

Abstract
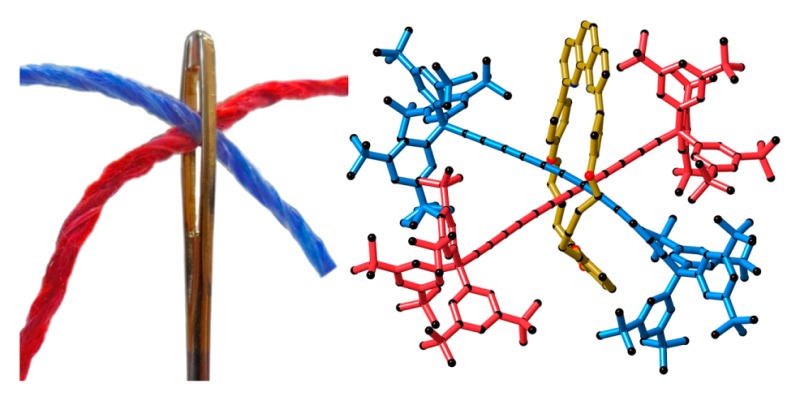
Active metal template Glaser coupling has been used to synthesize a series of rotaxanes consisting of a polyyne, with up to 24 contiguous sp-hybridized carbon atoms, threaded through a variety of macrocycles. Cadiot–Chodkiewicz cross-coupling affords higher yields of rotaxanes than homocoupling. This methodology has been used to prepare [3]rotaxanes with two polyyne chains locked through the same macrocycle. The crystal structure of one of these [3]rotaxanes shows that there is extremely close contact between the central carbon atoms of the threaded hexayne chains (C···C distance 3.29 Å vs 3.4 Å for the sum of van der Waals radii) and that the bond-length-alternation is perturbed in the vicinity of this contact. However, despite the close interaction between the hexayne chains, the [3]rotaxane is remarkably stable under ambient conditions, probably because the two polyynes adopt a crossed geometry. In the solid state, the angle between the two polyyne chains is 74°, and this crossed geometry appears to be dictated by the bulk of the “supertrityl” end groups. Several rotaxanes have been synthesized to explore gem-dibromoethene moieties as “masked” polyynes. However, the reductive Fritsch–Buttenberg–Wiechell rearrangement to form the desired polyyne rotaxanes has not yet been achieved. X-ray crystallographic analysis on six [2]rotaxanes and two [3]rotaxanes provides insight into the noncovalent interactions in these systems. Differential scanning calorimetry (DSC) reveals that the longer polyyne rotaxanes (C16, C18, and C24) decompose at higher temperatures than the corresponding unthreaded polyyne axles. The stability enhancement increases as the polyyne becomes longer, reaching 60 °C in the C24 rotaxane.
Introduction
Rotaxane formation provides a method for altering the chemical reactivity of a dumbbell-shaped molecule, without modifying its covalent structure, by locking it through the cavity of a macrocycle. The stability and photophysical behavior of many π-systems have been enhanced using this threading strategy.1−3 This approach is particularly appropriate for controlling the reactivity of extended polyynes and cumulenes, R—(C≡C)n—R and R2C=(C=C)n=CR2, since the only way to covalently modify these π-systems is to change the end groups (R), which becomes increasingly irrelevant as the system becomes longer, with increasing n. Recently, we4 and others5 reported the synthesis of polyyne rotaxanes using an active copper(I) template effect6 with a phenanthroline-based macrocycle.7 Here, we present a broad investigation of the synthesis, structure and properties of this new class of “insulated molecular wires”.
Polyynes have been studied extensively as analogues of carbyne8,9 and because of their unique electronic properties.10,11 Rotaxane formation has often been suggested as a strategy for improving the stability of linear carbon chains,1,8,12,13 but it is only recently that this effect has been demonstrated experimentally in polyynes14 and cumulenes.15 It has also been demonstrated that rotaxane formation can be used to modify the photophysical behavior of polyynes.16
Our strategy for the synthesis of polyyne rotaxanes is based on copper-mediated coupling of terminal oligoynes.17 Similar chemistry has been employed previously to prepare rotaxanes,18−20 catenanes,20−23 knots,24 and rotacatenanes,25 all linked with butadiyne (C4) moieties. Here we present the synthesis of rotaxanes with chains of 8–24 contiguous sp-hybridized carbon atoms and “supertrityl” (Tr*) end groups.26,27 Importantly, the yields of the rotaxanes have been improved by using Cadiot–Chodkiewicz cross-coupling19,22,28 and we have achieved remarkable selectivity by optimizing the coupling partners and the size of the macrocycle. Furthermore, rotaxanes with dibromoethene moieties have been synthesized as “masked” polyynes,29 although we have not yet achieved the reductive Fritsch–Buttenberg–Wiechell rearrangement of these rotaxanes to unmask the polyyne axles. Finally, differential scanning calorimetry confirms the hypothesis that rotaxane formation can indeed stabilize longer polyynes (C16–C24) relative to the corresponding naked polyynes.
The crystal structures of the new rotaxanes provide some insight into the origins of their thermal stability, in that the distances between neighboring polyyne chains are longer than for the corresponding naked polyynes,26 which is a key factor for solid-state polymerization.30 X-ray crystallographic analysis also reveals a wealth of information regarding dispersive interactions between the polyyne chains and the threaded macrocycles, based on a variety of CH/π and π/π interactions. The structure of a [3]rotaxane, with two C12 chains threaded through the same macrocycle, reveals a πsp/πsp interaction between the two hexayne chains, which are arranged in a crossed geometry.
This work establishes the synthetic methodology for preparing polyyne rotaxanes and provides a platform from which to create a broad range of functional carbon-rich materials.
Results and Discussion
Varying the Length of the Polyyne
Polyyne rotaxanes 2b–f·M1, with 8–24 contiguous sp-hybridized carbon atoms, were prepared using an active Cu(I)-template homocoupling strategy (Scheme 1, Table 1).4 In a typical procedure, the 1:1 complex of CuI and the phenanthroline-based macrocycle M1 (CuI·M1) reacted with a slight excess of the terminal polyyne 1b–f in THF at 60 °C in the presence of K2CO3 and iodine. Attempts to prepare the butadiyne-linked rotaxane 2a·M1 from the alkyne 1a were not successful; it seems that the Tr*-capped polyyne products need at least 8 sp-carbon atoms to provide space for the macrocycle. Preparation of a rotaxane with 32 sp-carbon atoms was also unsuccessful, and we observed complete decomposition of octayne 1g in the reaction mixture. This result indicates that the limit for the current coupling conditions has been reached at the stage of 2f·M1 with a C24 dodecayne dumbbell.
Scheme 1. Synthesis of a Series of Supertrityl End-Capped Rotaxanes with M1 Macrocycle via the Homo-Coupling.
Table 1. Summary of Synthesis of Tr*—(C≡C)n—Tr* Rotaxanes via Homo-Coupling (via Scheme 1)a.
| n | starting material | rotaxane product | yielda | reaction timeb |
|---|---|---|---|---|
| 2 | 1a | 2a·M1 | 0 | 48 h |
| 4 | 1b | 2b·M1 | 34% | 48 h |
| 6 | 1c | 2c·M1 | 32% | 24 h |
| 8 | 1d | 2d·M1 | 23% | 40 h |
| 10 | 1e | 2e·M1 | 15% | 36 h |
| 12 | 1f | 2f·M1 | 11% | 16 h |
| 16 | 1g | 2g·M1 | 0 | 48 h |
Yields for isolated rotaxanes based on amount of M1 starting material. Conditions: CuI·M1 (1.0 equiv; 5–10 mM), 1a–g (2.2 equiv), K2CO3 (4 equiv), I2 (1.1 equiv), THF, 60 °C.
Reaction times were judged by TLC.
The product yield for the series of rotaxanes 2b–f·M1 decreased with increasing length of the polyyne (Table 1), probably as a consequence of the lower stability of the longer terminal polyynes. Use of lower reaction temperatures suppresses decomposition of the starting materials, but does not increase the yield of rotaxane. In the synthesis of rotaxane 2f·M1, the reaction was stopped after 16 h, when TLC still showed traces of unreacted 1f, to minimize decomposition of the product. In all cases, the concentration of macrocycle M1 was 5–10 mM; increasing the concentration to 30 mM (for conversion of 1c to 2c·M1) did not noticeably alter the yield.
The copper-catalyzed cross-coupling of acetylenes, first reported by Cadiot and Chodkiewicz,28 has been successfully utilized for the synthesis of polyynes,9b,31 rotaxane-based shuttles19 and catenanes,22 but the reaction had not been explored for the synthesis of polyyne rotaxanes. We chose to test the synthesis of rotaxanes via Cadiot−Chodkiewicz coupling using triyne 1c and the corresponding bromotriyne 3 (prepared by AgNO3-catalyzed bromination of 1c with NBS).32 Equimolar amounts of triyne 1c and bromotriyne 3 were added to a solution of the CuI·M1 complex in THF, and the mixture was stirred under nitrogen at 60 °C. The reaction was complete after 4 h (monitored by TLC), and the corresponding rotaxane 2c·M1 was isolated in 38% yield (Table 2, entry 1). The yield of rotaxane from this cross-coupling reaction is slightly higher than that from homocoupling (32%). In the latter case, a slight excess of 1c (2.5 equiv) was used, thus in the next experiment we tested an excess of both 1c (1.2 equiv) and 3 (1.2 equiv), relative to the macrocycle. The reaction was complete after 4 h, and the rotaxane was isolated in 43% yield (Table 2, entry 2). We repeated this reaction keeping the amount and concentration of 1c constant (1.2 equiv) while varying the amount of 3 (1.3–1.5 equiv) and always obtained the rotaxane in good yields (47–53%, Table 2). Finally, we carried out the reaction at 20 °C using stoichiometric amounts of 1c and 3. The starting materials were consumed after 76 h, giving rotaxane 2c·M1 in 26% yield (Table 2, entry 5), while at 40 °C the reaction was complete after 24 h, and the product yield was 36% (Table 2, entry 4). The synthesis of hexayne rotaxanes via cross-coupling is more effective than homocoupling, and proceeds at lower temperatures (e.g., homocoupling failed at 40 °C). However, cross-coupling is not applicable to the synthesis of long polyyne rotaxanes due to the low stability of halogenated polyyne precursors.
Table 2. Optimization of the Synthesis of 2c·M1 Rotaxane via Cadiot–Chodkiewicz Cross-Couplinga.

