-length/KCM Tar-containing plasmids, or pKG116 were grown in tryptone broth, induced for 1 h with 2 μM sodium salicylate, washed in KEP buffer [10 mM potassium phosphate and 0.1 mM EDTA (pH 7.0)], and resuspended in tethering buffer (KEP buffer with 75 mM NaCl). Flagella were sheared in a Waring commercial blender for 15 s. Bacteria with sheared flagella were added to the anti-flagellin antibody (abcam, 1:500 final dilution), placed onto a microscope slide, and then visualized by dark-field microscopy. The rotation patterns of 50–100 cells were observed for 10 s each and recorded as exclusively CW or CCW, predominantly CW or CCW, or frequently reversing.
Methylation Status of Recombinant Tar Variants
E. coli (UU2612) +CheB/R expressing TarFO, TarSC, full-length Tar, Tar-KCM, and (UU2610) −CheB/R expressing full-length Tar were grown in Luria-Bertani broth and induced for 3.5 h with 2 μM sodium salicylate. Bacterial cells were washed in chemotaxis buffer [100 μM potassium-EDTA, 10 mM potassium phosphate (pH 7.0), 10 mM sodium lactate, 1 mM magnesium sulfate, and 1 mM ammonium sulfate] before protein synthesis was stopped with 500 μg/mL spectinomycin. Methylation was conducted as described by Kort et al.70 with modifications. Specifically, methylation was initiated by adding 10 μCi/mL l-[methyl-3H]methionine (82 Ci mmol–1, PerkinElmer), and reactions were stopped by adding formaldehyde. Tar/UU2612 methylation was also tested by adding 5 mM aspartate after l-[methyl-3H]methionine. After SDS–PAGE, gels were soaked for 30 min in Fluoro-hance (RPI Corp.), then dried, and exposed to an autoradiography film at −80 °C for at least 3 days. The steady-state expression level of each methylated protein was determined by Western blot using a 1:100000 anti-Tsr antibody dilution (generously provided by J. S. Parkinson).
Results
To develop a soluble MCP trimer-of-dimers mimetic, first the cytoplasmic kinase control module (KCM) of the E. coli aspartate receptor Tar was engineered into a single-chain module (TarSC) that included either (1) the entire KCM with the adaptation and protein interaction region (PIR), but not the unstructured C-terminal tail, or (2) only the PIR. In each case, the C-terminus of the first repeated module was joined to the N-terminus of another through a GASGGTG peptide linker (Table 1 and Figure 2). A foldon trimerization motif was then fused onto either the N-terminus or the C-terminus of TarSC. The length of the linker between the TarSC dimer and foldon motif was varied to optimize the stability of the variants and their trimerization. Two additional Gln substitutions were introduced into the adaptation region of each “subunit” to produce the “QQQQ” state, which mimics complete methylation (and hence maximal kinase activation). As an alternative strategy, a coiled-coil-based trimerization motif (LLVWEGDKRVYA)71 was also employed in substitution of the foldon, but the recombinant expressions of the coiled-coil variants were far inferior to those of the foldon fusions and were thus not pursued.
Table 1. TarFO Constructs Generated.
| methylation state | N- or C-foldon | first Tar KCM (residue numbers) | second Tar KCM (residue numbers) | |
|---|---|---|---|---|
| 1 | QEQE | n/a | 257–521 | 257–521 |
| 2 | QEQE | N | 257–521 | 257–521 |
| 3 | QEQE | N | 263–515 | 263–553 |
| 4 | QEQE | N | 263–515 | 263–515 |
| 5 | QEQE | N | GGGGG-263–515 | 263–515 |
| 6 | QEQE | N | 263–515 | 263–515-NWETF |
| 7 | QEQE | C | GGGGG-263–515 | 263–515 |
| 8 | QEQE | C | 263–515 | 263–515 |
| 9 | QEQE | C | 263–515 | 263–521 |
| 10 | QEQE | C | 257–521 | 263–515 |
| 11 | QEQE | C | 257–521 | 257–521 |
| 12 | QEQE | C | 257–521 | 257–515 |
| 13 | QQQQ | n/a | 257–521 | 257–521 |
| 14 | QQQQ | C | 257–521 | 257–515 |
| 15 | QQQQ | C | 257–521 | 257–521 |
| 16 | QQQQ | C | 257–521 | 257–528 |
| 17 | n/a | C | 353–424-GSAGTSG | 353–424-GASGSTG |
Figure 2.

Construction of recombinant Tar variants generated.
Oligomerization States of the TarFO Mimetics
Different variants of the trimer-of-dimer MCPs exhibited a range of expression levels and varied proteolytic sensitivities. Nevertheless, the expressed proteins generally assumed a trimeric association state (characterized by SEC and MALS) as well as a small percentage of higher-molecular weight (MW) aggregates (Figure 3). The most stable protein, TarFO 4Q 515 (number 14 in Table 1 and hereafter termed TarFO 4Q), produced the highest expression level and was hence subjected to further analysis. Subsequently, a shorter construct employing only the protein interaction region (PIR) of the receptor was also generated. The resulting TarFO short (number 17 in Table 1) was also well-behaved and investigated in more detail.
Figure 3.

Tar variant oligomerization by multi-angle light scattering (MALS). MALS–size-exclusion chromatography (SEC) data of the TarSC and TarFO variants. On the basis of molecular weight predictions, TarSC is monomeric and both TarFO variants produce trimeric species when injected at a concentration of 5 mg/mL. The TarFO short also shows a peak for 2 times the trimer MW. Note that the Tar variants likely have very different hydrodynamic properties, which will affect their elution positions on SEC. The molecular weights listed on each plot are based on averages across the elution peak.
MALS provides measurements of molecular weight (MW) by recording static light scattering from soluble samples at multiple angles. Coupling MALS measurements to SEC fractionation allows separate MW assessments for components of different size and hydrodynamic properties. Molecular weights were taken from averages across the peak elution. These MW estimates depend on accurate concentration measurements, which can be obtained from refractive index changes or molar absorbance, the latter with knowledge of the protein extinction coefficients. The MW estimates are also volume averages and susceptible to conformational variability. Particles with large dimensions (>15 nm) introduce angular dependencies to the light scattering that can affect the MW estimates if shapes are variable. MALS–SEC analysis indicates that the chromatographed mass of TarFO 4Q has an average MW of 168 kDa, roughly the expected molecular weight of the trimeric TarFO 4Q [3 × 59 = 177 kDa (Figure 3)]. The major peak for TarSC reflected a single-chain “dimer”, with a molecular weight of 72 kDa, which is somewhat elevated from the expected MW of 60 kDa. Although the TarFO short was more prone to aggregation, the major elution peak had the expected MW for a trimeric state at 63 kDa (Figure 3). A secondary peak at roughly twice this molecular weight was also observed for the TarFO short, which represents some dimerization of the trimeric species.
Shape of TarFO
SAXS data report on the overall shapes and conformational properties of biological macromolecules. For polymers, the shape of the Kratky plot {I(q)q2 vs q, where q represents the scattering vector [4πsin(θ)/λ]} will differentiate among globular structures [for which I(q)q2 attenuates at high q], rodlike shapes [for which I(q)q2 increases linearly with q], and Gaussian chains [for which I(q)q2 plateaus at high q].72,73 SAXS data indicate that dimeric KCMs and their single-chain counterparts produce structures characteristic of flexible rods in solution (Figure 4). In contrast, the TarFO 4Q and TarFO short produced globular structures. Calculated molecular envelopes for the TarFO variants generally fit the expected dimension of the respective species (Figure 4). The expanded width of the envelope relative to the tightly packed structural model based on fusion of the respective crystal structures reflects some flexibility of the helical subunits and perhaps a closer association of the trimerization motif with the top of the helical bundles.
