

Abstract
Proteolytic enzymes, released early in the course of an inflammatory response, hydrolyze fibronectin, producing fragments of the parent molecule that alter monocyte phenotype and migratory behavior. Here we test the hypothesis that macrophages, stimulated by the dominant 110–120 kd fibronectin fragments (FNf), as are found in lymphatic fluid draining sites of cardiac ischemia-reperfusion injury, produce factors that promote the survival of injured parenchymal cells. Rat splenic macrophages stimulated in vitro with purified FNf produced soluble factors that protected hypoxic rat cardiac myocytes from death by apoptosis. Addition of blocking antibodies specific for tumor necrosis factor-α (TNF-α), fibroblast growth factor-1 (FGF-1), insulin-like growth factor I (IGF-I), and leukemia inhibitory factor (LIF) partly reduced the protection against apoptosis provided to hypoxic cardiac myocytes by cell-free culture supernatants from FNf-stimulated macrophages. Complete blockade of this protection was achieved by a combination of antibodies specific for FGF-1, IGF-I, and LIF. Stimulation of human monocyte–derived macrophages in vitro with FNf significantly increased their output of TNF-α, FGF-1, IGF-I, and LIF. These results suggest that tissue degradation products, released in the early hours of an inflammatory response, stimulate tissue-infiltrating macrophages to protect injured but still viable parenchymal cells from death by apoptosis.
Keywords: apoptosis, cytokines, monocytes/macrophages, fibronectin, ischemia
Ischemia provokes an intense inflammatory response that initially exacerbates tissue injury (1). Much of the inflammatory damage is attributable to proteolytic enzymes (2) and other agents released by leukocytes that infiltrate formerly ischemic tissues during reperfusion (3). Some of the leukocytes, particularly monocyte-derived macrophages (4), also help to resolve and repair this injury (1, 5). Macrophages enter formerly ischemic tissue within 1–3 hrs after reperfusion (3), take up dead cells and debris within 24 hrs (4), and remodel the tissue matrix and stimulate production of collagen 3 or more days later (6). Although this repair may maintain organ integrity by scar formation, organ dysfunction may still result from extensive parenchymal cell death. Since macrophages have been shown to limit (7) as well as repair injury, we propose an additional role for them in preventing parenchymal cell loss. Our hypothesis is that during the early phases of an inflammatory response, monocyte-derived macrophages produce agents that protect parenchymal cells from death-inducing signals.
Our studies grew out of the observation that proteolytic enzymes, released during the first 5 hrs of an inflammatory response, generate fibronectin fragments that can activate tissue infiltrating monocyte-derived macrophages, altering their migratory behavior (4) and stimulating them to produce cytokines and other factors with the potential to influence parenchymal cell survival (8–11). We stimulated macrophages with 110–120 kd fibronectin fragments (FNf), the major fibronectin fragment in the lymphatic drainage of ischemic cardiac tissue (4), to identify the factors and their relevance to cardiac myocyte survival after hypoxic injury.
Materials and Methods
Materials and Reagents
Collagenase (Type 2) was obtained from Worthington Biochemical (Lakewood, NJ); pyrogen free human serum albumin (HSA) from Alpine Biologics, Inc. (Orangeburg, NJ); heparin from Elkins-Sinn, Inc. (Cherry Hill, NJ); human plasma fibronectin (FN) from Chemicon (Temecula, CA); mouse laminin and the 110 kD cell-binding fragment of human FN (FNf) from Upstate Biotechnology (Lake Placid, NY). Other chemicals came from Sigma Chemical Co. (St. Louis, MO). ITS (1.25 mg/ml insulin, 1.25 mg/ml transferrin, 1.25 μg/ml selenium) came from BioSource International (Camarillo, CA); Hanks’ basic salt solution (HBSS), Dulbecco’s PBS (DPBS), gentamicin, penicillin-streptomycin, distilled water (low endotoxin), and trypsin came from Gibco BRL (Grand Island, NY). Fetal bovine serum (FBS) was obtained from HyClone (Logan, UT). Endotoxin was undetectable by the Limulus amoebocyte assay (Associates of Cape Cod, Inc., Falmouth, MA) in these reagents. Joklik medium was reconstituted from powdered MEM and 2 g NaHCO3, 60 mM taurine, 17.5 mM creatine, 5 mM HEPES, and 0.1% human serum albumin; pH was adjusted with NaOH. Dulbecco’s modified Eagle’s medium (DMEM) base without glucose was reconstituted according to the manufacturer’s instructions. Media were sterilized by 0.2 μm filtration. Complete M199 was made by adding 0.4% (v/v) ITS, 100 U penicillin, 100 μg streptomycin, and 0.1% HSA. RPMI 1640 was used alone or after addition of 10% FBS and 10 μg/ml gentamicin. Media were warmed to 37°C and equilibrated with 5% CO2 in air before use. Glassware was baked at 220°C overnight to eliminate endotoxin.