| entry | 1c, equiv | 3, equiv | temp. | time | yielda |
|---|---|---|---|---|---|
| 1 | 1.0 | 1.0 | 60 °C | 4 h | 38% |
| 2 | 1.2 | 1.2 | 60 °C | 4 h | 43% |
| 3 | 1.2 | 1.5 | 60 °C | 4 h | 53% |
| 4 | 1.0 | 1.0 | 40 °C | 24 h | 36% |
| 5 | 1.0 | 1.0 | 20 °C | 76 h | 26% |
Yields calculated based on M1 (conc. 10 mM). Reaction conditions: CuI·M1 (1 equiv), K2CO3 (4 equiv), and O2-free THF.
Mechanistically, it is believed that Cadiot–Chodkiewicz coupling proceeds via oxidative addition of the bromoacetylene to a Cu-acetylide producing a Cu(III) intermediate, which undergoes reductive elimination affording the cross-coupled product.17,22,33 However, competing homocoupling of the alkynyl halide is often observed, via halogen-metal exchange,17,28,34 and it has been difficult to achieve high selectivity in cross-coupling reactions of polyynes. In some cases, selectivity can be obtained by careful choice of the amine base, solvent and reagent concentrations,35 or by applying a polymer-supporting technique.36 To test the selectivity of the cross-coupling reaction, the supertrityl diyne 1b (1.0 equiv) and bromotriyne 3 (1.1 equiv) were reacted with CuI·M1 complex (1.0 equiv) under the conditions described above (THF, 60 °C, 12 h). The reaction gave a mixture of two rotaxanes, as confirmed by the 1H NMR and MALDI spectra (Figure S15). The ratio of hexayne and pentayne rotaxanes (4:1) was estimated from 1H NMR spectrum. Thus, homocoupling of the bromotriyne 3 is indeed competitive with the desired heterocoupling reaction.
We envisioned that changing the coupling acetylene partners could provide higher selectivity in cross-coupling,28,34 so the synthesis of porphyrin–polyyne mixed rotaxanes was investigated. Porphyrin rotaxanes represent an important class of molecular machines and photoresponsive assemblies,37 and the macrocycle M1 has previously been used in the active-metal template homocoupling of meso-ethynyl-porphyrins affording porphyrin-capped rotaxanes.20 Thus, porphyrin 4 (1.0 equiv), supertrityl bromotriyne 3 (1.5 equiv) and CuI·M1 complex (1.0 equiv) were reacted in toluene/THF (2:1) under nitrogen at 60 °C to give rotaxane 5a·M1 in 19% yield (Scheme 2). To our surprise, the formation of unthreaded hexayne or bis-porphyrin rotaxanes was not observed by TLC analysis, 1H NMR, or MALDI spectra of the crude reaction mixture.
Scheme 2. Synthesis of Porphyrin–Polyyne [2] and [3]Rotaxanes.
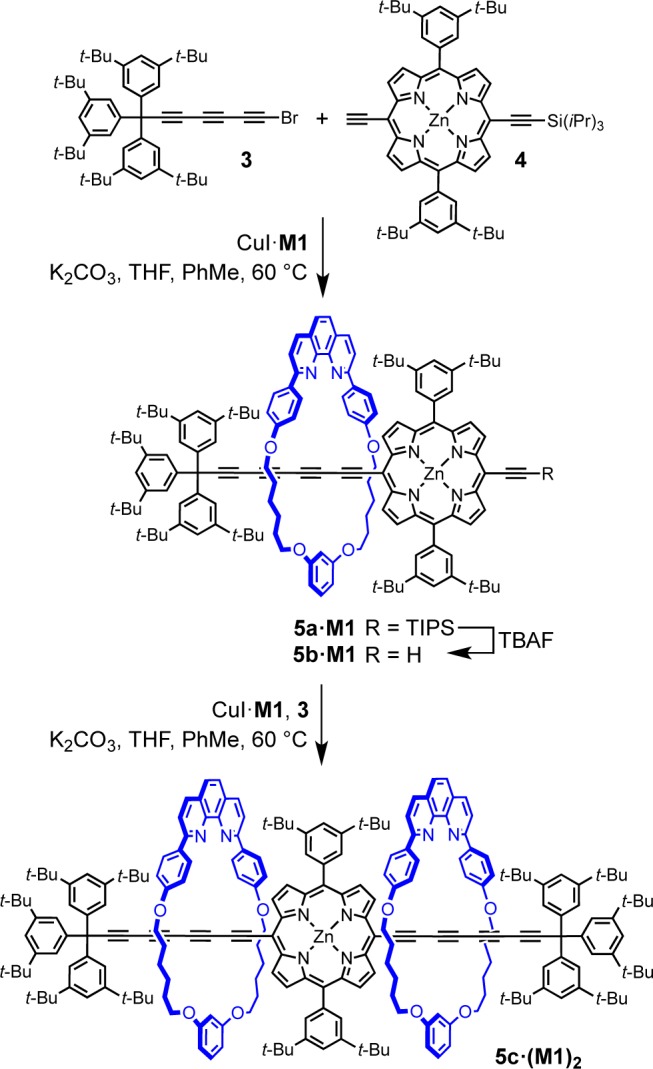
In rotaxane 5a·M1, the second meso position of the porphyrin can serve as a coupling partner after desilylation with TBAF. Thus, rotaxane 5b·M1 was subjected to conditions similar to that for 5a·M1 to give [3]rotaxane 5c·(M1)2 in 23% yield (Scheme 2). Again, the reaction was highly selective, affording the product without formation of 2c·M1 or bis(porphyrin)-capped rotaxanes.
Synthesis of “Masked” Polyyne Rotaxanes
One limitation of preparing rotaxanes by oxidative coupling of terminal and bromo-polyynes is the instability of the starting materials, and this encouraged us to explore alternative synthetic strategies. One approach is based on the use of masked oligoynes, in which the polyyne framework is assembled in a protected form.38 In the final step, the linear carbon chain is constructed via elimination of masking functional groups. Recently, the Fritsch–Buttenberg–Wiechell (FBW) rearrangement of carbene/carbenoid intermediates has evolved into a valuable synthetic methodology for the preparation of polyynes from geminal dihaloolefin-masked acetylene precursors.29 The basis of the FBW method is the treatment of 1,1-dibromo-2,2-dialkynylethenes with n-BuLi, which leads to the in situ formation of a carbenoid species, followed by 1,2-migration to yield the corresponding linear polyyne (Scheme 3).29 We are interested in utilizing gem-dibromoolefins as masked polyynes in the synthesis of rotaxanes. Compound 6a did not undergo homocoupling in the presence of the copper(I) complex of macrocycle M1. However, the reaction of 6a with the bromo-derivative 6b under cross-coupling conditions furnished the rotaxane 7a·M1 in 9% yield (Scheme 4). Increasing the length of the acetylenic axle dramatically improved the yield of this synthesis. Homocoupling of 6c, in the presence of CuI·M1 gave rotaxane 7b·M1 in 78% yield (Scheme 4). However, all attempts at FBW rearrangement of 7b·M1 were unsuccessful, despite testing a range of reaction conditions (Scheme 5 and Table S1); rotaxane 7b·M1 reacts with butyl lithium to give a complex mixture of products. It is not clear why this reaction fails, but it appears that the proximity of the phenanthroline macrocycle adversely affects the reactivity of the carbenoid intermediate, promoting alternative pathways.
Scheme 3. General Mechanism of FBW Rearrangement.
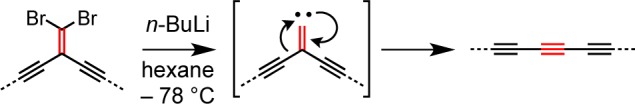
Scheme 4. Synthesis of Rotaxanes 7a·M1 and 7b·M1.
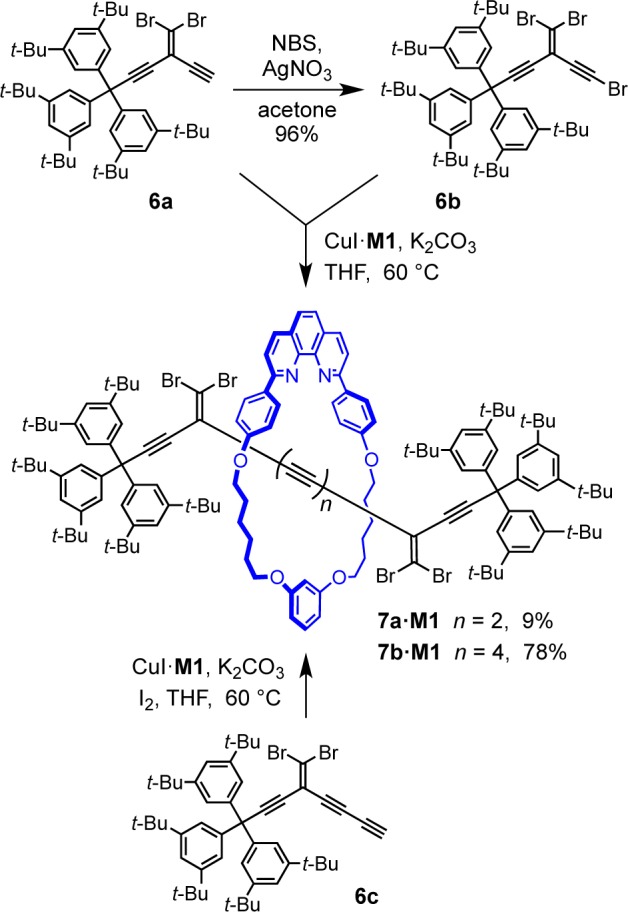
Scheme 5. Attempted Synthesis of Octayne Rotaxane 2d·M1 via FBW Reaction of 7b·M1.
