

Abstract
Objective:
To report novel disease and pathology due to HSPB8 mutations in 2 families with autosomal dominant distal neuromuscular disease showing both myofibrillar and rimmed vacuolar myopathy together with neurogenic changes.
Methods:
We performed whole-exome sequencing (WES) in tandem with linkage analysis and candidate gene approach as well as targeted next-generation sequencing (tNGS) to identify causative mutations in 2 families with dominant rimmed vacuolar myopathy and a motor neuropathy. Pathogenic variants and familial segregation were confirmed using Sanger sequencing.
Results:
WES and tNGS identified a heterozygous change in HSPB8 in both families: c.421A > G p.K141E in family 1 and c.151insC p.P173SfsX43 in family 2. Affected patients had a distal myopathy that showed myofibrillar aggregates and rimmed vacuoles combined with a clear neurogenic component both on biopsy and neurophysiologic studies. MRI of lower limb muscles demonstrated diffuse tissue changes early in the disease stage progressing later to fatty replacement typical of a myopathy.
Conclusion:
We expand the understanding of disease mechanisms, tissue involvement, and phenotypic outcome of HSPB8 mutations. HSPB8 is part of the chaperone-assisted selective autophagy (CASA) complex previously only associated with Charcot-Marie-Tooth type 2L (OMIM 60673) and distal hereditary motor neuronopathy type IIa. However, we now demonstrate that patients can develop a myopathy with histologic features of myofibrillar myopathy with aggregates and rimmed vacuoles, similar to the pathology in myopathies due to gene defects in other compounds of the CASA complex such as BAG3 and DNAJB6 after developing the early neurogenic effects.
Mutations in the small heat shock protein 22 gene (HSPB8, also called HSP22) located on chromosome 12q24.23 are associated with Charcot-Marie-Tooth type 2L (CMT2L) (OMIM 60673)1 and distal hereditary motor neuronopathy type IIa (dHMN2A).2 HSPB8 is part of the chaperone-assisted selective autophagy (CASA) complex, a vital part of the cellular protein quality control system in mechanically strained cells and tissues such as skeletal muscle, heart, and lung.3–5 HSPB8 has not been previously associated with a myopathy.
The CASA complex comprises the molecular chaperones HSPA8 and HSPB8 and the co-chaperones BAG3 and STUB1.5 In muscle, CASA has a specific role in maintenance of the Z-disk and protein turnover.6,7 CASA mediates degradation of the actin cross-linking protein filamin.3,5 If mechanical tension results in permanent unfolding of filamin, it is recognized by the CASA chaperone complex and leads to autophagic degradation of damaged proteins. Impairment of CASA in patients and animal models causes muscular dystrophy and cardiomyopathy.6–8 Specifically, mutations in the CASA component BAG3 cause myofibrillar myopathy. Furthermore, DNAJB6 (associated with limb girdle muscular dystrophy 1D [LGMD1D]) interacts with members of the CASA complex, including BAG3. Previous studies on LGMD1D suggest that its pathogenesis is also mediated by defective chaperone function leading to myofibrillar aggregates and rimmed vacuolar pathology.9
We report 2 families with HSPB8 mutations presenting with a novel distal neuromyopathy phenotype. Following the early neurogenic defect demonstrated in these families, the disease progresses to cause a myopathy in the later stage of the disease with myofibrillar aggregates and rimmed vacuolar pathology.
METHODS
Patient selection and evaluation.
We studied 2 families with a genetically undetermined dominant rimmed vacuolar distal myopathy and a length-dependent motor neuropathy. Clinical records and pathology were reviewed for the 2 families and the individuals were available for further examinations including muscle biopsy and neurophysiology testing.
Standard protocol approvals, registrations, and patient consents.
All family members gave written informed consent. This study was approved by the Northern Sydney Local Health District and the Sydney Children's Hospitals Network Human Research Ethics Committees and Helsinki University Hospital institutional review board (1010-349M, 10/CHW/45, and 195/13/03/00/11).
Histology and immunohistochemistry.