Isolation of Rat Cardiac Myocytes
Six- to 12-week-old Sprague-Dawley male rats (Charles River, Wilmington, MA), purchased under a protocol approved by Baylor College of Medicine and the Houston Veterans Affairs Medical Center, were anesthetized intraperitoneally with 0.6 ml of a 0.3:1:1 mixture of acepromazine (10 mg/ml):xylazine (20 mg/ml):ketamine (100 mg/ml). The hearts were removed under aseptic conditions, and cardiac myocytes were isolated using modifications of previously reported methods (12, 13). Isolated hearts were perfused with 1 U/ml heparin in Joklik medium followed by 0.7 mg/ml collagenase through autoclaved tubing (Masterflex Tygon LFL, L/S 13; Cole-Parmer, Vernon Hills, IL) at a rate of 12.6 ml/min by a peristaltic pump (Master Flex Easy Load II; Cole-Parmer). All perfusion fluids were kept at 38°C and equilibrated with 95% O2 plus 5% CO2 passed through a Vacushield 0.45 μm PTFE filter (Gelman Laboratory, Ann Arbor, MI). After perfusion, the ventricles were minced and further digested in a laminar flow hood. Cell suspensions were strained through a 210-μm mesh (Spectrum Laboratories, Inc., Rancho Dominguez, CA), then washed and placed on a 6% HSA cushion. CaCl2 was added stepwise to the sedimented cells to a final concentration of 100 mM. Fibroblasts were removed by adherence in T75 flasks (Falcon, Becton Dickinson, Franklin Lakes, NJ). This step results in >97% purity of myocytes (13). Myocytes were placed into complete M199 and 5 × 105/ml dispensed into 35-mm dishes (Falcon) coated with 10 μg laminin. After overnight culture, the myocytes were moved to glucose-free DMEM and placed at <10 torr PO2 (1 torr = 133.3 Pa = pressure of 1 mm mercury) for 3 hrs in an anaerobic workstation (Forma Scientific, Marietta, OH) perfused with 90% N2, 5% CO2, and 5% H2 (14). After the hypoxic interval, DMEM was replaced with complete M199. The glucose-free DMEM is also free of pyruvate, which prevents anaerobic metabolism and maximizes injury to cells deprived of oxygen. Normoxic control myocytes were also placed in DMEM for the same interval.
Assessment of Viability and Apoptosis
After 21 hrs reoxygenation, myocytes were detached by trypsinization and counted. Viability was assessed with calcein AM (LIVE/DEAD viability cytotoxicity assay; Molecular Probes, Eugene, OR) added in 1:1 M199/HBSS. To identify apoptotic cells, myocytes were fixed in 3% paraformaldehyde and permeabilized with TBST (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 0.1% Tween), after which 5% normal goat serum was added to block nonspecific antibody binding. The cells were stained with antibody to cleaved caspase 3 (Cell Signaling Technology, Beverly, MA) or control rabbit IgG. The secondary antibody was phycoerythrin-conjugated affinity-purified F(ab′)2 goat anti-rabbit Ig (BioSource). For flow cytometric analysis of the cardiac myocytes, a neutral density filter was inserted in front of the forward-angle light scatter detector (measuring size of the cells) of the Epics XL-MCL flow cytometer (Beckman Coulter, Miami, FL) to bring myocytes within the boundaries of the light scatter histogram. Gains on forward- and side-scatter detectors were set at one, voltages at 30, and events for both parameters were collected on log scales. Necrosis was assessed by measuring lactate dehydrogenase using a kit from Sigma (13). Annexin V binding was assessed using reagents from Becton Dickinson according to the supplier’s directions.
Isolation and Treatment of Rat Macrophages
Spleen cells were suspended in serum-free RPMI 1640 and distributed into 6-well culture plates coated with 60 μg/well FN. Nonadherent cells were removed after 2 hrs. Adherent macrophages were cultured in complete RPMI 1640 overnight and then washed to remove remaining nonadherent cells. Macrophages were scraped from the wells with rigid polypropylene cell lifters (Fisher, Pittsburgh, PA), resuspended in fresh medium, and counted. Purity ranged from 84%–92% by microscopy and by esterase staining. Macrophages were treated with 110 μg FNf per 106 cells for 90 mins in RPMI 1640 without serum (4) or held in RPMI 1640 for the same interval as an untreated control before washing and resuspension at 5 × 104/ml in complete M199 before co-culture with cardiac myocytes. Alternatively, filtered supernatants from macrophages cultured in M199 on laminin-coated dishes overnight were added to the cardiac myocytes.
Normal Donor Blood
Blood was obtained from volunteers under a protocol approved by the Institutional Review Boards of Baylor College of Medicine and the Houston Veterans Affairs Medical Center. Blood, anticoagulated with preservative-free heparin, was fractionated by Ficoll-Hypaque gradient centrifugation (Histopaque-1077; Sigma) to collect mononuclear cells that were then allowed to adhere in serum-free RPMI 1640 to plastic, six-well plates (Nunc; Nalge Nunc International, Denmark) coated with 60 μg/well mouse laminin. After 1 hr, nonadherent cells were removed and the remaining cells cultured for 24 hrs with or without addition of 110 μg FNf to each culture containing 107 original mononuclear leukocytes. Culture supernatants were harvested for measurement of cytokines and growth factors by enzyme-linked immunoabsorbent assay (ELISA) according to the manufacturer’s directions (R&D Systems, Minneapolis, MN).
