

Abstract
Rationale
The pathogenesis of insulin resistance involves dysregulated gene expression and function in multiple cell types including endothelial cells (ECs). Posttranscriptional mechanisms such as microRNA-mediated regulation of gene expression could affect insulin action by modulating EC function.
Objective
To determine whether microRNA-181b (miR-181b) affects the pathogenesis of insulin resistance by regulating EC function in white adipose tissue during obesity.
Methods and Results
MiR-181b expression was reduced in adipose tissue ECs of obese mice, and rescue of miR-181b expression improved glucose homeostasis and insulin sensitivity. Systemic intravenous delivery of miR-181b robustly accumulated in adipose tissue ECs, enhanced insulin-mediated Akt phosphorylation at Ser473, and reduced endothelial dysfunction, an effect that shifted macrophage polarization towards an M2 anti-inflammatory phenotype in epididymal white adipose tissue (eWAT). These effects were associated with increased eNOS and FoxO1 phosphorylation as well as nitric oxide activity in eWAT. In contrast, miR-181b did not affect insulin-stimulated Akt phosphorylation in liver and skeletal muscle. Bioinformatics and gene profiling approaches revealed that PHLPP2, a phosphatase that dephosphorylates Akt at Ser473, is a novel target of miR-181b. Knockdown of PHLPP2 increased Akt phosphorylation at Ser473 in ECs, and ‘phenocopied’ miR-181b’s effects on glucose homeostasis, insulin sensitivity, and inflammation of eWAT in vivo. Finally, ECs from diabetic subjects exhibited increased PHLPP2 expression.
Conclusions
Our data underscore the importance of adipose tissue EC function in controlling the development of insulin resistance. Delivery of miR-181b or PHLPP2 inhibitors may represent a new therapeutic approach to ameliorate insulin resistance by improving adipose tissue endothelial Akt-eNOS-NO signaling.
Keywords: microRNA-181b, insulin resistance, phosphatase, endothelial cells, adipose tissue, obesity
INTRODUCTION
Adipose tissue dysfunction, characterized by low grade inflammation, is considered to play a primary role in obesity-associated insulin resistance, which predisposes the majority of obese patients to the development of important chronic metabolic diseases including type 2 diabetes and cardiovascular diseases.1–4 Excessive caloric intake may expose tissues such as white adipose tissue to super-physiological levels of metabolic substrates and promote the development of low-grade inflammation.5 Inflamed white adipose tissue contains a range of leukocyte subsets including monocytes that preferentially differentiate toward M1 macrophages, and are associated with increased expression of proinflammatory cytokines.6, 7 Accumulating studies support the concept that chronic inflammation in white adipose tissue is critically involved in the pathogenesis of obesity-associated insulin resistance.1, 2, 4–6, 8–11 White adipose tissue is comprised of multiple cell types including not only adipocytes and leukocytes, but also endothelial cells (ECs). Cellular interactions emanating from white adipose tissue may control local and systemic homeostasis of cardiometabolic function. However, our understanding of the basic mechanisms linking EC dysfunction with the adipocyte response in insulin resistant states remains incompletely understood.
EC dysfunction is a common feature of type 2 diabetes and cardiovascular disease, and a hallmark of insulin resistance.12 The reciprocal relationships between insulin resistance and endothelial dysfunction are experimentally and clinically established.12–14 In both rodents and primates, EC activation is an early event that occurs prior to or in parallel with the development of impaired insulin signaling.15, 16 ECs of inflamed adipose tissue from obese subjects have adverse effects on insulin signaling in adipocytes such as reduced expression of phospho-Akt at Ser473, increased endoplasmic reticulum stress, and release of inflammatory mediators.17 On the other hand, EC function is subjected to the regulation by insulin-mediated signaling. For example, transient activation of endothelial PI3K/Akt signaling inhibits the expression of adhesion molecules involved in leukocyte rolling and adhesion to the vascular luminal wall.18 Insulin resistance also leads to EC dysfunction through increased circulating free fatty acids and hyperglycemia.19, 20 Indeed, ECs from visceral adipose tissue of obese mice or human subjects exhibit a marked inflammatory state with increased expression of chemokines, cytokines, and adhesion molecules.17, 21 Several studies have shown insulin signaling in ECs protect endothelial function and attenuates the progression of atherosclerosis.22–25 However, it remains unknown whether enhancing endothelial insulin signaling in adipose tissue may improve systemic insulin resistance.
Akt phosphorylation regulates many fundamental biological processes such as the insulin signaling cascade. Dysregulation of Akt phosphorylation often is involved in heart disease and diabetes.26, 27 Full Akt activity depends on the phosphorylation of residues Thr308 and Ser473, which can be repressed by the lipid phosphatase – PTEN and the protein phosphatase – PH domain leucine-rich repeat phosphatases isoform 2 (PHLPP2). PHLPP2 inactivates Akt signaling by specifically dephosphorylating Ser473 but not Thr308 of Akt.28, 29 It remains unknown whether altering the expression of PHLPP2 in ECs regulates Akt signaling, downstream substrates, insulin sensitivity, and glucose homeostasis.
MicroRNAs (miRNAs) are evolutionarily conserved small noncoding RNAs, which post-transcriptionally regulate gene expression by promoting mRNA degradation and/or inhibiting translation. It has been reported that miRNAs are differentially expressed in adipose tissue between lean and obese mice,30, 31 as well as lean and obese human subjects.32 We have shown that microRNA-181b (miR-181b) ameliorates NF-κB-mediated EC activation and vascular inflammation in mouse models of endotoxemia and atherosclerosis.33, 34 However, it remains unknown whether: 1) miR-181b expression is dysregulated in ECs of adipose tissue, and 2) increasing miR-181b expression in adipose tissue will ameliorate obesity-associated insulin resistance and inflammatory responses.
In this study, we examined the expression of miR-181b in adipose tissue ECs of obese mice and the role of miR-181b and PHLPP2 in modulating glucose homeostasis and insulin sensitivity. Our findings reveal that miR-181b improves insulin signaling and reduces inflammation and EC dysfunction in white adipose tissue by targeting endothelial PHLPP2 without altering hepatic steatosis or lipid profiles.
METHODS
Methods are provided in the Online Supplement.
RESULTS
MiR-181b is reduced by diabetic stimuli in endothelial cells
Differential expression of miRNAs has been observed in adipose tissue from lean and obese mice as well as humans, although miRNA expression in adipose tissue ECs has not been previously explored.30–32 To examine the expression of miR-181b, an anti-inflammatory microRNA that we previously identified in the macrovasculature33, 34, ECs or adipocytes were isolated as described21, 35–37 from eWAT of C57BL/6 mice fed a 60% HFD for 0, 3, 7, or 14 days followed by qPCR analysis (Figure 1A). We found miR-181b is the most dominantly expressed among miR-181 family members in adipose tissue ECs as demonstrated that its expression is 3.6-fold higher than that of miR-181a. MiR-181b expression was reduced by 33% and 36% after HFD for 7 and 14 days, respectively, while the expression of miR-181a and miR-181c was reduced by 53% and 67% after HFD for 14 days (Figure 1A). In contrast, the expression of miR-181 family members was not significantly changed in adipocytes after HFD for the indicated days (Figure 1A), and miR-181b expression was not reduced in liver or skeletal muscle ECs (Online Figure I A). However, miR-181b expression was reduced by 24%, 32%, 31%, and 33% in ECs in vitro after treatment with glucose, PMA, TNF-α, or palmitate (Figure 1B). In addition, we examined miR-181b expression in ECs of eWAT over the period of 12-weeks of HFD. MiR-181b expression was decreased by 42%, 84% and 66% in ECs isolated from eWAT of mice fed a HFD for 2, 6, and 12 weeks, respectively (Online Figure I B). Although the ratios of phosphorylated Akt / total Akt were not changed under basal conditions (Online Figure I C), insulin responsiveness was blunted as indicated by reduced phosphorylation of Akt in ECs isolated from eWAT of mice fed on HFD at 2 weeks (Online Figure I D). The expression of VCAM-1, ICAM-1, and E-selectin were increased in ECs from eWAT of mice fed a HFD for 2 weeks compared to chow-fed mice (Online Figure I E). These data demonstrate that the reduction of miR-181b in ECs is an early event during the development of insulin resistance, which was associated with ongoing inflammatory responses.
Figure 1. MiR-181b is reduced in response to inflammatory stimuli or hyperglycemia in endothelial cells.
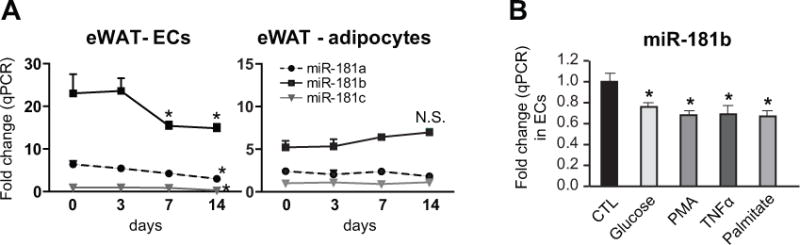
A, C57BL/6 mice were fed a high-fat diet as indicated (0, 3, 7, or 14 days). Endothelial cells (ECs) and adipocytes were isolated from eWAT for qPCR. The expression of miR-181 was normalized to small RNA U6 expression and compared to the expression of miR-181c in mice on chow that was subsequently set to a value of one, n=6 per group. B, ECs were cultured in the absence or presence of 30 mM D-glucose for 48 hours,100 nM PMA for 24 hours, 10 ng/ml TNF-α or 100 μM palmitate for 4 hours and harvested for qPCR of miR-181b, n=3 per group. Data show Mean ± SEM; *, P < 0.05. N.S., non-significant.
