

Abstract
Objective:
To identify the clinical and radiologic features that should raise suspicion for the pseudotumoral presentation of cerebral amyloid angiopathy–related inflammation (CAA-I).
Methods:
We retrospectively reviewed the characteristics of 5 newly diagnosed and 23 previously reported patients in whom the CAA-I imaging findings were initially interpreted as CNS neoplasms.
Results:
Most cases (85%) occurred in patients >60 years old. The clinical characteristics at presentation included subacute cognitive decline (50%), confusion (32%), focal deficits (32%), seizures (25%), and headaches (21%). Brain MRI demonstrated infiltrative white matter lesions that exhibited a loco-regional mass effect without parenchymal enhancement (93%). In general, these findings were interpreted as low-grade glioma or lymphoma. Eighteen patients (64%) underwent a biopsy, which was nondiagnostic in 4 patients (14%), and 6 patients (21%) underwent a surgical resection. The primary reason for the misinterpretation of the imaging findings was the absence of T2*-weighted gradient recalled echo (T2*-GRE) sequences on initial imaging (89%). When subsequently performed (39%), the T2*-GRE sequences demonstrated multiple characteristic cortical and subcortical microhemorrhages in all cases. Perfusion MRI and magnetic resonance spectroscopy (MRS), which were performed on a subset of patients, indicated markedly reduced relative cerebral blood flow and a normal metabolic ratio.
Conclusion:
The identification of one or several nonenhancing space-occupying lesions, especially in elderly patients presenting with cognitive impairment, should raise suspicion for the pseudotumoral presentation of CAA-I and lead to T2*-GRE sequences. Perfusion MRI and MRS appear to be useful techniques for the differential diagnosis of this entity.
Cerebral amyloid angiopathy (CAA), a common small vessel disease of the brain, is characterized by the progressive deposition of β-amyloid (Aβ) protein in the walls of small to medium-sized arteries, arterioles, and capillaries in the cerebral cortex and the overlying leptomeninges.1 The typical presentation of CAA is a spontaneous lobar intracerebral hemorrhage in an elderly patient. CAA can also manifest as cognitive impairment, dementia, or transient neurologic symptoms.1 T2*-weighted gradient-recalled echo (T2*-GRE) or susceptibility-weighted imaging (SWI) demonstrate extensive cortical microbleeds and lobar macrobleeds. A less frequent manifestation of CAA is CAA-related inflammation (CAA-I, also referred to as Aβ-related angiitis).2–6 CAA-I is thought to result from an inflammatory response to Aβ protein in the blood vessel walls. CAA-I can clinically and radiologically mimic a brain tumor.7,8 The aim of the present study was to report on 5 patients with a pseudotumoral presentation of CAA-I and to review the characteristics of 23 previously reported cases to identify the clinical and radiologic features that should lead to the suspicion of this diagnosis.
METHODS
We retrospectively reviewed the medical and radiologic records of 5 patients who were referred to our neuro-oncology department between 2008 and 2014 due to the suspicion of a brain tumor and in whom the final diagnosis was a pseudotumoral presentation of CAA-I. The criteria proposed by Chung et al.7 and modified Boston criteria were used for CAA-I and CAA diagnosis.9 Brain MRI scans were reviewed by two independent neuroradiologists for the presence and location of T2/fluid-attenuated inversion recovery (FLAIR) hyperintensity, T1 hypointensity, evidence of hemorrhage on T2*-GRE or SWI, the presence of parenchymal or meningeal postcontrast enhancement and mass effects, as well as abnormalities on diffusion-weighted imaging (DWI), proton magnetic resonance spectroscopy (1H-MRS), perfusion MRI, and magnetic resonance angiography (MRA). Previously reported cases of CAA with imaging findings initially interpreted as brain tumors were identified via PubMed searches from January 1970 to September 2014 using the terms CAA, CAA-related inflammation, neoplasm, and brain tumor. We retrieved all relevant articles and checked additional references quoted in these articles. We selected only the cases in which MRI descriptions were available.
RESULTS
Present case series.
The characteristics of our 5 patients are presented in table 1. The median age was 70 years. Four patients presented with subacute cognitive decline, which was associated with headaches and papilledema in one patient and with progressive hemiparesis in another patient. One patient presented with acute encephalopathy. All patients were referred to our department based on an initial standard MRI, which included precontrast and postcontrast T1 and T2/FLAIR sequences showing unilateral (n = 1) or bilateral (n = 4) asymmetric T2/FLAIR hyperintensities in the supratentorial white matter with a loco-regional mass effect, suggesting a differential diagnosis of a brain tumor (figure). In one case, involvement of the splenium of the corpus callosum was observed. After gadolinium injection, no parenchymal enhancement was detected. Leptomeningeal enhancement was present in 2 patients. Based on these initial findings, the suspected diagnosis was lymphoma in 2 patients, low-grade glioma in 2 patients, and gliomatosis cerebri in 1 patient.