Antibody Blocking Studies
Sterile blocking antibodies certified to contain less than 10 ng endotoxin per milligram of antibody protein were obtained from R&D Systems (Minneapolis, MN). Goat anti-rat TNF-α was used at 10 μg; rabbit anti-bovine FGF acid was used at 2.5 μg; goat anti-human IGF-I was used at 2.5 μg; and goat anti-mouse LIF was used at 0.5 μg/culture. Reactivity of the antibodies against rat factors was confirmed by immunoblots. Normal rabbit IgG, isolated from sterile serum by ammonium sulfate precipitation, was used as a control at 2.5 μg per culture.
Data Reporting and Statistics
To calculate the numbers of calcein bright rods per dish, the total number of cells harvested from each dish was multiplied by the fraction of bright events in the calcein analysis. Likewise, the number of nonapoptotic cells was determined by multiplying the total number of cells per dish by the fraction that were cleaved caspase-3 negative (formulas below):
Data from hypoxic cells were compared with those from cells maintained in 95% air plus 5% CO2. To facilitate comparisons between experiments, the numbers obtained with nonhypoxic controls in each experiment were set at 100%; experimental results were recorded as a percent of this control value. Data from multiple rats were pooled to yield the reported results. Paired t tests were used to measure statistical significance except where indicated.
Results
Assessment of Cardiac Myocyte Viability After Hypoxia
We used flow cytometric techniques to assess myocyte viability and apoptosis. Myocytes can be identified at the upper right of a light scatter histogram using a log scale for both forward-angle and side-angle parameters (Fig. 1A); myocytes were identified by live gating as the only events in the histogram that were fluorescence positive for calcein, a reagent identifying viable cells (Fig. 1B). Two peaks of calcein fluorescence were evident (Fig. 1B) on the four-decade log fluorescence scale. UV microscopy showed that the calcein-bright cells were rod shaped, which is the normal configuration of healthy, isolated cardiac myocytes. The calcein-dim cells were hypercontracted, a configuration assumed by injured and dying cardiac myocytes (15). Therefore, we counted calcein-bright events as indicating the presence of viable cells; that is, we only counted rod-shaped myocytes as healthy. Some injured myocytes also expressed the cleaved form of caspase 3, a marker of cells undergoing apoptosis (Fig. 1C). Cells that were negative for cleaved caspase 3 were then also used as an estimate of healthy nonapoptotic myocytes. The number of healthy myocytes was similar, whether assessed by calcein brightness or the absence of cleaved caspase 3. Release of lactate dehydrogenase (LDH), an indicator of cell necrosis, was not significantly different (paired t test) in normoxic cultures (196.6 ± 18.8 U/ml) and myocytes subjected to 3 hrs of hypoxia (245.6 ± 18 U/ml, n = 12). If myocytes are exposed to longer periods of hypoxia, LDH release occurs (13). In the current study, with the shorter interval of hypoxia, we only found myocytes with evidence of apoptotic injury.
Figure 1.

Analysis of viability and apoptosis. (A) Forward-angle light scatter (FS), an indicator of cell size, and side-angle light scatter (SS), an indicator of intracellular organelles, are shown on the y and x axes, respectively. Intact cells with calcein fluorescence (C) are encircled in the light scatter histogram, whereas debris is calcein-negative. (B) Control myocytes not given calcein produce background autofluorescence (left peak). By UV microscopy, weakly reactive cells under the middle peak were contracted (i.e., injured), whereas the strongly fluorescent cells under the far right peak were rod-shaped (i.e., viable; see Fig. 3 for appearance by microscopy). Myocytes were counted as viable if they were strongly calcein-positive. (C) After 6 hrs of hypoxia and 24 hrs of reoxygenation, many cells were stained with the anticleaved caspase 3 antibody. Background fluorescence was measured with an irrelevant IgG.
Macrophage Prevention of the Loss of Viable Myocytes After Hypoxia
The recovery of viable myocytes was significantly reduced by 3 hrs of hypoxia and 21 hrs reoxygenation, as compared with myocytes maintained under normoxic conditions for the same interval (negative controls). The decreased recovery was seen whether measured by calcein fluorescence (Fig. 2A) or by the frequency of myocytes negative for cleaved caspase 3 (Fig. 2B). Hypoxia caused a 53.7% decrease in live rods by the calcein assay (second bar from top, Fig. 2A). Similarly, hypoxia caused a 50.5% decrease in myocytes with no evidence of caspase 3 cleavage (second bar, Fig. 2B). All myocyte cultures were maintained in the same glucose- and pyruvate-free medium, whether normoxic or exposed to 3 hrs hypoxia. This ensured that any differential loss of viability was attributable to oxygen deprivation, not nutritional stress.
Figure 2.

Myocyte protective effects of macrophages. FNf-stimulated macrophages and FNf-stimulated macrophage culture supernatants enhanced survival of hypoxic cardiac myocytes. The percentage of live, calcein-positive, rod-shaped, nonapoptotic myocytes significantly increased when hypoxic, reoxygenated myocytes were incubated with FNf-stimulated macrophages (A and B, n = 5) or with culture supernatants from FNf-stimulated macrophages (C and D, n = 5; P < 0.01, asterisks, paired t test). Addition of untreated macrophages or untreated macrophage culture supernatants provided slight improvement but, except for the data shown in (panel B, P = 0.04, cross, paired t test), the improvement in survival was not statistically significant. Shaded bars = number of myocytes in cultures not subject to hypoxia or culture with macrophages or supernatants. Some values are greater than 100 as a result of normalization to normoxic myocyte cultures.