MiR-181b improves glucose tolerance and insulin sensitivity in a mouse model of diet-induced diabetes
Our prior observations indicate that miR-181b serves as an anti-inflammatory regulator in the macrovasculature. Based upon the reduced expression of miR-181b in the microvasculature of white adipose tissue, we hypothesize that rescue of miR-181b expression may delay the progression of inflammation and insulin resistance, and improve insulin sensitivity. To examine the effect of miR-181b systemic delivery on glucose homeostasis and insulin sensitivity, C57BL/6 mice were fed a 60% HFD for 12 weeks. After 6 weeks HFD, mice were treated with miR-181b mimics (181b-m) or miRNA negative control (NS-m) for 6 weeks (twice a week, i.v. 1nmol/injection) (Figure 2A). Insulin Tolerance Tests (ITT) and Glucose Tolerance Tests (GTT) were performed at week 5 and 6, respectively, after miR-181b treatment (Figure 2A). The body weights were significantly increased in HFD-fed mice, which were independent of miRNA treatments (Figure 2B). However, miR-181b treatment markedly improved glucose tolerance (Figure 2C and 2D) and insulin sensitivity (Figure 2E and 2F) compared with NS-m treatment. MiR-181b reduced the area under curves for ITT and GTT by 57% and 38%, respectively, compared to control mice (Figure 2D and 2F). These beneficial effects occurred independent of any changes in lipid profiles, fat mass, or plasma levels of insulin and free fatty acids (Online Table I). Taken together, these data demonstrate that miR-181b delivery is able to improve glucose homeostasis and insulin sensitivity.
Figure 2. Systemic delivery of miR-181b improves glucose tolerance and insulin sensitivity in diet-induced obese mice.
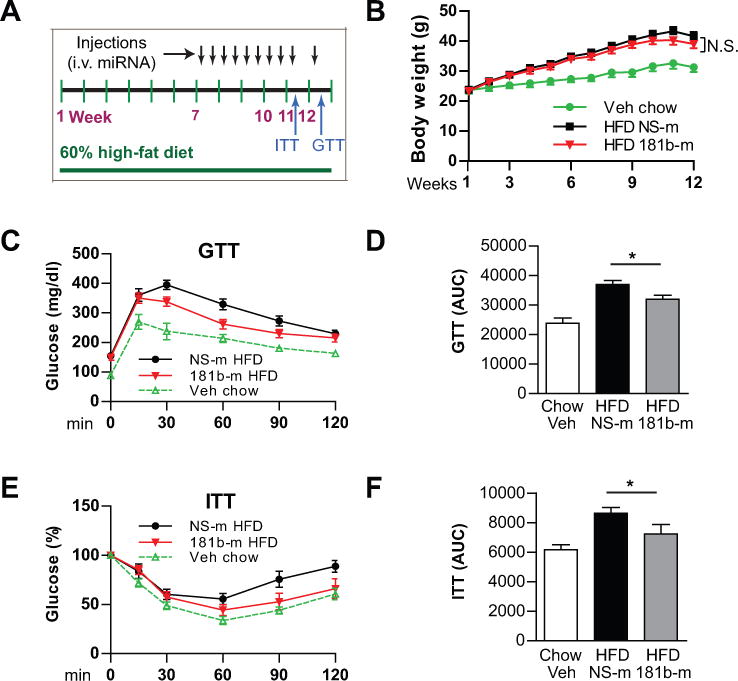
A, Schema of experimental procedures. C57BL/6J mice were fed a 60% high-fat diet (HFD) for 12 weeks. Six weeks after HFD, mice were treated with miR-181b (181b-m) or miRNA negative control (NS-m) for 6 weeks as indicated (i.v. 0.6 mg/kg). B, Body weights over time of mice on chow (injected with vehicle) or HFD (injected with NS-m or miR-181b). C and E, Blood glucose levels were measured on week 11 for the insulin tolerance test (ITT) and week 12 for the glucose tolerance test (GTT). Values were compared with basal glucose levels which were set as 100% for ITT. D and F, Area under the curve (AUC) of glucose and insulin tests was quantified. Mean ± SEM, n=7–9 mice per group; *, P<0.05.
MiR-181b reduces inflammation in epididymal white adipose tissue
We have shown that miR-181b ameliorates NF-κB-mediated EC activation and vascular inflammation in mouse models of endotoxemia and atherosclerosis.33, 34 This prompted us to examine the effect of miR-181b delivery on inflammation and EC dysfunction in obese mice. First, paraffin sections of eWAT or liver were stained for the macrophage marker Mac2. Macrophage content indicated by Mac2 staining was significantly increased in eWAT of mice after 12-week of HFD. However, macrophage accumulation in eWAT of miR-181b-treated mice was reduced by 60% compared to NS ctrl-treated mice (Figure 3A). The macrophage content in liver was not reduced by miR-181b delivery. Second, macrophage M1 and M2 markers were examined by qPCR. MiR-181b delivery reduced the expression of the macrophage M1 markers Tnfa, Il-1b, and Il12 by 48%, 51%, 39%, respectively, and increased the expression of M2 markers Mrc2, Mgl2, Fizz1, Ym1 by 43%, 33%, 60%, and 21%, respectively, in eWAT; while it exerted minimal effects on M1 and M2 markers of macrophage in liver (Figure 3B). Third, we observed that miR-181b delivery reduced ICAM-1 and VCAM-1 expression in eWAT from obese mice (Figure 3C), suggesting that EC dysfunction in eWAT in obese mice was ameliorated by miR-181b delivery. These effects of miR-181b treatment are associated with a 1.5-fold overexpression of miR-181b in eWAT and ~15-fold overexpression in ECs of eWAT (Online Figure II A). Systemic delivery of miR-181b could lead to more pronounced exogenous miR-181b expression in specific cell types within eWAT such as adipocytes or adipose tissue ECs. To test this, the expression of miR-181b in eWAT or ECs isolated from eWAT was examined after three consecutive injections of miRNA negative control (NS-m) or miR-181b. Three daily injections of miR-181b result in 105-fold expression of miR-181b in adipose tissue ECs, and 29-fold expression in eWAT (Online Figure II B). These data suggest that systemic delivery of miR-181b leads to predominant accumulation of exogenous miR-181b in adipose tissue ECs within eWAT, and reduces EC activation, macrophage accumulation, and inflammatory phenotype in white adipose tissue.
Figure 3. Systemic delivery of miR-181b reduces inflammation in epididymal fat.
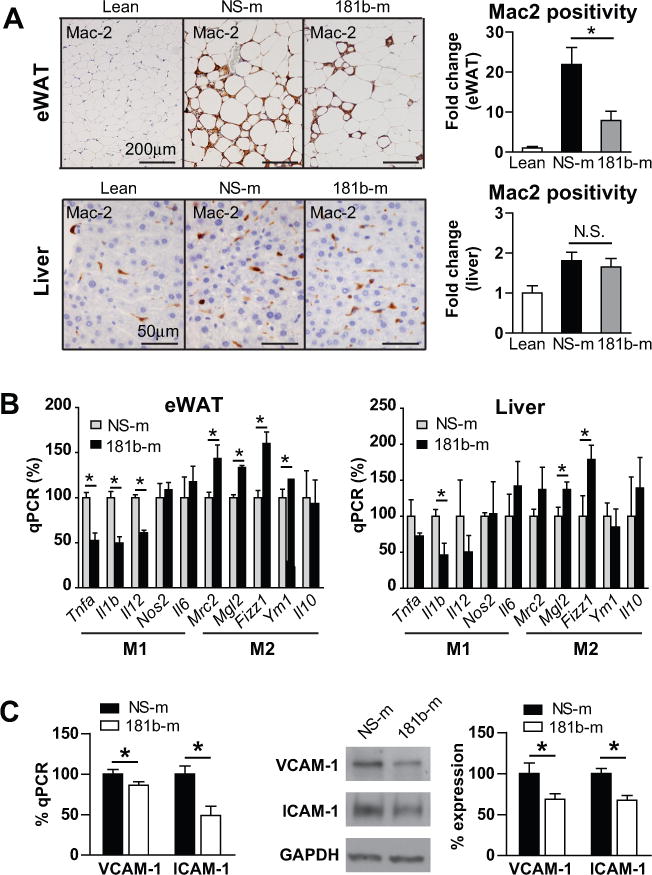
A, Paraffin sections of eWAT or liver were stained with Mac2, and the positive areas were quantified. B, qPCR of gene expression in epididymal white adipose tissue (eWAT) and liver; results were presented relative to those of miRNA negative control (NS-m) treated mice. C, qPCR and western blot analysis of VCAM-1 and ICAM-1 in eWAT. Mean ± SEM, n=6–9; *, P < 0.05.
MiR-181b does not directly inhibit the cell-intrinsic capacity of monocytes/macrophages to migrate, proliferate, or be activated
Macrophage infiltration, proliferation, and activation are all involved in the pathogenesis of obesity-induced insulin resistance.38, 39 Reduced macrophage content in eWAT could result from the direct effect of miR-181b on migratory and proliferative ability of monocytes/macrophages or by directly reducing EC activation and dysfunction. To explore any direct effects of miR-181b on monocyte migration, adoptive transfer of monocytes was conducted as previously described.40 Monocytes were isolated from HFD-fed mice treated with miR-181b or miRNA negative control (NS-m), labeled with PKH26, and injected into HFD-fed obese mice. Stromal vascular fractions were isolated from eWAT of the recipient mice two days later, and subjected to FACS analysis.41 As shown in Figure 4A, miR-181b overexpression (25-fold, data not shown) in monocytes does not significantly inhibit their migration into eWAT revealed by the percentage of PKH26-positive cells among CD11b and F4/80 double-positive cells [NS-m: (22.1 ± 5.1) %; 181b-m: (19.2 ± 4.0) %]. To examine the effect of miR-181b on macrophage proliferation, staining for Ki67 (cell division marker), F4/80 (macrophage marker), and DAPI (nuclear marker) was performed on paraffin sections of eWAT from mice. The percentages of proliferating macrophages are (13.9 ± 1.9) % and (12.5 ± 1.1) % in NS-m or miR-181b treated mice respectively, suggesting systemic delivery of miR-181b did not affect macrophage proliferation in eWAT (Figure 4B). We previously showed that miR-181b does not inhibit NF-κB activation in macrophages and NF-κB target gene expression in PBMCs.33 Consistently, miR-181b did not affect TNF-α, IL-1β, and COX-2 gene expression in PBMCs isolated from insulin-resistant mice (Figure 4C). Collectively, these data indicate that miR-181b does not directly regulate cell-intrinsic monocytes/macrophage function including migration, proliferation, and activation.