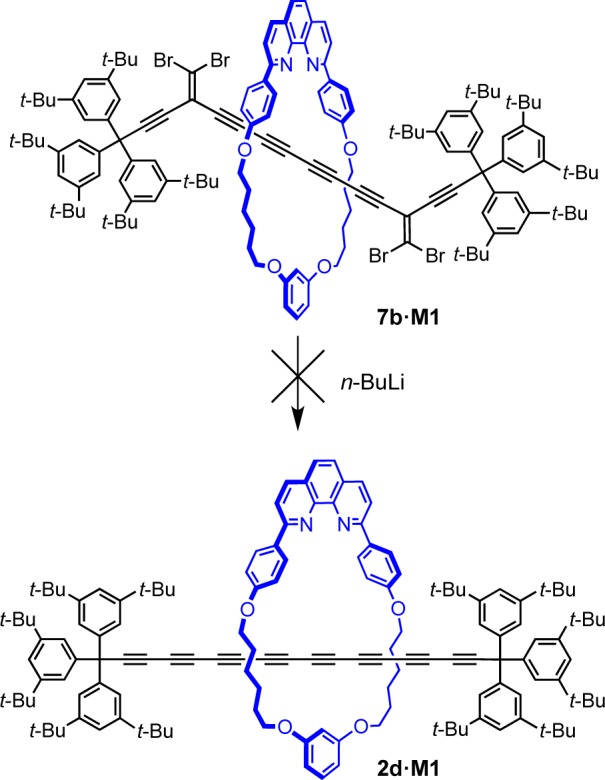
Varying the Size of the Macrocycle
The supertrityl end-group is large enough to prevent slippage of macrocycle M1, and we were interested to test whether it is an effective stopper for even larger macrocycles. However, use of smaller macrocycles is appealing because it should allow the synthesis of polyyne rotaxanes with simpler terminal groups. In many rotaxanes, the macrocycle protects the axle from the external environment,1,2,39 preventing enzymatic digestion,40 or chemical degradation,41 and in these cases a “tight” fit between macrocycle and thread is desirable. We tested a variety of macrocycles for the synthesis of hexayne rotaxanes (Scheme 6 and Table 3). The triyne 1c and bromotriyne 3 were chosen for rotaxane synthesis since a hexayne axle is long enough to eliminate steric interactions with the supertrityl end-group. The size of macrocycle M1 can easily be adjusted by changing the length of the alkyl bridge. Decreasing the size from C6 (M1) to C3 (M2) or C4 (M3) worked well, yielding hexayne rotaxanes 2c·M2 and 2c·M3 under cross-coupling conditions in 21% and 43% yields, respectively. Under homocoupling conditions, the same macrocycle gave lower yields and the rotaxane 2c·M2 was isolated in only 5% yield. A macrocycle with a C8 linker (M4)7 gave rotaxane 2c·M4 in 41% yield under cross-coupling (9% from homocoupling) but no rotaxane was isolated with the larger C10 macrocycle (M5),7 probably because this macrocycle slips off the dumbbell, as indicated by inspection of CPK models.
Scheme 6. Synthesis of Hexayne Rotaxanes with Different Macrocycles.
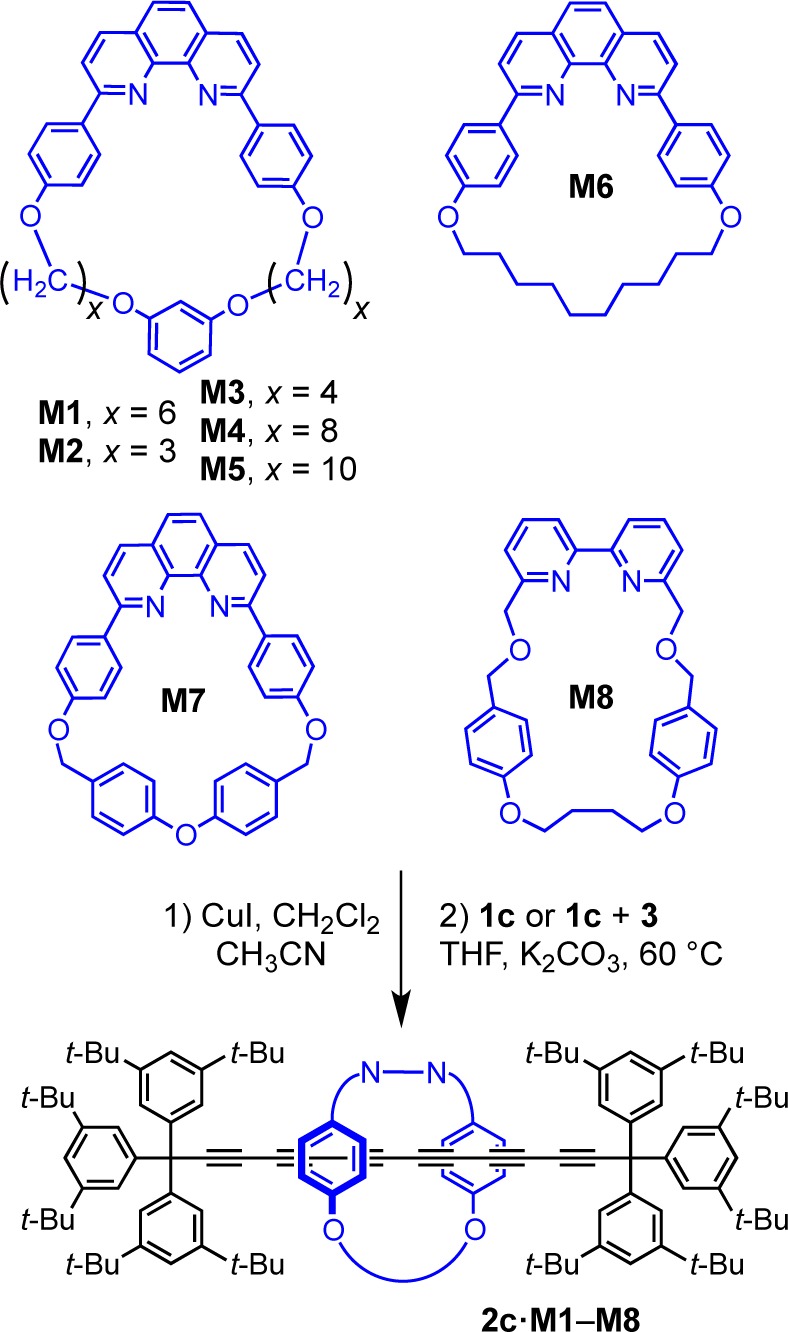
Table 3. Summary of the Syntheses of Rotaxanes 2c·M2–M8 Using Different Reaction Conditions (via Scheme 6).
| yield (reaction time)c |
|||
|---|---|---|---|
| macrocycle | rotaxane | homoa | crossb |
| M2 | 2c·M2 | 5% (50 h) | 21% (11 h) |
| M3 | 2c·M3 | 28% (48 h) | 43% (8 h) |
| M4 | 2c·M4 | 9% (42 h) | 41% (18 h)d |
| M5 | 2c·M5 | 0% (48 h) | 0% (48 h) |
| M6 | 2c·M6 | 17% (48 h) | 27% (6 h) |
| M7 | 2c·M7 | 23% (62 h) | 54% (12 h) |
| M8 | 2c·M8 | 23% (48 h) | 27% (4 h) |
Homocoupling reaction conditions: CuI·M, 1c (2.5 equiv), I2, K2CO3, THF, 60 °C.
Cross-coupling reaction conditions: CuI·M, 1c (1.1 equiv), 3 (1.5 equiv), K2CO3, THF, 60 °C.
Yields are calculated referring to macrocycles (c = 5–10 μM).
Reaction temperature: 50 °C.
The phenanthroline-based macrocycle M6 with a C10 alkyl strap gave the corresponding rotaxane 2c·M6 in 17% and 26% yields from homo- and cross-coupling, respectively. The macrocycle M7(15,42) with a more rigid p-tolyl ether framework gave 2c·M7 in 23% and 54% yield, for homo- and cross-coupling respectively (Table 3). Finally, a bipyridine-based macrocycle43 gave rotaxane 2c·M8 in 23% and 26% yield, from homo- or cross-coupling, respectively. Generally, small macrocycles reduce the yield of rotaxanes in both homo- and cross-coupling, except in the case of M7. This contrasts with the results obtained for the synthesis of rotaxanes via active template copper-catalyzed azide–alkyne cycloaddition reaction, where smaller macrocycles afforded better yields.43
Synthesis of [3]Rotaxanes with Two Threaded Polyyne Chains
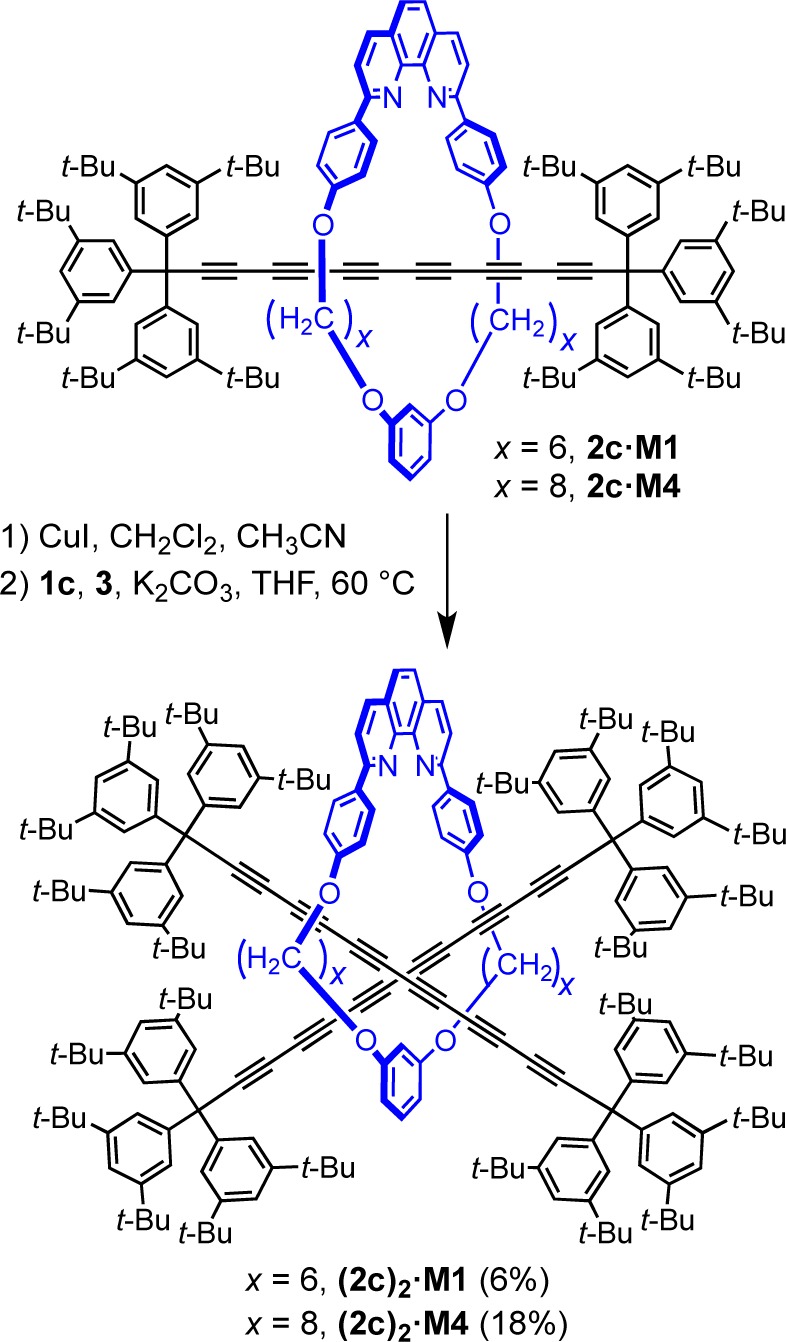
Rotaxanes with multiple axles passing through a single ring are very rare.44 The challenge in preparing this type of molecular architecture is to satisfy the structural demands of both components: The macrocycle may require more than one template site to assemble multiple-threads, and its cavity must be large enough to accommodate two axles, yet small enough to prevent the dethreading. Additionally, the macrocycle-thread interactions that direct the assembly process, must overcome steric hindrance between crowded dumbbell units. So far, single-macrocycle threaded [3]rotaxanes have been synthesized utilizing hydrophobic interactions,44a octahedral metal centers as templates,44b,44c hydrogen-bond formation between a thread and axles,44d and an active-metal templated acetylene homocoupling.44f The axles can be identical44b−44e or different44a and can be assembled using the same or different type of reactions for each step of the threading.
