

Abstract
SecA, a key component of bacterial Sec-dependent secretion pathway, is an attractive target for novel antimicrobial development. Through a combination of virtual screening and experimental exploration of surrounding chemical space, we identified a hit bistriazole SecA inhibitor, SCA-21, and studied a series of analogs by systematic dissections of the core scaffold. Evaluation of these analogs allowed us to establish an initial SAR in SecA inhibition. The best compounds in this group have potent inhibition activity of SecA-dependent protein-conducting channel activity and protein translocation activity at low to sub-μM concentrations. They also have MIC values against various strains of bacteria that are correlated with the SecA and protein translocation inhibition data. These compounds are effective against methicillin-resistant Staplylococcus aureus strains with various levels of efflux pumps, indicating the ability to null the effect of multiple-drug resistance with SecA inhibitors. Results from studies of drug affinity responsive target stability and protein pull-down assays are consistent with SecA as a target for these compounds.
Keywords: triazole, pyrimidine, SecA inhibitors, antimicrobials, protein secretion
Graphical Abstract
1. Introduction
Because of drug resistance issues, bacterial pathogens have re-emerged as a serious public health concern.[1] Thus, there is an urgent need to develop new antimicrobials, especially those with novel mechanisms of action. Common mechanisms of antibiotics include inhibition of essential enzymatic processes in the bacterium to interrupt replication, transcription, translation, or cell wall synthesis.[2] Recently, SecA, which is a key enzyme in protein translocation,[3] has drawn interests in antimicrobial development[4] because of its unique role in bacterial survival, virulence factor secretion, and membrane integration of certain efflux pumps, which are responsible for multi-drug resistance.[5]
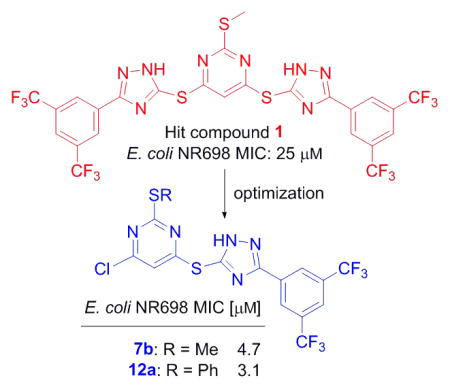
In both Gram-positive and Gram-negative bacteria, the majority of secretory proteins, including virulence factors, are translocated across the cytoplasmic membrane through the Sec-dependent pathway. SecA is the central component of Sec-dependent secretion pathway,[3e, 6] interacting with virtually all the other components of the system, acting as a molecular chaperone and motor, and providing the driving force for protein translocation.[7] In addition, SecA has been shown to form a protein-conducting channel in the presence of phospholipid bilayers.[8] SecA is required for bacterial viability and virulence, and is highly conserved in bacteria with no human counterpart.[9] Therefore, targeting SecA represents a unique strategy to combat bacterial pathogens with minimal human toxicity. In our previous studies, pyrimidine analogs were developed from virtual screening against an EcSecA crystal structure.[10] Through screening of related compounds, we have identified one bistriazole compound 1 (Fig. 1, SCA-21), which inhibits both the intrinsic ATPase activity and translocation ATPase activity of EcSecA, with an IC50 of about 30 μM. Through dissecting this hit, we designed and synthesized about 40 analogs, among which some (such as 7b and 12a) were found to potently inhibit SecA-dependent activities at high nM to low μM concentrations in an in vitro (vesicle) protein translocation study and an ion-channel oocyte assay. The inhibition of these functional activities correlates with the inhibition of bacterial growth.
Figure 1.

Optimization of the hit compound SCA-21
2. Results and Discussion
2.1 Chemistry
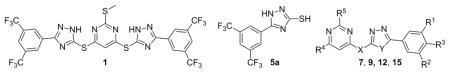
SCA-21 has a pyrimidine core conjugated to two substituted triazoles in a symmetric fashion via a thioether bond. It should be noted that these triazoles are 1,2,4-triazoles, which are different from the 1,2,3-triazoles synthesized through a [2+3] cycloadditon reaction between an organic azido compound and an alkyne. For improving its potency and understanding its SAR, we started to simplify the structure by dissecting the hit compound in half and removing part A. Figure 2 shows a general strategy of analog design. Specifically, the plan was to examine substitution effects in scaffold I, positional effect in scaffold IIA, the importance of the triazole ring in scaffold IIB, and the effect of conformational constraints around the triazole and pyrimidine axis in scaffold III.
Figure 2. Screening for inhibition of the ATPase activity of EcSecAN68.

The initial screening assays were carried out at 50 μM for all compounds.
For scaffold I, we were interested in modifying it by changing six different groups (R1, R2, R3, R4, R5 and X) (Fig. 1). Synthesis started with reacting commercially available benzoyl chloride 2 with hydrazinecarboamide 3, followed by self-condensation of 4 in the presence of 5% sodium hydroxide under reflux conditions to yield 5 (Scheme 1).[11] Subsequently, the triazole-pyrimidine analogs 7, 9, 12, and 15 were synthesized by reacting key intermediate 5 under weakly basic conditions with the appropriately substituted pyrimidines (Scheme 2–3).
Scheme 1.

a) THF, 0 ºC ~rt, overnight; b) 5% NaOH, reflux 5 h, 65–87% overall yields
Scheme 2.

a) K2CO3, acetone, rt, 2–3h, 35%–80%.
Scheme 3.

a) CS2, H2O/EtOH, reflux 5 h, 65%; b) K2CO3, acetone, rt, 2– 3 h, 80%.
Thus compounds 7, 9 and 12 were synthesized as described in Scheme 2. Series 7 has a methylthio ether at the 2-position, whereas series 9 does not. In series 12, the methylthio ether was replaced by either a larger thioether or an oxoether (Scheme 2).
Scheme 3 describes the synthesis of compound 15, which has scaffold I, but with an oxadiazole instead of a triazole linking the pyrimidine and the phenyl rings. Compound 15 was synthesized by first reacting commercially available 3, 5-bis(trifluoromethyl) benzohydrazide 13 with carbon disulfide to give 14.[12] Then reaction of 14 with 4, 6-dichloro-2-(methylthio)pyrimidine 6b, under weakly basic conditions (Scheme 2), gave compound 15.[13]
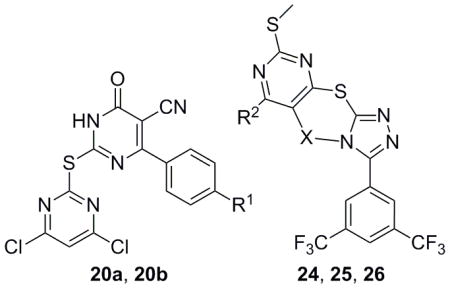
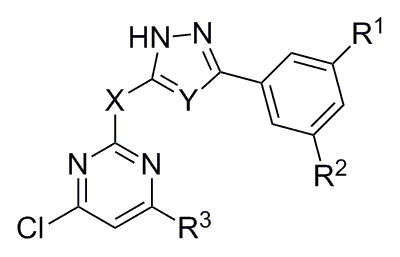
Analogs in scaffold II (Fig. 1) were designed to evaluate the effect of regioisomerism, i.e. whether the substitution is at 2 or 4 position of the pyrimidine ring (compounds 16, 17 and 18) as well as the importance of the triazole ring (compound 20). Scheme 4 describes the synthesis of compounds 16, 17, 18 and 20. Specifically, they were synthesized by the reaction of intermediate 5, 14 or 19 with 2, 4, 6-trichloro pyrimidine compounds under weakly basic conditions at room temperature (Scheme 4).[12]
Scheme 4.

a) K2CO3, acetone, rt, 2–3 h, 35%–80%.
Analogs in Class III (Fig. 1) were designed to examine the importance of conformational constraints, especially around the triazole and pyrimidine axis. Compound 22 was synthesized from commercially available 3, 5-bis(trifluoromethyl) benzohydrazide 21 by reaction with carbon disulfide under strongly basic conditions (Scheme 5).[12] Then reaction of 22 with 4, 6-dichloro-2-(methylthio) pyrimidine-5-carbaldehyde 23, gave compound 24. Compounds 25 and 26 were synthesized by reduction of 24 with varying equivalents of sodium borohydride at room temperature.[14]
Scheme 5.

a) CS2, NH2NH2, KOH/EtOH, 82%; b) DMF, 60 ºC, 4 h, 76%; c) NaBH4 (1 eq), EtOH, rt, 2 h, 82%; d) NaBH4 (10 eq), EtOH, rt, 2 h, 40%.
2.2 Biological evaluation
We first evaluated the inhibitory effect against the intrinsic ATPase activity of EcSecA N68, which is a truncated protein of E. coli SecA that lacks the inhibitory/regulatory C-terminus.[10b, 15] In previous publications, we discussed in detail the pros and cons of various assays in screening for SecA inhibition activities and the need to use more than one assay for validations.[4c, 16] Briefly, we recognize that this is not an ideal assay because EcSecA N68 may very well have a different conformation as compared to the membrane bound form in live bacteria, and the E. coli form may not represent the SecA from both Gram-positive and Gram-negative species. However, this assay allows for rapid screening of a large number of compounds and all the other assays are of low throughput.[10b, 15] In addition, we also used antimicrobial assays as a gate-keeper to make sure that we examine all compounds with potent antimicrobial activities, no matter what the results were from the EcSecA N68 assay. We used two representative strains for antimicrobial assays: a Gram-positive strain (Staphylococcus aureus 6538) and a Gram-negative strain (E. coli NR698)[4c, 10b, 17] with an outer membrane mutation resulting in increased drug permeability. Thus initial screening was conducted at 50 μM (Fig. 2). There were twenty two compounds that showed more than 50% inhibition at this concentration. Then these compounds were evaluated further at various concentrations to allow determination of IC50 values (Table 1–3). Several compounds (7b and 12a) from Class I showed significant inhibition of enzyme activities at low μM concentrations. It should be noted that the analog with pyrimidine ring (5a) removed showed reduced activity. Such results suggested that both triazole and pyrimidine rings are very important for inhibition activity. Changing the substitution position of triazole ring to the 2-position (Class II, Fig. 2) decreases the inhibition activity and conformationally constrained analogs (Class III, Fig. 2) did not yield active compounds either.
Table 1.