To evaluate whether macrophages affect the viability of hypoxic cardiac myocytes, we incubated adult rat splenic macrophages or macrophage culture supernatants with the myocytes. Addition of untreated macrophages to previously hypoxic myocytes (third bar, Fig. 2A and B) improved the recovery of viable myocytes, but the benefit was only marginally significant (P = 0.05, Wilcoxon signed rank test). However, addition of FNf-treated macrophages significantly increased the frequency of viable myocytes to levels comparable with those in normoxic cultures (fourth bars, Fig. 2A and B). Culture supernatants from untreated and FNf-treated macrophages were as effective as the cells themselves in protecting myocytes from hypoxia injury (compare Fig. 2C and D and Fig. 2A and B).
The effects of these macrophage culture supernatants on myocyte morphology are shown in Figure 3. Normoxic myocytes were rod shaped (Fig. 3A), whereas many hypoxic cells contracted (Fig. 3B). Supernatants from untreated macrophages protected some but not all myocytes from hypoxic injury (Fig. 3C). FNf-stimulated macrophage supernatants effectively protected most of the hypoxic myocytes with the result that almost all of the cells had a normal rod-like morphology (Fig. 3D).
Figure 3.
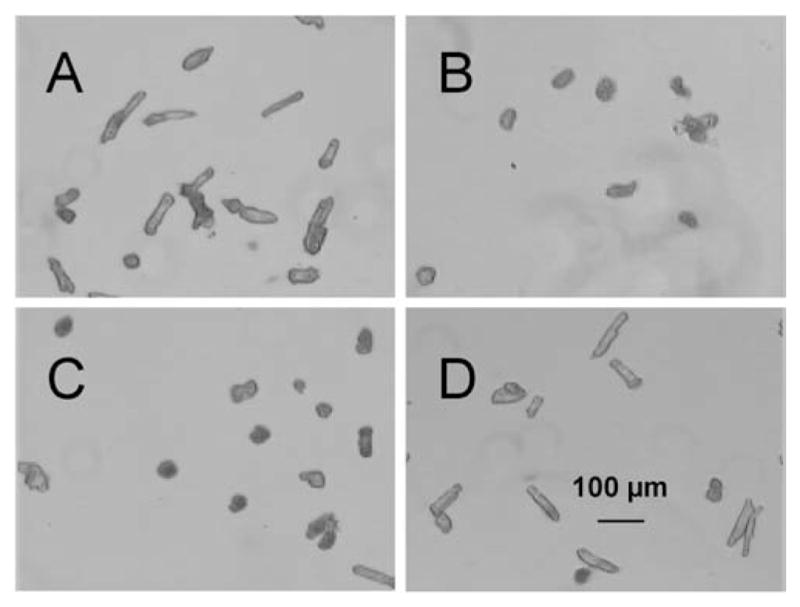
Microscopy of myocytes. Normoxic myocytes (A) exhibited typical rod morphology, whereas hypoxic myocytes (B) became contracted. Addition of untreated macrophage supernatant (C) preserved some of the myocytes in the rod configuration, but many were nevertheless contracted. Addition of supernatant from FNf-stimulated macrophages (D) maintained rod morphology in a majority of the myocytes. Magnification ×40.
Macrophage Factors that Protect Cardiac Myocytes Against Hypoxic Injury
Prior studies (16–21) have identified several cytokines and growth factors, most notably TNF-α, LIF, IGF-I, and FGF-1, which can protect myocytes from ischemic injury. We used blocking antibodies to determine whether these same agents also contributed to the protective effects of FNf-treated macrophages in vitro. As was seen for experiments shown in Figure 2, hypoxia without the addition of macrophages reduced the recovery of viable myocytes (Fig. 4, second bar compared with top bar in A and B). In the remaining experimental groups, macrophages treated with FNf were added to the myocyte cultures subjected to hypoxia, and antibodies were used to block the effects of the growth factors. Addition of FNf-treated macrophages with a control irrelevant antibody significantly increased myocyte survival to the same extent as we saw when FNf-treated macrophages were co-cultured with myocytes in the absence of added IgG (Fig. 2). Blocking antibodies to LIF, IGF-I, or FGF-1 significantly reduced the protection against cell death afforded by FNf-stimulated macrophages (P < 0.02, t test; Fig. 4A and B). To evaluate whether these factors had an additive effect on the survival of hypoxic myocytes, we combined blocking antibodies to the three most efficacious growth factors: LIF, IGF-I, and FGF-1 (Fig. 4C). Blocking all three factors effectively abolished the protection afforded by the FNf-treated macrophages (compare third and fourth bars in Fig. 4C).
Figure 4.

