

Summary
Integrated approaches using different in vitro methods in combination with bioinformatics can (i) increase the success rate and speed of drug development; (ii) improve the accuracy of toxicological risk assessment; and (iii) increase our understanding of disease. Three-dimensional (3D) cell culture models are important building blocks of this strategy which has emerged during the last years. The majority of these models are organotypic, i.e., they aim to reproduce major functions of an organ or organ system. This implies in many cases that more than one cell type forms the 3D structure, and often matrix elements play an important role. This review summarizes the state of the art concerning commonalities of the different models. For instance, the theory of mass transport/metabolite exchange in 3D systems and the special analytical requirements for test endpoints in organotypic cultures are discussed in detail. In the next part, 3D model systems for selected organs – liver, lung, skin, brain – are presented and characterized in dedicated chapters. Also, 3D approaches to the modeling of tumors are presented and discussed. All chapters give a historical background, illustrate the large variety of approaches, and highlight up- and downsides as well as specific requirements. Moreover, they refer to the application in disease modeling, drug discovery and safety assessment. Finally, consensus recommendations indicate a roadmap for the successful implementation of 3D models in routine screening. It is expected that the use of such models will accelerate progress by reducing error rates and wrong predictions from compound testing.
Keywords: organ-on-a-chip, organotypic, 3D models
1 Introduction
1.1 Background
Specific tissue function and physiology may be lost in 2D systems. 3D culture models were created to better mimic the in vivo situation and to maintain such functions (Haycock, 2011). The currently available models are mainly specific for single organs and have been established to study the liver, nervous system, skin, respiratory system, intestine and the heart, among others. Such models are already in use in biomedical research. However, they have the potential to be useful also in toxicological testing schemes and for drug discovery. Therefore, a workshop was organized by CAAT-Europe and partners (Alexandra Association, BASF SE, Beiersdorf AG, ECOPA and L’Oréal) to assemble experts for 3D models of the liver, brain, lung and skin and for using their potential for toxicological testing. Future improvements to each system, design of potential new systems and hurdles to regulatory acceptance were discussed. From these experts, authors were selected to assemble the chapters of this review. Overall recommendations of the workshop participants are presented with the aim of improving the quality of organotypic models and facilitating their application in toxicological testing and drug discovery as a viable alternative to the current animal testing paradigm.
Differences in artificial organ design are a result of the desired endpoint: for instance, a model for skin irritation can potentially be achieved using a simpler strategy than, perhaps, the more complex endpoint of developmental delay of the formation of the nervous system. However, both endpoints are important in testing of drug candidates and chemicals. In this report, we try to outline the wide variety of approaches. We also exemplify the different levels of sophistication and the broad range of applications of 3D models, using four major organ systems as examples.
1.2 Common themes
A most obvious common theme is cell choice and availability. Primary human cells can be difficult to come by, and they can be difficult to use in cell culture. Human stem cells and progenitor cells are an alternative option from the standpoint of sustainability; however, differentiation to the correct cell type must be highly reproducible and standardized. It is also important to ensure that the cells perform as differentiated adult cells do in human organs. The use of tumor cell lines is easier, but genetic changes in tumor cells may confound the results. Use of animal cells may appear like a step backwards, but they offer clear advantages in some cases, and they may allow investigation of species differences, and also allow some form of validation of 3D models in relation to the in vivo situation.
When discussing the transition from animal models to in vitro models, it is important to remember that test systems need not recapitulate every aspect of an animal model (or human disease), but they should provide predictive data for a particular question.
Another common theme is linked to technical limitations of model construction: at present, few models, such as the MCTS (multicellular tumour spheroids), have the potential to be produced at a large scale according to stringent standards required for toxicity testing and drug development. Mass production potential may be key for application of 3D cultures in safety testing and chemical and drug screening. Models requiring physical movement, e.g., lung models, may be more difficult to mass produce.
For each of the models, no matter how complex or simple, nutritional support, mass transport and monitoring (preferably online) are necessary (Fig. 1). In organotypic models, these all may become more complex. This important theme is therefore covered in a dedicated chapter on mass transport.
Fig. 1. Microenvironmental factors affecting cell behavior.
