

Abstract
The α7 nicotinic acetylcholine receptors (nAChRs) are uniquely sensitive to selective positive allosteric modulators (PAMs), which increase the efficiency of channel activation to a level greater than that of other nAChRs. Although PAMs must work in concert with “orthosteric” agonists, compounds such as GAT107 ((3aR,4S,9bS)-4-(4-bromophenyl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinoline-8-sulfonamide) have the combined properties of agonists and PAMs (ago-PAM) and produce very effective channel activation (direct allosteric activation (DAA)) by operating at two distinct sites in the absence of added agonist. One site is likely to be the same transmembrane site where PAMs like PNU-120596 function. We show that the other site, required for direct activation, is likely to be solvent-accessible at the extracellular domain vestibule. We identify key attributes of molecules in this family that are able to act at the DAA site through variation at the aryl ring substituent of the tetrahydroquinoline ring system and with two different classes of competitive antagonists of DAA. Analyses of molecular features of effective allosteric agonists allow us to propose a binding model for the DAA site, featuring a largely non-polar pocket accessed from the extracellular vestibule with an important role for Asp-101. This hypothesis is supported with data from site-directed mutants. Future refinement of the model and the characterization of specific GAT107 analogs will allow us to define critical structural elements that can be mapped onto the receptor surface for an improved understanding of this novel way to target α7 nAChR therapeutically.
Keywords: allosteric regulation, ion channel, nicotinic acetylcholine receptors (nAChR), receptor desensitization, Xenopus
Introduction
The α7 nicotinic acetylcholine receptor (nAChR)3 is a recognized target for both central nervous system disorders, such as Alzheimer disease and schizophrenia, and peripheral indications, such as inflammation and associated pain (1, 2). As with other nicotinic receptors, conventional thinking has focused on the function of α7 as a ligand-gated ion channel that is activated by the transmitter acetylcholine (ACh). However, this traditional perspective has been challenged and enlarged upon by numerous observations. First, α7 receptors can be activated by choline as well as ACh (3), removing it from the position of being strictly optimized for synaptic function and suggesting that it may respond to tissue factors, especially in the context of playing a role in the modulation of inflammation (4). Additional insights have come from the exploration of the rich pharmacology of this receptor, beginning with the identification of a variety of selective agonists and partial agonists that control the conformational dynamics of activation and desensitization in ways that are totally unlike the behavior of other nAChRs, such as those of the neuromuscular junction and autonomic ganglia that are clearly optimized for fast synaptic transmission (1, 5). To a large degree, the specializations of synaptic nAChR arise from the evolution of different types of subunits that create the ACh-binding site at the interface between α and non-α-type subunits. In contrast to synaptic-type nAChR, α7 receptors form as functional homopentameric receptors with five putative agonist-binding sites at the subunit interfaces (6). Activation of the α7 ion channel is most likely to occur with submaximal occupancy of the “orthosteric” agonist-binding sites (7), and even under optimal conditions, the channels activated by orthosteric agonists open with relatively low probability and for very brief periods of time compared with other nAChRs (8).
The richness of α7 nAChR pharmacology also comes from the identification of highly selective positive allosteric modulators (PAMs), which, to varying degrees, allow orthosteric agonists to overcome the factors that limit the channel's open probability (9). PNU-120596, one of the most efficacious of such agents, destabilizes one of the non-conducting states promoted by high levels of agonist occupancy and induces a novel conducting state that is long-lived and associated with protracted bursts of openings (10). Data suggest that the binding site for such PAMs is within the transmembrane domains of the receptor (11, 12).
As noted above, PAMs like PNU-120596 work synergistically with ligands at the orthosteric agonist-binding site. A new class of α7 receptor activators has also recently been discovered, which, in addition to being able to potentiate orthosteric activation, can directly activate the receptor's ion channel through an allosteric mechanism. The prototypical compound with such ago-PAM activity is GAT107, the active isomer of 4BP-TQS (13, 14), which is an analog of the PAM TQS. The PAM activity of TQS, as well as the direct allosteric activation (DAA) produced by GAT107, appears to rely on binding to the same transmembrane site as other PAMs of the PNU-120596 type. However, several lines of evidence suggest that the DAA produced by GAT107 also relies on the binding of the drug to a separate site in addition to the transmembrane PAM site (Fig. 1A, note that this figure is based on a previous model (13) but locates the DAA site in a position supported by our current data as discussed below). In this study we provide several additional lines of evidence supporting the hypothesis that DAA relies on the binding of GAT107 and active analogs to a unique site. A structural model suggests that this site is in the extracellular domain of the receptor extending into the vestibule of the ion channel. The properties of site-directed mutants support this model.
FIGURE 1.

Hypothetical model and proposed nomenclature for the multiple forms of GAT107 activity. A, schematic of an nAChR with proposed binding sites and examples of agents active at these sites. Previous work (13) has suggested that the activity of GAT107 arises from interactions at two separate sites on the receptor, a PAM site (P), common to other type II PAMs such as PNU-120596, and a unique site for direct allosteric activation (DAA) (D). Activity arising from these sites is further modulated by whether ACh or other agonists are bound to the orthosteric site (A). B, illustrated are three forms of GAT107 activity with proposed binding sites indicated above, observed with cells expressing human α7 and evaluated with two-electrode voltage clamp. Control applications of 100 μm ACh were followed by the application of the ago-PAM GAT107 and then another application of 100 μm ACh, with 3-min washes of Ringer's solution between applications. Inward currents activated during the application of 10 μm GAT107 indicate DAA, hypothetically arising from GAT107 effects at both the D and the P sites. As shown, after GAT107 was washed from the bath, there was a persistent “primed potentiation” of a subsequent response to ACh applied alone. This activity is hypothetically due to ACh binding at the A site and residual activity of GAT107 at the P site. The traces shown are the average response (black line) ± the S.E. (shaded band) calculated for each of the 10,500 points in the 210-s traces (acquired at 50 Hz). Prior to the averaging, the data from each cell were normalized by dividing the each value by the average peak amplitude of two prior ACh control responses from the same cell (the second ACh control response is shown in the figure). The scale bar indicates the normalized amplitude relative to the controls and in parentheses the conversion of this factor based on the averaged amplitude of the ACh controls. C, direct potentiation of 100 μm ACh-evoked responses co-applied with 10 μm GAT107. This form of activity is likely to be due to combined effects of binding at all three sites illustrated in the model. Average responses of five cells (± S.E. in shaded band) were normalized to their initial responses to ACh applied alone; the scale bar indicates 50 times the average peak current amplitude to ACh alone. D, original data for the five cells that were normalized and averaged for the traces shown in B. The control 100 μm ACh-evoked responses are shown in the left panel at an amplified scale. All traces are 210 s long. E, binding of GAT107 to both the D site and the P site appears to produce bursts similar to those recorded in the presence of ACh and PNU-120596 (10). The example shown was obtained from a cell-attached patch clamp recording from a cell stably expressing α7 and RIC-3 (8) with 10 μm GAT107 in the patch pipette. The period of frequent channel opening and closing shown was preceded and followed by several seconds without any channel activity, consistent with the burst arising from a single channel. The burst shown was 1.3 s in duration and contained 39 openings with average duration of 33 ms and average closed times between openings of 10 ms. An expansion of the single-channel currents from the middle of the burst are shown in the lower trace with the current levels associated with the closed (Ds) and open state (O′) indicated.
Materials and Methods
Commercial Reagents
Acetylcholine chloride (ACh), atropine, and other chemicals were purchased from Sigma. The methanethiosulfonate compounds 2-(aminocarbonyl)ethyl methanethiosulfonate, 2-(quinuclidinium)ethyl methanethiosulfonate (MTSEA), and carboxymethyl methanethiosulfonate were purchased from Toronto Research Chemicals Inc. (North York, Ontario, Canada). Fresh ACh stock solutions were made in Ringer's solution each day of experimentation. Stock solutions of the test drugs were made in Ringer's solution and kept at 4 °C and used within 2 days. Working solutions were prepared freshly at the desired concentration from the stored stock.
Compound Synthesis
The allosteric antagonists were synthesized using a three-component Povarov reaction by reacting the corresponding aldehyde, aniline, cyclopentadiene in acetonitrile in the presence of the catalyst indium chloride (15, 16). The substituted anilines and aldehydes were purchased commercially. The synthesis of 4-(2,3,5,6-tetramethylphenyl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinoline-8-sulfonamide (2,3,5,6MP-TQS) and 4-mesityl-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinoline-8-sulfonamide (2,4,6MP-TQS) has been previously reported (17), and their characterization has been confirmed, although we did see a slight improvement in the yield of 2,4,6MP-TQS with the in-house developed microwave-assisted synthesis procedure (15). All compounds have chemical purity of ≥98% as measured by HPLC.
4-(5-Bromothiophen-2-yl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinoline-8-sulfonamide (GAT154)
In a microwave vial, cyclopentadiene (3 eq) was added to a suspension of 5-bromothiophene-2-carboxaldehyde (1 eq), 4-aminosulfonamide (1 eq), and indium chloride (0.2 eq) in acetonitrile. The reaction vial was placed in a microwave synthesizer and irradiated to 100 °C for 15 min. The contents were added to aqueous (0.1 m) Na2CO3 solution (20 ml) and extracted with chloroform (3× at 30 ml). The combined organic layer was washed with water (20 ml) and brine (30 ml), dried over Na2SO4, and concentrated under reduced pressure. The residue was purified using flash chromatography with (EtOAc/hexanes = 40:60) or recrystallization using dichloromethane or acetone and hexanes (20:80). The reaction crude contained a mixture of diastereomers with <10% minor diastereomer. GAT154 was isolated as a pure cis diastereomer. 1H NMR (500 MHz, DMSO) δ 7.43 (d, J = 2.0 Hz, 1H); 7.34 (dd, J = 8.0 Hz, 2.0 Hz, 1H); 7.14 (d, J = 3.5 Hz, 1H); 7.00 (d, J = 4.0 Hz, 1H); 6.97 (s, 2H); 6.78 (d, J = 8.5 Hz, 1H); 6.49 (s, 1H); 5.92 to 5.87 (m, 1H); 5.69 - 5.64 (m, 1H); 4.88 (d, J = 3.0 Hz, 1H); 4.05 (d, J = 8.5 Hz, 1H); 2.93 (dq, J = 9.0 Hz, 2.0 Hz, 1H); 2.44 (qdd, J = 16.0 Hz, 9.0 Hz, 2.0 Hz, 1H); 2.00 (dd, J = 16.5 Hz, 9.0 Hz, 1H). MS m/z (M+ + 1) for C16H15BrN2O2S2 was 411.98.