Frozen muscle sections were processed for routine histochemical staining, including hematoxylin & eosin, modified Gomori trichrome, reduced nicotinamide adenine dinucleotide–tetrazolium reductase, and combined succinate dehydrogenase–cytochrome oxidase. For automated immunohistochemistry, antibodies against the following proteins were applied: myosin fast, myosin slow, fetal and neonatal myosin heavy chains (clones NCL-MHCf, NCL-MHCs, NCL-MHCd, and NCL-MHCn, Leica Biosystems, Newcastle, UK), MHC class I (M0736, Dako, Glostrup, Denmark), dystrophin (NCL-DYS2, Leica Biosystems), desmin (Biogenex, Fremont, CA), myotilin (Leica Biosystems), αB-crystallin (Leica Biosystems), LC3b (Cell Signaling Technology, Beverly, MA), p62 (Santa Cruz Biotechnology, Dallas, TX), TAR DNA-binding protein 43 (TDP-43) (Proteintech, Chicago, IL), and HSPB8 (Sigma-Aldrich, St. Louis, MO). Ventana Benchmark automated immunostainer with 3,3'-diaminobenzidine detection was used for immunohistochemistry (Ventana Medical Systems, Tucson, AZ). The sections were imaged on an Olympus BX50 microscope linked to a SPOT digital imaging camera (Diagnostic Instruments Inc., Sterling Heights, MI). After fixing of sections in 4% paraformaldehyde, further immunofluorescent analysis was performed on patient III-1 (family 1) using the primary antibodies HSPB8 ab79784 (Abcam, Cambridge, UK), DNAJB6 ab96539 (Abcam), myotilin 10731-1-AP (Proteintech), p62 P0067 (Sigma-Aldrich), BAG3 10599-1-AP (Proteintech), and TDP-43 clone 2E2-D3 (Sigma-Aldrich). Alexa-488/Alexa-546-conjugated secondary antibodies (Life Technologies, Carlsbad, CA; Thermo Fisher Scientific, Waltham, MA) were used for detection.
Next-generation sequencing and bioinformatic filtering.
Whole-exome sequencing (WES) was performed on family 1 (II-2 and III-2). Exome libraries were captured using hybridization with the Agilent (Wilmington, DE) SureSelect V2 and WES performed on an Illumina (San Diego, CA) HiSeq 2000 as previously described.10,11 FASTQ sequencing files were processed using Picard and jointly called with over 2,000 control samples using the GATK 3.0 Haplotype Caller. The data received had an average read-depth of >80 and 20-fold coverage in over 90% of targeted regions.
Filtering of variants was performed using xBrowse (http://atgu.mgh.harvard.edu/xbrowse), initially screening known myopathy genes (listed in the Neuromuscular Disorders Gene Table [December 2012; www.musclegenetable.fr]). Sequence variants were first filtered based on population frequency in the exome variant server (NHLBI GO Exome Sequencing Project, Seattle, WA [http://evs.gs.washington.edu/EVS/], March 2013), 1000 Genomes,12 and internal databases, with variants showing >1% frequency in any population discarded. Second, we considered only variants with predicted functional impact on coding regions (predicted missense, nonsense, and essential splice site single nucleotide polymorphisms, insertions or deletions [indels]). When a candidate neuromuscular disease gene was not identified, we searched for variants in all genes based on inheritance pattern.
Targeted next-generation sequencing (tNGS) was performed on the proband of family 2 as previously described13 using version 2 of the MYOcap gene panel that is targeted to the exons of 236 genes that are known or predicted to cause muscular dystrophy or myopathy.
Selection of candidate genes and confirmation of variants with Sanger sequencing.
Polyphen-2 pathogenicity prediction program was used to perform in silico analysis of missense variants (http://genetics.bwh.harvard.edu/pph2/). Variants were also correlated with patient phenotype and results of clinical investigations. Sanger sequencing was used to confirm all variants identified by WES and tNGS were truly present and to confirm familial segregation.
Linkage analysis.
Family 1 was genotyped for microsatellite markers D12S349, D12S385, D12S395, and D12S1666 spanning a region of 1.2 Mb around HSPB8. Fluorescently labeled PCR products were analyzed using ABI3730xl DNA Analyzer and GeneMapper v4.0 software (Applied Biosystems, Foster City, CA).
