

Abstract
The chaperome is a large and diverse protein machinery composed of chaperone proteins and a variety of helpers, such as the co-chaperones, folding enzymes and scaffolding and adapter proteins. Heat shock protein 90s and 70s (HSP90s and HSP70s), the most abundant chaperome members in human cells, are also the most complex. As we have learned to appreciate, their functions are context dependent and manifested through a variety of conformations that each recruit a subset of co-chaperone, scaffolding and folding proteins and which are further diversified by the post-translational modifications each carry, making their study through classic genetic and biochemical techniques quite a challenge. Chemical biology tools and techniques have been developed over the years to help decipher the complexities of the HSPs and this review will provide an overview of such efforts with focus on HSP90 and HSP70.
Introduction
The chaperome is a relatively new term to jointly refer to a group of cellular proteins dedicated to ensuring that the proteome is stable and structurally and functionally well-supported throughout their life cycle. These proteins include chaperone proteins, their helpers, the co-chaperones, as well as folding enzymes, scaffolding proteins, and adaptor proteins (Brehme et al., 2014, Finka and Goloubinoff, 2013, Taldone et al., 2014b). In humans, their numbers are in the hundreds. Heat shock proteins (HSPs), denoted as such because some members are heat inducible, are chaperones and the most abundant members of the chaperome in human cells. The HSPs are classified into families based on their molecular weights: the high molecular mass HSPs (≥ 100 kD), HSP90 (81 to 99kD), HSP70 (65–80kD), HSP60 (55 to 64 kD), HSP40 (35 to 54 kD), and the small HSPs (≤ 34 kD). Together these proteins are estimated to amount to 10% of the cellular mass, with HSP90s and HSP70s alone accounting for half of that. From a chemical biology perspective, these two abundant HSP families have received most of the interest and attention so far, and therefore will be the focus of this review.
In normal cells, different cellular comportments have dedicated networks of specific chaperones that occur and are active only within the context of the given location (Hartl et al., 2011, Kim et al., 2013). In humans, two major paralogs of HSP90, HSP90α and HSP90β, reside in the cytosol with a minor presence in the nucleus, while glucose-regulated protein 94 (GRP94) is found in the endoplasmic reticulum (ER) and tumor necrosis factor receptor-associated protein 1 (TRAP1) is located in the mitochondria. HSP70s family has more members, with nine genes encoding cytosolically expressed HSP70 proteins, one an ER protein (HSPA5 also known as BiP or GRP78) and one coding for a mitochondrial protein (HSPA9, also known as mortalin, GRP75, or mtHSP70). Of these, the most abundant HSP70s are the cytosolic HSC70 (encoded by HSPA8) and HSP70 (encoded by HSPA1A and HSPA1B) as well as BiP and mortalin (Daugaard et al., 2007).
All HSPs are flexible proteins composed of several domains. HSP90 for example has an N-terminal (N) domain, containing nucleotide binding sites, a middle (M) domain, which provides binding sites for client proteins, and a C-terminal (C) domain containing a dimerization motif. Additionally, all three domains are involved in binding an army of co-chaperones, proteins that assist the chaperone activity of HSP90. N and M domains are connected by an unstructured charged-linker region of significant but variable length, which provides conformational flexibility to HSP90s (Prodromou, 2012, Street et al., 2011). HSP70 is composed of an N-terminal nucleotide binding domain (NBD) and a C-terminal substrate-binding domain (SBD). The NBD binds to nucleotide and is further subdivided into two major lobes, with the ATP-binding cleft located between them. Likewise, the SBD is further subdivided into two major regions: a β-sandwich subdomain and an α-helical “lid.” The β-sandwich subdomain contains the hydrophobic groove that binds client proteins, while the lid regulates the kinetics of client binding. The NBD and SBD are connected by a short, hydrophobic linker, which mediates interdomain allostery (Mayer and Bukau, 2005, Kityk et al., 2012).
HSP70s and HSP90s are highly dynamic and flexible proteins and their conformation and activity is regulated at several levels; these include the ATPase cycle, association with conformation-specific cochaperones, and posttranslational modifications (PTMs). The chaperone function of HSP70 is based on an allosteric mechanism where binding and hydrolysis of ATP regulates chaperone conformation and substrate binding affinity (Mayer and Kityk, 2015). For HSP90, rather than dictating the conformational state, it is believed that ATP binding and hydrolysis only shift the equilibria between a pre-existing set of conformational states (Krukenberg et al., 2011). A yet another level of conformational and functional regulation comes from co-chaperones that may impact conformational equilibria and affect the kinetics of structural changes, ATP hydrolysis and substrate specificity, and from posttranslational modifications and client protein (substrate) binding (Mollapour and Neckers, 2012, Prodromou, 2012). For example, each co-chaperone and PTM is believed to have a set function. Cell division cycle 37 homolog (CDC37) is a cochaperone that is essential for the maturation of almost all HSP90-dependent kinases (Vaughan et al., 2008), HSP90-HSP70 organizing protein (HOP) is responsible for delivering substrates from HSP70 to HSP90 (Hernandez et al., 2002), AHA1 is an activator of HSP90 ATPase activity, HSP40 or DNAJ proteins stimulate ATP hydrolysis by HSP70 and provide substrate specificity (Kampinga and Craig, 2010) whereas CHIP, which has E3 ubiquitin ligase activity, promotes degradation of substrates bound by HSP70/HSC70 or HSP90 (Connell et al., 2001). Several nucleotide exchange factors, which include BAG and HSP110 proteins, regulate HSP70 function replacing ADP with ATP through opening of the N-terminal domain (Bracher and Verghese, 2015). Through these actions, the J proteins and NEFs coordinate ATP cycling and client loading onto HSP70. PTMs that have been documented for HSP90 and HSP70 include phosphorylation, acetylation, S-nitrosylation, oxidation, sumoylation and ubiquitination (Li et al., 2012). Although not fully explored, specific roles of PTMs are starting to emerge. For example, a tyrosine phosphorylation on HSP90 promotes recruitment of AHA1, stimulates ATPase activity, and regulates HSP90 chaperone cycle (Xu et al., 2012), a phosphorylation of the C-terminus of HSP70 appears to regulate co-chaperone binding (Muller et al., 2013), whereas enhanced methylation of an HSP70 lysine promotes cell proliferation (Cho et al., 2012).
Given these layers of structural and functional complexity, one may imagine that each cell contains a mix of HSPs differentiated by the interactor(s) (co-chaperones, adapters and substrates), the conformation the HSP adapts, and the cellular location it finds itself at. This complexity, as we will detail below, greatly influences the biological effect of a chemical tool and in turn, the phenotype one may observe when using any HSP-directed chemical tool in cellular or organismal models. For example one may imagine that a chemical tool of specificity for a subset of HSP conformations or complexes will lead to a phenotypic outcome distinct from another targeting a yet distinct pool of cellular HSP. This also means that the study of HSP90s and HSP70s requires a variety of experimental approaches and tools, and over the years many chemical tools, small molecules that bind HSPs and modulate one or more of their cellular functions, have been identified. This review will focus on describing small molecules tool compounds that have been used to advance our understanding of basic biology of HSP90s and HSP70s, discuss what constitutes a good tool compound, as well as what limitations the chemical biology tool developers are currently battling with.
HSP90 probes
Discovery of chemical tools for HSP90 and HSP70 has been driven by the therapeutic interest these two HSP families have received. Indeed, both HSP90 and HSP70 family members have been implicated in the pathology of numerous diseases such as cancer, neurodegenerative diseases, inflammation and infectious diseases (Sevin et al., 2015, Taldone et al., 2014b) therefore, it is of no surprise that many of the known chemical tools are drug candidates as well. This is particularly true for HSP90 (Figure 1A) (Patel et al., 2011). In addition to small molecule ligands, a variety of labeled versions of these ligands have been reported and used in numerous biological inquiries, as we shall detail below. These are appropriately labeled small molecules for investigations of the HSPs by classical biochemical and cell biology techniques such as fluorescent probes for microscopy and flow cytometry, biotinylated or solid-support attached probes for target identification, biased interactor validation or for non-biased HSP-interactome inquiries by mass spectrometry, and radiolabeled probes for use in target detection in vivo (Figure 1B,C).
Figure 1. HSP90 chemical probes.
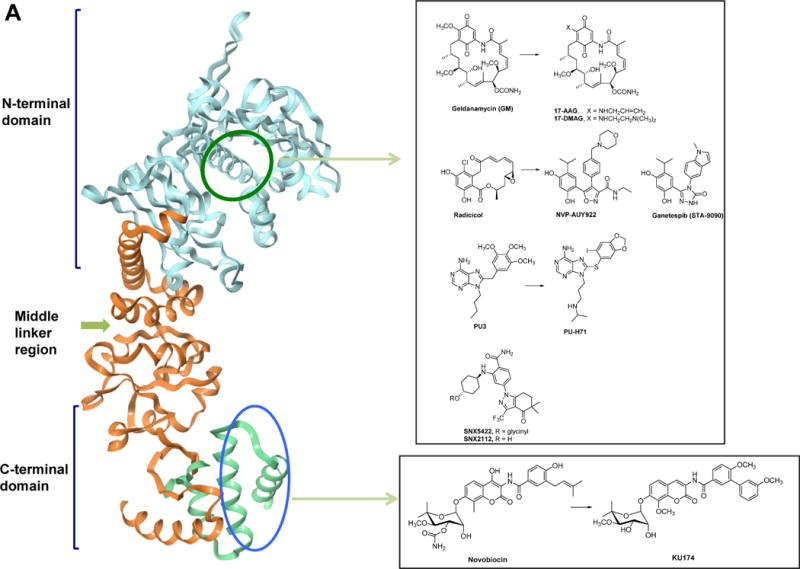

(A) Ribbon representation of the apo full-length monomer HtpG (E.coli HSP90, PDB: 2IOQ) and the reported mode of interaction for select HSP90 inhibitors. The chemical structure of each ligand is depicted on the right side. (B) Geldanamycin (GM)-derived HSP90 chemical probes. (C) Other HSP90 chemical probes.
Chemical biology had a significant impact on HSP90 research. In many ways, HSP90 became a prominent target for drug discovery because of chemical biology. How else anybody in their right mind would choose one of the most abundant proteins in unstressed cells, an ubiquitous molecular chaperone found in eubacteria and all branches of eukarya, and moreover, not particularly variable in expression between normal and diseased cells (ex. cancer cells), as a target in human disease? Indeed HSP90, discovered in the 1980s, has drawn little interest until early 1990s when the serendipitous discovery of a natural product, geldanamycin (GM) changed that state of affairs forever. GM was identified in a screen searching for compounds able to revert the phenotype of cells transfected with the v-src oncogene, but the target of intervention, like for many hits derived from phenotypic screens, remained a mystery (Whitesell et al., 1994). To solve it, Len Neckers and colleagues at NIH took a chemical biology approach (Neckers, 2006). Namely, they attached GM to a solid support and incubated the GM-beads with cellular homogenates and fished out one protein with a molecular mass of approximately 90 kD. Follow up experiments using western blotting with specific antibodies, and later with micro-sequencing protein material bound to radiolabeled affinity-tagged GM, revealed that the target for GM is HSP90. GM bound into the N-terminal regulatory pocket of HSP90 and by this, inhibited its function (Stebbins et al., 1997). While surprising in light of the available genetic data, low concentrations of this natural product were active on many cancer cells and induced differentiation, reduced cell proliferation and/or induced death, while being of no significant toxicity at such concentrations to normal cells (Neckers et al., 1999). Subsequent crystal structures of HSP90 in complex with GM, another natural product, radicicol, or with the regulatory nucleotides ATP and ADP have uncovered the unique characteristics of this pocket and its high potential for ligand design. Indeed, PU3 (Figure 1A) the first synthetic HSP90 ligand was designed to interact with the pocket taking clues from the structures of ATP-, ADP-and GM-bound HSP90 (Chiosis et al., 2001). Other N-terminal interactors soon followed (Figure 1A).