Effect of blocking antibodies. The percent of live, calcein-positive rods (A) and nonapoptotic myocytes (B) significantly decreased when the cells were exposed to hypoxia (shaded bar versus open bar). When hypoxic, reoxygenated myocytes were co-incubated with FNf-stimulated macrophages (crosshatched bars), and their viability increased if the activity of the FNf-treated macrophages was not blocked (control IgG, C). Blocking antibodies to tumor necrosis factor-alpha (TNF-α), leukemia inhibitory factor (LIF), insulin-like growth factor-I (IGF-I), or fibroblast growth factor-1 (FGF-1) partially inhibited the survival-enhancing effect of the macrophages. Measurements were expressed as a percent of normoxic control (white bar). Data are means ± SEM (n = 10). Statistical significance was estimated by the paired t test: Asterisk (*), P < 0.05. Actual P values for data in (A) are 10−6 for normoxic versus hypoxic, 0.05 for (C) versus TNF, 0.0005 for (C) versus LIF or IGF, and 0.002 for (C) versus FGF. Actual P values for (B) are 2 × 10−6 for normoxic versus hypoxic, 0.02 for (C) versus TNF, 0.0009 for (C) versus LIF, 0.0005 for (C) versus IGF, and 0.01 for (C) versus FGF. Blocking the activities of LIF, IGF-I, and FGF-1 with a combination of antibodies abolished the ability of FNf-stimulated macrophages to protect hypoxic cardiac myocytes (C). The shaded bar represents survival of nonhypoxic cells. The second bar down represents the survival of hypoxic myocytes without added macrophages. FNf-treated macrophages were added to myocytes in the remaining cultures. In the third set of cultures, an irrelevant control IgG was added to the myocyte/FNf-stimulated macrophage co-culture, whereas in the blocked cultures a mixture of anti-factor antibodies (fourth bar) was added. Data are means ± SEM (n = 4; asterisks indicate P = 0.03 for hypoxic myocytes without macrophages and P = 0.02 for cultures treated with antibodies to LIF + IGF-I + FGF-1 as compared with nonhypoxic myocytes).
Human Monocytes Produce the Same Factors Following FNf Stimulation
Unstimulated human monocytes adherent to laminin produced the same four factors: TNF-α, LIF, IGF-I, and FGF-1. When monocytes were exposed to FNf, the quantity of each of these factors increased significantly. Table 1 shows the quantities produced at the end of 24 hrs; however, levels in FNf-treated cultures were significantly elevated above unstimulated cultures after as little as 90 mins.
Table 1.
Factors Produced by Human Monocytes Stimulated with Fibronectin Fragments (FNf)a
| No treatment | FNf treatment | P value | |
|---|---|---|---|
| TNF-α | 11 ± 10 | 370 ± 60 | 0.0003 |
| LIF | 47 ± 2 | 56 ± 3 | 0.0003 |
| IGF-I | 295 ± 4 | 305 ± 4 | 0.001 |
| FGF-1 | 13 ± 1 | 16 ± 1 | 0.02 |
Monocytes adherent to laminin-coated plastic plates were stimulated with FNf, and the culture supernatant was harvested after 24 hrs. Factors were assayed by ELISA; data are means ± SEM expressed as picograms per milliliter (pg/ml) from nine different normal donors. Statistical analysis comparing the no treatment versus the FNf-treated cultures was done by paired t test. TNF-α, tumor necrosis factor-α; LIF, leukemia inhibitory factor; IGF-I, insulin-like growth factor-I; FGF-1, fibroblast growth factor-1.
Discussion
TNF-α, IGF-I, FGF-1, and LIF have previously been associated with protection of cardiac muscle cells from hypoxic or ischemia-reperfusion injury (16–18, 22–25). The novel, unifying concept brought out by the present report is that fragments of fibronectin, which are generated by proteolysis in the first minutes to hours after ischemic tissue injury (4), can stimulate macrophages to produce all of these agents. Blocking antibody studies suggest that each of these four agents, acting alone, significantly protects myocytes from apoptotic death and that FGF-1, IGF-I, and LIF may be additive in their effects. Both rat tissue macrophages and human monocytes made all the factors even without stimulation, perhaps as a result of adherence to the laminin substrate, but production was upregulated by FNf. Production by monocyte-derived macrophages indicates that leukocytes likely to enter newly injured tissue from the blood can be sources of these factors.
The multiplicity of factors produced as a result of FNf stimulation could reflect the fact that some are produced in a cascade. For example, IGF-I stimulates the production of TNF-α (26), which, in some cell types, induces LIF production (27). This diversity ensures that the injured myocytes receive effective anti-apoptotic signals through a variety of signaling pathways. TNF-α and IGF-I activate the phosphoinositol-3′-kinase/Akt pathway in cardiac myocytes (28, 29), but anti-apoptotic signals from IGF-I follow more than one signaling pathway (30, 31). To suppress apoptosis of cardiac myocytes, IGF-I also transmits signals through ERK- and MEK1-dependent pathways to activate the transcription factor CREB, which then induces expression of the anti-apoptotic factor Bcl-2 (31). TNF-α activates NF-κB, also via the MEK1 and I-κB kinase pathways, to induce production of the anti-apoptotic cytokine, interleukin-6 (32). FGF-2 activates ERK and MEK1 to protect cardiac myocytes from endotoxin-mediated apoptosis (33); it is not known if FGF-1 also acts through the same signaling pathway. LIF causes the phosphorylation of the signal transducer and activator of transcription 3 (STAT3) in cardiac myocytes (34), but STAT3 may also be phosphorylated via other kinase pathways (35). Many questions remain concerning the complex signaling pathways that protect cardiac myocytes against apoptosis, particularly whether altered balances of different pathways generate different outcomes (36).