Functional studies.
Plasmid constructs.
The coding region of human HSPB8 was cloned into pEF6/V5-His TOPO (Invitrogen, Carlsbad, CA). The K141E mutation was introduced to the wild-type HSPB8 construct by site-directed mutagenesis. Inducible constructs were made subcloning from pEF6 to pcDNA5/TO (Invitrogen). All constructs were verified by Sanger sequencing. The GFP-120Q-HTT construct was described earlier.14
Filter-trap assay.
The filter-trap assay was performed essentially as described.8 T-REx 293 cells were transfected with 875 ng inducible HSPB8 and 125 ng 120Q-HTT, induced after 4 hours and harvested 48 hours post transfection. Aggregation score ([aggregated/soluble]induced/[aggregated/soluble]uninduced) was calculated from the amounts of aggregated and soluble huntingtin in the uninduced and induced cells in each transfection pair as described.8
RESULTS
Patient characteristics.
Family 1.
The pedigree was consistent with an autosomal dominant inheritance pattern (figure 1). The proband (patient III-2), a 30-year-old man, born to nonconsanguineous Caucasian (English) parents with normal milestones, presented at age 22 years with progressive weakness predominantly affecting his distal lower limbs. He noted frequent falls when playing sports. On examination, there was bilateral pes cavus, atrophy of tarsal muscles, extensor digitorum brevis (EDB), and tibialis anterior bilaterally with predominantly distal weakness affecting ankle dorsiflexion (Medical Research Council [MRC] 3/5). Reflexes were absent at the ankles but sensation was intact. Nerve conduction studies (NCS) revealed a length-dependent axonal motor neuropathy predominantly affecting the lower limbs (table e-1 on the Neurology® Web site at Neurology.org). EMG findings showed features that were consistent with mixed myopathic and neurogenic pathology in the upper and lower limbs (table e-2).
Figure 1. Pedigrees of 2 unrelated families with HSPB8 mutations causing a distal myopathy.

Pedigrees of family 1 and family 2. The blue symbols represent unaffected members and red symbols represent affected members. *Patients with available DNA samples.
Previous laboratory investigations showed creatine kinase (CK) levels had ranged between 850 and 2,000 U/L (normal <250 U/L). Cardiac and respiratory investigations (including an ECG, echocardiogram, and standard respiratory function tests) were within normal limits. MRI of the lower limbs showed diffuse degenerative involvement predominantly affecting the distal lower limbs consistent with neurogenic changes (figure 2A). Genetic testing for DES, MHY7, FLNC, GNE, TPM2, VCP, MYH1-8, MYOT, MATR3, TCAP, TTN, TNNC1, TNNC2, TNNI1, TPM3, TIA1, and ZASP was performed and no pathogenic mutations were identified. Southern blot for facioscapulohumeral muscular dystrophy was also performed and did not detect any abnormal fragment.
Figure 2. Muscle MRI of the affected patients with HSPB8 variants.

(A) In patient III-2 (family 1), early diffuse degenerative changes are seen in the distal thigh muscles and in the lower legs, specifically in the gastrocnemius and peroneal muscles. (B) In patient II-2 (family 1), there is severe fatty degenerative replacement in the quadriceps muscles and milder in sartorius, gracilis, semimembranosus, and long head of biceps femoris. In the lower legs, there is generalized fatty replacement, although to a lesser extent in the left soleus and extensor digitorum longus. (C) In patient III-2 (family 2), fatty degenerative changes are present in the gluteus minimus and medius on the pelvic level, in the anterior compartment of the lower legs, and with more diffuse involvement in the thigh muscles.
The proband's 56-year-old mother (patient II-2) reported ankle pain since childhood. In her early 20s, she developed weakness of ankle dorsiflexion, eversion, and toe extension and flexion that progressed over the next 5 years to involve her proximal muscles. On examination, she had a waddling gait with bilateral foot drop, severe lower limb proximal weakness (MRC 2/5), and mobilized over short distances with a walker. Reflexes were present in the upper limbs and at the knees but absent at the ankles. Sensation was intact.
