

Abstract
Sirtuins are NAD+ dependent lysine deacylases involved in many regulatory processes such as control of metabolic pathways, DNA repair and stress response. Modulators of sirtuin activity are required as tools for uncovering the biological function of these enzymes and as potential therapeutic agents. Systematic discovery of such modulators is hampered by the lack of direct and continuous activity assays. The present study describes a novel continuous assay based on the increase of a fluorescence signal subsequent to sirtuin mediated removal of a fluorescent acyl chain from a modified TNFα-derived peptide. This substrate is well recognized by human sirtuins 1–6 and represents the best sirtuin 2 substrate described so far with a kcat/KM-value of 176 000 M−1s−1. These extraordinary substrate properties allow the first determination of Ki-values for the specific Sirt2 inhibitory peptide S2iL5 (600 nM) and for the quasi-universal sirtuin inhibitor peptide thioxo myristoyl TNFα (80 nM).
Reversible acylation of protein lysine residues is one of the most abundant posttranslational modifications (PTMs) involved in several cellular processes like metabolic regulation, cell cycle control and epigenetics1,2. Lysine acetylation is determined by the enzymatic activity of lysine acetyltransferases and lysine deacetylases. Recent studies detect alternative acylations as in vivo PTMs, including propionylations3,4, succinylations5,6, malonylations6,7, glutarylations8, crotonylations9, butyrylations3, 2-hydroxyisobutyrylations10, phosphoglycerations11 and myristoylations12. The generation of these PTMs is not fully understood, but it is evident that some of these acyl-transfers represent spontaneous reactions with acyl-CoAs or acylphosphates as acyl-donors forming stable amide bonds13,14,15,16,17. Removal of such acyl moieties from lysine side chains is catalyzed by either zinc ion dependent lysine deacetylases or by a conserved family of NAD+ dependent lysine deacylases, known as sirtuins. The mitochondrial sirtuin 5 (Sirt5) has over hundred-fold higher catalytic efficiency for succinylated and glutarylated lysine residues as compared to acetylated lysines7,8,18,19, whereas Sirt6 prefers long acyl chains, such as myristoylated lysine side chains20,21. Recently, it has been demonstrated that Sirt4 is able to remove lipoyl and biotinyl residues from lysine side chains both in vitro and in vivo22 while Sirt3 seems to be an in vivo decrotonylase, in addition to its established deacetylase function23. Furthermore, it is known that Sirt2 exhibits demyristoylase24,25 and depalmitoylase activity26. Sirtuin mediated deacetylations regulate several metabolic processes, such as fatty acid synthesis, glucose homeostasis and stress response27. Moreover, sirtuins are involved in diseases like diabetes, cancer and neurodegeneration27, making these enzyme attractive targets for pharmacological modulation. However, few compounds for sirtuin inhibition and activation are available, and the unavailability of sensitive and reliable assays also suitable for high-throughput screens has contributed to this lack of modulators28. In fact, assay artifacts contributed to a controversy about the general possibility to activate sirtuins, but more recent work involving robust yet time-demanding low-throughput mass spectrometry-based sirtuin assays confirmed the controversial Sirt1 activation and revealed the possibility to activate Sirt5 and Sirt629,30.
For the efficient development of sirtuin effectors, reliable and ideally continuous high-throughput assays are indispensable. Several existing, and in most cases discontinuous activity assays (reviewed in28,31) are based on the separation of products and substrates by HPLC/CE32,33,34, by mass spectrometry35,36 or spectrophotometric detection of one reaction component29,37,38,39. Nevertheless, continuous activity assays are known which couple the sirtuin reaction to either an additional enzymatic reaction38,40, a chemical reaction such as intramolecular transesterification41, an interaction with DNA42,43 or to fluorescence enhancement by aggregation-induced emission44,45.
For microtiter plate (MTP)-based assay formats the sirtuin reaction is currently coupled to enzymatic reactions either sensing the released nicotinamide38, the remaining NAD+ 46 or the deacylated peptide product40. One advantage of monitoring sirtuin-mediated release of nicotinamide is the compatibility with any substrate including proteins and also with any type of lysine acylation. However, the enzymatic cascade needed for signal generation, limits the linear range of the assay and makes it more sensitive to interference in compound tests as observed for GW5074, a Sirt5 inhibitor that also affects GDH activity47. Hubbard et al. substituted the last enzymatic step by a chemical reaction sensing ammonia allowing more accurate but discontinuous activity determinations29,48.
Assays sensing the deacylated product of the sirtuin reaction utilize the subsite specificity of proteases like Trypsin, which have a strong preference for positively charged side chains in the P1-position and thus do not cleave the acylated substrates of the sirtuin-mediated reaction. This principle has been introduced using peptidyl-7-amino-4-methyl-coumarin derivatives49.
Subsequent to deacylation of the peptidyl moiety the bond between the C-terminus of the peptidyl moiety and the amino-coumarin derivative is hydrolyzed by the helper protease hereby releasing the highly fluorescent 7-amino-4-methyl-coumarin.
This assay is very sensitive49 but makes use of sirtuin substrates with suboptimal KM values and it often has to be performed discontinuously because of the susceptibility of sirtuins to digestion by the helper protease. Appropriate substrates have been synthesized for assaying sirtuin isoforms activity against acetylated49, succinylated50, glutarylated8, adipoylated8 or myristoylated lysine residues51. The fluorophore replacing the C-terminal peptide part renders these substrates highly artificial and has been reported to cause artifacts in compound tests28,30,52,53. Improved substrates for Sirt1 and Sirt2 have been reported using FRET by introducing tetramethylrhodamine as a fluorophor and QSY-7 as a quencher at the N- and C-terminus, respectively, of a p53- derived peptide37. For Sirt5 and Sirt6 activity measurements the fluorophore/quencher pair Dabcyl and EDANS was used in a glutamate dehydrogenase derived peptide sequence54,55. Recently, we were able to show that use of 2-aminobenzoylamides as fluorophores and 3-nitrotyrosines as quenchers in a carbamoyl phosphate synthetase 1 derived peptide derivative allow sensitive detection of Sirt5 activity in a continuous format56.
Relatively high amounts (up to 4 μM) of sirtuin have been used in activity assays to correct for suboptimal substrate properties20. This limits the applicability of the Michaelis-Menten-equation, which is valid only if enzyme concentration is much lower than substrate concentration. Additionally, due to the high enzyme concentrations, reliable estimation of IC50- or Ki-values is difficult for inhibitors with affinities far below the enzyme concentration.
As previously established, sirtuins 1–6 are able to remove mid-chain acyl residues like octanoyl-, decanoyl- and myristoyl-moieties from lysine side chains in histone H3 derived model peptides20,57. Recently, using a similar histone H3 peptide substrate, it could be demonstrated that Sirt1-3 are able to remove myristoyl residues from lysine side chains25 and Sirt2 represents a very efficient demyristoylase24. This fact and inspection of several crystal structures of a myristoylated/thioxo myristoylated peptides in complex with Sirt6/Sirt2 (PDB IDs 3ZG6, 4R8M, 4Y6Q, 4Y6L)21,24,57 prompted us to test if the hydrophobic channel on the surfaces of Sirt621 and Sirt224,26,57 can accommodate small fluorophores, like 2-aminobenzoylamides, to create a continuous sirtuin activity assay.
If accepted by sirtuins, replacement of one amino acid residue within the TNFα derived substrate by a 3-nitrotyrosine residue as a quencher moiety should yield a peptide derivative increasing its fluorescence subsequent to sirtuin treatment in the presence of NAD+(Fig. 1).
Figure 1.

(a) Sirtuin-mediated deacylation reaction transfers fluorescently labeled acyl residues from lysine side chain to ADP-ribose. (b) Sirtuin-mediated deacylation reaction transfers quencher-containing acyl residue from lysine side chain to ADP-ribose. In both cases sirtuin activity causes an increase in the fluorescence signal. (Fl – fluorophore, Qu – quencher, ADPr – ADP ribose)
Results
We synthesized peptides derived from the TNFα sequence which are used as model substrates21,24,25 or inhibitors24,25 for different sirtuin isoforms (Figs 2 and 3). Nosyl-protection at one lysine residue for selective on-resin modification of this side chain and Fmoc-based solid phase peptide chemistry was employed. The peptide 1a is the best Sirt6 substrate described so far21.
