

Abstract
Diabetes is defined as a disease of hyperglycemic metabolic disorder caused by impaired insulin action or low insulin secretion, resulting in the occurrence of vascular complications. Based on this definition, diabetes therapy has long been oriented to correct hyperglycemia against the specific complications of diabetes. This definition has posed some difficulties, however, in understanding of the pathophysiology of this complicated disease and as such in the establishment of an effective treatment. With continuing efforts to explore the structural basis for diabetes onset and methodological development of immunohistochemistry, progressive decline of β‐cells is now established as a salient feature of type 2 diabetes. Accordingly, diabetes therapy has now turned out to protect β‐cells concurrently with the correction of hyperglycemia. Together with this effort, exploration of the means to regenerate β‐cells or to supply new β‐cells by, for example, induced pluripotential stem cells, are vigorously made with the search for the mechanism of β‐cell decline in diabetes. In the present review, we describe the advances in the islet pathology in type 2 diabetes with special reference to the dynamic alterations of islet endocrine cells in the milieu of maturation, obesity, aging and ethnic differences. The effect of amyloid deposition is also discussed. We hope it will help with understanding the pathophysiology of diabetes, and suggest the future direction of diabetes treatment.
Keywords: Islet, Pathology, Type 2 diabetes
Introduction
The description of pancreatic islets by Paul Langerhans1 was a milestone in the history of diabetes research, and contributed to the future discovery of insulin by Banting and Best2 in 1921. Until the 21st century, however, there had not been much information on the pathological changes of islets in type 2 diabetes, except for amyloid deposition reported by Opie in 19013. In contrast to the robust β‐cell loss with lymphocytic infiltration, called insulitis, detected in the pancreas of type 1 diabetes patients, findings on the islets of type 2 diabetes patients were not remarkable, and the distinction of type 1 diabetes from type 2 diabetes was often difficult, in part due to complex clinical manifestations. With the availability of accurate measurements of blood glucose4 and radioimmunoassay of insulin5, the profile of diabetes was first characterized by hyperglycemia and low insulin secretion6. Thus, diabetes was defined merely by clinical profile as a disease of chronic hyperglycemia as a result of impaired insulin action. No distinct pathological background underlying such a clinical profile was provided until the end of the 20th century. A brief history of the progress in islet pathology is summarized in Table 1.
Table 1.
History of islet pathology in human diabetes reported as original publications
| 1869 | Langerhans P. Discovery of pancreatic islets, Thesis, Gustav Lange1 |
| 1893 | Laguesse E. Named islet of Langerhans, Comp Rend Soc Biol 5: 622, 1893 |
| 1889 | von Mering, Minkowski O. Canine diabetes after pancreatectomy. Zbl Klin Med 10: 393, 1889 |
| 1901 | Opie EL. Hyaline changes in the islet of diabetes3 |
| 1921 | Banting FG, Best CH. Discovery of insulin2 |
| 1956 | Mclean N, Ogilvie RF. Islet area reduction in diabetes10 |
| 1965 | Gepts W. Insulitis in type 1 diabetes (Pathologic anatomy of the pancreas in human diabetes. Diabetes 14: 619–633, 1965) |
| 1977 | Saito K et al. Reduced islet area in type 1 and type 2 diabetes but no alteration of cell composition11 |
| 1982 | Stefan Y et al. No significant islet changes in European type 2 diabetes36 |
| 1983 | Rahier J et al. No significant changes in β cells in European type 2 diabetes37 |
| 1987 | Westermark P, Cooper GJ. Discovery of amylin in the hyalinized islets (Westermark P et al. Proc Natl Acad Sci USA 84: 3881–3885, 1987. Cooper GJ et al. Proc Natl Acad Sci USA 84: 8628–8632, 1987) |
| 2002 | Sakuraba H et al. Reduction of β cell mass in Japanese type 2 diabetes31 |
| 2003 | Butler A et al. Reduction of β cell volume density in American type 2 diabetes and subjects with impaired fasting glucose39 |
| 2003 | Yoon KH et al. Reduction of β cell volume density and increase in α cell volume density in Korean type 2 diabetes38 |
| 2007 | Ehses JA et al. Inflammation in the islet of type 2 diabetes. (Ehses JA et al. Diabetes 56: 2356–2370, 2007) |
| 2007 | Rahier J et al. Reduction of β cell volume density in European type 2 diabetes40 |
| 2007 | Henquin JC, Rahier J. Increase in α cell mass in European type 2 diabetes47 |
| 2014 | Mezza T et al. Transdifferentiation of islet endocrine cells in IGT49 |
| 2014 | Mizukami H et al. Involvement of oxidative stress, ER‐stress, and autophagy deficits in type 2 diabetic islets24 |
Diabetes is a heterogeneous disease and its classification is not straightforward7. Type 2 diabetes is mainly divided into the lean and obese type, but this separation is only provisional. Japanese type 2 diabetes is in general lean, and the average body mass index (BMI) is 22–25, whereas USA or European types are mostly obese with a BMI over 308, 9. It is therefore natural to consider that islet changes should be either primarily or secondarily different among various types of diabetes. It has been difficult to carry out accurate morphometric analysis on randomly distributed islets with a non‐uniform irregular shape. Immunohistochemical identification of islet endocrine cell type by use of high‐quality antibodies is essential to the precision of the data. There are some classic studies that disclosed reduced islet area in patients with type 2 diabetes compared with non‐diabetic patients, but the findings were not reproducible, because endocrine cells were determined by non‐specific histochemical methods, such as Gomori–Aldehyde–Fuchsin staining10, 11. It has only been recently possible to evaluate cell composition and cellular mass of islets in patients with diabetes on immunostained sections. With the availability of markers for cell growth or demise and related molecules, numerous findings of islet changes in type 2 diabetes have emerged. Nevertheless, systematic organization of dynamic alterations of islet endocrine cells seems to be required for a better understanding of the pathophysiology of diabetes. In the present review, we attempted to summarize recent key findings on islet pathology in type 2 diabetes, and we discuss their implication in the pathophysiology of diabetes and the treatment strategy.
Growth, Replication and Aging of β‐Cells
The genesis of pancreatic islets commences around the embryonic stage of 12 weeks of gestation. At 13–16 weeks of gestation, primitive endocrine precursor cells budding from the wall of the duct constitutes aggregates of cells, resulting in an initial primordium of islets (Figure 1)12. At 17–20 weeks of gestation, the direct connection between the islet and duct wall is lost, where the newly‐formed islets become isolated. At this stage, the islet consists mainly of the cells containing pancreatic polypeptide, somatostatin and glucagon, which derive from immature precursor cells. At 21–26 weeks of gestation, β‐cells distribute into the central area of the islets, which form typically for the islets seen in adults13. After birth, when meal intake starts, pancreatic exocrine tissues rapidly expand to increase the pancreatic weight, with its peak around 10–20 years of age. During maturation, β cells and islets also grow in size composed of replicated β‐cells by the age of 2 years. Occupancy of β‐cells in the pancreatic parenchyma (β‐cell volume density [Vβ]) reaches the adult level at the age of 5–10 years14. In the third decade of life, pancreatic weight becomes stable, almost being consistent until the 60s15. Thereafter, the pancreatic weight decreases slowly with progressive regression after the sixth decade of life.