Compounds that act on HSP90 through interaction with the C-terminal domain of HSP90 have also been uncovered (Hall et al., 2014). Some are related to novobiocin, such as KU-174 (Figure 1A). These compounds also destabilize HSP90 complexes in cancer cells, despite the fact that they engage a distinct domain and do not directly compete for binding to ATP/ADP. C-terminal binders such as the novobiocin-derived KU174 elicit and/or prefer an HSP90 conformation distinct from that induced by GM (Matts et al., 2011), and thus represent additional chemical tools for the investigation of the chaperome.
The contribution of chemical biology to chaperone biology continued after the discovery of HSP90 as the cellular target of GM. GM, and a variety of other HSP90 inhibitors developed and discovered over the years (Figure 1A), have been used as chemical tools to elucidate the biology of HSP90 first in cancer (Samant et al., 2012), and later on in neurodegenerative diseases (Carman et al., 2013), viral infections (Geller et al., 2012), and mechanisms of drug resistance (Whitesell et al., 2014). From these studies we came to understand the variety of cellular process that HSP90 regulates under different conditions of cellular stress.
Chemical biology approaches also shed light on functions of HSP90 in cancer cells other than regulation of protein stability and folding. For example, using chemical tools revealed that HSP90 may act as a scaffolding molecule to increase the competency of protein complexes involved in signaling and in transcription (Cerchietti et al., 2009, Moulick et al., 2011). Specifically, in chronic myeloid leukemia (CML), HSP90 was found to facilitate increased STAT5 signaling by binding to and influencing the conformation of STAT5 and by maintaining STAT5 in an active conformation directly within STAT5-containing transcriptional complexes (Moulick et al., 2011). In the cytosol, HSP90 binding to STAT5 modulated the conformation of the protein to alter STAT5 phosphorylation and dephosphorylation kinetics. In addition, HSP90 maintained STAT5 in an active conformation in STAT5-containing transcriptional complexes.
While the above studies led to the identification of important substrates of HSP90, commonly referred to as clients, it was clear that in order to understand the HSP90 interactome in cancer cells one needs to use large proteomic and genomic analyses combined with bioinformatic analyses. In general, tumor cells are found to be dependent on increased HSP90 function, likely needed to support their altered physiology and survival under stress conditions caused by the oncogenic transformation. But, because malignant evolution is characterized by alterations in a multitude of proteins and pathways, and no two tumors present an identical spectrum of defects, it is likely that addiction of cancer cells to HSP90 occurs in a tumor-specific manner and thus no two tumors will have an identical set of HSP90-sheltered proteins and protein networks. Recent investigations have used chemical biology methods to demonstrate the complexity of HSP90 sheltered networks, as we detail below.
In a classic, chemical perturbation approach, changes in the proteome of HeLa cervical cancer cells were analyzed upon treatment with the HSP90 inhibitor 17-(dimethylaminoethylamino)-17-demethoxygeldanamycin (17-DMAG; Figure 1A) using SILAC and high resolution, quantitative mass spectrometry (MS) (Sharma et al., 2012). This study found that the parts of proteome most sensitive to HSP90 inhibition fit the functional categories of “protein kinase activity” and “DNA metabolic processes”. The kinases regulated by HSP90 in HeLa cells were associated with the PI3K-AKT-, IL-6-, HER-, FAK-, Ephrin receptor-, ERK-MAPK-, NFκB-, inositol phosphate metabolism and G-protein coupled receptor-mediated networks. Additionally, processes such as “cellular response to DNA damage”, “DNA modification”, “response to DNA damage stimulus” and “regulation of transcription” that belong to the “DNA metabolic processes” group were also enriched.
This analysis painted a very broad picture of the far-reaching effect that inhibition of HSP90 has on the proteome. However, one thing to keep in mind is that proteins that changed upon HSP90 inhibition identified using SILAC/MS approach do not necessarily represent direct interactors of HSP90. For interactome identification, affinity-purification methods are more direct and more powerful. The challenge in affinity purification is finding a probe that preserves the endogenous HSP/protein complexes throughout the subsequent experimental steps (i.e. permeabilization, washes). In fact, initial attempts to apply such method to HSP90 were met with little success as an anti-HSP90 antibody or a solid support immobilized GM were ineffective in trapping HSP90–client protein complexes and the identified interactome was limited to abundant cellular proteins (Tsaytler et al., 2009).
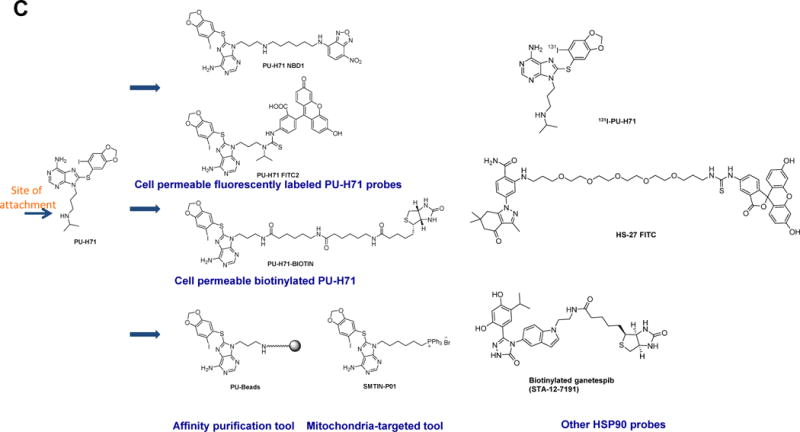
To develop an effective affinity-based method, Moulick et al took advantage of the fact that PU-H71 (Figure 1A) bound preferentially to the fraction of tumor HSP90 associated with altered client proteins and moreover, upon binding, locked HSP90 in an onco-client bound configuration (Moulick et al., 2011). Using PU-H71 derived beads, the PU-beads (Figure 1C), greatly facilitated the identification of tumor-associated protein clients by MS, providing tumor-by-tumor global insights into the HSP90-sheltered proteome, including in primary patient specimens. This chemoproteomics approach yielded some significant insights as it led to identification of the altered signalosome in chronic myeloid leukemia (CML), methyltransferase CARM1 as a transforming molecule in CML, and a novel mechanism of activation of STAT5 in CML. Later studies used the method to identify unanticipated combination therapies for cancer (Goldstein et al., 2015) and to elucidate that in virus-associated lymphomas, HSP90 regulates viral proteins, blocking latent and lytic viral functions (Nayar et al., 2013). Thus through chemical biology we came to learn that while HSP90 impacts numerous signaling pathways and cellular functions, the affected pathways do not overlap across all tumor types, nor is regulation of the signalosome behind HSP90 functions in all tumor types.
The tools mentioned above have been largely used to investigate the biology of the major HSP90, the cytosolic paralog. But as interest grew in the role of chaperone biology in diseases, the field started to appreciate that HSPs can change their cellular location. For example, in a variety of disease including cancer and infections diseases, cytosolic HSP90 was noted on the cell surface or translocated into the mitochondria (Alarcon et al., 2012, Altieri, 2013, Defee et al., 2011). Chemical biology again came to the rescue by providing tools to elucidate the functions of the errant chaperones.
First, a cell impermeable GM derivative, DMAG-N-oxide (Figure 1B), came along. Its use in the investigation of HSP90 revealed that an extracellular form of the protein (eHSP90) was expressed in certain cancers and that this cell surface HSP90 played an important role in modulating cancer cell migration (Tsutsumi et al., 2008). A cell-impermeable derivative of ganetespib (STA-12-7191; Figure 1C) was used as a less toxic alternative of DMAG-N-oxide and revealed that lysyl oxidase 2-like protein is one of the eHSP90 client proteins behind eHSP90’s cell migration-promoting role (Mccready et al., 2014). Other studies used DMAG-N-oxide to implicate eHSP90 in maturation of the cell surface enzyme matrix metalloproteinase-2 (Xu and Neckers, 2007), a key molecule in the invasion process of cancer cells. MMP2 can degrade the extracellular matrix, providing cancer cells with access to the vasculature and lymphatic system, allowing tumor dissemination (Tsutsumi and Neckers, 2007). A chemical tool was also used to implicate eHSP90 as a pro-inflammatory molecule in retinal pigment epithelial inflammatory responses (Qin et al., 2011). A cell-impermeable fluorescently labeled GM, GM-FITC (Figure 1B), was used to identify cell surface HSP90 by fluorescence microscopy (Imai et al., 2011). A combination of cell permeable and impermeable GM derivatives was used to show that in dendritic cells, cytosolic but not eHSP90 was essential for cross-priming of cell-associated antigen. Derivatives of SNX2112 (Figure 1A) that can carry optical or radioiodinated probes via a polyethyleneglycol tether, such as HS-27 FITC (Figure 1C), were used to recognize cells expressing eHSP90 and to demonstrate that an active cycling of the eHSP90 protein occurs at the plasma membrane (Barrott et al., 2013).
Another area of active research is developing probes that can inhibit the function of mitochondria-specific HSP90 chaperone. Here the main challenge has been figuring out how to deliver sufficient quantities of pan-HSP90 inhibitors to mitochondria without the inhibitors being trapped by binding to HSP90 that’s present in high concentrations in the cytosol. The Altieri group postulated that attaching GM to mitochondria targeting moieties such as TPP-OH (Kang et al., 2009) or tandem repeats of cyclic guanidinium (Kang et al., 2009), may mediate its fast mitochondrial targeting and achieve the rate of delivery that would escape sequestration by the cytosolic HSP90. These agents called Gamitrinibs (Figure 1B) were reported to accumulate in the mitochondria exhibiting a “mitochondriotoxic” mechanism of action, causing rapid tumor cell death (Kang et al., 2009). Combined with other tools, gamitrinibs were used to show that mitochondrial HSP90s confer cell death resistance to cancer cells by suppressing the mitochondria-initiated calcium-mediated interorganelle stress response (Park et al., 2014). They also highlighted that mitochondrial HSP90 chaperones, including TRAP-1, overcome metabolic stress and promote tumor cell metastasis by limiting the activation of the nutrient sensor AMPK and preventing autophagy (Caino et al., 2013). Additionally, mitochondrial HSP90 binds and stabilizes the electron transport chain Complex II subunit succinate dehydrogenase-B, maintaining cellular respiration under low-nutrient conditions, and contributing to hypoxia-inducible factor-1α-mediated tumorigenesis in patients carrying succinate dehydrogenase-B mutations (Chae et al., 2013). Use of gamitrinibs led to a mechanism involving TRAP-1 in metabolism and aging. Researchers found that “knockout” mice bred to lack the TRAP-1 protein compensated for this loss by switching to alternative cellular mechanisms for making energy in which cells consume more oxygen and metabolize more sugar (Lisanti et al., 2014). Very recently, another HSP90 inhibitor targeted to mitochondria, SMTIN-P01, was created by replacing the isopropyl amine of the PU-H71 with the mitochondria-targeting moiety triphenylphosphonium (Figure 1C) (Lee et al., 2015). We expect that SMTIN-P01 use will yield interesting insights into mitochondrial chaperome in the near future.
Perhaps the most important and exciting contribution of chemical biology to HSP90 chaperone biology was the realization that not all HSP90 is the same. The first glimpse came in 2003 when Kamal et al compared HSP90 found in normal and cancer cells. What they observed was that HSP90 behaved differently, despite being identical and not subject to specific mutational changes. While in normal cells HSP90 formed dynamic complexes and displayed low affinity for small molecule inhibitors, such as GM, in cancer cells the chaperone was found entirely in complexes and had high affinity and sensitivity to small molecule inhibitors (Kamal et al., 2003). In light of these observations, the distinct sensitivity of normal and cancer cells to GM and other HSP90 inhibitors could be explained by the difference in dynamics of HSP90 complexes.