Investigations revealed a CK that was only mildly elevated with the highest level recorded as 269 U/L (reference <250 U/L). Cardiorespiratory examination was normal. An echocardiogram and standard respiratory function tests were normal. NCS showed a predominantly lower limb axonal motor neuropathy (table e-1) and EMG demonstrated a diffuse myopathic process severely affecting the lower limbs (table e-3). T1-weighted MRI of the lower limbs showed extensive fatty replacement in the proximal and distal lower limbs with milder degenerative change in sartorius, gracilis, semimembranosus, long head of biceps femoris, left soleus, and extensor digitorum longus in the lower legs (figure 2B).
The sibling of the affected proband (patient III-3), aged 27 years, had reportedly been asymptomatic. However, neurophysiology findings were consistent with active and chronic neurogenic pathology affecting the distal lower limbs, consistent with axonal pathology (tables e-1 and e-4). In light of these findings, further genetic counseling and testing was offered to the patient.
Family 2.
The proband (patient III-2), a 62-year-old man, was born to nonconsanguineous Caucasian (French) parents with normal milestones. The family history revealed 4 other affected members (figure 1). The cousin (III-1) had been reviewed by a neurologist and the same phenotype was confirmed. At age 46 years, the proband underwent surgery for an acoustic neuroma. Postoperatively, he noticed deterioration in his balance. At the same time, he noticed muscle weakness in the distal lower limbs, which slowly progressed and led to an inability to walk on heels and high steppage gait, although at age 62 years he was still ambulant without aids. He had experienced cramps and fasciculations since early adulthood but had not had any sensory symptoms. At age 62 years, the proband had marked weakness of ankle dorsiflexion bilaterally and toe extension on the right (MRC 1–2/5). Hip flexion and arm abduction were mildly weak (MRC 4/5); otherwise, strength in all other limb muscles was normal. There was no facial or bulbar weakness. Muscle atrophy was observed in the scapular region with scapular winging (figure 3). Tibialis anterior was also symmetrically atrophied, whereas EDB was only atrophied on the right side. Sensation was intact. Reflexes were normal or brisk.
Figure 3. Scapular winging and muscle atrophy in proband of family 2 (III-2).
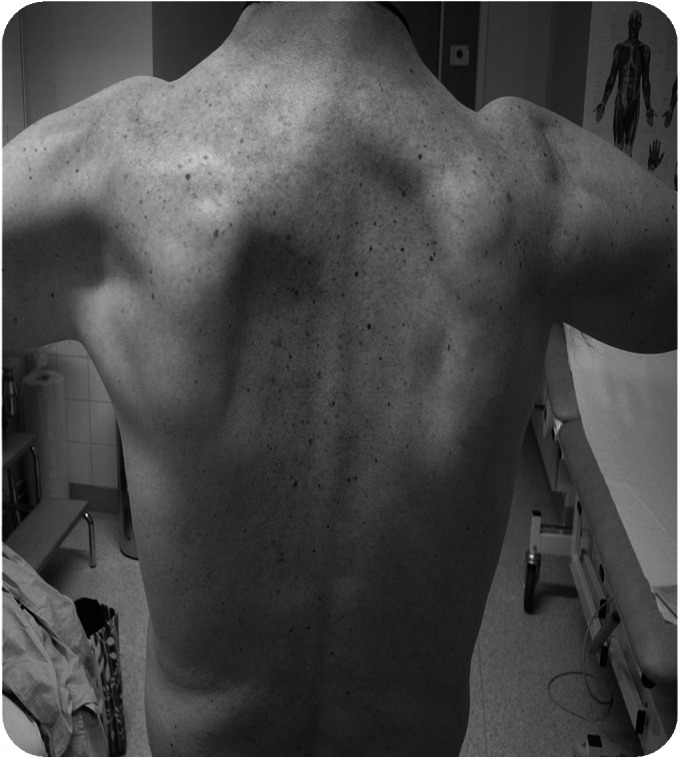
Asymmetrical upper limb proximal muscle atrophy was evident with scapular winging more pronounced on the left compared to the right.