Figure 1.
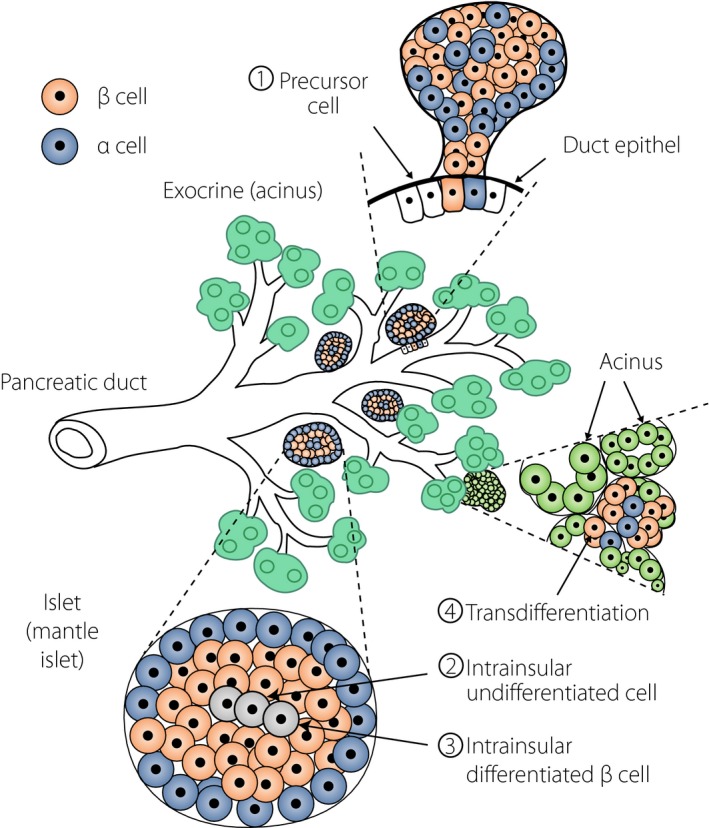
Development of new islets (islet neogenesis). New islets derive mostly from primitive precursor cells located in the ductal wall that form budding of aggregated endocrine cells as a primordium of islets (①). The islets grow in size and detach from the duct wall to become isolated. Within the islet, undifferentiated cells develop to differentiate into specific endocrine cell types (②), such as β‐cells or α‐cells, or others. Islet β‐cells can also replicate themselves to provide new β cells (③). Under metabolic stress or environmental stimuli, acinar cells can transdifferentiate into islet endocrine cells (④).
Cell replication potential or regeneration capacity is crucial for the development of the islet and for the adaptation of endocrine cells to environmental insults. Several methods are available for identifying the replicating cells. 3H‐tymidine uptake and bromo‐deoxy‐uridine tracing are used for the detection of deoxyribonucleic acid (DNA) duplication, but are only feasible for animal studies, except for some human cases treated with radioisotope‐labeled compounds before death, because markers need to be applied before death16, 17, 18, 19. On the formalin‐fixed paraffin sections, Ki67 protein (MIB‐1 index) and proliferating cell nuclear antigen (PCNA) can be applied to detect for replicating cells14, 20, 21. Ki67 is a nuclear antigen entering cell cycle (G1 to S‐ and G2 and M stage), but not necessarily indicating the actuarial cell growth22. PCNA is expressed only in a partial period of the cell cycle, and is not always reproducible for their positivity. Currently, Ki67 is most commonly used for quantification of replicating cells, but still requires caution for its implication.
The replication rate of β‐cells is well preserved during several years of the postnatal period at a rate of 3–4%. After 10 years‐of‐age, it is reduced to very low level at 0.05–0.1%, and remains low thereafter, at a rate of 0.01–0.1%14. Very recently, Sullivan et al.23 commented on the negative effects of delayed fixation on the Ki67 positivity of islet β‐cells, which in fact possess considerable potential for replication even in adults. Consistent with this contention, in our study of pancreases that were obtained within 5 h after death, the Ki67 positivity of islet cells was constantly 0.2–0.4% in patients either with or without diabetes15, 24, similar to the level detected in fresh pancreatic tissues. There was no significant difference in the Ki67 positivity between patients with diabetes (0.32 ± 0.12%) and without diabetes (0.41 ± 0.25%)24. Cnop et al.25 studied β‐cell life by examining the accumulation of lipofuscin granules by electron microscopic investigations. They found that the half‐life of β‐cells in 12‐month‐old rats was calculated to be approximately 30–60 days. In contrast to rats, the lifespan of human β‐cells was estimated to be more than 20 years. They further concluded that there was no apparent influence of obesity, insulin resistance or diabetes mellitus on the number of accumulated lipofuscin granules, which did not affect the replicating capacity of β‐cells25, 26. Hence, although still controversial, the lifespan of human β‐cells seems to be extremely long, but they still preserve replicating potential, in cases of pregnancy or hepatic disorders27, 28, 29.
The β‐cell replication depends on cell kinetics regulated by cyclin‐related molecules. The cell cycle for regeneration is suppressed by the molecules of p16, p26, p27 and cyclin D3, all of which are upregulated with increasing age, providing negative impacts on the regenerative potential for adult β‐cells20, 22. It is also of note that the expression of pancreatic and duodenal homeobox‐1 (PDX‐1), a key transcription factor for β‐cell differentiation, is reduced in adult β‐cells compared with young people14, 15. It was also shown that exposure to oxidative stress suppressed the expression of PDX‐1, which could in turn contribute to impaired insulin secretion and lowered replication of β‐cells30. In conjunction with these results, in the islet β‐cells of aged subjects, expression of PDX‐1 is reduced together with enhanced expression of markers of oxidative stress‐induced DNA injury, known as γH2AX or 8‐hydroxydeoxyguanosine24, 31.
Compensatory β‐Cell Hyperplasia in Obesity
Obesity causes insulin resistance that gives rise to hyperinsulinemia to maintain glucose homeostasis. In fact, markedly enhanced insulin secretion was a feature in USA and European obese subjects on glucose challenge9. To accommodate the augmented insulin secretion, there needs to be compensatory β‐cell hyperplasia in obese subjects32. In fact, a recent morphometric study33 disclosed a significant increase in β‐cell volume density in non‐diabetic obese subjects by approximately 1.5‐fold compared with lean non‐diabetic healthy Caucasians, whose β‐cell volume density increases in parallel with increasing BMI (Figure 2). With prolongation of such a period, it often becomes difficult for β‐cells to preserve continuous insulin secretion, eventually resulting in overt diabetes32. In contrast to obese Caucasians, there is no obvious evidence of hyperinsulinemia in Japanese subjects with obesity or impaired glucose tolerance (IGT)34. The difference in insulin secretion between Caucasians and Japanese might be relevant to the degree of obesity, because the average BMI in the former patients with type 2 diabetes exceeds 30, whereas that in the latter patients with type 2 diabetes or IGT is around 25, not sufficient to require compensatory β cell hyperplasia8, 15. Alternatively, β‐cell replicative capacity might be limited in obese Japanese. To support this contention, Kou et al.35 could not find a significant increase in β‐cell volume density in Japanese subjects with high BMI. In our own studies, there was no significant correlation between BMI and β‐cell volume density15. Because the number of evaluated Japanese subjects with high BMI is still small, it remains unclear whether there are ethnic differences in the replicating capacity of β‐cells between Caucasians and Japanese.