SAR tests for Class I compounds.
| |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Comp | SCA# | R1 | R2 | R3 | X | R4 | R5 | Y | IC50 (μM), EcSecAN68 | MIC (μM), E. coli NR698 | MIC (μM), S. aureus 6538 |
| 1 | SCA21 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | 25.0 | 25.0 | 12.5 |
| 5a | SCA126 | -CF3 | -CF3 | -H | -SH | N/A | N/A | -NH | 60.0 | >250.0 | >250.0 |
| 7a | SCA128 | -CF3 | -CF3 | -H | -S- | -CH3 | -SCH3 | -NH- | 25.0 | 33.3 | 18.8 |
| 7b | SCA107 | -CF3 | -CF3 | -H | -S- | -Cl | -SCH3 | -NH- | 30.0 | 4.7 | 3.1 |
| 7c | SCA123 | -CF3 | -CF3 | -H | -S- | -H | -SCH3 | -NH- | 95.0 | 50.0 | 62.5 |
| 7d | SCA117 | -CH3 | -CH3 | -H | -S- | -H | -SCH3 | -NH- | 65.0 | >250.0 | >250.0 |
| 7e | SCA106 | -CH3 | -CH3 | -H | -S- | -Cl | -SCH3 | -NH- | 30.0 | 50.0 | 50.0 |
| 7f | SCA153 | -F | -H | -H | -S- | -Cl | -SCH3 | -NH- | <50.0 | 200.0 | 175.0 |
| 7g | SCA155 | -F | -F | -H | -S- | -Cl | -SCH3 | -NH- | >100.0 | 50.0 | 41.6 |
| 7h | SCA159 | -OCH3 | -OCH3 | -H | -S- | -Cl | -SCH3 | -NH- | <50.0 | 100.0 | >250.0 |
| 7i | SCA161 | -H | -H | -CF3 | -S- | -Cl | -SCH3 | -NH- | <50.0 | 25.0 | 14.6 |
| 7j | SCA157 | -CF3 | -H | -H | -S- | -Cl | -SCH3 | -NH- | <50.0 | >250.0 | >250.0 |
| 9a | SCA111 | -CH3 | -CH3 | -H | -S- | -Cl | -H | -NH- | >200.0 | >250.0 | >250.0 |
| 9b | SCA110 | -CF3 | -CF3 | -H | -S- | -Cl | -H | -NH- | 100.0 | 35.4 | 18.8 |
| 12a | SCA112 | -CF3 | -CF3 | -H | -S- | -Cl | -SPh | -NH- | 20.0 | 3.1 | 1.8 |
| 12b | SCA113 | -CH3 | -CH3 | -H | -S- | -Cl | -SPh | -NH- | 50.0 | >250 | 12.5 |
| 12c | SCA114 | -CF3 | -CF3 | -H | -S- | -Cl | -OPh | -NH- | 9.0 | 8.3 | 3.1 |
| 12d | SCA130 | -CF3 | -CF3 | -H | -S- | -Cl | -S(CH2)4CH3 | -NH- | 50.0 | 6.3 | 1.6 |
| 12e | SCA138 | -CF3 | -CF3 | -H | -S- | -Cl | -S(CH2)4OH | -NH- | 25.0 | 43.8 | 25.0 |
| 12f | SCA156 | -F | -F | -H | -S- | -Cl | -SPh | -NH- | <50.0 | 31.3 | 20.8 |
| 12g | SCA158 | -CF3 | -H | -H | -S- | -Cl | -SPh | -NH- | <50.0 | 18.8 | 18.8 |
| 12h | SCA160 | -OCH3 | -OCH3 | -H | -S- | Cl | -SPh | -NH- | <50.0 | >250 | ≥100.0 |
| 12i | SCA162 | -H | -H | -CF3 | -S- | -Cl | -SPh | -NH- | <50.0 | 12.5 | 8.3 |
| 12j | SCA163 | -OCH3 | -H | -OH | -S- | -Cl | -SPh | -NH- | <50.0 | >250 | ≥250.0 |
| 12k | SCA154 | -F | -H | -H | -S- | -Cl | -SPh | -NH- | <50 | 83.3 | 175.0 |
| 15 | SCA133 | -CF3 | -CF3 | -H | -S- | -Cl | -SCH3 | -O- | 200 | >100 | >250.0 |
IC50: the concentration of compound which inhibit 50% of the ATPase activity of EcSecAN68. MIC: the lowest concentration of compound that inhibited the growth of bacteria after 24 hr incubation at 37 °C. E. coli NR698 (with an outer membrane mutation resulting in increased drug permeability) and S. aureus 6538 representing Gram-negative and Gram-positive bacteria to screen all compounds;
Table 3.
SAR tests for Class IIB & III compounds.
| |||||||
|---|---|---|---|---|---|---|---|
| Comp. | SCA# | R1 | Y | R2 | IC50 (μM), EcSecAN68 | MIC (μM), E coli NR698 | MIC (μM), S. aureus 6538 |
| 20a | SCA127 | Br | N/A | N/A | <50.0 | >250.0 | >250.0 |
| 20b | SCA137 | Et | N/A | N/A | <50.0 | >250.0 | >250.0 |
| 24 | SCA174 | N/A | -CH=N- | -Cl | <25.0 | 12.5 | 3.1 |
| 25 | SCA177 | N/A | -CH2-NH- | -Cl | 46.0 | >250.0 | >100.0 |
| 26 | SCA175 | N/A | -CH2-NH- | -H | 50.0 | >100.0 | >100.0 |
IC50: the concentration of compound which inhibit 50% of the ATPase activity of EcSecAN68. MIC: the lowest concentration of compound that inhibited the growth of bacteria after 24 hr incubation at 37°C
For Class I compounds, adding a chloro group at R4 position significantly increased inhibitory effect (7b, Table 1). Replacing one or both trifluro methyl groups at R1 and R2 positions with any other groups (7f–7i, Table 1) significantly decreased the bacteriostatic effect, suggesting that the two trifluoromethyl groups are important for inhibition. Changing triazole to oxadiazole in the structure decreased inhibition greatly, suggesting that the triazole group is very important (15, Table 1). Replacing methylthio group with hydrogen decreased inhibition effect and the methylthio group could be replaced with a phenylthio (9b, Table 1) or a phenoxy group (12c, Table 2) without significantly changing their inhibition effects.
Table 2.
SAR tests for Class IIA compounds.
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Comp. | SCA# | R1 | R2 | Y | X | R3 | IC50 (μM), EcSecAN68 | MIC (μM), E coli NR698 | MIC (μM), S. aureus 6538 |
| 16a | SCA124 | -CF3 | -CF3 | -NH- | -S- | -Cl | 50.0 | 12.5 | 6.3 |
| 16b | SCA116 | -CH3 | -CH3 | -NH- | -S- | -Cl | >50.0 | >164.0 | 100.0 |
| 16c | SCA139 | -CF3 | -CF3 | -NH- | -O- | -Cl | <50.0 | 250.0 | 37.5 |
| 16d | SCA152 | -H | -F | -NH- | -S- | -Cl | >100.0 | 200.0 | ≥100.0 |
| 16e | SCA131 | -CF3 | -CF3 | -NH- | -S- | -NHCH2Ph | >50.0 | ≥250.0 | 50.0 |
| 17 | SCA135 | -CF3 | -CF3 | -O- | -S- | -Cl | 70.0 | >250.0 | 18.8 |
| 18 | SCA140 | -CF3 | -CF3 | -NH- | -O- |
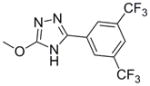
|
<50.0 | >250.0 | 250.0 |
IC50: the concentration of compound which inhibit 50% of the ATPase activity of EcSecAN68. MIC: the lowest concentration of compound that inhibited the growth of bacteria after 24 hr incubation at 37°C.
Among all the compounds, 7b (SCA-107) and 12a (SCA-112) are the two most potent analogs developed from this study. Thus we focused on these two compounds together with the original lead compound 1 for further evaluation. As discussed in previous studies,[4c, 15] inhibition effect on the soluble form of EcSecAN68 is a good initial screen. However, the inhibition potency in membrane-lipid environment using either an ion current channel activity assay or protein translocation assay seems to correlate more closely with antimicrobial activity.[8] Thus, we subjected 1, 7b, and 12a to such assays, and found that the IC50 values for 7b and 12a were around 1.5–2.0 μM in the membrane protein translocation assay using EcSecA and 2.0–4.8 μM in the SecA-liposomes translocation assay (Table 2). We also examined these SecA inhibitors using the channel activity assay in oocytes[8, 18] (Table 3). The results indicated that these compounds showed very effective inhibition of the ion-channel activity of EcSecA, with IC50 values around 1.3–2.4 μM (Table 3). We have also examined the effects of these compounds on the ion channel activity of SecA purified from other strains of bacteria and then reconstituted with liposomes or cognate membranes. Table 3 shows that these compounds were also very effective against SecA from both Gram-positive and Gram-negative bacteria with IC50 ranged from 0.7 to 2.6 μM. The inhibition potencies in the ion channel and protein translocation assays correlate very well with antimicrobial results (Tables 1–4 and the section below), once again demonstrating what we have reported earlier, i.e. (1) the EcSecAN68 assay is appropriate for the initial rapid screening and (2) the protein translocation and ion channel activity assays provide numerical values of inhibition activity that more closely parallel the antimicrobial efficacy. Corollary to these points is our examination of the effect of these three inhibitors on the full length SecA in solution (Table 4). As discussed previously,[15] the full length Sec has an inhibitory/regulatory C-terminus. In a non-membrane environment, the IC50 values of SecA inhibitors tend to be much higher than that obtained from the EcSecAN68, channel activity, and protein translocation assays. Thus the numerical IC50 values from the full-length SecA assay in solution need to be analyzed with the effect of the inhibitory/regulatory C-terminus taken into considerations.
Table 4.
Inhibition of various SecA ATPase and in vitro protein translocation activities of EcSecA
| Comp. | IC50 (μM), ATPase activity | IC50 (μM), translocation activity | |||
|---|---|---|---|---|---|
| EcSecA N68 | EcSecAa | EcSecA Tnb | Membranec | SecA-liposomesd | |
| 1 | 18.0 | 32.0 | 20.0 | 7.0 | ND |
| 7b | 30.0 | >200.0 | 28.0 | 1.5 | 2.0 |
| 12a | 20.0 | >200.0 | ND | 2.0 | 4.8 |
Intrinsic ATPase activity of EcSecA was the ATPase activity of full EcSecA without membrane and proOmpA;
translocation ATPase activity of EcSecA was the ATPase activity of EcSecA in presence of membrane (urea-washed E. coli BA13 membrane) and proOmpA;
In vitro translocation activity with membrane was determined by using OmpA-depleted and urea-washed 773 membranes and immunoblots to determine the translocation efficiency of proOmpA.