Investigations revealed normal echocardiogram and pulmonary function tests. NCS were normal, but EMG showed chronic neurogenic and myopathic changes in the lower limbs. CK was mildly elevated (369 U/L). T1-weighted MRI of the lower limbs showed extensive fatty degenerative changes in the anterior compartment of the lower legs, in gluteus minimus and medius, and some diffuse involvement in the posterior thigh muscles (figure 2C).
Next-generation sequencing identified a mutation in HSPB8 in families 1 and 2.
We initially searched for sequence variants in known myopathy genes that failed to reveal a candidate pathogenic gene that correlated with the phenotype. We then screened by inheritance pattern searching for de novo dominant maternally inherited pathogenic variants. Exome sequencing identified a single heterozygous change in exon 2 of HSPB8 (c.421A > G, p.K141E). We confirmed the mutation was truly present by Sanger sequencing and confirmed familial segregation. The same mutation in HSPB8 has previously been associated with dHMN2A/CMT2L but not with a myopathy.
tNGS revealed 2 possibly pathogenic heterozygous mutations in the proband of family 2: c.712A > G in SQSTM1 (according to transcript NM_003900), p.K238E and c.151insC in HSPB8 (according to transcript NM_014365), p.P173SfsX43. Both variants were confirmed by Sanger sequencing but only the variant in HSPB8 segregated correctly in the family. Mutation c.151insC p.P173SfsX43 has not been reported previously but it causes a frameshift in the last exon and transfers the stop codon forward, making the protein 18 amino acids longer, and is thus probably pathogenic.
Linkage analysis.
Linkage analysis was performed for family 1. The 3 affected patients, II-2, III-2, and III-3, with the HSPB8 mutation (c.421A > G p.K141E) shared the same haplotype. The haplotype was also present in I-1, II-4, and II-5 but without the mutation. This confirmed that the HSPB8 mutation was de novo in II-2 (table e-5).
Histology and immunohistochemistry.
Muscle biopsies of the left vastus lateralis (family 1, III-2) (figure 4, A–H) and tibialis anterior (family 2, III-2) (figure 4, I–P) showed dystrophic features. Fiber type grouping and small and larger group atrophies were compatible with a neurogenic component (figure e-1). There were increased internal nuclei and numerous rimmed vacuolar fibers, splitting, and cytoplasmic bodies. Moth-eaten fibers were noted on oxidative stains (figure e-1). In addition, some subsarcolemmal and non-rimmed vacuolation and sarcoplasmic masses were present. Myofibrillar aggregations were observed that were reactive for desmin, myotilin, αB-crystallin, and dystrophin. Vacuolated fibers were strongly positive for SMI-31, TDP-43, p62, and LC3b. In immunofluorescence analysis (family 1, III-2) (figure 4, D–H), HSPB8, DNAJB6, myotilin, and BAG3 showed moderate to strong colocalization with TDP-43 in the myofibrillar aggregates, and the rimmed vacuoles were strongly p62-positive. In addition, HSPB8 was diffusely increased in atrophic fibers (figure 4D).
Figure 4. Muscle histochemistry and immunohistochemistry of HSPB8-associated distal myopathy.

Panel shows histochemical and immunohistochemical analyses of patient III-2 (family 1): (A) Herovici, (B) α-B-crystallin, (C) LC3b, (D) HSPB8 (green)/TAR DNA-binding protein 43 (TDP-43) (red) double stain, (E) DNAJB6 (green)/TDP-43 (red), (F) myotilin (green)/TDP-43 (red), (G) p62 (green)/TDP-43 (red), (H) BAG3 (green)/TDP-43 (red); and patient III-2 (family 2): (I) Herovici, (J) α-B-crystallin, (K) LC3b, (L) myotilin, (M) SMI-31, (N) TDP-43, (O) DYS2, (P) p62. Magnification ×20 in 3,3'-diaminobenzidine/immunohistochemistry stainings (A–C, I–P) and ×40 in immunofluorescence stainings (D–H).
Loss of in vitro chaperone activity.