Figure 2.
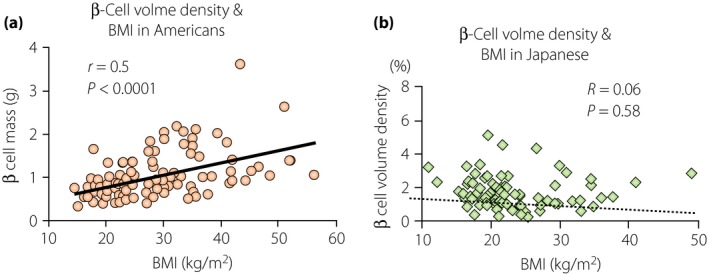
Ethnic differences in the relationship of β‐cell volume density with body mass index (BMI) between (a) USA patients and (b) Japanese subjects. (a) In USA subjects, there is a close correlation between β‐cell volume density and BMI, indicating compensatory hyperplasia in response to obesity in USA subjects33, (b) whereas this is not the case in Japanese subjects15. Thus, β‐cell plasticity in Japanese people is limited, accounting for susceptibility to diabetes in Japanese people.
β‐Cell Changes in Type 2 Diabetic Patients
Marked β‐cell loss with lymphocytic infiltration (insulitis) has been long known as a characteristic feature in type 1 diabetes, whereas no specific pathological changes in the islets were reported for type 2 diabetes, except for amyloid deposition, until the 21st century. With the introduction of immunohistochemistry for the identification of endocrine cells, two European studies by Stefan et al.36 and Rahier et al.37 were carried out on quantitative changes of islet endocrine cells in patients with type 2 diabetes. They could not find any significant difference in the β‐cell volume density (%; occupancy of β‐cells in pancreas parenchyma), β‐cell mass (g or mL; weight or volume obtained by multiplication of volume density by pancreas weight or volume) or β‐cell occupancy in the islets (%) between diabetic and non‐diabetic healthy control subjects. After these publications, there continued to be reports that could not show significant lesions in patients with type 2 diabetes, rendering the definition of diabetes as long‐term metabolic abnormalities caused by impaired insulin action.
From our survey of the pancreases obtained from Japanese patients with type 2 diabetes, we hinted that there must have been methodological problems for the measurement of β‐cell area in previous studies10, 11. We also considered that comparison of the data should be carefully matched for the age and site of the pancreas, because the tissues are confounded by senile atrophic or fibrotic changes in the elderly, and there is a topographic difference in the islet endocrine cell population from site to site of the pancreas. In our study on the body and tail of the pancreas obtained from fresh autopsy cases below the age 70 years, there was a specific reduction of β‐cell mass by 30% in patients with type 2 diabetes31. After this report, similar findings were presented from Korean patients with type 2 diabetes38 showing approximately 40% reduction of β‐cell volume density. In the same year, the results of USA diabetic patients were reported from a University of California, Los Angeles group, who divided diabetic patients into two groups of obese and lean patients, and compared the data with age‐ and weight‐matched non‐diabetic subjects (Figure 3)39. They found a significant reduction of β‐cell volume density in either the obese or lean type diabetic group by 63% and 41%, respectively. Furthermore, patients with impaired fasting glucose also showed 40% reduction of β‐cell volume density, showing that type 2 diabetes is characterized by a progressive decline of β‐cells from the prediabetic stage. Thereafter, the reduction of β‐cell volume density was confirmed in European people with diabetes40. From these lines of evidence, there emerged a world consensus that progressive β‐cell decline is a characteristic of islet pathology in type 2 diabetes (Figure 4)41, 42.
Figure 3.
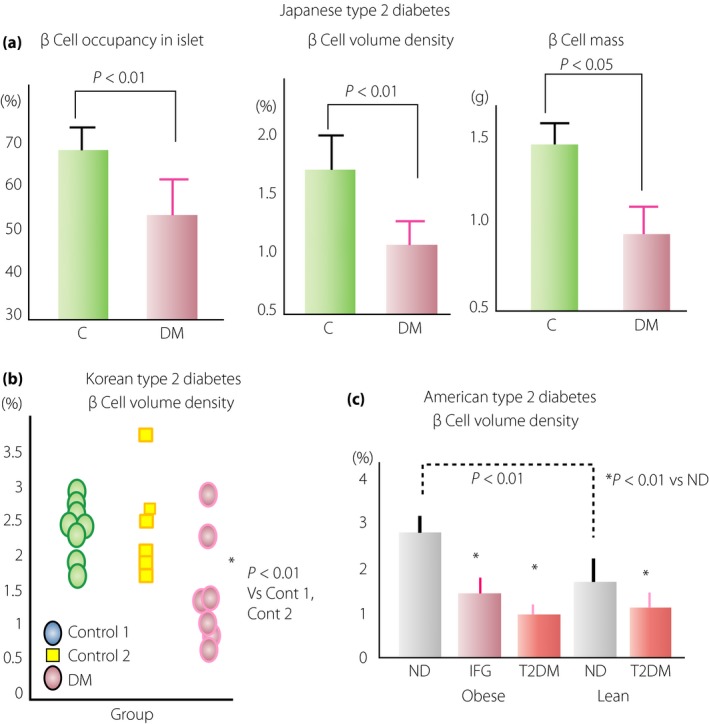
Reduced β‐cell volume density in people with type 2 diabetes mellitus (DM). (a) A specific reduction of β‐cell volume density and β‐cell mass was detected in Japanese patients with type 2 diabetes (lean) by morphometric analysis reported in 200231. There was no significant decrease in islet volume density, but β‐cell volume density and β‐cell mass were significantly reduced by 30% and 35%, respectively, in patients with type 2 diabetes compared with those in non‐diabetic (ND) subjects. (b) Reduced β‐cell volume density shown in Korean patients with type 2 diabetes reported in 200339, showing an approximately 40% decrease, similar to that in Japanese patients with type 2 diabetes, compared with non‐diabetic autopsy cases (control 1, □) or surgical cases (control 2, ○). (c) In the same year, the data of USA patients with type 2 diabetes were reported from University of California, Los Angeles, showing the reduction of β‐cell volume density39. In this report, both the obese type and lean type of USA patients with type 2 diabetes showed about 60% and 40% reduction of β‐cell volume density, respectively, compared with their respective weight‐matched non‐diabetic control subjects. Furthermore, they also found a significant reduction of β‐cell volume density in subjects with impaired fasting glucose (IFG). In addition, there was a significantly greater value of β‐cell volume density in non‐diabetic obese subjects compared with non‐diabetic lean subjects, indicating compensatory β‐cell hyperplasia in non‐diabetic obese subjects. C, control.