Then Moulick et al demonstrated that not all HSP90 in a cancer cell was inhibitor sensitive (Moulick et al., 2011). They used GM- and PU-beads (Figure 1C) and for comparison beads with an anti-HSP90 antibody (H9010) and demonstrated that although consecutive immunoprecipitation steps with H9010, but not with a nonspecific IgG, quantitatively depleted HSP90 from a cancer cell extract, sequential pull-downs with PU or GM beads removed only a limited fraction of total cellular HSP90. In MDA-MB-468 breast cancer cells, the combined PU-bead fractions represented ~20–30% of the total HSP90 pool. The PU-H71-depleted, remaining HSP90 fraction, although inaccessible to the small molecule, maintained affinity for H9010 leading the authors to conclude that a substantial fraction of HSP90 was still in a native conformation but less reactive with PU-H71. To exclude the possibility that changes in HSP90’s conformation in cell lysates make it unavailable for binding to immobilized PU-H71 but not to the antibody, the authors used radiolabeled 131I-PU-H71 (Figure 1C) to assess HSP90 in intact cancer cells. Binding of 131I-PU-H71 to HSP90 in several cancer cell lines saturated at a well-defined, although distinct, number of sites per cell; in MDA-MB-468 this amounted to 26.6–33.5% of the total HSP90, markedly similar to that obtained with PU-bead pull-downs in cell extracts. This confirmed that PU-H71 bound to approximately 30% of the total HSP90 pool in MDA-MB-468 cells and validated the use of PU-beads to efficiently isolate this pool. In K562 and other established t(9;22)+ CML cell lines, PU-H71 bound less than 25% of the total cellular HSP90 indicating that the expression of this HSP90 species was different among tumor cells.
To understand the nature of these HSP90 complexes, Moulick et al analyzed the associated proteins. Here, the authors choose to examine a cancer cell line that coexpressed the aberrant BCR-ABL protein, a constitutively active kinase, and its normal counterpart c-ABL (Moulick et al., 2011). These two ABL species are clearly separable by molecular weight and are therefore easily distinguishable by western blot. The analysis showed that while HSP90 interacted with both ABL and BCR-ABL, the PU-H71 HSP90 fraction was enriched in BCR-ABL. The fraction of HSP90 bound to BCR-ABL also associated with several co-chaperones, including HSP70, HSP40, HOP and HIP. Overall, PU-sensitive HSP90 interacted with the complement of altered proteins in a specific cancer and thus, Moulick et al named this HSP90 pool as “oncogenic HSP90”.
This landmark study used a variety of chemical tools to demonstrate that in cancer cells a dedicated pool of HSP90 binds a range of proteins with functional role in maintenance of the oncogenic state. This “oncogenic HSP90” regulates the altered proteins and protein networks and can be distinguished from the “housekeeping” chaperone, which represents the major fraction and retains functions similar to normal cells. However, the molecular basis for the “oncogenic HSP90” remains a mystery and much remains to be elucidated. The proteome changes that result in the expression of “oncogenic HSP90” are still not known, and the prevalence of the “oncogenic HSP90” in tumors is an open question. The importance of targeting “oncogenic HSP90 in cancer treatment is another open question that is being intensely pursued. Finding answers to these open questions is essential to better understand the role of HSP90 in cancer and for a better usage of its inhibitors in the clinic.
Another interesting question related to the mechanism of action for the inhibitors that target HSP90 is why HSP90 inhibitors that bind into the same pocket have different biological effects. For example, we know that solid support immobilized versions of several such inhibitors precipitate distinct HSP90 pools (Moulick et al., 2011, Prince et al., 2015). 17-AAG and NVP-AUY-922 but not PU-H71 (Figure 1A) augment the phenotype observed when cancer cells are treated with a compound that disrupts the HSP90-HOP interaction (Pimienta et al., 2011). Additionally, the tumor pharmacokinetic and pharmacodynamic properties as well as the therapeutic index of clinical HSP90 agents greatly differ (Jhaveri et al., 2014).
The Neckers group demonstrated that GM and PU-H71 select for overlapping but not identical subpopulations of total cellular HSP90, even though both inhibitors bind to an amino terminal nucleotide pocket and prevent N domain dimerization. For example, PU-H71 accesses a broader range of monomeric N domain conformations than GM and is less affected by HSP90 phosphorylation (Beebe et al., 2013). Using yeast genetics complemented in combination with chemical tools (i.e. small molecule ligands and a biotinylated version of ganetespib, Figure 1B) they demonstrated a role for posttranslational modifications on inhibitor interaction with HSP90. Here, asymmetric SUMOylation of a conserved lysine residue in the N domain of both yeast (K178) and human (K191) HSP90 facilitates both recruitment of the HSP90-activating cochaperone AHA1 and, unexpectedly, the binding of HSP90 inhibitors (Mollapour et al., 2014). Lastly, using a defined set of HSP90 conformational mutants, the authors found that some clients interact strongly with a single, ATP-stabilized HSP90 conformation, only transiently populated during the dynamic HSP90 chaperone cycle, while other clients interact equally with multiple HSP90 conformations. GM and STA9090 (ganetespib) were differentially able to access these HSP90 conformational states (Prince et al., 2015). Taldone et al used an FITC-labeled HSP90 probe coupled with fluorescence polarization and combined it with computational analyses to rationalize such differences at the structural level (Taldone et al., 2013a). They showed that while all clinically relevant HSP90 agents bind into the same pocket, they do insert into distinct subpockets of this binding site, and moreover, upon insertion some are more prone to induce a conformational change in the protein. This study also rationalized the paralog selectivity of these small molecules and defined not only the major binding modes that relay pan-paralog binding or, conversely, paralog selectivity, but also identified molecular characteristics that impart such features.
The view of HSPs that emerges from these studies is that HSP90 present in cells is a mixture of complexes, each with a distinct PTM states and in varied conformations. On the other hand, the view of small molecule modulators of HSP90 that we are converging on is that each small molecule may have a preference for one, or a subset of different HSP90 species over the other. Therefore, the phenotypes observed with one HSP inhibitor agent may not be identical with that seen with another despite the seemingly similar mode of binding. Thus in the eye of an HSP90 ligand, no HSP pool is ever the same. Conversely, in the eye of HSP90 no two HSP90 ligands ever are the same. The therapeutic consequences of this concept were recently discussed (Taldone et al., 2014b). But it is evident that such findings have clear implications for the use of HSP agents as chemical tools as well.
On one level, the take home message is simple: biologists need to learn how to choose the right tool for the type of the HSP90 question they want to answer and the chemists need to understand what specific biological questions are being asked in order to make the tools fit for the purpose. With their composition and stability likely to be dependent on endogenous conditions found in native tumors, these HSP90 species will resist investigation by current laboratory approaches that disrupt or engineer the cellular environment to facilitate analysis. Therefore, knowing where and how the small molecule tools act on HSP90s and their complexes can provide key insights into the biology of these chaperones that is not possible by genetic means. In fact, if chemical alterations such as post-translational modification, or biochemical changes via co-chaperone and adapter protein pre-recruitment, are the distinguishing characteristics of the “oncogenic HSP90” species, chemical biology definitely has an edge over other techniques to uncover and characterize them.
Our lab has spent considerable efforts to create chemical tools for the analysis and detection of the “oncogenic HSP90”. For example, an HSP90 cell-permeable probe that specifically and tightly interacts with “oncogenic HSP90” is favored for flow cytometry measurements of this species. Fixation/permeabilization methods used for the detection of intracellular antigens by flow cytometry may result in the destruction of the “oncogenic HSP90” complexes and of the cellular morphology and surface immunoreactivity, properties useful in flow cytometry for the characterization of cells in heterogeneous populations. We found two PU-H71 derivatives, PU-H71-FITC2 and PU-H71-NBD1 (Figure 1C), suitable for fluorescence-activated flow cytometry and fluorescence microscopy (Taldone et al., 2011). Thus these molecules serve as useful probes for studying “oncogenic HSP90” in heterogeneous live cell populations (Moulick et al., 2011). For example, confocal fluorescence microscopy of leukemia cells, but not normal cells, stained with PU-H71-FITC2 showed prominent intracellular localization. In these experiments, DAPI was used as a viability dye to discriminate between viable and non-viable cells. This dye is impermeable in live cells at the tested concentration, but permeates non-viable cells and binds specific regions of DNA. Thus these fluorescent derivatives of PU-H71 can be applied as probes for fluorescence-activated flow cytometry, as tools for monitoring real-time interaction of HSP90 with the target by fluorescence microscopy and provide simple means (i.e. mix cells with probe and measure fluorescence signal by flow cytometry) to quantify this species in a large spectrum of cancer cells.
To affinity purify the “oncogenic HSP90” complexes we developed the chemoproteomics method described above that uses the PU-beads (Figure 1C) for HSP90 purification in complex with its interactome (Moulick et al., 2011). We also searched for biotinylated analogues of PU-H71 that are capable of permeating cell membranes so as to enable the investigation of HSP90 complexes in live cells. The identified derivative PU-H71-biotin (Figure 1C) can isolate HSP90 through affinity purification and, as we show, represents a unique and useful tool to probe tumor HSP90 biology in live cells by affinity capture, flow cytometry and confocal microscopy (Taldone et al., 2013b).
HSP90-paralog selective probes
While much ink has been used to describe the function of HSP90 in disease, little remains known of its ER paralogue, GRP94 (Marzec et al., 2012). GRP94 is absent from most unicellular organisms (except Leishmania), thus, yeast genetics useful in deciphering HSP90 mechanisms cannot be applied to this chaperone. Knockdown of GRP94 in C. elegans by siRNA causes larval arrest, which makes this method not amenable to scoring the phenotype of site-directed mutants. Several studies of GRP94 have used mutant cell lines and gene-deficient mouse studies to shed light on GRP94 biology. But, assessing phenotypes, and in turn protein function in disease, in gene knockdown studies can offer only limited insights. On the other hand, the discovery of GRP94 specific inhibitors has been a challenge because of the high analogy in the ATP/ADP regulatory ligand-binding cavities among the four HSP90 paralogs. Thus, although of significant interest, both as therapeutics and as tools to dissect in a spatiotemporal manner the cell-specific effects and mechanisms associated with GRP94 in select phenotypes, the small molecule tools for GRP94 have been difficult to find. The first reported ligand with selectivity for GRP94 over cytosolic HSP90 was 5′-N-ethylcarboxamidoadenosine (NECA, Figure 2A), an adenosine A2 receptor antagonist which was discovered following a biochemical screen to identify additional cellular targets (Rosser and Nicchitta, 2000). The crystal structure GRP94-NECA indicated a conformational flexibility in the pocket domain of GRP94 to be at the root of selectivity; a distinct cavity adjacent to the adenine binding pocket that is not accessible in HSP90 was noted in GRP94 when bound to NECA (Soldano et al., 2003). While of little use as a chemical tool due to its target promiscuity, identification of NECA was proof that selective GRP94 inhibitors can be generated. The Blagg team created a NECA mimic, BnIm (Figure 2A), that was predicted to interact selectively with GRP94 and this compound (Duerfeldt et al., 2012, Hall et al., 2014) was used as a tool complementary to genetic approaches to elucidate the role of GRP94 in the regulation of the glaucoma-associated protein myocilin (Stothert et al., 2014). GRP94 chaperones mutant myocilin, triaging it through ERAD, a pathway incapable of efficiently handling the removal of mutant myocilin, leading to cell toxicity. When GRP94 was depleted, degradation of mutant myocilin was shunted away from ERAD toward a more robust clearance pathway for aggregation-prone proteins, the autophagy system (Suntharalingam et al., 2012).