By transient cotransfection of an aggregation prone protein (GFP-120Q-HTT) and HSPB8 constructs, a loss of ability to prevent aggregation was observed for the mutant HSPB8 protein (figure 5).
Figure 5. The K141E mutation alters the aggregation score compared to wild-type.
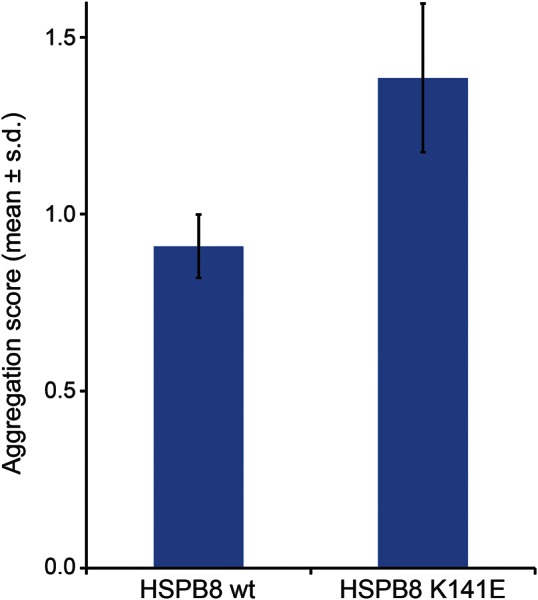
Transient cotransfection of an aggregation prone protein (GFP-120Q-HTT) and HSPB8 constructs results in a loss of ability to prevent aggregation in the mutant HSPB8 protein.
DISCUSSION
Next-generation sequencing identified the causative mutations in 2 families with autosomal dominant distal-onset myopathy and a length-dependent motor neuropathy. To date, myopathy has not been reported to be associated with HSPB8 mutations and we thus expand the organ involvement of HSPB8 mutations to include muscle tissue. In particular, the K141E mutation identified in family 1 has previously been associated only with a motor neuropathy CMT2L/dHMN2A.2,15 Muscle biopsy findings in both our families showed prominent rimmed vacuolar pathology and myofibrillar aggregates. The involvement of muscle tissue with mutated HSPB8 is not unexpected since HSPB8 is a direct ligand of DNAJB6, causing a similar pathology when mutated in LGMD1D.8 The K141E mutation alters the aggregation score compared to wild-type (figure 5) in a similar way as mutations in DNAJB6.8 Mutations in HSPB8 have also previously been shown to increase aggregation of 43Q-huntingtin in cell culture.4 Both HSPB8 and DNAJB6 are part of the CASA complex,5,8 suggesting that malfunction of CASA might be involved in both pathologies.
The disease onset in our families is similar to that previously reported in patients with K141 HSPB8 mutations.2,16 Clinically, the disease manifests as distal weakness affecting ankle dorsiflexion, eversion, and toe extension/flexion. In contrast to pure motor neuropathy, the muscle weakness progresses over the next 10–15 years to include proximal lower limb weakness with difficulties rising from the chair and a waddling gait in addition to the initial foot drop.
Neurophysiology studies demonstrated a length-dependent axonal motor neuropathy with EMG showing evidence of denervation in the distal lower limbs and myopathic changes proximally (tables e-2 through e-4). MRI of the lower limb muscles provided additional evidence for the dual tissue involvement: In the early stages of disease, there is diffuse neurogenic involvement of gastrocnemius, deep toe flexors, and peroneus. In the late stage of disease, as shown from the MRI of patient F1-II-2, there was progression to severe fatty degenerative (dystrophic) changes in proximal thigh muscles as well as in the lower legs (figure 2, A and B). The fatty degenerative change was more pronounced in the anterior compartment of the lower legs with the p.P173SfsX43 mutation (figure 2C).