In vitro translocation activity with SecA-liposomes was determined by using reconstituted SecA-only liposomes and immunoblots to determine the translocation efficiency of proOmpA. IC50 is the concentration of the compound that inhibits 50% of activity.
Thus far, the correlation between the inhibitory potency data from the channel activity and protein translocation assays and the antimicrobial data is consistent with the notion that the antimicrobial results (Tables 1–5) are largely due to SecA inhibition. To further examine this correlation, we conducted protein pull-down work. Thus we synthesized biotinylated 7b analog (SCA-256), which was used to carry out the protein pull-down assay (Fig. 4). Enzymatic assay showed that SCA-256 maintained a reasonable level of inhibitory activity of EcSecAN68 ATPase (IC50 ≈ 60 μM). From the Western blot results, it can be seen that SCA-256 could recognize and pull down SaSecA1 from whole cell lysates (Fig. 3). Such results demonstrated the specific interactions between SecA and the inhibitors, further supporting the notion that SaSecA1 is a target of this group of inhibitors. However, as with any protein pull down work, rarely only one protein is pulled down. This is no exception with SCA-256. There are many factors that could contribute to such results and still be consistent with SecA being the key target for the antimicrobial effects of these inhibitors. Furthermore, it should be noted that the channel activity and protein translocation with SecA-only liposomes clearly showed the specific inhibition of SecA functional activity.
Table 5.
Inhibition of ion-channel activity of various SecA-liposomes in oocytes
| IC50 (μM), ion-channel activity | |||||
|---|---|---|---|---|---|
|
| |||||
| Comp. | EcSecA | SaSecA1 | BaSecA1 | BsSecA | SpSecA |
| 1 | 2.4 | 1.6 | 1.5 | 2.6 | 1.0 |
| 7b | 1.6 | 0.6 | 0.7 | 2.1 | 0.7 |
| 12a | 1.3 | 1.0 | 1.0 | 2.3 | 1.3 |
Ion-channel activity of SecA-only-liposomes was determined by adding reconstituted SecA-liposome complex into oocytes and recording the current inducted by proOmpA. SaSecA1: Staphlocucus aureus SecA1; BaSecA1: Bacillus anthraces SecA1; BsSecA: Bacillus subtilis SecA, SpSecA1: Streptococcus pyogenes SecA1.
Figure 4. Ion-channel activity against EcSecA and SaSecA1.

Ion-channel activity of SecA-liposomes was determined by adding reconstitute SecA-liposome complex into oocytes and recording the current inducted by proOmpA. EcSecA: E. coli SecA; SaSecA1: Staphlocucus aureus SecA1.
Figure 3. Validation SaSecA1 as a drug target with pull down assay. (a).
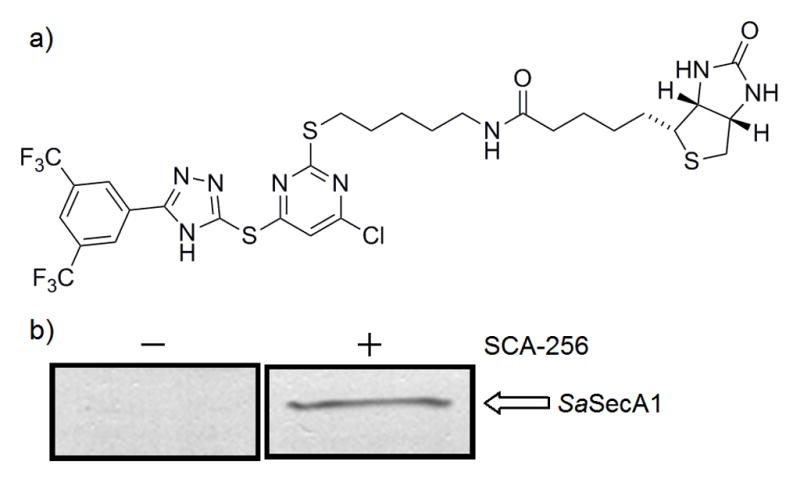
Structure of SCA-256. (b) Whole cell lysate of S. aureus ATCC 6538 was mixed with SCA-256 (biotinylated 7b analog) for 1 hr, then Streptavidin magnetic beads were used to pull out proteins interact with SCA-256. The interaction between SaSecA1 and SCA-256 was examined by Western blot with the cross-reacting EcSecA antibody (1:5000 dilution). Synthesis of SCA-256 is described in the Supplemental Information section.
To better understand the mechanism of inhibition in view of the difference on SecA ATPase activity and functional activities with lipid/membranes, we conducted inhibition kinetics studies of SecA-liposome channel activity using 7b (SCA-107) as a representative. Figure 4 shows that 7b (SCA-107) is a non-competitive inhibitor of EcSecA and SaSecA. Such results support the idea that the inhibitor binds not directly in the ATP binding pocket. Molecular modeling suggests that 7b (SCA-107) partially blocks the entry of ATP binding site (Fig. 5).
Figure 5. Molecular modeling of 7b (SCA-107) binding site in E. coli SecA. 6A.

Docking was done in SYBYL 2.0 using PDB (2FSG) with SecA dimers A and B. 7b binds to the interface of the A and B monomers, closer to A (6A), partially blocking the entrance to the ATP site. 6B: SCA-107 surrounded by amino acid residues.
To achieve an initial understanding of the possible binding site for these SecA inhibitors, docking studies were conducted using the SYBYL program. Figure 5A shows that SCA-107 resides at the interface of monomers A and B, slightly towards the A monomer; 6B shows the individual amino acid residues flanking SCA-107.
As discussed earlier, SecA is a conserved protein in bacteria. Thus there are good reasons to believe that SecA inhibitors can function as broad-spectrum antimicrobials. Our SecA inhibition data certainly support this notion. To further examine this aspect, we evaluated the inhibitory effect of 1 (SCA-21), 7b (SCA-107), and 12a (SCA-112) against various strains of bacteria (Table 4). The results show that these three inhibitors have excellent antimicrobial effects with MIC values between 0.8–6.3 μM, which are also comparable with the inhibition of the SecA-specific functions in channel activity and protein translocation assays (Table 3) and further substantiate the idea that the antimicrobial target is SecA. In addition to bacteriostatic effects, hit compound 1 also exhibit bactericidal effects with generally more than 2 log units reduction in CFU values at 25 μM against some S. aureus strains as listed in Table 7. Compound 7b decreased 2 log units of CFU of S. aureus Mu50 at 50 μM (Fig. 6).
Table 7.
Antimicrobial activities against S. aureus efflux strains
| Comp. | SCA# | WT 8325-4 | NorA− K1758 | NorA++ K2361 | MepA− K2908 | MepA++ K2068 | |
|---|---|---|---|---|---|---|---|
| Bacteriostatic effect, MIC (μM) | 1* | SCA21 | 8 | 8 | 8 | 8 | 6 |
| 7b | SCA107 | 2.1 | 3.1 | 3.1 | 2.1 | 3.1 | |
| 12a | SCA112 | 1.6 | 1.6 | 1.8 | 1.0 | 1.3 | |
|
| |||||||
| Bactericidal effect, log # of cell reduced | 1 | SCA21 | 3 | 3 | 4 | 2 | 3 |
Deletion (NorA−, MepA−) and over-expression (NorA++, MepA++) of NorA and MepA efflux pump strains, K1758, K2361, K2908, and K2068 are derivatives from S. aureus 8325-4. Bactericidal effects were determined by counting CFU after 1 hr treatment at 37°C with 25 μM.
the initial MIC tested for Compound 1 was from tube assay.
Figure 6. Bactericidal effect of 7b (SCA-107) against S. aureus Mu50.

Bactericidal effects were determined by counting CFU after 2 hr treatment with different concentration of 7b.
Some MRSA strains acquire multiple-drug resistance in addition to their resistance to methicilin. We further examined the efficacy of our inhibitors against one strain, Mu50, in comparison with clinically used antibiotics. The results show that 7b (SCA-107) (MIC: 0.75 μg/ml) and 12a (SCA-112) (MIC: 0.43 μg/ml) are more effective than most clinically used antibiotics including ampicillin (1000 μg/ml, >1300 fold), polymyxin B (31 μg/ml, >40 fold), erythromycin (1250 μg/ml, >1600 fold), tetracycline (63 μg/ml, >80 fold), kanamycin (1000 μg/ml, >1300 fold), rifampicin (1000 μg/ml, >1300 fold), norfloxacin (250 μg/ml, >330 fold), and even vancomycin (8 μg/ml, 10 fold for 7b, 18 fold for 12a), which is considered as the last resort drug for treating many of the drug-resistant strains of bacteria. These SecA inhibitors also exert bacteriacidal effects on a wide range of bacteria, including S. aureus Mu50, S. aureus N6538, and B. anthraces Sterne (Figure 6, and data not shown). We previously described two other classes of structurally different SecA inhibitors;[16b, 17b] and this novel class is the most effective in both bacteriostatic and bactericidal inhibition.
Compared against other known antimicrobial targets, one of the unique features of SecA is that this functions as a membrane protein. Thus it is possible to envision that inhibitors can access SecA without the need to accumulate in the intracellular compartment. One consequence of this ready accessibility of SecA is that efflux pumps are not expected to affect the potency of these SecA inhibitors. To assess this aspect, we examined various MRSA strains, which are known to possess multiple drug-resistance efflux pumps and thus render many drugs ineffective. The results showed that the inhibition of growth by these SecA inhibitors was independent of the level of the efflux pumps in these strains (Table 7). Such results are consistent with the notion that the SecA may be accessible by this class of inhibitors from the extracellular matrix. Thus SecA inhibitors may exert their effect without the need to accumulate in the intracellular compartment, and thus possess the ability to attenuate the effect of efflux pumps, a major mechanism for multidrug resistance.
All the evidence from inhibition of SecA-dependent protein translocation, channel activity kinetic studies, and anitimicrobial studies indicate that these SecA inhibitors exert their antimicrobial effect through inhibition of SecA functions in the membranes, and possess the ability to bypass/minimize the effects of efflux pumps.
2.3 Conclusions
In conclusion, we have synthesized and evaluated a new class of triazole-pyrimidine analogs as novel SecA inhibitors. Compounds 7b (SCA-107) and 12a (SCA-112) showed the most potent activity with IC50 and MIC values in the low to sub-μM range. In addition, results from target identification assays are consistent with SecA being a target. The inhibition of SecA-dependent channel activity and protein translocation correlates well with antimicrobial activity, further suggesting that these compounds exert their antimicrobial effect through SecA inhibition. However, this does not exclude the possibility of off-target effect in the antimicrobial assay. A unique feature of SecA is the fact that it is a widely conserved membrane protein, responsible for the secretion of virulence factors, and directly accessible from the extracellular matrix without the need for certain intraceullar concentrations. All these combined suggest that SecA inhibitors possess the potential of being broad-spectrum antimicrobials, can attenuate the pathogenicity of bacteria by directly inhibiting virulence factor production, and can overcome the effect of efflux pumps, which are responsible for multi-drug resistance.