Figure 4.
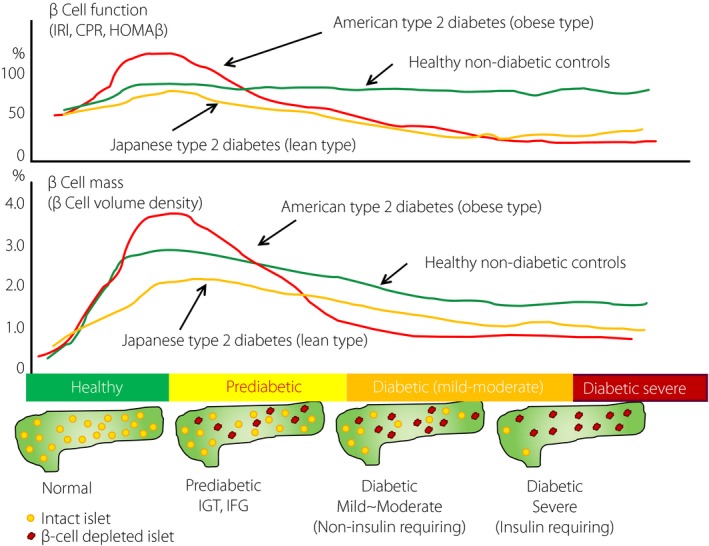
Natural history of type 2 diabetes and differences between USA obese type 2 diabetes patients and Japanese lean type 2 diabetes patients. In the prediabetic stage of impaired glucose tolerance (IGT) or before impaired fasting glucose (IFG), β‐cell function is augmented to compensate insulin resistance caused by obesity in USA people, whereas such compensation is obscure in Japanese people. Consistent with β‐cell function, β‐cell mass is increased to adapt to the increased demand in USA obese people with prediabetes. With progression to IFG and overt diabetes, progressive decline of β‐cells underlies the disease. In contrast to USA people, there is no obvious increase in β‐cell mass in Japanese people with prediabetes, and the β‐cell decline precedes long before overt diabetes. CPR, C‐peptide immunoreactivity; HOMA‐β, homeostasis model assessment of β‐cell function; IRΙ, immunoreactive insulin.
Can β‐Cell Decline Account for the Onset of Overt Diabetes?
It is natural to assume that the decrease in β‐cell volume density (β‐cell decline) by 30% detected in Japanese patients with type 2 diabetes contributes to the impaired insulin secretion. It seems to be impossible, however, to account for the onset of diabetes by β‐cell loss. The β‐cell volume density reflects merely the percentage area of insulin‐positive cells, not indicating the capacity of insulin secretion. By assuming that those β‐cells are able to secrete insulin, the β‐cell volume density multiplied by pancreas weight is called ‘functional β‐cell mass.’ In healthy animals that underwent partial pancreatectomy, the diabetic condition (hyperglycemia/glycosuria) manifests when the resected portion exceeds 65–70%43, 44. Thus, our body is protected from diabetes when the intact pancreas remains at least 30–35%, even if the rest of the pancreas is replaced by neoplasms or destroyed by inflammation or vascular occlusion. If this is the case, merely 30–35% loss of β‐cell volume density detected in Japanese patients with type 2 diabetes cannot account for the diabetes onset. It is thus likely that β‐cell dysfunction contributes to the onset of overt diabetes. It was also shown that the extent of β cell loss parallels with increased HbA1c values, indicating that severity of diabetes is relevant to β‐cell loss24. As the extent of β‐cell deficit is greater in obese USA patients with diabetes compared with lean diabetic patients, the surviving β‐cells might be strong enough to secrete more insulin in the former than the latter39.
Mechanisms of β Cell Loss in Diabetes
There have been a number of studies that explored the mechanism of β‐cell loss using experimental or spontaneously diabetic animal models or in vitro studies using isolated islets. As β‐cell mass is strictly regulated by the balance between cell death and regenerative capacity, either excessive cell death or poor replicating capacity results in the decrease in β‐cell mass. Based on molecular biological studies, β‐cell death in diabetic animal models has been attributed to oxidative stress and endoplasmic reticulum (ER) stress, as well as autophagy deficits30, 41. There is not much information, however, on which variables are most responsible for β‐cell loss in patients with type 2 diabetes. The reason for this is due in part to the difficulty in gaining access to fresh human pancreases or in carrying out longitudinal studies. Nevertheless, the advent of new markers for cell injury and the increase in study subjects will possibly provide valuable information for the process of β cell disappearance in type 2 diabetic islets24. We attempted to clarify to which extent the markers of oxidative stress, ER stress or autophagy deficits correlate with the decrease in β‐cell volume density in Japanese patients with type 2 diabetes. 8‐Hydroxydeoxyguanosine and γH2AX (phosphor S139 protein) were used as a marker of oxidative stress‐induced DNA injury, CCAAT‐enhancer binding protein‐β as ER stress and P62 as autophagy deficits by immunohistochemical analyses, respectively. We found enhanced expression of 8‐hydroxydeoxyguanosine‐positive and γH2AX‐positive cells in the islet of patients with diabetes, whereas they were almost nil in non‐diabetic healthy subjects, and the frequency of positive cells closely correlated with the reduced β‐cell volume density24, 31. In contrast, diabetic islets also showed enhanced expression of CCAAT‐enhancer binding protein‐β and P62‐positive cells, but there was no significant relationship between the positive rate of these markers and the extent of reduced β‐cell volume density24. Hierarchical cluster analysis showed that subjects positive for all three factors (γH2AX, CCAAT‐enhancer binding protein‐β, P62) showed the most severe loss of β‐cells, whereas subjects negative for these three factors showed only modest loss of β cells (Figure 5). Thus, it is conceivable that oxidative stress damage has the strongest impact on the β cell loss in Japanese patients with type 2 diabetes, and ER stress and autophagic deficits play additive roles in the decline of β‐cells24. In this case, there was no significant difference in the replicating capacity as measured by Ki67 index between patients with and without diabetes, and it seems likely that reduced β‐cell mass could be attributed to increased loss of β‐cells. Whether this contention can be applied to USA diabetic patients will require future investigations that compare the expressions of the aforementioned three markers simultaneously on the islet sections.
Figure 5.