Figure 2. Structures of synthesized substrates.

Figure 3. Structures of inhibitors.

1a and 2a were subjected to HPLC-based activity assay to assess their substrate properties and to determine if the quencher moiety is accepted by sirtuin 6. Negative controls without NAD+ under identical conditions yielded no conversion of substrates. The kinetic constants uncovered that the replacement of threonine in +1 position of the substrate by the quencher moiety 3-nitrotyrosine did not influence the turnover number and minimally disturbed the apparent affinity to the active site of Sirt6 as reflected by the almost comparable KM values for 1a (6 μM) and 2a (17 μM) (Supplementary Fig. S3).
Sirtuins 1–5 were also tested and 1a was shown to represent a universal sirtuin substrate with kcat/KM values in the range of 10 to 50 000 M−1s−1 (Supplementary Table S2). Therefore, we reasoned that use the fatty acid chain could be utilized as an attachment point for the very small fluorophore 2-amino-benzoylamide. Systematic variation of the distance (number of bonds) between the amide bond on the lysine side chain and the 2-amino-benzoylamide moiety (i.e. 3, 4, 4a) revealed good substrate properties for 3 only (Table 1). Peptide 4 was not a substrate for sirtuins 1 and 3–7 but showed some activity for Sirt2 in an HPLC based end-point activity assay. However, increasing the number of methylene groups to place the fluorophore to a different position yielded improvement in substrate properties for Sirt2 with an optimum for 3. Further elongation of the spacer resulted in >1000-fold decrease of substrate properties for Sirt2 (Table 1) and complete loss of activity for sirtuin isoforms 1 and 3–6. Substrate 3 represents a quasi-universal sirtuin substrate because it is recognized by isoforms 1–6. The development of 3 resulted in slightly decreased substrate properties for sirtuins 1, 3, 4, 5, and 6 as compared to 1a but interestingly yielded an improved substrate for Sirt2 with a specificity constant of 176 000 M−1s-1 representing the best Sirt2 substrate described so far (Table 2). Sirt7 was not able to recognize substrate 3 pointing to structural differences of the hydrophobic channel accommodating the acyl chain.
Table 1. Kinetic constants for 3, 4, 4a and 5 and Sirt2.
| Substrate | KM [μM] | 10−3xkcat [s−1] | kcat/KM [M−1s−1] | Distance (No. of bonds) |
|---|---|---|---|---|
| 4 | 17.7 ± 1.5 | 4.0 ± 0.1 | 224a | 6 |
| 4a | 1.2 ± 0.1 | 45.4 ± 1.8 | 38 600b | 8 |
| 3 | 0.1 ± 0.02 | 23.8 ± 0.8 | 176 000a | 11 |
| 5 | 15.3 ± 2.7 | 2.4 ± 0.1 | 156b | 13 |
ameasured using fluorescence spectrophotometer.
bmeasured using MTP fluorescence reader (see supporting information for details). Data are presented as mean ± s.d. (n = 2).
Table 2. Kinetic constants for 3 and Sirt1-6.
| Enzyme | KM [μM] | 10−3xkcat [s−1] | kcat/KM [M−1s−1] | c (Sirt) [nM] |
|---|---|---|---|---|
| Sirt1 | 0.7 ± 0.08 | 2.1 ± 0.1 | 287a | 500 |
| Sirt2 | 0.12 ± 0.02 | 23.8 ± 0.8 | 176 000a | 10 |
| Sirt3 | 3.3 ± 0.4 | 9.1 ± 0.4 | 2 800a | 100 |
| Sirt4 | 49.5 ± 7.5 | 0.4 ± 0.02 | 7a | 1000 |
| Sirt5 | 46.1 ± 7.2 | 3.2 ± 0.2 | 69b | 500 |
| Sirt6 | 23.5 ± 4.9 | 0.9 ± 0.1 | 39a | 500 |
ameasured using fluorescence spectrophotometer.
bmeasured using MTP fluorescence reader (see supporting information for details). Data are presented as mean ± s.d. (n = 2).
Sirtuin mediated transformation of 3 into 2b could be followed directly and continuously (λEx = 310 nm, λEm = 405 nm) using a fluorescence spectrometer (Supplementary Fig. S5). Without NAD+ in the presence of sirtuin enzyme or without sirtuin in the presence of NAD+ no significant change in fluorescence signal over time could be observed (Supplementary Fig. S5). This indicated that the observed fluorescence change results directly from sirtuin-mediated deacylation and not from unspecific interactions between NAD+ and/or sirtuin and 3.
The slope of change in fluorescence intensity is linearly dependent on the enzyme concentration (Supplementary Fig. S5). Progress curves at different concentrations were linear below 25% conversion of the substrate. We used a completely converted assay solution (controlled by LC-MS) for the generation of appropriate calibration curves (Supplementary Fig. S9). Additionally, we were able to demonstrate that the activity assay is compatible with 96- and 384-well microtiter plate-based equipment yielding Z´-factors of 0.85 for 3 at 25 μM concentration. Kinetic constants determined with either HPLC based assay or with the assay performed in both MTP fluorescence readers and spectrophotometers yielded comparable results (Supplementary Table S2).
Due to the relatively low kcat-values of the known substrates,
“classical” sirtuin activity assays are done in timeframes
between 30 and 120 min and at enzyme concentrations between 0.5 and
4 μM to generate sufficient signal changes. At these
conditions the basic assumption of the Michaelis-Menten-equation 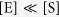 is not valid. Moreover, the high amount of enzyme prevents
the correct determination of Ki-values for sirtuin inhibitors with
affinities below half of the enzyme concentrations used. With substrate 3 we
were able to follow enzymatic activities down to 10 nM sirtuin
concentration (Supplementary Fig. S8).
We used a 96-well MTP fluorescence reader for the determination of the
Ki-values for different compounds (Fig. 3)
including inhibitors with high affinities to sirtuin isoforms (Table
3).
is not valid. Moreover, the high amount of enzyme prevents
the correct determination of Ki-values for sirtuin inhibitors with
affinities below half of the enzyme concentrations used. With substrate 3 we
were able to follow enzymatic activities down to 10 nM sirtuin
concentration (Supplementary Fig. S8).
We used a 96-well MTP fluorescence reader for the determination of the
Ki-values for different compounds (Fig. 3)
including inhibitors with high affinities to sirtuin isoforms (Table
3).
Table 3. Ki-values for different inhibitors.
| inhibitor | Enzyme | Ki (3) [μM] | Ki (NAD+) [μM] |
|---|---|---|---|
| NAM | Sirt3 | 93.0 ± 8.5 | 45.0 ± 14.2 |
| Sirt6 | 451.0 ± 60.7 | 415.0 ± 45.1 | |
| Ex-527 | Sirt6 | 100.0 ± 11.0 | n.d. |
| Quercetin | Sirt6 | 21.0 ± 3.4 | n.d. |
| S2iL5 | Sirt2 | 0.6 ± 0.2 | n.d. |
| 8 | Sirt2 | 0.08 ± 0.02 | n.d. |
| Sirt3 | 0.1 ± 0.02 | n.d. | |
| Sirt6 | 0.4 ± 0.1 | n.d. | |
| Sirt6a | 1.1 ± 0.1e | n.d. | |
| Sirt6b | 1.9 ± 0.2e | n.d. | |
| 9 | Sirt6c | 0.6 ± 0.1e | n.d. |
| Sirt6d | 1.7 ± 0.5e | n.d. | |
| 10 | Sirt2 | 0.3 ± 0.1 | n.d. |
| 11 | Sirt2 | 50.0 ± 9.9 | n.d. |
a2.5% cis isomer.
b25% cis isomer.
c72.4% cis isomer.
d29.7% cis isomer.
eIC50 value, n.d. not determined.