Figure 4.

Small-angle X-ray scattering (SAXS) plots and molecular envelopes of Tar variants. SAXS intensity (left) and Kratky plots (right) of TarSC, TarFO 4Q, and TarFO short at 1 mg/mL. A plateau in the Kratky plot at large values of q indicates a flexible rodlike structure,90−92 whereas a decrease in Iq2 indicates a globular polymer. Intensity data were smoothed and regularized in PRIMUS. Molecular envelopes calculated with ATSAS and Damaver are superimposed on models of the TarFO 4Q and TarFO short that were based on the fitting of cryo-EM electron density from native receptor arrays to known tightly packed KCM crystal structures6 and combining with crystal structures of the foldon species (right). Linkers between the foldon and KCM were modeled as extended chains but could be more compact. The SAXS envelopes are wider than the structures possibly because of movement in the helical regions.
The MCP Protein Interaction Regions (PIRs) Associate in TarFO
Pulsed-dipolar ESR spectroscopy (PDS) was used to monitor the interaction of the PIRs within TarFO. Nitroxide spin-labels were introduced at the very tip of the receptor by substituting Glu389 in the first MCP KCM repeat with cysteine and then reacting the variant with MTSSL to form nitroxide side chain R1. There are no other Cys residues in TarFO, and the native protein does not react with MTSSL. Because of the symmetry of the Tar variants, a label can potentially reside at six positions within the trimer. However, it seems likely that the linkage of the foldon to the C-terminus of the second KCM repeat will favor the labeled KCM at either the “inner” or “outer” position within the trimer, effectively yielding three spin-label positions per trimer related by 3-fold symmetry (Figure 5). If the receptor tips are associated, as in the membrane arrays, the intersubunit separations should produce distances in the range of ∼30–45 Å, depending on label conformational flexibility. Indeed, PDS measurements of spin-labeled TarFO reveal a wide but well-defined distance distribution for spin–spin separations of 28–35 Å with contributions also at ∼45 Å (Figure 5). The breadth of spin-separations observed is in the range bounded by reasonable conformer distributions of the spin-labels. Furthermore, if both inward and outward orientations of the TarSC units were possible, distances of ≤20 Å would be expected, and none are indicated. Thus, the receptor tips, which reside ∼200 Å from the trimerization motif, must be closely associated, and the single-chain KCMs have mostly fixed orientations within the trimer. The ∼45 Å spin–spin separations may represent some expansion or minor fraying of the tips, but even so, TarFO oligomerization is remarkably structured given that the TarSC units have little tendency to trimerize on their own at these concentrations (Figure 4).
Figure 5.

Pulsed-dipolar ESR spectroscopy (PDS) measurements of TarFO 4Q. (A) The spin-separation distance distribution P(r) and background-corrected time domain data (inset) for TarFO 4Q 515 E389C-R1 (50 μM) shows a broad distribution corresponding to a range of separation distances of the nitroxide spin-labels at the receptor tips. (B) Model of TarFO 4Q with closely associated interaction tips viewed from the tip toward the foldon. Distance separations bounded by plausible spin-label conformers generally agree with the distribution breadth (shown in panel A). If the labeled positions were to assume both inward- and outward-facing orientations within the trimers, short distances (≤20 Å) would be observed.
Effects of TarFO on CheA Kinase Activity
Autophosphorylation of CheA (Figure 6) was monitored by phospho-His (CheA-P) production after exposure to [γ-32P]ATP, gel electrophoresis, and phosphorimage analysis. Autophosphorylation activity was studied in the presence and absence of CheW and either TarSC, TarFO 4Q, or TarFO short (Figure 7). The intensity determined by phosphor analysis was relative to a CheA standard (30 s time point) within each individual gel. The 30 s time point discriminates differences in intensity across the range of activities observed. During CheA autophosphorylation, phosphate transfer equilibrates between the substrate histidine and ADP (Figure 6).74 Net His phosphorylation is then a first-order approach to this equilibrium whose end point depends on both forward and reverse rate constants (Figure 6). However, fast nucleotide exchange (approximately seconds)75−77 competes with the internal equilibration between phospho-His and ATP. Thus, under conditions of excess ATP, the reverse phosphorylation of ADP should be minimal. Nevertheless, ATP preparations can contain contaminating ADP in appreciable amounts and hence alter the end point of the autophosphorylation assay.53,75 Thus, we monitored loss of CheA-P after quenching with excess cold ATP. No appreciable change in CheA-P was seen over 30 min after chase with 2 mM cold ATP (note that this experiment also then measures the stability of CheA-P, whose t1/2 ≫ 30 min). Given these considerations, CheA autophosphorylation was treated as a first-order process and fit to the standard expression (see Experimental Procedures). In contrast, chasing free CheA with excess cold ADP (2 mM) favors the back reaction and thereby depletes CheA-P over the course of minutes (Figure 8B). There appears to be two phases to this response: a fast initial decay and then a slower prolonged decrease. The presence of CheW or Tar variants had minimal effect on the back reaction with ADP, but addition of the TarFO and TarSC species did appear to remove the fast phase of CheA-P depletion. In these and subsequent autophosphorylation reactions, proteins were left to complex for 10 min to 1 h with CheW and the Tar variants to facilitate complex formation. All reactions were run under conditions of excess ATP.
Figure 6.

CheA phosphotransfer reactions. After CheA autophosphorylation, the phosphate group can be transferred from the P1 histidine residue to CheY or back to ADP or undergo hydrolysis. Bimolecular binding of ATP to CheA is thought to be fast relative to autophosphorylation under conditions of excess ATP.75 Under steady-state conditions, when ATP and CheY are in excess and phosphotransfer from CheA-P to CheY is relatively fast, [CheY-P] = k1/k–p[CheA].
Figure 7.

CheA autophosphorylation in the presence of CheW and Tar variants or membrane arrays. (A) Phosphor image of a radioisotope PAGE gel of E. coli CheA autophosphorylation with receptor variants with or without CheW. All the receptors increase CheA activity only if CheW is present. E. coli CheA, CheW, and TarFO or TarSC (in a 1:1:6 subunit ratio, 2.5 μM CheA) were left to complex at 25 °C for 1 h prior to exposure to [γ-32P]ATP for 30 s. Top and bottom gels are shown at different imaging exposures to aid comparisons for the more active species. (B) PAGE gel comparing CheA activity with TarFO 4Q with and without CheY (40 μM) vs a membrane (Mem.) array comprised of CheA (2.5 μM), CheW (5 μM), and Tsr receptors (3.4 μM). All band intensities are scaled relative to a normalized free CheA control (30 s time point) present on each gel.
Figure 8.

Kinetics of CheA autophosphorylation with Tar variants. E. coli CheA, CheW, and TarFO 4Q and short or TarSC (in a 1:1:3 subunit ratio, 1 μM CheA note that the receptor subunit is a single-chain “dimer”) were allowed to complex at 25 °C for 1 h prior to exposure to [γ-32P]ATP for the indicated time points. Each data point represents an average over two to four assays. (A) CheA-P formation over time in the presence of CheW and Tar variants. The inset shows CheA-P buildup with TarFO short compared to CheA:CheW alone out to 30 min. Curves were fit to a first-order kinetic transition (see Experimental Procedures). (B) Addition of cold ADP to CheA and CheW with or without Tar variants after initial autophosphorylation with [γ-32P]ATP for 6 min. (C) Addition of cold ATP to CheA and CheW after incubation with [γ-32P]ATP for 6 min. (D) Transfer to CheY in the presence of CheA and CheW with or without TarFO 4Q and short. Error bars represent the standard error of the mean (SEM) calculated from three independent experiments (n = 3).
