

Abstract
Staphylococcus aureus (S. aureus) is a human pathogen that relies on the subversion of host phagocytes to support its pathogenic lifestyle. S. aureus strains can produce up to five beta‐barrel, bi‐component, pore‐forming leukocidins that target and kill host phagocytes. Thus, preventing immune cell killing by these toxins is likely to boost host immunity. Here, we describe the identification of glycine‐rich motifs within the membrane‐penetrating stem domains of the leukocidin subunits that are critical for killing primary human neutrophils. Remarkably, leukocidins lacking these glycine‐rich motifs exhibit dominant‐negative inhibitory effects toward their wild‐type toxin counterparts as well as other leukocidins. Biochemical and cellular assays revealed that these dominant‐negative toxins work by forming mixed complexes that are impaired in pore formation. The dominant‐negative leukocidins inhibited S. aureus cytotoxicity toward primary human neutrophils, protected mice from lethal challenge by wild‐type leukocidin, and reduced bacterial burden in a murine model of bloodstream infection. Thus, we describe the first example of staphylococcal bi‐component dominant‐negative toxins and their potential as novel therapeutics to combat S. aureus infection.
Keywords: bi‐component, dominant‐negative, leukocidin, pore‐forming toxin, Staphylococcus aureus
Subject Categories: Microbiology, Virology & Host Pathogen Interaction
Introduction
Staphylococcus aureus (S. aureus) is a significant human pathogen that can cause a range of diseases from mild (such as skin and soft tissue infections) to severe and life‐threatening infections including endocarditis, osteomyelitis, bacteremia, pneumonia, and sepsis 1. S. aureus produces a large array of virulence factors that thwart the host immune response to promote bacterial replication 2, 3. Among these factors, S. aureus strains produce a collection of bi‐component pore‐forming leukocidins 4. These toxins are secreted as two water‐soluble monomers (named S (slow)‐ or F (fast)‐subunit, based on their elution profiles in liquid chromatography) 4. The secreted S‐subunit monomer binds to proteinaceous cellular receptors on the plasma membrane of target host cell, causing the recruitment of the F‐subunit 5. Oligomerization of alternating S‐ and F‐subunits on the cellular membrane produces an octameric pre‐pore structure, followed by the insertion of the pre‐stem domains that assemble into a membrane‐piercing beta‐barrel structure that results in cell death via osmotic lysis 6, 7, 8. S. aureus strains associated with human infections can produce up to five bi‐component leukocidins: Panton‐Valentine leukocidin (PVL or LukSF‐PV), gamma hemolysin (two toxins, HlgAB and HlgCB), leukocidin AB (LukAB, also known as LukGH), and leukocidin ED (LukED), with the toxin subunits categorized as S‐subunits (LukS‐PV, HlgA, HlgC, LukA, and LukE) and F‐subunits (LukF‐PV, HlgC, LukB, and LukD) 4. These leukocidins target and kill leukocytes from both innate and adaptive host immune responses 4. High amino acid sequence homology is shared among S‐subunits and F‐subunits, with the exception of LukAB (> 65% homology versus ~30%) 9. The shared homology and similar cytotoxic effects on leukocytes previously suggested that these toxins were redundant. However, the identification of different proteinaceous cellular receptors targeted by these toxins on the surface of leukocytes challenges the notion of redundancy and provides an explanation for their cell and species tropism 10, 11, 12, 13, 14.
Development of new antibiotic treatments against S. aureus infections is hindered by the high rate of acquisition of antibiotic resistance by this bacterium. Furthermore, there is currently no effective vaccine against this pathogen 15, 16. Contributing to the challenge of therapeutic development is the assortment of virulence factors that S. aureus employs to subvert the host immune response 2, 3, 17. Thus, it is clear that a multivalent approach is needed to prevent S. aureus disease. Previous and current attempts have focused on the study of conserved surface antigens that are present in S. aureus strains, including capsular polysaccharides CP5 and CP8 18, 19, Protein A (SpA) 20, 21, iron‐scavenging proteins (IsdB) 22, 23, clumping factors (ClfA and ClfB) 24, 25, or secreted factors such as Hla, HlgC, or LukS‐PV 26, 27, 28, 29, 30, among others 26, 27, 28, 29, 30. Studies using surface‐bound molecules have either failed to translate into successful human clinical trials or are currently under preclinical or early stages of clinical trials (www.clinicaltrials.gov). Given the apparent lack of protection conferred by surface‐bound virulence factors already evaluated, emphasis has been shifting toward therapeutic targeting of alternative staphylococcal virulence factors, including secreted leukocidins 30, 31, 32, 33. As a result, studies involving secreted factors are currently being pursued and are still at early phases of human clinical trials 30, 34, 35, 36.
Here, we describe the identification of conserved glycine‐rich motifs that upon deletion render all five bi‐component leukocidins inactive against primary human neutrophils (polymorphonuclear leukocytes, herein referred to as PMNs). Remarkably, we found that the mutated toxins exert a dominant‐negative effect over their wild‐type (WT) counterparts and exhibit cross‐protective effects against other WT bi‐component leukocidins in vitro and in vivo. Mechanistic studies indicate that the inhibition of toxin activity by these dominant‐negative toxins occurs via competition for the receptor‐binding site and effective subunit recruitment, leading to the generation of defective pores. Taken together, our findings provide an alternative approach for the development of much needed anti‐S. aureus therapeutics.
Results
Stem domain deletions inactivate LukED
The S. aureus bi‐component leukocidins LukED, HlgAB, HlgCB, PVL, and LukAB target PMNs (Fig 1A). Importantly, these toxins can interact and form heterologous toxin pairs that also target and kill PMNs (Fig 1B). Of note, LukAB was not included due to its low solubility when purified as single subunits 37. Examination of the amino acid sequences of LukE and LukD revealed glycine‐rich motifs that localize to the stem domains of the monomers. Glycine residues lack side chains, and this property has been associated with increased torsion angles, particularly in polypeptide loops, providing increased flexibility. To elucidate the contribution of these motifs in LukED‐mediated killing of PMNs, the glycine‐rich motifs of 5–6 residues in length were deleted in both LukE and LukD (Fig 1C). The deleted amino acids from the mature toxin sequences were ∆112–117 (LukEmut1) and ∆126–131 (LukEmut2) from the S‐subunit LukE, and amino acids ∆130–134 (LukDmut) from the F‐subunit LukD. We found that when combined with its native subunit partner, the LukEmut1, LukEmut2, or LukDmut mutation resulted in an inactive toxin (Fig 1D). These data indicate that the glycine‐rich motifs within the stem domains of LukE and LukD are required for the formation of an active toxin.
Figure 1. Deletions of the glycine‐rich motifs of LukE and LukD inactivate the toxin and make the mutant LukED dominant negative.
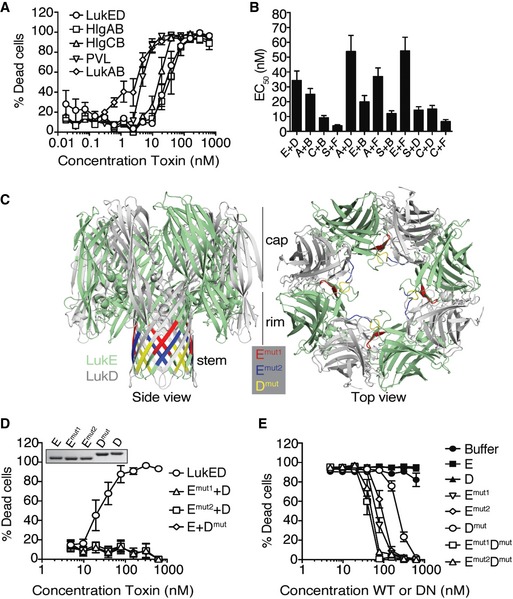
- Viability of primary human PMNs following treatment with purified recombinant bi‐component leukocidins measured using the viability dye CellTiter. Bars indicate mean ± SEM, with n = 5 donors.
- Half maximal effective concentrations (EC50) of native and heterologous toxin pairings on primary human PMNs measured using the viability dye CellTiter. E = LukE, D = LukD, A = HlgA, B = HlgB, C = HlgC, S = LukS‐PV, F = LukF‐PV. Bars indicate mean ± SEM, with n = 6 donors.
- Leukocidin octamer highlighting stem domain motifs deleted from LukE and LukD (LukEmut1, LukEmut2, and LukDmut), as shown from the side view (left) and top view (right) of the pore. The monomeric structures of LukE (3ROH) and LukD (4Q7G) were obtained from the Protein Data Bank, and superimposed and colored on the structure of the HlgA‐HlgB octameric pore (3BO7) using PyMOL. The location of where the deleted residues would be in the pore structure is shown from a side view on the left, and the location within an oligomeric pre‐pore structure is shown from a top view on the right.
- Viability of primary human PMNs following incubation with WT and mutant toxin pairings measured using the viability dye CellTiter. Insert: 2 μg per lane of purified recombinant toxins visualized by Coomassie Blue staining on a 12% SDS–PAGE gel. E = LukE, D = LukD, Emut1 = LukEmut1, Dmut = LukDmut. Bars indicate mean ± SEM, with n = 4 donors.
- Viability of primary human PMNs following simultaneous incubation with a lethal dose of WT LukED (78.12 nM per subunit) and a titration of WT or dominant‐negative (DN) toxin subunits measured using the viability dye CellTiter. E = LukE, D = LukD, Emut1 = LukEmut1, Emut2 = LukEmut2, Emut1Dmut = LukEmut1LukDmut, Emut2Dmut = LukEmut2LukDmut. Bars indicate mean ± SEM, with n = 6 donors.