Table 1.
Characteristics of our 5 patients with a pseudotumoral presentation of CAA-I
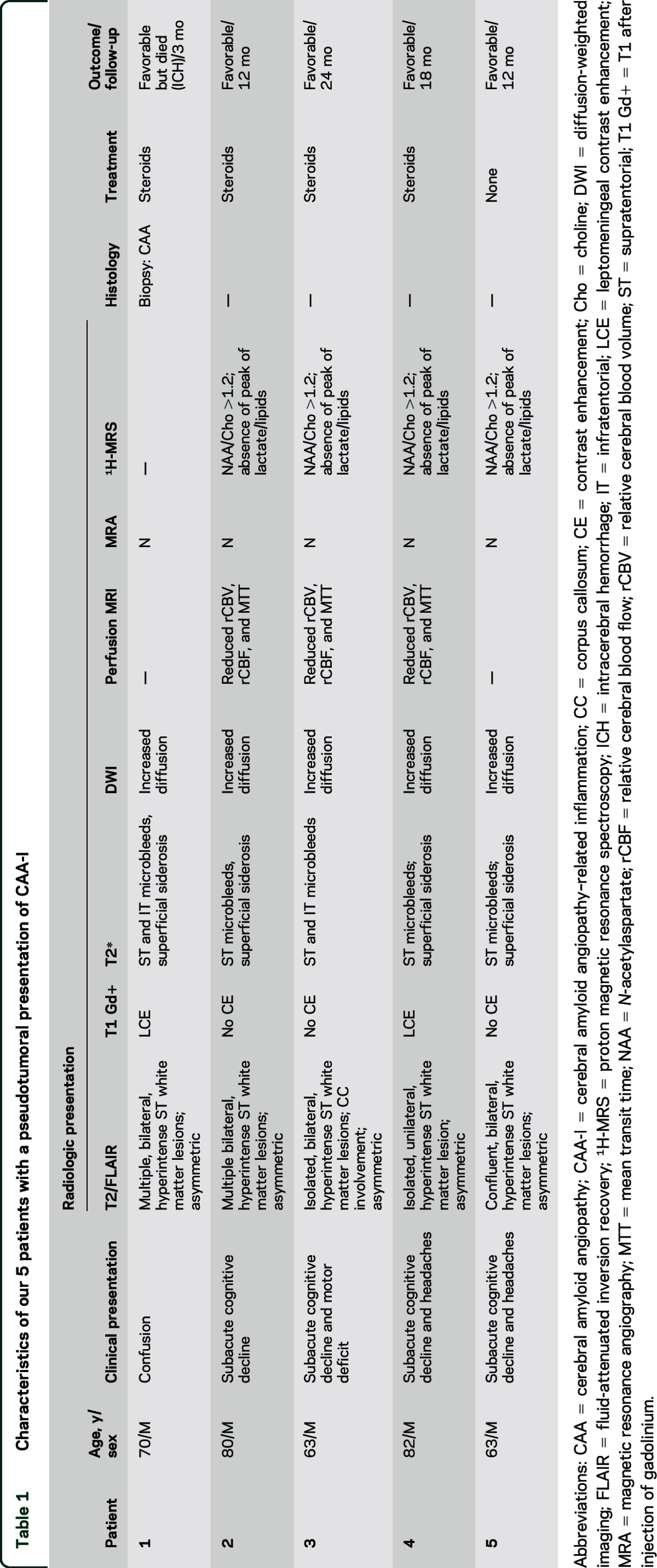
Figure. MRI findings at diagnosis and during follow-up.