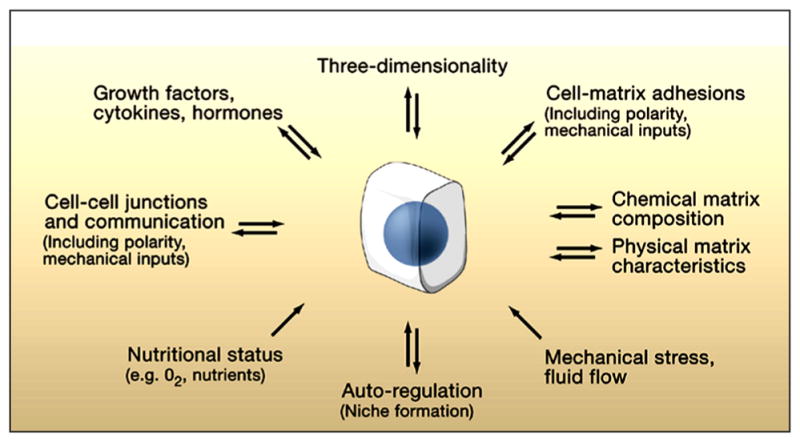
Numerous aspects of the microenvironment that change spatially and temporally may affect how accurately a 3D model reflects cellular behavior in vivo. Conversely, cells (center) can actively modify their local microenvironment. Reprinted with permission from Yamada and Cukierman (2007).
1.3 Mass transport
A basic requirement for the viability and functionality of a cell is continuous exchange (=bidirectional mass transport) with the adjacent environment. Cellular viability is maintained by a continuous supply of nutrients and oxygen as well as the removal of metabolic waste products (Hansmann et al., 2013). Additionally, biological information is transmitted, e.g., via diffusible substances. Spatial gradients and defined temporal changes result in patterns of signals and metabolites, which are essential for tissue development and maintenance (Fuchs et al., 2004; Votteler et al., 2010).
In vivo, physiological mass transport is maintained by active and passive processes on different scales (Aird, 2012; Pavelka and Roth, 2005). Systemically, macroscopic blood vessels transport oxygen, nutrients, hormones, carbon dioxide and catabolites. On a microscopic scale, in the capillaries, the mass exchange with the tissue is ensured across the endothelial cell layer. The endothelial layer can be fenestrated with pores to allow transit of large protein complexes (e.g., in the liver) or it can be very tight even to small molecules (e.g., in the bloodbrain barrier). For a modulated mass exchange across the cellular layer, different access routes are available, such as transport that is mediated by membrane proteins or receptors (Lum and Malik, 1994). In the tissue, a variety of biological and physical mechanisms contribute to the spatial distribution of the molecule (Datta, 2002). For instance, cells incorporate, metabolize or synthesize and secrete molecules. Additionally, a directed mass flux can be established by tissue-specific cell types (Pusch et al., 2011). In the cellular interspace, concentration gradients result in diffusion. Furthermore, molecules can be captured at binding sites of the extracellular matrix (Ruhrberg et al., 2002). Depending on the complexity of an in vitro construct, such as scaffold material composition or seeded cell type, the specific transport mechanisms can significantly determine tissue development (Hansmann et al., 2013).
Monitoring and analyzing the distribution of substances in a biological tissue is challenging (Langer and Muller, 2004). Alternatively, the chemical micro- and macro- environment in a tissue can be studied by computational modeling (Kalig et al., 2013; Kirouac et al., 2010). However, the validity of a mathematical model depends on the availability of input parameters. A simple mathematical model to predict the spatial and temporal concentration cA(x,t) of a substance A in a local environment is the diffusion equation. Consumption of the species can be introduced by a consumption term (Saltzman, 2004). The local concentration of the diffusible molecule is determined by its rate of diffusion DA and its rate of disappearance kA.
| (1) |
With the situation depicted in Figure 2, the initial and boundary conditions are:
Fig. 2. Schematic representation of the situation at the cell culture media/biological tissue interface of a tissue engineering construct.