When CheA is complexed with CheW and the TarFO variants, the level of autophosphorylation increases, but the progress curves had unexpected time dependencies. Under our conditions, CheW alone increases the level of CheA autophosphorylation somewhat at early time points (∼30 s). Nonetheless, the measured k1 value for first-order CheA–CheW autophosphorylation (1.2 min–1) is similar to that measured previously for CheA alone (1.5 min–1).75 The Tar variants without CheW generally inhibit autophosphorylation, but with CheW present, the level of autophosphorylation increases to levels that exceed those with only CheW (Figures 7 and 8 and Table 2). Maximal activation was observed when the proteins were allowed to complex at least 1 h prior to [γ-32P]ATP exposure. The level of autophosphorylation at <30 s appeared to increase upon addition of the Tar variants (Figures 7 and 8), but quantitative comparisons proved to be difficult at short times. Importantly, phosphorylated CheA-P accumulated much more in the presence of the variants, reaching plateau values that far exceeded that of the CheA–CheW complex alone (Figure 8 inset and Table 2). This reactivity is accentuated greatest by the TarFO short, which produces saturation levels of CheA-P ∼20 times greater than that of the CheA–CheW complex (Figure 8 and Table 2). All of the progress curves could be fit reasonably well to a first-order expression, with the resulting prefactors and rate constants dominated by the plateau behavior at long times (Table 2). The TarFO short showed some indication of a faster phase at <1.5 min, which was evident in the 30 s time points (Figure 7), but this was difficult to resolve in biexponental fits to the data. The initial rates of the time courses (k1A0) for the CheA–CheW complex alone, TarSC, TarFO 4Q, and TarFO short are all relatively similar, but the saturation levels of CheA-P (A0) are much different (Table 2).
Table 2. Kinetic Parameters for Autophosphorylation Assaysa.
| components | A0 (CheA-P relative) | k1 (min–1) | k1A0 (CheA-P min–1) | R2 |
|---|---|---|---|---|
| CheA and CheW | 0.99 ± 0.01 | 1.18 ± 0.05 | 1.2 | 0.99 |
| CheA, CheW, and TarSC 4Q | 8 ± 1 | 0.09 ± 0.02 | 0.7 | 0.98 |
| CheA, CheW, and TarFO 4Q | 6.7 ± 0.9 | 0.20 ± 0.06 | 1.3 | 0.95 |
| CheA, CheW, and TarFO short | 18 ± 1 | 0.073 ± 0.009 | 1.3 | 0.98 |
Prefactor and rate constant values from activity vs time fits to a first-order expression (see Experimental Procedures).
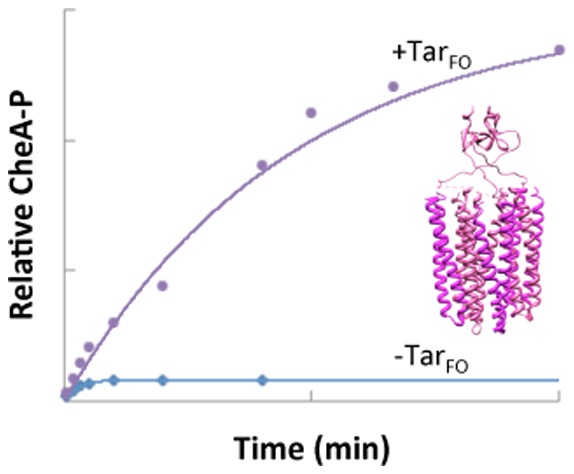
Notably, the amount of CheA-P produced with TarFO 4Q is commensurate with that observed in native membrane arrays that contain a similar amount of CheA (Figure 7B) and the serine receptor Tsr (membrane arrays were kindly provided by K. Piasta and J. Falke, University of Colorado). Although Tsr is a chemoreceptor different from Tar, the PIR regions of the two receptors are nearly identical in sequence. Previous studies of CheA activation on receptor binding gave changes in plateau CheA-P values on the order of 5–10-fold, but in these cases, CheA-P saturation usually occurred within a few minutes.39,44,53,74,78,79 Interestingly, the species with the largest prefactors have rate constants that are smaller than that for CheA–CheW alone (Table 2). This slower process likely represents conversion from an inactive to active form of CheA facilitated by the Tar variants. After conversion, fast autophosphorylation builds up CheA-P, which is then stable over the assay time course.
Although the Tar variants increase autophosphorylation yields, they do not activate subsequent CheY phosphotransfer to the extent observed in the membrane arrays (Figure 7B), where activity can change >100-fold.33,34,78,80 This is despite the fact that CheY completely dephosphorylates similar amounts of CheA-P in the membrane arrays or in complex with the foldon species (Figure 7). TarFO 4Q does produce a moderate increase in the level of phosphotransfer to CheY, but surprisingly, the TarFO short shows nearly no increase at all (Figure 8D). One possibility for this behavior is that exchange between the inactive and active forms of CheA, facilitated by the Tar variants, still greatly favors the inactive kinase, and thus, little CheA-P is available to overcome the instability of CheY-P. Alternatively, CheA may have the capability of being activated at two stages, one that increases the level of autophosphorylation and a second that increases the level of CheY phosphotransfer. The Tar variants then achieve only the former. To test this latter possibility, we measured CheY phosphorylation by CheA-P with TarFO short after CheA autophosphorylation for 30 min. After CheA-P had accumulated, we found rapid (<30 s) quantitative transfer of phosphate to CheY (Figure 2 of the Supporting Information). Furthermore, the amount of CheY-P formed after CheA autophosphorylation for 30 min decayed rapidly [t1/2 ∼ 30 s; k ∼ 1.3 min–1 (Figure 2 of the Supporting Information)]. Thus, phosphotransfer to CheY is not limited, but rather CheY-P hydrolysis outpaces rephosphorylation of CheA (see the legend of Figure 6). Higher concentrations of TarSC and TarFO do not further increase the level of CheA autophosphorylation or CheY phosphotransfer, probably because most of the CheA–CheW complex is complexed with the TarFO species under conditions of the assay (Figure 9) and more Tar variant may compete CheW away from CheA.39,81
Figure 9.

MALS–SEC traces of CheA–CheW complexes with Tar variants. Samples were allowed to form complexes at 25 °C for 1 h prior to injection with 1:1:6 CheA:CheW:TarSC and CheA:CheW:TarFO subunit ratios [note that the KCM units are single-chain “dimers” (Figure 2); hence, one subunit corresponds to a dimeric receptor tip]. Samples were run at a total protein concentration of 5 mg/mL. The TarFO complexes produced well-defined SEC elution profiles containing high-molecular weight complexes. The TarFO 4Q complex had a polydispersity greater than that of the TarFO short complex, which was mainly monodisperse within the complex peak. The TarFO short sample also contained free TarFO in keeping with a smaller receptor contribution to the complex than in the solution stoichiometry. In contrast, the TarSC shows only peaks corresponding to uncomplexed CheA dimer and TarSC. Cartoon representations of hypothetical complexes corresponding to the average molecular weights are shown.
Interactions of Tar Variants with the CheA–CheW Complex
MALS–SEC analysis was used to examine the interactions between the Tar variants and the CheA–CheW complex. Mixtures of TarSC and the CheA–CheW complex showed little complex formation on MALS–SEC, even after prior incubation at room temperature for 1 h. In contrast to TarSC, both TarFO 4Q and TarFO short produced large complexes that were reasonably monodisperse (Figure 9). In the case of TarFO short, the average MW corresponded to that of one CheA–CheW dimer and one TarFO short, whereas in the case of TarFO 4Q, the complex was larger and more heterogeneous and possibly contained at least two copies of TarFO, two copies of dimeric CheA, and four to eight CheWs. Other stoichiometries are also possible, but as only a single species forms, the complex should contain CheW, trimeric TarFO 4Q, and dimeric CheA, with a subunit excess of TarFO 4Q. MW and radius of gyration analyses of the elution peaks indicated primarily a single species for the CheA–CheW complexes with TarFO short and TarSC, but more heterogeneity for the TarFO 4Q complexes. The precise stoichiometry of the complexes cannot be obtained from MALS data alone, but nonetheless, the TarFO variants form soluble, nonaggregated complexes with CheA and CheW that are reasonably well-defined and can be separated via SEC.