(A) MRI findings of patient 1 to patient 5 are ordered from the top to the bottom. At diagnosis, axial T1 with gadolinium infusion and axial fluid-attenuated inversion recovery (FLAIR) sequences showed supratentorial white matter lesions with mass effects without parenchymal contrast enhancement; these lesions were associated with multiple cortical microbleeds on T2*-weighted gradient-recalled echo. Perfusion MRI on patients 2, 3, and 4 showed markedly reduced relative cerebral blood volume. At follow-up, axial T2* showed acute lobar hematoma in patient 1, and axial FLAIR showed a marked regression of the hyperintense lesions in patients 2 to 5. (B) Immunostaining for β-amyloid indicated amyloid deposits within leptomeningeal and cortical vessels in patient 1 (low magnification, ×100). Lower left corner: mild inflammatory infiltrates (arrows) around a vessel markedly thickened with amyloid (high magnification, ×400). (C) Proton magnetic resonance spectroscopy on patient 3 demonstrated a normal spectrum.
All patients subsequently underwent multimodal MRI. Extensive cortical and subcortical microbleeds were identified in all patients on T2*-GRE; these microbleeds were predominantly supratentorial (figure). Marginal siderosis was present in 4 patients. DWI showed areas of increased diffusion that corresponded to the T2/FLAIR signal abnormalities. Perfusion MRI was performed on 3 patients and did not reveal any signs of neoangiogenesis. In contrast, the relative cerebral blood volume (rCBV), relative cerebral blood flow (rCBF), and mean transit time (MTT) were markedly reduced in the T2/FLAIR signal abnormalities compared to the ipsilateral or contralateral normal-appearing white matter. 1H-MRS was performed on 4 patients and indicated a normal metabolic ratio and the absence of other pathologic peak. Time-of-flight MRA was also available in all cases and excluded stenosis in the great and middle-sized intracranial vessels. Fluorodeoxyglucose PET was performed on one patient and indicated hypometabolism in the areas of increased T2/FLAIR signal (patient 3). CSF analysis demonstrated isolated hyperproteinorachia in 4 patients; the cell count was normal in all patients, and none of the patients exhibited oligoclonal bands.
All of these findings were suggestive of tumefactive CAA-I. All patients met the criteria for the diagnosis of probable CAA-I according to Chung et al.7 In our index patient (patient 1), a cerebral and leptomeningeal biopsy was performed, which confirmed the diagnosis, demonstrating amyloid deposits within leptomeningeal and cortical vessels associated with foci of mild perivascular inflammation (figure). Perivascular inflammatory cells mostly consisted of monocyte/microglial cells (staining for CD68). T lymphocytes (staining for CD3) were also present, whereas B lymphocytes (staining for CD20) were absent. No biopsy was performed in the 4 subsequent patients. In 3 of these patients, however, CSF was analyzed for Aβ peptides 40 and 42, and the findings indicated low levels of these peptides, as typically observed in CAA.10 Two of these 3 patients were positive for the APOE ε4/ε4 genotype. Four patients (patients 1, 2, 3, and 4) were treated with a short course of steroids (2 mg/kg IV for 3 days followed by 1 mg/kg tapered over 4–6 weeks), which resulted in rapid clinical improvement in all patients. In 3 of these patients (patients 2, 3, and 4), MRI was performed 1 month after treatment onset, and the MRI results indicated a nearly complete resolution of the tumefactive T2 hyperintensities (figure). These 3 patients were clinically stable after a 12- to 24-month follow-up, and the imaging findings were also stable. Patient 1 died from a lobar hemorrhage 3 months after diagnosis. Patient 5 did not receive steroids and spontaneously improved over 3 months. In this patient, MRI performed 3 months after diagnosis indicated nearly complete resolution of the T2 hyperintensities (figure).
Analysis of the characteristics of patients with a pseudotumoral presentation of CAA-I.