4-(4-Bromothiophen-2-yl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinoline-8-sulfonamide (GAT155)
The titlecompound was synthesized according to the procedure used for GAT154 using cyclopentadiene (3 eq), 4-bromothiophene-2-carboxaldehyde (1 eq), 4-aminosulfonamide (1 eq), and indium chloride (0.2 eq) and isolated as a pure cis diastereomer. The reaction crude contained a mixture of diastereomers with <10% trans diastereomer. 1H NMR (500 MHz, CDCl3) δ 7.59 (d, J = 2.0 Hz, 1H); 7.53 (dd, J = 8.5 Hz, 2.0 Hz, 1H); 6.96 (d, J = 3.5 Hz, 1H); 6.84 (d, J = 4.5 Hz, 1H); 6.65 (d, J = 8.0 Hz, 1H); 5.91 to 5.85 (m, 1H); 5.76 to 5.70 (m, 1H); 4.90 (d, J = 3.0 Hz, 1H); 4.66 (s, 1H); 4.24 (s, 1H); 4.09 (d, J = 8.5 Hz, 1H); 3.02 (dq, J = 9.0 Hz, 3.5 Hz, 1H); 2.63 (ddd, J = 16.0 Hz, 9.5 Hz, 2.0 Hz, 1H); 2.16 (dd, J = 16.0 Hz, 8.0 Hz, 1H). MS m/z (M+ + 1) for C16H15BrN2O2S2 was 411.97.
4-Phenyl-N-propyl-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinoline-8-sulfonamide (GAT1324)
The title compound was synthesized according to the procedure used for GAT154 using cyclopentadiene (3 eq), benzaldehyde (1 eq), 4-amino-N-propylbenzenesulfonamide (1 eq), and indium chloride (0.2 eq). The reaction crude contained a mixture of diastereomers with <5% minor diastereomer. GAT1324 was isolated as a pure cis diastereomer in 60% yield. 1H NMR (500 MHz, DMSO) δ 7.45 (d, J = 7.5 Hz, 2H); 7.43 to 7.35 (m, 3H); 7.31 (d, J = 7.0 Hz, 1H); 7.28 (dd, J = 8.5 Hz, 2.5 Hz, 1H); 7.13 (t, J = 6.0 Hz, 1H); 6.82 (d, J = 8.5 Hz, 1H); 6.45 (s, 1H); 5.92 to 5.84 (m, 1H); 5.62 (d, J = 5.0 Hz, 1H); 4.65 (d, J = 3.0 Hz, 1H); 4.08 (d, J = 8.5 Hz, 1H); 3.00 to 2.90 (m, 1H); 2.63 (q, J = 7.5 Hz, 2H); 2.45 to 2.34 (m, 1H); 1.68 to 1.58 (m, 1H); 1.36 (sext, J = 7.5 Hz, 2H); 0.80 (t, J = 7.5 Hz, 3H); Rf: 0.40 (EtOAc/hexanes = 40:60). MS m/z (M+ + 1) for C21H24N2O2S was 386.16.
4-(4-Bromophenyl)-N-propyl-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinoline-8-sulfonamide (GAT1319)
The titlecompound was synthesized according to the procedure used for GAT154 using cyclopentadiene (3 eq), 4-bromobenzaldehyde (1 eq), 4-aminosulfonamide (1 eq), and indium chloride (0.2 eq). The reaction crude contained a mixture of diastereomers with <5% minor diastereomer. GAT1319 was isolated as a pure cis diastereomer. 1H NMR (500 MHz, DMSO) δ 7.59 (d, J = 8.5 Hz, 2H); 7.41 (d, J = 8.5 Hz, 2H); 7.38 (d, J = 2.0 Hz, 1H); 7.29 (dd, J = 8.5 Hz, 2.0 Hz, 1H); 7.14 (t, J = 5.5 Hz, 1H); 6.82 (d, J = 9.0 Hz, 1H); 6.45 (s, 1H); 5.93 to 5.84 (m, 1H); 5.61 (d, J = 5.0 Hz, 1H); 4.62 (d, J = 3.5 Hz, 1H); 4.08 (d, J = 9.0 Hz, 1H); 2.99 to 2.89 (m, 1H); 2.63 (q, J = 7.0 Hz, 2H); 2.39 to 2.29 (m, 1H; 1.69 to 1.59 (m, 1H); 1.37 (sext, J = 7.5 Hz, 2H); 0.80 (t, J = 7.5 Hz, 3H); Rf: 0.50 (EtOAc/hexanes = 40:60).; MS m/z (M+ + 1) for C21H23BrN2O2S, 448.06.
4-(4-(Bromomethyl)phenyl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinoline-8-sulfonamide (GAT904)
The title compound was synthesized according to the procedure used for GAT154 using cyclopentadiene (3 eq), 4-(bromomethyl)benzaldehyde (1 eq), 4-amino-N-propylbenzenesulfonamide (1 eq), and indium trichloride (0.2 eq), and isolated as a pure cis diastereomer exclusively. 1H NMR (500 MHz, DMSO) δ 7.46 (s, 4H); 7.43 (d, J = 2.0 Hz, 1H); 7.33 (dd, J = 8.5 Hz, 2.0 Hz, 1H); 6.96 (s, 2H); 6.79 (d, J = 8.0 Hz, 1H); 6.39 (s, 1H); 5.93 to 5.85 (m, 1H); 5.62 (d, J = 5.5 Hz, 1H); 4.78 (s, 2H); 4.64 (d, J = 8.5 Hz, 1H); 3.00 to 2.89 (m, 1H); 2.44 to 2.33 (m, 1H); 1.69 to 1.58 (m, 1H); Rf: 0.45 (EtOAc/hexanes = 40:60). MS m/z (M+ + 1) for C19H19BrN2O2S, 420.03.
4-(4-(3-Bromopropyl)phenyl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinoline-8-sulfonamide (GAT193)
The title compound was synthesized according to the procedure used for GAT154 using cyclopentadiene (3 eq), 4-(3-bromopropyl)benzaldehyde (1 eq), 4-aminobenzenesulfonamide (1 eq), and indium chloride (0.2 eq) The reaction crude contained a mixture of diastereomers with <5% minor diastereomer. GAT193 was isolated as a pure cis diastereomer. 1H NMR (500 MHz, DMSO) δ 7.43 (s, 1H); 7.37 (d, J = 8.5 Hz, 2H); 7.32 (dd, J = 9.0 Hz, 2.5 Hz, 1H); 7.24 (d, J = 8.5 Hz, 2H); 6.95 (s, 2H); 6.79 (d, J = 8.0 Hz, 1H); 6.34 (s, 1H); 5.91 to 5.85 (m, 1H); 5.62 (d, J = 5.0 Hz, 1H); 4.60 (d, J = 3.0 Hz, 1H); 4.06 (br d, J = 8.5 Hz, 1H); 2.92 (br q, J = 8.5 Hz, 1H); 2.72 (t, J = 7.0 Hz, 2H); 2.43 to 2.32 (m, 1H); 2.10 (quint, J = 7.0 Hz, 2H); 1.65 (dd, J = 16.0 Hz, 9.5 Hz, 1H); Rf: 0.60 (EtOAc/hexanes = 40:60). MS m/z (M+ +1) for C21H23BrN2O2S, 448.06.
Molecular Modeling
A homology model for the α7 nAChR was constructed using the humanized AChBP structure Protein Data Bank code 3SQ9 (18) as the starting structure. The initial monomeric model was created using SwissModel (19). The resulting structure was assembled into a pentamer using the matchmaker routine within Chimera (20). The structure was then relaxed by minimization with Amber 14 in a truncated octahedron of TIP3P water using the default PME method with a 10 Å cutoff. This preminimization utilized 5000 cycles of steepest decent followed by another 5000 cycles of conjugate gradient minimization. Each subunit then had ACh docked in and was reminimized as above but with the ACh molecules held fixed. This was repeated with the protein fixed and ACh, TIP3P water, and sodium counterions left unrestrained. Finally, the entire system was minimized with no restraints for 25,000 cycles of steepest decent followed by conjugate gradient minimization. The resulting structure was then subjected to a short 40-ps MD simulation for equilibration to 300 K at constant volume with weak restraint on the protein and then a 200-ps simulation at constant pressure until the density equilibrated to 1.03 g/ml. A subsequent 50-ns run at constant energy (NVE) was performed, over which time the system remained stable. A frame from this trajectory at 18.8 ns was minimized as above, and the bound ACh was extracted from the structure for docking using Glide (Schrodinger, Portland, OR) for docking at standard precision. Grids were calculated for a dimeric subunit, and during docking, ligand flexibility was allowed. Different poses were compared using the Glide Emodel score, which is more appropriate for ranking conformers than the Glide score. Molecular overlays for were prepared with the MatchMaker function within Chimera (20) in which the AChBP structure 2BYR (21) with bound methyllycaconitine (MLA) was superimposed on the α7 homology model to represent the approximate position of where MLA would bind in the α7 receptor.
Heterologous Expression of nAChRs in Xenopus laevis Oocytes
The human α7 nAChR clone was obtained from Dr. J. Lindstrom (University of Pennsylvania, Philadelphia, PA). The human resistance-to-cholinesterase 3 (RIC-3) clone, obtained from Dr. M. Treinin (Hebrew University, Jerusalem, Israel), was co-injected with α7 to improve the level and speed of α7 receptor expression without affecting the pharmacological properties of the receptors (22). Subsequent to linearization and purification of the plasmid cDNAs, cRNAs were prepared using the mMessage mMachine in vitro RNA transfection kit (Ambion, Austin, TX). Mutants were made as described previously (23). The cysteine mutants described (W149C, Y93C, and D101C) were made in a C116S pseudo wild-type Cys-null background to prevent the possible formation of spurious disulfides.
Oocytes were surgically removed from mature X. laevis frogs (Nasco, Ft. Atkinson, WI) and injected with appropriate nAChR subunit cRNAs as described previously (24). Frogs were maintained in the Animal Care Service facility of the University of Florida, and all procedures were approved by the University of Florida Institutional Animal Care and Use Committee. In brief, the frog was first anesthetized for 15–20 min in 1.5 liter of frog tank water containing 1 g of 3-aminobenzoate methanesulfonate buffered with sodium bicarbonate. The harvested oocytes were treated with 1.25 mg/ml collagenase (Worthington) for 2 h at room temperature in calcium-free Barth's solution (88 mm NaCl, 1 mm KCl, 2.38 mm NaHCO3, 0.82 mm MgSO4, 15 mm HEPES, and 12 mg/liter tetracycline, pH 7.6) to remove the follicular layer. Stage V oocytes were subsequently isolated and injected with 50 nl of 5–20 ng of nAChR subunit cRNA. Recordings were carried out 1–7 days after injection.