Effects of TarFO on CheA Conformation
The structure of the ternary complex between the T. maritima CheA–CheW complex and an inhibitory KCM from T. maritima receptor Tm14 (previously denoted MCPC, residues 41–254) has been previously investigated by nitroxide spin-labeling and PDS.64 The arrangements of the regulatory CheA P5 domains in this inhibitory complex were different from those predicted from the model derived from the ternary complex crystal structure and the ECT maps of intact receptor arrays, wherein CheA P5 and CheW polymerize into planar interlocking ring structures.6 Thus, we investigated the effect of TarFO 4Q on a spin-label reporter site of the T. maritima CheA–CheW P5 complex known to be sensitive to receptor binding (residue Q545C, T. maritima).64 A pull-down assay confirmed that TarFO 4Q and Tm14 bind to similar amounts of T. maritima P3P4P5 and CheW under conditions of the PDS experiments (Figure 10). Subsequent DEER measurements on the CheA–CheW complex spin-labeled at position 545 revealed that TarFO induces an ∼7 Å increase in the separation of the P5 domains relative to that observed with Tm14 (Figure 10A, B). In addition, the spin–spin distribution has greater breadth compared with that of the inhibitory receptor and retains a minor distance peak at the position seen with Tm14. The longer distances observed with TarFO 4Q are consistent with conversion to the more planar P5 arrangement needed to accommodate the larger trimeric assembly of receptors found in the membrane arrays. The broadness of the distribution may reflect flexibility in the absence of the interlocking ring structures inherent to the arrays.
Figure 10.

PDS distributions of CheA when bound to inhibitory or activating receptor mimetics. (A) Distance distributions (left) and background-corrected time domain data (right) of spin separations in T. maritima CheA P3P4P5 (Δ289) Q545C, CheW, and Tm14 KCM or TarFO 4Q (in a 1:2:3 subunit ratio, 38 μM CheA). (B) Structural prediction of the distance between the spin-labels in the inhibited form of CheA (red) and an active form with planar P5 domains (cyan). (C) Pull-down assay of T. maritima CheA Δ289 binding to CheW and receptor variants. The Δ289-His tag was pulled down with Ni2+-NTA beads in the presence of CheW and/or Tm14 or TarFO 4Q. The target protein coprecipitated with affinity-labeled CheA Δ289, without any nonspecific binding of the target to the Ni2+-NTA beads, produced the extent of protein pulled down. The interaction between Δ289 and CheW is similar in the presence of the two receptors, and the amount of CheA bound to the beads was highly reproducible in each experiment. T. maritima Tm14 and E. coli TarFO 4Q receptor are pulled down to a similar degree, and thus, both receptors interact with T. maritima CheA and CheW to produce the changes seen in CheA P3P4P5 Q545C PDS spectra.
Cellular Activities of TarFO
To test the function of TarFOin vivo, we introduced TarFO 4Q into an E. coli strain devoid of all other chemoreceptors and monitored flagellar rotation in a tethered cell assay. Of the two foldon species, TarFO 4Q was chosen for this assay because it shows the greatest ability to activate phosphotransfer to CheY. In the tethered cell assay, CheA inhibition causes CCW flagellar rotation, whereas CheA activation causes flagellar rotation in the opposite, CW direction. Flagellar rotation biases were determined for cell populations containing full-length membrane-incorporated Tar, the dimeric Tar KCM, TarSC, and TarFO either in the absence (−CheB/R, strain UU2610) or in the presence (+CheB/R, strain UU2612) of the methylation system (CheR and CheB). Introduction of full-length membrane-incorporated Tar causes a strongly CW phenotype in −CheB/R cells (UU2610) that adapts back toward CCW in the presence of the methylation system (+CheB/R, UU2612) (Figure 11). The Tar signaling domain alone (KCM) produces much less CW bias and is largely unaffected by the methylation system. The degree of CheA activity with KCM only is smaller than that seen with cytoplasmic receptor domains fused to an activating HAMP domain.47 In −CheB/R cells, both TarFO and TarSC similarly increase CW bias (Figure 11). For TarFO 4Q and TarSC CW, bias increases further in the presence of the methylation system to an extent that exceeds that of full-length Tar. This result is somewhat surprising because the methylation sites are encoded as all-Gln in each case, and hence, no further activation by CheR should be possible. Indeed, although all of the proteins were well expressed, TarSC or TarFO was not methylated in E. coli. The increased kinase activity of TarFO 4Q and TarSC 4Q in +CheB/R cells may reflect stabilization of the ternary complexes with TarFO and TarSC by binding of the methylation enzymes. Binding of CheR may favor more dynamic adaptation regions,48 which the TarFO may exaggerate in the absence of membrane association. Alternatively, CheB-mediated deamination may alter the TarFO conformation in a manner that produces an inverted response. Inversion effects have been observed with HAMP domain point mutations and generally speak to the conformational sensitivity of the signaling complex.47,82,83 Unfortunately, the QEQE versions of the TarFO were not sufficiently stable to be evaluated for their in vivo properties.
Figure 11.
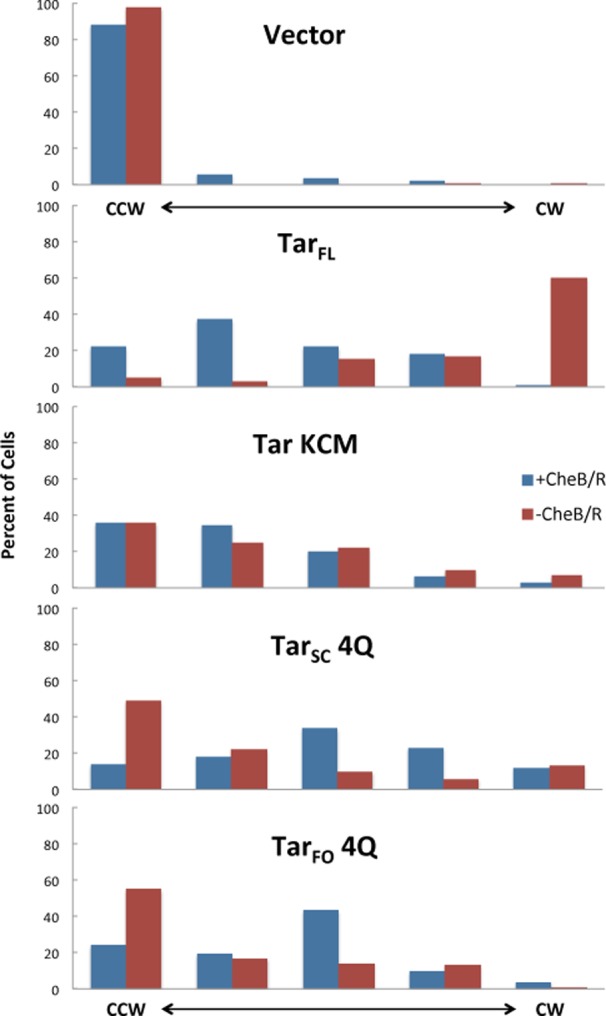
Flagellar rotation patterns of E. coli cells transformed with Tar variants. Rotation patterns were determined in the −CheB/R (UU2610) or +CheB/R (UU2612) cells. Both strains lacked all native chemoreceptors. Each histogram summarizes the behavior of rotating cells as exclusively CCW, predominantly CCW, frequently reversing with no overall directional bias, predominantly CW, and exclusively CW (from left to right, respectively).