3. Experimental Section
3.1 Chemistry
All chemical reagents used were purchased from Acros and Aldrich and used as received. 1H NMR and 13C NMR spectra were obtained on a Bruker 400 NMR spectrometer. Mass spectral analyses were performed by the Mass Spectrometry Facilities at Georgia State University. Molecular modeling work was conducted using SYBYL-X 2.0 and the standard procedures provided in the manual were used. Specifically, Surflex-dock was used to dock ligands into SecA crystal structure (PDB ID: 2FSG) using automatic docking. All standard parameters provided in SYBYL X 2.0 were employed for energy minimization of the crystal structure of SecA. AMBER7 FF99 force field along with the default parameters described in the manual was used and a maximum of 20 docking poses were generated for each molecule.
2-(3, 5-Bis(trifluoromethyl) benzoyl) hydrazinecarbothioamide (4a)
To a mixture of 3, 5-bis(trifluoromethyl) benzoyl chloride (1 g, 3.63 mmol) in 25 mL tetrahydrofuran was added hydrazinecarbothioamide (0.73 g, 7.98 mmol) slowly at 0–5 ºC. Then the temperature was allowed to go up to room temperature and the reaction was stirred overnight. Then the reaction was stopped with the addition of saturated sodium carbonate solution, and the mixture was extracted with ethyl acetate (50 mL × 3). The combined organic layers was washed with water and brine, and dried over Na2SO4. The solid was filtered off and the solvent was evaporated under reduced pressure to afford a crude product, which was used directly for the next step.
5-(3, 5-Bis(trifluoromethyl) phenyl)-4H-1, 2, 4-triazole-3-thiol (5a)
To the crude product 4a was added 5% sodium hydroxide solution (50 mL), and the mixture was heated at reflux for 5 hr. Then 2N hydrochloride acid was added to adjust the pH of the mixture to about 6–7. The white solid was filtered, washed with water for 3–5 times, and then dried under vacuum at room temperature to give 5a (423 mg, 65% overall for two steps). 1H-NMR (DMSO): δ 13.71 (s, 1H), 13.62 (s, 1H), 7.54 (s, 2H), 7.14 (s, 1H), 2.31 (s, 6H); 13C-NMR (DMSO): δ 168.1, 148.3, 131.8, 131.4, 128.3, 126.5, 124.7, 124.3, 122.0; ESI-MS: 314.3 [M+H]+.
5-(3, 5-Bis(trifluoromethyl) phenyl)-4H-1, 2, 4-triazol-3-amine (5b)
Synthesis of 5b followed the same procedure as for 5a in 64% yield. 1H-NMR (DMSO): δ 8.38 (s, 2H), 8.06 (s, 1H), 6.33 (s, 2H); 13C-NMR (DMSO): δ 158.6, 156.1, 135.0, 131.3, 131.0, 125.5, 125.0, 122.3, 121.9; ESI-MS: 297.0 [M+H]+.
5-(3, 5-Bis(trifluoromethyl) phenyl)-1H-1, 2, 4-triazol-3-ol (5c)
Synthesis of 5c followed the same procedure as for 5a in 56% yield. 1H-NMR (DMSO): δ 8.41 (s, 2H), 8.36 (s, 1H), 2.50 (s, 2H); 13C-NMR (DMSO): δ 165.1, 134.6, 131.3, 131.1, 130.7, 129.9, 127.4, 126.7, 124.7, 121.9; ESI-MS: 298.0 [M+H]+.
5-(3-Fluorophenyl)-1H-1, 2, 4-triazole-3-thiol (5d)
Synthesis of 5d followed the same procedure as for 5a in 66% yield. 1H-NMR (DMSO): δ 13.92 (s, 1H), 13.77 (s, 1H), 7.35–7.76(m, 4H); 13C-NMR (DMSO): δ 167.6, 163.9, 161.5, 149.5, 131.9, 128.0, 122.2, 118.0, 113.0; HRMS-ESI (+): Calc. for C8H7N3SF: 196.03, Found: 196.03 [M+H]+.
5-(3, 5-Difluorophenyl)-1H-1, 2, 4-triazole-3-thiol (5e)
Synthesis of 5e followed the same procedure as for 5a in 67% yield. 1H-NMR (DMSO): δ 8.34 (s, 2H), 8.32 (s, 1H); 13C-NMR (DMSO): δ 131.9, 131.6, 131.5, 127.6, 125.0, 124.6, 121.9.
5- (4-(Trifluoromethyl) phenyl)-1H-1, 2, 4-triazole-3-thiol (5h)
Synthesis of 5h followed the same procedure as for 5a in 72% yield. 1H-NMR (DMSO): δ 7.54 (s, 2H), 7.14 (s, 1H), 2.31 (s, 6H); 13C-NMR (DMSO): δ 167.3, 150.8, 138.7, 132.4, 125.7, 123.8.
4-((5-(3, 5-Bis(trifluoromethyl) phenyl)-4H-1, 2, 4-triazol-3-yl) thio)-6-methyl-2-(methylthio) pyrimidine (7a)
To compound 5a (70 mg, 0.34 mmol) in 10 ml acetone was added potassium carbonate (65.2 mg, 0.47 mmol) and 4,6-dichloro-2-(methylthio) pyrimidine (6a, 36.3 mg, 0.2mmol). The reaction mixture was stirred at room temperature overnight. Then the reaction was stopped with the addition of 4N HCl, and extracted with ethyl acetate (3 × 30 mL). The combined organic layers was washed with water and brine, dried over Na2SO4, and filtered. After purification with silica gel column chromatography (hexane: acetate 10:1), 7a (79 mg, 64%) was obtained. 1H-NMR (DMSO): δ 8.60 (s, 2H), 8.29 (s, 1H), 7.96 (s, 1H), 6.95 (s, 1H); 2.32 (s, 6H); 13C-NMR (DMSO): δ 171.4, 167.7, 162.7, 131.9, 131.6, 126.7, 124.8, 123.7, 122.1, 113.1; MS-ESI (+): Calc. for C16H11F6N5S2: 452.41, Found: 452.04 [M+H]+.
4-((5-(3, 5-Bis(trifluoromethyl) phenyl)-4H-1, 2, 4-triazol-3-yl) thio)-6-chloro-2-(methylthio)pyrimidine (7b)
Synthesis of 7b followed the same procedure as for 7a in 67% yield. 1H-NMR (DMSO): δ 15.50 (br s, 1H), 8.57 (s, 2H), 8.25 (s, 1H), 7.28 (s, 1H), 2.34 (s, 3H); 13C-NMR (DMSO): δ 172.7, 160.1, 131.8, 131.5, 131.2, 126.8, 124.8, 122.1, 113.0, 14.0; HRMS-ESI (+): Calc. for C15H8N5S2F6Cl: 471.9874 Found: 471.9856 [M+H]+.
4-((5-(3, 5-Bis(trifluoromethyl) phenyl)-4H-1, 2, 4-triazol-3-yl) thio)-2-(methylthio) pyrimidine (7c)
Synthesis of 7c followed the same procedure as for 7a in 62% yield. 1H-NMR (CDCl3): δ 13.34 (br, s, 1H), 8.60 (s, 2H), 8.39 (d, 1H, J = 5.6Hz), 7.93 (s, 1H), 7.01 (d, 1H, J = 5.6Hz), 2.57 (s, 3H); 13C-NMR (CDCl3): δ 173.4, 164.7, 160.1, 156.2, 132.4, 132.3, 132.0, 126.5, 124.5, 123.1, 121.8, 114.0, 14.2; ESI-MS: 438.0 [M+H]+; HRMS-ESI (+): Calc. for C15H10F6N5S2: 438.0282 Found: 438.0280 [M+H]+.
4-((5-(3, 5-Dimethylphenyl)-4H-1, 2, 4-triazol-3-yl) thio)-2-(methylthio) pyrimidine (7d)
Synthesis of 7d followed the same procedure as for 7a in 57% yield. 1H-NMR (DMSO): δ 8.78 (d, 1H, J = 5.2Hz), 8.60 (d, 1H, J = 5.2Hz), 7.92 (d, 1H, J = 5.2Hz), 7.62 (m, 2H), 7.11 (s, 1H), 2.61 (s, 3H), 2.34 (s, 6H); 13C-NMR (DMSO): δ 166.1, 160.5, 157.9, 138.4, 132.4, 124.4, 116.9, 21.3, 14.2, 13.9; ESI-MS (+): 330.1 [M+H]+; HRMS-ESI (+): Calc. for C15H16N5S2: 330.0847 Found: 330.0841 [M+H]+.
4-Chloro-6-((5-(3, 5-dimethylphenyl)-4H-1, 2, 4-triazol-3-yl) thio)-2-(methylthio) pyrimidine (7e)
Synthesis of 7e followed the same procedure as for 7a in 76% yield. 1H-NMR (DMSO): δ 15.06 (br s, 1H), 7.64 (s, 2H), 7.15 (s, 2H), 2.37 (s, 3H), 2.34 (s, 6H); 13C-NMR (DMSO): δ 172.6, 160.0, 138.8, 132.5, 124.4, 112.7, 60.2, 21.3, 14.1; HRMS-ESI (+): Calc. for C15H14N5S2Cl: 364.0457 Found: 364.0457 [M+H]+.
4-Chloro-6-((5-(3-fluorophenyl)-4H-1, 2, 4-triazol-3-yl) thio)-2-(methylthio) pyrimidine (7f)
Synthesis of 7f followed the same procedure as for 7a in 46% yield. 1H-NMR (DMSO): δ 7.87-7.85 (d, J=7.6 Hz,1H), 7.78-7.76 (d, J=9.2 Hz 1H), 7.62-7.56 (dd, J=7.6, 5.6 Hz 1H), 7.37-7.33 (t, J=7.6 Hz 1H), 7.23 (s, 1H), 2.50 (s, 3H), 2.34 (s, 3H); 13C-NMR (DMSO): δ 172.6, 170.8, 164.0, 161.5, 160.0, 131.9, 131.8, 122.7, 117.8, 117.6, 113.4, 113.1, 112.8, 14.04; ESI-MS (+): 354.0 [M+H]+.