To test the threading of two identical polyynes, the CuI·2c·M1 stoichiometric complex (1.0 equiv) was prepared and mixed with 1c (1.2 equiv) and 3 (1.6 equiv) in THF (Scheme 7). The oxygen-free reaction mixture was stirred for 36 h at 60 °C in the dark. After workup followed by silica and size-exclusion chromatography, the [3]rotaxane (2c)2·M1 was obtained in 6% yield, and 70% of the 2c·M1 rotaxane starting material was recovered. The product was characterized by mass spectrometry, NMR and UV–vis absorption spectroscopy, and the structure of the molecule was determined by X-ray crystallography. The [3]rotaxane (2c)2·M1 is stable as a crystalline solid at 4 °C, while at ambient conditions the yellow solid darkens slowly over weeks. To improve the yield of the double-threaded product, a larger macrocycle was tested, to reduce steric crowding. We were pleased to find that under similar reaction conditions, rotaxane 2c·M4, with a larger macrocycle, yields the corresponding [3]rotaxane (2c)2·M4 in 18% (Scheme 7). The stability of (2c)2·M4 is comparable to that of (2c)2·M1, and the solid discolors slowly under ambient conditions, but is stable indefinitely at −20 °C.
Scheme 7. Synthesis of (2c)2·M1 and (2c)2·M4 Polyyne [3]Rotaxanes via Cross-Coupling.
Spectroscopic Characterization of Rotaxanes
The polyyne rotaxanes were characterized by MALDI mass spectrometry, UV–vis absorption, and NMR spectroscopy. Threading does not significantly perturb the electronic structure of polyynes and the absorption spectra of the rotaxanes resemble the sum of the macrocycle and polyyne absorptions.4,26 For example, the absorption spectra of the rotaxanes 2b–f·M1 (Figure 1a) are essentially the sum of the spectra of their components, 2b–f and M1. The polyyne absorption bands in the rotaxanes are shifted to lower energy by about 4 nm, compared to the unthreaded analogs26 (Figure 1b).
Figure 1.
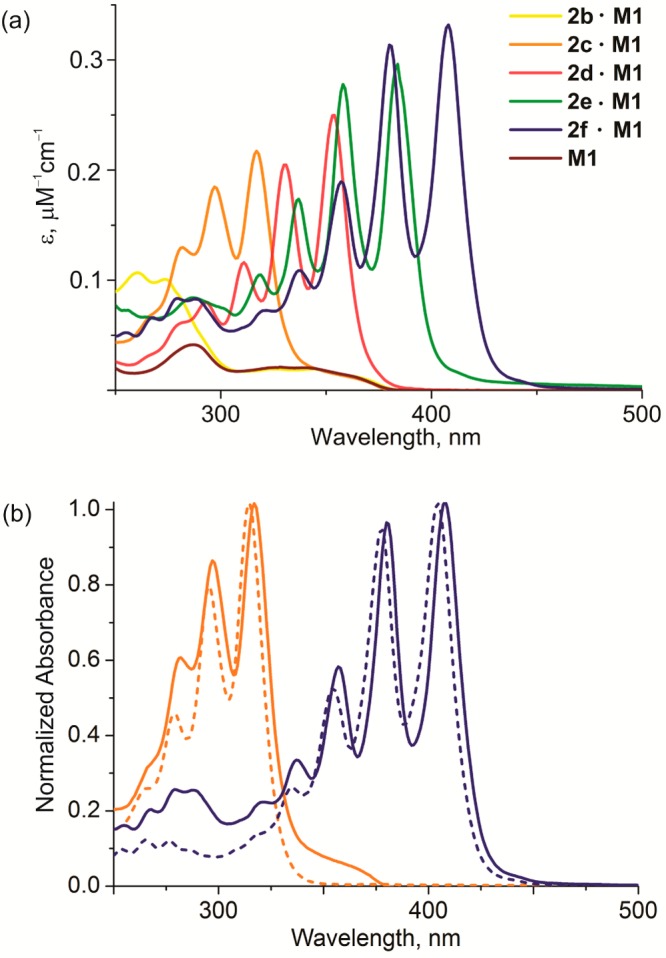
(a) The UV–vis absorption spectra of macrocycle M1 (brown line) and rotaxanes 2b–f·M1 in dichloromethane. (b) Normalized (at the highest absorption band) absorption spectra of 2c·M1 (orange) and 2f·M1 (blue) rotaxanes with their corresponding unthreaded polyynes (dashed lines) in dichloromethane.
In double-threaded [3]rotaxanes, the absorption in the polyyne region is double that of the parent rotaxanes, as expected. Normalization of the spectra of rotaxanes 2c·M1 and (2c)2·M1 at the absorption maximum (317 nm) shows that the second vibronic band at ∼297 nm is red-shifted by 1 nm and is slightly more intense in the [3]rotaxane (Figure 2).
Figure 2.
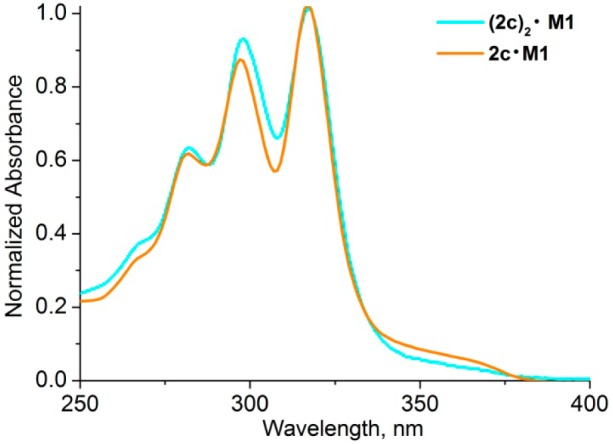
Comparison of absorption spectra of rotaxanes 2c·M1 and (2c)2·M1 (in dichloromethane) normalized at the absorption maximum at 317 nm.
The 1H NMR spectra of rotaxanes 2b–f·M1 reveal that the interactions between the supertrityl end groups and the macrocycle become weaker as the polyyne becomes longer (Figure 3). Upon threading, the chemical shift of proton Hf of the macrocycle resorcinol moiety increases, while those of protons Hh and Hg decrease. As the length of the polyyne chain is increased, these changes become insignificant. Thus, in rotaxane 2f·M1, resonances Hf, Hg, and Hh become almost identical to those from free macrocycle M1. The aromatic protons of the supertrityl end-group also move to higher chemical shift in response to polyyne elongation (H1 and H2, Figure 3).
Figure 3.
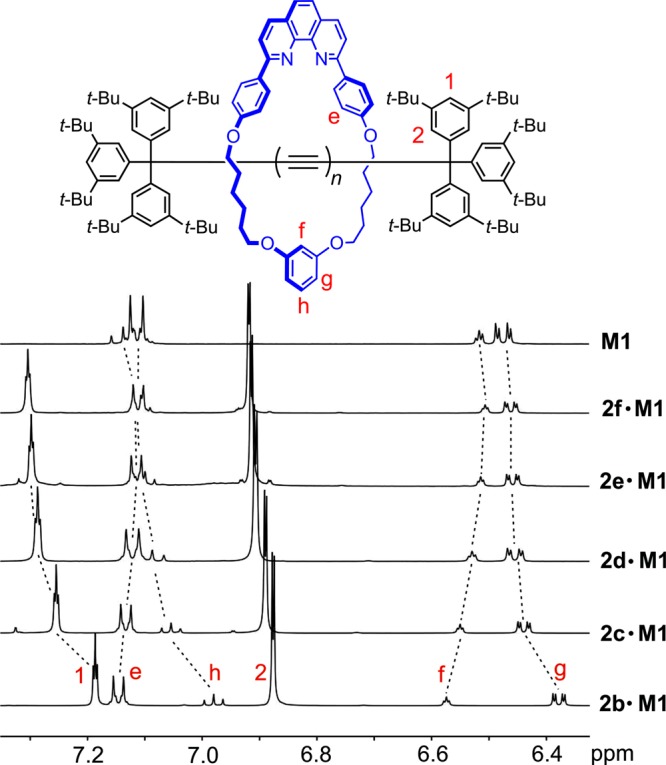
Partial 1H NMR spectra of rotaxanes 2b–f·M1, compared with that of the M1 macrocycle (CD2Cl2, 500 MHz, 298 K).
In the 1H NMR spectrum of the [3]rotaxane (2c)2·M1, protons labeled Hd, Hb, He, Hh, Hg, Hj, and Hi move to lower chemical shift, while the chemical shift of proton Hc increases, compared with the corresponding [2]rotaxane 2c·M1 (Figure 4). Resonances of the protons H1 and H2 from supertrityl group are unaffected. The two hexayne chains in (2c)2·M1 are undistinguishable by 1H and 13C NMR spectroscopy in CD2Cl2, both at 298 K and at 193 K (Figures S16 and S17).
Figure 4.