Discussion
A soluble chemoreceptor module that mimics the trimer-of-dimers assembly state found in membrane arrays was developed to study the effect of receptor trimerization on kinase activity and structure. Fusing KCM subunits together into a single-chain “dimer” and then trimerizing the resulting units with a surrogate foldon domain produce a surprisingly homogeneous and globular structure with associated PIRs. Additionally, conversion of the modification sites to all Gln (4Q) stabilizes the adaptation region by removing negative charge47,84,85 and improves expression levels and stability.
Structural Considerations of Soluble Receptor Mimics
Several approaches have been taken to study the interaction of CheA–CheW complexes with simplified receptor species. Leucine zipper dimerization and HAMP domains have been used to stabilize chemoreceptor cytoplasmic domains and form stable complexes with CheA.43−47 KCM fragments also show honeycomb structures when expressed with CheA and CheW and treated with osmolytes.37,49 KCM fragments templated on lipid bilayers produce hexagonal symmetries and give large degrees of CheA activation.39,41,86 Nanodisc-incorporated receptors show native-like behaviors and hence allow the assessment of different assembly states, but the quantities of activated complexes obtained are relatively small.1,32−34,87 All of these aforementioned CheA–CheW–receptor assemblies are heterogeneous, and many also involve lipid components. The TarFO variants developed here produce relatively homogeneous ternary complexes that bind CheA, increase the level of autophosphorylation, are relatively easy to manipulate, and can be studied by biophysical methods such as pulsed ESR spectroscopy, SAXS, single-particle EM, and crystallography. The globular nature of the TarFO from SAXS, MALS, and PDS distance restraints demonstrates the tendency for the PIRs to associate in the absence of other factors, provided that they are held in the proximity. Moreover, the receptor tips trimerize even though the foldon associates the KCM N-termini more closely than what is found in the natural arrays, where they become less ordered and spread apart as they approach the membrane.6 The trimeric nature of TarFO 4Q is also reflected in the conformation of bound CheA. Compared to the situation in which CheA binds a single KCM (e.g., Tm14), the P5 domains are spread farther apart across the CheA dimer interface when bound to TarFO 4Q to accommodate the larger trimeric species. The P5 domains also assume a planar arrangement, which is the conformation needed to template the hexagonal receptor arrays (Figure 1A). These results also agree with predictions from recent molecular dynamics simulations that implicate a planar arrangement of P5 as a preferred conformation for the kinase.88
Effects of Tar Variants on CheA Activity
Pull-down assays, PDS experiments, and activity assays all demonstrate that the TarFO species interact with CheA and CheW. The degree of CheA autophosphorylation observed with TarFO and CheW compares favorably with the degree of receptor-mediated CheA autophosphorylation in membrane arrays,39,44,53,74,78,79,89 but the kinetics differ. As in these other experiments, the TarFO species primarily increase the prefactor terms and hence the level of CheA-P at saturation. One explanation for such behavior is that receptors alter the position of the equilibrium (k1 and/or k–1) between forward phosphorylation of substrate His and reverse phosphorylation of ADP (Figure 6). However, in the presence of a saturating level of ATP, nucleotide exchange will outcompete the reverse reaction, and thus, His phosphorylation should proceed to completion. Furthermore, a 10-fold change in total CheA-P production would imply large changes to k1 and k–1 that are not indicated under conditions of cold ADP chase. A second rationale for the increase in peak autophosphorylation is that the fraction of kinase capable of autophosphorylation increases in the TarFO complexes; i.e., the CheA has an inactive and active form, and the receptor variants increase the level of access to an active conformation of the kinase. For free CheA, a large fraction of kinase is inactive, and this form exchanges slowly with an active fraction on the time scale of the experiment. Thus, autophosphorylation saturates quickly, but at low levels. The Tar variant complexes produce more active CheA, but exchange from the inactive form is also accelerated so that much greater levels of CheA-P accumulate. The rate constant for exchange is lower than that for autophosphorylation. The nature of this more active state in the TarFO complexes is an open question. The slow accumulation of CheA-P could indicate a small but accessible time-averaged fraction of the kinase in a fully active conformation, or a kinase that has been perturbed mostly but not completely toward the fully active conformation. The PDS distributions of Figure 10 suggest the latter because the reporter site conformations in the inactive and TarFO-bound states appear to be quite different. The reporter suggests that some inactive state is still represented in the TarFO complexes, but the majority of the kinase has changed to a conformation more inline with that expected for the receptor arrays. The lower activation numbers with TarSC likely reflect the higher entropic cost of assembling the active form, which is circumvented in TarFO by the preformed foldon trimer.
Unlike CheA autophosphorylation, net CheY phosphotransfer activity is not substantially increased by the TarFO variants. With all receptor preparations tested, CheY fully dephosphorylates CheA within tens of seconds, yet only with the membrane arrays do large amounts of CheY-P accumulate. One possibility for this difference is that the Tar variants do not lock in the phosphorylation-competent state of CheA to the same degree as do the membrane arrays. Exchange to the active form occurs more readily than with free CheA, but it is slower than CheY phosphotransfer and, importantly, CheY-P hydrolysis (Figure 6 and Figure 2 of the Supporting Information). With the Tar variants, the rate constant k1 largely represents this exchange rate. Consistent with this view, TarFO 4Q, which has the largest k1 value of the variants (Table 2), also shows the greatest increase in the level of CheY phosphotransfer (Figure 8D). The phospho-His is more labile in the activated form of membrane-associated CheA than in the inhibited state because of enhanced reaction with ADP.79 Interestingly, TarFO-associated CheA does not show this enhanced exchange with ADP, and if anything, the back reaction is diminished in these complexes. Thus, low phosphate exchange with ADP correlates with low phosphotransfer to CheY. Nonetheless, the TarFO 4Q variant does increase the level of CheY phosphotransfer to some extent, and this is reflected by increased CW-biased flagellar rotation with TarFO 4Q in the presence of the methylation system. Notably, the TarFO variants do not achieve the same degree of activation in vivo as Tar cytoplasmic domains that are fused to activating HAMP domains.47 Apparently, the specific conformation of each receptor dimer, set by HAMP, is important for achieving a high degree of CheA activation.
In conclusion, TarFO variants demonstrate that constrained receptor tips will trimerize and that these species will bind CheA and CheW to form defined complexes in the absence of membranes or other components. Thus, the Tar variants provide a useful tool for studying how receptor engagement affects the structure and activity of the CheA–CheW complex. Importantly, the trimer mimetics stimulate CheA autophosphorylation by increasing the fraction of active kinase and facilitating conversion from the inactive form. The active and inactive conformations of CheA when bound to these preformed trimers may represent the activity states modulated by a chemoattractant in native membrane arrays. Further investigations will be aimed at understanding what molecular features influence the stability of these states and hence the capability of the complexes to catalyze accumulation of phosphorylated CheY.
Acknowledgments
We thank Joanne Widom for assistance in constructing and cloning the Tar variants and the Cornell High Energy Synchrotron Source (CHESS) for access to data collection facilities. We are also grateful to Kene Piasta and Joseph Falke (University of Colorado) for providing samples of native chemoreceptor arrays.