LukED stem domain mutants behave as dominant‐negative toxins
Since the LukED mutants have no cytotoxic effects on PMNs, we hypothesized that these mutant toxins could exhibit a dominant‐negative effect by inhibiting LukED‐mediated lysis of PMNs. We found that as with the buffer control, incubation of single WT subunits (E or D) in the presence of a lethal dose of WT LukED did not protect PMNs from intoxication. In contrast, the addition of single toxin mutants LukEmut1, LukEmut2, and LukDmut protected PMNs against WT LukED (Fig 1E). Simultaneous addition of LukEmut1 and LukDmut2, or LukEmut2 and LukDmut, slightly potentiated the protective effect of these mutant toxins, with protection occurring at a ratio of 1:1 to 1:2 of WT to mutant toxin (75 nM of WT vs. 75–150 nM of mutant toxin when using a LD90 of WT LukED, or 46.9 nM of WT vs. 46.9–93.8 nM of mutant toxin when using a LD50 of WT LukED) (Figs 1E and EV1). Collectively, these data indicate that the LukE and LukD stem domain mutants behave as dominant‐negative toxins that neutralize WT LukED.
Figure EV1. Determination of the stoichiometry of WT to dominant‐negative LukED required for protection in human PMNs.
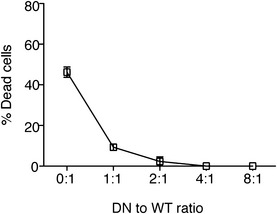
Viability of primary human PMNs in the presence of a LD50 (46.9 nM) of WT LukED and a titration of LukEmut1LukDmut evaluated using the metabolic dye CellTiter.
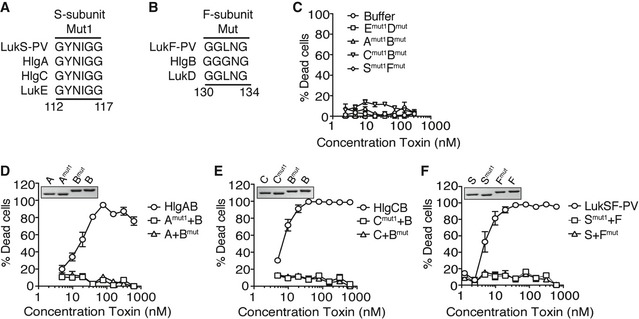
The glycine‐rich motifs within the stem domain are conserved in other bi‐component leukocidins
With the exception of LukAB, the other bi‐component pore‐forming leukocidins share over 60% sequence homology in both S‐ and F‐subunits 4, 9. Given the sequence conservation between the deleted glycine‐rich motifs and the overall amino acid homology between the leukocidins (Fig 2A and B), we generated the individual S‐ and F‐subunit deletions in HlgAB, HlgCB, and PVL (LukSF‐PV) that showed greater inhibitory effect against WT LukED (∆112–117, S‐subunit Mut1 and ∆130–134, F‐subunit Mut2) and tested whether the resulting mutant proteins could protect against their WT counterparts. As observed with the LukED mutants, deletion of these stem domain motifs in HlgAB, HlgCB, and PVL rendered the toxins inactive against PMNs (Fig 2C–F). Thus, the glycine‐rich motifs within the stem domains of the bi‐component leukocidins are required for toxin‐mediated death of PMNs.
Figure 2. The glycine‐rich motifs within the stem domain are required for the cytotoxic activity of other leukocidins.
-
A, BAmino acid sequences of toxin S‐subunits depicting (A) Mut1 (∆112‐117) and (B) Mut2 (∆130‐134) domains, based on ClustalW alignment.
-
CViability of primary human PMNs following incubation with mutant toxin pairings measured using the viability dye CellTiter. Bars indicate mean ± SEM, with n = 3 donors.
-
D–FViability of primary human PMNs following incubation with WT or mutant toxin pairings. Insert: 2 μg per lane of purified recombinant toxins visualized by Coomassie Blue staining using a 12% SDS–PAGE gel. Amut1 = HlgAmut1, Bmut = HlgBmut, Cmut1 = HlgCmut1, Smut1 = LukS‐PVmut1, Fmut = LukF‐PVmut. Bars indicate mean ± SEM, with n = 4 donors (D), n = 4 donors (E), and n = 5 donors (F).
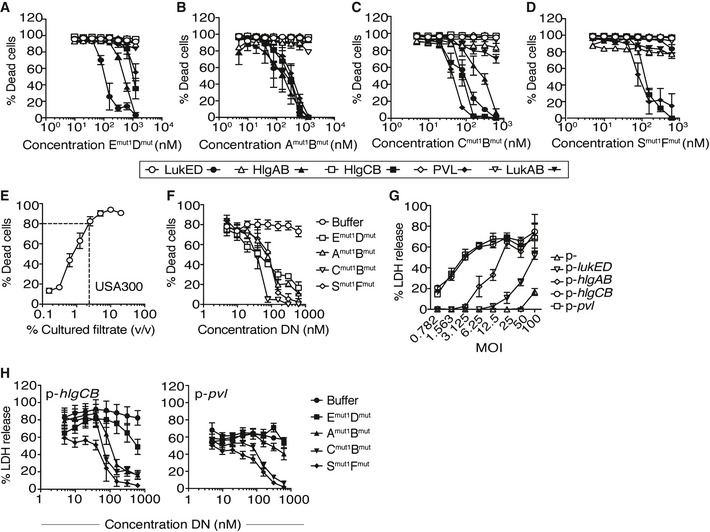
Dominant‐negative toxins exert cross‐neutralizing effects
Given that the majority of human‐associated S. aureus strains can produce between 4 and 5 of the bi‐component leukocidins, we tested whether the mutated toxins could inhibit other leukocidins. For these experiments, the inhibitory effects of the dominant‐negative toxins were evaluated against a lethal dose (LD90) of WT toxins in PMNs. We observed that the LukED mutants protected against all the toxins, albeit increased inhibition was observed against WT LukED and HlgAB (Fig 3A). Conversely, the HlgAB mutants were equally protective against all the WT toxins (Fig 3B). The HlgCB mutants protected PMNs against WT PVL and HlgCB and, at higher doses, against LukED and HlgAB (Fig 3C), while the PVL mutants only protected against WT PVL and HlgCB (Fig 3D). These results are in line with the recognition of specific proteinaceous cellular receptors on PMNs: LukED and HlgAB target CXCR1 and CXCR2 13, 14, whereas HlgCB and PVL target C5aR and C5L2 11, 14. Together, these data demonstrate that the mutant toxins show cross‐protection against various WT toxins and that this protection is dictated in part by receptor specificity.
Figure 3. Dominant‐negative toxins exhibit cross‐neutralizing effects over other WT leukocidins.
-
A–DViability of primary human PMNs following incubation with a LD90 of LukED (78.12 nM), HlgAB (78.12 nM), HlgCB (39.1 nM), or LukSF‐PV (19.53 nM) and the indicated dominant‐negative toxins measured using the viability dye CellTiter. Open white symbols indicate buffer treatment. Filled black symbols indicate treatment with dominant‐negative toxins. Emut1Dmut = LukEmut1LukDmut, Amut1Bmut = HlgAmut1HlgBmut, Cmut1Bmut = HlgCmut1HlgBmut, Smut1Fmut = LukS‐PVmut1LukF‐PVmut. Bars indicate mean ± SEM, with n = 5 donors.
-
EViability of primary human PMNs following incubation with filtered culture supernatants collected after 8 h from CA‐MRSA USA300 strain LAC measured using the viability dye CellTiter. Bars indicate mean ± SEM, with n = 6 donors.
-
FViability of primary human PMNs following incubation with 1.25% filtered culture supernatants and a titration of the indicated dominant‐negative (DN) toxins measured using the viability dye CellTiter. Emut1Dmut = LukEmut1LukDmut, Amut1Bmut = HlgAmut1HlgBmut, Cmut1Bmut = HlgCmut1HlgBmut, Smut1Fmut = LukS‐PVmut1LukF‐PVmut. Bars indicate mean ± SEM, with n = 3 donors.
-
GViability of primary human PMNs during a 3‐h ex vivo infection with the S. aureus Newman toxinless strain overproducing the indicated toxins using the CytoTox‐One Homogenous Membrane Integrity Assay to measure lactate dehydrogenase (LDH) release. Bars indicate mean ± SEM, with n = 3 donors.
-
HViability of primary human PMNs during a 4‐h ex vivo infection with a MOI of 50 of S. aureus Newman toxinless strain overproducing the indicated toxins in the presence of a titration of the DN toxins using the CytoTox‐One Homogenous Membrane Integrity Assay to measure lactate dehydrogenase (LDH) release. Emut1Dmut = LukEmut1LukDmut, Amut1Bmut = HlgAmut1HlgBmut, Cmut1Bmut = HlgCmut1HlgBmut, Smut1Fmut = LukS‐PVmut1LukF‐PVmut. Bars indicate mean ± SEM, with n = 6 donors.