The first product of the sirtuin reaction, nicotinamide (NAM), is known to be a non-competitive inhibitor with respect to both acylated peptide substrate and NAD+ cosubstrate by re-binding to the active site and attacking the sirtuin bound O-alkylimidate reforming NAD+. For Sirt6 an IC50 value of 2.2 mM was reported for NAM indicating that this isoform is not influenced by physiological NAM concentrations58
We determined the Ki-values of NAM for Sirt3 and Sirt6 to be 93 μM and 451 μM, respectively, using NAD+ at saturating conditions (Supplementary Fig. S13). Under peptide substrate saturating conditions Ki-values were found to be 45 μM and 415 μM, respectively (Supplementary Fig. S13). The Ki-value for NAM was lower than expected for Sirt6, but still higher than for other isoforms. Recently, it was shown that the IC50-values for NAM are dependent on the chemical nature of the acyl moiety and that different sirtuin isoforms have different acyl-dependent susceptibilities to NAM inhibition57. Our substrate closely resembles the physiological myristoyl substrate hence our value should reflect the sensitivity of this substrate modification. Recently, compounds Quercetin and Ex-527 were reported as Sirt6 inhibitors with inhibition of enzymatic activity of 52% and 56%, respectively, if used at 200 μM concentration58. We determined Ki-values for these two small molecules and found considerable non-competitive inhibition with respect to the peptide substrate (Table 3). The cyclic peptide derivative S2iL5, containing a trifluoroacetylated lysine side chain as a warhead for inhibition of sirtuin catalysis59, was claimed to be a Sirt2 specific inhibitor with affinities to the active site in the low nanomolar range as determined by isothermal calorimetric measurements60. Using 3 as substrate the determined Ki-value is 560 nM and the cyclic inhibitor behaved non-competitive for the peptide substrate (Supplementary Fig. S17). Replacement of the amide bond formed by the acyl chain and the ε-amino function of the lysine side chain by a thioxo amide bond transforms substrates into extremely slow substrates/inhibitors by generation of a stalled intermediate resembling sirtuin bi-substrate inhibitors61,62,63. Thioxo myristoylated and shortened derivatives of 1 were shown to be cell permeable inhibitors for Sirt6 with remarkable cross-reactivity to Sirt1-3 and reported IC50 values in the single digit micromolar range25. Due to the high sirtuin concentration used in this enzymatic assay (i.e. 1 μM Sirt6) we wondered if these values are too high, not properly reflecting the Ki. We determined the Ki-values of 8 for sirtuins 2, 3, and 6 using substrate 3 and 96-well-based readout (Table 3) and determined higher affinities to the sirtuins especially for Sirt2 with Ki-value of 80 nM. We were able to determine these Ki-values because in our case the enzyme concentration was about 100 times lower as compared to the assay proposed by He et al.25. Additionally, Sirt3 showed high affinity to 8. We were not able to determine the Ki-values if we pre-incubate the enzymes Sirt2 and Sirt3 (10 nM) with different concentrations of 8 in the presence of NAD+ for 30 min enabling formation of the “stalled” intermediate without competition with the substrate peptide. Starting the reaction by addition of 3 we observed complete inhibition with low nanomolar concentrations of 8 demonstrating that the pre-formed bi-substrate inhibitor has affinities to Sirt2 and Sirt3 in the very low nano- or picomolar range. The resulting non-competitive inhibition against peptide substrate was in accordance with the suggested model of bi-substrate like inhibitors for thioxo acylated derivatives (Supplementary Fig. S15).
Identification of small molecule modulators of sirtuin activity using 3 could be hampered by absorbance/fluorescence of the effectors in the range between 320 nm and 400 nm. Consequently, the known sirtuin activity modulator resveratrol could not be analyzed because of the high extinction coefficient in that range. Using HPLC-based activity assay we found no significant influence of resveratrol on Sirt1 mediated deacylation of 3 (Supplementary Fig. S4) To be able to analyze small molecules with absorption/fluorescence in the range of 2-aminobenzoylamide fluorescence, we exchanged the 2-aminobenzoylamide fluorophore by (4-N,N-dimethylamino-1,8-naphthalimido)-acetamide resulting in derivative 6 with fluorescence excitation at 471 nm64. Surprisingly, this fluorophore could be used in combination with the 3-nitro-L-tyrosine quencher despite non-optimal overlap of the spectra. Compound 6 was not a substrate for sirtuins 1, 3, 5 and 6 as determined by HPLC-based assays and was a weak substrate for Sirt2 with an about 500-fold lower kcat/KM-value as compared to 3. Interestingly, Sirt4 recognized 6 better than 3 resulting in an about 10-fold improved KM-value (Supplementary Table S2) indicative of differences in the flexibility of the hydrophobic channel accommodating the acyl chain between Sirt4 and the other sirtuin isoforms. However, these results showed that the development of substrates with different spectral properties is possible enabling the simultaneous detection of enzymatic activity using substrate mixtures. We analyzed kinetics for Sirt2 and Sirt4 using a mixture of substrates 3 and 6 using 290 nm for excitation of both fluorophores and recording fluorescence spectra over time (Supplementary Fig. S10 and S11). Furthermore development of isoform selective substrates should be possible by systematic variation of the size and position of the fluorophore in the acyl side chain.
Because of the obvious limitations for most of the sirtuins in accommodating bulkier fluorophores, we decided to create a small quencher moiety at this position closely related to the well-recognized 2-amino-benzoylamide residue. Addition of a nitro function in para-position to the amino-group of the 2-amino-benzoylamide moiety generated a very efficient quencher for more bulky fluorophores like 7-methoxy-coumaryl-L-alanines (Mca) or even (4-N,N-dimethylamino-1,8-naphthalimido)-L-alanines (Dma) (data not shown). We speculated that sirtuins are less sensitive for modifications within the peptide sequence and synthesized several derivatives of 1a characterized by substitutions of residues in different positions relative to the myristoylated lysine by either Dma (12, 13) or Mca (15, 16). Additionally, we attached the fluorophores to the N-terminus in form of appropriately substituted acetyl residues (14, 17). Analysis of sirtuin 2, 3, and 6 activity against these substrates using an HPLC-based assay revealed that all peptides are substrates but only 13 and 16 are well recognized (Supplementary Table S1). For solubility reasons (data not shown) we decided to combine the 7-methoxy-coumaryl-L-alanyl-residue with the 5-nitro-2-amino-benzoylamide quencher moiety resulting in 7. The substrate properties of 7 for Sirt2 and Sirt4 are superior to substrates described in the literature and similar to 3, demonstrating that fluorophore and quencher positions could be switched without influence on substrate properties (Supplementary Table S2 and Fig. 1).
Inspection of the published crystal structure of Sirt6 in complex with the myristoylated H3K9 substrate (PDB ID 3ZG6) and the respective electron density maps revealed that both conformations of the amide bond between the fatty acid and the lysine side chain amino function could be fitted, but the published coordinates are given in a conformation resembling the cis conformation of peptide bonds21. In the recently reported structures of Sirt2 complexed with a thioxo myristoylated inhibitor closely related to 8 (PDB ID 4Y6Q) or a thioxo myristoylated peptide derived from Histone H3 (PDB IDs 4Y6L and 4R8M) the conformation of the thioxo amide bond was in trans conformation24,57 which is the preferred conformation of secondary amide/thioxo amide bonds in aqueous solutions. We synthesized 8 in order to analyze if there is any isomer-specificity during binding to the active site of Sirt2 and Sirt6. Cis/trans isomerizations of secondary amide bonds are too fast compared to the time needed for “classical” sirtuin activity assays preventing such analyses. Our assay allowed enzymatic measurements within short time and the isomerization of thioxo amide bonds is slower at lower temperatures65. Moreover, the UV-absorption of the π-π* transition for the cis conformer of thioxo amide bond is slightly red-shifted enabling determination of cis/trans isomerization rates using UV-spectroscopy66. We determined the isomerization rate for 8, 10 and 11 at different temperatures subsequent to increasing the cis content in the photo-excited state using UV-light (Supplementary Figs S19–33)66,67. The re-equilibration to the ground state (nearly 100% trans-conformation) could be followed using UV-spectroscopy at 260 nm yielding activation parameters (Supplementary Figs S19–33). In order to analyze isomer-specific inhibition of Sirt2 by 8 we optimized the assay conditions to measure the enzymatic activity using 3 without significant cis/trans isomerization during the assay. The cis content of 8 is 2.5% in assay buffer and up to 25% in the photo-excited state as measured by HPLC (Supplementary Fig. S34). We determined the rate of re-equilibration for 8 and calculated the resulting cis content subsequent to different times of darkness (Supplementary Table S3). This setup enabled the determination of inhibition of Sirt2 mediated deacylation of 3 at different cis contents of 8 ranging from 2.5% to 25%. We found no significant difference in inhibitory effect depending on the cis content of 8 indicating an unexpected plasticity of the active site of Sirt2 to accommodate both conformations with similar affinities. This result indicated that the Sirt2/myristoyl-peptide complexes, which were modeled as in trans conformation24,57 would also be compatible with cis. IC50-values of Sirt6 were determined as 1.06 ± 0.12 μM and 1.87 ± 0.18 μM for 2.5% and 25% cis isomer of 8, respectively, pointing to a small preference for the cis conformation of the amide bond (Supplementary Fig. S36). Nevertheless, because of the suboptimal substrate properties of 3 for Sirt6 the assay duration was 30 min which allows significant re-equilibration of the photo-induced change of the cis/trans equilibrium. Therefore, we introduced an additional methyl group at the lysine nitrogen resulting in a tertiary thioxo amide 9 which is an inhibitor with similar affinities to the active site of Sirt6 as compared to 8 (Supplementary Table S4). The rate constant for the cis/trans isomerization of the tertiary thioxo amide bond of 9 (5.4 × 10−4 s−1 at 20 °C) was much slower than that of the secondary thioxo amide bond of 8 (Supplementary Fig. S35). HPLC analyses revealed a cis content of about 50% and there was no change detectable subsequent to photo-excitation at the π-π* transition of the tertiary thioxo amide bond. We tested several different organic solvents to change the cis content but found no sufficient differences (Supplementary Table S5). Therefore, the two isomers were separated by HPLC at low temperatures (4 °C).