We identified 23 previously reported patients with a pseudotumoral presentation of CAA-I8,11–23 (table e-1 on the Neurology® Web site at Neurology.org). In all patients, the imaging findings were initially interpreted as CNS neoplasm, which led to the performance of a biopsy (n = 17 [74%]) or surgical resection (n = 6 [26%]). T2*-GRE or SWI were performed prior to surgery in 2 patients and during follow-up in 4 patients. When specified, the initially suspected diagnoses included low-grade glioma, gliomatosis, lymphoma, or carcinomatous meningitis. In 21 of the 23 patients, the final diagnosis was made following histologic examination, which indicated CAA with (n = 12 [52%]) or without (n = 9 [39%]) signs of vascular inflammation. In 4 patients, the biopsy was nondiagnostic, which led to a repeated biopsy in one patient and a surgical resection in another patient.8,22 In the 2 other patients, the diagnosis was only made retrospectively when extensive microbleeds on T2*-GRE or SWI were observed on follow-up MRI.8
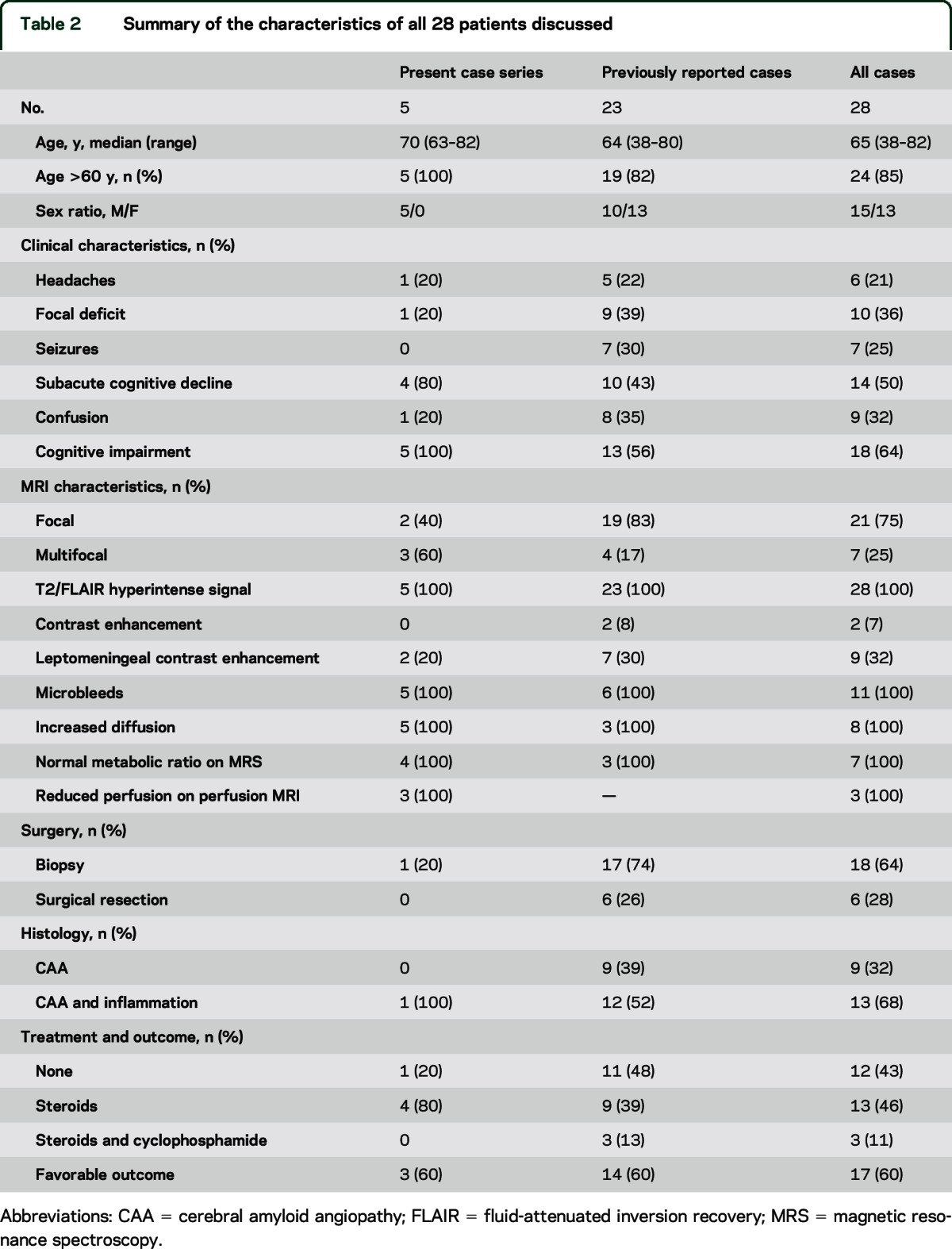
The characteristics of the patients from our series together with the previously reported cases are summarized in table 2. Most cases (n = 24 [85%]) occurred in patients aged >60 years. Two patients had a history of intracerebral hemorrhage. The clinical presentation was not specific and included a variable combination of subacute cognitive decline (n = 14 [50%]), confusion (n = 9 [32%]), focal deficits (n = 10 [36%]), seizures (n = 7 [25%]), or headaches (n = 6 [21%]). Nevertheless, approximately two-thirds of the patients exhibited acute or subacute cognitive impairment. The radiologic presentation was similar between patients. Standard MRI sequences demonstrated isolated (n = 22 [78%]) or multiple (n = 6 [22%]) T2/FLAIR tumefactive hyperintensities (n = 28 [100%]) without parenchymal postcontrast enhancement (n = 26 [93%]). Leptomeningeal contrast enhancement was observed in 11 cases (28%). The primary reason for the misinterpretation of the CAA-I imaging findings as CNS neoplasm was the absence of T2*-GRE sequences on initial imaging (n = 26 [89%]). When subsequently performed (n = 11 [39%]), these sequences demonstrated multiple characteristic cortical and subcortical microhemorrhages in all cases. When available, DWI and 1H-MRS indicated no restricted diffusion and normal spectra, respectively. After diagnosis, 15 patients (57%) received steroids with or without cyclophosphamide. The outcome was favorable in 17 patients (60%). The rate of favorable outcome was 78% in the patients who received steroids with or without cyclophosphamide and 58% in the patients who received no treatment (Fisher exact test, not significant). Among the 11 patients who underwent T2*-GRE sequences, 5 patients met the criteria for the diagnosis of definite CAA-I and 6 patients met those for the diagnosis of probable CAA-I.7 All of these patients met the modified Boston criteria for the diagnosis of probable CAA (5 patients with and 6 patients without supporting pathology).9
Table 2.