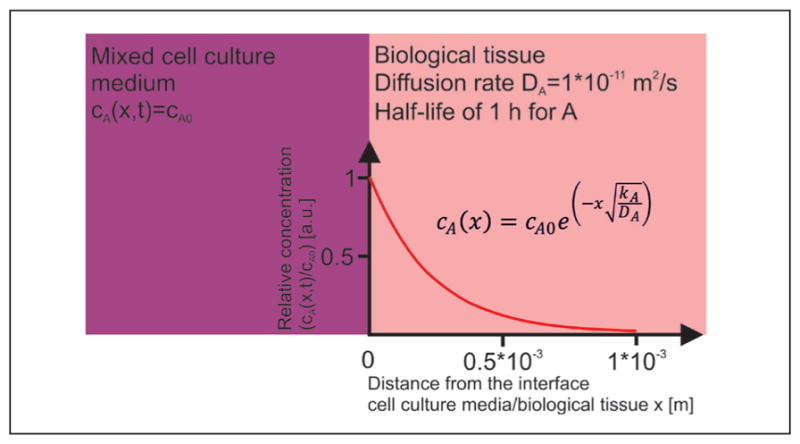
From the interface, substance A is distributed via diffusion and eliminated throughout the biological tissue.
The stirred cell culture medium provides a constant concentration CA0 at the cell culture medium/biological tissue interface. From this interface, substance A diffuses into the tissue and is eliminated equally throughout the construct. With the initial and boundary conditions, the steady-state solution of equation 1 becomes:
| (2) |
The parameters for the concentration gradient shown in Figure 1 are a diffusion rate of 1*10−11 m2/s and a half-life of 1 h.
From the interface, substance A is distributed via diffusion and eliminated throughout the biological tissue. The steady-state distribution of the concentration shows a strong decay along the distance from the source into the tissue.
Both parameters are within a realistic range for proteins (Colton et al., 1970). The diffusion-consumption model reveals a strong decay of the concentration with increasing distance from the cell culture medium/tissue interface and implicates areas devoid of substance A. When A is a secreted messenger molecule, the signaling range xr can be estimated when a threshold cmin in percent for its minimal required concentration is given:
| (3) |
This simple mathematical model provides a basic approach to describe necrotic cores in spheroids or the formation of niches (Saltzman, 2004).
When comparing mass transport in 2D and 3D cell culture environments, nutrients are provided homogenously to the cells in a 2D cell culture system. However, communication of a cell with cells outside of its neighborhood via soluble factors is restricted to diffusion in the liquid phase. The supernatant represents a reservoir for the accumulation of secreted messenger molecules (Fig. 3A). The concentration distribution is determined by the local secretion qS and local incorporation rate qI as well as the diffusivity DA of the messenger. Figure 3B depicts the distribution of a messenger molecule according to Figure 3A after 10 h. A protein is secreted with the secretion rate qS=1.78*10−16 mol/(s x m2), distributed with a diffusion rate of 1*10−11 m2/s and eliminated with qI=1.78*10−16 mol/(s x m2). The overall height and length of the supernatant is 0.5 mm and boundaries have no flux conditions; except at the x-axis where regions with secretion or elimination are highlighted. Basically, gradient formation along the cell layer is possible. However, the majority of the secreted factors diffuses into the supernatant and does not contribute to cellular communication. Additionally, the average level of the signaling factor can be increased over time and, thus, the formation of niches with cells that are not activated by the messenger is blocked. Moreover, when the cell culture medium is mixed, e.g., by stirring or convection, homogeneous concentrations of the messenger molecule are established. Thus, spatial and temporal information is lost.
Fig. 3. Cellular cross talk via soluble factors in a 2D system.

(A) Cells in a monolayer secrete and internalize signaling factor. The messenger is distributed in the supernatant. (B) Simulated distribution of the concentration of a signaling molecule in the supernatant. The molecule is secreted and eliminated along the x-axis by the cells. qS indicates secretion, qI internalization. The formation of a gradient at the cell/supernatant interface is possible.