Glossary
Abbreviations
- FPLC
fast protein liquid chromatography
- MCP
methyl-accepting chemotaxis protein
- MCPC
MCP cytoplasmic region
- KCM
kinase control module
- ESR
electron spin resonance
- PDS
pulsed-dipolar ESR spectroscopy
- DEER
double electron–electron resonance
- PIR
protein interaction region
- Tm14
T. maritima MCP TM0014
- Tris
2-amino-2-hydroxymethylpropane-1,3-diol
- FO
foldon trimerization motif
- SC
single-chain fused dimer
- CW
clockwise
- CCW
counterclockwise
- P1
CheA histidine phosphotransfer domain
- P2
CheA response regulator docking domain
- P3
CheA dimerization domain
- P4
CheA kinase domain
- P5
CheA regulatory domain
- CheA Δ289
domains P3P4P5
- EM
electron microscopy
- ECT
electron cryo-tomography
- DTT
dithiothreitol
- EDTA
ethylenediaminetetraacetic acid
- HEPES
2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid
- IPTG
β-d-thiogalactopyranosidase
- MALS
multiangle light scattering
- SAXS
small-angle X-ray scattering
- MTSSL
S-(2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl)methylmethanesulfonate
- PMSF
phenylmethanesulfonyl fluoride
- ADP
adenosine diphosphate
- GFB
gel-filtration buffer
- SEC
size-exclusion chromatography.
Supporting Information Available
Schematic for the cloning strategy of the TarSC and TarFO constructs and kinetics for CheY-P formation and decay after CheA-P buildup. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/bi501570n.
Author Contributions
A.R.G. and X.L. contributed equally to this work.
Support provided by National Institutes of Health Grants P41GM103521 (J.H.F.) and GM066775 (B.R.C.).
The authors declare no competing financial interest.
Supplementary Material
References
- Hazelbauer G. L.; Falke J. J.; Parkinson J. S. (2008) Bacterial chemoreceptors: High-performance signaling in networked arrays. Trends Biochem. Sci. 33, 9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazelbauer G. L.; Lai W.-C. (2010) Bacterial chemoreceptors: Providing enhanced features to two-component signaling. Curr. Opin. Microbiol. 13, 124–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberman L.; Berg H. C.; Sourjik V. (2004) Effect of chemoreceptor modification on assembly and activity of the receptor-kinase complex in Escherichia coli. J. Bacteriol. 186, 6643–6646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadhams G. H.; Armitage J. P. (2004) Making sense of it all: Bacterial chemotaxis. Nat. Rev. Mol. Cell Biol. 5, 1024–1037. [DOI] [PubMed] [Google Scholar]
- Briegel A.; Beeby M.; Thanbichler M.; Jensen G. J. (2011) Activated chemoreceptor arrays remain intact and hexagonally packed. Mol. Microbiol. 82, 748–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briegel A.; Li X.; Bilwes A. M.; Hughes K. T.; Jensen G. J.; Crane B. R. (2012) Bacterial chemoreceptor arrays are hexagonally packed trimers of receptor dimers networked by rings of kinase and coupling proteins. Proc. Natl. Acad. Sci. U.S.A. 109, 3766–3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briegel A.; Ortega D. R.; Tocheva E. I.; Wuichet K.; Li Z.; Chen S.; Müller A.; Iancu C. V.; Murphy G. E.; Dobro M. J.; Zhulin I. B.; Jensen G. J. (2009) Universal architecture of bacterial chemoreceptor arrays. Proc. Natl. Acad. Sci. U.S.A. 106, 17181–17186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P.; Bos E.; Heymann J.; Gnaegi H.; Kessel M.; Peters P. J.; Subramaniam S. (2004) Direct visualization of receptor arrays in frozen-hydrated sections and plunge-frozen specimens of E. coli engineered to overproduce the chemotaxis receptor Tsr. J. Microsc. (Oxford, U.K.) 216, 76–83. [DOI] [PubMed] [Google Scholar]
- Sourjik V.; Berg H. C. (2002) Receptor sensitivity in bacterial chemotaxis. Proc. Natl. Acad. Sci. U.S.A. 99, 123–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunten P.; Koshland D. E. (1991) Tuning the responsiveness of a sensory receptor via covalent modification. J. Biol. Chem. 266, 1491–1496. [PubMed] [Google Scholar]
- Borkovich K. A.; Alex L. A.; Simon M. I. (1992) Attenuation of sensory receptor signaling by covalent modification. Proc. Natl. Acad. Sci. U.S.A. 89, 6756–6760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aizawa S.-I.; Harwood C. S.; Kadner R. J. (2000) Signaling Components in Bacterial Locomotion and Sensory Reception. J. Bacteriol. 182, 1459–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G.; Weis R. M. (2000) Covalent Modification Regulates Ligand Binding to Receptor Complexes in the Chemosensory System of Escherichia coli. Cell 100, 357–365. [DOI] [PubMed] [Google Scholar]
- Sourjik V.; Berg H. C. (2004) Functional interactions between receptors in bacterial chemotaxis. Nature 428, 437–441. [DOI] [PubMed] [Google Scholar]
- Maddock J.; Shapiro L. (1993) Polar location of the chemoreceptor complex in the Escherichia coli cell. Science 259, 1717–1723. [DOI] [PubMed] [Google Scholar]
- Zhang P.; Khursigara C. M.; Hartnell L. M.; Subramaniam S. (2007) Direct visualization of Escherichia coli chemotaxis receptor arrays using cryo-electron microscopy. Proc. Natl. Acad. Sci. U.S.A. 104, 3777–3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briegel A.; Ding H. J.; Li Z.; Werner J.; Gitai Z.; Dias D. P.; Jensen R. B.; Jensen G. J. (2008) Location and architecture of the Caulobacter crescentus chemoreceptor array. Mol. Microbiol. 69, 30–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Hu B.; Morado D. R.; Jani S.; Manson M. D.; Margolin W. (2012) Molecular architecture of chemoreceptor arrays revealed by cryoelectron tomography of Escherichia coli minicells. Proc. Natl. Acad. Sci. U.S.A. 109, E1481–E1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ames P.; Parkinson J. S. (2006) Conformational suppression of inter-receptor signaling defects. Proc. Natl. Acad. Sci. U.S.A. 103, 9292–9297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gestwicki J. E.; Kiessling L. L. (2002) Inter-receptor communication through arrays of bacterial chemoreceptors. Nature 415, 81–84. [DOI] [PubMed] [Google Scholar]
- Hansen C. H.; Sourjik V.; Wingreen N. S. (2010) A dynamic-signaling-team model for chemotaxis receptors in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 107, 17170–17175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C.; Jackson M.; Lux R.; Khan S. (2001) Determinants of chemotactic signal amplification in Escherichia coli. J. Mol. Biol. 307, 119–135. [DOI] [PubMed] [Google Scholar]
- Li X.; Fleetwood A. D.; Bayas C.; Bilwes A. M.; Ortega D. R.; Falke J. J.; Zhulin I. B.; Crane B. R. (2013) The 3.2 Å Resolution Structure of a Receptor:CheA:CheW Signaling Complex Defines Overlapping Binding Sites and Key Residue Interactions within Bacterial Chemosensory Arrays. Biochemistry 52, 3852–3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falke J. J.; Hazelbauer G. L. (2001) Transmembrane signaling in bacterial chemoreceptors. Trends Biochem. Sci. 26, 257–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilwes A. M., Park S. Y., Quezada C. M., Simon M. I., and Crane B. R. (2003) Structure and Function of CheA, the Histidine Kinase Central to Bacterial Chemotaxis. In Histidine Kinases in Signal Transduction (Inouye M., Ed.) pp 48–74, Academic Press, San Diego. [Google Scholar]