Figure 4. MiR-181b does not inhibit the proliferation, migration, and activation of monocytes/macrophages.
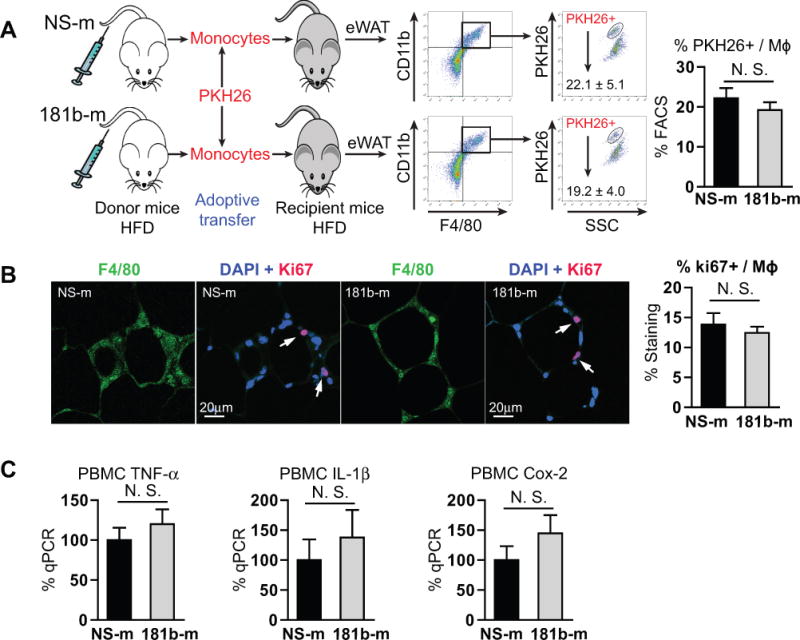
A, Paraffin sections of epididymal white adipose tissue (eWAT) were stained with F4/80, DAPI and Ki67. The percentages of Ki67 among F4/80 positive cells were calculated. Mean ± SEM, n=7–10 mice per group. B, Adoptive transfer of PKH26 labeled monocytes from obese mice overexpressing miRNA negative control (NS-m) or miR-181b, and FACS analysis of PKH26 positive cells among CD11b and F4/80 double positive cells in the eWAT of recipient obese mice. Mean ± SEM, n=4 mice per group. C, PBMCs were isolated from NS-m- or miR-181b-treated mice fed on a high-fat diet for qPCR analysis. Data show mean ± SEM, n=6 mice per group.
MiR-181b expression promotes glucose uptake in adipocytes in a paracrine manner
Since miR-181b does not directly regulate cell-intrinsic functions of monocytes/macrophages, the protective effects of miR-181b delivery on insulin signaling in eWAT may result from the direct effects of miR-181b on adipocytes or ECs. To assess this, glucose uptake experiments were performed in differentiated, mature 3T3-L1 adipocytes. Consistent with our hypothesis, miR-181b overexpression in 3T3-L1 adipocytes did not promote glucose uptake (Figure 5A). ECs of inflamed adipose tissue may have adverse effects on insulin signaling in adipocytes, possibly via paracrine effects.17 Since miR-181b attenuates endothelial inflammation in adipose tissues (Figure 3C), we reasoned that miR-181b improves insulin signaling in a paracrine manner by exerting protective effects in ECs. Indeed, the conditioned medium (Supe) from ECs overexpressing miR-181b markedly improved glucose uptake in adipocytes (Figure 5B). These data suggest that miR-181b overexpression in ECs may improve glucose uptake via paracrine mechanisms with adipocytes.
Figure 5. Glucose uptake in 3T3-L1 adipocytes is promoted in the presence of conditioned medium from endothelial cells overexpressing miR-181b.
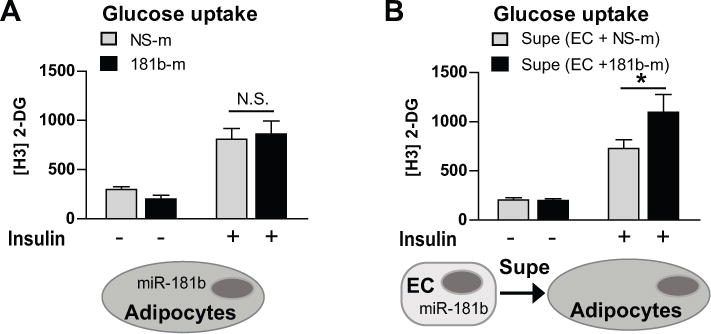
A, Adipocytes were transfected with miRNA negative control (NS-m) or miR-181b and glucose uptake was quantified. B, Adipocytes were cultured with conditioned medium from ECs overexpressing NS-m or miR-181b and glucose uptake was quantified. Mean ± SEM, n=4; *, P < 0.05. N.S., non-significant. Supe, supernatant.
MiR-181b delivery improves insulin signaling by increasing Akt phosphorylation in epididymal white adipose tissue
Because Akt phosphorylation is a central event in insulin signaling cascade, we examined the expression of phospho-Akt (Ser473 and Thr308) in eWAT, skeletal muscle, and liver. HFD mice were treated with miR-181b or NS ctrl mimics in an analogous manner as Figure 2 and stimulated in the presence or absence of insulin for an additional 10 min prior to tissue harvest. In insulin-stimulated miR-181b-treated mice, phospho-Akt (Ser473) was significantly increased 2.2-fold in eWAT, whereas there were no significant differences in skeletal muscle or liver (Figure 6A). Phospho-Akt (Thr-308) was not changed by miR-181b delivery compared with controls in all the tissues examined. Moreover, the effect of miR-181b delivery on upstream insulin signaling was examined in eWAT. Since inflammatory stimuli can blunt insulin action by affecting the phosphorylation of mediators in upstream insulin signaling such as insulin receptor beta (InR-β) and insulin receptor substrate 1 (IRS1),42–45 their phosphorylation status was examined by immunoprecipatation assays from eWAT lysates (Figure 6B). MiR-181b delivery did not affect the expression of phospho-InR-β (Tyr1162) and phospho-IRS1 (Ser307) in eWAT, suggesting upstream insulin signaling was not changed by miR-181b. Both FoxOs and eNOS are important molecules mediating insulin signaling events downstream of phospho-Akt in ECs.46–48 We found phospho-FoxO1 (Ser 256) is increased up to 2.0-fold in eWAT of miR-181b treated mice compared with controls in response to insulin (Figure 6B). Consistently, miR-181b overexpression increased Akt phosphorylation at Ser473 and reduced nuclear accumulation of FoxO1 at 10 min after insulin stimulation in ECs (Figure 6C). Similarly, miR-181b increased phospho-eNOS (Ser1176) by 1.7-fold (Figure 6D) and nitric oxide (NO) activity by 1.5-fold (Online Figure III) in eWAT compared to controls after insulin stimulation. In a separate experiment, obese mice were treated with miR-181b using the same dosing regimen as outlined in Figure 2, and ECs were isolated from eWATs to examine the effects of miR-181b delivery on insulin signaling and inflammation. Consistent with the effects of miR-181b on insulin signaling and inflammation in eWATs, we found miR-181b increased the phosphorylation of Akt by ~1.6-fold (Figure 6E) and reduced ICAM-1 expression by 43% in ECs isolated from eWATs (Online Figure IV). These data indicate that miR-181b delivery reduced insulin resistance and inflammation in ECs within eWATs. We also examined the expression of genes involved in thermogenesis and hepatic glucose production. The mRNA expression of UCP-1, PRDM16, and PGC-1α genes were not changed in brown fat and eWAT by miR-181b, suggesting that the thermogenic program is not affected by miR-181b delivery (Online Figure V A). To exclude any effects of miR-181b on hepatic glucose production, we examined miR-181b delivery on the expression of key metabolic enzymes involved in maintaining hepatic glucose homeostasis in liver from mice described in Figure 2. MiR-181b had no effect on mRNA expression for GS, GK, PK, GP, FBP1, G6P and PEPCK (Online Figure V B). Finally, miR-181b delivery had no effect on regulating lipid homeostasis in liver revealed by Oil Red O staining (Online Figure V C). These data suggest that systemic delivery of miR-181b improves glucose homeostasis and insulin sensitivity in a tissue-specific manner by promoting Akt phosphorylation and sensitizing insulin action in eWAT independent of any effects on insulin signaling in the liver and skeletal muscle, or lipid accumulation in the liver.
Figure 6. MiR-181b delivery improves Akt phosphorylation at Serine 473 in epididymal white adipose tissue.
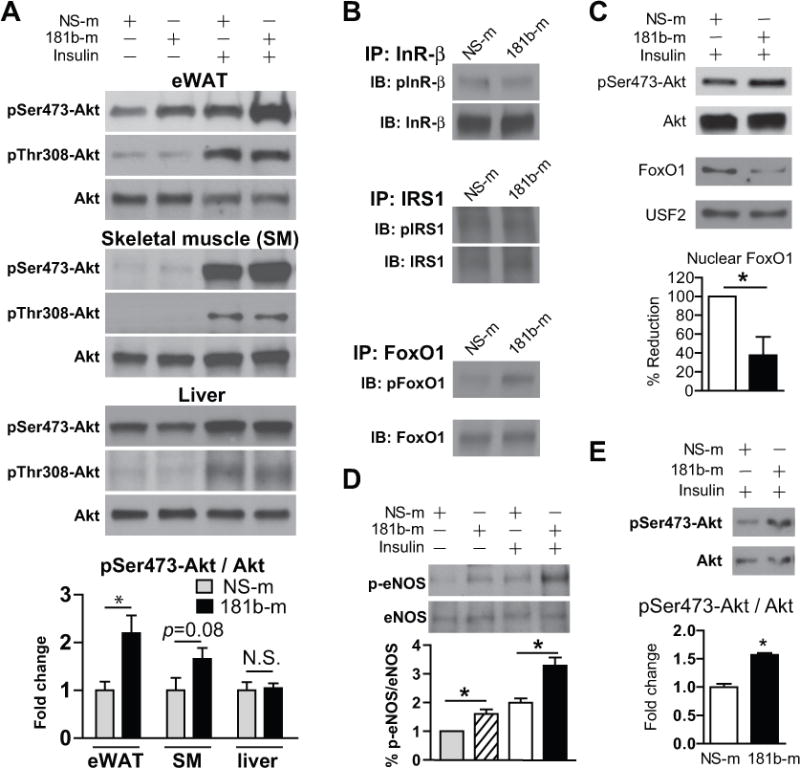
A, Western blot analysis of Akt phosphorylation at Serine 473 and Threonine 308, and quantifications of pSer473-Akt vs. total Akt in epididymal white adipose tissue (eWAT), skeletal muscle (SM), or liver from insulin-stimulated mice, n=6–9. B, Lysates of eWAT were used for immunoprecipitation and western blot analysis. C, MiRNA negative control (NS-m) or miR-181b-transfected HUVECs were treated with 100 nM insulin for 10 minutes. Western blot analysis of pSer473-Akt in total lysates, and total FoxO1 in nuclear fraction, n=3 independent experiments. D, Western blot analysis of eNOS phosphorylation at Serine 1176, and quantifications of pSer1176-eNOS vs. total eNOS in eWAT from insulin-stimulated mice, n=6–9. E, Western blot analysis of Akt phosphorylation at Serine 473 in ECs isolated from eWAT of obese mice treated with NS-m or miR-181b, n=6 mice / group. All values show Mean ± SEM; *, P < 0.05.