Nutrient supply is usually limiting in dense 3D multi-cellular constructs that show an increased need of nutrients and oxygen due to high cell numbers per volume. To ensure the maintenance of mass transport, the distance between a cell and a capillary vessel usually is no further than 200 μm in vivo (Novosel et al., 2011). When a region becomes supplied insufficiently, new vessels grow into the area by angiogenesis (Carmeliet, 2005). This process is controlled by a not yet fully understood spatial and temporal pattern of diffusible growth factors such as vascular endothelial growth factor (VEGF) and platelet-derived endothelial growth factor (PDEGF). In vivo, endothelial cells are responsive to these cues and begin to sprout into the area with low supply. In vitro, endothelial cells are also able to form vessel-like structures if seeded into 3D scaffolds such as Matrigel™ and collagen hydrogels (Donovan et al., 2001). However, no fully functional microcirculation system has been recreated, yet. A reason for the poor outcome is that the vessel-like structures are not perfused and, thus, the endothelial cells are cultured without shear stress, which is known to be a vital stimulus for the function of endothelial cells (Chien, 2007). In alternative approaches, endothelial cells are seeded into tubular structures, which are embedded in various scaffolds. These tubular structures can be connected to a fluidic system to generate a constant or pulsatile medium flow. The simplest method is to embed a needle into a hydrogel during gelation (Chrobak et al., 2006). More sophisticated systems use sugar derivatives to generate complex structures (Miller et al., 2012). The sugar can then be dissolved, which results in a branched interconnected system of perfusable channels. Alternatively, vascularized tissues/scaffolds can be generated by de-cellularization of xenogeneic tissues. In these biological scaffolds the structures of the former vascular system are still present and can be connected to fluidic systems (Scheller et al., 2013).
2 3D models of the liver
The liver plays a central role in detoxification processes, metabolism of carbohydrates, lipids and proteins as well as biotransformation of endogenous and exogenous substances. The main biotransformation processes, catalyzed by cytochrome P450 (CYP), take place in the liver tissue (Brandon et al., 2003; Coon et al., 1992; Hewitt et al., 2001). Liver functionality is tightly linked to the assembly of specialized cell types, the majority of which are hepatocytes, embedded in a complex 3-dimensional structure made up of so-called lobules. Liver lobules are the smallest functional units of the liver. The hepatocytes are polarized and at their apical domains form an interconnected tubular structure, the bile canaliculi, which are the smallest branches of the biliary tree. The basolateral-apical polarization of the hepatocytes is essential to maintain the directed uptake and efflux of endogenous and exogenous substances, determining the metabolic capacity of the hepatocyte. Often drug-induced hepatotoxic effects result from long-term exposure. Biotransformation of compounds usually results in non-toxic and more soluble metabolites, however, occasionally, more toxic metabolites may be formed causing hepatotoxicity (e.g., paracetamol overdose toxicity; James et al., 2003). Hepatocytes require their 3-dimensional polarized structure to maintain liver-specific functionality. Therefore, cell culture technologies that maintain liver-specific metabolism and functionality over the long-term are necessary to allow chronic liver toxicity testing on human hepatocytes. Accordingly, the in vitro organotypic models of liver should allow measurement of various endpoints of physiological relevance in order to mimic the situation in the liver in vivo. Figure 4 shows some exemplary 3D liver cell cultivation techniques.
Fig. 4. 3D liver cell cultivation techniques.

(A) Polarized hepatocytes in collagen sandwich cultures (reprinted with permission from Tuschl, 2009). (B) 3D membrane bioreactors with perfusion system (reprinted with permission from Zeilinger et al., 2011) (C) Stirrer bioreactor for 3D aggregates (reprinted with permission from Tostoes et al., 2012). (D) Hanging drop liver organoids (reprinted with permission from Mueller et al., 2013). (E) 3D polystyrene scaffold culture (sc-scaffold, grey arrow indicates microvilli) (reprinted with permission from Bokhari, 2007). (F) A microfluidic device ensuring perfusion in multi-well plate format (reprinted with permission from Domansky, 2010).
2.1 Cell sources for in vitro liver models
In vitro liver models include various methods of using the liver cells in basic research as well as in toxicity screenings. The source of the cells and the cultivation techniques are determining factors in the application of these in vitro liver models.
Traditionally, mammalian liver cells (e.g., primary rodent or porcine hepatocytes) are used in biotransformation and toxicity screenings. However, species-specific differences in xenobiotic metabolism necessitate the use of human hepatocytes in preclinical pharmaceutical development (Hewitt et al., 2001; Lauer et al., 2009; Uehara et al., 2008). In particular, human cells most faithfully replicate actual human pharmaco- (and toxico-) kinetics and drug (chemical) metabolism. However, current methods mainly use either fractions of cells or non-adherent hepatocytes in short-term studies: Cultured primary human hepatocytes can provide the whole range of liver-specific metabolism, including transporter proteins and human-relevant CYPs. However, simple 2D culture methods have been shown to quickly lose functionality. Comparative studies with primary human hepatocytes in 2D show a faster decrease of cells with the differentiated hepatic phenotype, leading to lower production of urea and albumin and loss of CYP expression, than hepatocytes in 3D systems.