Figure 2. HSP90 paralog selective probes.
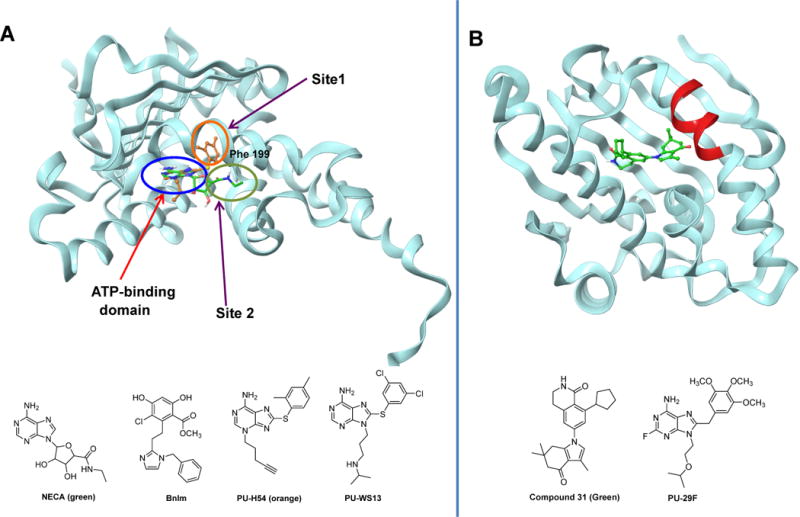
(A) Ribbon view of the N-terminal domain of GRP94 (Canis lupus familiaris, PDB:3O2F) in complex with the PU-WS13 derivative PU-H54 (orange). The distinction between the binding site of NECA (green) and PU-H54 is shown through the overlay of PU-H54 with NECA bound to GRP94 (PDB: 1U2O). (B) Ribbon representation of the N-terminal domain of HSP90α in complex with compound 31 (Homo sapiens, PDB:4O0B. Also shown is the HSP90α/β selective inhibitor PU29F. The chemical structure of each ligand is depicted below each protein structure.
Our approach to developing paralog-specific inhibitors of HSP90s tapped into the substantial conformational changes that are associated with HSP90 function (Patel et al., 2013), and the insight that the pocket available for ligand binding may change depending on the conformational states. By interacting with the protein at different sites, inhibitors may sample non-overlapping conformations of the protein or freeze the protein in distinct conformations. For example, this can manifest at the local level, when ligand binding causes specific rearrangements in a particular domain or subdomain, or more global, when the effects of ligand binding are felt throughout the protein, for example like in the case of major conformational changes noted for the HSP90s. Using a screening approach we identified ligands, such as PUH54 and PU-WS13 (Figure 2A) that strongly preferred GRP94 (100-fold selectivity) over the cytosolic HSP90. Mechanistically, these ligands bind to a novel allosteric pocket in GRP94 and “freeze” it in a unique conformation not accessible by the cytosolic HSP90. This pocket is not identical to that proposed for NECA and BnIm (Patel et al., 2015). The screen also identified ligands selective for the cytosolic HSP90α (PU29F, Figure 2A), thus providing an important toolset to investigate the biological roles of the two HSP90s in endogenous systems. We confirmed that the toolset retained paralog selectivity in cells and then used it to uncover a novel mechanism for the regulation of the cancer promoting protein HER2 (Patel et al., 2013). Specifically, we identified cell-specific, proteome-alteration driven regulation of HER2 by distinct HSP90 paralogs. These effects were previously unknown and unanticipated although HER2 is a widely-studied HSP90 client protein, providing evidence of the power of chemical biology when addressing biological question in the endogenous, un-engineered biological model. The use of these tools also provided the first recorded instance of hierarchical use of the chaperome machinery in cancer. It showed that to avoid “over-capacitance” (Rutherford and Lindquist, 1998, Sangster et al., 2004) in the face of an increased demand from the altered proteome, the cancer cells react not by overexpression, but rather by co-opting, in a hierarchical order, other chaperome members. The GRP94 agent, PU-WS13, was also used to investigate the role of GRP94 in multiple myeloma (Hua et al., 2013), in chaperoning integrins to promote metastasis (Hong et al., 2013) and regulating Toll-like receptor 9 proteolytic processing and conformational stability (Brooks et al., 2012). The Stamos team used a structure-based drug design strategy to optimize a benzolactam series of HSP90α/β inhibitors to achieve >1000-fold selectivity versus GRP94 and TRAP1 (Compound 31, Figure 2B). They used the agents to demonstrate a role for cytosolic HSP90 in promoting the clearance of mutant huntingtin protein and to propose cytosolic HSP90 as a potential target in Huntington’s disease (Ernst et al., 2014).
It turns out, GRP94, like its cytosolic relative, has many aspects to its personality. We were intrigued by the previous observation of Duerfeldt et al that BnIm lacked antiproliferative effects against the HER2-overexpressing SKBr3 cells (Duerfeldt et al., 2012). This contrasted with our findings and those of others that inhibition of GRP94 by PU-WS13 or knockdown of GRP94 by siRNA led to growth impairment in these cells (Li et al., 2015b, Patel et al., 2013). In SKBr3 cells GRP94 translocates to the plasma membrane where it’s needed to maintain the transforming ability of HER2 kinase. While both BnIm and PU-WS13 bind in a GRP94 pocket that partly overlaps with that occupied by ATP, as mentioned, the pockets occupied by the two synthetic ligands are not identical (Figure 2A). We synthesized BnIm and confirmed that under identical experimental conditions, indeed PU-WS13 but not BnIm altered HER2 (Patel et al., 2015), even when we used a concentration of BnIm as high as 100 μM, a concentration recently shown to inhibit the chaperoning of myocilin by GRP94 (Stothert et al., 2014). Through these data we cannot conclusively attribute the lack of effect for BnIm in SKBr3 cancer cells to its distinct binding interaction with GRP94 but we favor an explanation that diverse binding modes may enable each compound to sample a diverse range of conformations resulting in different phenotypes. Other mechanisms such as inability of BnIm to access GRP94 at the plasma membrane might also account for the phenotypic differences. Nonetheless, these results demonstrate that the effect of small molecule inhibition of GRP94 is context dependent and suggests that conclusions about the mechanism and the activity of GRP94 should not be derived based on the use of singular experimentation mode. We highly recommend chemical tool and genetic approaches to be used in parallel.
HSP70 probes
Another major chaperome member is HSP70. HSP70 chemical tool discovery is a yet developing field, and relatively young when compared to similar efforts for HSP90, explaining the relative lower frequency of cases where such agents have been used, in a proper fashion, as chemical tools. However, HSP70 is a promising target in a variety of diseases (Pratt et al., 2015, Sherman and Gabai, 2015) and we expect that the HSP70 chemical tool landscape will exponentially change in the next few years.
So far, the modulation of HSP70 activity has been achieved through inhibitors that bind the protein at multiple sites (Figure 3). Initial efforts to develop HSP70 inhibitors through ATP-competitive small molecules were proven challenging. The hydrophilic nature of the ATP binding pocket, made the discovery of high-affinity, drug-like ligands, difficult. Nonetheless, chemical tools need not be drugs. VER-155008 (Figure 3A) (Massey et al., 2010), an ATP-competitive modulator was created by structure-based design to act on HSP70 via insertion into The ATP-binding pocket. A Vernalis team used a collection of adenosine analogs and screened them against HSP70. A hit molecule was co-crystallized with HSP70 to confirm its binding into the ATP-binding cleft. Structure-guided medicinal chemistry led to VER-155008, a pan-HSP70 ligand. The Vernalis team used a similar approach to discover inhibitors with specificity for GRP78, the ER-resident member of the HSP70 family (Macias et al., 2011).
Figure 3. HSP70 chemical probes.
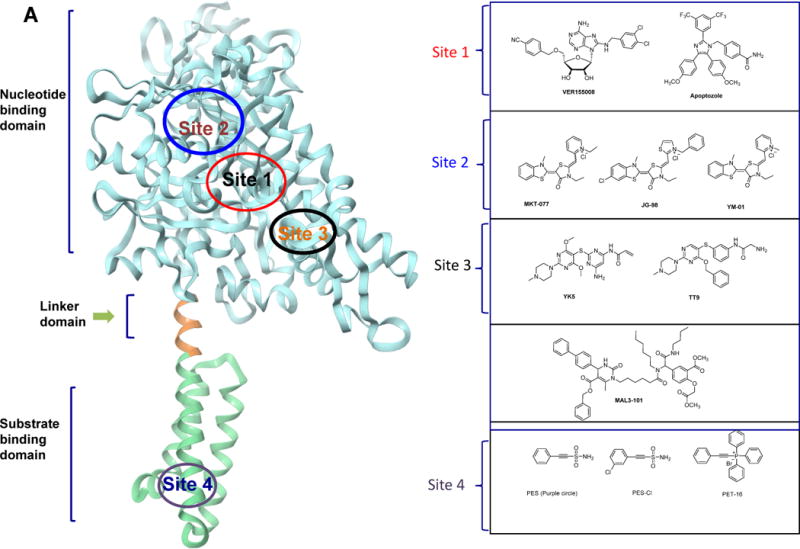
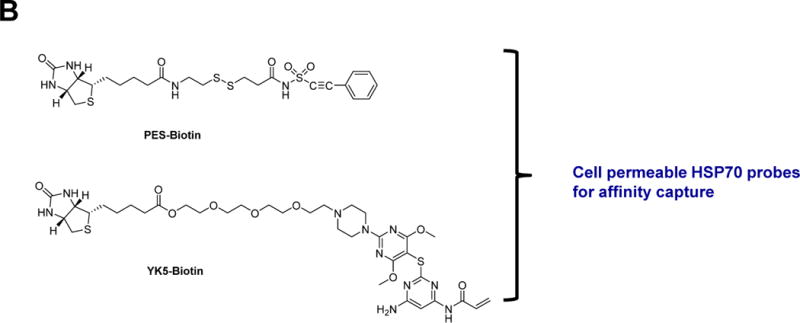
(A) Ribbon representation of the HSP70 homolog DnaK (E.coli, PDB:4B9Q) and demonstrated or proposed mode of interaction for select HSP70 inhibitors. The chemical structure of each ligand is depicted on the right side. (B) HSP70 inhibitor-derived chemical probes
The pan-HSP70 compound VER-155008 has biological activity in cells and was used in subsequent investigations into the biology of HSP70. For example, it was used to show a role for HSP70 in cancer cell migration through HSP70’s ability to bind the protein cross-linking enzyme, tissue transglutaminase. It highlighted an unconventional role for HSP70 in the localization of a key regulatory protein at the leading edges of cancer cells and the important consequences that this holds for their ability to migrate (Boroughs et al., 2011). Others used VER155008 to investigate stress granule formation (Bounedjah et al., 2014), the pathogenicity and virulence of Entamoeba histolytica (Santos et al., 2015) to confirm a role for HSP70 in the infection cycle of baculoviruses. When VER-155008 was added early in infection, the synthesis of viral proteins, genome replication and the production of budded virions were markedly inhibited by the chemical tool indicating the dependence of virus reproduction on HSP70 (Lyupina et al., 2014). Other studies used VER155008 to provide a link between HSP70 and the NEK protein kinase family in mitotic progression (O’regan et al., 2015).