We were able to enrich the faster migrating cis isomer to 72.4% and the trans isomer to 70.3% (Fig. 4). The frozen isomers (−70 °C) were stable for several days (Supplementary Fig. S39).
Figure 4. Enrichment of cis and trans isomers of 9 using HPLC.
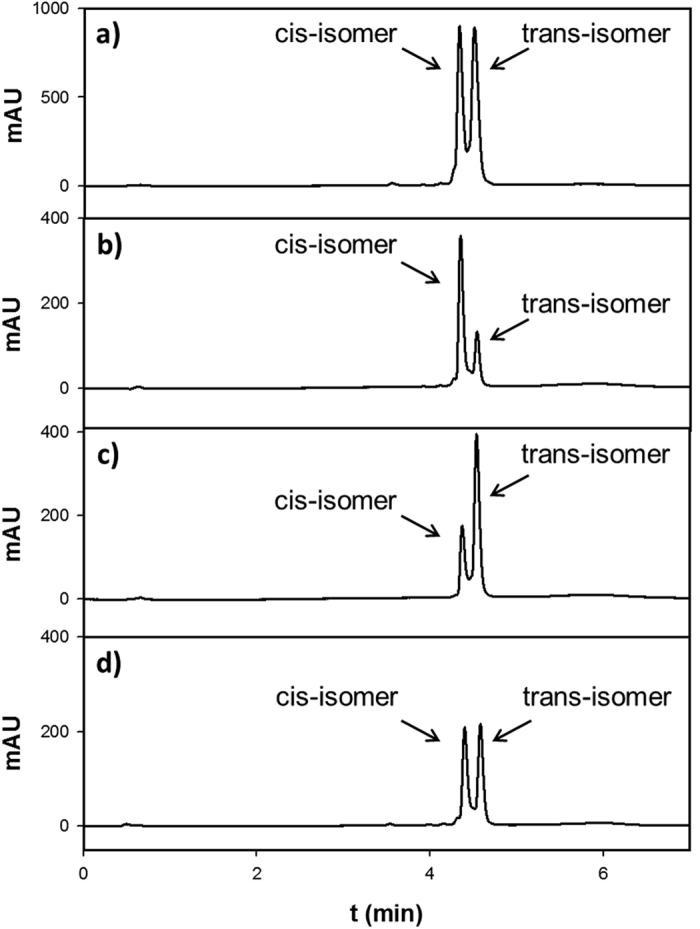
(a) 50 mM solution of 9 equilibrated at RT for 24 h. (b,c) Fractions collected by HPLC. (d) Aliquot of fraction (b) was equilibrated at 20 °C for 24 h.
Determination of inhibition of Sirt6 by 9 using samples with different cis content showed minimal preference for the cis isomer (IC50 values of 0.6 μM and 1.7 μM for 72.4% and 29.7% of cis of 9, respectively Supplementary Fig. S38). These results again demonstrated the plasticity within the active site of sirtuins, at least for Sirt6, enabling both isomers to bind with similar affinities. Inspection of the electron density maps of PDB 4R8M and 3ZG6 suggest that there is sufficient space around the lysine side chain amide/thioxo amide bond to fit both isomers. Recently, Sirt6 coordinates of 3ZG6 were re-refined by Denus lab and it was established that the myristoylated peptide should be in a trans conformation regarding the amide bond between the lysine side chain and the acyl moiety57.
Here we present a continuous sirtuin activity assay allowing convenient measurement of highly accurate data. The sensitivity of the activity assay enables the reliable determination of Ki-values for inhibitors with affinities below 100 nM. Because of the demonstrated compatibility with 384-well MTP readout we expect that this assay principle will find widespread application in drug discovery projects. Additionally, the superior substrate properties of 3 allow the investigation of isomer specificity in the binding of inhibitors to the active site of sirtuins enlarging the portfolio of tools in sirtuin research.
Methods
Chemicals and general methods
All chemicals were purchased from Sigma (Saint Louis, USA) if not denoted otherwise. Rink amide MBHA resin was obtained from Iris Biotech (Marktredwitz, Germany). 9-fluorenylmethoxy-carbonyl- (Fmoc) protected amino acid derivatives and O-(Benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU) were purchased from Merck (Darmstadt, Germany). Trifluoroacetic acid (TFA) was obtained from Roth (Karlsruhe, Germany).
For HPLC separations solvents consisting of water (solvent A) and ACN (solvent B), both containing 0.1% TFA, were used. Analytical runs were performed on an Agilent 1100 HPLC (Boeblingen, Germany) with a quaternary pump, a well-plate autosampler and a variable wavelength detector. Separations were performed on a 3.0 × 50 mm reversed phase column (Phenomenex Kinetex XB C-18, 2.6 μm) with a flow-rate of 0.6 mL/min. A Merck-Hitachi High Speed LC system (Darmstadt, Germany) with a Merck Hibar Li Chrospher® RP-8 column (250–25 mm, 5 μm) was used for preparative separations (flow-rate: 8 mL/min). Eluted compounds were analyzed by MALDI mass spectrometry. NMR spectroscopy was carried out using Varian Gemini 2000 spectrometer in deuterated chloroform.
Synthesis of Fmoc-Lys(Nosyl)-OH (N-α-(9-Fluorenylmethyloxycarbonyl)-N-ε-(2-nitrobenzenesulfonyl)-L-lysine)
The solution of L-lysine hydrochloride (20 mmol) and NaHCO3 (20 mmol) in 20 mL of H2O was combined with CuSO4·5H2O (10 mmol) solution in 40 mL of H2O. The vigorously stirred purple solution was cooled in an ice bath and a solution of 2-nitrobenzenesulfonyl chloride (30 mmol) in acetone (60 mL) was added. Next solid NaHCO3 (75 mmol) was added in portions over 1 hour. The stirred reaction mixture was left in a melting ice bath overnight. The blue precipitate was filtered, subsequently washed with H2O, ethanol and diethyl ether (Et2O). After air drying yield of the complex was 87%. To the copper complex of ε-nosyl lysine (5 mmol) a solution of ethylenediaminetetraacetic acid (EDTA) disodium salt (6.5 mmol) in 40 ml of H2O was added. This suspension was stirred and heated at 70–80 °C until no blue complex was left and then cooled to room temperature. Afterwards, solid NaHCO3 (10.5 mmol) was added to the formed suspension of ε-nosyl lysine and followed by solution of Fmoc-N-hydoxysuccinimide ester (Fmoc-OSu) (10.5 mmol) in 30 mL of acetone. The mixture was stirred vigorously overnight, diluted with 250 mL of 1% solution of NaHCO3 and extracted with Et2O (3 × 100 mL). Ether washings were back extracted with diluted NaHCO3 solution and discarded. Combined aqeous phases were acidified with 10% HCl and extracted with dichloromethane (DCM) (3 × 50 mL). Combined organic phases were washed with water and dried over Na2SO4. Solvent was evaporated to afford target compound as white foam. Yield: 91%.