We previously found that fibronectin fragments regulate the ability of monocytes to migrate through injured tissue (4). Evidence that FNf also induce monocyte-derived macrophages to produce agents that protect parenchymal cells against apoptosis expands its homeostatic role and indicates that, even at the outset, host responses to tissue injury may produce agents that help to limit that injury. For example, a conventional view of the host inflammatory response to ischemic injury is that when tissue infiltrating neutrophils degranulate and release reactive oxygen intermediates, proteases, and other degradative enzymes, they cause more tissue injury than would have occurred in their absence (5). Although this may be true, the present studies suggest that by causing proteolysis of fibronectin, the inflammatory response also generates critical negative regulatory signals that help parenchymal cells survive.
The macrophage’s role in tissue injury has been considered to be that of a late-entering phagocyte that ingests parenchymal cell and neutrophil debris, and then calls in fibroblasts to remodel tissue and form a scar. With the discovery that monocytes enter damaged tissue as early as neutrophils (3), and that their constitutive production of factors that promote parenchymal cell survival is quickly upregulated by FNf, it is clear that a role for tissue preservation soon after the onset of injury must be added to the macrophage repertory. That fibronectin fragments enhance this role may explain the finding that a lack of plasma fibronectin exacerbates the extent of injury after reperfusion of the ischemic brain (37). Sakai et al. (37) found that the presence of plasma fibronectin decreased the number of apoptotic cells in the infarcted areas, and they postulated that neurons may respond directly to fibronectin by upregulating anti-apoptotic cell proteins such as Bcl-2. Our data suggest an additional possibility: stimulation of macrophages by fibronectin fragments induces the prompt release of diverse factors that enhance parenchymal cell survival.
Acknowledgments
We thank Pamela Fritz and Christine Norman-Tiner for technical assistance. We also wish to thank Blase Carabello, MD, for support and encouragement.
This work was supported by the Department of Veterans Affairs and the American Heart Association, Texas Affiliate, and by grants RO1-HL58515, -AG19327, -AI46285, -MH63035, and -HL42550 from the National Institutes of Health.
References
- 1.Rossen RD, Mann DL. Myocardial inflammation. In: Rich RR, editor. Clinical Immunology: Principles and practice. 2. New York: Mosby; 2001. pp. 80.1–80.15. [Google Scholar]
- 2.Lindsey M, Wedin K, Brown MD, Keller C, Evans AJ, Smolen J, Burns AR, Rossen RD, Michael LH, Entman ML. Matrix-dependent mechanism of neutrophil-mediated release and activation of matrix metalloproteinase 9 in myocardial ischemia/reperfusion. Circulation. 2001;103:2181–2187. doi: 10.1161/01.cir.103.17.2181. [DOI] [PubMed] [Google Scholar]
- 3.Birdsall HH, Green DM, Trial J, Youker KA, Burns AR, McKay CR, LaRosa GJ, Hawkins HK, Smith CW, Michael LH, Entman ML, Rossen RD. Complement C5a, TGF-1, and MCP-1, in sequence, induce migration of monocytes into ischemic myocardium within the first 1 to 5 hours following reperfusion. Circulation. 1997;95:684–692. doi: 10.1161/01.cir.95.3.684. [DOI] [PubMed] [Google Scholar]
- 4.Trial J, Baughn RE, Wygant JN, McIntyre BW, Birdsall HH, Youker KA, Evans A, Entman ML, Rossen RD. Fibronectin fragments modulate monocyte VLA-5 expression and monocyte migration. J Clin Invest. 1999;104:419–430. doi: 10.1172/JCI4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Entman ML, Smith CW. Postperfusion inflammation: a model for reaction to injury in cardiovascular disease. Cardiovasc Res. 1994;28:1301–1311. doi: 10.1093/cvr/28.9.1301. [DOI] [PubMed] [Google Scholar]
- 6.Weihrauch D, Arras M, Zimmerman R, Schaper J. Importance of monocytes/macrophages and fibroblasts for healing of micronecroses in porcine myocardium. Mol Cell Biochem. 1995;147:13–19. doi: 10.1007/BF00944778. [DOI] [PubMed] [Google Scholar]
- 7.Boyle MP, Weisman HF. Limitation of infarct expansion and ventricular remodeling by late reperfusion. Circulation. 1993;88:2872–2883. doi: 10.1161/01.cir.88.6.2872. [DOI] [PubMed] [Google Scholar]