Two-electrode Voltage Clamp Electrophysiology
Experiments were conducted using OpusXpress 6000A (Molecular Devices, Union City, CA) (24). Both the voltage and current electrodes were filled with 3 m KCl. Oocytes were voltage-clamped at −60 mV. The oocytes were bath-perfused with Ringer's solution (115 mm NaCl, 2.5 mm KCl, 1.8 mm CaCl2, 10 mm HEPES, and 1 μm atropine, pH 7.2) at 2 ml/min. To evaluate the effects of experimental compounds compared with ACh-evoked responses of various nAChR subtypes expressed in oocytes, control responses were defined as the average of two initial applications of ACh made before test applications. The solutions were applied from a 96-well plate via disposable tips, and the test compounds were applied alone, co-applied with ACh, or co-applied with GAT107. Unless otherwise indicated, drug applications were 12 s in duration followed by a 181-s washout period. A typical recording for each oocyte constituted two initial control applications of ACh, one or more experimental compound applications, and then a follow-up control application of ACh to determine primed potentiation of the ACh-evoked responses. The control ACh concentration was 60 μm for wild-type α7 unless otherwise indicated. The responses were calculated as both peak current amplitudes and net charge, as described previously (25). The average responses of the two initial ACh controls (60 μm unless otherwise indicated) from each cell were used for normalization. Statistical analyses were conducted based on unpaired t test comparisons of the normalized net-charge responses of groups of replicate oocytes.
Data were collected at 50 Hz, filtered at 20 Hz, and analyzed by Clampfit 9.2 or 10.0 (Molecular Devices) and Excel (Microsoft, Redmond, WA). Data were expressed as means ± S.E. from at least four oocytes for each experiment and plotted by KaleidaGraph 3.0.2 (Abe Beck Software, Reading, PA). Type II PAMs produce extremely large increases (>100,000-fold) in the single-channel currents of a small fraction of the receptors (≤1%), so they are intrinsically variable in amplitude and duration (10), making it difficult to identify truly “representative” responses. Therefore, we displayed multicell averages for comparisons of these complex responses. The averages of normalized data were calculated using an Excel (Microsoft) template for each of the 10,500 points in each of the 210-s traces (acquired at 50 Hz). Following subtraction of the basal holding current, data from each cell, including the ACh controls, were normalized by dividing each point by the peak of the ACh control from the same cell. The normalized data were then averaged, and standard errors of the mean (S.E.) for the multicell averages were calculated on a point-by-point basis. The dark lines in the figures represent the average normalized currents, and the shaded areas the range of the S.E. Scale bars in the figures of averaged traces reflect the scaling factor relative to the average peak current amplitude of the ACh controls (con.) used for the normalization procedures. Also provided are those values converted to current amplitudes, based on the scaling factors and average ACh controls.
Cell-attached Patch Clamp Electrophysiology
Cell-attached records were obtained from A7R3HC10 cells and HEK293 cells stably expressing human α7 and human RIC-3, as described previously (8). Cells were cultured in Dulbecco's modification of Eagle's medium (with 4.5 g/liter glucose, l-glutamine, and sodium pyruvate; Mediatech) supplemented with 10% heat-inactivated fetal bovine serum, 0.45 mg/ml geneticin-selective antibiotic (G418 sulfate; Gibco), and 0.015 mg/ml hygromycin B (Invitrogen), at 37 °C with 5% CO2. During passaging, cells were dissociated with 1 mm EDTA in calcium- and magnesium-free Hanks' balanced saline solution (Gibco) to avoid damaging the α7 nAChRs expressed on the cell surface.
Prior to recording, A7R3HC10 cells were plated onto 12-mm glass coverslips (Thermo Fisher Scientific) that had been previously coated with 0.1 mg/ml poly-d-lysine (Sigma) at 37 °C for ∼5 min. Patch pipettes were created from borosilicate glass (outer diameter 1.5 mm, inner diameter 0.86 mm; Sutter Instruments), pulled using a Flaming/Brown micropipette puller (Model P-97; Sutter Instruments), fire-polished to 4–7 megohms, filled with solution, and briefly dipped in Sigmacote (Sigma) just before recording. Cells in the recording chamber were bathed in an external solution containing (in mm) 165 NaCl, 5 KCl, 2 CaCl2, 10 glucose, 5 HEPES, and 0.001 atropine, and pH-adjusted to ∼7.3 using NaOH. Pipettes contained GAT107 dissolved in external solution.
Recordings were performed at room temperature, using a Multiclamp 700B amplifier and digitized with a Digidata 1440A and Clampex 10.2 software (Molecular Devices). Data were low-pass filtered to 10 kHz using the amplifier Bessel filter and sampled at 100 kHz.
Results
As described previously (13), the ago-PAM GAT107 has three forms of activity illustrated in Fig. 1, B and C. When applied alone, it evokes large currents associated with DAA. Although these responses decay more slowly than ACh-evoked responses, they persist only as long as the drug is free in solution, suggesting that the DAA site is solvent-accessible and that this activity is in part diffusion-limited (26). The second form of activity is a long lasting “primed” positive allosteric modulation (primed potentiation), which is apparent when ACh (or other orthosteric agonist) is applied alone following an application of GAT107 (Fig. 1B; see Fig. 1D for the original data from single cells). This substantial enhancement of ACh-evoked responses is relatively stable, such that it will persist for multiple sequential agonist applications with relatively little rundown (13). Presumably, this long-lasting effect is a function of a slow off-rate for the bound PAM. The third type of activity, direct potentiation, and the largest enhancement of ACh-evoked responses is produced when GAT107 and ACh are co-applied (Fig. 1C). However, this form of activation appears to shift the conformational equilibrium of the receptors toward PAM-insensitive desensitized (Di) states (10) so that even primed potentiation of subsequent ACh responses is minimal (13).
Hypothetical energy landscapes for the various conditions of ligand binding to α7 have previously been described (13). The binding of orthosteric agonists alone promote only rare and brief openings, although orthosteric agonists with the PAMs also bound produce occasional protracted bursts of openings occurring from a PAM-sensitive desensitized state (Ds) (10). Shown in Fig. 1E is a single-channel recording of α7 receptor currents in the presence of 10 μm GAT107. Bursting activity was observed that was similar to that reported for ACh and PNU-120596 (10).
Several lines of evidence support the hypothesis that the three forms of GAT107 activity illustrated in Fig. 1, B and C, arise from its effects at two distinct binding sites as follows: a specific site for direct allosteric activation and a PAM site that is shared by other PAMs, which do not produce DAA (11). As noted above, one line of evidence is the difference in the kinetics of the DAA and the potentiating effects of GAT107. We have also previously reported that DAA is sensitive to MLA, a selective competitive antagonist of α7 nAChR, whereas primed potentiation is not (13). The hypothesis that the PAM site for GAT107 is related to that for other efficacious PAMs, such as PNU-120596, is supported by the observation that transmembrane domain mutant M254L that reduces PNU-120596 effects (11) also reduces the PAM activity of 4BP-TQS, the racemic form of GAT107 (14). We hypothesize that although GAT107 has activity at two separate sites, to detect the activity at the DAA site, GAT107 must also work at the PAM site or the PAM site needs to be occupied by another PAM, as described next.
Low concentrations of GAT107 produce relatively little DAA. To test the hypothesis that DAA is coupled to, and dependent on, concomitant PAM activity, we pre-applied the PAM, PNU-120596, prior to applying 3 μm GAT107 (Fig. 2, A–C). As shown in Fig. 2D, pre-applications of 10 or 30 μm PNU-120596 significantly (p < 0.001) increased DAA by GAT107 with no effect on the primed potentiation of subsequent ACh-evoked responses.
FIGURE 2.

Amplification of DAA by a low concentration of GAT107 by pre-application of the type II PAM PNU-120596. A, averaged net-charge responses of α7-expressing cells (n = 7) to the application of 3 μm GAT107 alone and the primed potentiation of subsequent ACh responses, with (dark blue with blue shading (lower traces) or without (black with tan shading representing the S.E., upper traces) 30-s pre-applications of 30 μm PNU-120596. Each trace is 210 s long, and the vertical scales are relative to the peak currents of the initial 60 μm ACh controls. B, overlay of the 3 μm GAT107 responses from A at an expanded time scale. C, overlay of the ACh-evoked responses that followed the 3 μm GAT107 responses from A, shown at an expanded time scale. D, averaged normalized net-charge responses to 3 μm GAT107 with pre-applications of control Ringer's solution or different concentrations of PNU-120596. The 10 or 30 μm PNU-120596 pre-applications significantly increased (p < 0.001) the responses to 3 μm GAT107 (DAA) compared with control. There were no significant effects on the subsequent GAT107-primed ACh responses. Data were obtained from seven cells for the 0, 3, and 30 μm PNU-120596 concentrations and eight cells with the 10 μm PNU-120596 concentration. **, p < 0.01.
Several lines of evidence also support the hypothesis that the DAA site is distinct from the orthosteric agonist site (13, 14). To further test this hypothesis, we utilized the “gatekeeper” cysteine mutant L119C (23). This mutation (made in the cysteine-null pseudo wild-type C116S) places a reactive cysteine in the complementary surface of the ACh-binding site where reaction with a suitable sulfhydryl reagent such as MTSEA can block ACh-evoked activation. As shown in Fig. 3, although MTSEA with wild type has no significant effects (despite the free cysteine at residue 116), reaction of MTSEA with C116S,L119C profoundly reduces primed potentiation that requires ACh binding (p < 0.00001) but has no effect on DAA by GAT107.
FIGURE 3.

Differential effects of sulfhydryl modification of the gatekeeper residue L119C (23) on the primed potentiation of ACh-evoked response and the DAA produced by GAT107. The average traces were prepared as described (see under “Materials and Methods”) from either wild-type α7 (A) or the double mutant α7C116S,L119C (B). The C116S mutation provided a Cys-null pseudo wild-type for the characterization of sulfhydryl modification at specific single residues. Cells were pretreated with either 5-min static bath applications of control Ringer's solution or 2 mm MTSEA made up in Ringer's solution. The MTSEA treatments had no significant effects on the wild-type cells and no effects on the GAT107 responses of C116S,L119C cells, but it significantly reduced the normalized net-charge responses of the C116S,L119C GAT107-primed ACh responses (p < 0.00001).