The reduction of endogenous miR-181b by inflammatory stimuli such as TNF-α could affect insulin signaling in ECs. Indeed, TNF-α treatment reduced insulin-induced Akt phosphorylation in ECs (Online Figure VI A) as previously described.49 MiR-181b inhibition reduced insulin-induced Akt phosphorylation, which was potentiated in the presence of TNF-α treatment (Online Figure VI A). Furthermore, miR-181b overexpression rescued the reduction of insulin-induced Akt phosphorylation by TNF-α treatment (Online Figure VI B). The data imply that the early reduction of endogenous miR-181b in ECs of eWAT is likely involved in the pathogenesis of endothelial insulin resistance in eWATs during the development of obesity.
MiR-181b targets PHLPP2
MiR-181b could improve insulin signaling by directly regulating a target intrinsic to this pathway, or by suppressing target genes of other inter-related signaling pathways. Our previous studies demonstrated that miR-181b can repress importin-α3, a protein involved in the nuclear translocation of NF-κB (p65/p50), by binding to its 3′-UTR.33, 34 MiR-181b could inhibit adipose tissue inflammation by reducing importin-α3 expression in ECs. We examined NF-κB p65 nuclear accumulation in adipose tissue ECs. Surprisingly, nuclear p65 expression was not significantly induced by HFD-feeding, and miR-181b delivery did not reduce p65 nuclear accumulation in adipose tissue ECs (Online Figure VII). A combined strategy using bioinformatics and microarray gene chip profiling was taken to identify potential miR-181b direct targets. Genes reduced by miR-181b overexpression identified by microarray gene chip analysis (GEO database accession no. GSE35030)34 that are also predicted as direct targets of miR-181b by different algorithms will be of interest as potential miR-181b targets. Six algorithms predicted 1280 mouse and human orthologous genes as miR-181b direct targets (Figure 7A). Among 4190 genes identified by microarray gene chip assay with more than 1.2-fold reduction by miR-181b overexpression, 306 genes were also found in the list of 1280 genes predicted by in silico algorithms50 as miR-181b direct targets (Figure 7A). Gene ontology analysis of these 306 genes revealed that the targets of miR-181b encode regulators of biological processes such as protein amino acid phosphorylation, phosphorylation, intracellular signaling cascade, protein serine/threonine kinase activity, among others (Figure 7B). We focused on phosphatases since miR-181b likely promotes Akt phosphorylation at Serine 473 by reducing the expression of a phosphatase. Several serine/threonine phosphatases have been shown to regulate insulin signaling, and are involved in the pathogenesis of insulin resistance.29, 51, 52 Among the combined list of 306 genes was Pleckstrin homology domain leucine-rich repeat protein phosphatase (PHLPP) isozyme2 (PHLPP2) which can directly de-phosphorylate and inactivate Akt.28, 29 Indeed, we found miR-181b reduced PHLPP2 protein expression in ECs (Figure 7C). To verify that miR-181b directly targets PHLPP2, we performed Argonaute2 (AGO2) micro-ribonucleoprotein IP (miRNP-IP) studies to assess whether PHLPP2 mRNA is enriched in the RNA-induced silencing complex (RISC) following miR-181b overexpression. An approximately 4-fold enrichment of PHLPP2 mRNA was observed after AGO2 miRNP-IP in the presence of miR-181b, as compared with that with the miRNA-negative control (Figure 7D). Furthermore, miR-181b reduced the PHLPP2 3′-UTR activity by 54% (Figure 7E). To show the specificity of miR-181b on PHLPP2 3′UTR, the seed sequence in pcDNA3.1-miR-181b construct was mutated. MiR-181b mutant lost the ability to inhibit the activity of PHLPP2 3′UTR as shown in Figure 7E. Importantly, pharmacological inhibition53 or siRNA knockdown of PHLPP2 promoted insulin-induced Akt phosphorylation in ECs (Figure 7F). To examine whether miR-181b targets PHLPP2 in vivo, PHLPP2 expression was examined in lysates of eWAT, skeletal muscle, and liver from obese mice treated with miR-181b (twice a week for 6 weeks). Systemic delivery of miR-181b reduced PHLPP2 protein expression in eWAT, but not in skeletal muscle or liver (Figure 7G and Online Figure VIII A). Because PHLPP2 expression is significantly lower in liver (nearly undetectable) than in eWAT and skeletal muscle, PHLPP2 may play a minimal role in regulating insulin signaling in liver (Online Figure VIII B). Moreover, miR-181b treatment (twice a week for 6 weeks) reduced PHLPP2 expression by 55% in ECs isolated from eWAT of obese mice (Figure 7H). These results identify PHLPP2 as a bona fide direct target of miR-181b. PHLPP2 expression was significantly increased in ECs of eWAT from mice fed a HFD for 2, 6, or 12 weeks (Online Figure VIII C), suggesting a counter-regulation of the miR-181b target PHLPP2 during the development of insulin resistance. In addition to PHLPP2, we examined the effects of miR-181b on other potential phosphatases including INPP5E, PPM1A, CTDSPL, and PPAR2B, which are predicted as miR-181b targets among 306 genes. MiR-181b overexpression reduced the expression of CTDSPL and PPAP2B by 41% and 56%, respectively, in HUVECs (Online Figure VIII D). In contrast, miR-181b overexpression had no effects on the expression of PPM1A and INPP5E. However, siRNA-mediated knockdown of CTDSPL or PPAP2B did not promote the phosphorylation of Akt at Serine 473 in response to insulin in HUVECs (Online Figure VIII E), suggesting CTDSPL and PPAR2B did not mediate miR-181b’s effect on insulin-induced Akt phosphorylation. It has been reported that miR-181b reduces IGF-1R expression in tumor cells, and inhibits VEGF-induced PI3K-Akt signaling;54 in contrast, in T cells it may reduce PTEN expression, thereby promoting PI3K-Akt signaling.55 Therefore, we examined whether miR-181b regulates Akt phosphorylation by reducing IGF-1R and PTEN expression in ECs. As shown in Online Figure VIII F, miR-181b overexpression did not reduce PTEN and IGF-1R expression in ECs, and also had no effect on PTEN expression in eWAT. These data suggest a cell-specific regulation of PTEN and IGF-1R expression by miR-181b. Collectively, these results identify PHLPP2 as a direct target of miR-181b that may mediate miR-181b’s effects on the insulin signaling pathway.
Figure 7. MiR-181b targets PHLPP2.
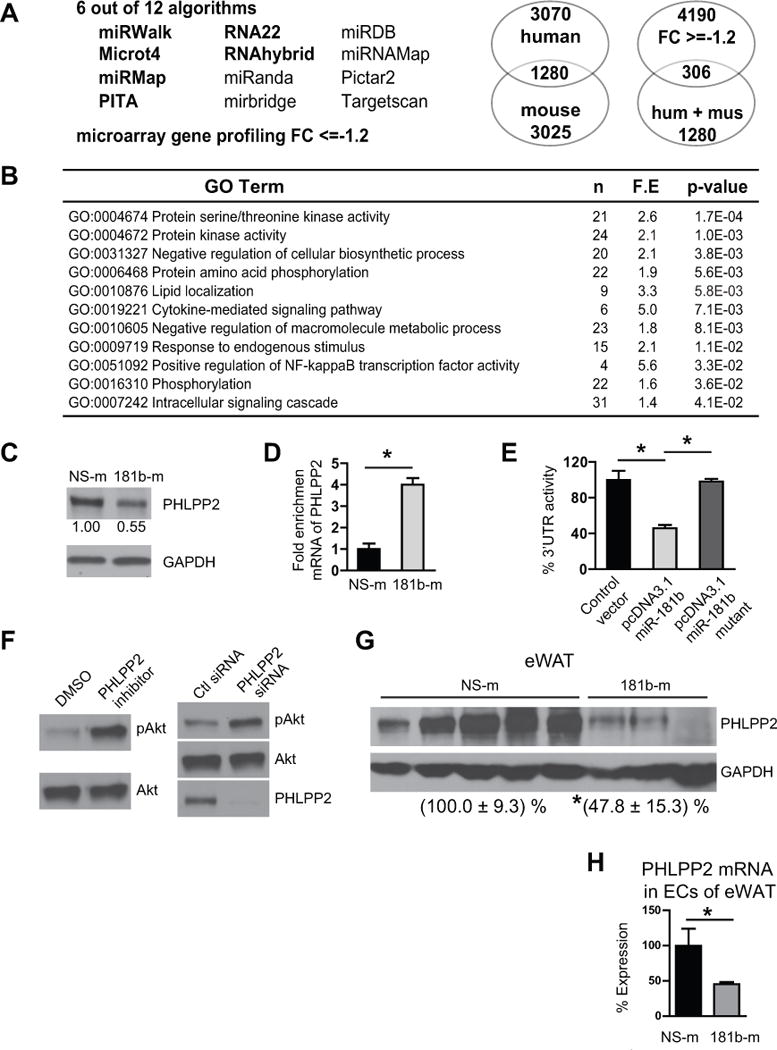
A, Bioinformatic approach predicts miR-181b direct targets among genes reduced by miR-181b overexpression in ECs identified by microarray gene chip profiling. FC, fold-change. B, Gene ontology analysis of 306 genes identified in A. C, Western blot analysis of endothelial cells (ECs) transfected with 10 nM miRNA negative control (NS-m) or 181b-m. D, miRNP-IP analysis of enrichment of PHLPP2 mRNA in HUVECs transfected with NS-m or 181b-m, n=2 independent experiments. E, Luciferase reporter assay of PHLPP2 3′UTR in the presence of pcDNA3.1(+), pcDNA3.1(+)-miR-181b, and pcDNA3.1-miR-181b mutant in HUVECs, n=3 independent experiments. F, Pharmacological inhibition or siRNA knockdown of PHLPP2 increases pSer473-Akt in ECs in response to 100 nM insulin at 10 minutes. G, Western blot analysis of PHLPP2 expression in eWAT, n= 3 – 5. H, PHLPP2 expression was detected in ECs isolated from epididymal white adipose tissue (eWAT) of obese mice treated with NS-m or miR-181b using the dosing regimen as outlined in Figure 2, n=6 mice per group. All values showmean ± SEM; *, P < 0.05.