Compared to the conventional maintenance in 2D culture methods, the three-dimensional cultivation of hepatic cells using extracellular matrix scaffolds or microcarriers, or the hollow-fibre based perfused 3D four-compartment bioreactor, offers improved conditions for long-term preservation of cell integrity and functionality by supporting cell-cell contacts and signal exchange. It has been shown that 3D-culture enables prolonged cultivation of primary human hepatocytes, which in turn allows longer-term (greater than 3 week) studies to be performed. Cultivation in a perfused bioreactor provides a continuous mass exchange of culture media and decentralized controlled oxygenation. The cells are supported by aggregation to other cells. This provides an excellent scaffold to maintain a physiological environment for in vitro cultures allowing intercellular communication.
2.2 Benefits and uses of 3D organotypic liver models
In vitro, the 3-dimensional structure of hepatocytes can be maintained by controlling the physical interactions between the cells. The current gold standard to maintain hepatocyte polarization and functionality is the sandwich culture, in which cells are grown in between two layers of extracellular matrix constituents such as collagen. Other approaches to coax hepatocytes into the third dimension make use of either solid scaffold materials such as polystyrene scaffolds or de-cellularized tissues, hydrogels such as collagen type-I or alginate embedding, or self-assembling of hepatocytes into spheroids. In the majority of the 3D cell culture approaches, improved liver specific functionality and metabolism over longer time periods is achieved as compared to classical monolayer cultures. In order to further improve exchange of nutrients, oxygenation and waste removal perfused bioreactor systems are being developed to grow hepatocytes.
2.3 Disadvantages of 3D organotypic liver models
The cell source plays a pivotal role for the predictive power of in vitro liver models. The decision whether to use primary cell sources or cell lines, single type models or co-cultures depends strongly on the purpose and the end point of the assay as well as the number of data points required. Due to species-specific divergences of the xenobiotic metabolism, primary human hepatocytes are considered as gold standard. Genetic and metabolic variations of the donors impact the response towards substances, representing the diversity of the human population. However, the availability and lifetime of freshly isolated primary human hepatocytes (PHH) is limited. Recently, it was reported that use of suitable cultivation medium ensures long-term viability and functionality of PHH for application in chronic toxicity studies (Mueller et al., 2012). Human liver cell lines such as HepG2 and Hep2/C3A have been used for a long time in toxicological profiling. However, many of these cell lines show only poor, or fractional, hepatic enzyme expression. Also, normal energy metabolism can be largely different from that of primary cells (Latta et al., 2000). This limits the use of these cell lines in metabolism mediated toxicity studies. However, the HepG2 cell line has been used in studies for parent compound toxicity, cell polarity or chemotherapy resistance (Hoekstra et al., 1999; Mueller et al., 2011a; Noor et al., 2009; O’Brien et al., 2006). A promising cell source is represented by the HepaRG cell line, which shows a high comparability to primary human liver cells regarding metabolic activities (Andersson et al., 2012; Guillouzo et al., 2007; Kanebratt and Andersson, 2008; Lubberstedt et al., 2011). Nevertheless, a recent publication demonstrated that in contrast to primary hepatocytes the sensitivity of cell line-based models to detect substance toxicity is below that of primary human hepatocytes (Gerets et al., 2012). Another promising source of human hepatocytes can be seen in human embryonic stem cell (hESC) derived hepatocytes (Mandenius et al., 2011). However, ethical issues with the use of hESC derived cells limit their general acceptance. Next generation cell sources for developing in vitro liver toxicity models will be derived from induced pluripotent stem cells (iPSC) (Shtrichman et al., 2013). Since iPSC are generated by reprogramming of cells from adult tissue, they can be derived from multiple individuals and thus allow covering diverse genetic backgrounds. Moreover, due to their expansion capacity they allow performing repeated experiments using the same cell line over time.