1H NMR (400 MHz, CDCl3) δ ppm: 8.15 – 8.06 (m, 1H), 7.85 – 7.78 (m, 1H), 7.75 (d, J = 7.5 Hz, 2H), 7.71 – 7.63 (m, 2H), 7.59 (d, J = 7.3 Hz, 2H), 7.38 (t, J = 7.4 Hz, 2H), 7.29 (t, J = 7.4 Hz, 2H), 5.48 (t, J = 5.8 Hz, 1H), 5.41 (d, J = 7.8 Hz, 1H), 4.48 – 4.29 (m, 3H), 4.21 (t, J = 6.6 Hz, 1H), 3.16 – 3.0 (m, 2H), 1.94 – 1.34 (m, 6H). 13C NMR (100 MHz, CDCl3) δ ppm: 176.0, 156.1, 148.0, 143.8, 143.7, 141.3, 133.5, 132.7, 131.0, 127.7, 127.1, 125.3, 125.1, 120.0, 67.2, 53.3, 47.1, 43.3, 31.6, 28.9, 21.9.
Synthesis of carboxymethyl dithiomyristoate
Carboxymethyl dithioester was prepared in accordance to Leon et al.68. To the solution of myristic acid (4 mmol), HBTU (4 mmol) and N,N-diisopropylethylamine (DIPEA) (8 mmol) in DCM (30 mL) was added piperidine (4.2 mmol). After 4 hours reaction mixture was diluted with water and extracted with DCM. Extracts were washed with diluted HCL, diluted NaHCO3 and with water, dried over Na2SO4 and DCM was evaporated. To the residue toluene (10 mL) was added followed by Lawesson’s reagent (2 mmol). Reaction was heated at 106 °C for 3.5 hours. Solvent was evaporated and the residue flash-chromatographed (silica gel, ethyl acetate (EtOAc)/petr.ether 1:9) (Yield: 74%).
The solution of N-thiomyristoyl piperidine (3 mmol) and bromoacetic acid (3.2 mmol) in 7 mL of dried DMF was left at room temperature overnight. The solution was saturated with dry H2S for 30 min and DMF was evaporated in vacuo. Flash-chromatography of the residue (silica gel, DCM/AcOH 10:0.05) afforded the pure product as a yellow solid (Yield: 25%).
Synthesis of methyl 3-[(methylthio)thiocarbonyl]propanoate
A solution of succinic anhydride (50 mmol) and piperidine (50 mmol) in EtOAc (10 mL) was refluxed for 10 min. On the next day, precipitated product was filtered, washed with EtOAc and air dried (Yield: 90%).
N,N-pentamethylenesuccinamic acid (10 mmol) was refluxed in anhydrous MeOH (15 mL) containing 2 drops of H2SO4 for 4 hours. Solvent was evaporated and residual oil dissolved in EtOAc, washed with NaHCO3 solution, water and dried over Na2SO4. Evaporation of EtOAc afforded product as a colorless oil (Yield: 84%).
Methyl N,N-pentamethylenesuccinamate (8.4 mmol) and Lawesson’s reagent (5.1 mmol) were refluxed in tetrahydrofuran (THF) (10 mL) for 1 hour. THF was evaporated in vacuum and the residue was flash-chromatographed (silica gel, EtOAc/petr.ether 1:5). Yield of slightly yellowish oil 86%.
To a solution of Methyl 3-(N,N-pentamethylenethiocarbamoyl)propanoate (2 mmol) in anhydrous THF (8 mL) MeI (10 mmol)was added. The reaction was conducted for 48 h in darkness Yellow-colored THF was decantated, the crystals were briefly washed with dry THF and dissolved in dried DMF (3 mL). Dried H2S was bubbled into solution for 2 h and mixture was left at 0 °C for 24 h. After addition of H2O (100 mL), product was extracted with EtOAc, washed several times with water, brine and dried over Na2SO4. Evaporation of EtOAc in vacuo gave the crude product as a yellow oil (Yield: 81.5%).
Synthesis of TNFα peptide derivatives
The peptide Ac-EALPKK(NS)XGG-NH2 (X = T, Y(NO2) or Mcm) was synthesized by standard manual solid-phase-peptide synthesis using Fmoc-protected amino acid derivatives. Rink amide MBHA resin was treated with N,N-dimethylformamide (DMF) at room temperature (RT) for 10 min. The Fmoc-protecting group was removed with 20% piperidine in DMF (2 × 10 min). After washing with DMF (5 × 5 min) the resin was incubated with 4 eq of amino acid derivative, 4 eq HBTU and 8 eq of N,N-diisopropylethylamine (DIPEA) in DMF at RT (60 min). The N-terminus was modified with 4 eq acetic anhydride and 8 eq DIPEA in DCM (60 min). Nosyl-group was cleaved using 5 eq 1,8-Diazabicyclo[5.4.0]undec-7-en (DBU) and 5 eq thiophenol in DMF (2 × 90 min). Afterwards the resin was washed with DMF. Free lysine side chain was modified on-resin with HBTU (4 eq.), DIPEA (8 eq.) in DCM/DMF mixure (1:1) and myristic acid (1a, 2a), 6-(Fmoc-amino)-caproic acid and N-Boc-anthranilic acid (4, 5) or 8-(Fmoc-amino)-octanoic acid and N-Boc-anthranilic acid (4a). For 3 free lysine residue was acylated with 11-azidoundecanoic acid69 according to the method used for peptides 1a and 1b. The resin was treated with a solution of triphenylphosphine (5 eq) in tetrahydrofuran (THF)/H2O (95:5) for several days (small portions of resin were taken for the test cleavage and MS-analysis). After washing N-Boc-anthranilic acid was coupled by the standard method (see peptides 1a and 2b). 6 and 7 were prepared like peptide 3 with (4-N,N-dimethylamino-1,8-naphthalimid)-acetic acid64 (6)or 2-amino-5-nitrobenzoic acid (7) instead of N-Boc-anthranilic acid. For 8 the resin bound peptide was incubated with the solution of carboxymethyl dithiomyristoate (3 eq) and DIPEA (3 eq) in DMF for 3 h and cleaved as described in general procedure. For 9 an an ice-cooling prepared solution of 5 eq of triphenylphosphine, 5 eq of diethyl azodicarboxylate (DIAD) and 10 eq of dried MeOH in dried DCM was added to the resin-bound fully protected peptide with the ε-Nosyl-protected lysine (Mitsunoby reaction). After one hour of incubation the resin was washed 5 times with DMF. Nosyl-group was removed as described above and resin was treated with a solution of carboxymethyl dithiomyristoate (3 eq) and DIPEA (3 eq) in DMF for 3 h.
Peptides 10 and 11 are based on a CPS1-peptide (Bz-GVLKEYGV-NH2). To a DMF solution of the CPS1 peptide, ethyl dithioacetate (1.2 eq) and triethylamine (5 eq) (10) or methyl 3-[(methylthio)thiocarbonyl]propanoate (1.1 eq) and triethylamine (5 eq) (11) were added. Reaction mixture was stirred for 3–5 h. For 11 1 M NaOH (6 eq) was added and stirring continued for another 2 h.
The cyclic peptide inhibitor S2iL5 was synthesized by standard Fmoc-based solid phase peptide synthesis as described by Yamagata et al.60.
All 3-nitrotyrosine containing peptides (2a, 2b, 3, 4, 4a, 5 and 6) require an additional piperidine treatment step (20% piperidine in DMF, 2 × 10 min) to remove acyl-group from 3-nitrotyrosine before cleavage.
The resin was washed with DCM (5 × 4 min), methanol (3 × 4 min) and DCM again and treated with TFA/H2O (98:2) (2 × 60 min). Combined TFA solutions were evaporated in vacuum and re-dissolved in ACN/H2O solution (50:50). HPLC purification and subsequent lyophilization yielded pure peptides.
Expression and purification of human sirtuins
Sirt1, Sirt2, Sirt3, Sirt5 and Sirt6 were expressed and purified as described before26,30,70,71.
To obtain the expression plasmid of human (His)6-SUMO-Sirt4(29–314), the respective DNA fragment was PCR-amplified using gene-specific primers from the plasmid pET101-Sirt4, which carries the Sirt4 gene, and cloned into the BsaI, XbaI sites of pE-SUMO yielding the plasmid pE-SUMO-Sirt4(29–314).