- 8.Clark RA, Wikner NE, Doherty DE, Norris DA. Cryptic chemotactic activity of fibronectin for human monocytes resides in the 120-kDa fibroblastic cell-binding fragment. J Biol Chem. 1988;263:12115–12123. [PubMed] [Google Scholar]
- 9.Beezhold DH, Personius C. Fibronectin fragments stimulate tumor necrosis factor secretion by human monocytes. J Leukoc Biol. 1992;51:59–64. doi: 10.1002/jlb.51.1.59. [DOI] [PubMed] [Google Scholar]
- 10.Tremble PM, Damsky CH, Werb Z. Fibronectin fragments, but not intact fibronectin, signalling through the fibronectin receptor induce metalloproteinase gene expression in fibroblasts. Matrix. 1992;1(Suppl):212–214. [PubMed] [Google Scholar]
- 11.Forsyth CB, Pulai J, Loeser RF. Fibronectin fragments and blocking antibodies to alpha2beta1 and alpha5beta1 integrins stimulate mitogen-activated protein kinase signaling and increase collagenase 3 (matrix metalloproteinase 13) production by human articular chondrocytes. Arthritis Rheum. 2002;46:2368–2376. doi: 10.1002/art.10502. [DOI] [PubMed] [Google Scholar]
- 12.Ford DA, Rovetto MJ. Rat cardiac myocyte adenosine transport and metabolism. Am J Physiol. 1987;252:H54–H63. doi: 10.1152/ajpheart.1987.252.1.H54. [DOI] [PubMed] [Google Scholar]
- 13.Sun L, Chang J, Kirchhoff SR, Knowlton AA. Activation of HSF and selective increase in heat-shock proteins by acute dexamethasone treatment. Am J Physiol Heart Circ Physiol. 2000;278:H1091–H1097. doi: 10.1152/ajpheart.2000.278.4.H1091. [DOI] [PubMed] [Google Scholar]
- 14.Kirchhoff SR, Gupta S, Knowlton AA. Cytosolic heat shock protein 60, apoptosis, and myocardial injury. Circulation. 2002;105:2899–2904. doi: 10.1161/01.cir.0000019403.35847.23. [DOI] [PubMed] [Google Scholar]
- 15.Nakano M, Mann DL, Knowlton AA. Blocking the endogenous increase in HSP 72 increases susceptibility to hypoxia and reoxygenation in isolated adult feline cardiocytes. Circulation. 1997;95:1523–1531. doi: 10.1161/01.cir.95.6.1523. [DOI] [PubMed] [Google Scholar]
- 16.Kurrelmeyer KM, Michael LH, Baumgarten G, Taffet GE, Peschon JJ, Sivasubramanian N, Entman ML, Mann DL. Endogenous tumor necrosis factor protects the adult cardiac myocyte against ischemic-induced apoptosis in a murine model of acute myocardial infarction. Proc Natl Acad Sci U S A. 2000;97:5456–5461. doi: 10.1073/pnas.070036297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buerke M, Murohara T, Skurk C, Nuss C, Tomaselli K, Lefer AM. Cardioprotective effect of insulin-like growth factor I in myocardial ischemia followed by reperfusion. Proc Natl Acad Sci U S A. 1995;92:8031–8035. doi: 10.1073/pnas.92.17.8031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cuevas P, Reimers D, Carceller F, Martinez-Coso V, Redondo-Horcajo M, Saenz de Tejada I, Gimenez-Gallego G. Fibroblast growth factor-1 prevents myocardial apoptosis triggered by ischemia reperfusion injury. Eur J Med Res. 1997;2:465–468. [PubMed] [Google Scholar]
- 19.Eddy LJ, Goeddel DV, Wong GHW. Tumor necrosis factor—a pretreatment is protective in a rat model of myocardial ischemia-reperfusion injury. Biochem Biophys Res Commun. 1992;184:1056–1059. doi: 10.1016/0006-291x(92)90698-k. [DOI] [PubMed] [Google Scholar]
- 20.Negoro S, Kunisada K, Funamoto M, Darville MI, Eizirik DL, Osugi T, Izumi M, Oshima Y, Nakaoka Y, Hirota H, Kishimoto T, Yamauchi-Takahara K. Activation of signal transducer and activator of transcription 3 protects cardiomyocytes from hypoxia/reoxygenation-induced oxidative stress through the upregulation of manganese superoxide dismutase. Circulation. 2001;104:979–981. doi: 10.1161/hc3401.095947. [DOI] [PubMed] [Google Scholar]
- 21.Wang F, Seta Y, Baumgarten G, Engel DJ, Sivasubramanian N, Mann DL. Functional significance of hemodynamic overload-induced expression of leukemia-inhibitory factor in the adult mammalian heart. Circulation. 2001;103:1296–1302. doi: 10.1161/01.cir.103.9.1296. [DOI] [PubMed] [Google Scholar]
- 22.Arras M, Strasser R, Mohri M, Doll R, Eckert P, Schaper W, Schaper J. Tumor necrosis factor-alpha is expressed by monocyte/macrophages following cardiac microembolization and is antagonized by cyclosporine. Basic Res Cardiol. 1998;93:97–107. doi: 10.1007/s003950050069. [DOI] [PubMed] [Google Scholar]