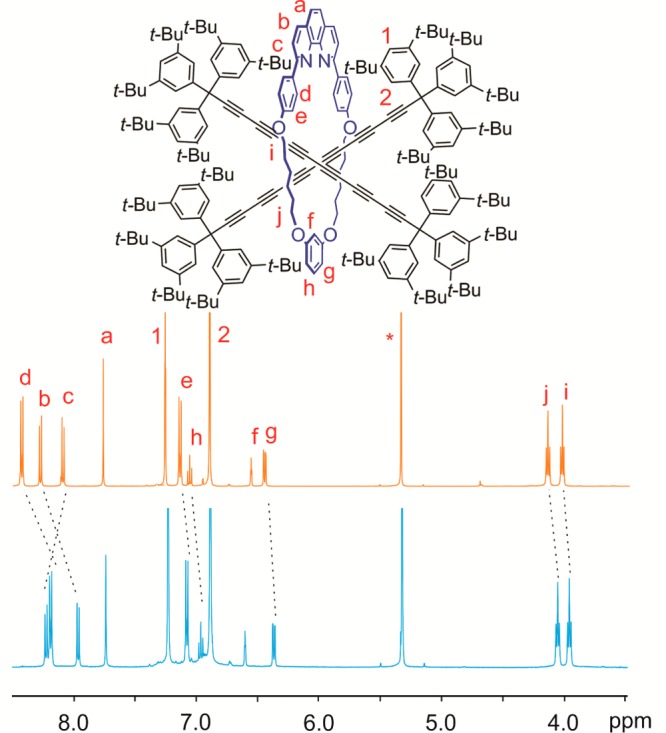
Comparison of 1H NMR spectra of rotaxanes 2c·M1 (orange) and (2c)2·M1 (blue). Asterisk denotes the solvent peak (500 MHz, CD2Cl2, 298 K).
Thermal Stability
It was hypothesized decades ago that mechanical encapsulation via rotaxination should stabilize extended polyynes.8,12,13 While remarkable stabilization for cumulene rotaxanes was demonstrated recently,15 the thermal stability of polyyne rotaxanes was a key aspect that we sought to address in this study. The thermal stabilities of rotaxanes 2c–f·M1 and the corresponding polyynes 2c–f were compared by differential scanning calorimetry (DSC), by heating the samples to 400 °C at 10 °C/min under an atmosphere of nitrogen (Table 4), and observing the exothermic thermal decomposition. Naked polyyne dumbbells show a sharp decrease in thermal stability with increasing chain length,26 whereas there is less change in stability for the rotaxanes.
Table 4. Comparison of the Decomposition Temperature (Peak) of Tr*—(C≡C)n—Tr* Polyyne Rotaxanes and Corresponding Free Polyynes from DSC Analysis.
| decomposition
temperature |
||
|---|---|---|
| n | rotaxane | polyyne26 |
| 6 | 2c·M1, 287 °C | 2c, 323 °C |
| 8 | 2d·M1, 291 °C | 2d, 275 °C |
| 10 | 2e·M1, 234 °C | 2e, 220 °C |
| 12 | 2f·M1, 228 °C | 2f, 168 °C |
Rotaxane 2c·M1 decomposes at a lower temperature than the bare hexayne 2c,26 which is probably because the rotaxane melts (melting point: 216 °C; dec 287 °C), whereas the dumbbell decomposes without melting (dec 323 °C). Here the threaded macrocycle serves to reduce the symmetry of the polyyne and to disrupt crystal-packing interactions, reducing the melting point and consequently reducing the thermal stability. However, with further increase of the polyyne length, all of the compounds undergo thermal decomposition without melting and the rotaxanes become more stable than the corresponding naked dumbbells. The difference in stability increases with increasing polyyne chain length. The greatest enhancement in thermal stability is observed for 2f·M1, as DSC shows a decomposition peak at 228 °C, while for the free dumbbell 2f, the decomposition peak occurs at 168 °C (Figure 5). As the polyyne chain gets longer, steric shielding of the carbon chain by the supertrityl groups decreases, providing an opportunity for the macrocycle to act as an additional shield and suppress the polyyne degradation.
Figure 5.
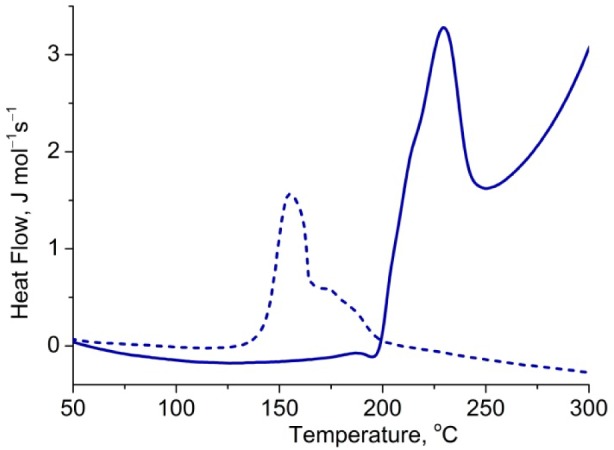
DSC traces of dodecayne 2f (dash line) and the corresponding rotaxane 2f·M1 (solid line). Heating: 10 °C/min.
X-ray Crystallography
Here we report crystal structures45 of polyyne rotaxanes 2c·M2, 2c·M6, 2c·M7 and 2d·M1, dibromolefin rotaxane 7a·M1, porphyrin rotaxane 5a·M1, and [3]rotaxanes 5c·(M1)2 and (2c)2·M1. Full crystallographic details regarding crystal growth, data collection, analysis, and crystal packing are given in the SI. Crystallographic data (excluding structure factors) have been deposited with the Cambridge Crystallographic Data Centre (CCDC 1437276–1437283) and can be obtained via www.ccdc.cam.ac.uk/data_request/cif. The crystal structures of rotaxane 2c·M1 and the free hexayne 2c have been published previously4,26 and are included in the discussion for comparison. Selected parameters are summarized in Table 5.
Table 5. Summary of Crystallographic Data of 2c·M1, 2c·M2, 2c·M6, 2c·M7, and 2d·M1 Rotaxanesa.
| compound | avg. ∠Csp—C≡C (deg) | ∠φ (deg)b | BLA (Å) | avg. Csp—Csp (Å) | avg. C≡C(Å) | ref. |
|---|---|---|---|---|---|---|
| 2c·M1 | 177.8(11) | 171.8(1) | 0.143(9) | 1.357(5) | 1.214(5) | (4) |
| 2c·M2 | 177.6(14) | 174.1(1) | 0.148(14) | 1.358(9) | 1.207(8) | c |
| 2c·M6 | 174.7(21) | 168.1(1) | 0.160(8) | 1.343(15) | 1.219(16) | c |
| 2c·M7 | 175.8(20) | 164.5(1) | 0.140(15) | 1.355(6) | 1.211(7) | c |
| 2d·M1 | 177.0(19) | 172.0(1) | 0.143(11) | 1.356(7) | 1.211(5) | c |
| 2c | 177.0(14) | 180d | 0.143(8) | 1.359(5) | 1.208(7) | (26) |
For comparison data for 2c·M1 rotaxane and free hexayne 2c also are presented.
φ is the angle between two terminal sp-carbons and the centroid of the central C—C bond of polyyne chain.
This work.
2c occupies a position across a crystallographic inversion center resulting in φ = 180°.
X-ray crystallography studies of polyynes provide insights into the structures of these molecules and into their noncovalent interactions.46 For example, an analysis of the structures of a series of t-butyl-capped polyynes47 indicated that infinitely long polyynes reach a saturation in the bond-length-alternation (BLA), which implies that carbyne has alternating single and triple bonds.48 This result is in agreement with data from the electronic absorption spectra of Tr*-capped polyynes which predict a finite optical bandgap (Eg = 2.56 eV) for carbyne.26
It is expected that threading will affect the conformation and packing of a polyyne. For example, the unthreaded hexayne 2c is centrosymmetric,26 whereas in all hexayne rotaxanes the macrocycle breaks the symmetry. Thus, the hexayne chain in 2c·M2 is slightly bent in a helical shape and the average C≡C—C(sp) angle is 177.6(14)°. Similarly, in rotaxanes 2c·M6 and 2c·M7 the hexayne axles are semihelical and S-shaped, with average C≡C—C(sp) angles of 175(2)° and 176(2)°, respectively. In 2d·M1, the octayne chain is slightly curved in a helical fashion and the average C≡C—C(sp) angle is 177.0(19)° (Figure 6). Only three X-ray structures of octaynes have been reported previously.9a,47,49
Figure 6.

X-ray crystal structures of rotaxanes with noncovalent interactions (green lines) between macrocycle and dumbbell. (a) 2d·M1; d(CH/Csp): a: 2.738 Å; b: 2.839 Å; c: 2.828 Å. (b) 2c·M6; d(CH/Csp): a: 2.813 Å; b: 2.670 Å; c: 2.817 Å; d: 2.827 Å. (c) 2c·M7 with highlighted Carene/Csp contacts: d(Carene/Csp): a: 3.276 Å; b: 3.193 Å; c: 3.296 Å; d: 3.307 Å; e: 3.373. (d) 2c·M2 with highlighted Carene/Csp contacts: a: 3.26 Å; b: 3.40 Å; c: 3.36 Å. (e) (2c)2·M1 [3]rotaxane. (f) Carbon–carbon bond lengths and BLA values of hexayne chains in (2c)2·M1 (A and B) and 2c·M1. Errors are estimated at 3σ probability. Unrelated hydrogen atoms and solvents are omitted for clarity.
The angle between two terminal sp-carbon atoms and the centroid of the central C—C bond of the polyyne chain (φ) is a useful parameter for quantifying the bending of a polyyne (Table 5).16 In this family of polyyne rotaxanes, the axle in 2c·M7 exhibits the most bending with φ = 165(1)°, however, 2c·M6 has the smallest average C≡C–C(sp) angle (175(2)°). All the polyyne [2]rotaxanes have mean BLAs of approximately 0.14–0.15 Å (Table 5), with the exception of 2c·M6 (BLA = 0.160(8) Å); it is not clear whether the unusually high BLA in 2c·M6 is a result of conformational adjustment due to the threaded macrocycle or other effects (such as packing interactions).
The cylindrical π-systems of polyynes can become involved in CH/πsp interactions. Traditionally, CH/πarene interactions have received more attention due to their abundance in biology,50 whereas CH/πsp interactions are rarely discussed,51 and the few existing reports focus on terminal alkynes rather than polyynes. We have analyzed the intermolecular CH/πsp and πarene/πsp interactions in polyynes having four or more triple bonds from the Cambridge Structural Database (CSD),52 and herein we compare these data with results for polyyne rotaxanes. The following discussion of CH/πsp interactions offers, to the best of our knowledge, the first rigorous analysis of this secondary bonding motif.