The decision whether to use primary cell sources, cell lines and single type or co-culture models depends strongly on the biological question and the endpoint of the assay as well as on the number of data points required (fit for purpose principle).
2.4 Technologies used to generate 3D organotypic liver models
Current methods mainly use either fractions of cells or non-adherent hepatocytes in short-term studies. Cultured primary human hepatocytes can provide the whole range of liver-specific metabolism, including transporter proteins and human-relevant CYPs. However, hepatocytes maintained in simple 2D culture have been shown to dedifferentiate and rapidly lose functionality (Godoy et al., 2009; Gomez-Lechon et al., 1998). Compared to the conventional maintenance in 2D culture systems, the three-dimensional cultivation of hepatic cells offers improved conditions for long-term preservation of cell integrity and functionality by supporting cell-cell contacts and signal exchange. Various methods of 3D cultivation have been reported using extracellular matrix scaffolds (Dunn et al., 1989; Marion et al., 2007), microcarriers (Miranda et al., 2009; Ringel et al., 2005) or the hollow-fibre based perfused 3D four-compartment bioreactor (Gerlach et al., 2003) to name a few. Primary human hepatocytes cultured in 3D bioreactors showed a better maintenance of the differentiated hepatic phenotype, along with a higher stability of urea and albumin production and CYP expression, as compared with conventional 2D cultures (Hoffmann et al., 2012). An excellent review on advances in 2D and 3D liver systems has been recently published (Godoy et al., 2013).
Collagen-sandwich cultures
The simplest “quasi 3D” system to maintain hepatocyte polarization and functionality is the sandwich culture, in which cells are grown in between two layers of extracellular matrix constituents such as collagen (Marion et al., 2007) or Matrigel™ (Ogimura et al., 2011). Collagen sandwich cultures have been in use since more than two decades, have been shown to maintain differentiated hepatic phenotype (Dunn et al., 1991) and have been applied in toxicity and drug-drug interaction studies (Tuschl et al., 2009). PHH in sandwich cultures show comparable transporter expression to human liver tissue (Schaefer et al., 2012). Sandwich cultures do show differences as compared to 3D culture and in vivo liver (Lu et al., 2012b). Other similar cultures with embedded hepatocytes, including the hydrogels such as Matrigel™, collagen, gelatine and peptide hydrogels, have been reported (see review by Godoy et al., 2013).
3D bioreactors
3D liver bioreactors were initially developed for clinical use as bioartificial liver (BAL). Many such devices for in vitro applications have been reported (Mueller et al., 2011b; Darnell et al., 2011; Hoekstra et al., 2011; Tostoes et al., 2012; Zeilinger et al., 2011). Cultivation in a perfused bioreactor provides a continuous mass exchange of culture media and decentralized controlled oxygenation. The cells are supported by aggregation to other cells. This provides an excellent scaffold to maintain a physiological environment for in vitro cultures allowing intercellular communication. It has been shown that 3D-culture enables prolonged cultivation of primary human hepatocytes, which in turn allows longer-term (>3 weeks) studies to be performed (Zeilinger et al., 2011). A major advantage of this system with respect to its use in pre-clinical toxicity assays is that it allows for cultivation under serum-free conditions (Lubberstedt et al., 2012).
In addition, in these hollow-fibre bioreactors, the differentiation of human embryonic stem cells towards hepatic cells as a potential alternative cell source for studies on hepatic cells is demonstrated by a significant up-regulation of genes related to liver functions (Miki et al., 2011) and enhanced hepatic differentiation in the 3D four-compartment bioreactor system compared to conventional 2D cultures (Sivertsson et al., 2013).
Multicellular spheroid cultures
3D multicellular liver spheroids can be produced in scaffold free systems in multiwell plates by the self-assembling of hepatocytes into aggregates (Brophy et al., 2009; Kelm and Fussenegger, 2004; Kelm et al., 2003) or in stirrer bioreactors (Miranda et al., 2009). Hepatic spheroids form liver like structures within and are viable for long periods of time exhibiting good functionality. These are well suited for high throughput applications. The size of the spheroids is a critical parameter as large spheroids may result in oxygen and nutrient limitation in the centre leading to necrotic cores (Lin and Chang, 2008) or in stirrer bioreactors (Brophy et al., 2009; Kelm and Fussenegger, 2004; Kelm et al., 2003). Hepatic spheroids form liver like structures within and are viable for long periods of time exhibiting good functionality. These are well suited for high throughput applications. The size of the spheroids is a critical parameter as large spheroids may result in oxygen and nutrient limitation in the centre leading to necrotic cores (Lin et al., 2008; Lin and Chang, 2008).