Selective Antagonists of DAA
Two compounds (2,3,5,6MP-TQS and 2,4,6MP-TQS, Fig. 4A) have recently been reported that are structurally related to TQS, the PAM on which 4BP-TQS and GAT107 were based. These compounds lack PAM or allosteric agonist activity and were reported to selectively antagonize DAA by 4BP-TQS (17). These allosteric antagonists, also referred to as “silent allosteric modulators” (17), were reported to be ineffective at antagonizing PAM activity or activation by ACh. This suggests that they are suitable probes for the DAA site, although the structural basis for their antagonist activity is not entirely clear. They were designed as part of a series of compounds to test the significance of methyl group number and position on the TQS phenyl ring, but the methylation patterns had the effect of influencing the product stereochemistry during synthesis so that these compounds were obtained as trans diastereomers.
FIGURE 4.
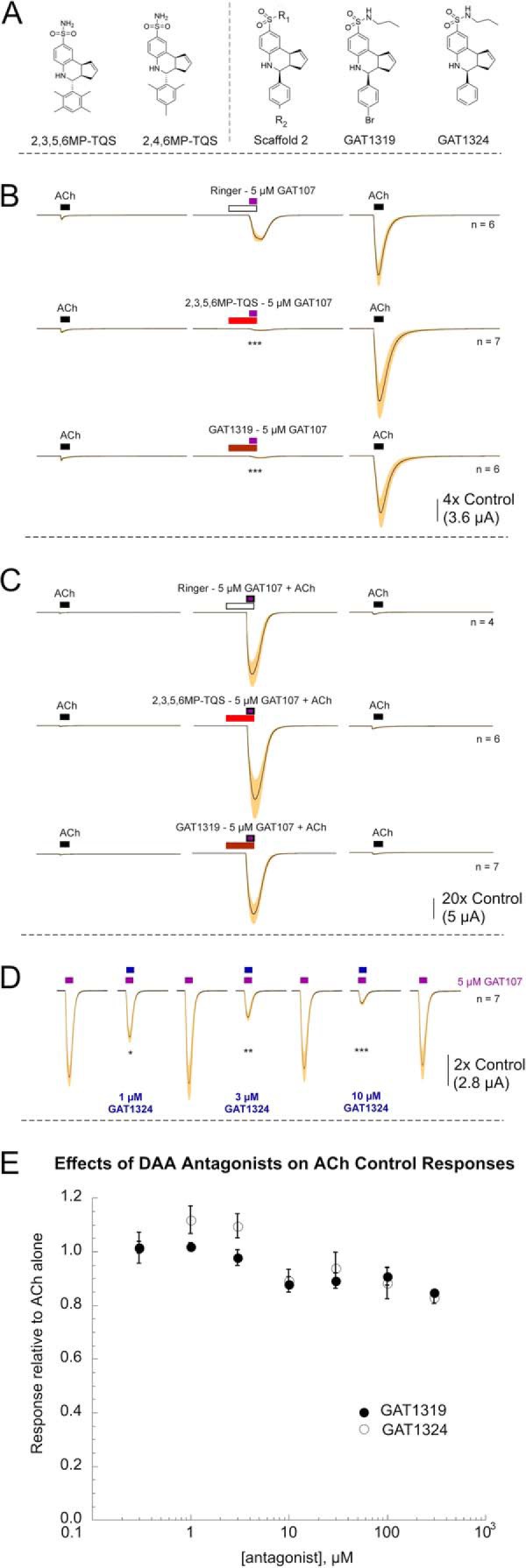
Specific antagonists of DAA. A, structure of two previously reported selective antagonists of DAA (17) and an alternative scaffold for a novel class of antagonists based on modification of the sulfonamide group (R1) with two representative compounds of this class. GAT1319 differs from GAT107 only in the R1 modification, whereas GAT1324 also has a modification at R2. B, averaged data showing the effects of 50 μm 2,3,5,6MP-TQS or GAT1319 pre-applied for 60 s and then co-applied with 5 μm GAT107 on the DAA by GAT107 without effects on subsequent primed potentiation. C, lack of effect of the allosteric antagonists on the directly potentiated responses to ACh co-applied with GAT107. D, concentration-dependent effects of GAT1324 co-application with 5 μm GAT107 on the inhibition of DAA. Statistical significance of normalized net charge responses compared with untreated controls as indicated: *, p < 0.05; **, p < 0.01; ***, p < 0.001. E, average 60 μm ACh control net charge responses measured when ACh was co-applied with either GAT1319 or GAT1324 (± S.E., n = 8 for the GAT1324 data and n = 7 for the GAT1319 results). Data are calculated relative to the average of two initial applications of 60 μm ACh alone.
We have identified several new additional DAA antagonists based on the TQS scaffold (Fig. 4A). These rely on modifications to the sulfonamide (R1), with or without the bromo group (X = H, Br). Two examples from this group are shown, GAT1319 and GAT1324. Like the methyl-TQS compounds, these antagonists inhibit DAA (Fig. 4B) with no significant effects on either primed (Fig. 4B) or direct potentiation (Fig. 4C). The antagonism of DAA was concentration-dependent and reversible (Fig. 4D). At concentrations less than 1 mm, these compounds had little or no effect on activation by 60 μm ACh applied alone (Fig. 4E). The results further underscore the idea that the DAA site is distinct from both the orthosteric agonist and PAM sites.
GAT107 Brings Receptors Not Activatable by Orthosteric Agonists to Life
Further evidence that DAA can be decoupled from orthosteric activation has come from the identification α7 mutants such as Y93C, which are insensitive to activation by ACh but are fully activatable by GAT107 (13); we refer to such mutants as non-orthosterically activatable receptors (NOARs). These mutant receptors are inactive to ACh or other orthosteric agonists but can be rendered active when stimulated with GAT107. Mutations of the tryptophan critical for orthosteric activation (27) (Trp-149 in the human sequence and Trp-148 in the rat sequence) also result in NOARs that are activatable by GAT107. This was previously reported for rat α7W148F (14), and similar effects are seen with human α7W149C and α7W149Y (α7W149C data shown in Fig. 5A). We have identified another family of NOARs associated with the modification of the C-loop vicinal cysteines (Cys-190 and Cys-191) (28) that are essential for orthosteric activation. ACh responses are profoundly disrupted if the disulfide bond between these residues is chemically reduced (29) or prevented by mutation of either one of the cysteine pairs. We determined that C190S, C190P, C191S, and C191P mutants were NOARs that were readily activated by GAT107 (data for C190S shown in Fig. 5A). Although it could be argued that the NOAR mutations render the receptors better able to facilitate conversion of the PAM-site-bound receptor to a conductive state, the alternative and more tenable hypothesis is that the GAT107 activation of these NOARs is accomplished by binding to the same DAA site as in the wild-type α7. This hypothesis is supported by the observation that the GAT107 activation of the NOARs is sensitive to the same two classes of allosteric antagonists (Fig. 5B) that are effective on wild-type α7 GAT107 responses. Moreover, whereas the NOARs are activated by GAT107, they are not activated by PNU-120596.
FIGURE 5.

Mutants of α7 that are insensitive to ACh-evoked orthosteric activation but directly activatable by GAT107 (NOAR). A, tryptophan 149 is a key residue for α7 activation by ACh (27), and likewise orthosteric activation by ACh relies on the patency of the disulfide between the vicinal cysteines 190 and 191. Shown are examples of representative mutations at these sites that destroy orthosteric activation by ACh but leave the receptors competent for DAA by GAT107. Lacking internal ACh controls, the averaged responses shown are directly scaled in μA. Typical non-potentiated ACh controls of wild-type type α7 range from 100 nA to 2–3 μA, depending on interval after injection (average ≈440 nA 2–4 days post-injection). Note that GAT107 produced negligible primed potentiation of subsequent ACh responses. B, DAA of ACh-insensitive mutants is sensitive to the selective antagonists shown in Fig. 4. Statistical significance on normalized net charge responses compared with untreated controls as indicated: **, p < 0.01; ***, p < 0.001.
Probative Compounds for the DAA Site
We have identified several small differences in the structure of GAT107 analogs that profoundly alter the profile of the resulting compounds in such a way as to be potentially useful for characterization of the DAA site of the α7 nAChR. Two compound pairs of particular interest are shown in Fig. 6, A and B. In the upper panel of Fig. 6A are two compounds with a thiophene ring substituted for the phenyl of GAT107. The disposition of the bromine atom relative to the sulfur in the thiophene is not only critical for DAA but also regulates primed potentiating activity (Fig. 6A). The efficacy for direct potentiation was also reduced for GAT155 compared with GAT154 (Fig. 6C).
FIGURE 6.

Molecular switches in the structure of GAT107 analogs. A, differences in the disposition of the bromo group profoundly affect the activity profiles of GAT154, a very active ago-PAM, compared with GAT155, a relatively weak PAM with no activity as an allosteric agonist. B, bromo-alkyl probes control DAA. Whereas GAT904 has strong DAA and weak primed potentiation, GAT193 has little if any DAA but retains primed potentiation. The data shown in both panels are averaged raw data (± S.E.) normalized to each cell's respective ACh controls, as described (see under “Materials and Methods”). C, efficacy for direct potentiation of 60 μm ACh-evoked responses by GAT155 or GAT154. The net charge responses (± S.E., n = 5) relative to the average of two initial applications of 60 μm ACh alone. Point-wise comparison at the various concentrations indicated greater potentiation by GAT154 than by GAT155 (**, p < 0.01; ***, p < 0.001). D, GAT904 acts as a direct potentiator of ACh-evoked currents. Shown is the average (±S.E.) responses of six oocytes expressing human α7 to the co-application of 100 μm GAT904 and 60 μm ACh. Peak currents were ∼170-fold higher than those of the ACh controls.
A couple of interesting effects were observed when the bromine was displaced from the phenyl group of GAT107 with one or three methylene spacers (Fig. 7B). GAT904 is an effective allosteric activator and also acts as a direct potentiator (Fig. 6D); however, it was relatively ineffective at producing primed potentiation, either due to relatively reversible PAM-site binding or due to preferential induction of PAM-insensitive desensitization (10). The further distance between the bromine atom and phenyl ring in GAT193 is associated with the loss of activity as an allosteric agonist, but primed potentiation was observed and was larger than that seen with GAT904 (Fig. 6B).