The polyyne π-systems and macrocycles interact in the solid state via CH/Csp and C/Csp short contacts (Figure 6). The phenoxyl groups of the macrocycles interact with the polyyne π-system and the CH/Csp distances are within the range of values found in CSD (2.65–2.75 Å; see SI for details). Additionally, in rotaxane 2c·M6, the alkyl strap of the macrocycle makes weak contacts with the polyyne π-system (c and d contacts in Figure 6b). In 2c·M7, the shortest CH/Csp distance is 2.702 Å, which is significantly shorter than the mean value found for tetraynes and longer homologues (2.82(7) Å, SI). In addition to the many CH/πsp interactions, the phenoxyl groups of the compact macrocycle M7 interact with the hexayne π-system through πarene/πsp interactions (Figure 6c). A search of the CSD revealed that πarene/πsp interactions are rare in comparison to CH/πsp interactions (14 vs 120 observed short contacts, respectively). Interestingly, the closest Carene/Csp contact in 2c·M7 (Figure 6c: 3.193 Å) is the shortest distance compared to other molecules found in the CSD (mean value for Carene/Csp contacts: 3.36(4) Å). Once the macrocycle cavity becomes smaller (M7, M2) the Carene/Csp interactions become significant, in addition to CH/Csp contacts (Figure 6).
Another consequence of mechanical encapsulation is the larger distances between neighboring polyynes. For instance, the inter-sp-chain distance for 2c·M7 is about 9.7 Å, for 2c·M6 it is 11.6 Å and for 2c·M2 it is 12.9 Å, much longer than the shortest distance between neighboring molecules of free hexayne 2c (8.1 Å).26 These distances are far from that required for topochemical polymerization of the polyynes (ca. 4 Å).30 While this result is not surprising for rotaxinated polyynes, the crystallographic data clearly illustrate the protective role of the threaded macrocycles.
In the [3]rotaxane (2c)2·M1, two hexayne chains are arranged in a crossed geometry (Figure 6e), and the macrocycle M1 sits around the middle of the two hexayne chains (designated as A and B). The angle between chains A and B is 74° (measured as a torsional angle between the quaternary sp3 carbons of the Tr* end groups and the centroids between pairs of these quaternary carbon atoms). In the solid state, the molecule is chiral, due to the helical arrangement of the two hexayne chains inside the cavity of the macrocycle, but the crystal is racemic (P-1). The closest contact between the two hexayne chains is 3.290 Å (C6A–C6B), which is less than the sum of van der Waals radii of two sp-carbon atoms (3.4 Å).53 Few crystal structures with πsp/πsp interaction are known54 and in all cases the alkyne chains adopt a crossed alignment, which may prevent significant orbital overlap between the two close-lying alkynes, conferring stability to the compounds.
The two hexayne chains in (2c)2·M1 have the same curvature within error: the average ∠C—C≡C angles are 176(2)° and 176(3)° for chains A and B, respectively. In chain A, the average BLA is 0.150(11) Å and in chain B it is 0.175(44) Å, which is surprisingly high (Figure 6f). As a general rule, in extended polyynes, BLA gradually decreases toward the middle of the chain.47 For example, in rotaxane 2c·M1 the C≡C triple bonds get longer and the single C—C bonds get shorter toward the middle of the chain, as depicted in Figure 6f (red line). In (2c)2·M1 both chains deviate from this trend, especially C—C single bonds at C6, the position where polyyne chains form a van der Waals contact. This aberration is clearly reflected in BLA values of A and B chains of (2c)2·M1 (Figure 6f). To our knowledge, this is the first time that such a long single bond has been found in the middle of a hexayne chain.
In rotaxane 7a·M1, the macrocycle sits on top of one dibromoethene moiety, while the second dibromoethene moiety points in the opposite direction with a slight twist (torsion angle 141.85(8)°, Figure 7a). Both central triple bonds have a similar length (1.202 Å), and the length of the single bond between them is 1.39 Å. The macrocycle interacts with the axle through several CH/π short contacts formed between the π-system of the axle and alkyl chain of the macrocycle. In addition, a bromine atom of the second dibromoethene moiety forms van der Waals contacts with the aromatic π system of the resorcinol part of the macrocycle (Figure 7a).
Figure 7.
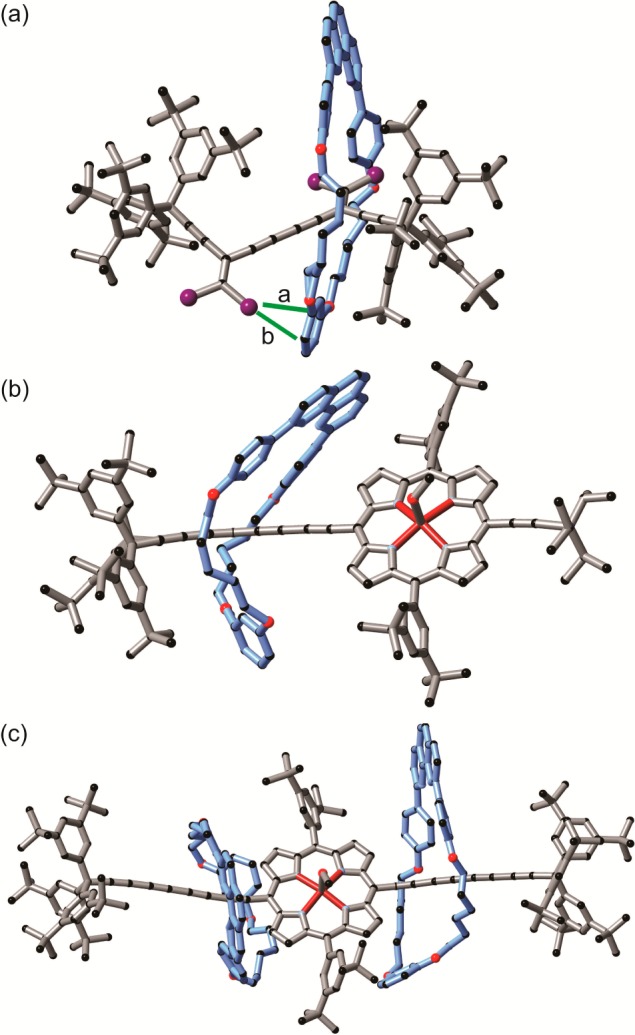
X-ray crystal structure of rotaxanes 7a·M1 (a), 5a·M1 (b), and 5c·(M1)2 (c). Caryl/Br short contacts in 7a·M1 are highlighted with green lines. a: 3.33 Å; b: 3.37 Å.
In the porphyrin rotaxanes 5a·M1 and 5c·(M1)2, one molecule of methanol is coordinated to the Zn center (Figure 7b,c). In both cases, the tetrayne chains are slightly arced, with an average ∠C—C≡C angle of 176.3(13)° in 5a·M1.
Conclusions
We have demonstrated that polyyne rotaxanes, with up to 24 sp-hybridized carbon atoms in the axle, can be prepared by active-metal templating using a variety of macrocycles. Through DSC analysis, we showed that the macrocycle in polyyne rotaxanes mechanically protects the carbon chain, proving the protective effect of molecular encapsulation and offering a valuable design motif toward the future study of carbyne analogs. It is amazing that the dodecayne rotaxane 2f·M1 is stable to >220 °C. The utilization of small macrocycles in polyyne rotaxane synthesis allows the mechanical insulation of polyynes with smaller, functionally diverse end-groups. We have shown that Cadiot–Chodkiewicz cross-coupling of polyynes is a suitable strategy for the preparation of topologically complex polyyne rotaxanes. We prepared polyyne rotaxanes “masked” with gem-dibromoethene moieties. We were conscious that the gem-dibromoethene moieties themselves could act as stoppers for miniature threaded macrocycles, reducing the necessity of bulky end-groups. However, despite efforts, we have not yet been able to carry out the final carbenoid rearrangement of the dibromoethene groups in the rotaxinated systems. Through a number of X-ray crystallographic structure determinations, we detail a plethora of intermolecular noncovalent interactions within the mechanical bond of the rotaxanes. The character of the polyyne and macrocycle interaction depends on the structure and the size of the macrocycle. Interestingly, these intermolecular interactions significantly perturb the BLA in the polyyne chains of the [3]rotaxane (2c)2·M1.
Acknowledgments
We thank Steve M. Goldup (University of Southampton) for providing a sample of macrocycle M8 and Fabian Fritze (University of Erlangen) for help with optimization of cross-coupling conditions. We thank the European Research Council (grant 320969) for support, and the EPSRC UK Mass Spectrometry Facility at Swansea University for mass spectra. L.D.M. acknowledges an Oxford University Raffy Manoukian scholarship. Funding is gratefully acknowledged from the University of Erlangen-Nuremberg, the Deutsche Forschungsgemeinschaft (DFG–SFB 953, “Synthetic Carbon Allotropes” and DFG grant “Rotaxane Protected Polyynes and Cumulenes).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.5b12049.
The authors declare no competing financial interest.