Other 3D cultivations
Other approaches to coax hepatocytes into the third dimension make use of either solid scaffold materials such as polystyrene scaffolds (Bokhari et al., 2007; Burkard et al., 2012; Schutte et al., 2011b) or de-cellularized tissues (Linke et al., 2007).
3D hepatic cultures also have been used in microfluidic devices in multiwell or chip based systems for controlled medium flow and microenvironment (Domansky et al., 2010; Powers et al., 2002; Schutte et al., 2011a). Co-culture of hepatic spheroids with endothelial cells has been reported (Schimek et al., 2013). A recent review discusses the application of microfluidic devices especially in multiorgan systems (Materne et al., 2013).
2.5 Applications of 3D organotypic liver models
Two recently published reviews summarize the applications of 3D liver models (Godoy et al., 2013; Mueller et al., 2013a). 3D liver models can be applied in toxicological screenings as well as in the studies of drug metabolism and pharmacokinetics as discussed below.
2.6 Toxicology research
Due to prolonged viability and functionality the 3D liver models have great potential in toxicological research especially for long-term studies. Both bioreactors and spheroid cultures have been used in toxicity applications.
The liver spheroids formed in stirrer bioreactors have been proposed to be suitable for toxicity studies (Leite et al., 2012; Miranda et al., 2010; Tostoes et al., 2011). The hanging drop liver spheroids have been extensively characterised for liver specific functions (Michailov et al., 2004) and applied in the assessment of toxicity (Gunness et al., 2013). These spheroid cultures have also been applied for screening of a number of hepatotoxic drugs and in the repeated dose toxicity of chlorpromazine (Mueller et al., 2013b).
2.7 Drug metabolism and pharmacokinetics
Predicting in vivo hepatic clearance using in vitro metabolism assays is an inherent part in drug development to determine half-life, bioavailability, dose and dosing regimens (Obach, 2011). Especially challenging are low-clearance compounds, which are becoming more and more important in drug development (Di et al., 2012). Current gold standard models such as human liver microsomes and suspension culture of human hepatocytes usually have a too short assay window leading to imprecisions in the prediction of human clearance, half-life and dose. Due to improved long-term metabolic competence of the 3D organotypic hepatic cultures, these can be applied in drug metabolism and pharmacokinetic studies, especially to determine the metabolic profile of low-clearance compounds (Butler et al., 2013; Darnell et al., 2011; Nyberg et al., 1994; Zeilinger et al., 2002).
2.8 Limitations of 3D organotypic liver models
Although 3D organotypic liver cultures show definite advantages in long-term maintenance of hepatic functions, depending on the system there are limitations to their universal application as with any other system. There are several technical challenges in addition to limitations in applications. For example, the perfused 3D bioreactors provide the option for long-term studies in a serum-free culture environment. They have been miniaturized (Zeilinger et al., 2011) and even allow for kinetic biochemical assays (Mueller et al., 2013b). Nevertheless, current 3D bioreactor systems for the liver are, from a model perspective, quite large, and since the reorganization of the cells requires a critical cell mass, the extent of miniaturization is limited. As such, application in high throughput platforms is very limited. The requirement of high cell numbers also limits the number of parallel bioreactors from the same donor in case of PHH. Another factor limiting miniaturization possibilities is the clear necessity of complex mass-transport to provide the appropriate nutrients and remove waste at the appropriate time. This is a common problem in 3D models and will be discussed in more detail in a later section. Most of the perfused bioreactor systems are closed systems and therefore allow only endpoint sampling of the tissue material, e.g., for immunohistological investigations. This also means that the injected cell mass needs a minimum quality since dead cells are also injected into the system and cannot be removed. The binding of lipophilic drugs is another problem. The various perfused bioreactor systems require technical expertise and experience.