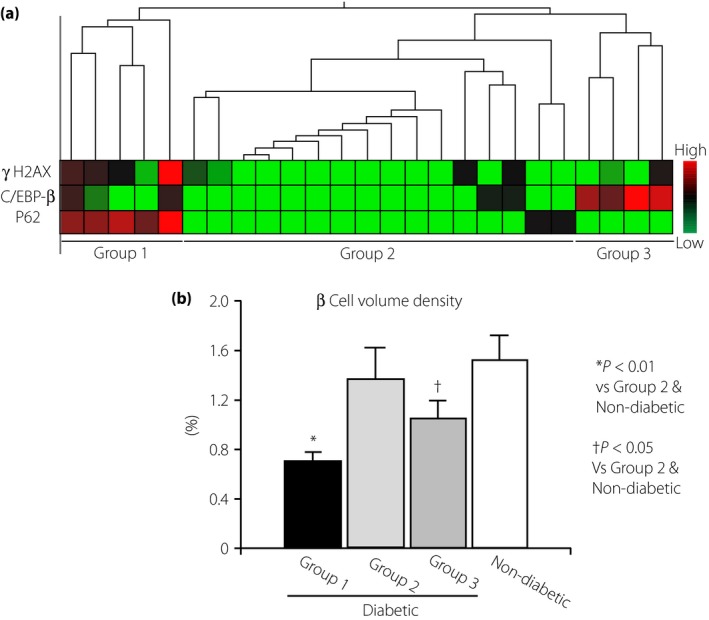
Implication of oxidative stress (OS), endoplasmic reticulum (ER) stress and autophagy deficits in β‐cell decline in patients with type 2 diabetes24. (a) By hierarchy cluster analysis, the impact of OS, ER stress and autophagy deficits on the β‐cell deficits were evaluated by the expression levels of γH2AX as OS‐induced deoxyribonucleic acid damage marker, CCAAT‐enhancer binding protein‐β (C/EBP‐β) as ER stress marker and P62 as autophagy deficit marker, respectively. (b) The results showed three groups: group 1 consists of subjects who were positive for all three factors; group 2 consists of subjects negative for all three factors; and group 3 consists of subjects positive for one or two of the factors. The extent of β‐cell decline was marked in the order of group 1, 3 and 2. In particular, expression of γH2AX was significantly correlated with the reduced β‐cell volume density.
Increase in α‐Cells in Diabetes as a Bi‐Hormonal Disorder
A new contention that diabetes should be regarded as a bi‐hormonal disorder with imbalance of insulin and glucagon has emerged45, 46. In fact, diabetic patients show hyperglucagonemia, which is one of the critical factors that make diabetes treatment difficult. Although the results are still controversial, some autopsy studies showed an increase in α cell volume density in Korean and European patients with type 2 diabetes compared with non‐diabetic subjects38, 47. In our study published in 2002, there was a trend toward an increase in α‐cell volume density in 15 patients with type 2 diabetes compared with non‐diabetic controls, but the difference did not reach a statistical difference31. However, by increasing the number of study participants to 47 patients with diabetes, we could show a significant increase in α‐cell volume density in diabetic group24. There was a reciprocal relationship between increased α‐cell and decreased β‐cell volume density. As there was no significant increase in Ki67‐positive α‐cells, the increase was not simply accounted for by accelerated replication of α cells.
Islet Remodeling and Transdifferentiation of Endocrine Cells
It has been increasingly clear that islet endocrine cells once differentiated are not fixed, but change dynamically into different cell types under metabolic stress48, 49. Such cellular alteration is called transdifferentiation or de‐differentiation (Figure 6). Because there is no significant decrease in islet volume density despite marked β‐cell loss in type 2 diabetic islets, the increase in α‐cell volume density might be explained by transdifferentiation of β‐cells into α‐cells. In fact, Mezza et al.49 suggested that β‐cell hyperplasia encountered in human pancreases obtained from IGT patients who had undergone pancreatectomy is ascribed to transdifferentiation from α‐cells49. In an experimental model, Talchai et al. proposed that suppression of nuclear expression of FOXO1, a β‐cell transcription factor, under metabolic stress hampered the β‐cell differentiation, and instead promoted them to transdifferentiate to α, δ or pancreatic polypeptide cells, resulting in the decrease in β‐cell volume density49. It remains unclear, however, to what extent the altered population of endocrine cells is present in type 2 diabetic islets or whether there is a difference in the regulation of differentiation between IGT and type 2 diabetes. Thus, dynamic alterations of islet endocrine cells are currently hot targets for research, but the precise mechanisms are largely yet to be clear. It appears to be certain that the islet changes we observe in type 2 diabetes are not simply explained by cell death or cell replication, because apoptosis or cell replication rate is much less frequent than previously reported, and the implication of transdifferentiation (metaplasia) might become more important50. It would be possible to develop new therapeutic means for diabetes to target the α‐cell increase or hyperglucagonemia by future investigations.
Figure 6.
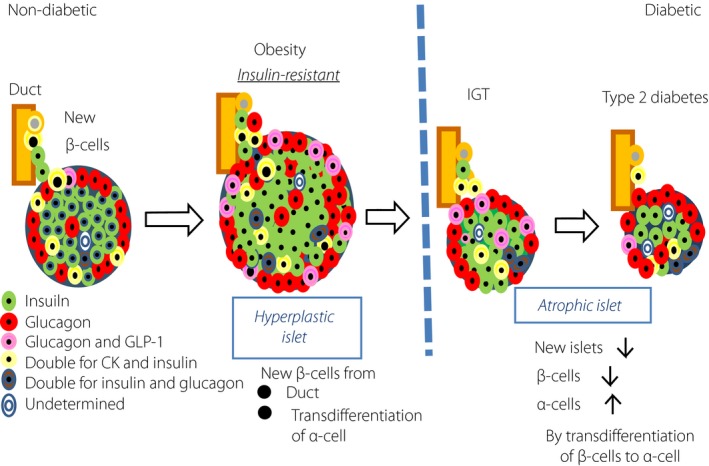
Emerging concept of islet endocrine cell transdifferentiation (metaplasia)50. There is remodeling of islet endocrine cells in obese or diabetic subjects. In healthy control subjects, islet β‐cells undergo compensatory hyperplastic changes in response to obesity. For this background, it is proposed that increased β‐cells derive from α‐cells by transdifferentiation in obese insulin‐resistant subjects. In contrast, with continuing metabolic stress, transcription factor FOXO1 is suppressed to translocate to nuclei, resulting in promoting transdifferentiation of α‐cells to β‐cells48. Such transdifferentiation of β‐cells to α‐cells are supposed to contribute to decreased β‐cell volume density in subjects with impaired glucose tolerance (IGT) and type 2 diabetes49. CK, cytokeratin; GLP‐1, glucagon‐like peptide‐1.
Role of Amyloid in β‐Cell Decline
Amyloid deposition has been considered for a long time as the most characteristic pathological feature in the islets of type 2 diabetes since Opie first described it3. Nevertheless, it has not been determined whether it is merely a by‐product or the cause of the β‐cell loss in diabetes51, 52. It is intriguing that more than 80% of USA patients with type 2 diabetes have islet amyloid deposition, but just 27% of Japanese patients with type 2 diabetes are positive for amyloid, showing the ethnic difference in amyloid deposition in type 2 diabetes53. The difference in the prevalence of amyloid deposition might be relevant to obesity or insulin resistance, which characterizes the clinical features of USA patients with diabetes. Furthermore, our study confirmed that the BMI of amyloid‐positive cases in Japanese patients with type 2 diabetes was greater (24.7 ± 0.7) compared with the value of amyloid‐free subjects (22.4 ± 0.6; Figure 7). Consistent with this result, Chinese patients with type 2 diabetes showed a greater BMI in the amyloid‐rich islet group compared with those without amyloid54.
Figure 7.