Dominant‐negative toxins protect human PMNs from S. aureus cytotoxicity
Bi‐component leukocidins are secreted into the extracellular milieu where they are part of a large number of proteins that are produced by S. aureus that interact with and subvert leukocytes. To evaluate whether the mutated leukocidins were also able to block native leukocidins in more complex and physiological settings, we first tested their inhibitory activity toward culture supernatants from the current community‐acquired methicillin‐resistant S. aureus (CA‐MRSA) clone prevalent in the United States, pulsed‐field gel electrophoresis type USA300 represented by strain LAC 38, 39. As shown previously 40, PMNs were highly susceptible to filtered culture supernatants from USA300 grown to stationary phase in rich medium (Fig 3E). To test the effect of the mutant toxins, we utilized a dose of culture supernatant that killed ~80–90 percent (LD80–90) of PMNs against a titration of mutant toxins. Remarkably, all the dominant‐negative leukocidins protected against the cytotoxicity exhibited by USA300 culture filtrates (Fig 3F).
To evaluate whether the mutated leukocidins also protect from S. aureus infection, we first challenged PMNs with a S. aureus strain that lacked all the leukocidins 13. We had to use a leukocidin minus strain since LukAB is highly potent during ex vivo infection of PMNs, masking any contribution of the other toxins 41. The S. aureus strain lacking all leukocidins (empty vector, p−) was severely impaired in killing PMNs at all the different multiplicity of infection (MOI) tested (Fig 3G), consistent with the notion that leukocidins are responsible for targeting and killing PMNs. In contrast, expression of lukED, pvl, hlgAB, or hlgCB in the leukocidin minus strain rescued the cytotoxicity of the strain toward PMNs, albeit at different levels (Fig 3G). In these experiments, PVL and HlgCB exhibited the highest activity (Fig 3G) and as such were used for the dominant‐negative experiments. PMNs were then infected with S. aureus producing HlgCB or PVL at a MOI of 50 in the presence of increasing concentrations of the dominant‐negative toxins and PMN lysis monitored. We observed that the dominant‐negative leukocidins also protected from ex vivo infection (Fig 3H), with the HlgCB and PVL dominant‐negative proteins exhibiting the greatest protection.
LukAB dominant‐negative mutants protect against S. aureus infection
LukAB shares minimal homology with the other leukocidins 5. However, upon examination of the mature protein sequences of LukA and LukB and the crystal structure of the LukAB octamer (PDB 4TW1), we identified di‐glycine motifs that also mapped to the subunits’ stem domains (Fig 4A). Since LukAB shows increased stability when purified as a dimer 37, we generated a plasmid that expressed lukAB containing both deletions together (hence producing LukABmut1 and LukABmut2). Unfortunately, we were unable to generate purified proteins due to poor stability of the mutated proteins upon purification. However, the mutated proteins were produced and secreted by S. aureus (Fig 4B). Thus, we evaluated the ability of the LukAB mutants to block S. aureus‐mediated lysis of PMNs. To this end, we introduced the expression plasmids carrying the mutated lukAB loci into WT S. aureus, which resulted in co‐production of WT and mutated LukAB toxins (Fig 4C), and evaluated whether the isogenic strains exhibit reduced lysis of PMNs. Remarkably, we found that the LukAB mutants significantly protected PMNs against S. aureus infection ex vivo (Fig 4D). Taken together, these data demonstrate that the dominant‐negative leukocidins protect against intoxication and infection of PMNs.
Figure 4. Deletions of the glycine‐rich motifs within the stem domain of LukAB protect against S. aureus infection.
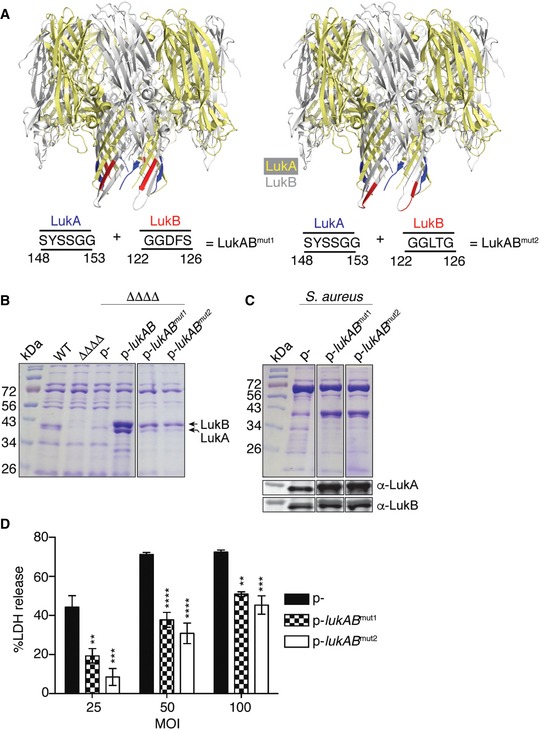
- LukAB octamers (4TW1) highlighting the location of where the deleted residues would be in the pore structure (left: LukABmut1, right: LukABmut2), as shown from the side view of the pore. The octameric LukAB structure was obtained from the Protein Data Bank and the motifs labeled using PyMOL.
- Exoproteome analysis of LukAB mutants produced by S. aureus strain Newman in a toxinless background. Samples were electrophoresed on a 12% SDS–PAGE gel and visualized by staining with Coomassie Blue. A representative of two experiments is shown.
- Exoproteome and immunoblot analyses of wild‐type S. aureus strain Newman transformed with empty plasmid (p−) or with plasmids containing the mutated lukAB loci. A representative of two independent experiments is shown.
- Viability of primary human PMNs during a 2‐h ex vivo infection with the indicated S. aureus strains using the CytoTox‐One Homogenous Membrane Integrity Assay to measure lactate dehydrogenase (LDH) release. Bars indicate mean ± SEM, with n = 3. Statistical significance is indicated as follows: MOI of 100: **P = 0.0026 for p− vs. LukABmut1 and ***P = 0.0003 for p− vs. LukABmut2; MOI of 50: ****P < 0.0001 for p− vs. LukABmut1 and ****P < 0.0001 for p− vs. LukABmut2; MOI of 25: ***P = 0.0007 for p− vs. LukABmut1 and ****P < 0.0001 for p− vs. LukABmut2 using two‐way ANOVA with Tukey's post hoc test correction for multiple comparisons.
Dominant‐negative toxins block the formation of active pores by WT toxins
To evaluate the mechanism by which dominant‐negative toxins inhibit WT toxins, we first tested whether the mutated toxins bound to the surface of PMNs. For this, we incubated PMNs in the presence of green‐fluorescent protein (GFP)‐fused LukE (78.12 nM) against a titration of either unlabeled WT or dominant‐negative mutant, and evaluated the loss of fluorescence using flow cytometry (Fig 5A). Both WT LukE and LukEmut1 equally competed binding of GFP‐LukE in a dose‐dependent and comparable level (Fig 5B). Similarly, HlgA and HlgAmut1 competed GFP‐LukE from the surface of PMNs, albeit to a lesser degree than WT LukE and LukEmut1. In contrast, HlgC and LukS‐PV were incapable of competing with GFP‐LukE for the receptor‐binding site on PMNs (Fig 5B), a finding in line with the receptor targeting specificity of these toxins 11, 13, 14, 42, 43.
Figure 5. Dominant‐negative toxins act by forming defective pores on the target host cell plasma membrane.
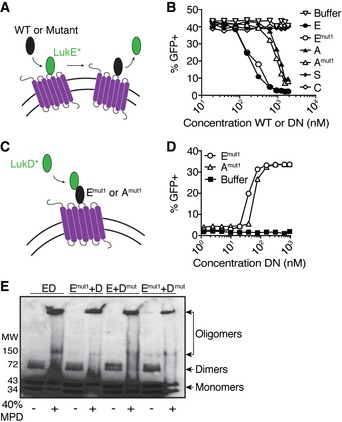
- Schematic depicting competition assay using GFP‐LukE and unlabeled WT or mutant toxins.
- Competition assay using GFP‐LukE (312.5 nM) and a titration of unlabeled WT or dominant‐negative (DN) toxin subunits evaluated by flow cytometry on the surface of primary human PMNs. E = LukE, Emut1 = LukEmut1, A = HlgA, Amut1 = HlgAmut1, S = LukS‐PV, C = HlgC. Data are presented as total fluorescence emitted from PMNs. A representative of three independent experiments each done with PMNs isolated from different donors is shown.
- Schematic depicting recruitment assay using GFP‐LukD and unlabeled WT or dominant‐negative toxins.
- Recruitment assay using GFP‐LukD (156.3 nM) and a titration of unlabeled dominant‐negative (DN) toxin subunits evaluated by flow cytometry on the surface of primary human PMNs. Emut1 = LukEmut1, Amut1 = HlgAmut1. Data are presented as total fluorescence emitted from GFP‐positive PMNs. A representative of three independent experiments each done with PMNs isolated from different donors is shown.
- Immunoblot using anti‐His antisera to detect dimer and oligomer formation by purified polyhistidine (6xHis)‐tagged WT or dominant‐negative LukED toxins in the absence or presence of 2‐methyl‐2,4‐pentanediol (MPD, 40% v/v). Emut1 = LukEmut1, Dmut = LukDmut. A representative of two independent experiments is shown.
Next, we tested whether the mutant LukE could recruit WT LukD. To this end, PMNs were incubated with multiple doses of unlabeled LukEmut1 followed by the addition of GFP‐LukD (156.3 nM) and fluorescence levels evaluated by flow cytometry. LukEmut1 was indeed able to recruit GFP‐LukD to the surface of PMNs, as indicated by the dose‐dependent and saturable fluorescence (Fig 5D). Similar results were observed when using HlgAmut1 (Fig 5D).