- Gosink K. K.; Zhao Y. M.; Parkinson J. S. (2011) Mutational Analysis of N381, a Key Trimer Contact Residue in Tsr, the Escherichia coli Serine Chemoreceptor. J. Bacteriol. 193, 6452–6460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K. K.; Yokota H.; Kim S.-H. (1999) Four-helical-bundle structure of the cytoplasmic domain of a serine chemotaxis receptor. Nature 400, 787–792. [DOI] [PubMed] [Google Scholar]
- Falke J. J.; Piasta K. N. (2014) Architecture and signal transduction mechanism of the bacterial chemosensory array: Progress, controversies, and challenges. Curr. Opin. Struct. Biol. 29, 85–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sourjik V. (2004) Receptor clustering and signal processing in E. coli chemotaxis. Trends Microbiol. 12, 569–576. [DOI] [PubMed] [Google Scholar]
- Ames P.; Studdert C. A.; Reiser R. H.; Parkinson J. S. (2002) Collaborative signaling by mixed chemoreceptor teams in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 99, 7060–7065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studdert C. A.; Parkinson J. S. (2004) Crosslinking snapshots of bacterial chemoreceptor squads. Proc. Natl. Acad. Sci. U.S.A. 101, 2117–2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boldog T.; Grimme S.; Li M.; Sligar S. G.; Hazelbauer G. L. (2006) Nanodiscs separate chemoreceptor oligomeric states and reveal their signaling properties. Proc. Natl. Acad. Sci. U.S.A. 103, 11509–11514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M.; Hazelbauer G. L. (2011) Core unit of chemotaxis signaling complexes. Proc. Natl. Acad. Sci. U.S.A. 108, 9390–9395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M.; Khursigara C. M.; Subramaniam S.; Hazelbauer G. L. (2011) Chemotaxis kinase CheA is activated by three neighbouring chemoreceptor dimers as effectively as by receptor clusters. Mol. Microbiol. 79, 677–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piasta K. N.; Ulliman C. J.; Slivka P. F.; Crane B. R.; Falke J. J. (2013) Defining a Key Receptor–CheA Kinase Contact and Elucidating Its Function in the Membrane-Bound Bacterial Chemosensory Array: A Disulfide Mapping and TAM-IDS Study. Biochemistry 52, 3866–3880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard A. M.; Bilwes A. M.; Crane B. R. (2009) The Structure of a Soluble Chemoreceptor Suggests a Mechanism for Propagating Conformational Signals. Biochemistry 48, 1936–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briegel A.; Ladinsky M. S.; Oikonomou C.; Jones C. W.; Harris M. J.; Fowler D. J.; Chang Y.-W.; Thompson L. K.; Armitage J. P.; Jensen G. J. (2014) Structure of bacterial cytoplasmic chemoreceptor arrays and implications for chemotactic signaling. eLife 3, e02151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ames P.; Parkinson J. S. (1994) Constitutively signaling fragments of Tsr, the E. coli serine chemoreceptor. J. Bacteriol. 176, 6340–6348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asinas A. E.; Weis R. M. (2006) Competitive and cooperative interactions in receptor signaling complexes. J. Biol. Chem. 281, 30512–30523. [DOI] [PubMed] [Google Scholar]
- Besschetnova T. Y.; Montefusco D. J.; Asinas A. E.; Shrout A. L.; Antommattei F. M.; Weis R. M. (2008) Receptor density balances signal stimulation and attenuation in membrane-assembled complexes of bacterial chemotaxis signaling proteins. Proc. Natl. Acad. Sci. U.S.A. 105, 12289–12294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrout A. L.; Montefusco D. J.; Weis R. M. (2003) Template-Directed Assembly of Receptor Signaling Complexes. Biochemistry 42, 13379–13385. [DOI] [PubMed] [Google Scholar]
- Seeley S. K.; Weis R. M.; Thompson L. K. (1996) The Cytoplasmic Fragment of the Aspartate Receptor Displays Globally Dynamic Behavior. Biochemistry 35, 5199–5206. [DOI] [PubMed] [Google Scholar]
- Francis N. R.; Wolanin P. M.; Stock J. B.; DeRosier D. J.; Thomas D. R. (2004) Three-dimensional structure and organization of a receptor/signaling complex. Proc. Natl. Acad. Sci. U.S.A. 101, 17480–17485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levit M. N.; Stock J. B. (2002) Receptor Methylation Controls the Magnitude of Stimulus-Response Coupling in Bacterial Chemotaxis. J. Biol. Chem. 277, 36760–36765. [DOI] [PubMed] [Google Scholar]
- Wolanin P. M.; Baker M. D.; Francis N. R.; Thomas D. R.; DeRosier D. J.; Stock J. B. (2006) Self-assembly of receptor/signaling complexes in bacterial chemotaxis. Proc. Natl. Acad. Sci. U.S.A. 103, 14313–14318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochran A. G.; Kim P. S. (1996) Imitation of Escherichia coli Aspartate Receptor Signaling in Engineered Dimers of the Cytoplasmic Domain. Science 271, 1113–1116. [DOI] [PubMed] [Google Scholar]
- Airola M. V.; Sukomon N.; Samanta D.; Borbat P. P.; Freed J. H.; Watts K. J.; Crane B. R. (2013) HAMP domain conformers that propagate opposite signals in bacterial chemoreceptors. PLoS Biol. 11, e1001479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samanta D.; Borbat P. P.; Dzikovski B.; Freed J. H.; Crane B. R. (2015) Bacterial chemoreceptor dynamics correlate with activity state and are coupled over long distances. Proc. Natl. Acad. Sci. U.S.A. 112, 2455–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briegel A.; Wong M. L.; Hodges H. L.; Oikonomou C. M.; Piasta K. N.; Harris M. J.; Fowler D. J.; Thompson L. K.; Falke J. J.; Kiessling L. L.; Jensen G. J. (2014) New Insights into Bacterial Chemoreceptor Array Structure and Assembly from Electron Cryotomography. Biochemistry 53, 1575–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhardwaj A.; Walker-Kopp N.; Wilkens S.; Cingolani G. (2008) Foldon-guided self-assembly of ultra-stable protein fibers. Protein Sci. 17, 1475–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habazettl J.; Reiner A.; Kiefhaber T. (2009) NMR Structure of a Monomeric Intermediate on the Evolutionarily Optimized Assembly Pathway of a Small Trimerization Domain. J. Mol. Biol. 389, 103–114. [DOI] [PubMed] [Google Scholar]
- Tao Y.; Strelkov S. V.; Mesyanzhinov V. V.; Rossmann M. G. (1997) Structure of bacteriophage T4 fibritin: A segmented coiled coil and the role of the C-terminal domain. Structure 5, 789–798. [DOI] [PubMed] [Google Scholar]
- Ninfa E. G.; Stock A.; Mowbray S.; Stock J. (1991) Reconstitution of the bacterial chemotaxis signal transduction system from purified components. J. Biol. Chem. 266, 9764–9770. [PubMed] [Google Scholar]
- Skou S.; Gillilan R. E.; Ando N. (2014) Synchrotron-based small-angle X-ray scattering of proteins in solution. Nat. Protoc. 9, 1727–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen S. S.; Møller M.; Gillilan R. E. (2012) High-throughput biological small-angle X-ray scattering with a robotically loaded capillary cell. J. Appl. Crystallogr. 45, 213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen S. S.; Toft K. N.; Snakenborg D.; Jeppesen M. G.; Jacobsen J. K.; Vestergaard B.; Kutter J. P.; Arleth L. (2009) BioXTAS RAW, a software program for high-throughput automated small-angle X-ray scattering data reduction and preliminary analysis. J. Appl. Crystallogr. 42, 959–964. [Google Scholar]
- Konarev P. V.; Volkov V. V.; Sokolova A. V.; Koch M. H. J.; Svergun D. I. (2003) PRIMUS: A Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 36, 1277–1282. [Google Scholar]