The protein was overexpressed in E. coli BL21 (DE3) cells at 18 °C. The purification of the protein was performed using affinity chromatography on Ni-NTA resin in 10 mM Tris-HCl, pH 7.5, 0.5 M NaCl. The matrix-bound (His)6-SUMO-Sirt4(29–314) was eluted by imidazole in the buffer and further purified by gel filtration in 10 mM HEPES, pH 7.8, 150 mM KCl, 1.5 mM MgCl2, and stored at −20 °C for use.
pQE-80L (Qiagen, Valencia, Ca) His-tagged Sirt6 (1–355) was transformed into the competent E. coli strain, BL21 DE3 and overexpressed at room temperature. For purification a nickel resin affinity chromatography was used in 50 mM NaPO4 pH 7.2, 250 mM NaCl, 5 mM imidazol, 1mM BME and eluted by 250 mM imidazol in the buffer. Furthermore, Sirt6 was purified secondarily via a HiTrap SP-Sepharose Fast Flow column (GE Healthcare) using a linear gradient from 50–750 mM NaCl in 50 mM NaPO4 pH 7.2, 1 mM BME. Afterwards fractions containing purified Sirt6 were pooled, concentrated and dialyzed into 50 mM Tris, pH 8.0 (4 °C), 150 mM NaCl, 100 μM TCEP and 5% (w/v) glycerol and stored at −70 °C.
HPLC based activity assay
For the determination of kinetic constants for all sirtuin mediated reactions solutions containing 20 mM TRIS/HCl pH 7.8, 150 mM NaCl, 5 mM MgCl2 (assay-buffer), 500 μM NAD+ and varying substrate concentrations (0.5–100 μM) were used. Deacylation was started by adding human sirtuin to reach a final concentration of 0.01–0.5 μM. Enzyme-catalyzed reaction was stopped using TFA (1% final concentration) after 1 min to 180 min of incubation at 37 °C depending on substrate reactivity. The cleavage rate of the different TNFα peptide derivatives was analyzed using analytical reversed phase HPLC. 40 to 80 μl of compounds or reaction solutions were injected and separated using a linear gradient from 5% to 95% solvent B within 6min. The product and substrate peaks were quantified using absorbance at 220 nm or 365 nm (absorption of 3-Nitrotyrosyl moiety). The peak areas were integrated and converted to initial velocity rates calculated from the ratio of product area to total peak area. Linear regression of conversions plotted against time yielded reaction rates in μM/min (relative conversion below 20% of substrate). Non-linear regression according to Michaelis-Menten of the reaction rates at different substrate concentrations yielded KM- and kcat-values using the program SigmaPlot 8 (Systat Software, San Jose, USA). All measurements were done in duplicates.
Continuous fluorescence assay
The fluorescence measurements were performed on a Hitachi F-4500 fluorescence spectrophotometer (Tokyo, Japan) at λEx = 310 nm and λEm = 405 nm (slitEx = 5 nm, slitEm = 2.5 nm, PMT = 700 V for 3, 4 and 5 as well as slitEx = 10 nm, slitEm = 10 nm, PMT = 950 V for 4a). Each reaction mixture contained assay-buffer, 0.5 mM NAD+ and various peptide concentrations (0.1–100 μM) and was preincubated for 5 minutes at 37 °C. The reaction was started by adding human sirtuin (0.1–0.5 μM) and observed for 5–10 minutes. Product formation could be monitored by increase of relative fluorescence. This signal was converted into product concentration via calibration lines. The slope of the linear regression of product formation against time yielded the reaction velocity rates in μM/s. KM and kcat were obtained by non-linear regression according to Michaelis-Menten. All measurements were done in duplicates. For determination of reaction velocity rates in μM/s calibration lines were necessary. Therefore a reaction mixture was prepared, containing assay-buffer, 2 μM Sirt2, 500 μM NAD+ and 100 μM of 3, 4, 4a or 5 was incubated overnight at 37 °C. The reaction mixture was analyzed with HPLC, to control if the entire peptide substrate was turned to product. Additionally the mixture was diluted (0.1–25 μM) and measured with Hitachi F-4500 fluorescence spectrophotometer at the same conditions as described above.
The microtiter plate fluorescence measurements were performed on a Tecan Infinite M200 microplate reader (Maennedorf, Switzerland) at λEx = 320 nm and λEm = 408 nm (lag time 9 μs, integration time 20 μM, gain 160, 170 or 182). The reactions (total volume 100 μl) were measured in black low-binding 96- well microtiter plates (NUNC). Assay-buffer, 500 μM NAD+ and 0.07–200 μM peptide substrate were pre-incubated at 37 °C for 5 min. The reaction was started by adding human sirtuin (0.01–0.5 μM). The signals were converted into product concentration via calibration lines and the resulting data were evaluated as described above (single fluorescence measurement). The determination for the kinetic constants of NAD+ was performed in the same way, except that the peptide concentration was fixed (5, 25 or 200 μM) and the NAD+ concentration was varied (10–1500 μM). All measurements were done in duplicates. The reaction mixture for the calibration lines was prepared as described for the single fluorescence measurements. After complete turnover of peptide substrate 3, the solution was diluted (0.2–20 μM) and measured on Tecan infinite M200 microplate reader at λEx = 320 nm and λEm = 408 nm (lag time 9 μs, integration time 20 μM, gain 182 (G182), 170 (G170) and 160 (G160)) Ki values of the inhibitors were determined by recording kcat and KM values for 3 in the presence of varying inhibitor concentrations (0.01–600 μM). The resulting plots were analyzed by a competitive inhibition and non-competitive inhibition model using the program Sigma Plot 8. The linear regression of the apparent KM-values against the corresponding inhibitor concentration yielded the inhibitor constant Ki for competitive inhibition. The Ki for non-competitive inhibition was determined by linear regression of 1/apparent Vmax against the corresponding inhibitor concentration. The negative Ki value can be determined as intersection with the X-axis from these plots.
Photo-induced change of cis content of thioxo peptides
Excitation experiments of thioxo peptides were done in a cuvette at 254 nm under stirring with a UV-lamp (UV handheld lamp, Carl Roth). For irradiation a distance of 5 cm between cuvette and UV-lamp was chosen. UV-spectra were recorded between 230 and 325 nm using a spectrophotometer (Specord M500).
For determination of temperature dependent cis/trans isomerization a 50 μM solution of thioxo peptide was incubated for 10 min at different temperatures (10–70 °C). UV-spectra were recorded at ground state (GS) and after irradiation at 254 nm (irradiation time 45 s to 5 min) at photostationary state (PSS). Several UV-spectra over time were recorded to determine rates of cis/trans isomerization. Using a differential spectrum (UV spectrum GS – UV spectrum PSS) activation parameter and isomerization velocity could be examined.
The cis/trans content of a 50 μM thioxo peptide solution was changed by 5 min irradiation at 254 nm and the resulting solution was analysed by HPLC. Additionally, several solvents were tested to enhance cis content. As solvents H2O, acetic acid, TFA, trifluoro ethanol (TFE), 0.5 M LiCl in H2O/ethanol (EtOH)/TFE, methanol (MeOH), formic acid, N-methyl pyrolidon (NMP), DMF, Dimethylsulfoxid (DMSO) and tetrahydrofuran (THF) were chosen. Cis content was determined via HPLC of a 500 μM solution of 9.
For the separation of isomers, 5–6 mg of 9 were dissolved in 50% ACN and equilibrated overnight. For better separation HPLC-solvents were cooled down to 4 °C and a linear gradient of 45% solvent B to 55% solvent B in 70 min was used. Eluted fractions were immediately frozen in liquid nitrogen. HPLC-based determination of cis content was done directly after preparative separation.
The examination of the isomer specific inhibition of 8, 9, 10 and 11 was examined via HPLC using reaction solutions composed of 500 μM NAD+, 30 μM peptide, 0.5 μM sirtuin and 0.5–40 μM inhibitor in GS, PSS or GS* in assay buffer. After 30 min incubation at 20 °C reaction was stopped using 10% TFA solution. Inhibitor solutions were irradiated at 254 nm for 5 min. Separated isomers were applied in concentrations from 1 to 10 μM. The influence of cis content on sirtuin inhibition using fluorescence spectrometer was determined with 500 μM NAD+, 5 μM 3, 0.1 μM Sirt2 and 0.1 μM 9. Reactions were done at 20 °C with 9 in GS and PSS (after 5 min irradiation at 254 nm). 9 in PSS was applied immediately after irradiation (transfer time ~5 s) and started directly by adding sirtuin. Reactions were measured within 1 min to avoid re-isomerization.