HSPB8 mutations have previously only been associated with neuropathies. Three mutations have been reported with a missense change at the K141 residue: c. 421A > G (K141E), c.423 G > C (K141N), and c.423 G > T (K141N). The lysine residue is located in the α-crystallin domain that is a highly conserved region within the heat shock protein superfamily. Mutations in the α-crystallin region have been shown to decrease its chaperone activity17 and the decreased chaperone activity has been suggested as a mechanism for the peripheral neuropathy.4 In our families with both mutations, there was clear evidence of a myofibrillar autophagic myopathy including rimmed vacuoles and protein aggregates positive for a range of proteins associated with myofibrillar myopathy such as desmin, myotilin, and α-B-crystallin. These aggregates also contain HSPB8 and other CASA complex partners DNAJB6 and BAG3. The autophagic component, as highlighted by reactivity for LC3, p62, TDP-43, and SMI-31, is the cellular response to abnormal misfolded proteins due to malfunction and defect in chaperone protein quality control and turnover, and therefore also emphasizes the defective chaperone activity as a disease mechanism. The muscle pathology was markedly identical with the novel c.151insC p.P173SfsX43 mutation, suggesting a similar molecular dysfunction of the chaperone activity.
We have identified 2 families with mutations in HSPB8 causing a dual involvement of a peripheral motor neuropathy and a rimmed vacuolar myofibrillar myopathy, presenting with distal weakness that progresses to involve the proximal muscles. This is consistent with the functional data on HSPB8 as a key component of the Z-disk-associated CASA complex with previously documented importance in several myopathies showing similar myofibrillar and autophagic pathology.7,8,18 This report brings new light into the function of HSPB8 and the clinical consequences of mutations to include a distinct myopathy that predominates later in the disease course. In the previously reported patients with K141 mutations, a relatively unusual marked progression to significant proximal weakness was also reported clinically15 and on muscle MRI.16 We recommend that families with a dominant rimmed vacuolar myofibrillar myopathy and motor neuropathy should be screened for HSPB8 mutations.
Supplementary Material
GLOSSARY
- CASA
chaperone-assisted selective autophagy
- CK
creatine kinase
- CMT2L
Charcot-Marie-Tooth type 2L
- dHMN2A
distal hereditary motor neuronopathy type IIa
- EDB
extensor digitorum brevis
- LGMD1D
limb-girdle muscular dystrophy 1D
- MRC
Medical Research Council
- NCS
nerve conduction studies
- TDP-43
TAR DNA-binding protein 43
- tNGS
targeted next-generation sequencing
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
R. Ghaoui: study concept, acquisition of data, analysis and interpretation, draft of the manuscript. J. Palmio: acquisition of data, analysis and interpretation, draft of the manuscript. M. Lek: acquisition of data, analysis and interpretation. M. Needham: acquisition of data. A. Evilä: acquisition of data, analysis and interpretation. P. Hackman: acquisition of data, analysis and interpretation. P.-H. Jonson: acquisition of data, analysis and interpretation. S. Penttilä: acquisition of data, analysis and interpretation. A. Vihola: acquisition of data, analysis and interpretation. S. Huovinen: acquisition of data. M. Lindfors: acquisition of data. R.L. Davis: acquisition of data. L. Waddell: acquisition of data, analysis and interpretation. S. Kaur: acquisition of data. C. Yiannikas: acquisition of data. K.N. North: study concept, acquisition of data. D.G. MacArthur: acquisition of data, analysis and interpretation. N.F. Clarke: acquisition of data. C.M. Sue: study concept, acquisition of data, critical revision of the manuscript for important intellectual content. B. Udd: study concept, acquisition of data, critical revision of the manuscript for important intellectual content.
STUDY FUNDING
This project was supported by Australian National Health and Medical Research Council (NHMRC) grants: APP1074954 (R. Ghaoui), APP1031893 and APP1022707 (K.N. North and N.F. Clarke), APP1035828 (N.F. Clarke), APP1008433 (C.M. Sue), APP1037797 (R.L. Davis), and Muscular Dystrophy New South Wales (R. Ghaoui). Exome sequencing was supported by grants from the National Human Genome Research Institute of the NIH (Medical Sequencing Program grant U54 HG003067 to the Broad Institute principal investigator, E. Lander). Also supported by Juselius Foundation, Finnish Academy, and the Folkhalsan Foundation (B. Udd).