4-Chloro-6-((5-(3, 5-difluorophenyl)-4H-1, 2, 4-triazol-3-yl) thio)-2-(methylthio) pyrimidine (7g)
Synthesis of 7g followed the same procedure as for 7a in 47% yield. 1H-NMR (DMSO): δ 7.67-7.65 (m, 1H), 7.44 (m, 1H), 7.26 (s, 1H), 2.33 (S, 3H).
4-Chloro-6-((5-(3, 5-dimethoxyphenyl)-4H-1, 2, 4-triazol-3-yl) thio)-2-(methylthio) pyrimidine (7h)
Synthesis of 7h followed the same procedure as for 7a in 59% yield. 1H-NMR (DMSO): δ 15.01(s, 1H), 7.16 (s, 3H), 6.61 (s, 1H), 3.80(s, 6H), 2.49 (s, 3H); 13C-NMR (DMSO): δ 172.5, 161.3, 160.0, 104.4, 102.9, 60.2, 55.8, 14.5; ESI-MS (+): 396.03 [M+H]+.
4-Chloro-2-(methylthio)-6-((5-(4-(trifluoromethyl) phenyl)-4H-1, 2, 4-triazol-3-yl) thio) pyrimidine (7i)
Synthesis of 7i followed the same procedure as for 7a in 52% yield. 1H-NMR (DMSO): δ 8.22-8.20 (d, J=8.0Hz, 2H), 7.90-7.88 (d, J=8.0Hz, 2H), 7.26 (s, 1H), 2.32 (s, 3H); 13C-NMR (DMSO): δ 172.6, 170.4, 160.0, 130.8, 130.5, 128.8, 126.5, 126.4, 123.3, 112.9, 14.9.
4-Chloro-2-(methylthio)-6-((5-(3-(trifluoromethyl) phenyl)-4H-1, 2, 4-triazol-3-yl) thio) pyrimidine (7j)
Synthesis of 7j followed the same procedure as for 7a in 39% yield. 1H-NMR (DMSO): δ 8.46-7.85 (m, 5H), 2.51 (s, 3H).
4-Chloro-6-((5-(3, 5-dimethylphenyl)-4H-1, 2, 4-triazol-3-yl) thio) pyrimidine (9a)
Synthesis of 9a followed the same procedure as for 7a in 57% yield. 1H-NMR (DMSO): δ 15.07 (br s, 1H), 8.84 (s, 1H), 7.66 (s, 2H), 7.53 (s, 1H), 7.18 (s, 1H), 2.35 (s, 6H); 13C-NMR (DMSO): δ 160.4, 158.7, 138.86, 124.4, 118.0, 21.3; ESI-MS (+): 318.0 [M+H]+.
4-((5-(3, 5-Bis(trifluoromethyl) phenyl)-4H-1, 2, 4-triazol-3-yl) thio)-6-chloropyrimidine (9b)
Synthesis of 9b followed the same procedure as for 7a in 47% yield. 1H-NMR (DMSO): δ 15.52 (br s, 1H), 8.85 (s, 1H), 8.61 (s, 2H), 8.31 (s, 1H), 7.66 (s, 1H); 13C-NMR (DMSO): δ 160.6, 158.8, 132.1, 131.8, 131.5, 131.1, 126.8, 124.8, 124.0, 122.1, 118.3; ESI-MS: 426.0 [M+H]+.
4, 6-Dichloro-2-(phenylthio) pyrimidine (11a)
To a mixture of thiophenol (0.28 ml, 2.73 mmol), potassium carbonate (565 mg, 4.1 mmol) and 5 mL acetone in a 10 mL round bottom flask was added 10, 2, 4, 6-trichloropyrimidine (500 mg, 2.73 mmol) at room temperature. The mixture was kept stirring overnight. Then the solid was filtered off, and the reaction mixture was concentrated by evaporation under vacuum. The residue was purified by silica gel column chromatography (eluent, hexane: ethyl acetate= 25:1) to give 11a (600 mg), which are directly used for the next step without purification.
4-((5-(3, 5-Bis(trifluoromethyl) phenyl)-4H-1, 2, 4-triazol-3-yl) thio)-6-chloro-2-(phenylthio) Pyrimidine (12a)
To a mixture of 5a (470 mg, 1.5 mmol), potassium carbonate (276 mg, 2 mmol) and acetone (5mL) in a 10 mL round bottom flask, was added 11a (300 mg) at room temperature. The reaction was kept stirring at room temperature overnight. Then the solid portion was filtered off and the residue after solvent evaporation was purified by silica gel column chromatograph (eluent hexane: ethyl acetate= 25:1 to 10:1) to give 12a (478 mg, 51% for two steps). 1H-NMR (DMSO): δ 15.34 (br, s, 1H), 8.58 (s, 2H), 8.29 (s, 1H), 7.44 (m, 3H), 7.20 (m, 2H), 7.11 (m, 1H); 13C-NMR (DMSO): δ 171.9, 160.2, 135.1, 132.1, 131.8, 131.5, 129.7, 127.7, 126.8, 124.9, 124.0, 122.2, 114.1; ESI-MS (+): 534.0 [M+H]+.
4-Chloro-6-((5-(3, 5-dimethylphenyl)-4H-1, 2, 4-triazol-3-yl) thio)-2-(phenylthio) pyri-midine (12b)
Synthesis of 12b followed the same procedure as for 12a in 56% yield. 1H-NMR (CD3OD): δ 7.56 (m, 2H), 7.38 (m, 2H), 7.17 (m, 5H), 2.40 (s, 6H); 13C-NMR (CD3OD): δ 172.6, 170.8, 160.0, 138.8, 138.6, 134.8, 132.1, 129.1, 128.7, 127.8, 126.5, 123.9, 123.8, 112.8, 20.1; ESI-MS: 426.1 [M+H]+; ESI-MS (+): 426.1 [M+H]+.
4-((5-(3, 5-Bis(trifluoromethyl) phenyl)-4H-1, 2, 4-triazol-3-yl) thio)-6-chloro-2-phenoxy-pyrimidine (12c)
Synthesis of 12c followed the same procedure as for 12a in 43% yield. 1H-NMR (DMSO): δ 15.52 (br, s, 1H), 8.56 (m, 3H), 7.49 (m, 7H); 13C-NMR (DMSO): δ 170.5, 163.5, 161.8, 158.9, 152.2, 151.9, 131.8, 131.4, 130.5, 129.8, 126.9, 125.8, 124.8, 124.1, 122.1, 121.8, 112.7, 107.7, 103.1; ESI-MS: 518.0 [M+H]+; ESI-MS (+): 518.0 [M+H]+.
4-((5-(3, 5-Bis(trifluoromethyl) phenyl)-4H-1, 2, 4-triazol-3-yl) thio)-6-chloro-2-(hexylthio) pyrimidine (12d)
Synthesis of 12d followed the same procedure as for 12a in 36% yield. 1H-NMR (CDCl3): δ 8.61 (s, 2H), 7.94 (s, 1H), 7.03 (s, 1H), 3.15-3.11(t, J=7.6, 7.2 Hz, 2H), 1.75-1.70 (m, 2H), 1.43-1.27 (m, 6H), 0.95-0.92 (m, 3H); 13C-NMR (CDCl3): δ 173.9, 165.7, 160.5, 132.4, 132.1, 132.0, 126.4, 124.5, 123.1, 121.8, 117.8, 113.1, 31.5, 30.9, 30.8, 30.1, 28.3, 22.1, 13.8; ESI-MS: 528.1 [M+H]+.
4-((4-((5-(3, 5-Bis(trifluoromethyl) phenyl)-4H-1, 2,4-triazol-3-yl) thio)-6-chloropyri- midin-2-yl) thio) butan-1-ol (12e)
Synthesis of 12e followed the same procedure as for 12a in 33% yield. 1H-NMR (CDCl3): δ 8.59 (s, 2H), 7.94 (s, 1H), 6.96 (s, 1H), 3.76-3.73 (m, 2H), 2.94-2.90 (m, 2H), 1.73-1.62 (m. 4H); 13C-NMR (CDCl3): δ 173.3, 167.1, 160.4, 132.5, 132.2, 131.9, 127.1, 124.4, 123.3, 121.7, 113.0, 62.2, 31.1, 30.8, 25.5; HRMS-ESI (+): Calc. for C18H14ClF6N5OS2: 530.0316 Found: 530.0318 [M+H]+.
4-Chloro-6-((5-(3, 5-difluorophenyl)-4H-1, 2, 4-triazol-3-yl) thio)-2-(phenylthio) pyrimidine (12f)
Synthesis of 12f followed the same procedure as for 12a in 59% yield. 1H-NMR (CDCl3): δ 7.56-7.43 (m, 7H), 6.50 (s, 2H); 13C-NMR (CDCl3): δ 178.6, 164.5, 164.4, 162.3, 161.8, 158.3, 135.6, 131.0, 130.4, 126.0, 121.6, 108.6, 105.2.
4-Chloro-2-(phenylthio)-6-((5-(3-(trifluoromethyl) phenyl)-4H-1, 2, 4-triazol-3-yl) thio) pyrimidine (12g)
Synthesis of 12g followed the same procedure as for 12a in 41% yield. 1H-NMR (CDCl3): δ 8.15-8.13 (d, J = 7.6 Hz, 1H), 7.14-7.12 (d, J = 8.0 Hz, 1H), 7.72-7.59 (t, J = 7.6, 8.0 Hz, 1H), 7.64-7.44 (d, J = 8.8, 7.2 Hz, 2H), 7.21-7.14 (m, 3H), 6.34 (s, 1H); 13C-NMR (CDCl3): δ 176.5, 159.2, 135.5, 131.5, 131.2, 130.6, 129.8, 128.8, 125.7, 123.6, 123.5, 122.4, 111.3.
4-Chloro-6-((5-(3, 5-dimethoxyphenyl)-4H-1, 2, 4-triazol-3-yl) thio)-2-(phenylthio) pyrimidine (12h)
Synthesis of 12h followed the same procedure as for 12a in 45% yield. 1H-NMR (CDCl3): δ 7.56-7.55 (d, J=4.4 Hz, 2H), 7.26 (m, 5H), 7.06 (s, 2H), 3.83 (s, 6H); 13C-NMR (CDCl3): δ 176.1, 161.2, 159.2, 135.3, 130.6, 130.1, 126.3, 111.1, 104.4, 102.9, 55.6; HRMS-ESI (+): Calc. for C20H16ClN5O2S2: 458.0526, Found: 458.0567 [M+H]+.