PHLPP2 knockdown improves glucose tolerance and insulin sensitivity in diet-induced diabetic mice
To examine whether PHLPP2 knockdown will phenocopy miR-181b’s effects to improve glucose homeostasis, insulin sensitivity, and reduce adipose tissue dysfunction in vivo, C57BL/6 obese mice were therapeutically treated with PHLPP2 siRNAs or negative control siRNAs for 6 weeks (twice a week, i.v. 1nmol/injection) (Figure 8A). PHLPP2 siRNAs injection did not affect the body weights of mice compared with control siRNAs (Figure 8B). However, GTT and ITT studies revealed that PHLPP2 knockdown significantly improved glucose tolerance (Figure 8C and 8D) and insulin sensitivity (Figure 8E and 8F) by 19% and 20%, respectively, compared to mice treated with control siRNAs controls (Figure 8D and 8F). The effect of PHLPP2 knockdown on ITT and GTT was associated with a ~1.5-fold increase of Akt phosphorylation at Ser473 in eWAT but not in liver (Figure 8G). Macrophage content indicated by Mac-2 staining was also reduced by 58% in eWAT of PHLPP2 siRNA treated mice (Figure 8H). In a separate experiment, three daily injections of PHLPP2 siRNA lead to a 72% and 12% reduction of PHLPP2 mRNA expression in adipose tissue ECs and adipocytes, respectively, suggesting that the majority of systemically delivered siRNAs accumulate in adipose tissue ECs (Figure 8I). Interestingly, PHLPP2 expression revealed by immunostaining was increased by 33% in ECs isolated from diabetic patients compared with control subjects (Figure 8J and Online Figure IX). PHLPP2 inhibition may represent a new therapeutic approach to reduce insulin resistance by improving vascular EC function within eWAT. These data suggest that knockdown of PHLPP2 improves glucose tolerance and insulin sensitivity, reduces adipose tissue endothelial inflammation, and inhibits macrophage accumulation.
Figure 8. Systemic delivery of PHLPP2 siRNA improves glucose tolerance and insulin sensitivity in diet-induced obese mice.
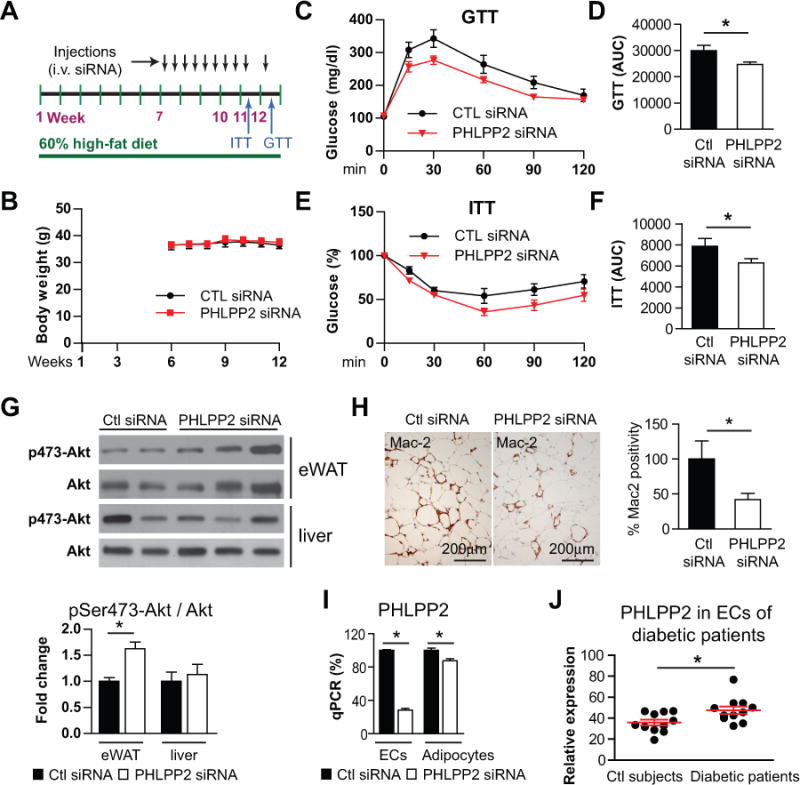
A, Schema of experimental procedure. C57BL/6J mice were fed a 60% high-fat diet (HFD) for 12 weeks. Six weeks after HFD, mice were treated with negative control siRNA or PHLPP2 siRNA for the subsequent 6 weeks as indicated (i.v. 0.6 mg/kg). B, Body weights over time of mice injected with control siRNAs or PHLPP2 siRNAs respectively. C and E, Blood glucose levels were measured on week 11 for the insulin tolerance test (ITT) and week 12 for the glucose tolerance test (GTT). Values were compared with basal glucose levels which were set as 100% for ITT. D and F, Area under the curves (AUC) for ITT and GTT were quantified. G, Western blot analysis of Akt and pSer473-Akt in epididymal white adipose tissue (eWAT) and liver. H, Paraffin sections of eWAT were stained with Mac2, and the positive areas were quantified. B–H, n=6–7. I, Mice were injected with negative control or PHLPP2 siRNAs three times on consecutive days, and the expression of PHLPP2 was examined in endothelial cells (ECs) or adipocytes isolated from eWAT, n=3 mice per group. J, Quantification of PHLPP2 protein expression in ECs freshly isolated from control and diabetic subjects, n=11 subjects each group. All data show mean ± SEM; *, P<0.05.
DISCUSSION
A number of miRNAs have been identified as regulators of obesity-induced insulin resistance. For example, miR-802, miR-143, and miR-103/7 all regulate glucose metabolism and insulin sensitivity in obesity.56–58 It remains unknown whether miRNAs affect insulin sensitivity by regulating EC function within eWAT. In the present study, we have discovered that: (1) the expression of miR-181b is reduced in white adipose tissue ECs of HFD mice, which can be rescued by miR-181b systemic delivery; (2) miR-181b delivery improves glucose homeostasis and insulin sensitivity associated with reduced EC activation, macrophage infiltration, and inflammatory phenotype in eWAT; (3) miR-181b had no direct effects on regulating monocyte/macrophage activation, proliferation, or recruitment in vivo; (3) miR-181b targets PHLPP2, a phophatase that dephosphorylates Akt at Ser473 in ECs; and (4) siRNA-mediated knockdown of PHLPP2 phenocopies miR-181b’s effect on glucose homeostasis, insulin sensitivity, and eWAT macrophage accumulation. We demonstrated that enhanced insulin signaling in adipose ECs exerts beneficial effects and promotes glucose uptake in adipocytes in a paracrine manner, and importantly, improves systemic glucose homeostasis and insulin sensitivity without altering hepatic steatosis or lipid profiles.
Several studies have been performed by other groups to uncover the causal relationships among vascular endothelial function, inflammation, and metabolic insulin resistance. For example, vascular inflammation (increased phosphorylation of IκBα and ICAM-1 expression) and insulin resistance (reduced phosphorylation of Akt and eNOS) are detectable in aortas of mice fed a HFD for 1 week,15 which precedes the onset of peripheral insulin resistance in liver, skeletal muscle, and adipose tissue. Another group showed systemic insulin resistance occurred in mice fed a HFD for three days revealed by GTT and hyperinsulinemic-euglycemic clamp studies.59 However, the relationship between EC function and endothelial insulin resistance within eWAT remains unclear. We examined Akt phoshporylation in ECs from eWATs of chow- and HFD-fed mice for 2 weeks. Our data suggest that ECs within eWAT display an insulin-resistant and inflammatory phenotype in mice after 2-week HFD (Online Figure I D and E). The reduction of miR-181b in ECs of eWAT was detected as early as 1 week after HFD (Figure 1A), suggesting that the reduction of miR-181b in ECs and associated endothelial activation was an early event during the development of insulin resistance.
Endothelial eNOS-NO signaling is impaired in ECs of visceral fat in obese patients.60 Activation of eNOS by insulin through Akt in ECs is important for limiting obesity-induced insulin resistance and inflammation.61 The production of NO by activated eNOS inhibits NF-κB activity, decreases cytokine-induced endothelial activation, and shifts macrophage polarization towards an M2 anti-inflammatory phenotype.62–64 MiR-181b delivery increased the phosphorylation of Akt and eNOS, increased NO activity (Online Figure III), and decreased FoxO activity and ICAM-1 expression in eWATs (Figure 6). In ECs of eWATs, miR-181b also increased Akt phosphorylation and decreased ICAM-1 expression (Figure 6E and Online Figure IV). In contrast, miR-181b delivery had no intrinsic effects on macrophage activation, infiltration, and proliferation (Figure 4). However, miR-181b delivery reduced macrophage accumulation and shifted M1 macrophages to an M2 anti-inflammatory phenotype in eWATs (Figure 3A). These data indicate that miR-181b improved insulin resistance in eWATs by primarily improving endothelial Akt-eNOS-NO signaling through targeting PHLPP2, an effect reducing endothelial ICAM-1 expression, EC-leukocyte interactions, and favorably shifting M1 to M2 macrophages thereby generating an anti-inflammatory milieu in eWAT. This anti-inflammatory milieu in eWATs generated by miR-181b may also promote glucose uptake in adipocytes through a paracrine manner (Figure 5).