Supplementary Material
References
- a Anderson S.; Anderson H. L. Angew. Chem., Int. Ed. Engl. 1996, 35, 1956–1959. 10.1002/anie.199619561. [DOI] [Google Scholar]; b Cacialli F.; Wilson J. S.; Michels J. J.; Daniel C.; Silva C.; Friend R. H.; Severin N.; Samorì P.; Rabe J. P.; O’Connell M. J.; Taylor P. N.; Anderson H. L. Nat. Mater. 2002, 1, 160–164. 10.1038/nmat750. [DOI] [PubMed] [Google Scholar]; c Frampton M. J.; Anderson H. L. Angew. Chem., Int. Ed. 2007, 46, 1028–1064. 10.1002/anie.200601780. [DOI] [PubMed] [Google Scholar]
- a Arunkumar E.; Forbes C. C.; Smith B. D. Eur. J. Org. Chem. 2005, 2005, 4051–4059. 10.1002/ejoc.200500372. [DOI] [Google Scholar]; b Arunkumar E.; Fu N.; Smith B. D. Chem. - Eur. J. 2006, 12, 4684–4690. 10.1002/chem.200501541. [DOI] [PubMed] [Google Scholar]; c Baumes J. M.; Gassensmith J. J.; Giblin J.; Lee J.-J.; White A. G.; Culligan W. J.; Leevy W. M.; Kuno M.; Smith B. D. Nat. Chem. 2010, 2, 1025–1030. 10.1038/nchem.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terao J.; Tanaka Y.; Tsuda S.; Kambe N.; Taniguchi M.; Kawai T.; Saeki A.; Seki S. J. Am. Chem. Soc. 2009, 131, 18046–18047. 10.1021/ja908783f. [DOI] [PubMed] [Google Scholar]
- Movsisyan L. D.; Kondratuk D. V.; Franz M.; Thompson A. L.; Tykwinski R. R.; Anderson H. L. Org. Lett. 2012, 14, 3424–3426. 10.1021/ol301392t. [DOI] [PubMed] [Google Scholar]
- a Weisbach N.; Baranová Z.; Gauthier S.; Reibenspies J. H.; Gladysz J. A. Chem. Commun. 2012, 48, 7562–7564. 10.1039/c2cc33321j. [DOI] [PubMed] [Google Scholar]; b Sahnoune H.; Baranová Z.; Bhuvanesh N.; Gladysz J. A.; Halet J.-F. Organometallics 2013, 32, 6360–6367. 10.1021/om400709q. [DOI] [Google Scholar]; c Baranová Z.; Amini H.; Bhuvanesh N.; Gladysz J. A. Organometallics 2014, 33, 6746–6749. 10.1021/om501026u. [DOI] [Google Scholar]
- Crowley J. D.; Goldup S. M.; Lee A. L.; Leigh D. A.; McBurney R. T. Chem. Soc. Rev. 2009, 38, 1530–1541. 10.1039/b804243h. [DOI] [PubMed] [Google Scholar]
- Saito S.; Nakazono K.; Takahashi E. J. Org. Chem. 2006, 71, 7477–7480. 10.1021/jo060829h. [DOI] [PubMed] [Google Scholar]
- Diederich F. Nature 1994, 369, 199–207. 10.1038/369199a0. [DOI] [Google Scholar]
- a Eisler S.; Slepkov A. D.; Elliott E.; Luu T.; McDonald R.; Hegmann F. A.; Tykwinski R. R. J. Am. Chem. Soc. 2005, 127, 2666–2676. 10.1021/ja044526l. [DOI] [PubMed] [Google Scholar]; b Chalifoux W. A.; Tykwinski R. R. C. R. Chim. 2009, 12, 341–358. 10.1016/j.crci.2008.10.004. [DOI] [Google Scholar]; c Tykwinski R. R. Chem. Rec 2015, 15, 1060–1074. 10.1002/tcr.201500018. [DOI] [PubMed] [Google Scholar]
- a Wang C.; Batsanov A. S.; Bryce M. R.; Martin S.; Nichols R. J.; Higgins S. J.; García-Suárez V. M.; Lambert C. J. J. Am. Chem. Soc. 2009, 131, 15647–15654. 10.1021/ja9061129. [DOI] [PubMed] [Google Scholar]; b Moreno-García P.; Gulcur M.; Manrique D. Z.; Pope T.; Hong W.; Kaliginedi V.; Huang C.; Batsanov A. S.; Bryce M. R.; Lambert C.; Wandlowski T. J. Am. Chem. Soc. 2013, 135, 12228–12240. 10.1021/ja4015293. [DOI] [PubMed] [Google Scholar]; c Gulcur M.; Moreno-García P.; Zhao X.; Baghernejad M.; Batsanov A. S.; Hong W.; Bryce M. R.; Wandlowski T. Chem. - Eur. J. 2014, 20, 4653–4660. 10.1002/chem.201304671. [DOI] [PubMed] [Google Scholar]
- Cretu O.; Botello-Mendez A. R.; Janowska I.; Pham-Huu C.; Charlier J.-C.; Banhart F. Nano Lett. 2013, 13, 3487–3493. 10.1021/nl4018918. [DOI] [PubMed] [Google Scholar]
- Simpkins S. M. E.; Kariuki B. M.; Cox L. R. J. Organomet. Chem. 2006, 691, 5517–5523. 10.1016/j.jorganchem.2006.07.021. [DOI] [Google Scholar]
- Huuskonen J.; Buston J. E. H.; Scotchmer N. D.; Anderson H. L. New J. Chem. 1999, 23, 1245–1252. 10.1039/a906403f. [DOI] [Google Scholar]
- Schrettl S.; Contal E.; Hoheisel T.; Fritzsche M.; Balog S.; Szilluweit R.; Frauenrath H. Chem. Sci. 2015, 6, 564–574. 10.1039/C4SC03154G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franz M.; Januszewski J. A.; Wendinger D.; Neiss C.; Movsisyan L. D.; Hampel F.; Anderson H. L.; Görling A.; Tykwinski R. R. Angew. Chem., Int. Ed. 2015, 54, 6645–6649. 10.1002/anie.201501810. [DOI] [PubMed] [Google Scholar]
- Movsisyan L. D.; Peeks M. D.; Greetham G. M.; Towrie M.; Thompson A. L.; Parker A. W.; Anderson H. L. J. Am. Chem. Soc. 2014, 136, 17996–18008. 10.1021/ja510663z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siemsen P.; Livingston R. C.; Diederich F. Angew. Chem., Int. Ed. 2000, 39, 2632–2657. 10.1002/1521-3773(20000804)39:15<2632::AID-ANIE2632>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Saito S.; Takahashi E.; Nakazono K. Org. Lett. 2006, 8, 5133–5136. 10.1021/ol062247s. [DOI] [PubMed] [Google Scholar]
- Berná J.; Goldup S. M.; Lee A.-L.; Leigh D. A.; Symes M. D.; Teobaldi G.; Zerbetto F. Angew. Chem., Int. Ed. 2008, 47, 4392–4396. 10.1002/anie.200800891. [DOI] [PubMed] [Google Scholar]
- Langton M. J.; Matichak J. D.; Thompson A. L.; Anderson H. L. Chem. Sci. 2011, 2, 1897–1901. 10.1039/c1sc00358e. [DOI] [Google Scholar]
- Dietrich-Buchecker C. O.; Khemiss A.; Sauvage J.-P. J. Chem. Soc., Chem. Commun. 1986, 1376–1378. 10.1039/c39860001376. [DOI] [Google Scholar]
- Goldup S. M.; Leigh D. A.; Long T.; McGonigal P. R.; Symes M. D.; Wu J. J. Am. Chem. Soc. 2009, 131, 15924–15929. 10.1021/ja9070317. [DOI] [PubMed] [Google Scholar]
- Sato Y.; Yamasaki R.; Saito S. Angew. Chem., Int. Ed. 2009, 48, 504–507. 10.1002/anie.200804864. [DOI] [PubMed] [Google Scholar]
- Carina R. F.; Dietrich-Buchecker C.; Sauvage J.-P. J. Am. Chem. Soc. 1996, 118, 9110–9116. 10.1021/ja961459p. [DOI] [Google Scholar]
- Hayashi R.; Wakatsuki K.; Yamasaki R.; Mutoh Y.; Kasama T.; Saito S. Chem. Commun. 2014, 50, 204–206. 10.1039/C3CC47425A. [DOI] [PubMed] [Google Scholar]
- Chalifoux W. A.; Tykwinski R. R. Nat. Chem. 2010, 2, 967–971. 10.1038/nchem.828. [DOI] [PubMed] [Google Scholar]
- Khuong T.-A. V.; Zepeda G.; Ruiz R.; Khan S. I.; Garcia-Garibay M. A. Cryst. Growth Des. 2004, 4, 15–18. 10.1021/cg034144b. [DOI] [Google Scholar]
- Chodkiewicz W.; Cadiot P. C. R. Hebd. Seances Acad. Sci. 1955, 241, 1055–1057. [Google Scholar]
- a Eisler S.; Tykwinski R. R. J. Am. Chem. Soc. 2000, 122, 10736–10737. 10.1021/ja005557t. [DOI] [Google Scholar]; b Jahnke E.; Tykwinski R. R. Chem. Commun. 2010, 46, 3235–3249. 10.1039/c003170d. [DOI] [PubMed] [Google Scholar]; c Knorr R. Chem. Rev. 2004, 104, 3795–3849. 10.1021/cr030616h. [DOI] [PubMed] [Google Scholar]
- a Baughman R. H.; Yee K. C. Macromol. Rev. 1978, 13, 219–239. 10.1002/pol.1978.230130104. [DOI] [Google Scholar]; b Enkelmann V. Chem. Mater. 1994, 6, 1337–1340. 10.1021/cm00044a035. [DOI] [Google Scholar]; c Xiao J.; Yang M.; Lauher J. W.; Fowler F. W. Angew. Chem., Int. Ed. 2000, 39, 2132–2135. 10.1002/1521-3773(20000616)39:12<2132::AID-ANIE2132>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]; d Zhao Y.; Luu T.; Bernard G. M.; Taerum T.; McDonald R.; Wasylishen R. E.; Tykwinski R. R. Can. J. Chem. 2012, 90, 994–1014. 10.1139/v2012-075. [DOI] [Google Scholar]
- a Utesch N. F.; Diederich F. Org. Biomol. Chem. 2003, 1, 237–239. 10.1039/b210184j. [DOI] [PubMed] [Google Scholar]; b Gibtner T.; Hampel F.; Gisselbrecht J.-P.; Hirsch A. Chem. - Eur. J. 2002, 8, 408–432. 10.1002/1521-3765(20020118)8:2<408::AID-CHEM408>3.3.CO;2-C. [DOI] [PubMed] [Google Scholar]; c Gulia N.; Ejfler J.; Szafert S. Tetrahedron Lett. 2012, 53, 5471–5474. 10.1016/j.tetlet.2012.07.141. [DOI] [Google Scholar]
- Hofmeister H.; Annen K.; Laurent H.; Wiechert R. Angew. Chem., Int. Ed. Engl. 1984, 23, 727–729. 10.1002/anie.198407271. [DOI] [Google Scholar]
- Zhang G.; Yi H.; Zhang G.; Deng Y.; Bai R.; Zhang H.; Miller J. T.; Kropf A. J.; Bunel E. E.; Lei A. J. Am. Chem. Soc. 2014, 136, 924–926. 10.1021/ja410756b. [DOI] [PubMed] [Google Scholar]
- Shi W.; Luo Y.; Luo X.; Chao L.; Zhang H.; Wang J.; Lei A. J. Am. Chem. Soc. 2008, 130, 14713–14720. 10.1021/ja8049436. [DOI] [PubMed] [Google Scholar]