Apoptozole (Figure 3A) was discovered in a phenotypic screen searching for apoptosis triggering compounds (Williams et al., 2008). A solid-support immobilized apoptozole was used to pinpoint HSP70 as the potential target of apoptozole in cells; the full-length protein and the ATPase domain bound to the resin, but the SBD did not. Mechanistic studies showed that apoptozole blocks the interaction of HSP70 with APAF-1 (apoptotic peptidase activating factor 1) without interfering with its binding to ASK1 (apoptosis signal-regulating kinase 1), JNK (c-Jun N-terminal kinase), BAX and AIF (apoptosis-inducing factor) (Ko et al., 2015). It restored the chloride channel activity of mutant cystic fibrosis transmembrane conductance regulator (CFTR) by promoting its membrane trafficking, suggesting its potential use, when utilized in concert with complementary methods, as a tool to dissect disease mechanism associated with cystic fibrosis (Cho et al., 2011).
Garrido and team identified a domain in the apoptosis-inducing factor that interacts with the SBD of HSP70 and designed a peptidomimetic, ADD70 that disrupts HSP70/AIF binding (Schmitt et al., 2006). They used ADD70 to show that its transfection in cancer cell elicited CD8+ T cell sequestration, supporting a role of this HSP70 axis in regulating an anti-tumor immune response.
Other efforts focused on allosteric inhibitors, a path that provided a variety of HSP70 probes, and hopefully, leads for drug development purposes. For example, Fewell et al screened for compounds that act on the ability of J proteins to stimulate HSP70’s ATPase activity; from a panel of dihydropyrimidines, they discovered MAL3-101 (Figure 3A) (Fewell et al., 2004). MAL3-101 and derivatives bind to the NBD of HSP70 in a region previously implicated in J protein interactions (Wisen et al., 2010). MAL3-101 showed cellular activity in Merkel cell carcinoma cell lines (Adam et al., 2014) and multiple myeloma (Braunstein et al., 2011) indicating a role for HSP70 in these cancers. MAL3-101 was also used as a chemical probe to investigate the role of HSP70 in the anti-apoptotic phenotype of small cell lung carcinoma (Rodina et al., 2007).
2-Phenylethynesulfonamide (PES or pifithrin-μ, PFTμ, Figure 3A)) was originally discovered out of a screen for molecules that activate p53-mediated apoptosis (Leu et al., 2009). Use of a biotinylated PES (Figure 3B) analog indicated HSP70 as a target of PES in cell homogenates. Co-crystal structures of HSP70 with a PES analog, PET-16 (Figure 3A), located a hydrophobic groove in the substrate binding domain of HSP70, as the site of ligand interaction. PES disrupts binding of HSP70 to p53, and blocks the interactions of HSP70 with some of its co-chaperones, including CHIP, certain J proteins (e.g., DjB1), and Bag1. PES was used to investigate a role for HSP70 in regulating the autophagy-lysosome and other protein clearance systems (Leu et al., 2009, Leu et al., 2011b) and to demonstrate a particular relevance for this mechanism in specific primary effusion lymphoma cells (Granato et al., 2013) and pancreatic cancer cells (Monma et al., 2013). PES-mediated inhibition of HSP70 also indirectly altered the activities of HSP90 (Leu et al., 2011a). The use of PES uncovered a novel pro-survival function of BAX (Mattiolo et al., 2014), whereas that of a PES derivative, PES-Cl (Figure 3A), indicated a potential role for HSP70 in anaphase promoting complex activity (Balaburski et al., 2013).
The rhodocyanine MKT-077 (Figure 3A) came out from a phenotypic screen in which antiproliferative activity was the read-out (Chiba et al., 1998, Koya et al., 1996). Its activity is HSP70 mediated and it was later found to insert itself into an allosteric site in HSP70 located in the nucleotide binding domain (Rousaki et al., 2011). Although this pocket is adjacent to the ATP-binding cleft, MKT-077 does not compete for binding to the nucleotide. MKT-077 was rapidly metabolized in vivo, which limited its use as a chemical probe in vivo. The Gestwicki lab improved upon this liability by creating derivative JG-98 (Figure 3A) (Li et al., 2015a). This derivative disrupted HSP70 interaction with its cochaperone (Li et al., 2015a). The BAG3/HSP70 complex was recently postulated to regulate several cancer-related signaling networks (Colvin et al., 2014). Another MKT-077 derivative YM-01 (Figure 3A) was used in models of neurodegenerative diseases. To investigate the link between HSP70 and tau, the Dickey team tested YM-01 in a tauopathy model to show that it reduced tau levels in HeLa cells overexpressing tau as well as endogenous tau in primary neurons from the mutant tau mouse model rTg4510 (Abisambra et al., 2013). Others used YM-01 to investigate the role of HSP70 in polyglutamine androgen receptor regulation, a toxic protein behind spinobulbar muscular atrophy (Wang et al., 2013).
We took a structure-based design approach. First, through computational analyses we identified a novel allosteric pocket on HSP70 (Rodina et al., 2013) that exists only in the ADP-bound HSP70 conformation. Then, by mental design and chemical intuition, and taking advantage of a reactive cysteine, C267, located deep inside the pocket, we designed a new chemical entity, never made by man or nature, to interact with such pocket, YK5 (Figure 3A). A biotinylated YK5 (Figure 3B) in concert with western blot, silver staining and mass spectrometry, was used to demonstrate HSP70 is the target of YK5 in cancer cells. When inside the pocket, YK5 reacts with C267 to form a covalent bond. Consequent structure-activity relationship studies improved the enthalpy of binding and led to reversible inhibitors (TT9; Figure 3A) as potent as YK5 (Taldone et al., 2014a). In cancer cells, both YK5 and TT9 altered the formation of a functional HSP70-HSP90 machinery and promoted the degradation of several of its onco-clients. Rodina et al took advantage of YK5 to design and develop an affinity purification chemical toolset for potential use in the investigation of the endogenous HSP70-interacting proteome in cancer (Rodina et al., 2014). A cell permeable YK5-biotin (Figure 3B) locked HSP70 in complex with onco-client proteins and effectively isolated HSP70 complexes for identification through biochemical techniques. Its capture ability was superior to that of widely used immunopurification tools, such as the BB70 antibody. Application of the chemical biology tools provided insights into the complex roles played by HSP70 in maintaining a multitude of cell-specific malignancy-driving proteins.
Conclusions
Stresses imposed on the human cell by specific imbalances and alterations in the proteome may lead to disease. The stresses may have genetic or environmental origin, or a combination of both, and ultimately manifest themselves in altering the proteome, which in turn perturbs the chaperome (Taldone et al., 2014b). As we are starting to appreciate, chemical modifications, such as PTMs, as well as biochemical modifications, such as co-chaperone, scaffold and adapter protein recruitment and binding, physically alter the chaperome to modify its function. In addition to activating the chaperome, these modifications contribute to the altered cellular location of its members as noted in numerous diseases. Together, all these operate simultaneously to provide cells with a balanced number of chaperome species that are best primed to assist the need of a specific proteome.
The complexity of the chaperome explains the limitations of classical approaches, i.e. genetic and biochemical, towards understanding stress, both as it relates to the chaperome and to the proteome it regulates. Most such methods treat the chaperome as a monolithic entity and thus, are unable to tackle the acknowledged contribution of protein conformation, protein complex components and PTMs to the activity of these proteins. For example, by not differentiating between the housekeeping and the stress species, genetic manipulations silencing the HSPs are often lethal, and therefore not deeply informative (Taldone et al., 2014b). Alternatively, due to the feedback synthesis of one closely related family member after the knock-down of another, such studies may often lead to no observable phenotypes (Daugaard et al., 2007). Cellular manipulations that are often conducted to investigate the function of a protein and its potential interactors, i.e. by transfection of mutants, tagged proteins, or overexpression systems, are also bound to lead to “false positives” for HSPs. This is of no surprise as the chaperome is the “buffer” of cellular stress, and such manipulations, which lead to proteome stress, are likely to impose artificial interactions on HSPs with the transfected proteins. Furthermore, these chaperome complexes are likely to be cell- and type-specific, and thus their study in an engineered cellular system may lead to an understanding that fits the model of study, but is far from the reality of the endogenous cell.
What is needed then is a diverse chemical toolset to inquire into the function and of as many HSP complexes and conformation as possible. Small molecules are sensors of protein thermodynamics and thus best primed to “see”, “capture” and “understand” the many faces of the chaperome species. The complexity of the chaperome as we described here also tells us that HSPs need a toolset rather than one specific tool to inquire into their biology and their role in disease. Genetic, biochemical and chemical biology methods should be used hand-in-hand to complement each other and provide the global, comprehensive insights that are needed to move this complex field forward. Only through such deep understanding will we be finally able to use this complexity to our advantage and be finally successful in co-opting it into the successful treatment of diseases.
Finally, when developing and implementing such tools, testing for in cell selectivity should be a priority. The cellular abundance and distinct cellular location of these chaperome complexes suggests that it is insufficient to prove selectivity in vitro, at the biochemical level, as it is likely that it will not translate into the same selectivity in cell based studies. Therefore, all in vitro selectivity profiles and on target activity need to be carefully re-evaluated and verified when using such chemical tools in cells. Lastly, robust pharmacodynamic studies, to validate on-target activity, should accompany any tool development aimed for use at organismal level.
It is true that first-generation probes, such as the majority of HSP70 currently are, likely have off-target activities, nonetheless these probes might still be useful provided that the off-target activities are appreciated in the experimental design and the findings resulted from such investigations are validated by complementary methods. Unfortunately little such complementary validation has been carried out so far and we propose a change in this practice.
To conclude, much has been gained and much more remains to be discovered about the complex cellular machinery, the chaperome. Substantial contributions to this knowledge have come through the use of sophisticated chemical biology tools and methods and we foresee that this contribution will continue to increase over the next years. We should better promote these tools and make them available to our cell biology and translational colleagues as only through these open ways of communication we can combine our synergistic expertise and make the big advancements needed for true transformative knowledge.