To evaluate whether the dominant‐negative mutant toxins can form oligomers, we incubated purified monomers in the presence of 2‐methyl‐2,4‐pentanediol, which has been shown previously to promote oligomer formation in solution 6, 7, 8, 32, 44, 45. We detected the formation of dimers and oligomers indistinguishable between WT LukED and the different combinations of WT and mutated LukE and LukD subunits (Fig 5E).
Altogether, these studies suggest that the mutated toxins exhibit a dominant‐negative effect by blocking binding to the target receptor, as well as by recruiting compatible toxin subunits and assembling into defective pores on the cell membrane.
Dominant‐negative toxins protect in vivo
We next tested the therapeutic potential of the dominant‐negative toxins in vivo in an intoxication model. Administration of purified WT leukocidins intravenously into mice revealed that PVL and HlgCB had no effect on the health of mice, which is consistent with the human cell tropism of these two toxins 11, 14, 42, 46. In contrast, LukED and HlgAB caused rapid signs of morbidity in mice (Fig 6A). Next, we evaluated the prophylactic inhibitory activity of the dominant‐negative toxins in vivo. Dominant‐negative toxins were administered intravenously 1 h prior to challenge with WT LukED or HlgAB, and mice were monitored for signs of morbidity. We found that the LukEmut1LukDmut, HlgAmut1HlgBmut, HlgCmut1HlgBmut toxins protected against intoxication with WT LukED (Fig 6B) or HlgAB (Fig 6C). In contrast, no protection was observed with the buffer control or the LukSmut1LukFmut toxins. Moreover, we found that while pre‐treatment with LukEmut1LukDmut for up to 5 h protected mice from LukED intoxication (Fig EV2A and B), mice pre‐treated 24 h prior to challenge were not protected (Fig EV2C and D).
Figure 6. Dominant‐negative toxins are active in vivo .
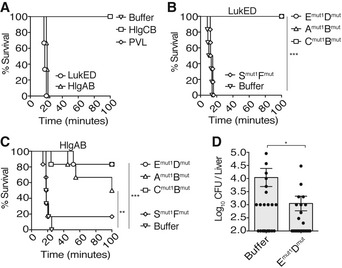
- Survival of Swiss‐Webster mice following intravenous administration of purified recombinant toxins: LukED (10 μg per subunit), HlgAB (5 μg per subunit), HlgCB (35 μg per subunit), and LukSF‐PV/PVL (35 μg per subunit). Buffer: n = 2 mice; LukED: n = 3 mice; HlgAB: n = 3 mice; HlgCB: n = 2 mice; PVL: n = 3. A representative of two independent experiments is shown.
- Survival of Swiss‐Webster mice following prophylactic intravenous administration of dominant‐negative mutants, followed by intravenous challenge with a lethal dose of WT LukED. Emut1Dmut = LukEmut1LukDmut, Amut1Bmut = HlgAmut1HlgBmut, Cmut1Bmut = HlgCmut1HlgBmut, Smut1Fmut = LukS‐PVmut1LukF‐PVmut. n = 6 mice per treatment. ***P = 0.0005 for buffer vs. Emut1Dmut, buffer vs. Amut1Bmut, and buffer vs. Cmut1Bmut. P = 0.1050 (not significant, ns) for buffer vs. Smut1Fmut. Statistics performed using log‐rank (Mantel–Cox) test. A representative of two independent experiments is shown.
- Survival of Swiss‐Webster mice following prophylactic intravenous administration of dominant‐negative mutants, followed by intravenous challenge with a lethal dose of WT HlgAB. Emut1Dmut = LukEmut1LukDmut, Amut1Bmut = HlgAmut1HlgBmut, Cmut1Bmut = HlgCmut1HlgBmut, Smut1Fmut = LukS‐PVmut1LukF‐PVmut. n = 6 mice per treatment. ***P = 0.0004 for buffer vs. Emut1Dmut and buffer vs. Amut1Bmut. **P = 0.0015 for buffer vs. Cmut1Bmut. P = 0.7955 (not significant, ns) for buffer vs. Smut1Fmut. Statistics performed using log‐rank (Mantel–Cox) test. A representative of two independent experiments is shown.
- Bacterial burden 96 h post‐infection in the liver of mice infected systemically with ˜1.5 × 107 CFU of S. aureus strain Newman and intravenously treated with buffer or LukEmut1LukDmut (Emut1Dmut, 10 μg per subunit) at t = 0, 24, and 48 h.p.i. Bars indicate mean ± SEM, with n = 20 mice (buffer) and n = 19 mice (Emut1Dmut2) combined from two independent experiments. *P = 0.0303 by unpaired, parametric, two‐tailed Student's t‐test.
Figure EV2. Evaluation of the activity of dominant‐negative toxins in vivo .
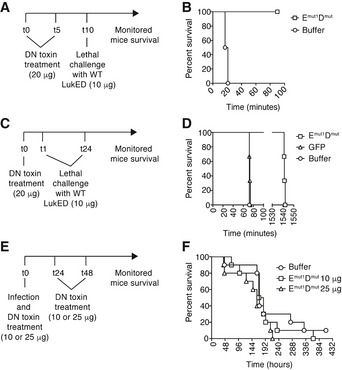
- Experimental scheme to determine “half‐life” of prophylactic LukEmut1LukDmut. Mice were challenged with wild‐type LukED at 0 and 5 h post‐treatment with the dominant‐negative toxins. t, time in hours.
- Survival of Swiss‐Webster mice after treatment with LukEmut1LukDmut or buffer as indicated in (A). n = 2 for buffer and n = 3 for LukEmut1LukDmut.
- Experimental scheme to determine “half‐life” of prophylactic LukEmut1LukDmut, after lethal challenge with wild‐type LukED 1 and 24 h post‐treatment with the dominant‐negative toxins. t, time in hours.
- Survival of Swiss‐Webster mice after challenge with wild‐type LukED 1 and 24 h post‐treatment with LukEmut1LukDmut, GFP (negative control protein), or buffer, as indicated in (C). n = 3 per condition.
- Experimental scheme to determine whether the LukEmut1LukDmut dominant‐negative toxins protect from in vivo infection when injected at 0, 24, and 48 h post‐intravenous infection. t, time in hours.
- Survival of Swiss‐Webster mice after simultaneous prophylaxis and infection with LukEmut1LukDmut, followed by treatment at 24 or 48 h post‐infection, as indicated in (E). n = 10 per condition.
Next, we evaluated the inhibitory potential of the dominant‐negative LukEmut1LukDmut toxin during in vivo infection. Mice were infected with 3.7 × 107 CFU of S. aureus strain Newman, systemically administered LukEmut1LukDmut at 0, 24, and 48 h post‐infection, and monitored for signs of acute infection for 18 days (Fig EV2E). Three separate cohorts of mice were used to treat with two different doses of LukEmut1LukDmut (10 or 25 μg per subunit per mouse) or buffer. We observed no difference between mice treated with buffer or LukEmut1LukDmut at the two different doses tested (Fig EV2F). The lack of protection is likely due to the poor half‐lives of the dominant‐negative proteins in vivo (Fig EV2C and D).
Lastly, we tested the therapeutic potential of the dominant‐negative toxins in ameliorating bacterial burden in tissues during infection. S. aureus strain Newman (~1.5 × 107 CFU) was simultaneously administered with LukEmut1LukDmut (10 μg per subunit) intravenously into mice and bacterial burden in infected livers enumerated 96‐h post‐infection. We found that LukEmut1LukDmut protected mice from S. aureus infection resulting in a ~1‐log reduction in bacterial burden compared to the buffer‐treated mice (Fig 6D).
Discussion
Staphylococcus aureus continues to be a formidable foe to humans. Despite this, concerted efforts by numerous groups to identify suitable S. aureus virulence factors to be utilized as vaccine components have been unsuccessful in clinical trials. Bi‐component leukocidins are a family of highly related toxins that contribute to the pathobiology of S. aureus. Blocking the leukocidins is likely to improve host‐mediated immunity as the leukocytes in charge of detecting and killing the pathogen will be protected from the lytic action of these toxins. While deciphering the molecular means by which LukED targets and kills host cells, we identified conserved, short glycine‐rich motifs within the stem domains of the S‐ and F‐subunits that are required for toxin activity. Remarkably, toxins lacking these motifs behaved as dominant‐negative toxins and neutralized the cytolytic activity of leukocidins in vitro, ex vivo, and in vivo when employed as either a prophylactic or therapeutic treatment against S. aureus systemic infection.
Similar to dominant‐negative forms of the B. anthracis protective antigen (PA) subunit of anthrax toxin 47 and the Helicobacter pylori vacuolating cytotoxin (VacA) 48 , the S. aureus dominant‐negative leukocidins form hetero‐oligomers with WT subunits that assemble into a defective pore in the host cell plasma membrane. While the stoichiometry of the PA dominant‐negative toxin occurs at a 7:1 ratio (WT to dominant‐negative toxin) 47, and dominant‐negative forms of VacA show stoichiometries between 5:1 to 7:1 (WT to dominant‐negative toxin) 48, the mutated leukocidins show an approximate stoichiometry of 1:1 to 1:2 (WT to dominant‐negative toxin). Our findings are consistent with the growing notion that dominant‐negative toxins could be generated by exploiting the mechanism by which pore‐forming toxins assemble into active pores.