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Tang B, Zhao GH, Luo W, et al. Small heat-shock protein 22 mutated in autosomal dominant Charcot-Marie-Tooth disease type 2L. Hum Genet 2005;116:222–224. [DOI] [PubMed] [Google Scholar]
- 2.Irobi J, Van Impe K, Seeman P, et al. Hot-spot residue in small heat shock protein 22 causes distal motor neuropathy. Nat Genet 2004;36:597–601. [DOI] [PubMed] [Google Scholar]
- 3.Ulbricht A, Eppler FJ, Tapia VE, et al. Cellular mechanotransduction relies on tension-induced and chaperone-assisted autophagy. Curr Biol 2013;23:5. [DOI] [PubMed] [Google Scholar]
- 4.Carra S, Sivilotti M, Chavez Zobel AT, Lambert H, Landry J. HSBP8, a small heat shock protein mutated in human neuromuscular disorders, has in vivo chaperone activity in cultured cells. Hum Mol Genet 2005;14:1659–1669. [DOI] [PubMed] [Google Scholar]
- 5.Arndt V, Dick N, Tawo R, et al. Chaperone-assisted selective autophagy is essential for muscle maintenance. Curr Biol 2010;20:143–148. [DOI] [PubMed] [Google Scholar]
- 6.Homma S, Iwasaki M, Shelton GD, Engvall E, Reed JC, Takayama S. BAG3 deficiency results in fulminant myopathy and early lethality. Am J Pathol 2006;169:761–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Selcen D, Muntoni F, Burton BK, et al. Mutation in BAG3 causes severe dominant childhood muscular dystrophy. Ann Neurol 2009;65:83–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sarparanta J, Jonson PH, Golzio C, et al. Mutations affecting the cytoplasmic functions of the co-chaperone DNAJB6 cause limb-girdle muscular dystrophy. Nat Genet 2012;44:450–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sarparanta J, Jonson PH, Luque H, Udd B. LGMD1D mutations impair the antiaggregation activity of DNAJB6. Neuromuscul Disord 2011;21:668. [Google Scholar]
- 10.Menezes MP, Waddell LB, Lenk GM, et al. Whole exome sequencing identifies three recessive FIG4 mutations in an apparently dominant pedigree with Charcot-Marie-Tooth disease. Neuromuscul Disord 2014;24:666–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ghaoui R, Cooper ST, Lek M, et al. Use of whole-exome sequencing for diagnosis of limb-girdle muscular dystrophy: outcomes and lessons learned. JAMA Neurol 2015;72:1424–1432. [DOI] [PubMed] [Google Scholar]
- 12.McVean GA, et al. An integral map of genetic variation from 1,092 human genomes. Nature 2012;491:56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Evila A, Udd B, Hackman P. G.P.20: a targeted next-generation sequencing panel for diagnostic use in primary myopathies. Neuromuscul Disord 2014;24:800. [DOI] [PubMed] [Google Scholar]
- 14.Hasholt L, Abell K, Norremolle A, Nellemann C, Fenger K, Sørensen SA. Antisense downregulation of mutant huntingtin in a cell model. J Gene Med 2003;5:528–538. [DOI] [PubMed] [Google Scholar]
- 15.Timmerman V, Raeymaekers P, Nelis E, et al. Linkage analysis of distal hereditary motor neuropathy type II (distal HMNII) in a single pedigree. J Neurol Sci 1992;109:41–48. [DOI] [PubMed] [Google Scholar]
- 16.Nakhro K, Park JM, Kim YJ, et al. A novel Lys141Thr mutation in small heat shock protein 22 (HSPB8) gene in Charcot-Marie-Tooth disease type 2L. Neuromuscul Disord 2013;23:656–663. [DOI] [PubMed] [Google Scholar]
- 17.Muchowski PJ, Wu GJ, Liang JJ, Adam ET, Clark JI. Site-directed mutations within the core “alpha-crystallin” domain of the small heat-shock protein, human alpha B-crystallin, decrease molecular chaperone functions. J Mol Biol 1999;289:397–411. [DOI] [PubMed] [Google Scholar]
- 18.Udd B. Molecular biology of distal muscular dystrophies-sarcomeric proteins on top. Biochim Biophys Acta 2007;1772:145–158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}