Acknowledgments
G.C. is funded by R01 CA172546, R01 CA155226, P01 CA177575, U01 AG032969 and P50 CA192937 and the David Rubenstein Center for Pancreatic Cancer Research. H.J.P. is a Susan G. Komen for the Cure fellow.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abisambra J, Jinwal UK, Miyata Y, Rogers J, Blair L, Li X, Seguin SP, Wang L, Jin Y, Bacon J, et al. Allosteric heat shock protein 70 inhibitors rapidly rescue synaptic plasticity deficits by reducing aberrant tau. Biol Psychiatry. 2013;74:367–74. doi: 10.1016/j.biopsych.2013.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adam C, Baeurle A, Brodsky JL, Wipf P, Schrama D, Becker JC, Houben R. The Hsp70 modulator mal3-101 inhibits merkel cell carcinoma. PloS one. 2014;9:e92041. doi: 10.1371/journal.pone.0092041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alarcon SV, Mollapour M, Lee MJ, Tsutsumi S, Lee S, Kim YS, Prince T, Apolo AB, Giaccone G, Xu W, et al. Tumor-intrinsic and tumor-extrinsic factors impacting Hsp90- targeted therapy. Curr Mol Med. 2012;12:1125–41. doi: 10.2174/156652412803306729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altieri DC. Mitochondrial Hsp90s and tumor cell metabolism. Autophagy. 2013;9:244–5. doi: 10.4161/auto.22527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaburski GM, Leu JI, Beeharry N, Hayik S, Andrake MD, Zhang G, Herlyn M, Villanueva J, Dunbrack RL, Jr, Yen T, et al. A modified Hsp70 inhibitor shows broad activity as an anticancer agent. Mol Cancer Res. 2013;11:219–29. doi: 10.1158/1541-7786.MCR-12-0547-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrott JJ, Hughes PF, Osada T, Yang XY, Hartman ZC, Loiselle DR, Spector NL, Neckers L, Rajaram N, Hu F, et al. Optical and radioiodinated tethered Hsp90 inhibitors reveal selective internalization of ectopic Hsp90 in malignant breast tumor cells. Chem Biol. 2013;20:1187–97. doi: 10.1016/j.chembiol.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beebe K, Mollapour M, Scroggins B, Prodromou C, Xu W, Tokita M, Taldone T, Pullen L, Zierer BK, Lee MJ, et al. Posttranslational modification and conformational state of heat shock protein 90 differentially affect binding of chemically diverse small molecule inhibitors. Oncotarget. 2013;4:1065–74. doi: 10.18632/oncotarget.1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boroughs LK, Antonyak MA, Johnson JL, Cerione RA. A unique role for heat shock protein 70 and its binding partner tissue transglutaminase in cancer cell migration. J Biol Chem. 2011;286:37094–107. doi: 10.1074/jbc.M111.242438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bounedjah O, Desforges B, Wu TD, Pioche-Durieu C, Marco S, Hamon L, Curmi PA, Guerquin-Kern JL, Pietrement O, Pastre D. Free mrna in excess upon polysome dissociation is a scaffold for protein multimerization to form stress granules. Nucleic Acids Res. 2014;42:8678–91. doi: 10.1093/nar/gku582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracher A, Verghese J. GrpE, Hsp110/Grp170, Hspbp1/sil1 and bag domain proteins: Nucleotide exchange factors for Hsp70 molecular chaperones. Subcell Biochem. 2015;78:1–33. doi: 10.1007/978-3-319-11731-7_1. [DOI] [PubMed] [Google Scholar]
- Braunstein MJ, Scott SS, Scott CM, Behrman S, Walter P, Wipf P, Coplan JD, Chrico W, Joseph D, Brodsky JL, et al. Antimyeloma effects of the heat shock protein 70 molecular chaperone inhibitor mal3-101. J Oncol. 2011;2011:232037. doi: 10.1155/2011/232037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brehme M, Voisine C, Rolland T, Wachi S, Soper JH, Zhu Y, Orton K, Villella A, Garza D, Vidal M, et al. A chaperome subnetwork safeguards proteostasis in aging and neurodegenerative disease. Cell Rep. 2014;9:1135–50. doi: 10.1016/j.celrep.2014.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks JC, Sun W, Chiosis G, Leifer CA. Heat shock protein gp96 regulates toll-like receptor 9 proteolytic processing and conformational stability. Biochem Biophys Res Commun. 2012;421:780–4. doi: 10.1016/j.bbrc.2012.04.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caino MC, Chae YC, Vaira V, Ferrero S, Nosotti M, Martin NM, Weeraratna A, O’connell M, Jernigan D, Fatatis A, et al. Metabolic stress regulates cytoskeletal dynamics and metastasis of cancer cells. J Clin Invest. 2013;123:2907–20. doi: 10.1172/JCI67841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carman A, Kishinevsky S, Koren J, 3rd, Lou W, Chiosis G. Chaperone-dependent neurodegeneration: A molecular perspective on therapeutic intervention. J Alzheimers Dis Parkinsonism. 2013:2013. doi: 10.4172/2161-0460.S10-007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerchietti LC, Lopes EC, Yang SN, Hatzi K, Bunting KL, Tsikitas LA, Mallik A, Robles AI, Walling J, Varticovski L, et al. A purine scaffold Hsp90 inhibitor destabilizes bcl-6 and has specific antitumor activity in bcl-6-dependent b cell lymphomas. Nat Med. 2009;15:1369–76. doi: 10.1038/nm.2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chae YC, Angelin A, Lisanti S, Kossenkov AV, Speicher KD, Wang H, Powers JF, Tischler AS, Pacak K, Fliedner S, et al. Landscape of the mitochondrial Hsp90 metabolome in tumours. Nat Commun. 2013;4:2139. doi: 10.1038/ncomms3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba Y, Kubota T, Watanabe M, Otani Y, Teramoto T, Matsumoto Y, Koya K, Kitajima M. Selective antitumor activity of mkt-077, a delocalized lipophilic cation, on normal cells and cancer cells in vitro. J Surg Oncol. 1998;69:105–10. doi: 10.1002/(sici)1096-9098(199810)69:2<105::aid-jso11>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Chiosis G, Timaul MN, Lucas B, Munster PN, Zheng FF, Sepp-Lorenzino L, Rosen N. A small molecule designed to bind to the adenine nucleotide pocket of Hsp90 causes her2 degradation and the growth arrest and differentiation of breast cancer cells. Chem Biol. 2001;8:289–99. doi: 10.1016/s1074-5521(01)00015-1. [DOI] [PubMed] [Google Scholar]
- Cho HJ, Gee HY, Baek KH, Ko SK, Park JM, Lee H, Kim ND, Lee MG, Shin I. A small molecule that binds to an ATPase domain of hsc70 promotes membrane trafficking of mutant cystic fibrosis transmembrane conductance regulator. J Am Chem Soc. 2011;133:20267–76. doi: 10.1021/ja206762p. [DOI] [PubMed] [Google Scholar]
- Cho HS, Shimazu T, Toyokawa G, Daigo Y, Maehara Y, Hayami S, Ito A, Masuda K, Ikawa N, Field HI, et al. Enhanced Hsp70 lysine methylation promotes proliferation of cancer cells through activation of aurora kinase b. Nat Commun. 2012;3:1072. doi: 10.1038/ncomms2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colvin TA, Gabai VL, Gong J, Calderwood SK, Li H, Gummuluru S, Matchuk ON, Smirnova SG, Orlova NV, Zamulaeva IA, et al. Hsp70-bag3 interactions regulate cancer-related signaling networks. Cancer Res. 2014;74:4731–40. doi: 10.1158/0008-5472.CAN-14-0747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connell P, Ballinger CA, Jiang J, Wu Y, Thompson LJ, Hohfeld J, Patterson C. The co-chaperone Chip regulates protein triage decisions mediated by heat-shock proteins. Nat Cell Biol. 2001;3:93–6. doi: 10.1038/35050618. [DOI] [PubMed] [Google Scholar]
- Daugaard M, Rohde M, Jaattela M. The heat shock protein 70 family: Highly homologous proteins with overlapping and distinct functions. FEBS Lett. 2007;581:3702–10. doi: 10.1016/j.febslet.2007.05.039. [DOI] [PubMed] [Google Scholar]
- Defee MR, Qin Z, Dai L, Toole BP, Isaacs JS, Parsons CH. Extracellular Hsp90 serves as a co-factor for nf-kappab activation and cellular pathogenesis induced by an oncogenic herpesvirus. Am J Cancer Res. 2011;1:687–700. [PMC free article] [PubMed] [Google Scholar]
- Duerfeldt AS, Peterson LB, Maynard JC, Ng CL, Eletto D, Ostrovsky O, Shinogle HE, Moore DS, Argon Y, Nicchitta CV, et al. Development of a Grp94 inhibitor. J Am Chem Soc. 2012;134:9796–804. doi: 10.1021/ja303477g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst JT, Neubert T, Liu M, Sperry S, Zuccola H, Turnbull A, Fleck B, Kargo W, Woody L, Chiang P, et al. Identification of novel Hsp90alpha/beta isoform selective inhibitors using structure-based drug design. Demonstration of potential utility in treating cns disorders such as Huntington’s disease. J Med Chem. 2014;57:3382–400. doi: 10.1021/jm500042s. [DOI] [PubMed] [Google Scholar]
- Fewell SW, Smith CM, Lyon MA, Dumitrescu TP, Wipf P, Day BW, Brodsky JL. Small molecule modulators of endogenous and co-chaperone-stimulated Hsp70 ATPase activity. J Biol Chem. 2004;279:51131–40. doi: 10.1074/jbc.M404857200. [DOI] [PubMed] [Google Scholar]
- Finka A, Goloubinoff P. Proteomic data from human cell cultures refine mechanisms of chaperone-mediated protein homeostasis. Cell Stress Chaperones. 2013;18:591–605. doi: 10.1007/s12192-013-0413-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geller R, Taguwa S, Frydman J. Broad action of Hsp90 as a host chaperone required for viral replication. Biochim Biophys Acta. 2012;1823:698–706. doi: 10.1016/j.bbamcr.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein RL, Yang SN, Taldone T, Chang B, Gerecitano J, Elenitoba-Johnson K, Shaknovich R, Tam W, Leonard JP, Chiosis G, et al. Pharmacoproteomics identifies combinatorial therapy targets for diffuse large b cell lymphoma. J Clin Invest. 2015:2015. doi: 10.1172/JCI80714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granato M, Lacconi V, Peddis M, Lotti LV, Di Renzo L, Gonnella R, Santarelli R, Trivedi P, Frati L, D’orazi G, et al. Hsp70 inhibition by 2-phenylethynesulfonamide induces lysosomal cathepsin d release and immunogenic cell death in primary effusion lymphoma. Cell Death Dis. 2013;4:e730. doi: 10.1038/cddis.2013.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall JA, Forsberg LK, Blagg BS. Alternative approaches to Hsp90 modulation for the treatment of cancer. Future Med Chem. 2014;6:1587–605. doi: 10.4155/fmc.14.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl FU, Bracher A, Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. Nature. 2011;475:324–32. doi: 10.1038/nature10317. [DOI] [PubMed] [Google Scholar]
- Hernandez MP, Sullivan WP, Toft DO. The assembly and intermolecular properties of the Hsp70-Hop-Hsp90 molecular chaperone complex. J Biol Chem. 2002;277:38294–304. doi: 10.1074/jbc.M206566200. [DOI] [PubMed] [Google Scholar]