A study by Adhikari et al reported the use of antibodies raised against a LukS‐PV mutant and their protective efficacy against purified canonical and non‐canonical toxin pairs 49. The toxins used across their study were generated and purified from E. coli, which can potentially affect the activity of the toxins and could explain the differences in activity compared to the S. aureus‐purified toxins used in our study. Moreover, the purified HlgAB in the Adhikari et al's study was not acutely lethal, which also disagrees with our findings (Fig 5A and C). Recently, a study by Rouha et al also used purified toxins from E. coli, which exhibited in vivo activities different to the proteins described here 30. Thus, care should be taken when expressing and purifying these toxins, and it seems that for maximal activity and stability, these toxins are best if purified from S. aureus.
The bi‐component pore‐forming leukocidins are present in the large majority of clinical isolates 4, and target phagocytes, including antigen‐presenting cells 4, 10, 41, 50, 51, 52, 53. As the first line of defense against bacterial infection, and as a critical component for the initiation of adaptive immunity, it is crucial to prevent death of these sentinels to enhance host immunity. Our studies provide insight into an alternative approach to combat S. aureus using dominant‐negative toxins to protect these leukocytes. These toxins could exert protection by a dual mode that combines direct blocking of toxins in vivo while also serving as antigens for the generation of neutralizing antibodies. The potential use of mutant toxins as inhibitors of WT toxin activity has been demonstrated in S. aureus with a non‐toxigenic form of Hla similar to the known Hla mutant H35L, H35A 54. Thus, it is plausible to generate proteins with small mutations that neutralize WT toxin activity while maintaining their structural assembly for the benefit of generating neutralizing antibodies. Further studies are required to elucidate the immunogenicity of these dominant‐negative toxins in the host and their potential synergism with other therapies currently being evaluated.
Materials and Methods
Ethics statement
Blood was obtained from healthy, de‐identified, consenting adult donors as buffy coats from the New York Blood Center. All experiments involving animals were reviewed and approved by the Institutional Animal Care and Use Committee of New York University and were performed according to guidelines from the National Institutes of Health (NIH), the Animal Welfare Act, and US Federal Law.
Isolation of primary human PMNs
PMNs were separated from erythrocytes by splitting blood into three equal parts, then mixing 1:1 with sterile, endotoxin‐free 0.9% sodium chloride (Baxter) + 3% dextran (Dextran 500; Pharmacosmos) at room temperature (RT), mixing gently by inverting the tubes, and incubating at room temperature for 25 min. The top fraction containing the PMNs and peripheral blood mononuclear cells (PBMCs) was centrifuged for 10 min at 805 g, RT, and the volume of the resulting pellets brought up to 30 ml with 1× PBS (Cellgro) and centrifuged again as before. The pellets were resuspended each in 17 ml of Hank's balanced salt solution (HBSS; Gibco), and the HBSS/cell mixture was filtered through a 70‐μm nylon cell strainer (Corning) into tubes containing 12.5 ml Ficoll (Ficoll‐Paque PLUS; GE). The samples were centrifuged for 30 min at 805 g, RT, without brake and the supernatants removed. Pellets of each donor were combined and transferred into a new tube, where the volume was brought up to 30 ml with 1× PBS and centrifuged for 10 min at 805 g, RT; 9 ml of ACK lysing buffer (Gibco) was added to lyze contaminating erythrocytes, resuspended, and incubated for 2–3 min at RT, followed by the addition of 1 ml of 10× PBS (Cellgro). The samples were centrifuged for 8 min at 394 g, RT, to pellet the PMNs. The cells were gently resuspended in 10 ml media and filtered through a 70‐μm nylon cell strainer into a new tube, followed by the dilution of PMNs in 10–40 ml of media. Purity of PMNs was evaluated at 90 to 95% by flow cytometry (LSRII with FACSDiva software; BD).
Generation of dominant‐negative mutant toxins
Isogenic dominant‐negative mutant strains were generated by overlap extension PCR of mutant DNA fragments (see Tables EV1 and EV2 for primer and bacterial strain information). S. aureus strain Newman genomic DNA (gDNA) was used as template to generate the LukED, HlgAB, HlgCB mutants. S. aureus strain LAC gDNA was used as template to generate the LukSF‐PV mutants. All PCR were performed using Phusion High‐Fidelity Polymerase (Thermo Scientific).
LukED
To generate WT lukE, gDNA was amplified using primers VJT629 and VJT1114. For lukD, gDNA was amplified using primers VJT1152 and VJT1153. For lukE mut1, gDNA was amplified with primers VJT629 and VJT686, and VJT685 and VJT1114. For lukE mut2, gDNA was amplified using primers VJT629 and VJT688, and VJT687 and VJT1114. For lukD mut, gDNA was amplified with primers VJT1152 and VJT692, and VJT691 and VJT1153. To obtain the final PCR product for the lukE mutants, each DNA fragment per mutant was included in a PCR containing the flanking primers VJT629 and VJT1114. To obtain the final PCR product for the lukD mutants, each DNA fragment per mutant was included in a PCR containing the flanking primers VJT1152 and VJT1153.
HlgAB
To generate hlgA, gDNA was amplified using primers VJT635 and VJT1209. For hlgB, gDNA was amplified using primers VJT1156 and VJT1157. For hlgA mut1, gDNA was amplified with primers VJT635 and VJT1241, and VJT1209 and VJT1240. For hlgB mut, gDNA was amplified using primers VJT1156 and VJT1249, and VJT1248 and VJT1157. To obtain the final PCR product for hlgA mut1, each DNA fragment was included in a PCR containing the flanking primers VJT635 and VJT1209. To obtain the final PCR product for hlgB mut, each DNA fragment was included in a PCR containing the flanking primers VJT1156 and VJT1157.
HlgCB
To generate hlgC, gDNA was amplified with primers VJT631 and VJT1210. For hlgC mut1, gDNA was amplified with primers VJT631 and VJT1245, and VJT1244 and VJT1210. To obtain the final PCR product for hlgC mut1, each DNA fragment was included in a PCR containing the flanking primers VJT631 and VJT1210. See above to generate hlgB and hlgB mut.
LukSF‐PV
To generate lukS‐PV, gDNA was amplified using primers VJT1208 and VJT1117. For lukF‐PV, gDNA was amplified with primers VJT1154 and VJT1155. For lukS mut1, gDNA was amplified with primers VJT1208 and VJT1251, and VJT1250 and VJT1117. For lukF mut, gDNA was amplified using primers VJT1154 and VJT1255, and VJT1254 and VJT1155. To obtain the final PCR product for lukS mut1, each DNA fragment was included in a PCR containing the flanking primers VJT1208 and VJT1117. To obtain the final PCR product for lukF mut, each DNA fragment was included in a PCR containing the flanking primers VJT1154 and VJT1155.
LukAB mutants
To generate lukAB mut1, gDNA was amplified with primers VJT544 and VJT1257, VJT1256 and VJT1452, and VJT1453 and VJT543, and the fragments were annealed by sewing overlap extension PCR. To generate lukAB mut2, gDNA was amplified with primers VJT544 and VJT1257, VJT1256 and VJT1454, and VJT1455 and VJT543, and the fragments were annealed by sewing overlap extension PCR. To obtain the final PCR products for the lukAB mut1 and lukAB mut2 mutants, the DNA fragments were included in a PCR containing the flanking primers VJT544 and VJT543.
All amplicons were cloned into pOS1‐PlukAB‐lukA .s.s.‐6xHis as previously described 12 using BamHI and PstI restriction sites, producing N‐terminal 6xhistidine (His)‐tagged toxin mutants that were transformed into competent E. coli DH5α, selected for and propagated in Luria–Bertani (LB) media supplemented with ampicillin (100 μg ml−1). The plasmid was purified and electroporated into competent S. aureus strain RN4220 and clones selected for in tryptic soy agar (TSA) media supplemented with chloramphenicol (10 μg ml−1), followed by electroporation into S. aureus strain Newman ΔΔΔΔ (ΔlukED ΔhlgACB::tet ΔlukAB::spec Δhla::ermC) 13.
Toxin purification from S. aureus
To purify WT and dominant‐negative toxins, S. aureus Newman ΔΔΔΔ strains harboring plasmids with the mutant sequences were grown overnight in 5 ml tryptic soy broth (TSB) supplemented with chloramphenicol (10 μg ml−1) at 37°C with 180 rpm shaking, then subcultured the following day at a 1:100 dilution in TSB supplemented with chloramphenicol (10 μg ml−1), and incubated for 5 h at 37°C with 180 rpm shaking. The cultures were centrifuged for 10 min at 4,424 g, 4°C, and the supernatants were filter‐sterilized through a 0.22‐μm filter (Corning). The filtrates were incubated in the presence of a final concentration of 10 mM imidazole and nickel‐nitriloacetic acid (Ni‐NTA) agarose resin equilibrated with 10 mM imidazole (Fisher) in 1× Tris‐buffered saline (TBS; Cellgro), and incubated for 30 min at 4°C while nutating. The filtrates were flowed through a column by gravity flow, the column was washed with 25 mM imidazole, and the nickel‐NTA‐bound toxins were eluted using 500 mM imidazole. When required, the toxins were concentrated using concentrator columns (Ultra‐15 Centrifugal Filter Units 10,000 NMWL, 15‐ml volume capacity; EMD Millipore Amicon) before measuring protein concentration using absorbance at 280 nm with a NanoDrop (Thermo Scientific). All holotoxin concentrations are represented per subunit unless indicated otherwise.