4-Chloro-2-(phenylthio)-6-((5-(4-(trifluoromethyl)phenyl)-4H-1, 2, 4-triazol-3-yl) thio) pyrimidine (12i)
Synthesis of 12i followed the same procedure as for 12a in 59% yield. 1H-NMR (DMSO): δ 8.22-8.20 (d, J=8.0Hz, 2H), 7.90-7.88 (d, J=8.0Hz, 2H), 7.26 (s, H), 2.32 (s, 3H); 13C-NMR (DMSO): δ 172.6, 170.4, 160.0, 130.8, 130.5, 128.8, 128.4, 125.5, 123.0, 111.8, 14.0.
3-(5-((6-Chloro-2-(phenylthio) pyrimidin-4-yl) thio)-4H-1, 2, 4-triazol-3-yl)-5-methoxy phenol (12j)
Synthesis of 12j followed the same procedure as for 12a in yield 31% yield. 1H-NMR (DMSO): δ 7.47-7.45 (d, J = 7.2 Hz, 2H), 7.21-7.19 (d, J = 7.2 Hz, 2H), 7.11-7.09 (d, J = 7.2 Hz, 1H), 6.95 (s, 2H), 6.54 (s, 1H), 6.07 (s, 1H), 3.81 (s, 3H); 13C-NMR (DMSO): δ 175.7, 160.3, 159.4, 158.6, 135.6, 131.9, 125.9, 110.8, 108.6, 104.1, 103.1, 58.8.
3-(5-((6-Chloro-2-(phenylthio) pyrimidin-4-yl) thio)-4H-1, 2, 4-triazol-3-yl)-5-fluorophenol (12k)
Synthesis of 12k followed the same procedure as for 12a in 54% yield. 1H-NMR (CDCl3): δ 7.68 (m, 1H), 7.62-7.60 (d, J=9.2 Hz, 1H), 7.43-7.39 (t, J=9.2, 7.2 Hz, 3H), 7.23-7.13 (m, 4H), 6.32 (s, 1H); 13C-NMR (CDCl3): δ 176.4, 169.3, 164.1, 159.2, 135.5, 130.7, 130.1, 126.1, 122.2, 122.1, 117.7, 117.4, 113.8, 113.6, 111.2; HRMS-ESI (+): Calc. for C18H11ClN5S2: 416.0208, Found: 416.0207 [M+H]+.
5-(3, 5-Bis(trifluoromethyl) phenyl)-1, 3, 4-oxadiazole-2-thiol (14)
Synthesis of 14 followed the same procedure as for 5a in 69% yield. 1H-NMR (CDCl3): δ 11.7 (s, 1H), 8.44 (s, 2H), 8.09 (s, 1H); 13C-NMR (CDCl3): δ 178.1, 158.8, 133.0, 126.4, 125.8, 124.4, 123.8, 121.1; HRMS-ESI (+): Calc. for C10H4F6N2OS: 315.0021, Found: 315.0027 [M+H]+.
2-(3, 5-Bis(trifluoromethyl) phenyl)-5-((6-chloro-2-(methylthio) pyrimidin-4-yl) thio)-1, 3, 4-oxadiazole (15)
Synthesis of 15 followed the same procedure as for 7a in 58% yield. 1H-NMR (CDCl3): δ 8.57 (s, 2H), 8.12 (s, 1H), 7.21 (s, 1H), 2.35 (s, 3H); 13C-NMR (CDCl3): δ 174.2, 165.5, 165.1, 161.0, 158.3, 133.4, 133.1, 127.0, 125.9, 123.9, 121.2, 113.0, 14.2; ESI-MS: 472.8 [M+H]+; HRMS-ESI (+): Calc. for C15H8ClF6N4OS2: 472.9719, Found: 472.9722 [M+H]+.
2-((3-(3, 5-Bis(trifluoromethyl) phenyl)-1H-1, 2, 4-triazol-5-yl) thio)-4, 6-dichloropyrimidine(16a)
Synthesis of 16a followed the same procedure as for 7a in 78% yield. 1H-NMR (CDCl3): δ 8.59 (s, 2H), 7.95 (s, 1H), 7.37 (s, 1H); 13C-NMR (CDCl3): δ 169.2, 162.1, 160.1, 132.5, 132.2, 131.8, 126.55, 124.4, 123.4, 121.7, 119.0, 116.7; ESI-MS: 461.9 [M+H]+; HRMS-ESI (+): Calc. for C14H5Cl2F6N5S: 459.9637, Found: 459.9638 [M+H]+.
4, 6-Dichloro-2-((3-(3, 5-dimethylphenyl)-1H-1, 2, 4-triazol-5-yl) thio) pyrimidine (16b)
Synthesis of 16b followed the same procedure as for 7a in 74% yield. 1H-NMR (DMSO): δ 15.1 (s, 1H), 7.64 (s, 2H), 7.52 (s, 1H), 7.17 (s, 1H), 2.35 (s, 6H); 13C-NMR (DMSO): δ 174.8, 161.4, 158.9, 157.5, 151.3, 136.8, 132.6, 128.0, 124.5, 118.3, 21.3; ESI-MS: 352.1 [M+H]+; HRMS-ESI (+): Calc. for C14H12Cl2N5OS: 352.0184, Found: 352.0190 [M+H]+.
4, 6-Dichloro-2-((3-(3, 5-dimethylphenyl)-1H-1, 2, 4-triazol-5-yl) oxy) pyrimidine (16c)
Synthesis of 16c followed the same procedure as for 7a in 64% yield. 1H-NMR (CDCl3): δ 8.56 (s, 2H), 8.11 (s, 1H), 7.66 (s, 1H); 13C-NMR (CDCl3): δ 167.6, 165.6, 162.7, 160.4, 157.4, 133.4, 133.0, 127.1, 125.9, 124.9, 123.9, 121.2, 117.0; ESI-MS: 445.3 [M+H]+.
4, 6-Dichloro-2-((5-(3-fluorophenyl)-4H-1, 2, 4-triazol-3-yl) thio) pyrimidine (16d)
Synthesis of 16d followed the same procedure as for 7a in 71% yield. 1H-NMR (CDCl3): δ 7.77-7.75 (d, J=7.2 Hz, 1H), 7.70-7.68 (d, J=9.2 Hz, 1H), 7.45-7.39 (dd, J=8.4, 7.2 Hz, 1H), 7.26 (s, 1H), 7.19-7.14 (m, 1H); 13C-NMR (CDCl3): δ 171.8, 164.1, 161.7, 130.9, 129.0, 122.2, 118.1, 117.8, 116.3, 113.8, 113.6; HRMS-ESI (+): Calc. for C12H7Cl2FN5S: 341.9784, Found: 341.9783 [M+H]+.
N-Benzyl-2-((3-(3, 5-bis(trifluoromethyl) phenyl)-4H-1, 2, 4-triazol-5-yl) thio)-6-chloro- pyrimidin-4-amine (16e)
Synthesis of 16e followed the same procedure as for 7a in 45% yield. 1H-NMR (CDCl3): δ 7.55 (s, 3H), 7.42-7.27 (m, 6H), 6.75 (s, 1H), 2.39 (s, 2H); 13C-NMR (CDCl3): δ 198.4, 143.5, 134.4, 130.5, 128.9, 128.2, 128.1, 127.6, 127.5, 127.1, 126.9, 126.6, 27.5.
2-(3, 5-Bis(trifluoromethyl) phenyl)-5-((4, 6-dichloropyrimidin-2-yl) thio)-1, 3, 4-oxadiazole (17)
Synthesis of 17 followed the same procedure as for 7a in 62% yield. 1H-NMR (CDCl3): δ 8.57 (s, 2H), 8.12 (s, 1H), 7.66 (s, 1H); 13C-NMR (CDCl3): δ 167.6, 165.6, 162.7, 160.5, 157.4, 133.4, 127.2, 125.9, 124.9, 123.9, 121.2, 117.0; HRMS-ESI (+): Calc. for C14H5Cl2F6N4OS: 460.9445, Found: 460.9465 [M+H]+.
2-((3-(3, 5-Bis(trifluoromethyl) phenyl)-1H-1, 2, 4-triazol-5-yl) oxy)-4-((5-(3, 5-bis(trifluoro methyl) phenyl)-4H-1, 2, 4-triazol-3-yl) oxy)-6-chloropyrimidine (18)
Synthesis of 18 followed the same procedure as for 7a in 65% yield. 1H-NMR (CDCl3): δ 8.53 (s, 4H), 8.12 (s, 2H); 13C-NMR (CDCl3): δ 167.3, 165.5, 160.3, 157.8, 133.3, 133.0, 127.2, 125.9, 124.9, 123.9, 121.2, 14.1.
4-(4-Bromophenyl)-2-((4, 6-dichloropyrimidin-2-yl) thio)-6-oxo-1, 6-dihydropyrimidine-5-carbonitrile (20a)
Synthesis of 20a followed the same procedure as for 7a in 45% yield. 1H-NMR (CDCl3): δ 7.82-7.80 (d, J = 8.4 Hz, 2H), 7.66 (d, J = 8.4 Hz, 2H), 7.56 (s, 1H); HRMS-ESI (+): Calc. for C15H7BrCl2N5OS: 453.8855, Found: 453.8932 [M+H]+.
2-((4, 6-Dichloropyrimidin-2-yl) thio)-4-(4-ethylphenyl)-6-oxo-1, 6-dihydropyrimidine-5-carbonitrile (20b)
Synthesis of 20b followed the same procedure as for 7a in 52% yield. 1H-NMR (CDCl3): δ 7.93 (s, 1H), 7.72-7.70 (d, J= 7.6 Hz, 2H), 7.43-7.41 (d, J= 7.6 Hz, 2H), 7.38 (s, 1H), 2.80-2.74 (m, 2H), 1.31-1.24 (m, 3H).
4-Amino-5-(3, 5-bis(trifluoromethyl) phenyl)-4H-1, 2, 4-triazole-3-thiol (22)
To a mixture of 21, 3, 5-bis(trifluoromethyl)benzohydrazide (2.7 g, 10 mmol) and carbon disulphide (1.14 g, 15 mmol) in 20 mL of absolute ethanol was added KOH (0.84 g, 15 mmol) pellets and the reaction was kept at reflux for 6 hr. The solvent was evaporated under vacuum to afford potassium dithiocarbazates as white solid, which was treated with hydrazine monohydrate (1.6 mL, 25 mmol) in water and then heated at reflux for 4 hr. The color of the reaction mixture changed to green with the evolution of hydrogen sulfide gas (lead acetate paper test and odor). Then the reaction mixture was cooled to room temperature, diluted with water (100 mL), and acidified with concentrated hydrochloric acid. The resulting precipitate was filtered, washed thoroughly with cold water, and recrystallized from ethanol to give compound 22 as white solid (2.62 g, 82%). 1H-NMR (DMSO): δ 14.2 (s, 1H), 8.69 (s, 2H), 8.31 (d, J = 7.6 Hz, 1H), 5.86 (s, 3H). HRMS-ESI calcd for C10H7F6N4S: 329.0290, found: 329.0294[M + H]+.