- Hong F, Liu B, Chiosis G, Gewirth DT, Li Z. Alpha7 helix region of alphai domain is crucial for integrin binding to endoplasmic reticulum chaperone gp96: A potential therapeutic target for cancer metastasis. J Biol Chem. 2013;288:18243–8. doi: 10.1074/jbc.M113.468850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua Y, White-Gilbertson S, Kellner J, Rachidi S, Usmani SZ, Chiosis G, Depinho R, Li Z, Liu B. Molecular chaperone gp96 is a novel therapeutic target of multiple myeloma. Clin Cancer Res. 2013;19:6242–51. doi: 10.1158/1078-0432.CCR-13-2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai T, Kato Y, Kajiwara C, Mizukami S, Ishige I, Ichiyanagi T, Hikida M, Wang JY, Udono H. Heat shock protein 90 (Hsp90) contributes to cytosolic translocation of extracellular antigen for cross-presentation by dendritic cells. Proc Natl Acad Sci USA. 2011;108:16363–8. doi: 10.1073/pnas.1108372108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhaveri K, Ochiana SO, Dunphy MP, Gerecitano JF, Corben AD, Peter RI, Janjigian YY, Gomes-Dagama EM, Koren J, 3rd, Modi S, et al. Heat shock protein 90 inhibitors in the treatment of cancer: Current status and future directions. Expert Opin Investig Drugs. 2014;23:611–28. doi: 10.1517/13543784.2014.902442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamal A, Thao L, Sensintaffar J, Zhang L, Boehm MF, Fritz LC, Burrows FJ. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature. 2003;425:407–10. doi: 10.1038/nature01913. [DOI] [PubMed] [Google Scholar]
- Kampinga HH, Craig EA. The Hsp70 chaperone machinery: J proteins as drivers of functional specificity. Nat Rev Mol Cell Biol. 2010;11:579–92. doi: 10.1038/nrm2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang BH, Plescia J, Song HY, Meli M, Colombo G, Beebe K, Scroggins B, Neckers L, Altieri DC. Combinatorial drug design targeting multiple cancer signaling networks controlled by mitochondrial Hsp90. J Clin Invest. 2009;119:454–64. doi: 10.1172/JCI37613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YE, Hipp MS, Bracher A, Hayer-Hartl M, Hartl FU. Molecular chaperone functions in protein folding and proteostasis. Annu Rev Biochem. 2013;82:323–55. doi: 10.1146/annurev-biochem-060208-092442. [DOI] [PubMed] [Google Scholar]
- Kityk R, Kopp J, Sinning I, Mayer MP. Structure and dynamics of the ATP-bound open conformation of Hsp70 chaperones. Mol Cell. 2012;48:863–74. doi: 10.1016/j.molcel.2012.09.023. [DOI] [PubMed] [Google Scholar]
- Ko SK, Kim J, Na DC, Park S, Park SH, Hyun JY, Baek KH, Kim ND, Kim NK, Park YN, et al. A small molecule inhibitor of ATPase activity of Hsp70 induces apoptosis and has antitumor activities. Chem Biol. 2015;22:391–403. doi: 10.1016/j.chembiol.2015.02.004. [DOI] [PubMed] [Google Scholar]
- Koya K, Li Y, Wang H, Ukai T, Tatsuta N, Kawakami M, Shishido, Chen LB. Mkt-077, a novel rhodacyanine dye in clinical trials, exhibits anticarcinoma activity in preclinical studies based on selective mitochondrial accumulation. Cancer Res. 1996;56:538–43. [PubMed] [Google Scholar]
- Krukenberg KA, Street TO, Lavery LA, Agard DA. Conformational dynamics of the molecular chaperone Hsp90. Q Rev Biophys. 2011;44:229–55. doi: 10.1017/S0033583510000314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Park HK, Jeong H, Lim J, Lee AJ, Cheon KY, Kim CS, Thomas AP, Bae B, Kim ND, et al. Development of a mitochondria-targeted Hsp90 inhibitor based on the crystal structures of human trap1. J Am Chem Soc. 2015;137:4358–67. doi: 10.1021/ja511893n. [DOI] [PubMed] [Google Scholar]
- Leu JI, Pimkina J, Pandey P, Murphy ME, George DL. Hsp70 inhibition by the small-molecule 2-phenylethynesulfonamide impairs protein clearance pathways in tumor cells. Mol Cancer Res. 2011a;9:936–47. doi: 10.1158/1541-7786.MCR-11-0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leu JIJ, Pimkina J, Frank A, Murphy ME, George DL. A small molecule inhibitor of inducible heat shock protein 70. Mol Cell. 2009;36:15–27. doi: 10.1016/j.molcel.2009.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leu JIJ, Pimkina J, Pandey P, Murphy ME, George DL. Hsp70 inhibition by the small-molecule 2-phenylethynesulfonamide impairs protein clearance pathways in tumor cells. Mol Cancer Res. 2011b;9:936–947. doi: 10.1158/1541-7786.MCR-11-0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Soroka J, Buchner J. The Hsp90 chaperone machinery: Conformational dynamics and regulation by co-chaperones. Biochim Biophys Acta. 2012;1823:624–35. doi: 10.1016/j.bbamcr.2011.09.003. [DOI] [PubMed] [Google Scholar]
- Li X, Colvin T, Rauch JN, Acosta-Alvear D, Kampmann M, Dunyak B, Hann B, Aftab BT, Murnane M, Cho M, et al. Validation of the Hsp70-bag3 protein-protein interaction as a potential therapeutic target in cancer. Mol Cancer Ther. 2015a;14:642–8. doi: 10.1158/1535-7163.MCT-14-0650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Sun L, Hou J, Gui M, Ying J, Zhao H, Lv N, Meng S. Cell membrane gp96 facilitates her2 dimerization and serves as a novel target in breast cancer. Int J Cancer. 2015b;137:512–24. doi: 10.1002/ijc.29405. [DOI] [PubMed] [Google Scholar]
- Lisanti S, Tavecchio M, Chae YC, Liu Q, Brice AK, Thakur ML, Languino LR, Altieri DC. Deletion of the mitochondrial chaperone TRAP-1 uncovers global reprogramming of metabolic networks. Cell Rep. 2014;8:671–7. doi: 10.1016/j.celrep.2014.06.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyupina YV, Orlova OV, Abaturova SB, Beljelarskaya SN, Lavrov AN, Mikhailov VS. Egress of budded virions of autographa californica nucleopolyhedrovirus does not require activity of spodoptera frugiperda Hsp/hsc70 chaperones. Virus Res. 2014;192:1–5. doi: 10.1016/j.virusres.2014.08.002. [DOI] [PubMed] [Google Scholar]
- Macias AT, Williamson DS, Allen N, Borgognoni J, Clay A, Daniels Z, Dokurno P, Drysdale MJ, Francis GL, Graham CJ, et al. Adenosine-derived inhibitors of 78 kda glucose regulated protein (Grp78) ATPase: Insights into isoform selectivity. J Med Chem. 2011;54:4034–41. doi: 10.1021/jm101625x. [DOI] [PubMed] [Google Scholar]
- Marzec M, Eletto D, Argon Y. Grp94: An Hsp90-like protein specialized for protein folding and quality control in the endoplasmic reticulum. Biochim Biophys Acta. 2012;1823:774–87. doi: 10.1016/j.bbamcr.2011.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massey AJ, Williamson DS, Browne H, Murray JB, Dokurno P, Shaw T, Macias AT, Daniels Z, Geoffroy S, Dopson M, et al. A novel, small molecule inhibitor of hsc70/Hsp70 potentiates Hsp90 inhibitor induced apoptosis in hct116 colon carcinoma cells. Cancer Chemother Pharmacol. 2010;66:535–45. doi: 10.1007/s00280-009-1194-3. [DOI] [PubMed] [Google Scholar]
- Mattiolo P, Barbero-Farran A, Amigo J, Ripamonti M, Ribas J, Boix J. Cell death induced by 2-phenylethynesulfonamide uncovers a pro-survival function of bax. Cancer Lett. 2014;354:115–21. doi: 10.1016/j.canlet.2014.07.037. [DOI] [PubMed] [Google Scholar]
- Matts RL, Brandt GE, Lu Y, Dixit A, Mollapour M, Wang S, Donnelly AC, Neckers L, Verkhivker G, Blagg BS. A systematic protocol for the characterization of Hsp90 modulators. Bioorg Med Chem. 2011;19:684–92. doi: 10.1016/j.bmc.2010.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer MP, Bukau B. Hsp70 chaperones: Cellular functions and molecular mechanism. Cell Mol Life Sci. 2005;62:670–84. doi: 10.1007/s00018-004-4464-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer MP, Kityk R. Insights into the molecular mechanism of allostery in Hsp70s. Front Mol Biosci. 2015;2:58. doi: 10.3389/fmolb.2015.00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mccready J, Wong DS, Burlison JA, Ying W, Jay DG. An impermeant ganetespib analog inhibits extracellular Hsp90-mediated cancer cell migration that involves lysyl oxidase 2-like protein. Cancers. 2014;6:1031–46. doi: 10.3390/cancers6021031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollapour M, Bourboulia D, Beebe K, Woodford MR, Polier S, Hoang A, Chelluri R, Li Y, Guo A, Lee MJ, et al. Asymmetric Hsp90 n domain sumoylation recruits aha1 and ATP-competitive inhibitors. Mol Cell. 2014;53:317–29. doi: 10.1016/j.molcel.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollapour M, Neckers L. Post-translational modifications of Hsp90 and their contributions to chaperone regulation. Biochim Biophys Acta. 2012;1823:648–55. doi: 10.1016/j.bbamcr.2011.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monma H, Harashima N, Inao T, Okano S, Tajima Y, Harada M. The Hsp70 and autophagy inhibitor pifithrin-mu enhances the antitumor effects of trail on human pancreatic cancer. Mol Cancer Ther. 2013;12:341–51. doi: 10.1158/1535-7163.MCT-12-0954. [DOI] [PubMed] [Google Scholar]
- Moulick K, Ahn JH, Zong H, Rodina A, Cerchietti L, Gomes Dagama EM, Caldas-Lopes E, Beebe K, Perna F, Hatzi K, et al. Affinity-based proteomics reveal cancer-specific networks coordinated by Hsp90. Nat Chem Biol. 2011;7:818–26. doi: 10.1038/nchembio.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller P, Ruckova E, Halada P, Coates PJ, Hrstka R, Lane DP, Vojtesek B. C-terminal phosphorylation of Hsp70 and Hsp90 regulates alternate binding to co-chaperones Chip and Hop to determine cellular protein folding/degradation balances. Oncogene. 2013;32:3101–10. doi: 10.1038/onc.2012.314. [DOI] [PubMed] [Google Scholar]
- Nayar U, Lu P, Goldstein RL, Vider J, Ballon G, Rodina A, Taldone T, Erdjument-Bromage H, Chomet M, Blasberg R, et al. Targeting the Hsp90-associated viral oncoproteome in gammaherpesvirus-associated malignancies. Blood. 2013;122:2837–47. doi: 10.1182/blood-2013-01-479972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neckers L. Chaperoning oncogenes: Hsp90 as a target of geldanamycin. Handb Exp Pharmacol. 2006:259–77. doi: 10.1007/3-540-29717-0_11. [DOI] [PubMed] [Google Scholar]
- Neckers L, Schulte TW, Mimnaugh E. Geldanamycin as a potential anti-cancer agent: Its molecular target and biochemical activity. Invest New Drugs. 1999;17:361–73. doi: 10.1023/a:1006382320697. [DOI] [PubMed] [Google Scholar]
- O’regan L, Sampson J, Richards MW, Knebel A, Roth D, Hood FE, Straube A, Royle SJ, Bayliss R, Fry AM. Hsp72 is targeted to the mitotic spindle by nek6 to promote k-fiber assembly and mitotic progression. J Cell Biol. 2015;209:349–58. doi: 10.1083/jcb.201409151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HK, Lee JE, Lim J, Kang BH. Mitochondrial Hsp90s suppress calcium-mediated stress signals propagating from mitochondria to the er in cancer cells. Mol Cancer. 2014;13:148. doi: 10.1186/1476-4598-13-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel HJ, Modi S, Chiosis G, Taldone T. Advances in the discovery and development of heat-shock protein 90 inhibitors for cancer treatment. Expert Opin Drug Discov. 2011;6:559–587. doi: 10.1517/17460441.2011.563296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel HJ, Patel PD, Ochiana SO, Yan P, Sun W, Patel MR, Shah SK, Tramentozzi E, Brooks J, Bolaender A, et al. Structure-activity relationship in a purine-scaffold compound series with selectivity for the endoplasmic reticulum Hsp90 paralog Grp94. J Med Chem. 2015;58:3922–43. doi: 10.1021/acs.jmedchem.5b00197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel PD, Yan P, Seidler PM, Patel HJ, Sun W, Yang C, Que NS, Taldone T, Finotti P, Stephani RA, et al. Paralog-selective Hsp90 inhibitors define tumor-specific regulation of her2. Nat Chem Biol. 2013;9:677–84. doi: 10.1038/nchembio.1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pimienta G, Herbert KM, Regan L. A compound that inhibits the Hop-Hsp90 complex formation and has unique killing effects in breast cancer cell lines. Mol Pharm. 2011;8:2252–61. doi: 10.1021/mp200346y. [DOI] [PubMed] [Google Scholar]