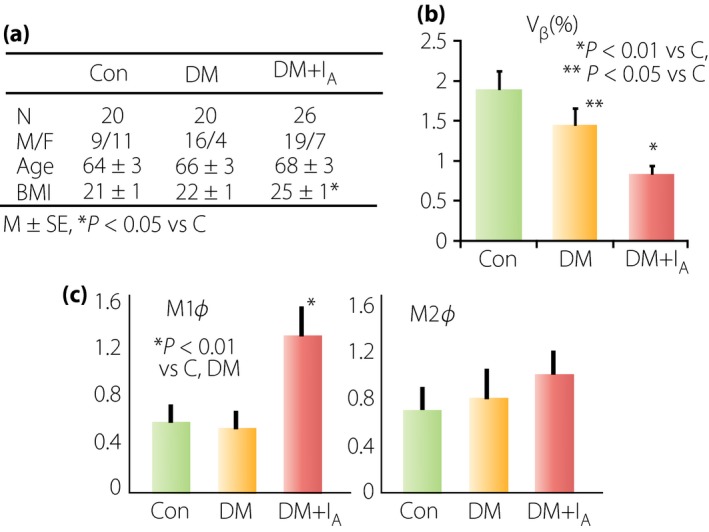
Implication of amyloid deposition in the islets of Japanese patients with type 2 diabetes53. (a) The group of diabetic patients with amyloid deposition (DM + IA) showed a greater body mass index (BMI) than that in the group of patients with diabetes without amyloid deposition (DM). (b) There was a more severe reduction of β‐cell volume density (vβ) in diabetic patients with amyloid deposition compared with those in diabetic patients without amyloid. (c) Within the islets with amyloid, there was a marked infiltration of macrophages. It is of note that M1 type (pro‐inflammatory) macrophages were significantly increased in diabetic islets with amyloid, whereas there was no difference in M2 type (anti‐inflammatory) macrophages in the islets between the amyloid‐rich group and amyloid‐free group. Thus, it is likely that amyloid promotes β‐cell loss through exerting inflammatory processes in the islets. Con, control.
Recent experimental studies showed β‐cell toxicity of amyloid fibrils or amyloid oligomers52, 55. It was also shown that impaired degradation of amyloid molecules causes ER stress in β‐cells, resulting in β‐cell damage and death56, although the data are still controversial57. Induction of autophagy deficit is also raised for β‐cell death by amyloid clearance processes58. After these proposals, the extent of amyloid deposition was found to be parallel with the degree of β‐cell reduction in USA patients with type 2 diabetes59. In our study, we also found greater reduction of β‐cell volume density in the amyloid‐rich diabetic group compared with the amyloid‐free diabetic group, so that amyloid deposition facilitated β‐cell loss in type 2 diabetes53. In addition, there was an enhanced expression of γH2AX as oxidative stress‐induced DNA damage in amyloid‐rich islets, showing that amyloid elicited oxidative stress with enhancement of β‐cell damage, resulting in augmented reduction of β‐cells53.
Pro‐Inflammatory Reactions in Islets
There is accumulating evidence that pro‐inflammatory reactions in the islet are closely related to amyloid deposition and β‐cell loss60, 61, 62. In fact, there is robust inflammatory cell infiltration in the islets of patients with type 2 diabetes53. In this setting, M1 type macrophages positive for CD68 and induced nitric oxide synthase as pro‐inflammatory cells are predominant in amyloid‐rich islets. In contrast, M2 type macrophages positive for CD163 and CD204 as anti‐inflammatory cells are not increased in amyloid‐rich islets. Amyloid deposition thus activates inflammatory reaction, producing inflammatory substances to release inflammasomes, such as Nlrp3, which in turn stimulates production of interleukin‐1β to damage β‐cells (Figure 8)63, 64. A randomized clinical trial of interleukin‐1β receptor antagonist for type 2 diabetic patients showed improvement of glucose intolerance65. Apparently, more precise and longer‐term studies on the relationship between islet inflammation and β‐cell loss are required for future clinical application.
Figure 8.
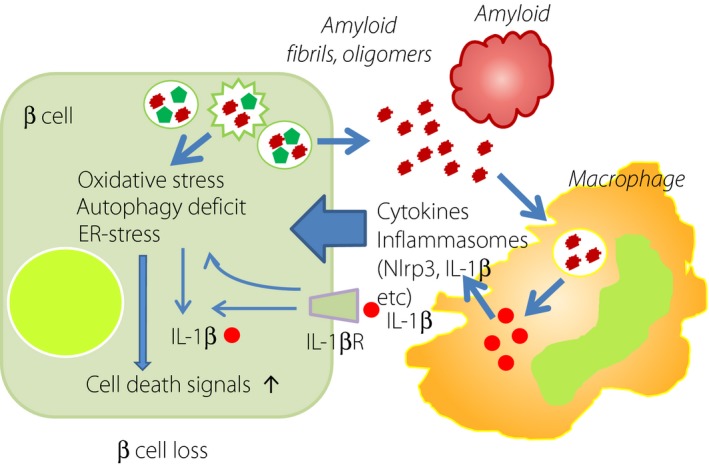
Pro‐inflammatory activation and amyloid deposition in the islet of type 2 diabetes. In the islet of type 2 diabetes, there is an increase in macrophage infiltration particular to pro‐inflammatory type 1. Amyloid deposition accelerates macrophage migration, and the release of cytokines and inflammasomes, such as Nlrp3 and interleukin‐1β (IL‐1β). IL‐1β in turn elicits β‐cell injury to disturb insulin secretion. Amyloid fibrils and oligomers are toxic to β‐cells, and augment the cell death pathway through oxidative stress, endoplasmic reticulum (ER) stress or autophagy deficits. IL‐1βR, interleukin‐1β receptor.
Future Perspective
With advancement of diabetic pathology, it is now evident that progressive decline of β‐cell mass and/or β‐cell volume density underlies pathophysiology of type 2 diabetes. With this background, diabetes therapy has been redirected from the tight blood glucose control for preventing diabetic complications toward the protection of β‐cells or promotion of β‐cell regeneration. We are now endowed with multiple choices for diabetes therapy from incretin‐related agents to sodium‐glucose transporter 2 inhibitor. Non‐injectable insulin might also be readily used for preserving the integrity of islets in the near future. In accordance with this paradigm shift of diabetes treatment, research activities are now oriented to the exploration of the mechanism of β‐cell death and β‐cell regeneration. Evidently, β‐cells are found to be more plastic than previously thought. Potentials for transdifferentiation and reversibility of β‐cells should be vigorously elucidated, and could be a target of the future treatment of diabetes. We are currently expecting the clinical application of induced pluripotential stem cell therapy to replace the damaged pancreas for a complete recovery from diabetes. It might not be necessary, however, if alternate means to promote a self‐repairing system of the islet can be developed. To this end, much more information on the dynamic pathology and associated molecular mechanisms in the islet is essential.
Diclosure
The authors declare no conflict of interest.
Acknowledgments
The authors are grateful to previous collaborators for their continuous efforts and technical excellence that allowed them to publish their findings. They also acknowledge the grant support for their studies on the islet changes in diabetes from the Japanese Ministry of Science, Culture, Education and Sports, and the Japanese Ministry of Health and Welfare.