Summary of the characteristics of all 28 patients discussed
DISCUSSION
The identification of intra-axial, tumefactive, non-neoplastic neurologic disorders that can mimic brain tumors is of utmost importance in the preoperative setting.24 In the present study, we demonstrate that CAA-I can incorrectly suggest a brain tumor and that to identify this condition, T2*-GRE or SWI should be performed during the diagnostic workup of brain masses, especially in elderly patients presenting with subacute cognitive decline and no parenchymal postcontrast enhancement. In centers that already include T2*-GRE/SWI sequences in their standard MRI protocols, these sequences should be actively reviewed by the clinicians evaluating patients with suspected brain tumors. Centers that do not include those sequences should strongly consider adding them.
The clinical and radiologic presentations of tumefactive CAA-I were not specific. Nevertheless, most patients were elderly (85%) and exhibited cognitive impairment (65%) and patchy or confluent hyperintense T2/FLAIR lesions without contrast enhancement on MRI (93%). The 2 most frequently suspected diagnoses were low-grade glioma and lymphoma. However, older age at onset, cognitive impairment, and extensive vasogenic edema are unusual features in low-grade gliomas, while the absence of parenchymal contrast enhancement is rare in cerebral lymphomas. Therefore, tumefactive CAA-I should be considered as a differential diagnosis of suspected low-grade glioma in elderly patients and of suspected lymphomas when no contrast enhancement is present. Less frequently, tumefactive CAA-I suggested gliomatosis cerebri (due to extensive leukoencephalopathy) and carcinomatous meningitis (due to marked leptomeningeal contrast enhancement). Careful examination of FLAIR sequences usually disclosed a mass effect that was less pronounced than it is in brain tumors. However, the primary reason for the misinterpretation of CAA-I imaging findings as CNS neoplasm was the absence of T2*-GRE/SWI sequences on initial imaging. When subsequently performed, these sequences demonstrated multiple cortical and subcortical microbleeds in all cases; these microbleeds were predominantly supratentorial. Marginal siderosis was also frequently identified based on these sequences. Perfusion MRI was only performed on 3 patients; however, it consistently showed a reduction in the main perfusion parameters (rCBV, rCBF, and MTT). This finding is interesting because it supports the results of a previous study that used PET-MRI in a mouse model, which showed that Aβ deposition is accompanied by a decrease in rCBF. In this model, the loss of perfusion correlated with the increase in the Aβ plaque burden but not with the number of microbleeds.25 Thus, perfusion MRI appears to serve as a promising technique for the diagnosis and even the follow-up of CAA-I. 1H-MRS did not indicate any specific signs of CAA-I; however, it facilitated the suspicion of a non-neoplastic disorder because it showed unremarkable spectra. In addition to brain tumors, the differential diagnosis of CAA-I includes infections (e.g., progressive multifocal leukoencephalopathy, fungal disease, tuberculosis, infectious endocarditis), immune-related disorders (e.g., acute disseminated encephalomyelitis, neurosarcoidosis, primary CNS vasculitis), and posterior reversible leukoencephalopathy/reversible vasoconstriction syndrome.6,7
Mass-like lesions have been reported in approximately 15%–25% of patients with CAA-I.6,26 The clinical and radiologic characteristics of the patients in our series were similar to those reported in CAA-I except the fact that in our series the lesions seemed to be more frequently focal (75%) and more frequently associated with mass effect (100%).6,7 CAA-I is thought to result from an inflammatory and immune reaction directed against the Aβ deposits in blood vessel walls.1 This hypothesis is supported by the occurrence of cases of subacute meningoencephalitis displaying characteristics similar to CAA-I in patients who received vaccinations to the Aβ42 form of Aβ or monoclonal anti-amyloid antibodies as a potential therapy for Alzheimer disease.27–30 The recent identification anti-Aβ autoantibodies in CAA-I patients also supports the hypothesis of an autoimmune reaction against cerebrovascular Aβ.31 Diagnostic criteria for CAA-I have been proposed.7 The diagnosis of CAA-I is considered as probable if all following criteria are met: (1) acute or subacute onset of symptoms; (2) 40 years of age or older; (3) at least one of the following clinical features: headache, mental status or behavioral change, focal neurologic signs, and seizures; (4) MRI showing patchy or confluent