- 23.Kluge A, Zimmermann R, Munkel B, Mohri M, Sack S, Schaper J, Schaper W. Insulin-like growth factor I is involved in inflammation linked angiogenic processes after microembolization in porcine heart. Cardiovasc Res. 1995;29:407–415. [PubMed] [Google Scholar]
- 24.Kuwabara K, Ogawa S, Matsumoto M, Koga S, Clauss M, Pinsky DJ, Lyn P, Leavy J, Witte L, Joseph-Silverstein J, Furie MB, Torcia G, Cozzolino F, Kamada T, Stern DM. Hypoxia-mediated induction of acidic/basic fibroblast growth factor and platelet-derived growth factor in mononuclear phagocytes stimulates growth of hypoxic endothelial cells. Proc Natl Acad Sci U S A. 1995;92:4606–4610. doi: 10.1073/pnas.92.10.4606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zou Y, Takano H, Mizukami M, Akazawa H, Qin Y, Toko H, Sakamoto M, Minamino T, Nagai T, Komuro I. Leukemia inhibitory factor enhances survival of cardiomyocytes and induces regeneration of myocardium after myocardial infarction. Circulation. 2003;108:748–753. doi: 10.1161/01.CIR.0000081773.76337.44. [DOI] [PubMed] [Google Scholar]
- 26.Renier G, Clement I, Desfaits A-C, Lambert A. Direct stimulatory effect of insulin-like growth factor-I on monocyte and macrophage tumor necrosis factor-a production. Endocrinology. 1996;137:4611–4618. doi: 10.1210/endo.137.11.8895324. [DOI] [PubMed] [Google Scholar]
- 27.Hamilton JA, Waring PM, Filonzi EL. Induction of leukemia inhibitory factor in human synovial fibroblasts by IL-1 and tumor necrosis factor-a. J Immunol. 1993;150:1496–1502. [PubMed] [Google Scholar]
- 28.Wu W, Lee W, Wu Y, Chen D, Liu T, Sharma PM, Wang PH. Expression of constitutively active phosphatidylinositol 3 kinase inhibits activation of caspase 3 and apoptosis of cardiac muscle cells. J Biol Chem. 2000;275:40113–40119. doi: 10.1074/jbc.M004108200. [DOI] [PubMed] [Google Scholar]
- 29.Condorelli G, Morisco C, Latronico MVG, Claudio PP, Dent P, Tsichlis P, Condorelli G, Frati G, Drusco A, Croce CM, Napoli C. TNF-a signal transduction in rat neonatal cardiac myocytes: definition of pathways generating from the TNF-a receptor. FASEB J. 2002;16:1732–1737. doi: 10.1096/fj.02-0419com. [DOI] [PubMed] [Google Scholar]
- 30.Parrizas M, Saltiel AR, LeRoith D. Insulin-like growth factor I inhibits apoptosis using the phosphatidylinositol 3′-kinase and mitogen-activated protein kinase pathways. J Biol Chem. 1997;272:154–161. doi: 10.1074/jbc.272.1.154. [DOI] [PubMed] [Google Scholar]
- 31.Mehrhof FB, Muller FU, Bergmann MW, Li P, Wang Y, Schmitz W, Dietz R, von Harsdorf R. In cardiomyocyte hypoxia, insulin-like growth factor-I-induced antiapoptotic signaling requires phosphatidylinositol-3-OH-kinase-dependent and mitogen-activated protein kinase-dependent activation of the transcription factor cAMP response element-binding protein. Circulation. 2001;104:2088–2094. doi: 10.1161/hc4201.097133. [DOI] [PubMed] [Google Scholar]
- 32.Craig R, Larkin A, Mingo AM, Thuerauf DJ, Andrews C, McDonough PM, Glembotski CC. p38 MAPK and NF-kB collaborate to induce inter5leukin-6 gene expression and release. J Biol Chem. 2000;275:23814–23824. doi: 10.1074/jbc.M909695199. [DOI] [PubMed] [Google Scholar]
- 33.Iwai-Kanai E, Hasegawa K, Fujita M, Araki M, Yanazume T, Adachi S, Sasayama S. Basic fibroblast growth factor protects cardiac myocytes from iNOS-mediated apoptosis. J Cell Physiol. 2002;190:54–62. doi: 10.1002/jcp.10036. [DOI] [PubMed] [Google Scholar]
- 34.Tone E, Kunisada K, Fujio Y, Matsui H, Negoro S, Oh H, Kishimoto T, Yamauchi-Takihara K. Angiotensin II interferes with leukemia inhibitory factor-induced STAT3 activation in cardiac myocytes. Biochem Biophys Res Commun. 1998;253:147–150. doi: 10.1006/bbrc.1998.9767. [DOI] [PubMed] [Google Scholar]
- 35.Ihle JN. The STAT family in cytokine signaling. Curr Opin Cell Biol. 2001;13:211–217. doi: 10.1016/s0955-0674(00)00199-x. [DOI] [PubMed] [Google Scholar]
- 36.Wang PH. Roads to survival: insulin-like growth factor-1 signaling pathways in cardiac muscle. Circ Res. 2001;88:552–554. doi: 10.1161/01.res.88.6.552. [DOI] [PubMed] [Google Scholar]
- 37.Sakai T, Johnson KJ, Murozono M, Sakai K, Magnuson MA, Weiloch T, Cronberg T, Isshiki A, Erickson HP, Fassler R. Plasma fibronectin supports neuronal survival and reduces brain injury following transient focal cerebral ischemia but is not essential for skin-wound healing and hemostasis. Nature Med. 2001;7:324–330. doi: 10.1038/85471. [DOI] [PubMed] [Google Scholar]