J Diabetes Investig 2016; 7: 155–165
References
- 1. Langerhans P. Beiträge zur mikroskopischen Anatomie der Bauchspeicheldruse. Med. Dissertation, Thesis, Gustav Lange, Berlin, 1869. [Google Scholar]
- 2. Banting FG, Best CH. Pancreatic extracts. J Lab Clin Med 1922; 7: 464–472. [PubMed] [Google Scholar]
- 3. Opie EL. On the relation of diabetes mellitus to lesions of the pancreas. Hyaline degeneration of the islands of Langerhans. J Exp Med 1901; 5: 527–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tompsett SL. The determination of blood sugar: critical analysis of the reduction of alkaline copper by glucose and other substances. Biochem J 1930; 24: 1148–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yalow RS, Berson SA. Immunoassay of endogenous plasma insulin in man. J Clin Invest 1960; 39: 1157–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Berson SA, Yalow RS. Immunochemical distinction between insulins with identical amino‐acid sequences. Nature 1961; 30: 1392–1393. [DOI] [PubMed] [Google Scholar]
- 7. Deckers JG, Schellevis FG, Fleming DM. WHO diagnostic criteria as a validation tool for the diagnosis of diabetes mellitus: a study in five European countries. Eur J Gen Pract 2006; 12: 108–113. [DOI] [PubMed] [Google Scholar]
- 8. Sone H, Ito H, Ohashi Y, et al Japan Diabetes Complication Study Group. Obesity and type 2 diabetes in Japanese patients. Lancet 2003; 361: 85. [DOI] [PubMed] [Google Scholar]
- 9. Hsu WC, Boyko EJ, Fujimoto WY, et al Pathophysiologic differences among Asians, native Hawaiians, and other Pacific Islanders and treatment implications. Diabetes Care 2012; 35: 1189–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mclean N, Ogilvie RF. Quantitative estimation of the pancreatic islet tissue in diabetic subjects. Diabetes 1955; 4: 367–376. [DOI] [PubMed] [Google Scholar]
- 11. Saito K, Yaginuma N, Takahashi T. Differential volumetry of A, B and D cells in the pancreatic islets of diabetic and nondiabetic subjects. Tohoku J Exp Med 1979; 129: 273–283. [DOI] [PubMed] [Google Scholar]
- 12. Peters J, Jűrgensen A, Klőppel G. Ontogeny, differentiation and growth of the endocrine pancreas. Virchows Arch 2000; 436: 527–538. [DOI] [PubMed] [Google Scholar]
- 13. Gregg BE, Moore PC, Demozay D, et al Formation of a human β‐cell population within pancreatic islets is set early in life. J Clin Endocrinol Metab 2012; 97: 3197–3206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Meier JJ, Butler AE, Saisho Y, et al β‐Cell replication is the primary mechanism subserving the postnatal expansion of β‐cell mass in humans. Diabetes 2008; 57: 1584–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mizukami H, Takahashi K, Inaba W, et al Age‐associated changes of islet endocrine cells and the effects of body mass index in Japanese. J Diabetes Investig 2014; 5: 38–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hulinky I, Hulinska H, Silink M. DNA synthesis in cultured neonatal rat islets—a comparison of two methods. Diabetes Res Clin Pract 1995; 27: 119–126. [DOI] [PubMed] [Google Scholar]
- 17. Francis RJ, Southgate JL, Wilkin TJ, et al Expression of an islet regenerating (reg) gene in isolated rat islets: effects of nutrient and non‐nutrient growth factors. Diabetologia 1992; 35: 238–242. [DOI] [PubMed] [Google Scholar]
- 18. Koyama M, Wada R, Sakuraba H, et al Accelerated loss of islet βcells in sucrose‐fed Goto‐Kakizaki rats, a genetic model of non‐insulin‐dependent diabetes mellitus. Am J Pathol 1998; 153: 537–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Perl S, Kushner JA, Buchholz BA, et al Significant human β‐cell turnover is limited to the first three decades of life as determined by in vivo thymidine analog incorporation and radiocarbon dating. J Clin Endocrinol Metab 2010; 95: E234–E239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kőhler CU, Olewinski M, Tannapfel A, et al Cell cycle control of β‐cell replication in the prenatal and postnatal human pancreas. Am J Physiol Endocrinol Metab 2011; 300: E221–E230. [DOI] [PubMed] [Google Scholar]
- 21. Meier JJ, Kohler CU, Alkhtib B, et al β Cell development and turnover during prenatal life in humans. Eur J Endocrinol 2010; 162: 559–568. [DOI] [PubMed] [Google Scholar]
- 22. Georgia S, Bhushan A. p27 Regulates the transition of β‐cells from quiescence to proliferation. Diabetes 2006; 55: 2950–2956. [DOI] [PubMed] [Google Scholar]
- 23. Sullivan BA, Hollister‐Lock J, Bonner‐Weir S, et al Reduced Ki67 staining in the postmortem state calls into question past conclusions about the lack of turnover of adult human beta cells. Diabetes 2015; 64: 1698–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mizukami H, Takahashi K, Inaba W, et al Involvement of oxidative stress, endoplasmic reticulum stress, and autophagy deficit in β cell decline in Japanese type 2 diabetic patients. Diabetes Care 2014; 37: 1966–1974. [DOI] [PubMed] [Google Scholar]
- 25. Cnop M, Hughes SJ, Igoillo‐Esteve M, et al The long lifespan and low turnover of human islet β cells estimated by mathematical modelling of lipofuscin accumulation. Diabetologia 2010; 53: 321–330. [DOI] [PubMed] [Google Scholar]
- 26. Cnop M, Igollo‐Esteve M, Hughes SJ, et al Longevity of human islet a‐ and β‐cells. Diabetes Obes Metab 2011; 13(Suppl 1): 39–46. [DOI] [PubMed] [Google Scholar]
- 27. Rieck S, Kaestner KH. Expansion of β‐cell mass in response to pregnancy. Trends Endocrinol Metab 2010; 21: 151–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hughes E, Huang C. Participation of Akt, menin, and p21 in pregnancy‐induced β‐cell proliferation. Endocrinology 2011; 152: 847–855. [DOI] [PubMed] [Google Scholar]
- 29. Klőppel G, Anlauf M, Raffel A, et al Adult diffuse nesidioblastosis: genetically or environmentally induced? Hum Pathol 2008; 39: 3–8. [DOI] [PubMed] [Google Scholar]
- 30. Kaneto H, Matsuoka TA. Down‐regulation of pancreatic transcription factors and incretin receptors in type 2 diabetes. World J Diabetes 2013; 4: 263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sakuraba H, Mizukami H, Yagihashi N, et al Reduced β‐cell mass and expression of oxidative stress‐related DNA damage in the islet of Japanese Type II diabetic patients. Diabetologia 2002; 45: 85–96. [DOI] [PubMed] [Google Scholar]
- 32. Rhodes CJ. Type 2 diabetes‐a matter of β‐cell life and death? Science 2005; 307: 380–384. [DOI] [PubMed] [Google Scholar]