- a Brandsma L.Preparative Acetylene Chemistry; Elsevier: Amsterdam, 1988; Chap. 10, pp 219–227. [Google Scholar]; b Peng H.; Xi Y.; Ronaghi N.; Dong B.; Akhmedov N. G.; Shi X. J. Am. Chem. Soc. 2014, 136, 13174–13177. 10.1021/ja5078365. [DOI] [PubMed] [Google Scholar]
- Montierth J. M.; DeMario D. R.; Kurth M. J.; Schore N. E. Tetrahedron 1998, 54, 11741–11748. 10.1016/S0040-4020(98)83035-4. [DOI] [Google Scholar]
- a Durola F.; Heitz V.; Reviriego F.; Roche C.; Sauvage J.-P.; Sour A.; Trolez Y. Acc. Chem. Res. 2014, 47, 633–645. 10.1021/ar4002153. [DOI] [PubMed] [Google Scholar]; b Faiz J. A.; Heitz V.; Sauvage J.-P. Chem. Soc. Rev. 2009, 38, 422–442. 10.1039/B710908N. [DOI] [PubMed] [Google Scholar]
- a Orita A.; Otera J. Chem. Rev. 2006, 106, 5387–5412. 10.1021/cr050560m. [DOI] [PubMed] [Google Scholar]; b Rubin Y.; Kahr M.; Knobler C. B.; Diederich F.; Wilkins C. L. J. Am. Chem. Soc. 1991, 113, 495–500. 10.1021/ja00002a017. [DOI] [Google Scholar]; c Tobe Y.; Fujii T.; Naemura K. J. Org. Chem. 1994, 59, 1236–1237. 10.1021/jo00085a003. [DOI] [Google Scholar]; d Simpkins S. M. E.; Weller M. D.; Cox L. R. Chem. Commun. 2007, 4035–4037. 10.1039/b707681a. [DOI] [PubMed] [Google Scholar]
- a Eelkema R.; Maeda K.; Odell B.; Anderson H. L. J. Am. Chem. Soc. 2007, 129, 12384–12385. 10.1021/ja074997i. [DOI] [PubMed] [Google Scholar]; b Yau C. M. S.; Pascu S. I.; Odom S. A.; Warren J. E.; Klotz E. J. F.; Frampton M. J.; Williams C. C.; Coropceanu V.; Kuimova M. K.; Phillips D.; Barlow S.; Brédas J.-L.; Marder S. R.; Millar V.; Anderson H. L. Chem. Commun. 2008, 2897–2899. 10.1039/b802728e. [DOI] [PubMed] [Google Scholar]; c Winn J.; Pinczewska A.; Goldup S. M. J. Am. Chem. Soc. 2013, 135, 13318–13321. 10.1021/ja407446c. [DOI] [PubMed] [Google Scholar]
- a Cheetham A. G.; Hutchings M. G.; Claridge T. D. W.; Anderson H. L. Angew. Chem., Int. Ed. 2006, 45, 1596–1599. 10.1002/anie.200504064. [DOI] [PubMed] [Google Scholar]; b Fernandes A.; Viterisi A.; Coutrot F.; Potok S.; Leigh D. A.; Aucagne V.; Papot S. Angew. Chem., Int. Ed. 2009, 48, 6443–6447. 10.1002/anie.200903215. [DOI] [PubMed] [Google Scholar]; c Schneider H.-J.; Agrawal P.; Yatsimirsky A. K. Chem. Soc. Rev. 2013, 42, 6777–6800. 10.1039/c3cs60069f. [DOI] [PubMed] [Google Scholar]
- a Craig M. R.; Hutchings M. G.; Claridge T. D. W.; Anderson H. L. Angew. Chem., Int. Ed. 2001, 40, 1071–1074. 10.1002/1521-3773(20010316)40:6<1071::AID-ANIE10710>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]; b Gassensmith J. J.; Baumes J. M.; Smith B. D. Chem. Commun. 2009, 6329–6338. 10.1039/b911064j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco-Rodriguez A. M.; Towrie M.; Collin J. P.; Záliš S.; Vlćek A. Jr. Dalton Trans. 2009, 3941–3949. 10.1039/b820748h. [DOI] [PubMed] [Google Scholar]
- Lahlali H.; Jobe K.; Watkinson M.; Goldup S. M. Angew. Chem., Int. Ed. 2011, 50, 4151–4155. 10.1002/anie.201100415. [DOI] [PubMed] [Google Scholar]
- a Klotz E. J. F.; Claridge T. D. W.; Anderson H. L. J. Am. Chem. Soc. 2006, 128, 15374–15375. 10.1021/ja0665139. [DOI] [PubMed] [Google Scholar]; b Prikhod’ko A. I.; Durola F.; Sauvage J.-P. J. Am. Chem. Soc. 2008, 130, 448–449. 10.1021/ja078216p. [DOI] [PubMed] [Google Scholar]; c Prikhod’ko A.; Sauvage J.-P. J. Am. Chem. Soc. 2009, 131, 6794–6807. 10.1021/ja809267z. [DOI] [PubMed] [Google Scholar]; d Lee C. F.; Leigh D. A.; Pritchard R. G.; Schultz D.; Teat S. J.; Timco G. A.; Winpenny R. E. P. Nature 2009, 458, 314–318. 10.1038/nature07847. [DOI] [PubMed] [Google Scholar]; e Cheng H. M.; Leigh D. A.; Maffei F.; McGonigal P. R.; Slawin A. M. Z.; Wu J. J. Am. Chem. Soc. 2011, 133, 12298–12303. 10.1021/ja205167e. [DOI] [PubMed] [Google Scholar]; f Yamashita Y.; Mutoh Y.; Yamasaki R.; Kasama T.; Saito S. Chem. - Eur. J. 2015, 21, 2139–2145. 10.1002/chem.201405090. [DOI] [PubMed] [Google Scholar]
- In general, low temperature single crystal diffraction data were collected using I19(EH1) at Diamond Light Source.55 Data were collected using CrystalClear (Rigaku Inc., 2009) and reduced using CrysAlisPro (Oxford Diffraction/Agilent, 2011). The structures were solved using SuperFlip56 and refined using full-matrix least-squares within the CRYSTALS software suite.57 Where the structure contained large solvent-accessible voids comprising diffuse electron density, the discrete Fourier transforms of the void regions were treated as contributions to the A and B parts of the calculated structure factors using PLATON/SQUEEZE58 integrated with the CRYSTALS software. Hydrogen atoms were positioned geometrically and refined in CRYSTALS.57c For structure 2c·M7, data were collected using an Oxford Diffraction/Agilent SuperNova. The structure was solved using ShelXS program and refined with ShelXL.59
- a Szafert S.; Gladysz J. A. Chem. Rev. 2003, 103, 4175–4206. 10.1021/cr030041o. [DOI] [PubMed] [Google Scholar]; b Szafert S.; Gladysz J. A. Chem. Rev. 2006, 106, PR1–PR33. 10.1021/cr068016g. [DOI] [PubMed] [Google Scholar]
- Chalifoux W. A.; MacDonald R.; Ferguson M. J.; Tykwinski R. R. Angew. Chem., Int. Ed. 2009, 48, 7915–7919. 10.1002/anie.200902760. [DOI] [PubMed] [Google Scholar]
- a Chernick E. T.; Tykwinski R. R. J. Phys. Org. Chem. 2013, 26, 742–749. 10.1002/poc.3160. [DOI] [Google Scholar]; b Liu M.; Artyukhov V. I.; Lee H.; Xu F.; Yakobson B. I. ACS Nano 2013, 7, 10075–10082. 10.1021/nn404177r. [DOI] [PubMed] [Google Scholar]
- Mohr W.; Stahl J.; Hampel F.; Gladysz J. A. Chem. - Eur. J. 2003, 9, 3324–3340. 10.1002/chem.200204741. [DOI] [PubMed] [Google Scholar]
- a Castellano R. K.; Diederich F.; Meyer E. A. Angew. Chem., Int. Ed. 2003, 42, 1210–1250. 10.1002/anie.200390319. [DOI] [PubMed] [Google Scholar]; b Nishio M. CrystEngComm 2004, 6, 130–158. 10.1039/b313104a. [DOI] [Google Scholar]; c Schneider H.-J. Acc. Chem. Res. 2015, 48, 1815–1822. 10.1021/acs.accounts.5b00111. [DOI] [PubMed] [Google Scholar]
- a Braga D.; Grepioni F.; Tedesco E. Organometallics 1998, 17, 2669–2772. 10.1021/om971096h. [DOI] [Google Scholar]; b Robinson J. M. A.; Kariuki B. M.; Gough R. J.; Harris K. D. M.; Philp D. J. Solid State Chem. 1997, 134, 203–206. 10.1006/jssc.1997.7646. [DOI] [Google Scholar]; c McAdam C. J.; Cameron S. A.; Hanton L. R.; Manning A. R.; Moratti S. C.; Simpson J. CrystEngComm 2012, 14, 4369–4383. 10.1039/c2ce25267h. [DOI] [Google Scholar]
- Allen F. H. Acta Crystallogr., Sect. B: Struct. Sci. 2002, 58, 380–388. 10.1107/S0108768102003890. [DOI] [PubMed] [Google Scholar]
- Rowland R. S.; Taylor R. J. Phys. Chem. 1996, 100, 7384–7391. 10.1021/jp953141+. [DOI] [Google Scholar]
- a Owen G. R.; Gauthier S.; Weisbach N.; Hampel F.; Bhuvanesh N.; Gladysz J. A. Dalton Trans. 2010, 39, 5260–5271. 10.1039/c002041a. [DOI] [PubMed] [Google Scholar]; b Tykwinski R. R.; Kendall J.; McDonald R. Synlett 2009, 13, 2068–2075. 10.1055/s-0029-1217706. [DOI] [Google Scholar]
- Nowell H.; Barnett S. A.; Christensen K. E.; Teat S. J.; Allan D. R. J. Synchrotron Radiat. 2012, 19, 435–441. 10.1107/S0909049512008801. [DOI] [PubMed] [Google Scholar]
- Palatinus L.; Chapuis G. J. Appl. Crystallogr. 2007, 40, 786–790. 10.1107/S0021889807029238. [DOI] [Google Scholar]
- a Betteridge P. W.; Carruthers J. R.; Cooper R. I.; Prout K.; Watkin D. J. J. Appl. Crystallogr. 2003, 36, 1487. 10.1107/S0021889803021800. [DOI] [Google Scholar]; b Parois P.; Cooper R. I.; Thompson A. L. Chem. Cent. J. 2015, 9, 30. 10.1186/s13065-015-0105-4. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Cooper R. I.; Thompson A. L.; Watkin D. J. J. Appl. Crystallogr. 2010, 43, 1100–1107. 10.1107/S0021889810025598. [DOI] [Google Scholar]
- Spek A. L. J. Appl. Crystallogr. 2003, 36, 7–13. 10.1107/S0021889802022112. [DOI] [Google Scholar]
- Sheldrick G. M. Acta Crystallogr., Sect. A: Found. Crystallogr. 2008, A64, 112–122. 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.