Cytotoxicity assays
To evaluate the viability of primary human PMNs to the leukocidins in the presence or absence of dominant‐negative toxins in vitro, PMNs were seeded at 2 × 105 cells per well in Roswell Park Memorial Institute (RPMI without phenol red; Gibco) media supplemented with 10% heat‐inactivated fetal bovine serum (FBS, Gemini Bio‐Products) in the presence of WT, dominant‐negative toxins, or both (as indicated) for 1 h at 37°C + 5% CO2. To measure cell viability, the metabolic dye CellTiter (Promega) was added at a final concentration of 10% per well and incubated for 2–3 h at 37°C + 5% CO2. Absorbance at 492 nm was measured using an EnVision 2103 Multilabel Reader (PerkinElmer).
Collection of S. aureus culture supernatants for human PMN intoxication
To collect culture supernatants from CA‐MRSA USA300, an overnight culture of LAC was grown in TSB at 37°C with 180 rpm shaking, then subcultured the following day at a 1:100 dilution into TSB, and grown for 8 h at 37°C with 180 rpm shaking. At this time point, the contribution of LukAB to USA300 secretome‐mediated killing of PMN is minimal due to the lack of LukAB in the culture filtrates 43. The culture was centrifuged for 15 min at 3,220 g, 4°C, and the culture supernatant was filter‐sterilized using a 0.22‐μm filter and stored at −80°C before use.
Evaluation of S. aureus exoprotein profiles
To collect culture filtrates to evaluate the exoprotein profiles from S. aureus, overnight cultures of each strain were grown in Roswell Park Memorial Institute (RPMI) media supplemented with 1% casamino acids (RPMI+CAS) in the presence of 10 μg ml−1 of chloramphenicol when required. The overnight cultures were grown at 37°C with 180 rpm shaking, then subcultured the following day at a 1:100 dilution into RPMI + CAS, and grown for 5 h at 37°C with 180 rpm shaking. The cultures were centrifuged at 4°C for 15 min and 3,220 g, and the supernatants were filter‐sterilized with a 0.22‐μm filter, followed by incubation on ice for 30 min. To precipitate proteins in the culture filtrate, 100% trichloroacetic acid (TCA) was added to a final concentration of 10% v/v and incubated overnight at 4°C. The samples were then centrifuged for 15 min at 3,220 g and 4°C, and the resulting protein pellets were washed in 100% ethanol for 1 h at 4°C, then centrifuged for 15 min at 3,220 g, 4°C, and dried overnight. The dried pellets were resuspended in 15 μl of 8 M urea and incubated for 30 min at room temperature, followed by the addition of 15 μl 2× TCA‐SDS sample buffer and boiled for 10 min. To visualize the secreted proteins, 15 μl was run on a 12% SDS–PAGE gel and stained with Coomassie Blue. EZ‐Run Prestained Protein Molecular Marker (Fisher) was used as a molecular weight marker.
Ex vivo human PMN infection
Staphylococcus aureus Newman toxinless strains (ΔΔΔΔ) were used that overexpress the indicated toxin loci under the control of the lukAB promoter as previously described 13, 37.
Plasmids encoding lukAB mut1 or lukAB mut2 derived from S. aureus RN4220 were electroporated into S. aureus strain Newman and validated by exoprotein profile and immunoblot analyses.
To infect primary human PMNs ex vivo, overnight cultures of each S. aureus Newman strain were grown in TSB at 37°C with 180 rpm shaking, then subcultured the following day at a 1:100 dilution into TSB, and grown for 5 h at 37°C with 180 rpm shaking. The cultures were centrifuged at 4°C for 5 min and 3,220 g, washed once in 1× PBS, and centrifuged again at 4°C for 5 min and 3,220 g. The bacterial pellets were resuspended in 1× PBS and normalized to 1.0 × 109 CFU ml−1 using a Genesys 20 Spectrophotometer (OD600 nm). PMNs were resuspended in RPMI media supplemented with 10% heat‐inactivated FBS, followed by the addition of the normalized bacteria at a final MOI per well of 100, 50, and 25 on a 96‐well flat bottom plate (Corning). The PMNs were infected for 3 or 4 h (for infections using Newman ΔΔΔΔ strains), or 2 h (for infections using WT Newman strains containing the plasmids encoding for the dominant negative LukAB proteins) at 37°C + 5% CO2 in the presence or absence of dominant‐negative proteins as indicated. The plates were centrifuged for 5 min at 387 g, 4°C; 25 μl of the infection supernatants was transferred into a plate containing 25 μl of CytoTox‐One Homogenous Membrane Integrity Assay (Promega) in black 96‐well plates to measure lactate dehydrogenase (LDH) release. LDH release was quantified per manufacturer's instructions using an EnVision 2103 Multilabel Reader (PerkinElmer).
Binding and competition assays
For binding/competition assays, primary human PMNs were seeded at 5 × 104 cells/well and a titration of unlabeled single subunits was added to the cells, immediately followed by the addition of GFP‐LukE (300 nM) and incubated for 30 min on ice. Cells were centrifuged for 5 min at 387 g, 4°C, washed once in FACS buffer (1× PBS + 2% heat‐inactivated FBS + 0.05% sodium azide), centrifuged again for 5 min at 387 g, 4°C, followed by fixing in 50 μl FACS fixation buffer (1× PBS + 2% heat‐inactivated FBS + 2% paraformaldehyde + 0.05% sodium azide). Fluorescence of cell‐bound toxins was analyzed using flow cytometry (LSRII with FACSDiva software; BD), where data are shown as the percentage of total GFP‐positive cells.
Recruitment assays
Primary human PMNs were seeded at 5 × 104 cells/well and a titration of unlabeled LukEmut1 or HlgAmut1 (or buffer as a negative control) was added to the cells and incubated for 30 min on ice, followed by the addition of GFP‐LukD (156.3 nM) for 30 min on ice. Cells were then centrifuged for 5 min at 387 g, 4°C, washed on ice in FACS buffer (1× PBS + 2% heat‐inactivated FBS + 0.05% sodium azide), centrifuged again for 5 min at 387 g, 4°C, followed by fixing in 50 μl FACS fixation buffer (1× PBS + 2% heat‐inactivated FBS + 2% paraformaldehyde + 0.05% sodium azide). Fluorescence of cell‐bound toxins was analyzed using flow cytometry (LSRII with FACSDiva software; BD), where data are shown as the percentage of total GFP‐positive cells.
Immunoblot assays
To detect toxin oligomers, toxin subunits were normalized to 0.44 mg ml−1, mixed 1:1 with the corresponding toxin subunit, and the volume brought up to 18 μl with 1× PBS. To favor the formation of oligomers in solution, 12 μl of 2‐methyl‐2,4‐pentanediol (MPD; Sigma) was added to a final concentration of 40% (v/v) per sample and incubated overnight at 4°C 6, 7, 8, 32, 44, 45. The following day, 4× SDS sample buffer was added to a final concentration of 1×, without boiling the sample, and 1 μg in a volume of 20 μl of each sample per well was electrophoresed on a 4–15% SDS–PAGE gel (Mini Protean TGX gels; Bio‐Rad) without boiling. The proteins were transferred to a 0.45‐μm nitrocellulose membrane (Bio‐Rad) for 1 h at 1 A and the membrane was blocked in blocking buffer (1× PBS + 0.01% Tween‐20 + 5% powdered milk + 0.05% sodium azide) for 1 h at RT while nutating, followed by the addition of mouse anti‐His primary antibody (1:3,000; Cell Sciences) to detect 6xHis‐tagged proteins and incubated overnight at 4°C while nutating. The membrane was rinsed thrice in wash buffer (1× PBS + 0.01% Tween‐20 + 0.05% sodium azide) the next day, followed by the addition of goat anti‐mouse HRP‐conjugated secondary antibody in 1× PBS + 0.01% Tween‐20 + 5% powdered milk + 0.05% sodium azide for 1 h at RT while nutating. The membrane was then washed thrice as before, followed by the addition of SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Scientific) for protein detection.
To detect LukA and LukB in strains used for ex vivo infections, the same experimental procedure as above was used, but with the following modifications: 5 μl of TCA‐treated samples was electrophoresed on a 12% SDS–PAGE gel. Rabbit anti‐LukA (1:5,000) and rabbit anti‐LukB (1:1,000) polyclonal antibodies in 1× PBS + 0.01% Tween‐20 + 5% powdered milk + 0.05% sodium azide were used as primary antibodies. Alexa Fluor 680‐conjugated goat anti‐rabbit IgG (1:25,000; Life Technologies) was used as secondary antibody, and the membranes were visualized using an Odyssey Infrared Imaging System (LI‐COR Biosciences).
Murine in vivo intoxications
Age‐matched (4‐ to 5‐week‐old) Swiss‐Webster female mice (Taconic; USA) were mixed together, then randomly allocated across experimental groups. All injections were done in a final volume of 100 μl. Time to acute intoxication was recorded after sacrifice of mice displaying signs of morbidity, including ruffled fur, hunched posture, paralysis, inability to walk, or inability to consume food or water. Similar results were obtained with Swiss‐Webster mice from Harlan laboratories (USA).
To evaluate the effects of the bi‐component leukocidins in vivo, mice were anesthetized by inhalation of isoflurane gas (2%) and the toxins administered systemically at the indicated doses via retro‐orbital injection. Mice were monitored for signs of morbidity and mortality, after which the mice were sacrificed and the time to acute morbidity was recorded.
To evaluate protection in vivo, mice were anesthetized by inhalation of isoflurane gas (2%) and the dominant‐negative toxins administered systemically at the indicated toxins via retro‐orbital injection, followed by retro‐orbital administration of a lethal dose of WT toxin at the indicated time post‐treatment (0, 1, 5, and 24 h). Mice were monitored as indicated above.