- 33. Saisho Y, Elashoff D, Butler AE, et al β‐cell mass and turnover in humans. Effects of obesity and aging. Diabetes Care 2013; 36: 111–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yabe D, Kuroe A, Watanabe K, et al Early phase glucagon and insulin secretory abnormalities, but not incretin secretion, are similarly responsible for hyperglycemia after ingestion of nutrients. J Diabetes Complications 2015; 29: 413–421. [DOI] [PubMed] [Google Scholar]
- 35. Kou K, Saisho Y, Satoh S, et al Change in β‐cell mass in Japanese nondiabetic obese individuals. J Clin Endocrinol Metab 2013; 98: 3724–3730. [DOI] [PubMed] [Google Scholar]
- 36. Stefan Y, Orci L, Malaisse‐Lagae F, et al Quantitation of endocrine cell content in the pancreas of nondiabetic and diabetic humans. Diabetes 1982; 31: 694–700. [DOI] [PubMed] [Google Scholar]
- 37. Rahier J, Goebbels RM, Henquin JC. Cellular composition of the human diabetic pancreas. Diabetologia 1983; 24: 366–371. [DOI] [PubMed] [Google Scholar]
- 38. Yoon KH, Ko SH, Cho JH, et al Selective β‐cell loss and α‐cell expansion in patients with type 2 diabetes mellitus in Korea. J Clin Endocrinol Metab 2003; 88: 2300–2308. [DOI] [PubMed] [Google Scholar]
- 39. Butler AE, Janson J, Bonner‐Weir S, et al β‐Cell deficit and increased β‐cell apoptosis in humans with type 2 diabetes. Diabetes 2003; 52: 102–110. [DOI] [PubMed] [Google Scholar]
- 40. Rahier J, Guiot Y, Goebbels RM, et al Pancreatic β‐cell mass in European subjects with type 2 diabetes. Diabetes Obes Metab 2008; 10(Suppl 4): 32–42. [DOI] [PubMed] [Google Scholar]
- 41. Leibowitz G, Kaiser N, Cerasi E. β‐Cell failure in type 2 diabetes. J Diabetes Investig 2011; 2: 82–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yagihashi S. Clinical staging of type 2 diabetes. The time has come. J Diabetes Investig 2012; 3: 1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Matveyenko AV, Veldhuis JD, Butler PC. Mechanisms of impaired fasting glucose and glucose intolerance induced by an approximate 50% pancreatectomy. Diabetes 2006; 55: 2347–2356. [DOI] [PubMed] [Google Scholar]
- 44. Menge BA, Tannapfel A, Belyaev O, et al Partial pancreatectomy in adult humans does not provoke β‐cell regeneration. Diabetes 2008; 57: 142–149. [DOI] [PubMed] [Google Scholar]
- 45. Unger RH, Orci L. Paracrinology of islets and the paracrinopathy of diabetes. Proc Natl Acad Sci USA 2010; 107: 16009–16012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Unger RH, Cherrington AD. Glucagonocentric restructuring of diabetes: a pathophysiologic and therapeutic makeover. J Clin Invest 2012; 122: 4–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Henquin JC, Rahier J. Pancreatic α cell mass in European subjects with type 2 diabetes. Diabetologia 2011; 54: 1720–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Talchai C, Xuan S, Lin HV, et al Pancreatic β cell dedifferentiation as a mechanism of diabetic β cell failure. Cell 2012; 150: 1223–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mezza T, Muscogiuri G, Sorice GP, et al Insulin resistance alters islet morphology in nondiabetic humans. Diabetes 2014; 63: 994–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yagihashi S, Mizukami H, Inaba W. Quo vadis: where have the β‐cells gone? J Diabetes Investig 2015; 6: 393–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Clark A, Nilsson MR. Islet amyloid: a complication of islet dysfunction or an aetiological factor in Type 2 diabetes? Diabetologia 2004; 47: 157–169. [DOI] [PubMed] [Google Scholar]
- 52. Westermark P. Amyloid in the islets of Langerhans: thoughts and some historical aspects. Ups J Med Sci 2011; 116: 81–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kamata K, Mizukami H, Inaba W, et al Islet amyloid with macrophage migration correlates with augmented β‐cell deficits in type 2 diabetic patients. Amyloid 2014; 21: 191–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhao HL, Lai FM, Tong PC, et al Prevalence and clinicopathological characteristics of islet amyloid in Chinese patients with type 2 diabetes. Diabetes 2003; 52: 2759–2766. [DOI] [PubMed] [Google Scholar]
- 55. Last NB, Rhoades E, Miranker AD. Islet amyloid polypeptide demonstrates a persistent capacity to disrupt membrane integrity. Proc Natl Acad Sci USA 2011; 108: 9460–9465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Huang CJ, Lin CY, Haataja L, et al High expression rates of human amyloid polypeptide induce endoplasmic reticulum stress mediated β‐cell apoptosis, a characteristic of humans with type 2 but not type 1 diabetes. Diabetes 2007; 56: 2016–2017. [DOI] [PubMed] [Google Scholar]
- 57. Hull RL, Zraika S, Udayasankar J, et al Amyloid formation in human IAPP transgenic mouse islets and pancreas, and human pancreas, is not associated with endoplasmic reticulum stress. Diabetologia 2009; 52: 1102–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rivera JF, Gurlo T, Daval M, et al Human‐IAPP disrupts the autophagy/lysosomal pathway in pancreatic β‐cells: protective role of p62‐positive cytoplasmic inclusions. Cell Death Differ 2011; 18: 415–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jűrgens CA, Toukatly MN, Fligner CL, et al β‐Cell loss and β‐cell apoptosis in human type 2 diabetes are related to islet amyloid deposition. Am J Pathol 2011; 178: 2632–2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. de Koning EJ, van den Brand JJ, Mott VL, et al Macrophages and pancreatic islet amyloidosis. Amyloid 1998; 5: 247–254. [DOI] [PubMed] [Google Scholar]
- 61. Donath MY, Dalmas E, Sauter NS, et al Inflammation in obesity and diabetes.: islet dysfunction and therapeutic opportunity. Cell Metab 2013; 17: 860–872. [DOI] [PubMed] [Google Scholar]
- 62. Eguchi K, Manabe I. Macrophages and islet inflammation in type 2 diabetes. Diabetes Obes Metab 2013; 63: 1698–1711. [DOI] [PubMed] [Google Scholar]
- 63. Westwell‐Roper CY, Ehses JA, Verchere CB. Resident macrophages mediate islet amyloid polypeptide‐induced islet IL‐1β production and beta cell dysfunction. Diabetes 2014; 63: 1698–1711. [DOI] [PubMed] [Google Scholar]
- 64. Masters SL, Dunne A, Subramanian SL, et al Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL‐1β in type 2 diabetes. Nat Immunol 2010; 11: 897–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Larsen CM, Faulenbach M, Vaag A, et al Interleukin‐1‐receptor antagonist in type 2 diabetes mellitus. N Engl J Med 2007; 356: 1517–1526. [DOI] [PubMed] [Google Scholar]