T2 or FLAIR hyperintensity, which is typically asymmetric, with or without mass effects and with or without leptomeningeal or parenchymal enhancement; (5) evidence of preexisting CAA on SWI, such as multiple cortical or subcortical hemorrhages or microhemorrhages or recent or previous lobar hemorrhage; and (6) the absence of neoplastic, infectious, or other causes of symptoms. The diagnosis of CAA-I is considered as definite if, in addition to these criteria, a histopathologic examination indicates perivascular, transmural, or intramural inflammation and amyloid deposition within vessels of the affected area in the cortex and the leptomeninges. The extent of inflammation is variable, ranging from a mild perivascular nondestructive inflammation (as in patient 1) to a vasculitic transmural, granulomatous, inflammatory infiltration.26 However, likely because of the segmental inflammatory involvement of cerebral vessels, inflammatory changes are not always identified, even in typical cases.26
The identification of CAA-I in the preoperative setting is important to avoid futile surgical resections and nondiagnostic biopsies (28% and 14% of the patients in the present study, respectively). Histologic confirmation of the diagnosis requires both cortical and leptomeningeal material (otherwise, amyloid deposition within vessels might not be identified). Whereas definite diagnosis can only be achieved by histopathologic examination, it has been proposed that selected patients with typical CAA-I characteristics and no evidence for another cause of symptoms may be managed without brain biopsy.4,7 However, T2*-GRE microbleeds are not specific for CAA-I and the diagnostic criteria for CAA-I (especially the specificity of neuroimaging findings) remain to be validated. In addition, in a condition for which the natural history is poorly understood, lack of biopsy may confound progress in understanding the relationship between CAA and vascular inflammation.26 In the future, the noninvasive diagnosis of CAA-I might be facilitated by the identification of anti-Aβ autoantibodies in CSF31 and by the development of PET32 or MRI33 techniques that enable the visualization of amyloid deposition.
The optimal treatment for CAA-I remains to be defined.7 Rapid clinical and radiologic responses have been reported using steroids alone or in conjunction with other immunosuppressive drugs such as cyclophosphamide.7 However, spontaneous improvement has also been reported. In the present case series, the rate of favorable outcome tended to be higher among the patients who received steroids (with or without cyclophosphamide) than among those who received no treatment (78% vs 58%). In our experience, a short course of steroids was sufficient to elicit rapid and persistent clinical and radiologic improvement in 3 of our 4 patients. Nevertheless, relapse upon treatment withdrawal has been reported; therefore, the optimal treatment duration remains to be determined.7
In addition to its retrospective design and its small size, limitations of the present study include the limited number of patients who underwent advanced MRI techniques and the absence of histologic examination in all of the patients.
Supplementary Material
GLOSSARY
- Aβ
β-amyloid
- CAA
cerebral amyloid angiopathy
- CAA-I
CAA-related inflammation
- DWI
diffusion-weighted imaging
- FLAIR
fluid-attenuated inversion recovery
- 1H-MRS
proton magnetic resonance spectroscopy
- MRA
magnetic resonance angiography
- MTT
mean transit time
- rCBF
relative cerebral blood flow
- rCBV
relative cerebral blood volume
- SWI
susceptibility-weighted imaging
- T2*-GRE
T2*-weighted gradient-recalled echo
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
Dr. Ronsin: design and conceptualization of the study, data analysis and interpretation. Dr. Deiana: data acquisition, analysis and interpretation. Dr. Geraldo: data acquisition, analysis and interpretation. Dr. Durand-Dubief: data analysis and interpretation. Dr. Thomas-Maisonneuve: data acquisition, analysis and interpretation. Dr. Formaglio: data analysis and interpretation. Dr. Desestret: data acquisition, analysis and interpretation. Dr. Meyronet: data acquisition, analysis and interpretation. Pr. Nighoghossian: data analysis and interpretation, manuscript revision regarding intellectual content. Pr. Berthezène: data analysis and interpretation, manuscript revision regarding intellectual content. Pr. Honnorat: design and conceptualization of the study, data analysis and interpretation, manuscript revision regarding intellectual content. Dr. Ducray: design and conceptualization of the study, data analysis and interpretation, manuscript revision regarding intellectual content.