FIGURE 7.

Transient priming of ACh evoked responses by an extended application of GAT904. After obtaining control ACh responses (the second of the two controls is shown), 10 μm GAT904 was added to the bath and allowed to incubate without washout for 30 min. As shown, the direct activation produced by GAT904 was transient even in the continued presence of the drug. The GAT904 was washed out for 3 min, and 60 μm ACh was applied at 4-min intervals. There was substantial priming of the first ACh application and much less of subsequent ACh applications.
The properties of GAT904 were further probed with an additional protocol (Fig. 7). An extended application of 10 μm was made (30 min in a static bath) to saturate the putative PAM-binding site. This was followed by three applications of ACh. During the 30-min application, the response to GAT904 decayed to a very low level of steady-state activation. Subsequently there was only a single period of large primed potentiation of ACh following the GAT904 exposure, and the other two ACh responses were not significantly different from the original control. These data suggest that the PAM activity of GAT904, unlike that of GAT107, is readily reversible.
Molecular Modeling of GAT107/α7 Binding Interactions
Docking of GAT107 and related compounds to the extracellular domain of the receptor allowed us to identify two putative binding sites for DAA activity (Fig. 8A). One is in a shallow groove that partially overlaps the C-loop, and the other is in a deep pocket in the vestibule of the receptor. The best pose in the vestibule site was ∼4.7 kcal/mol lower in energy (Emodel score) than the best pose in the alternative binding site. We favored the vestibule binding site for further analysis based on the following considerations. First, GAT compounds are uncharged and relatively nonpolar, whereas the orthosteric binding site is specialized for the binding of cationic species. Second, NOARs reveal that a non-functional orthosteric binding site (under the C-loop) is still able to respond to GAT107 with DAA activity, arguing that it is binding elsewhere. Third, the allosteric antagonist compounds discussed above inhibit DAA but do not interfere with ACh binding, again pointing to a site other than the orthosteric site. Finally, we note that Spurny et al. (30) identified a vestibular binding pocket analogous to the one we describe here in their studies of the humanized acetylcholine-binding protein, which is homologous to the α7 extracellular domain. Taken together, these results prompt us to consider the vestibular binding site in more detail.
FIGURE 8.

Modeling the binding site for DAA. A, two distinct binding sites for docking GAT107. A dimeric unit from the α7 homology model is shown. Glide docking of GAT107 into the model identified a vestibular pocket and C-loop binding site. The space-filling models shown represent the top two poses at each of the two sites. B, putative binding site for DAA by GAT107 as identified by docking. The side chains of Arg-99 and Asp-101 are shown. This view is from the “top” of the extracellular domain, looking down into the vestibule. C, vestibule binding site for DAA with a molecular surface color-coded for amino acid polarity. Blue corresponds to positive charge, red to negative charge, and white is neutral. This is a view from inside the vestibule with residues from other subunits removed for clarity. D, same view as in C but with GAT107 bound. The black arrow indicates the position of the sulfonamide group on GAT107. E, comparison of GAT193 versus GAT107 in their vestibule binding modes. GAT193 is colored in light blue; the bromine atoms in each molecule are colored light green. The view is from inside the vestibule. F, composite overlay model showing MLA bound to α7 under the C-loop (pink) interacting with Gln-57 and Trp-55 (yellow), which are on a β-strand that lines the proposed DAA site harboring a bound GAT107.
The putative vestibular binding pocket (Fig. 8B) is accessible from the vestibule of the extracellular domain, with its mouth at the interface of two subunits, and the bulk of the pocket extending into the interior of a receptor subunit in a region found within the β-sandwich. The key amino acids presenting side chains in this hydrophobic pocket are Leu-56, Met-58, Lys-87, Ile-90, Leu-92, Phe-100, Ala-102, Tyr-118, and Pro-120. Although this pocket is largely non-polar, its mouth is quite polar (Fig. 8C), with Tyr-95 and Thr-103 having close proximity to the charged residues Arg-99 and Asp-101. Interestingly, the positions of Arg-99 and Asp-101 are such that in adjacent subunits Arg-99 of one subunit is potentially able to interact with Asp-101 of the next subunit (Fig. 8B). The deep nonpolar pocket is large enough to easily accommodate a bound GAT107 molecule (Fig. 8D), with the non-polar bromophenyl group buried in its core, and the sulfonamide residue proximate to the Arg-99/Asp-101 residues. It is noteworthy that in addition to GAT107, we find GAT154, -155, and -904 all have a preference for binding into the vestibular pocket in a mode similar to that of GAT107. The exception to this is GAT193, which features a bromopropyl group instead of the bromo group found on the phenyl ring of GAT107. The best poses for GAT193 placed it at about a 90° angle to GAT107's binding mode and did not insert into the vestibule binding pocket (Fig. 8E). The Glide Escore for binding GAT193 was ∼25 kcal/mol higher in energy than for binding GAT107, suggesting that it is weakly bound in this mode. In some of our docking poses we identified a “flipped” pose for the GAT compounds, with the polar sulfonamide buried in the hydrophobic core and the bromophenyl group placed in the water-exposed mouth of the vestibule. We consider this type of pose less likely, given the mismatch in polarity and generally worse Glide Escores for the flipped arrangement, although they were typically within a few kilocalories/mol. Rather than attempt to divine meaningful differences from docking scores that are within a few kilocalories/mol of each other, ongoing work will continue to probe for the binding mode and provide verification for the docking models via mutagenesis. One observation we have made from examination of this model is that the bound GAT107 may communicate with the amino acid residues that constitute the −-strand of the orthosteric agonist-binding site (Fig. 8F). Also, the proposed interactions of the sulfonamide with the Arg-99/Asp-101 side chains appear to be a potential way to modulate communication from one subunit to another. We thus sought to obtain experimental support for the idea that interactions of the sulfonamide group with the Arg-99/Asp-101 grouping are important for function of GAT107 and that mutation of Asp-101 could negatively impact ago-PAM function.
Asp-101 as a Regulator of α7 Activation
Mutation of Asp-101 to alanine reduced the potency of both ACh (Fig. 9A) for orthosteric activation and GAT107 for DAA (Fig. 9B). However, as shown in Fig. 9, C and D, although there was negligible DAA of α7D101A by 10 μm GAT107 compared with that produced by 100 μm, the potentiating activity, both primed and direct, was equivalent at the two GAT107 concentrations when the ACh concentrations were adjusted to compensate for the increase in EC50 value (100 μm ACh for wild-type and 1 mm ACh for D101A).
FIGURE 9.

DAA, primed potentiation, and direct potentiation of wild-type α7 and the D101A mutant. A, potency for ACh activation of α7D101A reduced, as shown by the net-charge concentration-response curves, which indicate an EC50 of ≈1 mm ACh for the mutant compared with 30 μm ACh for the wild type (25). Hence, for this mutant, 1 mm ACh was used as a control for normalization. Each point is the average response of n = 7 for wild-type and n = 4 for D101A (± S.E.), normalized to the ACh maximum. B, reduced potency and efficacy of the D101A mutant compared with wild-type α7 for DAA by GAT107. Shown are the normalized net-charge values for n = 7 for wild-type and n = 6 for D101A (± S.E.) for each condition. C, PAM activity of GAT107 is relatively unaffected by the D101A mutation compared with DAA by 10 μm GAT107 (***, p < 0.001). D, efficacy of GAT107 for DAA of α7D101A is increased at higher concentrations whereasthe potentiating activity remains comparable with the lower concentration shown in C.
Covalent Modification of a Residue at Position 101
We tested whether covalent modification of a cysteine in position Asp-101 by reactive sulfhydryl reagents would be detectable as a change in the activity profile of GAT107. Three different reagents were used, MTSEA (positively charged) and compounds 2-(aminocarbonyl)ethyl methanethiosulfonate (uncharged) andcarboxymethyl methanethiosulfonate (negatively charged). Only MTSEA produced a significant reduction in the DAA (p < 0.001 determined by paired t tests between GAT response prior to and following MTSEA treatment; Fig. 10, A and B). There were no significant effects on the primed potentiated ACh responses by any of the reagents (based on unpaired t-tests of ACh responses normalized to the initial GAT107 responses from the same cells). To test whether the partial reduction of the GAT107 responses by MTSEA treatment represented an incomplete reaction with the sulfhydryl reagent, the experiment was repeated with the MTSEA increased from 2 to 10 mm. Essentially the same results were obtained (Fig. 10C).
FIGURE 10.

α7D101C mutant is an atypical NOAR. A, D101C mutation produces a loss of orthosteric ACh responses that is reversible with primed potentiationby GAT107. In this regard D101C is unlike other NOAR. Mutants such as those illustrated in Fig. 5, which are unresponsive to ACh but activatable by GAT107, typically retain that ACh insensitivity even under the influence of a PAM (note the lack of primed potentiation in Fig. 5A). B, sulfhydryl modification of α7D101C by MTSEA selectively affects DAA by GAT107 without affecting primed potentiation. Cells (n = 7) were first stimulated with 30 μm GAT107 and then incubated for 5 min in control (Ringer) or 2 mm of one of the sulfhydryl reagents shown. The net-charge responses to subsequent applications of 30 μm GAT107 or 1 mm ACh were normalized relative to the initial responses to 30 μm GAT107 prior to the sulfhydryl treatment. C, partial blockade of D101C responses to GAT107 with 5-min applications of either 2 or 10 mm MTSEA. Responses are the net charge values following the MTSEA treatments relative to pre-application responses (n = 8 ± S.E.).
Discussion
The α7 nAChR has become an extremely interesting and actively pursued drug target (31). This process began with the identification of the subtype-selective partial agonist GTS-21 (32) followed by a wide variety of other structurally diverse orthosteric agonists (5). Interestingly, although GTS-21 has relatively low ion channel efficacy (33, 34), it is still being actively investigated for its activity in models of cognitive dysfunction (35–37) and, more recently, inflammatory diseases (38–43). Of course, even a partial agonist with low channel activation efficacy may still be quite effective at inducing structural changes associated with the desensitized conformations of the receptor (44), and this has led to the hypothesis that α7 function, especially in non-neuronal cells incapable of ion-channel signaling, may be due to the ability of orthosteric ligands to induce global changes in receptor structure that can be transmitted to the receptor's protein interactome (45, 46) and accomplish metabotropic type signaling (47–49). The potential importance for signaling through non-conducting states is also supported by the identification of silent agonists (50) such as NS6740 (51, 52), which has been shown to be a very effective analgesic in several models of inflammatory or neuropathic pain (53), although its primary effect on channel activity is desensitization.