Murine in vivo infections
Age‐matched (4‐ to 5‐week‐old) Swiss‐Webster female mice (Harlan Laboratories) were mixed together, then randomly allocated across experimental groups to minimize cage or litter bias. To examine the survival of mice treated with different doses of LukEmut1LukDmut during bloodstream infection, mice were anesthetized intraperitoneally with 300 μl Avertin (2,2,2‐tribromoethanol dissolved in tert‐Amyl alcohol and diluted to a final concentration of 2.5% v/v in 0.9% sterile saline), followed by systemic (retro‐orbital) injection of 3.7 × 107 colony‐forming units (CFU) of S. aureus strain Newman and LukEmut1LukDmut administered simultaneously systemically via retro‐orbital injection at 0 h, followed by additional LukEmut1LukDmut administration at 24 and 48 h at the indicated doses. Mice were monitored for signs of morbidity and mortality, after which the mice were sacrificed and the time to acute morbidity was recorded.
To evaluate protection by LukEmut1LukDmut in vivo, age‐matched (4‐ to 5‐week‐old) Swiss‐Webster female mice (Harlan Laboratories) were mixed together, then randomly allocated across experimental groups to minimize cage or litter bias. Mice were anesthetized intraperitoneally with 300 μl Avertin, followed by systemic (retro‐orbital) injection of ~1.5 × 107 colony‐forming units (CFU) of S. aureus strain Newman and systemic injection of 10 μg per subunit of LukEmut1LukDmut. The same dose of LukEmut1LukDmut was also administered 24 and 48 h post‐infection, for a total of three treatments; 96 h post‐infection, mice were euthanized and the livers were harvested and homogenized in 1 ml of 1× PBS, followed by tenfold serial dilutions in 1× PBS, and the dilutions were plated in TSA plates. The plates were incubated overnight at 37°C to quantify bacterial burden.
Graphical and statistical analyses
Analyses of flow cytometric data were performed using FlowJo (Tree Star Software). Statistical significance was determined using Prism 6.0 g (GraphPad Software), with two‐way ANOVA with Tukey's post hoc test for multiple comparisons, log‐rank (Mantel–Cox) test, or Student's t‐test with SEM as indicated.
Author contributions
TRR and VJT conceived and designed experiments. TRR and AL generated mutants. TRR performed experiments and analyzed data. DBL performed structural analysis. FA contributed technical expertise. VJT supervised the project. TRR and VJT wrote the manuscript.
Conflict of interest
VJT, FA, and TRR are listed as inventors on patent applications filed by New York University School of Medicine, which are currently under commercial license to Janssen Biotech, Inc. The other authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Review Process File
Acknowledgements
We thank Rita Chan and Aidan O'Malley (NYU School of Medicine) for the purification of primary human PMNs and members of the Torres Laboratory for comments on the manuscript. This work was supported by grants from the National Institutes of Health (T32 AI007180 and F31 AI112290 to TRR; T32 AI007180 and F32 AI098395 to FA; and R01 AI099394 and R01 AI105129 to VJT). VJT is a Burroughs Wellcome Fund Investigator in the Pathogenesis of Infectious Diseases.
EMBO Reports (2016) 17: 428–440
See also: D Parker & A Prince (March 2016)
References
- 1. Chambers HF, Deleo FR (2009) Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat Rev Microbiol 7: 629–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nizet V (2007) Understanding how leading bacterial pathogens subvert innate immunity to reveal novel therapeutic targets. J Allergy Clin Immunol 120: 13–22 [DOI] [PubMed] [Google Scholar]
- 3. Spaan AN, Surewaard BG, Nijland R, van Strijp JA (2013) Neutrophils versus Staphylococcus aureus: a biological tug of war. Annu Rev Microbiol 67: 629–650 [DOI] [PubMed] [Google Scholar]
- 4. Alonzo F 3rd, Torres VJ (2014) The bicomponent pore‐forming leucocidins of Staphylococcus aureus . Microbiol Mol Biol Rev 78: 199–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. DuMont AL, Torres VJ (2014) Cell targeting by the Staphylococcus aureus pore‐forming toxins: it's not just about lipids. Trends Microbiol 22: 21–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yamashita K, Kawai Y, Tanaka Y, Hirano N, Kaneko J, Tomita N, Ohta M, Kamio Y, Yao M, Tanaka I (2011) Crystal structure of the octameric pore of staphylococcal gamma‐hemolysin reveals the beta‐barrel pore formation mechanism by two components. Proc Natl Acad Sci USA 108: 17314–17319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yamashita D, Sugawara T, Takeshita M, Kaneko J, Kamio Y, Tanaka I, Tanaka Y, Yao M (2014) Molecular basis of transmembrane beta‐barrel formation of staphylococcal pore‐forming toxins. Nat Commun 5: 4897 [DOI] [PubMed] [Google Scholar]
- 8. Badarau A, Rouha H, Malafa S, Logan DT, Hakansson M, Stulik L, Dolezilkova I, Teubenbacher A, Gross K, Maierhofer B et al (2015) Structure‐function analysis of heterodimer formation, oligomerization, and receptor binding of the Staphylococcus aureus bi‐component toxin LukGH. J Biol Chem 290: 142–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yoong P, Torres VJ (2013) The effects of Staphylococcus aureus leukotoxins on the host: cell lysis and beyond. Curr Opin Microbiol 16: 63–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Alonzo F 3rd, Kozhaya L, Rawlings SA, Reyes‐Robles T, DuMont AL, Myszka DG, Landau NR, Unutmaz D, Torres VJ (2013) CCR5 is a receptor for Staphylococcus aureus leukotoxin ED. Nature 493: 51–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Spaan AN, Henry T, van Rooijen WJ, Perret M, Badiou C, Aerts PC, Kemmink J, de Haas CJ, van Kessel KP, Vandenesch F et al (2013) The staphylococcal toxin Panton‐Valentine Leukocidin targets human C5a receptors. Cell Host Microbe 13: 584–594 [DOI] [PubMed] [Google Scholar]
- 12. DuMont AL, Yoong P, Day CJ, Alonzo F 3rd, McDonald WH, Jennings MP, Torres VJ (2013) Staphylococcus aureus LukAB cytotoxin kills human neutrophils by targeting the CD11b subunit of the integrin Mac‐1. Proc Natl Acad Sci USA 110: 10794–10799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Reyes‐Robles T, Alonzo F 3rd, Kozhaya L, Lacy DB, Unutmaz D, Torres VJ (2013) Staphylococcus aureus leukotoxin ED targets the chemokine receptors CXCR1 and CXCR2 to kill leukocytes and promote infection. Cell Host Microbe 14: 453–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Spaan AN, Vrieling M, Wallet P, Badiou C, Reyes‐Robles T, Ohneck EA, Benito Y, de Haas CJ, Day CJ, Jennings MP et al (2014) The staphylococcal toxins gamma‐haemolysin AB and CB differentially target phagocytes by employing specific chemokine receptors. Nat Commun 5: 5438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pier GB (2013) Will there ever be a universal Staphylococcus aureus vaccine? Hum Vaccin Immunother 9: 1865–1876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. David MZ, Daum RS (2010) Community‐associated methicillin‐resistant Staphylococcus aureus: epidemiology and clinical consequences of an emerging epidemic. Clin Microbiol Rev 23: 616–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Thammavongsa V, Kim HK, Missiakas D, Schneewind O (2015) Staphylococcal manipulation of host immune responses. Nat Rev Microbiol 13: 529–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Park S, Gerber S, Lee JC (2014) Antibodies to Staphylococcus aureus serotype 8 capsular polysaccharide react with and protect against serotype 5 and 8 isolates. Infect Immun 82: 5049–5055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fattom AI, Sarwar J, Ortiz A, Naso R (1996) A Staphylococcus aureus capsular polysaccharide (CP) vaccine and CP‐specific antibodies protect mice against bacterial challenge. Infect Immun 64: 1659–1665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim HK, Emolo C, DeDent AC, Falugi F, Missiakas DM, Schneewind O (2012) Protein A‐specific monoclonal antibodies and prevention of Staphylococcus aureus disease in mice. Infect Immun 80: 3460–3470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kim HK, Cheng AG, Kim HY, Missiakas DM, Schneewind O (2010) Nontoxigenic protein A vaccine for methicillin‐resistant Staphylococcus aureus infections in mice. J Exp Med 207: 1863–1870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kuklin NA, Clark DJ, Secore S, Cook J, Cope LD, McNeely T, Noble L, Brown MJ, Zorman JK, Wang XM et al (2006) A novel Staphylococcus aureus vaccine: iron surface determinant B induces rapid antibody responses in rhesus macaques and specific increased survival in a murine S. aureus sepsis model. Infect Immun 74: 2215–2223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stranger‐Jones YK, Bae T, Schneewind O (2006) Vaccine assembly from surface proteins of Staphylococcus aureus . Proc Natl Acad Sci USA 103: 16942–16947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vernachio J, Bayer AS, Le T, Chai YL, Prater B, Schneider A, Ames B, Syribeys P, Robbins J, Patti JM (2003) Anti‐clumping factor A immunoglobulin reduces