3-(3, 5-Bis(trifluoromethyl)phenyl)-7-chloro-9-(methylthio) pyrimido[5, 4-f][1, 2,4]triazolo [3, 4-b][1, 3, 4] thiadiazepine (24)
A mixture of compound 22 (328 mg, 1 mmol) and compound 23, 4, 6-dichloro-2-(methylthio)pyrimidine-5-carbaldehyde (223 mg, 1 mmol) in DMF 6 mL was heated at 60 ºC for 4 hr. After cooling to room temperature, 20 mL water was added. The precipitate was filtered off and the resulting solid was further purified by flash chromatography (hexane/EtOAc 1: 1) to afford 24 (380 mg, 76%). 1H-NMR (DMSO): δ 8.88 (s, 1H), 8.50 (s, 2H), 8.38 (s, 1H), 2.63 (s, 3H). HRMS (ESI) calcd for C16H8ClF6N6S2: 496.9839, found: 496.9828[M + H]+.
3-(3, 5-Bis(trifluoromethyl) phenyl)-7-chloro-9-(methylthio)-5, 6-dihydropyrimido[5, 4-f][1, 2, 4] triazolo[3, 4-b][1, 3, 4] thiadiazepine (25)
To a suspension of compound 24 (50 mg, 0.1 mmol) in absolute ethanol (2 mL) was added sodium borohydride (3.8 mg, 0.1 mmol) in one portion with stirring. The yellow solution formed was kept at room temperature for 2 hr. The solvent was removed under reduced pressure, and the residue was washed with water and filtered. The resulting solid was collected to give compound 25 (41 mg, 82%). 1H-NMR (CDCl3): δ 8.72 (s, 2H), 8.03 (s, 1H), 6.27 (t, J = 6.2 Hz, 1H), 4.52 (d, J = 6.2 Hz, 1H), 2.51 (s, 3H). HRMS (ESI) calcd for C16H10ClF6N6S2: 498.9996, found: 498.9991[M + H]+.
3-(3, 5-Bis(trifluoromethyl) phenyl)-9-(methylthio)-5, 6, 7, 8-tetrahydropyrimido[5, 4-f][1, 2, 4] triazolo[3, 4-b][1, 3, 4] thiadiazepine (26)
To a suspension of compound 24 (50 mg, 0.1 mmol) in absolute ethanol (2 mL) Sodium borohydride (38 mg, 1 mmol) was added in portions with stirring. The yellow solution formed was kept at room temperature for 2 hr. The solvent was removed under reduced pressure, and the residue was washed with water, and then filtered. The resulting solid was collected to give compound 26 (20 mg, 40%). 1H-NMR (CDCl3/CD3OD 1/1): δ 8.72 (s, 2H), 8.02 (s, 1H), 3.69 (d, J = 8.2 Hz, 4H), 2.51 (s, 3H). HRMS (ESI) calcd for C16H13F6N6S2: 467.0542, found: 467.0551[M + H]+.
3.2 Biology
Bacterial strains and culture condition
E. coli NR698 (MC4100 imp4213),[19] an outer membrane leaky mutant strain, was kindly provided by Thomas J Silhavy of Princeton University. S. aureus ATCC strains 6538 and 35556 were from the American Type Culture collection. S. aureus strains Mu50, Mu3, and N315 were kindly provided by C.-D. Lu, and Newman strain from Z. Eichenbaum of Georgia State University. Five efflux pump-related S. aureus strains 8325-4, K1758 (NorA−), K2361 (NorA++), K2908 (MepA−), K2068 (MepA++)[1d, 20] were kindly provided by GW Kaatz at Wayne State University School of Medicine. B. subtilis 168 and B. anthracis Sterne were lab stocks. All strains were grown on Luria-Bertani (LB) broth or agar plates at 37 °C.
Chemical compounds
DMSO was purchased from Sigma. The initial 38 compounds for screening including hit compound 1 were purchased from Maybridge.
Protein purification
E. coli SecA, SpSecA and BsSecA were purified as described.[21] EcSecAN68, a truncated mutant of EcSecA containing the N-terminal catalytic domain, was purified as previously described.[16b, 21b] SasecA1 gene was amplified from S. aureus ATCC35556 and cloned into pET-21d with 6X his-tag at C-teriminal. BasecA1 gene was amplified from B. anthracis Sterne, and cloned into pET-20b with 6X his-tag at C-terminal. SaSecA1 and BaSecA1 were purified with HisTrap affinity column and Superdex-200 column (GE healthcare).
Liposome and membrane preparation
The preparation of liposomes is as described previously.[8] Briefly, E. coli total lipids (Avanti) were dried and re-suspended in 150 mM KCl solution, and sonicated in an ice water bath until the solution became clear, usually for 3–5 mins, which resulted in an average size of liposomes about 130 nm. The liposome solutions were aliquoted, and stored at −80 °C before use. The liposomes were frozen and thawed once for the experiments. The preparation of SecA-depleted native membranes from BA13 is as previously described.[18a, 22] The BA13 cells were grown at 42 °C to deplete the SecA, and the cells were collected, and lysed by French Press as described. E. coli 773 is an ompA-deleted strain in lab stock; the membrane vesicles were prepared from sucrose gradients as described.[23]
ATPase assays for EcSecAN68 and EcSecA
ATPase activities were determined by malachite green colorimetric assay as described previously.[15, 16b] Translocation ATPase was assayed in the presence of SecA-depleted membranes and pOmpA precursors. IC50 is the concentration of the compound that inhibits 50% of ATPase activities.
In vitro translocation activity of EcSecA
In vitro translocation activity of EcSecA was determined by using OmpA-depleted 773 membranes and SecA-liposome as described previously.[24] Briefly, Reconstituted liposomes, SecA and pOmpA were added into the translocation buffer[24] separately before reaction. Liposomes at 12 μg or 4.5 μg of OmpA-depleted 773 membranes with various amounts of SecA were used in the 0.1 ml reaction mixtures. The reactions were at 37 °C for 45 min, and the translocation mixtures were processed by immunoblots as previously described.[24] IC50 is the concentration of the compound that inhibits 50% of in vitro translocation activity.
Ion-channel activity of SecA-liposomes in oocytes
Liposomes were prepared as described previously.[9, 15, 16b, 24–25] Oocytes were obtained from live frog Xenopus laevis (Xenopus Express, Inc) and injected with sample mixtures as described previously.[18b, 24] Briefly, 50 nl sample mixtures containing 120 ng liposomes, 120 ng SecA, 14 ng proOmpA, 2 mM ATP, 1 mM Mg(OAC)2, and different concentrations of inhibitors were injected into oocytes (average volume: 500 nl) using Nanoject II injector. The voltage clamp adapted from an electrophysiological method was utilized to measure the opening of protein conducting channels as described previously.[18b, 24] After injection, the oocytes were incubated at 23 C for 3 hr, then the ion current was recorded continuously for 1 min. The inward currents and outward currents were recorded to measure the net currents for channel opening. IC50 is the concentration of the compound that inhibits 50% ion-channel activities of SaSecA1.
Bacteriostatic effect
Bacteriostatic effects were tested according to the guidelines of the Clinical and Laboratory Standards Institute.[26] This assay was performed in a 96-well microtiter plate in triplicates in three separate experiments as described previously at 37 °C with shaking at 250 rpm for 24 hrs. MIC is the lowest concentration of inhibitor at which cells were not able to grow.
Bactericidal effect
Bactericidal effect was determined as described previously.[16b] Log-phase growing cells (OD600 ≈ 0.5) were mixed with different concentrations of inhibitors, and then incubated in an Eppendorf Thermonmixer R (Brinkmann instruments) at 37 °C with shaking (1,000 rpm) for 1 or 2 hr. Cultures were serially diluted with LB broth and spreaded on LB plates, and incubated at 37°C overnight for colony forming units (CFU). Bactericidal effect was determined by the reduction of CFU as described previously.
Pull down assay
S. aureus ATCC 6538 was grown in LB medium at 37 °C overnight. Then cells were harvested by centrifugation, washed with TBST buffer (0.1 M Tris-HCl pH 7.0, 0.15 M NaCl, 0.1% Tween-20) and re-suspended with TBST buffer containing EDTA-free cocktail protease inhibitors. The cells were broken with French Press at 10,000 psi. Unbroken cells were removed by 5,000 rpm centrifugation. Whole cell lysates were treated with PureProteome Streptavidin Magnetic Beads (from Millipore) at 4 °C for 1 hr to remove non-specific binding. The supernatant was mixed beads with or without 400 μM SCA-256 at 4 °C for 1 hr. The beads were washed with TBST buffer for 3 times and were mixed with SDS sample buffer, and boiled for 15 min. Cross-reacting EcSecA antibody (lab stock) was used to detect SaSecA1 by Western blots.
Supplementary Material
Table 6.
Bacteriostatic effects against various bacterial strains, MIC (μM)
| Comp. | E. coli NR698 | B. subtilis 168 | B. anthracis Sterne | S. aureus 6538 | S. aureus Mu50 | S. aureus N315 | S. aureus Mu3 | S. aureus Newman |
|---|---|---|---|---|---|---|---|---|
| 1 | 25 | 6.3 | 6.3 | 12.5 | ND | ND | ND | ND |
| 7b | 5.2 | 1.6 | 3.1 | 3.1 | 1.6 | 0.8 | 3.1 | 1.6 |
| 12a | 3.1 | 0.8 | 1.6 | 1.8 | 0.8 | 0.8 | 1.6 | ND |
Log-phase cells were diluted to OD600 ≈ 0.05, followed by incubation with varying concentrations of compounds for 16 hours at 37 °C. MIC (μM) refers to minimal concentration at which cell growth was inhibited.
Acknowledgments
Financial support by the National Institutes of Health (AI104168) and Fellowships of Molecular Basis of Diseases Program at Georgia State University (J.S., A.S.C., Y.H.H., and W.C.) is gratefully acknowledged.