Z’ factor analysis
The Z′ factor is a dimensionless, simple statistic parameter for high-throughput screening assays72. It is defines as the ratio of separation band to signal dynamic range of the assay and used the signal variation at the two extremes of the activity range (0 and 100% activity).
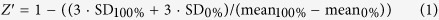 |
Additional Information
How to cite this article: Schuster, S. et al. A continuous sirtuin activity assay without any coupling to enzymatic or chemical reactions. Sci. Rep. 6, 22643; doi: 10.1038/srep22643 (2016).
Supplementary Material
Acknowledgments
We would like to thank the proteomics facility of the Martin-Luther University Halle-Wittenberg, especially Dr. Angelika Schierhorn and Christina Gersching, for recording of the mass spectra. We thank Norbert Grillenbeck and Barbara Korge for technical assistance. We thank Maximilian Buettner and Dr. Miriam Arbach for critical reading of the manuscript. This study was funded by the BMBF research grant ProNet-T3.
Footnotes
The authors declare no competing financial interests.
Author Contributions M.S., S.S. and C.R. designed the assay and wrote the manuscript. S.S., C.R. and M.M. wrote supplementary information. S.S., M.M. and P.Z. synthesized peptides. S.S. and C.R. performed kinetic analysis with HPLC-based assay. S.S. and Z.S. performed kinetic measurements with direct fluorescence assay. P.Z. and C.R. performed photo-induced cis/trans isomerization of thioxo peptides. P.Z. and C.R. expressed and purified Sirt5, C.K. and C.S. Sirt1, Sirt2, Sirt3 and Sirt6. C.S.-F. performed expression and purification of Sirt4 and M.O.H. of Sirt7.
References
- Choudhary C., Weinert B. T., Nishida Y., Verdin E. & Mann M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat Rev Mol Cell Biol 15, 536–550 (2014). [DOI] [PubMed] [Google Scholar]
- Zhao S. et al. Regulation of cellular metabolism by protein lysine acetylation. Science 327, 1000–1004 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y. et al. Lysine propionylation and butyrylation are novel post-translational modifications in histones. Mol Cell Proteomics 6, 812–819 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrity J., Gardner J. G., Hawse W., Wolberger C. & Escalante-Semerena J. C. N-lysine propionylation controls the activity of propionyl-CoA synthetase. J Biol Chem 282, 30239–30245 (2007). [DOI] [PubMed] [Google Scholar]
- Zhang Z. et al. Identification of lysine succinylation as a new post-translational modification. Nat Chem Biol 7, 58–63 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z. et al. Lysine succinylation and lysine malonylation in histones. Mol Cell Proteomics 11, 100–107 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng C. et al. The first identification of lysine malonylation substrates and its regulatory enzyme. Mol Cell Proteomics 10, M111 012658 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan M. et al. Lysine glutarylation is a protein posttranslational modification regulated by SIRT5. Cell Metab 19, 605–617 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan M. et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 146, 1016–1028 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai L. et al. Lysine 2-hydroxyisobutyrylation is a widely distributed active histone mark. Nat Chem Biol 10, 365–370 (2014). [DOI] [PubMed] [Google Scholar]
- Moellering R. E. & Cravatt B. F. Functional lysine modification by an intrinsically reactive primary glycolytic metabolite. Science 341, 549–553 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson F. T., Bursten S. L., Fanton C., Locksley R. M. & Lovett D. H. The 31-kDa precursor of interleukin 1 alpha is myristoylated on specific lysines within the 16-kDa N-terminal propiece. Proc Natl Acad Sci USA 90, 7245–7249 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simic Z., Weiwad M., Schierhorn A., Steegborn C. & Schutkowski M. The epsilon-Amino Group of Protein Lysine Residues Is Highly Susceptible to Nonenzymatic Acylation by Several Physiological Acyl-CoA Thioesters. Chembiochem 16, 2337–2347 (2015). [DOI] [PubMed] [Google Scholar]
- Wagner G. R. & Payne R. M. Widespread and enzyme-independent Nepsilon-acetylation and Nepsilon-succinylation of proteins in the chemical conditions of the mitochondrial matrix. J Biol Chem 288, 29036–29045 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert B. T. et al. Acetyl-phosphate is a critical determinant of lysine acetylation in E. coli. Mol Cell 51, 265–272 (2013). [DOI] [PubMed] [Google Scholar]
- Kuhn M. L. et al. Structural, kinetic and proteomic characterization of acetyl phosphate-dependent bacterial protein acetylation. PLoS One 9, e94816 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avalos J. L., Boeke J. D. & Wolberger C. Structural basis for the mechanism and regulation of Sir2 enzymes. Mol Cell 13, 639–648 (2004). [DOI] [PubMed] [Google Scholar]
- Du J. et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 334, 806–809 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J. et al. SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol Cell 50, 919–930 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman J. L., Baeza J. & Denu J. M. Activation of the protein deacetylase SIRT6 by long-chain fatty acids and widespread deacylation by mammalian sirtuins. J Biol Chem 288, 31350–31356 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H. et al. SIRT6 regulates TNF-alpha secretion through hydrolysis of long-chain fatty acyl lysine. Nature 496, 110–113 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathias R. A. et al. Sirtuin 4 is a lipoamidase regulating pyruvate dehydrogenase complex activity. Cell 159, 1615–1625 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao X. et al. Identification of ‘erasers’ for lysine crotonylated histone marks using a chemical proteomics approach. Elife 3, e02999 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng Y. B. et al. Efficient demyristoylase activity of SIRT2 revealed by kinetic and structural studies. Sci Rep 5, 8529 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B., Hu J., Zhang X. & Lin H. Thiomyristoyl peptides as cell-permeable Sirt6 inhibitors. Org Biomol Chem 12, 7498–7502 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moniot S., Schutkowski M. & Steegborn C. Crystal structure analysis of human Sirt2 and its ADP-ribose complex. J Struct Biol 182, 136–143 (2013). [DOI] [PubMed] [Google Scholar]
- Haigis M. C. & Guarente L. P. Mammalian sirtuins—emerging roles in physiology, aging, and calorie restriction. Genes Dev 20, 2913–2921 (2006). [DOI] [PubMed] [Google Scholar]
- Schutkowski M., Fischer F., Roessler C. & Steegborn C. New assays and approaches for discovery and design of Sirtuin modulators. Expert Opin Drug Discov 9, 183–199 (2014). [DOI] [PubMed] [Google Scholar]
- Hubbard B. P. et al. Evidence for a common mechanism of SIRT1 regulation by allosteric activators. Science 339, 1216–1219 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertz M. et al. A molecular mechanism for direct sirtuin activation by resveratrol. PLoS One 7, e49761 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y. et al. A mini-review on Sirtuin activity assays. Biochem Biophys Res Commun 3, 459–466 (2015). [DOI] [PubMed] [Google Scholar]
- Fan Y. & Scriba G. K. Electrophoretically mediated microanalysis assay for sirtuin enzymes. Electrophoresis 31, 3874–3880 (2010). [DOI] [PubMed] [Google Scholar]
- Tanner K. G., Landry J., Sternglanz R. & Denu J. M. Silent information regulator 2 family of NAD- dependent histone/protein deacetylases generates a unique product, 1-O-acetyl-ADP-ribose. Proc Natl Acad Sci USA 97, 14178–14182 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roessler C. et al. Chemical probing of the human sirtuin 5 active site reveals its substrate acyl specificity and Peptide-based inhibitors. Angew Chem Int Ed Engl 53, 10728–10732 (2014). [DOI] [PubMed] [Google Scholar]