the duration of methicillin‐resistant Staphylococcus aureus bacteremia in an experimental model of infective endocarditis. Antimicrob Agents Chemother 47: 3400–3406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schaffer AC, Solinga RM, Cocchiaro J, Portoles M, Kiser KB, Risley A, Randall SM, Valtulina V, Speziale P, Walsh E et al (2006) Immunization with Staphylococcus aureus clumping factor B, a major determinant in nasal carriage, reduces nasal colonization in a murine model. Infect Immun 74: 2145–2153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sampedro GR, DeDent AC, Becker RE, Berube BJ, Gebhardt MJ, Cao H, Bubeck Wardenburg J (2014) Targeting Staphylococcus aureus alpha‐toxin as a novel approach to reduce severity of recurrent skin and soft‐tissue infections. J Infect Dis 210: 1012–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kennedy AD, Bubeck Wardenburg J, Gardner DJ, Long D, Whitney AR, Braughton KR, Schneewind O, DeLeo FR (2010) Targeting of alpha‐hemolysin by active or passive immunization decreases severity of USA300 skin infection in a mouse model. J Infect Dis 202: 1050–1058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Adhikari RP, Karauzum H, Sarwar J, Abaandou L, Mahmoudieh M, Boroun AR, Vu H, Nguyen T, Devi VS, Shulenin S et al (2012) Novel structurally designed vaccine for S. aureus alpha‐hemolysin: protection against bacteremia and pneumonia. PLoS ONE 7: e38567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Delfani S, Mohabati Mobarez A, Imani Fooladi AA, Amani J, Emaneini M (2015) Protection of mice against Staphylococcus aureus infection by a recombinant protein ClfA‐IsdB‐Hlg as a vaccine candidate. Med Microbiol Immunol 205: 47–55 [DOI] [PubMed] [Google Scholar]
- 30. Rouha H, Badarau A, Visram ZC, Battles MB, Prinz B, Magyarics Z, Nagy G, Mirkina I, Stulik L, Zerbs M et al (2015) Five birds, one stone: neutralization of alpha‐hemolysin and 4 bi‐component leukocidins of Staphylococcus aureus with a single human monoclonal antibody. MAbs 7: 243–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Aman MJ, Adhikari RP (2014) Staphylococcal bicomponent pore‐forming toxins: targets for prophylaxis and immunotherapy. Toxins (Basel) 6: 950–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Karauzum H, Adhikari RP, Sarwar J, Devi VS, Abaandou L, Haudenschild C, Mahmoudieh M, Boroun AR, Vu H, Nguyen T et al (2013) Structurally designed attenuated subunit vaccines for S. aureus LukS‐PV and LukF‐PV confer protection in a mouse bacteremia model. PLoS ONE 8: e65384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Laventie BJ, Rademaker HJ, Saleh M, de Boer E, Janssens R, Bourcier T, Subilia A, Marcellin L, van Haperen R, Lebbink JH et al (2011) Heavy chain‐only antibodies and tetravalent bispecific antibody neutralizing Staphylococcus aureus leukotoxins. Proc Natl Acad Sci USA 108: 16404–16409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fowler VG, Allen KB, Moreira ED, Moustafa M, Isgro F, Boucher HW, Corey GR, Carmeli Y, Betts R, Hartzel JS et al (2013) Effect of an investigational vaccine for preventing Staphylococcus aureus infections after cardiothoracic surgery: a randomized trial. JAMA 309: 1368–1378 [DOI] [PubMed] [Google Scholar]
- 35. Rupp ME, Holley HP Jr, Lutz J, Dicpinigaitis PV, Woods CW, Levine DP, Veney N, Fowler VG Jr (2007) Phase II, randomized, multicenter, double‐blind, placebo‐controlled trial of a polyclonal anti‐Staphylococcus aureus capsular polysaccharide immune globulin in treatment of Staphylococcus aureus bacteremia. Antimicrob Agents Chemother 51: 4249–4254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Moustafa M, Aronoff GR, Chandran C, Hartzel JS, Smugar SS, Galphin CM, Mailloux LU, Brown E, Dinubile MJ, Kartsonis NA et al (2012) Phase IIa study of the immunogenicity and safety of the novel Staphylococcus aureus vaccine V710 in adults with end‐stage renal disease receiving hemodialysis. Clin Vaccine Immunol 19: 1509–1516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. DuMont AL, Yoong P, Liu X, Day CJ, Chumbler NM, James DB, Alonzo F 3rd, Bode NJ, Lacy DB, Jennings MP et al (2014) Identification of a crucial residue required for Staphylococcus aureus LukAB cytotoxicity and receptor recognition. Infect Immun 82: 1268–1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Diep BA, Gill SR, Chang RF, Phan TH, Chen JH, Davidson MG, Lin F, Lin J, Carleton HA, Mongodin EF et al (2006) Complete genome sequence of USA300, an epidemic clone of community‐acquired meticillin‐resistant Staphylococcus aureus . Lancet 367: 731–739 [DOI] [PubMed] [Google Scholar]
- 39. Deleo FR, Otto M, Kreiswirth BN, Chambers HF (2010) Community‐associated meticillin‐resistant Staphylococcus aureus . Lancet 375: 1557–1568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Voyich JM, Braughton KR, Sturdevant DE, Whitney AR, Said‐Salim B, Porcella SF, Long RD, Dorward DW, Gardner DJ, Kreiswirth BN et al (2005) Insights into mechanisms used by Staphylococcus aureus to avoid destruction by human neutrophils. J Immunol 175: 3907–3919 [DOI] [PubMed] [Google Scholar]
- 41. DuMont AL, Nygaard TK, Watkins RL, Smith A, Kozhaya L, Kreiswirth BN, Shopsin B, Unutmaz D, Voyich JM, Torres VJ (2011) Characterization of a new cytotoxin that contributes to Staphylococcus aureus pathogenesis. Mol Microbiol 79: 814–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Spaan AN, Schiepers A, de Haas CJ, van Hooijdonk DD, Badiou C, Contamin H, Vandenesch F, Lina G, Gerard NP, Gerard C et al (2015) Differential Interaction of the Staphylococcal Toxins Panton‐Valentine Leukocidin and gamma‐Hemolysin CB with Human C5a Receptors. J Immunol 195: 1034–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. DuMont AL, Yoong P, Surewaard BG, Benson MA, Nijland R, van Strijp JA, Torres VJ (2013) Staphylococcus aureus elaborates leukocidin AB to mediate escape from within human neutrophils. Infect Immun 81: 1830–1841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gouaux JE, Braha O, Hobaugh MR, Song L, Cheley S, Shustak C, Bayley H (1994) Subunit stoichiometry of staphylococcal alpha‐hemolysin in crystals and on membranes: a heptameric transmembrane pore. Proc Natl Acad Sci USA 91: 12828–12831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Song L, Hobaugh MR, Shustak C, Cheley S, Bayley H, Gouaux JE (1996) Structure of staphylococcal alpha‐hemolysin, a heptameric transmembrane pore. Science 274: 1859–1866 [DOI] [PubMed] [Google Scholar]
- 46. Loffler B, Hussain M, Grundmeier M, Bruck M, Holzinger D, Varga G, Roth J, Kahl BC, Proctor RA, Peters G (2010) Staphylococcus aureus Panton‐Valentine leukocidin is a very potent cytotoxic factor for human neutrophils. PLoS Pathog 6: e1000715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sellman BR, Mourez M, Collier RJ (2001) Dominant‐negative mutants of a toxin subunit: an approach to therapy of anthrax. Science 292: 695–697 [DOI] [PubMed] [Google Scholar]
- 48. Vinion‐Dubiel AD, McClain MS, Czajkowsky DM, Iwamoto H, Ye D, Cao P, Schraw W, Szabo G, Blanke SR, Shao Z et al (1999) A dominant negative mutant of Helicobacter pylori vacuolating toxin (VacA) inhibits VacA‐induced cell vacuolation. J Biol Chem 274: 37736–37742 [DOI] [PubMed] [Google Scholar]
- 49. Adhikari RP, Kort T, Shulenin S, Kanipakala T, Ganjbaksh N, Roghmann MC, Holtsberg FW, Aman MJ (2015) Antibodies to S. aureus LukS‐PV attenuated subunit vaccine neutralize a broad spectrum of canonical and non‐canonical bicomponent leukotoxin pairs. PLoS ONE 10: e0137874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Alonzo F 3rd, Benson MA, Chen J, Novick RP, Shopsin B, Torres VJ (2012) Staphylococcus aureus leucocidin ED contributes to systemic infection by targeting neutrophils and promoting bacterial growth in vivo. Mol Microbiol 83: 423–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Melehani JH, James DB, DuMont AL, Torres VJ, Duncan JA (2015) Staphylococcus aureus Leukocidin A/B (LukAB) Kills Human Monocytes via Host NLRP3 and ASC when Extracellular, but Not Intracellular. PLoS Pathog 11: e1004970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Perret M, Badiou C, Lina G, Burbaud S, Benito Y, Bes M, Cottin V, Couzon F, Juruj C, Dauwalder O et al (2012) Cross‐talk between S. aureus leukocidins‐intoxicated macrophages and lung epithelial cells triggers chemokine secretion in an inflammasome‐dependent manner. Cell Microbiol 14: 1019–1036 [DOI] [PubMed] [Google Scholar]
- 53. Holzinger D, Gieldon L, Mysore V, Nippe N, Taxman DJ, Duncan JA, Broglie PM, Marketon K, Austermann J, Vogl T et al (2012) Staphylococcus aureus Panton‐Valentine leukocidin induces an inflammatory response in human phagocytes via the NLRP3 inflammasome. J Leukoc Biol 92: 1069–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Liang X, Yan M, Ji Y (2009) The H35A mutated alpha‐toxin interferes with cytotoxicity of staphylococcal alpha‐toxin. Infect Immun 77: 977–983 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Table EV2
Review Process File