- Pratt WB, Gestwicki JE, Osawa Y, Lieberman AP. Targeting Hsp90/Hsp70-based protein quality control for treatment of adult onset neurodegenerative diseases. Annu Rev Pharmacol Toxicol. 2015;55:353–71. doi: 10.1146/annurev-pharmtox-010814-124332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prince TL, Kijima T, Tatokoro M, Lee S, Tsutsumi S, Yim K, Rivas C, Alarcon S, Schwartz H, Khamit-Kush K, et al. Client proteins and small molecule inhibitors display distinct binding preferences for constitutive and stress-induced Hsp90 isoforms and their conformationally restricted mutants. PloS one. 2015;10:e0141786. doi: 10.1371/journal.pone.0141786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prodromou C. The ‘active life’ of Hsp90 complexes. Biochim Biophys Acta. 2012;1823:614–23. doi: 10.1016/j.bbamcr.2011.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin S, Ni M, Wang X, Maurier-Mahe F, Shurland DL, Rodrigues GA. Inhibition of rpe cell sterile inflammatory responses and endotoxin-induced uveitis by a cell-impermeable Hsp90 inhibitor. Exp Eye Res. 2011;93:889–97. doi: 10.1016/j.exer.2011.10.002. [DOI] [PubMed] [Google Scholar]
- Rodina A, Patel PD, Kang Y, Patel Y, Baaklini I, Wong MJ, Taldone T, Yan P, Yang C, Maharaj R, et al. Identification of an allosteric pocket on human Hsp70 reveals a mode of inhibition of this therapeutically important protein. Chem Biol. 2013;20:1469–80. doi: 10.1016/j.chembiol.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodina A, Taldone T, Kang Y, Patel PD, Koren J, 3rd, Yan P, Dagama Gomes EM, Yang C, Patel MR, Shrestha L, et al. Affinity purification probes of potential use to investigate the endogenous Hsp70 interactome in cancer. ACS chem biol. 2014;9:1698–705. doi: 10.1021/cb500256u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodina A, Vilenchik M, Moulick K, Aguirre J, Kim J, Chiang A, Litz J, Clement CC, Kang Y, She Y, et al. Selective compounds define Hsp90 as a major inhibitor of apoptosis in small-cell lung cancer. Nat Chem Biol. 2007;3:498–507. doi: 10.1038/nchembio.2007.10. [DOI] [PubMed] [Google Scholar]
- Rosser MF, Nicchitta CV. Ligand interactions in the adenosine nucleotide-binding domain of the Hsp90 chaperone, Grp94. I. Evidence for allosteric regulation of ligand binding. J Biol Chem. 2000;275:22798–805. doi: 10.1074/jbc.M001477200. [DOI] [PubMed] [Google Scholar]
- Rousaki A, Miyata Y, Jinwal UK, Dickey CA, Gestwicki JE, Zuiderweg ER. Allosteric drugs: The interaction of antitumor compound mkt-077 with human Hsp70 chaperones. J Mol Biol. 2011;411:614–32. doi: 10.1016/j.jmb.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford SL, Lindquist S. Hsp90 as a capacitor for morphological evolution. Nature. 1998;396:336–42. doi: 10.1038/24550. [DOI] [PubMed] [Google Scholar]
- Samant RS, Clarke PA, Workman P. The expanding proteome of the molecular chaperone Hsp90. Cell cycle. 2012;11:1301–8. doi: 10.4161/cc.19722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangster TA, Lindquist S, Queitsch C. Under cover: Causes, effects and implications of Hsp90-mediated genetic capacitance. BioEssays: news and reviews in molecular, cellular and developmental biology. 2004;26:348–62. doi: 10.1002/bies.20020. [DOI] [PubMed] [Google Scholar]
- Santos F, Nequiz M, Hernandez-Cuevas NA, Hernandez K, Pineda E, Encalada R, Guillen N, Luis-Garcia E, Saralegui A, Saavedra E, et al. Maintenance of intracellular hypoxia and adequate heat shock response are essential requirements for pathogenicity and virulence of entamoeba histolytica. Cell Microbiol. 2015;17:1037–51. doi: 10.1111/cmi.12419. [DOI] [PubMed] [Google Scholar]
- Schmitt E, Maingret L, Puig PE, Rerole AL, Ghiringhelli F, Hammann A, Solary E, Kroemer G, Garrido C. Heat shock protein 70 neutralization exerts potent antitumor effects in animal models of colon cancer and melanoma. Cancer Res. 2006;66:4191–7. doi: 10.1158/0008-5472.CAN-05-3778. [DOI] [PubMed] [Google Scholar]
- Sevin M, Girodon F, Garrido C, De Thonel A. Hsp90 and Hsp70: Implication in inflammation processes and therapeutic approaches for myeloproliferative neoplasms. Mediators Inflamm. 2015;2015:970242. doi: 10.1155/2015/970242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma K, Vabulas RM, Macek B, Pinkert S, Cox J, Mann M, Hartl FU. Quantitative proteomics reveals that Hsp90 inhibition preferentially targets kinases and the DNA damage response. Mol Cell Proteomics. 2012;11:M111014654. doi: 10.1074/mcp.M111.014654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman MY, Gabai VL. Hsp70 in cancer: Back to the future. Oncogene. 2015;34:4153–61. doi: 10.1038/onc.2014.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldano KL, Jivan A, Nicchitta CV, Gewirth DT. Structure of the n-terminal domain of Grp94. Basis for ligand specificity and regulation. J Biol Chem. 2003;278:48330–8. doi: 10.1074/jbc.M308661200. [DOI] [PubMed] [Google Scholar]
- Stebbins CE, Russo AA, Schneider C, Rosen N, Hartl FU, Pavletich NP. Crystal structure of an Hsp90-geldanamycin complex: Targeting of a protein chaperone by an antitumor agent. Cell. 1997;89:239–50. doi: 10.1016/s0092-8674(00)80203-2. [DOI] [PubMed] [Google Scholar]
- Stothert AR, Suntharalingam A, Huard DJ, Fontaine SN, Crowley VM, Mishra S, Blagg BS, Lieberman RL, Dickey CA. Exploiting the interaction between Grp94 and aggregated myocilin to treat glaucoma. Hum Mol Genet. 2014;23:6470–80. doi: 10.1093/hmg/ddu367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Street TO, Lavery LA, Agard DA. Substrate binding drives large-scale conformational changes in the Hsp90 molecular chaperone. Mol Cell. 2011;42:96–105. doi: 10.1016/j.molcel.2011.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suntharalingam A, Abisambra JF, O’leary JC, 3rd, Koren J, 3rd, Zhang B, Joe MK, Blair LJ, Hill SE, Jinwal UK, Cockman M, et al. Glucose-regulated protein 94 triage of mutant myocilin through endoplasmic reticulum-associated degradation subverts a more efficient autophagic clearance mechanism. J Biol Chem. 2012;287:40661–9. doi: 10.1074/jbc.M112.384800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taldone T, Gomes-Dagama EM, Zong H, Sen S, Alpaugh ML, Zatorska D, Alonso-Sabadell R, Guzman ML, Chiosis G. Synthesis of purine-scaffold fluorescent probes for heat shock protein 90 with use in flow cytometry and fluorescence microscopy. Bioorg Med Chem Lett. 2011;21:5347–52. doi: 10.1016/j.bmcl.2011.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taldone T, Kang Y, Patel HJ, Patel MR, Patel PD, Rodina A, Patel Y, Gozman A, Maharaj R, Clement CC, et al. Heat shock protein 70 inhibitors. 2. 2,5′-thiodipyrimidines, 5-(phenylthio)pyrimidines, 2-(pyridin-3-ylthio)pyrimidines, and 3-(phenylthio)pyridines as reversible binders to an allosteric site on heat shock protein 70. J Med Chem. 2014a;57:1208–24. doi: 10.1021/jm401552y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taldone T, Ochiana SO, Patel PD, Chiosis G. Selective targeting of the stress chaperome as a therapeutic strategy. Trends Pharmacol Sci. 2014b;35:592–603. doi: 10.1016/j.tips.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taldone T, Patel PD, Patel M, Patel HJ, Evans CE, Rodina A, Ochiana S, Shah SK, Uddin M, Gewirth D, et al. Experimental and structural testing module to analyze paralogue-specificity and affinity in the Hsp90 inhibitors series. J Med Chem. 2013a;56:6803–18. doi: 10.1021/jm400619b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taldone T, Rodina A, Dagama Gomes EM, Riolo M, Patel HJ, Alonso-Sabadell R, Zatorska D, Patel MR, Kishinevsky S, Chiosis G. Synthesis and evaluation of cell-permeable biotinylated pu-h71 derivatives as tumor Hsp90 probes. Beilstein J Org Chem. 2013b;9:544–56. doi: 10.3762/bjoc.9.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsaytler PA, Krijgsveld J, Goerdayal SS, Rudiger S, Egmond MR. Novel Hsp90 partners discovered using complementary proteomic approaches. Cell Stress Chaperones. 2009;14:629–38. doi: 10.1007/s12192-009-0115-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutsumi S, Neckers L. Extracellular heat shock protein 90: A role for a molecular chaperone in cell motility and cancer metastasis. Cancer Sci. 2007;98:1536–9. doi: 10.1111/j.1349-7006.2007.00561.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutsumi S, Scroggins B, Koga F, Lee MJ, Trepel J, Felts S, Carreras C, Neckers L. A small molecule cell-impermeant Hsp90 antagonist inhibits tumor cell motility and invasion. Oncogene. 2008;27:2478–87. doi: 10.1038/sj.onc.1210897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan CK, Mollapour M, Smith JR, Truman A, Hu B, Good VM, Panaretou B, Neckers L, Clarke PA, Workman P, et al. Hsp90-dependent activation of protein kinases is regulated by chaperone-targeted dephosphorylation of cdc37. Mol Cell. 2008;31:886–95. doi: 10.1016/j.molcel.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang AM, Miyata Y, Klinedinst S, Peng HM, Chua JP, Komiyama T, Li X, Morishima Y, Merry DE, Pratt WB, et al. Activation of Hsp70 reduces neurotoxicity by promoting polyglutamine protein degradation. Nat Chem Biol. 2013;9:112–8. doi: 10.1038/nchembio.1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitesell L, Mimnaugh EG, De Costa B, Myers CE, Neckers LM. Inhibition of heat shock protein Hsp90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: Essential role for stress proteins in oncogenic transformation. Proc Natl Acad Sci USA. 1994;91:8324–8. doi: 10.1073/pnas.91.18.8324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitesell L, Santagata S, Mendillo ML, Lin NU, Proia DA, Lindquist S. Hsp90 empowers evolution of resistance to hormonal therapy in human breast cancer models. Proc Natl Acad Sci USA. 2014;111:18297–302. doi: 10.1073/pnas.1421323111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DR, Ko SK, Park S, Lee MR, Shin I. An apoptosis-inducing small molecule that binds to heat shock protein 70. Angew Chem Int Ed Engl. 2008;47:7466–7469. doi: 10.1002/anie.200802801. [DOI] [PubMed] [Google Scholar]
- Wisen S, Bertelsen EB, Thompson AD, Patury S, Ung P, Chang L, Evans CG, Walter GM, Wipf P, Carlson HA, et al. Binding of a small molecule at a protein-protein interface regulates the chaperone activity of Hsp70-Hsp40. ACS chem biol. 2010;5:611–22. doi: 10.1021/cb1000422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Mollapour M, Prodromou C, Wang S, Scroggins BT, Palchick Z, Beebe K, Siderius M, Lee MJ, Couvillon A, et al. Dynamic tyrosine phosphorylation modulates cycling of the Hsp90-p50(cdc37)-aha1 chaperone machine. Mol Cell. 2012;47:434–43. doi: 10.1016/j.molcel.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Neckers L. Targeting the molecular chaperone heat shock protein 90 provides a multifaceted effect on diverse cell signaling pathways of cancer cells. Clin Cancer Res. 2007;13:1625–9. doi: 10.1158/1078-0432.CCR-06-2966. [DOI] [PubMed] [Google Scholar]