- Fischer F. et al. Sirt5 deacylation activities show differential sensitivities to nicotinamide inhibition. PLoS One 7, e45098 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rye P. T., Frick L. E., Ozbal C. C. & Lamarr W. A. Advances in label-free screening approaches for studying sirtuin-mediated deacetylation. J Biomol Screen 16, 1217–1226 (2011). [DOI] [PubMed] [Google Scholar]
- Marcotte P. A. et al. Fluorescence assay of SIRT protein deacetylases using an acetylated peptide substrate and a secondary trypsin reaction. Anal Biochem 332, 90–99 (2004). [DOI] [PubMed] [Google Scholar]
- Smith B. C., Hallows W. C. & Denu J. M. A continuous microplate assay for sirtuins and nicotinamide-producing enzymes. Anal Biochem 394, 101–109 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfson N. A., Pitcairn C. A., Sullivan E. D., Joseph C. G. & Fierke C. A. An enzyme-coupled assay measuring acetate production for profiling histone deacetylase specificity. Anal Biochem 456, 61–69 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegener D., Wirsching F., Riester D. & Schwienhorst A. A fluorogenic histone deacetylase assay well suited for high-throughput activity screening. Chem Biol 10, 61–68 (2003). [DOI] [PubMed] [Google Scholar]
- Baba R., Hori Y., Mizukami S. & Kikuchi K. Development of a fluorogenic probe with a transesterification switch for detection of histone deacetylase activity. J Am Chem Soc 134, 14310–14313 (2012). [DOI] [PubMed] [Google Scholar]
- Minoshima M., Matsumoto T. & Kikuchi K. Development of a fluorogenic probe based on a DNA staining dye for continuous monitoring of the histone deacetylase reaction. Anal Chem 86, 7925–7930 (2014). [DOI] [PubMed] [Google Scholar]
- Han Y. et al. Time-Resolved Luminescence Biosensor for Continuous Activity Detection of Protein Acetylation-Related Enzymes Based on DNA-Sensitized Terbium(III) Probes. Anal Chem 87, 9179–9185 (2015). [DOI] [PubMed] [Google Scholar]
- Dhara K., Hori Y., Baba R. & Kikuchi K. A fluorescent probe for detection of histone deacetylase activity based on aggregation-induced emission. Chem Commun (Camb) 48, 11534–11536 (2012). [DOI] [PubMed] [Google Scholar]
- Wang Y., Chen Y., Wang H., Cheng Y. & Zhao X. Specific Turn-On Fluorescent Probe with Aggregation-Induced Emission Characteristics for SIRT1 Modulator Screening and Living-Cell Imaging. Anal Chem 10, 5046–5049 (2015). [DOI] [PubMed] [Google Scholar]
- Liu Y., Gerber R., Wu J., Tsuruda T. & McCarter J. D. High-throughput assays for sirtuin enzymes: a microfluidic mobility shift assay and a bioluminescence assay. Anal Biochem 378, 53–59 (2008). [DOI] [PubMed] [Google Scholar]
- Suenkel B., Fischer F. & Steegborn C. Inhibition of the human deacylase Sirtuin 5 by the indole GW5074. Bioorg Med Chem Lett 23, 143–146 (2013). [DOI] [PubMed] [Google Scholar]
- Hubbard B. P. & Sinclair D. A. Measurement of sirtuin enzyme activity using a substrate-agnostic fluorometric nicotinamide assay. Methods Mol Biol 1077, 167–177 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howitz K. T. et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 425, 191–196 (2003). [DOI] [PubMed] [Google Scholar]
- Madsen A. S. & Olsen C. A. Substrates for efficient fluorometric screening employing the NAD-dependent sirtuin 5 lysine deacylase (KDAC) enzyme. J Med Chem 55, 5582–5590 (2012). [DOI] [PubMed] [Google Scholar]
- Hu J., He B., Bhargava S. & Lin H. A fluorogenic assay for screening Sirt6 modulators. Org Biomol Chem 11, 5213–5216 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeberlein M. et al. Substrate-specific activation of sirtuins by resveratrol. J Biol Chem 280, 17038–17045 (2005). [DOI] [PubMed] [Google Scholar]
- Pacholec M. et al. SRT1720, SRT2183, SRT1460, and resveratrol are not direct activators of SIRT1. J Biol Chem 285, 8340–8351 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Huang W., You L., Xie T. & He B. A FRET-based assay for screening SIRT5 specific modulators. Bioorg Med Chem Lett 25, 1671–1674 (2015). [DOI] [PubMed] [Google Scholar]
- Li Y. et al. A FRET-based assay for screening SIRT6 modulators. Eur J Med Chem 96, 245–249 (2015). [DOI] [PubMed] [Google Scholar]
- Roessler C., Tuting C., Meleshin M., Steegborn C. & Schutkowski M. A Novel Continuous Assay for the Deacylase Sirtuin 5 and Other Deacetylases. J Med Chem 58, 7217–7223 (2015). [DOI] [PubMed] [Google Scholar]
- Feldman J. L. et al. Kinetic and Structural Basis for Acyl-Group Selectivity and NAD Dependence in Sirtuin-Catalyzed Deacylation. Biochemistry 19, 3037–3050 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokkonen P. et al. Studying SIRT6 regulation using H3K56 based substrate and small molecules. Eur J Pharm Sci 63, 71–76 (2014). [DOI] [PubMed] [Google Scholar]
- Smith B. C. & Denu J. M. Acetyl-lysine analog peptides as mechanistic probes of protein deacetylases. J Biol Chem 282, 37256–37265 (2007). [DOI] [PubMed] [Google Scholar]
- Yamagata K. et al. Structural basis for potent inhibition of SIRT2 deacetylase by a macrocyclic peptide inducing dynamic structural change. Structure 22, 345–352 (2014). [DOI] [PubMed] [Google Scholar]
- Fatkins D. G., Monnot A. D. & Zheng W. Nepsilon-thioacetyl-lysine: a multi-facet functional probe for enzymatic protein lysine Nepsilon-deacetylation. Bioorg Med Chem Lett 16, 3651–3656 (2006). [DOI] [PubMed] [Google Scholar]
- Smith B. C. & Denu J. M. Mechanism-based inhibition of Sir2 deacetylases by thioacetyl-lysine peptide. Biochemistry 46, 14478–14486 (2007). [DOI] [PubMed] [Google Scholar]
- Fatkins D. G. & Zheng W. Substituting N(epsilon)-thioacetyl-lysine for N(epsilon)-acetyl-lysine in peptide substrates as a general approach to inhibiting human NAD(+)-dependent protein deacetylases. Int J Mol Sci 9, 1–11 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loving G. & Imperiali B. A versatile amino acid analogue of the solvatochromic fluorophore 4-N,N-dimethylamino-1,8-naphthalimide: a powerful tool for the study of dynamic protein interactions. J Am Chem Soc 130, 13630–13638 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J., Micheau J. C., Vargas C. & Schiene-Fischer C. cis/trans photoisomerization of secondary thiopeptide bonds. Chemistry 10, 6093–6101 (2004). [DOI] [PubMed] [Google Scholar]
- Wildemann D. et al. A nearly isosteric photosensitive amide-backbone substitution allows enzyme activity switching in ribonuclease s. J Am Chem Soc 129, 4910–4918 (2007). [DOI] [PubMed] [Google Scholar]
- Frank R., Jakob M., Thunecke F., Fischer G. & Schutkowski M. Thioxylation as One-Atom-Substitution Generates a Photoswitchable Element within the Peptide Backbone We thank Dr. Peter Bayer for the NMR investigations and Dirk Wildemann for his assistance with the peptide syntheses. This work was supported by the Deutsche Forschungsgemeinschaft, the Fond der Chemischen Industrie, the Boehringer-Ingelheim-Stiftung, and the Land Sachsen-Anhalt. Angew Chem Int Ed Engl 39, 1120–1122 (2000). [PubMed] [Google Scholar]
- Leon N. H. New compounds: synthesis of carboxymethyl carbodithioates. J Pharm Sci 65, 146–148 (1976). [DOI] [PubMed] [Google Scholar]
- Gubbens J. et al. Photocrosslinking and click chemistry enable the specific detection of proteins interacting with phospholipids at the membrane interface. Chem Biol 16, 3–14 (2009). [DOI] [PubMed] [Google Scholar]
- Schlicker C., Boanca G., Lakshminarasimhan M. & Steegborn C. Structure-based development of novel sirtuin inhibitors. Aging (Albany NY) 3, 852–872 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu D. et al. A SIRT7-dependent acetylation switch of GABPbeta1 controls mitochondrial function. Cell Metab 20, 856–869 (2014). [DOI] [PubMed] [Google Scholar]
- Zhang J. H., Chung T. D. & Oldenburg K. R. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen 4, 67–73 (1999). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.