Our results demonstrate that miR-181b delivery improves insulin signaling in epididymal fat but not in liver or skeletal muscle. There are at least two reasons that may account for this tissue-specific effect: 1) miRNAs may confer target and functional specificity in different cell types or tissues.65, 66 MiR-181b may target PHLPP2 in eWAT but not liver and skeletal muscle (Figure 7G and Online Figure VIII A); 2) tissue- and cell-specific accumulation of exogenous miR-181b. Although miR-181b delivery leads to its highest overexpression in liver, exogenous miR-181b is enriched higher in adipose ECs than in liver or skeletal muscle ECs (Online Figure II B). A fenestrated, discontinuous endothelium in liver may cause this differential accumulation of exogenous miR-181b. Furthermore, expression of the miR-181b target PHLPP2 is minimally expressed in liver compared to eWAT, a finding that may account for the lack of miR-181b’s effects on insulin signaling in liver. Interestingly, improved insulin signaling in liver ECs potentiated hepatic insulin resistance,67 suggesting potential phenotype functional differences of liver ECs vs eWAT ECs.
NF-κB mediated vascular inflammation participates in the pathogenesis of obesity-associated insulin resistance.15, 68, 69 We previously demonstrated that miR-181b reduces acute and chronic vascular inflammation by targeting endothelial importin-α3 and NF-κB signaling.33, 34 While we anticipated that miR-181b could improve insulin sensitivity by reducing NF-κB-mediated EC activation and inflammation; surprisingly, we found that nuclear accumulation of NF-κB p65 is not significantly induced in white adipose tissue ECs after 12 weeks HFD, and miR-181b delivery had no inhibitory effect (Online Figure VII), suggesting that miR-181b targets different gene(s) than importin-α3 in adipose tissue ECs. Indeed, using a combination of bioinformatics and gene microarray profiling studies, our studies revealed that miR-181b directly targets PHLPP2 in ECs. As miRNAs are known to regulate several targets, our study cannot rule out the possibility that miR-181b targets additional gene(s) that mediate its beneficial effects on insulin sensitivity and glucose homeostasis.
In our study, epididymal fat mass in miR-181b treated HFD mice was not reduced, an effect suggesting that miR-181b delivery does not reduce the capacity of white adipocytes/adipose tissue to store lipids in the context of over-nutrition. Consistent with this, the level of circulating triglycerides and liver lipid content were not significantly different between conditions. Furthermore, adipocytes incubated with conditioned medium from ECs overexpressing miR-181b exhibited markedly improved insulin resistance, an effect highlighting the potential importance of EC-adipocyte interactions in regulating the insulin resistant state. Our data also suggest that miR-181b is able to reduce adipose tissue EC inflammation, leukocyte accumulation, and improve systemic glucose homeostasis and insulin sensitivity without potentiating the development of hepatic steatosis.
In conclusion, our study in HFD mice demonstrates that miR-181b expression is reduced in response to HFD-induced obesity and that rescue of miR-181b in the microvasculature of eWAT is sufficient to improve glucose homeostasis and insulin sensitivity. MiR-181b delivery decreases inflammation in adipose tissue ECs in eWAT by targeting the phosphatase PHLPP2, an effect that increases phospho-AKT (Ser473) to improve insulin signaling. MiR-181b-mediated effects were selective for ECs of eWAT because miR-181b overexpression in adipocytes did not promote glucose uptake. These data indicate that strategies aimed at improving microvascular EC function in visceral fat in general and restoring miR-181b expression (or inhibition of PHLPP2 expression) in adipose tissue ECs in particular, may provide the basis for the rationale design of novel therapies for insulin resistance and its attendant cardiovascular complications.
Supplementary Material
Novelty and Significance.
What Is Known?
Obesity-associated insulin resistance is a major risk factor for cardiovascular disease.
Endothelial dysfunction contributes significantly to the pathogenesis of obesity-associated insulin resistance.
We previously showed that the microRNA-181b (miR-181b) inhibits NF-κB-mediated endothelial activation in response to acute (endotoxemia) and chronic (atherosclerosis) inflammation. MiR-181b reduces expression of importin-α3, a protein critical for NF-κB translocation from cytoplasm to nucleus. However, the role of miRNA-181b in obesity-associated insulin resistance has not been examined.
What New Information Does This Article Contribute?
MiR-181b expression in adipose tissue endothelial cells (ECs) is reduced early after high-fat diet in C57BL/6 mice, coincident with the onset of insulin resistance. Systemic intravenous delivery of miR-181b significantly improves glucose homeostasis and insulin sensitivity.
MiR-181b reduces adipose EC inflammation, inhibits macrophage infiltration, and shifts M1 to anti-inflammatory M2 macrophages in epididymal white adipose tissue (eWAT). MiR-181b is identified as a new regulator of EC-adipocyte interactions in white adipose tissue, thereby controlling insulin resistance and inflammation.
Mechanistically, miR-181b targets PHLPP2, a phosphatase that dephosphorylates Akt, leading to increased Akt phosphorylation at Serine 473 and improved EC insulin signaling. PhiosphoAkt in turn phosphorylate eNOS and increases nitric oxide activity in eWAT but not liver and skeletal muscle.
Knockdown of PHLPP2 mimics miR-181b’s effects including increased Akt phosphorylation in ECs and eWAT, reduced macrophage content in eWAT, and improved glucose homeostasis and insulin sensitivity.
Insulin resistance resulting from obesity commonly predisposes to the development of type 2 diabetes mellitus and cardiovascular disease. Chronic low-grade inflammation initiated in adipose tissue is a hallmark of obesity, which has been recognized as a key step in the development of insulin resistance. However, our understanding of adipose tissue EC dysfunction contributing to adipose tissue inflammation and insulin resistance remains inadequate. Results from this study will advance our understanding of miRNA-mediated regulation of adipose tissue EC function in the pathogenesis of adipose tissue insulin resistance and inflammation, which could provide novel and effective therapeutic strategies to manage a range of cardiovascular disease states that are exacerbated by insulin resistance.
Acknowledgments
We thank Dr. Alexandra Newton (University of California, San Diego) for providing the PHLPP2 inhibitors.
SOURCES OF FUNDING
This work was supported by the National Institutes of Health (HL115141, HL117994, and GM115605 to M.W.F.), the Arthur K. Watson Charitable Trust (to M.W.F.), the Dr. Ralph & Marian Falk Medical Research Trust, Bank of America, N.A. Trustee (to M.W.F.), a State Scholarship Fund of the China Scholarship Council (to J.B.L.), a Jonathan Levy Research Fund (to M.W.F.), American Heart Association (SDG#15SDG25400012 to X.H.S) and a Boston Nutrition Obesity Research Center Pilot and Feasibility award to X.H.S. under NIH/NIDDK P30KD046200.
Nonstandard Abbreviations and Acronyms
- EC
endothelial cell
- HFD
high-fat diet
- MiRNA
microRNA
- miR-181b
microRNA-181b
- 3′UTR
3′-untranslated region
- PBMCs
peripheral blood mononuclear cells
- NS-m
miRNA negative control mimics
- 181b-m
microRNA-181 mimics
- HUVECs
human umbilical vein endothelial cells
- GTT
glucose tolerance test
- ITT
insulin tolerance test
- Ewat
epididymal white adipose tissue
- PHLPP2
PH domain and leucine rich repeat protein phosphatase 2
Footnotes
Subject Terms:
Endothelium/Vascular Type/Nitric Oxide
Cell Signaling/Signal Transduction
Gene Expression and Regulation
DISCLOSURES
None.