A major breakthrough in the development of α7-directed pharmacotherapies was the discovery of the subtype-selective PAMs such as PNU-120596 (9, 54), which seem to work upon a form of desensitization that is unique to α7 and connect the desensitized state to a novel conducting state with protracted bursts of channel openings (10). Although PNU-120596 in combination with an orthosteric agonist can sporadically induce an extremely large increase in the opening of single channels, it also has the effect of stabilizing alternative desensitized non-conducting conformations and has similar efficacy as the desensitizing agent NS6740 in the same pain models (55–57). The discovery of the ago-PAM 4BP-TQS (14) and the characterization of its active isomer GAT107 (13) has now brought us to a further threshold for advancing α7 drug development.
The localization of the binding site for orthosteric agonists at the extracellular interface between subunits was first predicted by site-directed mutagenesis experiments (58) and later confirmed with crystal structures of the invertebrate AChBP (59, 60). Models based on these structures were further refined through the development of an α7/AChBP chimera (18).
The binding sites for PAMs of other Cys-loop receptors, such as GABAA and heteromeric nAChR, have been found at subunit interfaces homologous to, but distinct from, the orthosteric ligand-binding sites (54). These sites are not present in homomeric α7 nAChR, because the potential for an orthosteric binding site is present at every subunit interface (6). Studies of α7 and related chimeras have instead indicated that the binding sites for efficacious PAMs like PNU-120596 are most likely located within the hydrophobic transmembrane domains (11, 12). The PAM activity associated with 4BP-TQS and GAT107 is hypothesized to arise from binding to a site related to that of PNU-120596, due to effects of mutations in the region (14). We have shown however that the DAA of GAT107 can be separated from its PAM activity, and most appear to arise from binding to a novel solvent-accessible site, probably in the extracellular domain.
Case for a Two-site Binding Model for Ago-PAMs
The observation that certain members of the TQS family of compounds as exemplified by GAT107 are ago-PAMs with multiple modes of activity toward the α7 nAChR raises two intriguing questions we seek to answer. First, we would like to identify the molecular features in these compounds that confer the unique ability to directly activate α7, unlike traditional type II PAMs, which require an orthosteric agonist to exert their effect. Second, we would like to identify the site for DAA and define structural features of the receptor that are uniquely associated with the DAA activity. This is critical to distinguish ago-PAMs from PAMs within the TQS family and will permit the development of ago-PAMs as therapeutic agents. Earlier reports of the DAA by 4BP-TQS and related compounds considered that allosteric agonism and PAM activity arose at the same transmembrane site (14, 61). We believe that the available data support the hypothesis that requires a secondary alternative binding site to produce DAA activity. One of the key observations of Millar and co-workers (14) was that mutation M253L in the transmembrane region that abolish PAM activity also abolished DAA activity (referred to as “agonist activation” in that work). This result was taken as evidence that the transmembrane region was the binding site for DAA activity, but an equally valid interpretation of this result is that DAA activity at the alternative binding site requires concomitant binding of the PAM site. So, if binding to the PAM site is precluded (e.g. via mutation in the transmembrane binding region), no DAA activity will be observed.
Separation of DAA from PAM Activity
Previously, we have reported a number of observations that indicate there is an alternative binding site for allosteric activation of GAT107 that is not in the membrane (13). One key point noted was rapid washout kinetics associated with DAA but not PAM activity. The rapid washout kinetic data for DAA activity (but not potentiation activity) supports the two-site binding model in the following way. If binding for DAA activity were in the same site as for PAM activity, washout of the compound would necessarily involve loss of both DAA and the primed potentiation activity, which was not observed in our experiments. Indeed, with the two-site model, the data accounting for washout of bound ligand from the DAA site without any loss of potentiation activities as long as the PAM site remained bound. We are unable to propose a reasonable model for the behavior of ago-PAMs that features a single binding site in the transmembrane region. Additional support for a two-site model follows from the application of allosteric antagonists (Fig. 4) that differ from GAT107 in the substitution of the phenyl ring. The tetra- and trimethyl TQS compounds shown in Fig. 4 are incompetent as PAMs, lacking in the ability to perform primed or direct potentiation, but inhibit DAA (17). Similarly, the new N-propyl sulfonamides GAT1319 and GAT1324 are not able to produce direct potentiation but are effective antagonists of DAA. Either 2,3,5,6MP-TQS or GAT1319 were able to suppress DAA by GAT107 without effect on primed potentiation. These same compounds did not antagonize the directly potentiated responses to ACh when co-applied with GAT107. This pattern of behavior is inconsistent with a single binding site model in the transmembrane region but does agree with a two-site model. Note that in these two series, the modifications that result in converting a TQS family member to an allosteric antagonist occur at either end of the TQS framework (and include a ring stereochemistry change). These observations suggest that binding at the DAA site is tolerant of either cis or trans geometry between the cyclopentenyl ring fusion and the aromatic substituent of the tetrahydroquinoline ring. We have found that mono-alkyl substitution of the sulfonamide can abolish all three modes of ago-PAM behavior but may still retain the ability to bind competitively at the DAA site. Docking of GAT1324 shows that it was readily accommodated in the vestibule binding pocket, with the best vestibule pose placing the N-propyl sulfonamide in the proximity of Arg-99 and Asp-101, with the alkyl portion of the Arg-99 side chain, although this remains speculative at this time. By virtue of its activity, it is apparent from the data that the N-alkyl substituent has not impeded the ability to bind at the DAA site. The results imply that a free amino group at the sulfonamide is required for DAA, and perhaps the alkyl substitution interferes with the conformational itinerary that is part of the DAA process.
DAA-binding Site for Ago-PAMs Is Distinct from the Orthosteric Agonist-binding Site
The L119C mutation and its subsequent labeling with thiol reagents (Fig. 3) severely impacted this site's ability to bind ACh, but it did not impact DAA by GAT107. To further characterize the DAA site, we utilized known and new TQS compounds with antagonist activity for DAA, in conjunction with mutations made in the orthosteric binding site. GAT1319 and GAT1324 have also been useful for analysis of the NOAR data shown in Fig. 5. These α7 mutants are effectively dead to activation by ACh in the orthosteric site, and they can be directly activated by GAT107, and GAT1319 or GAT1324 still serve as antagonists of DAA as they do for the wild-type receptor, providing still further support for the idea that the DAA site is distinct from the orthosteric site.
We suggest that the DAA site may be capable of communication with the orthosteric agonist-binding site. MLA, a competitive antagonist at the orthosteric site, was observed to also interfere with GAT107 DAA, which is intriguing. Inspection of the AChBP crystal structures in MLA-bound form (Protein Data Bank code 2BYR) (21) reveals that Tyr-55 and Gln-57 (Trp-55 and Gln-57 in α7) make intimate contact with the bound MLA. Furthermore, these same residues sit on a β-strand that lines the vestibular pocket that we identified as a putative binding site for the DAA mode of GAT107 (Fig. 8F). We hypothesize that the binding of MLA in the orthosteric site may be negatively coupled with binding of GAT107 in the vestibule pocket via interactions through the β-strand on which Trp-55 and Gln-57 reside. Although beyond the scope of this study, it would also be of interest to bind GAT107 to the humanized AChBP for structural studies to investigate this hypothesis. Trp-55 and perhaps Gln-57 appear to be important for mediating communication between orthosteric and DAA-binding sites. Specifically, mutations of Trp-55 decouple activity of compounds at the transmembrane PAM site, including TQS and PNU-120596, from the strict requirement of co-activation at the orthosteric site or, for GAT107, from activity at the DAA site (13). Although direct activation of the wild-type receptor by GAT107 was sensitive to MLA, activation of W55F by GAT107 was insensitive to MLA.
Homology models of the α7 receptor and docking techniques have been invaluable for inquiry into binding interactions and the mechanism of the receptor (62, 63). In this study, we built a homology model to investigate the putative vestibule binding pocket that may mediate DAA. Although point-to-point correlations with this model and docked ligands are relatively course-grained in the information we can obtain, a few observations bear mention. The internal surface of this pocket is relatively non-polar, with few charged residues as exemplified in Fig. 8, C and D, which color code the molecular surface on the basis of charge. Our preferred docked pose for GAT107 places the relatively non-polar TQS ring system and phenyl substituent deeply into this pocket. Furthermore, the polar sulfonamide appears to be well poised to interact with the Arg-99/Asp-101 salt bridge, which effectively delineates the opening to the DAA pocket as a polar region. The loss of potency for GAT107 with D101A supports the idea that loss of the Asp-101 carboxylate may have removed a hydrogen bond between it and the sulfonamide amino group. We observed that the D101C mutation was rendered less active upon treatment with MTSEA, possibly because the replacement of the Asp-101 residues with the positively charged amino group conferred from MTSEA would have introduced additional positive charge density in the region of Arg-99 with unfavorable structural consequences, or it might have been less effective in binding and recognizing the sulfonamide group due to removal of a putative hydrogen bond acceptor. Work is in progress to identify additional mutations that are suitable for probing the binding interactions at the mouth of this pocket.
Substituent Effects Control the Mode of Activity
As discussed previously by Gill-Thind et al. (17), it is possible to tune TQS compounds for activity. In this work, we show profound contrasts in DAA activity by virtue of the location of a critical bromine atom with respect to the aromatic ring. The data from Fig. 6 in which the phenyl ring of GAT107 has been replaced with a bromothiophene (top) or the 4-bromophenyl replaced with 4-bromomethylphenyl or 4-(3-(bromopropyl))-phenyl yielded some important insight into the requirements for both DAA, as well as the potentiating functions of these molecules. Furthermore, the data from experiments presented in Fig. 6 provide a clear demonstration of how molecules in this class may be tuned for a desired activity with α7. The results for the bromothiophene compounds GAT154 and GAT155 demonstrate that the placement of the bromine atom is absolutely critical for DAA and PAM activity. GAT154 is capable of both DAA and primed potentiation, whereas GAT155 produces no direct activation and is less effective as a PAM (Fig. 6A). This suggests an important point-to-point interaction with the bromo group that may minimally mediate potentiation in the PAM site, and it is absolutely required for DAA. Unfortunately, although both compounds can be docked into the vestibular binding pocket, the poses obtained do not shed functional light on how the position of the bromine influences activity. The data in Fig. 6B further extend the idea of tuning activity that is complementary to the all-or-none activity we report in Fig. 6A for GAT154 and GAT155. GAT904, with a bromomethyl functionality on the phenyl ring, is capable of effective DAA activity, but it shows very little primed potentiation. In contrast, GAT193 places the bromo group further from the aryl ring on a flexible propyl linker, and it is inactive for DAA but is still able to mediate primed potentiation. We consider that GAT904 may have enhanced kinetics for off-rate from, or suboptimal binding interactions in, the PAM site. GAT193 can clearly bind to the PAM site, but it may not be able to effectively bind at the DAA site. This is strongly supported in the docking studies, which show that GAT193 does not adopt the same family of poses that GAT107, GAT154, or GAT155 do (Fig. 8A). The results demonstrate that it is possible to tune members of the TQS family for selective PAM behavior, ago-PAM behavior, DAA, and inhibition of DAA. Further studies in this series are underway to deduce the point-to-point interactions with the substituents in the TQS rings to optimize potency and selectivity for one or more of these different aspects of ago-PAM behavior.