References
- 1.a) Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J. Clin Infect Dis. 2009;48:1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]; b) CDC. 2013 [Google Scholar]; c) Fernández L, Gooderham WJ, Bains M, McPhee JB, Wiegand I, Hancock REW. Antimicrob Agents Chemother. 2010;54:3372–3382. doi: 10.1128/AAC.00242-10. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Kaatz GW, McAleese F, Seo SM. Antimicrob Agents Chemother. 2005;49:1857–1864. doi: 10.1128/AAC.49.5.1857-1864.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Chopra I, Schofield C, Everett M, O’Neill A, Miller K, Wilcox M, Frère JM, Dawson M, Czaplewski L, Urleb U, Courvalin C. Lancet Infect Dis. 2008;8:133–139. doi: 10.1016/S1473-3099(08)70018-5. [DOI] [PubMed] [Google Scholar]
- 2.a) Briken V. Curr Drug Targets. 2008;9:150–157. doi: 10.2174/138945008783502449. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Cao H, Baldini RL, Rahme LG. Annu Rev Phytopathol. 2001;39:259–284. doi: 10.1146/annurev.phyto.39.1.259. [DOI] [PubMed] [Google Scholar]
- 3.a) Gold VAM, Whitehouse S, Robson A, Collinson I. Biochem J. 2013;449:695–705. doi: 10.1042/BJ20121314. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Or E, Boyd D, Gon S, Beckwith J, Rapoport T. J Biol Chem. 2005;280:9097–9105. doi: 10.1074/jbc.M413947200. [DOI] [PubMed] [Google Scholar]; c) Economou A. Expert Opin Ther Targets. 2001;5:141–153. doi: 10.1517/14728222.5.2.141. [DOI] [PubMed] [Google Scholar]; d) Abe A. Kansenshogaku Zasshi. 2009;83:94–100. doi: 10.11150/kansenshogakuzasshi.83.94. [DOI] [PubMed] [Google Scholar]; e) Lee VT, Schneewind O. Genes Dev. 2001;15:1725–1752. doi: 10.1101/gad.896801. [DOI] [PubMed] [Google Scholar]; f) Scott JR, Barnett TC. Annu Rev Microbiol. 2006;60:397–423. doi: 10.1146/annurev.micro.60.080805.142256. [DOI] [PubMed] [Google Scholar]
- 4.a) Segers K, Klaassen H, Economou A, Chaltin P, Anné J. Anal Biochem. 2011;413:90–96. doi: 10.1016/j.ab.2011.02.012. [DOI] [PubMed] [Google Scholar]; b) Hsieh YH, Huang Y, Jin J, Yu L, Yang H, Jiang C, Wang B, Tai PC. Biochem Biophys Res Commun. 2014;454:308–312. doi: 10.1016/j.bbrc.2014.10.070. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Jin J, Cui J, Chaudhary AS, Hsieh Y, Damera K, Zhang H, Yang H, Wang B, Tai PC. Bioorg Med Chem. 2015 doi: 10.1016/j.bmc.2015.1009.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Chaudhary AS, Chen W, Jin J, Tai PC, Wang B. Future Med Chem. 2015;7:989–1007. doi: 10.4155/fmc.15.42. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Huang YJ, Wang H, Gao FB, Li M, Yang H, Wang B, Tai PC. ChemMedChem. 2012;7:571–577. doi: 10.1002/cmdc.201100594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Schmidt MG, Rollo EE, Grodberg J, Oliver DB. J Bacteriol. 1988;170:3404–3414. doi: 10.1128/jb.170.8.3404-3414.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Magiorakosa AP, Srinivasanb A, Careyb RB, Carmelic Y, Falagasd ME, Giskef CG, Harbarthg S, Hindlerh JF, Kahlmeteri G, Olsson–Liljequistj B, Patersonk DL, Ricel LB, Stellingm J, Struelensa MJ, Vatopoulosn A, Weberb JT, Monneta DL. Clin Microbiol Infect. 2012;8:268–281. doi: 10.1111/j.1469-0691.2011.03570.x. [DOI] [PubMed] [Google Scholar]; c) Siboo IR, Chaffin DO, Rubens CE, Sullam PM. J Bacteriol. 2008;190:6188–6196. doi: 10.1128/JB.00300-08. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Rigel NWBM. Mol Microbiol. 2008;69:291–302. doi: 10.1111/j.1365-2958.2008.06294.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Chitlaru T, Gat O, Gozlan Y, Ariel N, Shafferman A. J Bacteriol. 2006;188:3551–3571. doi: 10.1128/JB.188.10.3551-3571.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sibbald MJ, Ziebandt AK, Engelmann S, Hecker M, de Jong A, Harmsen HJ, Raangs GC, Stokroos I, Arends JP, Dubois JY, van Dijl JM. Microbiol Mol Biol Rev. 2006;70:755–788. doi: 10.1128/MMBR.00008-06. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Walz A, Mujer CV, Connolly JP, Alefantis T, Chafin R, Dake C, Whittington J, Kumar SP, Khan AS, DelVecchio VG. Proteome Sci. 2007;5:11. doi: 10.1186/1477-5956-5-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Oliver DB. Mol Microbiol. 1993;7:159–165. doi: 10.1111/j.1365-2958.1993.tb01107.x. [DOI] [PubMed] [Google Scholar]; b) Eichler J, Rinard K, Wickner W. J Biol Chem. 1998;273:21675–21681. doi: 10.1074/jbc.273.34.21675. [DOI] [PubMed] [Google Scholar]; c) Driessen AJ, Fekkes P, van der Wolk JP. Curr Opin Microbiol. 1998;1:216–222. doi: 10.1016/s1369-5274(98)80014-3. [DOI] [PubMed] [Google Scholar]
- 8.Hsieh YH, Zhang H, Lin BR, Cui N, Na B, Yang H, Jiang C, Sui SF, Tai PC. J Biol Chem. 2011;286:44702–44709. doi: 10.1074/jbc.M111.300111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rapoport TA, Jungnickel B, Kutay U. Annu Rev Biochem. 1996;65:271–303. doi: 10.1146/annurev.bi.65.070196.001415. [DOI] [PubMed] [Google Scholar]
- 10.a) Li M, Huang YJ, Tai PC, Wang B. Biochem Biophys Res Commun. 2008;368:839–845. doi: 10.1016/j.bbrc.2008.01.135. [DOI] [PubMed] [Google Scholar]; b) Chen W, Huang YJ, Gundala SR, Yang H, Li M, Tai PC, Wang B. Bioorg Med Chem. 2010;18:1617–1625. doi: 10.1016/j.bmc.2009.12.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Rivera NR, Balsells J, Hansen KB. Tetrahedron Lett. 2006;47:4889–4891. [Google Scholar]; b) Plech T, Wujec M, Siwek A, Kosikowska U, Malm A. Eur J Med Chem. 2011;46:241–248. doi: 10.1016/j.ejmech.2010.11.010. [DOI] [PubMed] [Google Scholar]; c) Saha AK, Kumar RR, Devakumar C. J Heterocycl Chem. 2010;47:838–845. [Google Scholar]
- 12.Patil SS, Jadhav RP, Patil AA, Patil SV, BVD J Chem Pharm Res. 2010;4:38–51. [Google Scholar]
- 13.Salerno L, Guerrera F, Modica M, Romeo G, Pittala V, Siracusa MA, Mereghetti I, Cagnotto A, Mennini T. Med Chem. 2007;3:551–560. doi: 10.2174/157340607782360281. [DOI] [PubMed] [Google Scholar]
- 14.Pardeshi SP, Patil SV, Patil R, Bobade VD. J Chem Pharm Res. 2014;6:675–681. [Google Scholar]
- 15.Huang YJ, Wang H, Gao FB, Li M, Yang H, Wang B, Tai PC. ChemMedChem. 2012;7:571–577. doi: 10.1002/cmdc.201100594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.a) Chaudhary AS, Chen W, Jin J, Tai PC, Wang B. Future Med Chem. 2012;7:989–1007. doi: 10.4155/fmc.15.42. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Cui J, Jin J, Hsieh YH, Yang H, Ke B, Damera K, Tai PC, Wang B. ChemMedChem. 2013;8:1384–1393. doi: 10.1002/cmdc.201300216. [DOI] [PubMed] [Google Scholar]
- 17.a) Jin J, Cui J, Chaudhary AS, Hsieh Y, Damera K, Zhang H, Yang H, Wang B, Tai PC. bioorganic and medicinal chemistry. doi: 10.1016/j.bmc.2015.09.0272015. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chaudhary AS, Jin J, Chen W, Tai PC, Wang B. Bioorg Med Chem. 2015;23:105–117. doi: 10.1016/j.bmc.2014.11.017. [DOI] [PubMed] [Google Scholar]
- 18.a) Lin BR, Hsieh YH, Jiang C, Tai PC. J Membrane Biol. 2012;245:747–757. doi: 10.1007/s00232-012-9477-8. [DOI] [PubMed] [Google Scholar]; b) Lin BR, Gierasch LM, Jiang C, Tai PC. J Membr Biol. 2006;214:103–113. doi: 10.1007/s00232-006-0079-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ruiz N, Falcone B, Kahne D, Silhavy TJ. Cell. 2005;121:307–317. doi: 10.1016/j.cell.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 20.Kaatz GW, Moudgal VV, Seo SM. J Antimicrob Chemother. 2002;50:833–838. doi: 10.1093/jac/dkf224. [DOI] [PubMed] [Google Scholar]
- 21.a) Cabelli RJ, Dolan KM, Qian LP, Oliver DB. J Biol Chem. 1991;266:24420–24427. [PubMed] [Google Scholar]; b) Chen X, Brown T, Tai PC. J Bacteriol. 1998;180:527–537. doi: 10.1128/jb.180.3.527-537.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Chen X, Xu H, Tai PC. J Biol Chem. 1996;271:29698–29706. doi: 10.1074/jbc.271.47.29698. [DOI] [PubMed] [Google Scholar]
- 22.Osborne RS, Silhavy TJ. EMBO J. 1993;12:3391–3398. doi: 10.1002/j.1460-2075.1993.tb06013.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.a) Yang YB, Lian J, Tai PC. J Bacteriol. 1997;179:7386–7393. doi: 10.1128/jb.179.23.7386-7393.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Yang YB, Yu N, Tai PC. J Biol Chem. 1997;272:13660–13665. doi: 10.1074/jbc.272.21.13660. [DOI] [PubMed] [Google Scholar]
- 24.Hsieh YH, Zhang H, Lin BR, Cui N, Na B, Yang H, Jiang C, Sui SF, Tai PC. J Biol Chem. 2011;286:44702–44709. doi: 10.1074/jbc.M111.300111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang HW, Chen Y, Yang H, Chen X, Duan MX, Tai PC, Sui SF. Proc Natl Acad Sci USA. 2003;100:4221–4226. doi: 10.1073/pnas.0737415100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.CLSI. 2011 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.