- Franke D.; Svergun D. I. (2009) DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J. Appl. Crystallogr. 42, 342–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkov V. V.; Svergun D. I. (2003) Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 36, 860–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozin M. B.; Svergun D. I. (2001) Automated matching of high- and low-resolution structural models. J. Appl. Crystallogr. 34, 33–41. [Google Scholar]
- Erbse A. H.; Falke J. J. (2009) The Core Signaling Proteins of Bacterial Chemotaxis Assemble To Form an Ultrastable Complex. Biochemistry 48, 6975–6987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borbat P. P., and Freed J. H. (2013) Pulse Dipolar Electron Spin Resonance: Distance Measurements. In Structural Information from Spin-Labels and Intrinsic Paramagnetic Centres in the Biosciences (Timmel C. R., and Harmer J. R., Eds.) pp 1–82, Springer, Berlin. [Google Scholar]
- Borbat P. P., and Freed J. H. (2007) Measuring Distances by Pulsed Dipolar ESR Spectroscopy: Spin-Labeled Histidine Kinases. In Methods in Enzymology (Simon M. I., Crane B. R., and Crane A. M., Eds.) pp 52–116, Academic Press, San Diego. [DOI] [PubMed] [Google Scholar]
- Bhatnagar J.; Borbat P. P.; Pollard A. M.; Bilwes A. M.; Freed J. H.; Crane B. R. (2010) Structure of the Ternary Complex Formed by a Chemotaxis Receptor Signaling Domain, the CheA Histidine Kinase, and the Coupling Protein CheW As Determined by Pulsed Dipolar ESR Spectroscopy. Biochemistry 49, 3824–3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeschke G.; Polyhach Y. (2007) Distance measurements on spin-labelled biomacromolecules by pulsed electron paramagnetic resonance. Phys. Chem. Chem. Phys. 9, 1895–1910. [DOI] [PubMed] [Google Scholar]
- Sale K.; Song L. K.; Liu Y. S.; Perozo E.; Fajer P. (2005) Explicit treatment of spin labels in modeling of distance constraints from dipolar EPR and DEER. J. Am. Chem. Soc. 127, 9334–9335. [DOI] [PubMed] [Google Scholar]
- Chiang Y. W.; Borbat P. P.; Freed J. H. (2005) The determination of pair distance distributions by pulsed ESR using Tikhonov regularization. J. Magn. Reson. 172, 279–295. [DOI] [PubMed] [Google Scholar]
- Chiang Y. W.; Borbat P. P.; Freed J. H. (2005) Maximum entropy: A complement to Tikhonov regularization for determination of pair distance distributions by pulsed ESR. J. Magn. Reson. 177, 184–196. [DOI] [PubMed] [Google Scholar]
- Slocum M. K.; Parkinson J. S. (1985) Genetics of methyl-accepting chemotaxis proteins in Escherichia coli: Null phenotypes of the tar and tap genes. J. Bacteriol. 163, 586–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kort E. N.; Goy M. F.; Larsen S. H.; Adler J. (1975) Methylation of a membrane protein involved in bacterial chemotaxis. Proc. Natl. Acad. Sci. U.S.A. 72, 3939–3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolfson D. N.; Alber T. (1995) Predicting oligomerization states of coiled coils. Protein Sci. 4, 1596–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambo R. P.; Tainer J. A. (2011) Characterizing flexible and intrinsically unstructured biological macromolecules by SAS using the Porod-Debye law. Biopolymers 95, 559–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammel M. (2012) Validation of macromolecular flexibility in solution by small-angle X-ray scattering (SAXS). Eur. Biophys. J. 41, 789–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surette M. G.; Levit M.; Liu Y.; Lukat G.; Ninfa E. G.; Ninfa A.; Stock J. B. (1996) Dimerization Is Required for the Activity of the Protein Histidine Kinase CheA That Mediates Signal Transduction in Bacterial Chemotaxis. J. Biol. Chem. 271, 939–945. [DOI] [PubMed] [Google Scholar]
- Tawa P.; Stewart R. C. (1994) Kinetics of CheA Autophosphorylation and Dephosphorylation Reactions. Biochemistry 33, 7917–7924. [DOI] [PubMed] [Google Scholar]
- Stewart R. C. (2005) Analysis of ATP Binding to CheA Containing Tryptophan Substitutions near the Active Site. Biochemistry 44, 4375–4385. [DOI] [PubMed] [Google Scholar]
- Eaton A. K.; Stewart R. C. (2010) Kinetics of ATP and TNP-ATP Binding to the Active Site of CheA from Thermotoga maritima. Biochemistry 49, 5799–5809. [DOI] [PubMed] [Google Scholar]
- Borkovich K. A.; Kaplan N.; Hess J. F.; Simon M. I. (1989) Transmembrane signal transduction in bacterial chemotaxis involves ligand-dependent activation of phosphate group transfer. Proc. Natl. Acad. Sci. U.S.A. 86, 1208–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borkovich K. A.; Simon M. I. (1990) The dynamics of protein phosphorylation in bacterial chemotaxis. Cell 63, 1339–1348. [DOI] [PubMed] [Google Scholar]
- Stewart R. C.; Van Bruggen R. (2004) Association and dissociation kinetics for CheY interacting with the P2 domain of CheA. J. Mol. Biol. 336, 287–301. [DOI] [PubMed] [Google Scholar]
- Gegner J. A.; Graham D. R.; Roth A. F.; Dahlquist F. W. (1992) Assembly of an MCP receptor, CheW, and kinase CheA complex in the bacterial chemotaxis signal transduction pathway. Cell 70, 975–982. [DOI] [PubMed] [Google Scholar]
- Zhou Q.; Ames P.; Parkinson J. S. (2009) Mutational analyses of HAMP helices suggest a dynamic bundle model of input-output signalling in chemoreceptors. Mol. Microbiol. 73, 801–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q.; Ames P.; Parkinson J. S. (2011) Biphasic control logic of HAMP domain signalling in the Escherichia coli serine chemoreceptor. Mol. Microbiol. 80, 596–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surette M. G.; Stock J. B. (1996) Role of α-Helical Coiled-coil Interactions in Receptor Dimerization, Signaling, and Adaptation during Bacterial Chemotaxis. J. Biol. Chem. 271, 17966–17973. [DOI] [PubMed] [Google Scholar]
- Starrett D. J.; Falke J. J. (2005) Adaptation mechanism of the aspartate receptor: Electrostatics of the adaptation subdomain play a key role in modulating kinase activity. Biochemistry 44, 1550–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weis R. M.; Hirai T.; Chalah A.; Kessel M.; Peters P. J.; Subramaniam S. (2003) Electron Microscopic Analysis of Membrane Assemblies Formed by the Bacterial Chemotaxis Receptor Tsr. J. Bacteriol. 185, 3636–3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M.; Hazelbauer G. L. (2014) Selective allosteric coupling in core chemotaxis signaling complexes. Proc. Natl. Acad. Sci. U.S.A. 111, 15940–15945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X. Q.; Vu A.; Lee K.; Dahlquist F. W. (2012) CheA-Receptor Interaction Sites in Bacterial Chemotaxis. J. Mol. Biol. 422, 282–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borkovich K. A. (1991) Coupling of receptor function to phosphate-transfer reactions in bacterial chemotaxis. Methods Enzymol. 200, 205–214. [DOI] [PubMed] [Google Scholar]
- Putnam C. D.; Hammel M.; Hura G. L.; Tainer J. A. (2007) X-ray solution scattering (SAXS) combined with crystallography and computation: Defining accurate macromolecular structures, conformations and assemblies in solution. Q. Rev. Biophys. 40, 191–285. [DOI] [PubMed] [Google Scholar]
- Mertens H. D. T.; Svergun D. I. (2010) Structural characterization of proteins and complexes using small-angle X-ray solution scattering. J. Struct. Biol. 172, 128–141. [DOI] [PubMed] [Google Scholar]
- Svergun D. I.; Koch M. H. J. (2003) Small-angle scattering studies of biological macromolecules in solution. Rep. Prog. Phys. 66, 1735. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.