References
- 1.Ouchi N, Parker JL, Lugus JJ, Walsh K. Adipokines in inflammation and metabolic disease. Nature reviews. Immunology. 2011;11:85–97. doi: 10.1038/nri2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annual review of immunology. 2011;29:415–445. doi: 10.1146/annurev-immunol-031210-101322. [DOI] [PubMed] [Google Scholar]
- 3.Wilson PW, D’Agostino RB, Sullivan L, Parise H, Kannel WB. Overweight and obesity as determinants of cardiovascular risk: The framingham experience. Archives of internal medicine. 2002;162:1867–1872. doi: 10.1001/archinte.162.16.1867. [DOI] [PubMed] [Google Scholar]
- 4.Guilherme A, Virbasius JV, Puri V, Czech MP. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nature reviews. Molecular cell biology. 2008;9:367–377. doi: 10.1038/nrm2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Odegaard JI, Chawla A. Pleiotropic actions of insulin resistance and inflammation in metabolic homeostasis. Science. 2013;339:172–177. doi: 10.1126/science.1230721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annual review of physiology. 2010;72:219–246. doi: 10.1146/annurev-physiol-021909-135846. [DOI] [PubMed] [Google Scholar]
- 7.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW., Jr Obesity is associated with macrophage accumulation in adipose tissue. The Journal of clinical investigation. 2003;112:1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arkan MC, Hevener AL, Greten FR, Maeda S, Li ZW, Long JM, Wynshaw-Boris A, Poli G, Olefsky J, Karin M. Ikk-beta links inflammation to obesity-induced insulin resistance. Nature medicine. 2005;11:191–198. doi: 10.1038/nm1185. [DOI] [PubMed] [Google Scholar]
- 9.Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nature reviews. Immunology. 2011;11:98–107. doi: 10.1038/nri2925. [DOI] [PubMed] [Google Scholar]
- 10.Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. The Journal of clinical investigation. 2011;121:2111–2117. doi: 10.1172/JCI57132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Solinas G, Karin M. Jnk1 and ikkbeta: Molecular links between obesity and metabolic dysfunction. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2010;24:2596–2611. doi: 10.1096/fj.09-151340. [DOI] [PubMed] [Google Scholar]
- 12.Kim JA, Montagnani M, Koh KK, Quon MJ. Reciprocal relationships between insulin resistance and endothelial dysfunction: Molecular and pathophysiological mechanisms. Circulation. 2006;113:1888–1904. doi: 10.1161/CIRCULATIONAHA.105.563213. [DOI] [PubMed] [Google Scholar]
- 13.Muniyappa R, Sowers JR. Role of insulin resistance in endothelial dysfunction. Reviews in endocrine & metabolic disorders. 2013;14:5–12. doi: 10.1007/s11154-012-9229-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Del Turco S, Gaggini M, Daniele G, Basta G, Folli F, Sicari R, Gastaldelli A. Insulin resistance and endothelial dysfunction: A mutual relationship in cardiometabolic risk. Current pharmaceutical design. 2013;19:2420–2431. doi: 10.2174/1381612811319130010. [DOI] [PubMed] [Google Scholar]
- 15.Kim F, Pham M, Maloney E, Rizzo NO, Morton GJ, Wisse BE, Kirk EA, Chait A, Schwartz MW. Vascular inflammation, insulin resistance, and reduced nitric oxide production precede the onset of peripheral insulin resistance. Arteriosclerosis, thrombosis, and vascular biology. 2008;28:1982–1988. doi: 10.1161/ATVBAHA.108.169722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chadderdon SM, Belcik JT, Bader L, Kirigiti MA, Peters DM, Kievit P, Grove KL, Lindner JR. Pro-inflammatory endothelial activation detected by molecular imaging in obese non-human primates coincides with the onset of insulin resistance and progressively increases with duration of insulin resistance. Circulation. 2013 doi: 10.1161/CIRCULATIONAHA.113.003645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pellegrinelli V, Rouault C, Veyrie N, Clement K, Lacasa D. Endothelial cells from visceral adipose tissue disrupt adipocyte functions in a three-dimensional setting: Partial rescue by angiopoietin-1. Diabetes. 2014;63:535–549. doi: 10.2337/db13-0537. [DOI] [PubMed] [Google Scholar]
- 18.Aljada A, Saadeh R, Assian E, Ghanim H, Dandona P. Insulin inhibits the expression of intercellular adhesion molecule-1 by human aortic endothelial cells through stimulation of nitric oxide. The Journal of clinical endocrinology and metabolism. 2000;85:2572–2575. doi: 10.1210/jcem.85.7.6677. [DOI] [PubMed] [Google Scholar]
- 19.Geraldes P, Hiraoka-Yamamoto J, Matsumoto M, Clermont A, Leitges M, Marette A, Aiello LP, Kern TS, King GL. Activation of pkc-delta and shp-1 by hyperglycemia causes vascular cell apoptosis and diabetic retinopathy. Nature medicine. 2009;15:1298–1306. doi: 10.1038/nm.2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maloney E, Sweet IR, Hockenbery DM, Pham M, Rizzo NO, Tateya S, Handa P, Schwartz MW, Kim F. Activation of nf-kappab by palmitate in endothelial cells: A key role for nadph oxidase-derived superoxide in response to tlr4 activation. Arteriosclerosis, thrombosis, and vascular biology. 2009;29:1370–1375. doi: 10.1161/ATVBAHA.109.188813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Villaret A, Galitzky J, Decaunes P, Esteve D, Marques MA, Sengenes C, Chiotasso P, Tchkonia T, Lafontan M, Kirkland JL, Bouloumie A. Adipose tissue endothelial cells from obese human subjects: Differences among depots in angiogenic, metabolic, and inflammatory gene expression and cellular senescence. Diabetes. 2010;59:2755–2763. doi: 10.2337/db10-0398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Duplain H, Burcelin R, Sartori C, Cook S, Egli M, Lepori M, Vollenweider P, Pedrazzini T, Nicod P, Thorens B, Scherrer U. Insulin resistance, hyperlipidemia, and hypertension in mice lacking endothelial nitric oxide synthase. Circulation. 2001;104:342–345. doi: 10.1161/01.cir.104.3.342. [DOI] [PubMed] [Google Scholar]
- 23.Kubota T, Kubota N, Kumagai H, Yamaguchi S, Kozono H, Takahashi T, Inoue M, Itoh S, Takamoto I, Sasako T, Kumagai K, Kawai T, Hashimoto S, Kobayashi T, Sato M, Tokuyama K, Nishimura S, Tsunoda M, Ide T, Murakami K, Yamazaki T, Ezaki O, Kawamura K, Masuda H, Moroi M, Sugi K, Oike Y, Shimokawa H, Yanagihara N, Tsutsui M, Terauchi Y, Tobe K, Nagai R, Kamata K, Inoue K, Kodama T, Ueki K, Kadowaki T. Impaired insulin signaling in endothelial cells reduces insulin-induced glucose uptake by skeletal muscle. Cell metabolism. 2011;13:294–307. doi: 10.1016/j.cmet.2011.01.018. [DOI] [PubMed] [Google Scholar]
- 24.Tsuchiya K, Tanaka J, Shuiqing Y, Welch CL, DePinho RA, Tabas I, Tall AR, Goldberg IJ, Accili D. Foxos integrate pleiotropic actions of insulin in vascular endothelium to protect mice from atherosclerosis. Cell metabolism. 2012;15:372–381. doi: 10.1016/j.cmet.2012.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rask-Madsen C, Li Q, Freund B, Feather D, Abramov R, Wu IH, Chen K, Yamamoto-Hiraoka J, Goldenbogen J, Sotiropoulos KB, Clermont A, Geraldes P, Dall’Osso C, Wagers AJ, Huang PL, Rekhter M, Scalia R, Kahn CR, King GL. Loss of insulin signaling in vascular endothelial cells accelerates atherosclerosis in apolipoprotein e null mice. Cell metabolism. 2010;11:379–389. doi: 10.1016/j.cmet.2010.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yano T, Ferlito M, Aponte A, Kuno A, Miura T, Murphy E, Steenbergen C. Pivotal role of mtorc2 and involvement of ribosomal protein s6 in cardioprotective signaling. Circulation research. 2014;114:1268–1280. doi: 10.1161/CIRCRESAHA.114.303562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vasudevan KM, Garraway LA. Akt signaling in physiology and disease. Current topics in microbiology and immunology. 2010;347:105–133. doi: 10.1007/82_2010_66. [DOI] [PubMed] [Google Scholar]
- 28.Brognard J, Sierecki E, Gao T, Newton AC. Phlpp and a second isoform, phlpp2, differentially attenuate the amplitude of akt signaling by regulating distinct akt isoforms. Molecular cell. 2007;25:917–931. doi: 10.1016/j.molcel.2007.02.017. [DOI] [PubMed] [Google Scholar]
- 29.Gao T, Furnari F, Newton AC. Phlpp: A phosphatase that directly dephosphorylates akt, promotes apoptosis, and suppresses tumor growth. Molecular cell. 2005;18:13–24. doi: 10.1016/j.molcel.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 30.Oger F, Gheeraert C, Mogilenko D, Benomar Y, Molendi-Coste O, Bouchaert E, Caron S, Dombrowicz D, Pattou F, Duez H, Eeckhoute J, Staels B, Lefebvre P. Cell-specific dysregulation of microrna expression in obese white adipose tissue. The Journal of clinical endocrinology and metabolism. 2014:jc20134259. doi: 10.1210/jc.2013-4259. [DOI] [PubMed] [Google Scholar]
- 31.Chartoumpekis DV, Zaravinos A, Ziros PG, Iskrenova RP, Psyrogiannis AI, Kyriazopoulou VE, Habeos IG. Differential expression of micrornas in adipose tissue after long-term high-fat diet-induced obesity in mice. PloS one. 2012;7:e34872. doi: 10.1371/journal.pone.0034872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ortega FJ, Moreno-Navarrete JM, Pardo G, Sabater M, Hummel M, Ferrer A, Rodriguez-Hermosa JI, Ruiz B, Ricart W, Peral B, Fernandez-Real JM. Mirna expression profile of human subcutaneous adipose and during adipocyte differentiation. PloS one. 2010;5:e9022. doi: 10.1371/journal.pone.0009022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun X, He S, Wara AK, Icli B, Shvartz E, Tesmenitsky Y, Belkin N, Li D, Blackwell TS, Sukhova GK, Croce K, Feinberg MW. Systemic delivery of microrna-181b inhibits nuclear factor-kappab activation, vascular inflammation, and atherosclerosis in apolipoprotein e-deficient mice. Circulation research. 2014;114:32–40. doi: 10.1161/CIRCRESAHA.113.302089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun X, Icli B, Wara AK, Belkin N, He S, Kobzik L, Hunninghake GM, Vera MP, Blackwell TS, Baron RM, Feinberg MW. Microrna-181b regulates nf-kappab-mediated vascular inflammation. The Journal of clinical investigation. 2012;122:1973–1990. doi: 10.1172/JCI61495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arts CH, Heijnen-Snyder GJ, Joosten PP, Verhagen HJ, Eikelboom BC, Sixma JJ, de Groot PG. A novel method for isolating pure microvascular endothelial cells from subcutaneous fat tissue ideal for direct cell seeding. Laboratory investigation; a journal of technical methods and pathology. 2001;81:1461–1465. doi: 10.1038/labinvest.3780360. [DOI] [PubMed] [Google Scholar]