STUDY FUNDING
No targeted funding reported.
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Charidimou A, Gang Q, Werring DJ. Sporadic cerebral amyloid angiopathy revisited: recent insights into pathophysiology and clinical spectrum. J Neurol Neurosurg Psychiatry 2012;83:124–137. [DOI] [PubMed] [Google Scholar]
- 2.Eng JA, Frosch MP, Choi K, Rebeck GW, Greenberg SM. Clinical manifestations of cerebral amyloid angiopathy-related inflammation. Ann Neurol 2004;55:250–256. [DOI] [PubMed] [Google Scholar]
- 3.Scolding NJ, Joseph F, Kirby PA, et al. Abeta-related angiitis: primary angiitis of the central nervous system associated with cerebral amyloid angiopathy. Brain 2005;128:500–515. [DOI] [PubMed] [Google Scholar]
- 4.Kinnecom C, Lev MH, Wendell L, et al. Course of cerebral amyloid angiopathy-related inflammation. Neurology 2007;68:1411–1416. [DOI] [PubMed] [Google Scholar]
- 5.Salvarani C, Brown RD, Jr, Calamia KT, et al. Primary central nervous system vasculitis: comparison of patients with and without cerebral amyloid angiopathy. Rheumatology 2008;47:1671–1677. [DOI] [PubMed] [Google Scholar]
- 6.Danve A, Grafe M, Deodhar A. Amyloid beta-related angiitis: a case report and comprehensive review of literature of 94 cases. Semin Arthritis Rheum 2014;44:86–92. [DOI] [PubMed] [Google Scholar]
- 7.Chung KK, Anderson NE, Hutchinson D, Synek B, Barber PA. Cerebral amyloid angiopathy related inflammation: three case reports and a review. J Neurol Neurosurg Psychiatry 2011;82:20–26. [DOI] [PubMed] [Google Scholar]
- 8.Kotsenas AL, Morris JM, Wald JT, Parisi JE, Campeau NG. Tumefactive cerebral amyloid angiopathy mimicking CNS neoplasm. AJR Am J Roentgenol 2013;200:50–56. [DOI] [PubMed] [Google Scholar]
- 9.Linn J, Halpin A, Demaerel P, et al. Prevalence of superficial siderosis in patients with cerebral amyloid angiopathy. Neurology 2010;74:1346–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Verbeek MM, Kremer BP, Rikkert MO, Van Domburg PH, Skehan ME, Greenberg SM. Cerebrospinal fluid amyloid beta(40) is decreased in cerebral amyloid angiopathy. Ann Neurol 2009;66:245–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karbowniczek A, Wierzba-Bobrowicz T, Mendel T, Nauman P. Cerebral amyloid angiopathy manifested as a brain tumour: clinical and neuropathological characteristics of two cases. Folia Neuropathol 2012;50:194–200. [PubMed] [Google Scholar]
- 12.Andrade GC, Silveira RL, Pinheiro N, Jr, Rocha EM, Pittella JE. Cerebral amyloid angiopathy presenting as a brain tumor: case report [in Portuguese]. Arq Neuropsiquiatr 2006;64:153–156. [DOI] [PubMed] [Google Scholar]
- 13.Safriel Y, Sze G, Westmark K, Baehring J. MR spectroscopy in the diagnosis of cerebral amyloid angiopathy presenting as a brain tumor. AJNR Am J Neuroradiol 2004;25:1705–1708. [PMC free article] [PubMed] [Google Scholar]
- 14.Vandermissen B, Salmon I, Hildebrand J. Recurrent nonhemorrhagic mass lesion due to cerebral amyloid angiopathy. J Neurol 2003;250:239–240. [DOI] [PubMed] [Google Scholar]
- 15.Tamargo RJ, Connolly ES, Jr, McKhann GM, et al. Clinicopathological review: primary angiitis of the central nervous system in association with cerebral amyloid angiopathy. Neurosurgery 2003;53:136–143; discussion 143. [DOI] [PubMed] [Google Scholar]
- 16.Schwab P, Lidov HG, Schwartz RB, Anderson RJ. Cerebral amyloid angiopathy associated with primary angiitis of the central nervous system: report of 2 cases and review of the literature. Arthritis Rheum 2003;49:421–427. [DOI] [PubMed] [Google Scholar]