What has newly emerged from these studies is a clear indication that the α7 nAChR has an additional and potentially druggable site, with molecules working at this DAA site allowing fine-tuning over a broad range of pharmacological responses. We and others are actively pursuing the further development and potential applications of these compounds. With the multitude of potential therapeutic indications proposed for α7, from Alzheimer disease to arthritis, it is exciting to consider that there is a new drug profile available to augment those already available. We may consider a range of drugs from full agonists to silent agonists, as well as the type I and type II PAMs, and now add ago-PAMs such as GAT107, which are uniquely able to both activate and modulate, and in so doing find improved matches between targets and therapeutics.
Author Contributions
A. R. K. and G. U. C. performed synthetic chemistry. C. S. performed electrophysiology experiments. G. A. T. designed syntheses. R. L. P. designed and analyzed electrophysiology experiments. N. A. H. performed molecular modeling. N. A. H., G. A. T., and R. L. P. wrote the manuscript.
Acknowledgment
Some OpusXpress experiments were conducted by Shehd Abdullah Abbas Al Rubaiy.
This work was supported by National Institutes of Health Grants GM57481 (to R. L. P.) and EY024717 (to G. A. T.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- nAChR
- nicotinic acetylcholine receptor
- PAM
- positive allosteric modulator
- DAA
- direct allosteric activation
- ACh
- acetylcholine
- MTSEA
- 2-(quinuclidinium)ethyl methanethiosulfonate
- 2,3,5,6MP-TQS
- 4-(2,3,5,6-tetramethylphenyl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinoline-8-sulfonamide
- 2,4,6MP-TQS
- 4-mesityl-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinoline-8-sulfonamide
- MLA
- methyllycaconitine
- NOAR
- non-orthosterically activatable receptor
- AChBP
- acetylcholine-binding protein
- 4BP-TQS
- 4-(4-bromophenyl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinoline-8-sulfonamide
- ago-PAM
- allostericagonist-PAM
- TQS
- 4-(naphthalen-2-yl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinoline-8-sulfonamide.
References
- 1. Papke R. L. (2014) Merging old and new perspectives on nicotinic acetylcholine receptors. Biochem. Pharmacol. 89, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rosas-Ballina M., and Tracey K. J. (2009) Cholinergic control of inflammation. J. Intern. Med. 265, 663–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Papke R. L., Bencherif M., and Lippiello P. (1996) An evaluation of neuronal nicotinic acetylcholine receptor activation by quaternary nitrogen compounds indicates that choline is selective for the α7 subtype. Neurosci. Lett. 213, 201–204 [DOI] [PubMed] [Google Scholar]
- 4. de Jonge W. J., and Ulloa L. (2007) The α7 nicotinic acetylcholine receptor as a pharmacological target for inflammation. Br. J. Pharmacol. 151, 915–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Horenstein N. A., Leonik F. M., and Papke R. L. (2008) Multiple pharmacophores for the selective activation of nicotinic α7-type acetylcholine receptors. Mol. Pharmacol. 74, 1496–1511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Palma E., Bertrand S., Binzoni T., and Bertrand D. (1996) Neuronal nicotinic α7 receptor expressed in Xenopus oocytes presents five putative binding sites for methyllycaconitine. J. Physiol. 491, 151–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Williams D. K., Stokes C., Horenstein N. A., and Papke R. L. (2011) The effective opening of nicotinic acetylcholine receptors with single agonist-binding sites. J. Gen. Physiol. 137, 369–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Williams D. K., Peng C., Kimbrell M. R., and Papke R. L. (2012) The intrinsically low open probability of α7 nAChR can be overcome by positive allosteric modulation and serum factors leading to the generation of excitotoxic currents at physiological temperatures. Mol. Pharmacol. 82, 746–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hurst R. S., Hajós M., Raggenbass M., Wall T. M., Higdon N. R., Lawson J. A., Rutherford-Root K. L., Berkenpas M. B., Hoffmann W. E., Piotrowski D. W., Groppi V. E., Allaman G., Ogier R., Bertrand S., Bertrand D., and Arneric S. P. (2005) A novel positive allosteric modulator of the α7 neuronal nicotinic acetylcholine receptor: in vitro and in vivo characterization. J. Neurosci. 25, 4396–4405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Williams D. K., Wang J., and Papke R. L. (2011) Investigation of the molecular mechanism of the α7 nAChR positive allosteric modulator PNU-120596 provides evidence for two distinct desensitized states. Mol. Pharmacol. 80, 1013–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Young G. T., Zwart R., Walker A. S., Sher E., and Millar N. S. (2008) Potentiation of α7 nicotinic acetylcholine receptors via an allosteric transmembrane site. Proc. Natl. Acad. Sci. U.S.A. 105, 14686–14691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bertrand D., Bertrand S., Cassar S., Gubbins E., Li J., and Gopalakrishnan M. (2008) Positive allosteric modulation of the α7 nicotinic acetylcholine receptor: ligand interactions with distinct binding sites and evidence for a prominent role of the M2-M3 segment. Mol. Pharmacol. 74, 1407–1416 [DOI] [PubMed] [Google Scholar]
- 13. Papke R. L., Horenstein N. A., Kulkarni A. R., Stokes C., Corrie L. W., Maeng C. Y., and Thakur G. A. (2014) The activity of GAT107, an allosteric activator and positive modulator of α7 nicotinic acetylcholine receptors (nAChR), is regulated by aromatic amino acids that span the subunit interface. J. Biol. Chem. 289, 4515–4531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gill J. K., Savolainen M., Young G. T., Zwart R., Sher E., and Millar N. S. (2011) Agonist activation of α7 nicotinic acetylcholine receptors via an allosteric transmembrane site. Proc. Natl. Acad. Sci. U.S.A. 108, 5867–5872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kulkarni A. R., and Thakur G. A. (2013) Microwave-assisted expeditious and efficient synthesis of cyclopentene ring-fused tetrahydroquinoline derivatives using three-component Povarov reaction. Tetrahedron Lett. 54, 6592–6595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Thakur G. A., Kulkarni A. R., Deschamps J. R., and Papke R. L. (2013) Expeditious synthesis, enantiomeric resolution and enantiomer functional characterization of (4-(4-bromophenyl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinoline-8-sulfonamide (4BP-TQS) an allosteric agonist–positive allosteric modulator of α7 nAChR. J. Med. Chem. 56, 8943–8947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gill-Thind J. K., Dhankher P., D'Oyley J. M., Sheppard T. D., and Millar N. S. (2015) Structurally similar allosteric modulators of α7 nicotinic acetylcholine receptors exhibit five distinct pharmacological effects. J. Biol. Chem. 290, 3552–3562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li S. X., Huang S., Bren N., Noridomi K., Dellisanti C. D., Sine S. M., and Chen L. (2011) Ligand-binding domain of an α7-nicotinic receptor chimera and its complex with agonist. Nat. Neurosci. 14, 1253–1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Biasini M., Bienert S., Waterhouse A., Arnold K., Studer G., Schmidt T., Kiefer F., Cassarino T. G., Bertoni M., Bordoli L., and Schwede T. (2014) SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 42, W252–W258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 21. Hansen S. B., Sulzenbacher G., Huxford T., Marchot P., Taylor P., and Bourne Y. (2005) Structures of Aplysia AChBP complexes with nicotinic agonists and antagonists reveal distinctive binding interfaces and conformations. EMBO J. 24, 3635–3646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Halevi S., Yassin L., Eshel M., Sala F., Sala S., Criado M., and Treinin M. (2003) Conservation within the RIC-3 gene family. Effectors of mammalian nicotinic acetylcholine receptor expression. J. Biol. Chem. 278, 34411–34417 [DOI] [PubMed] [Google Scholar]
- 23. Papke R. L., Stokes C., Williams D. K., Wang J., and Horenstein N. A. (2011) Cysteine accessibility analysis of the human α7 nicotinic acetylcholine receptor ligand-binding domain identifies L119 as a gatekeeper. Neuropharmacology 60, 159–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Papke R. L., and Stokes C. (2010) Working with OpusXpress: methods for high volume oocyte experiments. Methods 51, 121–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Papke R. L., and Porter Papke J. K. (2002) Comparative pharmacology of rat and human α7 nAChR conducted with net charge analysis. Br. J. Pharmacol. 137, 49–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Papke R. L. (2010) Tricks of perspective: insights and limitations to the study of macroscopic currents for the analysis of nAChR activation and desensitization. J. Mol. Neurosci. 40, 77–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Williams D. K., Stokes C., Horenstein N. A., and Papke R. L. (2009) Differential regulation of receptor activation and agonist selectivity by highly conserved tryptophans in the nicotinic acetylcholine receptor binding site. J. Pharmacol. Exp. Ther. 330, 40–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kao P. N., Dwork A. J., Kaldany R. J., Silver M. L., Wideman J., Stein S., and Karlin A. (1984) Identification of two α-subunit half-cystines specifically labeled by an affinity reagent for the acetylcholine binding site. J. Biol. Chem. 259, 1162–1165 [PubMed] [Google Scholar]