- 36.Kajimoto K, Hossen MN, Hida K, Ohga N, Akita H, Hyodo M, Hida Y, Harashima H. Isolation and culture of microvascular endothelial cells from murine inguinal and epididymal adipose tissues. Journal of immunological methods. 2010;357:43–50. doi: 10.1016/j.jim.2010.03.011. [DOI] [PubMed] [Google Scholar]
- 37.van Beijnum JR, Rousch M, Castermans K, van der Linden E, Griffioen AW. Isolation of endothelial cells from fresh tissues. Nature protocols. 2008;3:1085–1091. doi: 10.1038/nprot.2008.71. [DOI] [PubMed] [Google Scholar]
- 38.Tateya S, Kim F, Tamori Y. Recent advances in obesity-induced inflammation and insulin resistance. Frontiers in endocrinology. 2013;4:93. doi: 10.3389/fendo.2013.00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McNelis JC, Olefsky JM. Macrophages, immunity, and metabolic disease. Immunity. 2014;41:36–48. doi: 10.1016/j.immuni.2014.05.010. [DOI] [PubMed] [Google Scholar]
- 40.Oh DY, Morinaga H, Talukdar S, Bae EJ, Olefsky JM. Increased macrophage migration into adipose tissue in obese mice. Diabetes. 2012;61:346–354. doi: 10.2337/db11-0860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cho KW, Morris DL, Lumeng CN. Flow cytometry analyses of adipose tissue macrophages. Methods in enzymology. 2014;537:297–314. doi: 10.1016/B978-0-12-411619-1.00016-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Werner ED, Lee J, Hansen L, Yuan M, Shoelson SE. Insulin resistance due to phosphorylation of insulin receptor substrate-1 at serine 302. The Journal of biological chemistry. 2004;279:35298–35305. doi: 10.1074/jbc.M405203200. [DOI] [PubMed] [Google Scholar]
- 43.Aguirre V, Werner ED, Giraud J, Lee YH, Shoelson SE, White MF. Phosphorylation of ser307 in insulin receptor substrate-1 blocks interactions with the insulin receptor and inhibits insulin action. The Journal of biological chemistry. 2002;277:1531–1537. doi: 10.1074/jbc.M101521200. [DOI] [PubMed] [Google Scholar]
- 44.Kahn CR, White MF. The insulin receptor and the molecular mechanism of insulin action. The Journal of clinical investigation. 1988;82:1151–1156. doi: 10.1172/JCI113711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.White MF, Shoelson SE, Keutmann H, Kahn CR. A cascade of tyrosine autophosphorylation in the beta-subunit activates the phosphotransferase of the insulin receptor. The Journal of biological chemistry. 1988;263:2969–2980. [PubMed] [Google Scholar]
- 46.Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by akt-dependent phosphorylation. Nature. 1999;399:601–605. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- 47.Symons JD, McMillin SL, Riehle C, Tanner J, Palionyte M, Hillas E, Jones D, Cooksey RC, Birnbaum MJ, McClain DA, Zhang QJ, Gale D, Wilson LJ, Abel ED. Contribution of insulin and akt1 signaling to endothelial nitric oxide synthase in the regulation of endothelial function and blood pressure. Circulation research. 2009;104:1085–1094. doi: 10.1161/CIRCRESAHA.108.189316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kops GJ, Burgering BM. Forkhead transcription factors: New insights into protein kinase b (c-akt) signaling. J Mol Med (Berl) 1999;77:656–665. doi: 10.1007/s001099900050. [DOI] [PubMed] [Google Scholar]
- 49.Hermann C, Assmus B, Urbich C, Zeiher AM, Dimmeler S. Insulin-mediated stimulation of protein kinase akt: A potent survival signaling cascade for endothelial cells. Arteriosclerosis, thrombosis, and vascular biology. 2000;20:402–409. doi: 10.1161/01.atv.20.2.402. [DOI] [PubMed] [Google Scholar]
- 50.Dweep H, Gretz N. Mirwalk2.0: A comprehensive atlas of microrna-target interactions. Nature methods. 2015;12:697. doi: 10.1038/nmeth.3485. [DOI] [PubMed] [Google Scholar]
- 51.Xiao L, Gong LL, Yuan D, Deng M, Zeng XM, Chen LL, Zhang L, Yan Q, Liu JP, Hu XH, Sun SM, Liu J, Ma HL, Zheng CB, Fu H, Chen PC, Zhao JQ, Xie SS, Zou LJ, Xiao YM, Liu WB, Zhang J, Liu Y, Li DW. Protein phosphatase-1 regulates akt1 signal transduction pathway to control gene expression, cell survival and differentiation. Cell death and differentiation. 2010;17:1448–1462. doi: 10.1038/cdd.2010.16. [DOI] [PubMed] [Google Scholar]
- 52.Xu E, Schwab M, Marette A. Role of protein tyrosine phosphatases in the modulation of insulin signaling and their implication in the pathogenesis of obesity-linked insulin resistance. Reviews in endocrine & metabolic disorders. 2014;15:79–97. doi: 10.1007/s11154-013-9282-4. [DOI] [PubMed] [Google Scholar]
- 53.Sierecki E, Sinko W, McCammon JA, Newton AC. Discovery of small molecule inhibitors of the ph domain leucine-rich repeat protein phosphatase (phlpp) by chemical and virtual screening. Journal of medicinal chemistry. 2010;53:6899–6911. doi: 10.1021/jm100331d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shi ZM, Wang XF, Qian X, Tao T, Wang L, Chen QD, Wang XR, Cao L, Wang YY, Zhang JX, Jiang T, Kang CS, Jiang BH, Liu N, You YP. Mirna-181b suppresses igf-1r and functions as a tumor suppressor gene in gliomas. RNA. 2013;19:552–560. doi: 10.1261/rna.035972.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Henao-Mejia J, Williams A, Goff LA, Staron M, Licona-Limon P, Kaech SM, Nakayama M, Rinn JL, Flavell RA. The microrna mir-181 is a critical cellular metabolic rheostat essential for nkt cell ontogenesis and lymphocyte development and homeostasis. Immunity. 2013;38:984–997. doi: 10.1016/j.immuni.2013.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kornfeld JW, Baitzel C, Konner AC, Nicholls HT, Vogt MC, Herrmanns K, Scheja L, Haumaitre C, Wolf AM, Knippschild U, Seibler J, Cereghini S, Heeren J, Stoffel M, Bruning JC. Obesity-induced overexpression of mir-802 impairs glucose metabolism through silencing of hnf1b. Nature. 2013;494:111–115. doi: 10.1038/nature11793. [DOI] [PubMed] [Google Scholar]
- 57.Jordan SD, Kruger M, Willmes DM, Redemann N, Wunderlich FT, Bronneke HS, Merkwirth C, Kashkar H, Olkkonen VM, Bottger T, Braun T, Seibler J, Bruning JC. Obesity-induced overexpression of mirna-143 inhibits insulin-stimulated akt activation and impairs glucose metabolism. Nature cell biology. 2011;13:434–446. doi: 10.1038/ncb2211. [DOI] [PubMed] [Google Scholar]
- 58.Trajkovski M, Hausser J, Soutschek J, Bhat B, Akin A, Zavolan M, Heim MH, Stoffel M. Micrornas 103 and 107 regulate insulin sensitivity. Nature. 2011;474:649–653. doi: 10.1038/nature10112. [DOI] [PubMed] [Google Scholar]
- 59.Lee YS, Li P, Huh JY, Hwang IJ, Lu M, Kim JI, Ham M, Talukdar S, Chen A, Lu WJ, Bandyopadhyay GK, Schwendener R, Olefsky J, Kim JB. Inflammation is necessary for long-term but not short-term high-fat diet-induced insulin resistance. Diabetes. 2011;60:2474–2483. doi: 10.2337/db11-0194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Karki S, Farb MG, Ngo DT, Myers S, Puri V, Hamburg NM, Carmine B, Hess DT, Gokce N. Forkhead box o-1 modulation improves endothelial insulin resistance in human obesity. Arteriosclerosis, thrombosis, and vascular biology. 2015;35:1498–1506. doi: 10.1161/ATVBAHA.114.305139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Handa P, Tateya S, Rizzo NO, Cheng AM, Morgan-Stevenson V, Han CY, Clowes AW, Daum G, O’Brien KD, Schwartz MW, Chait A, Kim F. Reduced vascular nitric oxide-cgmp signaling contributes to adipose tissue inflammation during high-fat feeding. Arteriosclerosis, thrombosis, and vascular biology. 2011;31:2827–2835. doi: 10.1161/ATVBAHA.111.236554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.De Caterina R, Libby P, Peng HB, Thannickal VJ, Rajavashisth TB, Gimbrone MA, Jr, Shin WS, Liao JK. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. The Journal of clinical investigation. 1995;96:60–68. doi: 10.1172/JCI118074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Peng HB, Libby P, Liao JK. Induction and stabilization of i kappa b alpha by nitric oxide mediates inhibition of nf-kappa b. The Journal of biological chemistry. 1995;270:14214–14219. doi: 10.1074/jbc.270.23.14214. [DOI] [PubMed] [Google Scholar]
- 64.Lee WJ, Tateya S, Cheng AM, Rizzo-DeLeon N, Wang NF, Handa P, Wilson CL, Clowes AW, Sweet IR, Bomsztyk K, Schwartz MW, Kim F. M2 macrophage polarization mediates anti-inflammatory effects of endothelial nitric oxide signaling. Diabetes. 2015;64:2836–2846. doi: 10.2337/db14-1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bartel DP. Micrornas: Target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pan D, Mao C, Quattrochi B, Friedline RH, Zhu LJ, Jung DY, Kim JK, Lewis B, Wang YX. Microrna-378 controls classical brown fat expansion to counteract obesity. Nature communications. 2014;5:4725. doi: 10.1038/ncomms5725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tsuchiya K, Accili D. Liver sinusoidal endothelial cells link hyperinsulinemia to hepatic insulin resistance. Diabetes. 2013;62:1478–1489. doi: 10.2337/db12-1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim F, Pham M, Luttrell I, Bannerman DD, Tupper J, Thaler J, Hawn TR, Raines EW, Schwartz MW. Toll-like receptor-4 mediates vascular inflammation and insulin resistance in diet-induced obesity. Circulation research. 2007;100:1589–1596. doi: 10.1161/CIRCRESAHA.106.142851. [DOI] [PubMed] [Google Scholar]
- 69.Hasegawa Y, Saito T, Ogihara T, Ishigaki Y, Yamada T, Imai J, Uno K, Gao J, Kaneko K, Shimosawa T, Asano T, Fujita T, Oka Y, Katagiri H. Blockade of the nuclear factor-kappab pathway in the endothelium prevents insulin resistance and prolongs life spans. Circulation. 2012;125:1122–1133. doi: 10.1161/CIRCULATIONAHA.111.054346. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.