- 17.Oide T, Tokuda T, Takei Y, Takahashi H, Ito K, Ikeda S. Serial CT and MRI findings in a patient with isolated angiitis of the central nervous system associated with cerebral amyloid angiopathy. Amyloid 2002;9:256–262. [DOI] [PubMed] [Google Scholar]
- 18.De Broucker T, Henin D, Claquin G, et al. Cerebral amyloid angiopathy presenting as a pseudotumor: 2 cases with spontaneously favorable outcomes [in French]. Rev Neurol (Paris) 2000;156:859–863. [PubMed] [Google Scholar]
- 19.Polivka M, Vallat AV, Woimant F, et al. Cerebral amyloid angiopathy (CAA) with presentation as a brain inflammatory pseudo-tumour. Clin Exp Pathol 1999;47:303–310. [PubMed] [Google Scholar]
- 20.Fountain NB, Eberhard DA. Primary angiitis of the central nervous system associated with cerebral amyloid angiopathy: report of two cases and review of the literature. Neurology 1996;46:190–197. [DOI] [PubMed] [Google Scholar]
- 21.Ortiz O, Reed L. Cerebral amyloid angiopathy presenting as a nonhemorrhagic, infiltrating mass. Neuroradiology 1996;38:449–452. [DOI] [PubMed] [Google Scholar]
- 22.Osumi AK, Tien RD, Felsberg GJ, Rosenbloom M. Cerebral amyloid angiopathy presenting as a brain mass. AJNR Am J Neuroradiol 1995;16:911–915. [PMC free article] [PubMed] [Google Scholar]
- 23.Mandybur TI, Balko G. Cerebral amyloid angiopathy with granulomatous angiitis ameliorated by steroid-cytoxan treatment. Clin Neuropharmacol 1992;15:241–247. [DOI] [PubMed] [Google Scholar]
- 24.Omuro AM, Leite CC, Mokhtari K, Delattre JY. Pitfalls in the diagnosis of brain tumours. Lancet Neurol 2006;5:937–948. [DOI] [PubMed] [Google Scholar]
- 25.Maier FC, Wehrl HF, Schmid AM, et al. Longitudinal PET-MRI reveals beta-amyloid deposition and rCBF dynamics and connects vascular amyloidosis to quantitative loss of perfusion. Nat Med 2014;20:1485–1492. [DOI] [PubMed] [Google Scholar]
- 26.Salvarani C, Hunder GG, Morris JM, Brown RD, Jr, Christianson T, Giannini C. Abeta-related angiitis: comparison with CAA without inflammation and primary CNS vasculitis. Neurology 2013;81:1596–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ferrer I, Boada Rovira M, Sanchez Guerra ML, Rey MJ, Costa-Jussa F. Neuropathology and pathogenesis of encephalitis following amyloid-beta immunization in Alzheimer's disease. Brain Pathol 2004;14:11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO. Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: a case report. Nat Med 2003;9:448–452. [DOI] [PubMed] [Google Scholar]
- 29.Orgogozo JM, Gilman S, Dartigues JF, et al. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology 2003;61:46–54. [DOI] [PubMed] [Google Scholar]
- 30.Sperling R, Salloway S, Brooks DJ, et al. Amyloid-related imaging abnormalities in patients with Alzheimer's disease treated with bapineuzumab: a retrospective analysis. Lancet Neurol 2012;11:241–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Piazza F, Greenberg SM, Savoiardo M, et al. Anti-amyloid beta autoantibodies in cerebral amyloid angiopathy-related inflammation: implications for amyloid-modifying therapies. Ann Neurol 2013;73:449–458. [DOI] [PubMed] [Google Scholar]
- 32.Baron JC, Farid K, Dolan E, et al. Diagnostic utility of amyloid PET in cerebral amyloid angiopathy-related symptomatic intracerebral hemorrhage. J Cereb Blood Flow Metab 2014;34:753–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Viola KL, Sbarboro J, Sureka R, et al. Towards non-invasive diagnostic imaging of early-stage Alzheimer's disease. Nat Nanotechnol 2015;10:91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.