- 29. Wang J., Horenstein N. A., Stokes C., and Papke R. L. (2010) Tethered agonist analogs as site-specific probes for domains of the human α7 nicotinic acetylcholine receptor that differentially regulate activation and desensitization. Mol. Pharmacol. 78, 1012–1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Spurny R., Debaveye S., Farinha A., Veys K., Vos A. M., Gossas T., Atack J., Bertrand S., Bertrand D., Danielson U. H., Tresadern G., and Ulens C. (2015) Molecular blueprint of allosteric binding sites in a homologue of the agonist-binding domain of the α7 nicotinic acetylcholine receptor, Proc. Natl. Acad. Sci. U.S.A. 112, E2543–E2552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bertrand D., Lee C. H., Flood D., Marger F., and Donnelly-Roberts D. (2015) Therapeutic potential of α7 nicotinic acetylcholine receptors. Pharmacol. Rev. 67, 1025–1073 [DOI] [PubMed] [Google Scholar]
- 32. de Fiebre C. M., Meyer E. M., Henry J. C., Muraskin S. I., Kem W. R., and Papke R. L. (1995) Characterization of a family of anabaseine-derived compounds reveals that the 3-(4)-dimethylaminocinnamylidine derivative (DMAC) is a selective agonist at neuronal nicotinic α7/[125I]α-bungarotoxin receptor subtypes. Mol. Pharmacol. 47, 164–171 [PubMed] [Google Scholar]
- 33. Briggs C. A., Anderson D. J., Brioni J. D., Buccafusco J. J., Buckley M. J., Campbell J. E., Decker M. W., Donnelly-Roberts D., Elliott R. L., Gopalakrishnan M., Holladay M. W., Hui Y., Jackson W. J., Kim D. J., Marsh K. C., et al. (1997) Functional characterization of the novel nicotinic receptor ligand GTS-21 in vitro and in vivo. Pharmacol. Biochem. Behav. 57, 231–241 [DOI] [PubMed] [Google Scholar]
- 34. Meyer E. M., Kuryatov A., Gerzanich V., Lindstrom J., and Papke R. L. (1998) Analysis of 40H-GTS-21 selectivity and activity at human and rat a7 nicotinic receptors. J. Pharmacol. Exp. Ther. 287, 918–925 [PubMed] [Google Scholar]
- 35. Kong F. J., Ma L. L., Zhang H. H., and Zhou J. Q. (2015) α7 nicotinic acetylcholine receptor agonist GTS-21 mitigates isoflurane-induced cognitive impairment in aged rats. J. Surg. Res. 194, 255–261 [DOI] [PubMed] [Google Scholar]
- 36. Lewis A. S., Mineur Y. S., Smith P. H., Cahuzac E. L., and Picciotto M. R. (2015) Modulation of aggressive behavior in mice by nicotinic receptor subtypes. Biochem. Pharmacol. 97, 488–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cannon C. E., Puri V., Vivian J. A., Egbertson M. S., Eddins D., and Uslaner J. M. (2013) The nicotinic α7 receptor agonist GTS-21 improves cognitive performance in ketamine impaired rhesus monkeys. Neuropharmacology 64, 191–196 [DOI] [PubMed] [Google Scholar]
- 38. Hu Y., Liu R., Li J., Yue Y., Cheng W., and Zhang P. (2014) Attenuation of collagen-induced arthritis in rat by nicotinic α7 receptor partial agonist GTS-21. BioMed. Res. Int. 2014, 325875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wu S., Zhao H., Luo H., Xiao X., Zhang H., Li T., and Zuo X. (2014) GTS-21, an α7-nicotinic acetylcholine receptor agonist, modulates Th1 differentiation in CD4 T cells from patients with rheumatoid arthritis. Exp. Ther. Med. 8, 557–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yue Y., Liu R., Cheng W., Hu Y., Li J., Pan X., Peng J., and Zhang P. (2015) GTS-21 attenuates lipopolysaccharide-induced inflammatory cytokine production in vitro by modulating the Akt and NF-κB signaling pathway through the α7 nicotinic acetylcholine receptor. Int. Immunopharmacol. 15, 1–6 [DOI] [PubMed] [Google Scholar]
- 41. Kox M., van Velzen J. F., Pompe J. C., Hoedemaekers C. W., van der Hoeven J. G., and Pickkers P. (2009) GTS-21 inhibits pro-inflammatory cytokine release independent of the Toll-like receptor stimulated via a transcriptional mechanism involving JAK2 activation. Biochem. Pharmacol. 78, 863–872 [DOI] [PubMed] [Google Scholar]
- 42. Pavlov V. A., Ochani M., Yang L. H., Gallowitsch-Puerta M., Ochani K., Lin X., Levi J., Parrish W. R., Rosas-Ballina M., Czura C. J., Larosa G. J., Miller E. J., Tracey K. J., and Al-Abed Y. (2007) Selective α7-nicotinic acetylcholine receptor agonist GTS-21 improves survival in murine endotoxemia and severe sepsis. Crit. Care Med. 35, 1139–1144 [DOI] [PubMed] [Google Scholar]
- 43. Chatterjee P. K., Al-Abed Y., Sherry B., and Metz C. N. (2009) Cholinergic agonists regulate JAK2/STAT3 signaling to suppress endothelial cell activation. Am. J. Physiol. Cell Physiol. 297, C1294–1306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Papke R. L., Kem W. R., Soti F., López-Hernández G. Y., and Horenstein N. A. (2009) Activation and desensitization of nicotinic α7-type acetylcholine receptors by benzylidene anabaseines and nicotine. J. Pharmacol. Exp. Ther. 329, 791–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Paulo J. A., Brucker W. J., and Hawrot E. (2009) Proteomic analysis of an α7 nicotinic acetylcholine receptor interactome. J. Proteome Res. 8, 1849–1858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mulcahy M. J., Blattman S. B., Barrantes F. J., Lukas R. J., and Hawrot E. (2015) Resistance to inhibitors of cholinesterase 3 (Ric-3) expression promotes selective protein associations with the human α7-nicotinic acetylcholine receptor interactome. PLoS ONE 10, e0134409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Stokes C., Treinin M., and Papke R. L. (2015) Looking below the surface of nicotinic acetylcholine receptors. Trends Pharmacol. Sci. 36, 514–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kabbani N., Nordman J. C., Corgiat B. A., Veltri D. P., Shehu A., Seymour V. A., and Adams D. J. (2013) Are nicotinic acetylcholine receptors coupled to G proteins? Bioessays 35, 1025–1034 [DOI] [PubMed] [Google Scholar]
- 49. King J. R., Nordman J. C., Bridges S. P., Lin M. K., and Kabbani N. (2015) Identification and characterization of a G protein-binding cluster in α7 nicotinic acetylcholine receptors. J. Biol. Chem. 290, 20060–20070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Papke R. L., Chojnacka K., and Horenstein N. A. (2014) The minimal pharmacophore for silent agonism of α7 nAChR. J. Pharmacol. Exp. Ther. 350, 665–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Briggs C. A., Grønlien J. H., Curzon P., Timmermann D. B., Ween H., Thorin-Hagene K., Kerr P., Anderson D. J., Malysz J., Dyhring T., Olsen G. M., Peters D., Bunnelle W. H., and Gopalakrishnan M. (2009) Role of channel activation in cognitive enhancement mediated by α7 nicotinic acetylcholine receptors. Br. J. Pharmacol. 158, 1486–1494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Thomsen M. S., and Mikkelsen J. D. (2012) The α7 nicotinic acetylcholine receptor ligands methyllycaconitine, NS6740 and GTS-21 reduce lipopolysaccharide-induced TNF-α release from microglia. J. Neuroimmunol. 251, 65–72 [DOI] [PubMed] [Google Scholar]
- 53. Papke R. L., Bagdas D., Kulkarni A. R., Gould T., AlSharari S. D., Thakur G. A., and Damaj M. I. (2015) The analgesic-like properties of the α7 nAChR silent agonist NS6740 is associated with nonconducting conformations of the receptor. Neuropharmacology 91, 34–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Williams D. K., Wang J., and Papke R. L. (2011) Positive allosteric modulators as an approach to nicotinic acetylcholine receptor-targeted therapeutics: advantages and limitations. Biochem. Pharmacol. 82, 915–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Alsharari S. D., Freitas K., and Damaj M. I. (2013) Functional role of α7 nicotinic receptor in chronic neuropathic and inflammatory pain: studies in transgenic mice. Biochem. Pharmacol. 86, 1201–1207 [DOI] [PubMed] [Google Scholar]
- 56. Freitas K., Carroll F. I., and Damaj M. I. (2013) The antinociceptive effects of nicotinic receptors α7-positive allosteric modulators in murine acute and tonic pain models. J. Pharmacol. Exp. Ther. 344, 264–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Freitas K., Ghosh S., Ivy Carroll F., Lichtman A. H., and Imad Damaj M. (2013) Effects of α7 positive allosteric modulators in murine inflammatory and chronic neuropathic pain models. Neuropharmacology 65, 156–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Corringer P. J., Le Novère N., and Changeux J. P. (2000) Nicotinic receptors at the amino acid level. Annu. Rev. Pharmacol. Toxicol. 40, 431–458 [DOI] [PubMed] [Google Scholar]
- 59. Sixma T. K., and Smit A. B. (2003) Acetylcholine binding protein (AChBP): a secreted glial protein that provides a high-resolution model for the extracellular domain of pentameric ligand-gated ion channels. Annu. Rev. Biophys. Biomol. Struct. 32, 311–334 [DOI] [PubMed] [Google Scholar]
- 60. Celie P. H., van Rossum-Fikkert S. E., van Dijk W. J., Brejc K., Smit A. B., and Sixma T. K. (2004) Nicotine and carbamylcholine binding to nicotinic acetylcholine receptors as studied in AChBP crystal structures. Neuron 41, 907–914 [DOI] [PubMed] [Google Scholar]
- 61. Gill J. K., Dhankher P., Sheppard T. D., Sher E., and Millar N. S. (2012) A series of α7 nicotinic acetylcholine receptor allosteric modulators with close chemical similarity but diverse pharmacological properties. Mol. Pharmacol. 81, 710–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Parthiban M., Rajasekaran M. B., Ramakumar S., and Shanmughavel P. (2009) Molecular modeling of human pentameric α(7) neuronal nicotinic acetylcholine receptor and its interaction with its agonist and competitive antagonist. J. Biomol. Struct. Dyn. 26, 535–547 [DOI] [PubMed] [Google Scholar]
- 63. Bisson W. H., Westera G., Schubiger P. A., and Scapozza L. (2008) Homology modeling and dynamics of the extracellular domain of rat and human neuronal nicotinic acetylcholine receptor subtypes α4β2 and α7. J. Mol. Model. 14, 891–899 [DOI] [PubMed] [Google Scholar]