2D models can be easily adapted to metabolomics and flux analyses (Klein et al., 2013; Niklas et al., 2009; Strigun et al., 2011a,b, 2012). Although some 3D models can be very well adapted to “-omics” application, most of these have their own advantages and disadvantages. In case of 3D hollow fibre bioreactors, large perfusion volumes limit the use of expensive 13C substrates for flux analysis. As for the 3D spheroids, there are many advantages. However, in this case very low volumes make the various analyses, such as transcriptomics and proteomics, difficult. In addition, the readouts are from a few thousand cells and therefore the analysis methods have to be optimized for sensitivity.
2.9 Challenges and future focus of 3D liver models
Evaluating liver toxicity is crucial in the assessment of the safety of drugs, chemical substances, nutritional supplements and cosmetics. Current state-of-the-art in vitro and in vivo technologies have shown limitations in accurately predicting human hepatotoxicity. Next generation in vitro liver models will have to reflect the liver architecture and cell composition more closely to allow for more precise toxicological profiling.
Complexity of liver (non-parenchymal cells)
Liver is a very complex organ and includes in addition to hepatocytes (80%) various non-parenchymal cells (endothelial, stellate and Kupffer cells). It is known that the non-parenchymal cells (NPCs) also play a role in hepatotoxicity. Various 3D co-cultures of hepatocytes with NPCs have been reported, e.g., models for vascularized liver (Inamori et al., 2009) with co-cultured spheroids or in-chip based systems (Messner et al., 2013; Powers et al., 2002). However, this area needs further development for toxicological purposes, e.g., in case of immunity mediated toxicity and other pathophysiology such as fibrosis.
Complexity of liver (zonation)
Since the liver performs a wide range of diverse functions, the hepatocytes show a large heterogeneity and plasticity of functions. This is referred to as metabolic zonation in the liver (Gebhardt and Jonitza, 1991; Jungermann and Kietzmann, 1996). In case of isolated hepatocytes the zonation is lost and it would be of tremendous interest if this zonation can be restored in any of the 3D cultures. This will pave the way to use in vitro 3D organotypic liver models to study in vivo liver functions such as lipid and bile acid metabolism as well as glucose homeostasis. However, in case of toxicity studies, this may not be a critical point as all isolated hepatocytes may be synchronized by induction of drug metabolizing enzymes and transporters.
2.10 Implications of use of 3D liver models for the future
3D organotypic liver models exist for more than two decades. However, the paradigm of drug testing is only now moving from 2D cultures to 3D liver cultures with advances in technology. This has become possible with better accessibility to techniques of 3D cultivation and at the same time technological progress with respect to miniaturization and modern perfusion systems, allowing the idea of “liver on a chip” to become a reality. For specific purposes 2D cultivation may still be suitable. However, 3D organotypic cultivation must lead to in vivo relevant systems biology in vitro. In addition, 3D organotypic liver cultures will allow better in vitro to in vivo extrapolation of pharmacokinetics of drug/compounds. It is expected that the 3D organotypic liver models will greatly improve the prediction of effects and bioavailability. Another exciting future direction is the use of 3D liver cultures in multi-organ chips in a “body-on-a-chip” approach. 3D organotypic liver cultures will also be valuable in mechanistic studies for DILI (drug induced liver injury) and may one day allow completely replacing of the use of animals for specific testing purposes.
3 3D models of the nervous system
In vitro models have contributed a lot to the elucidation of the modes of action of neurotoxicants. Moreover, they have been useful to establish rank orders of potency of various compounds, once their mechanism has been understood. In contrast to this, cell culture-based predictions of neurotoxicity, starting without prior knowledge, have been difficult. This is in part due to the vast complexity of the nervous system, in particular the brain, spinal cord and peripheral nervous system, and to the associated large number of molecular targets and possible pathways of toxicity that may become relevant. The use of more complex models may improve this situation, as their underlying basic biological functions may be used as readouts, without necessitating exact knowledge of the upstream modes of action of potential toxicants. 3D models are expected to contribute to this progress by allowing the modelling of more biological processes that are essential for the functioning of the brain and nervous systems.
3.1 What are the key issues to be modelled by 3D nervous system models?
Three fundamentally different aspects are important for the models: 1) cell type and composition; 2) maturity of the cells and 3) structural organization of cells and their matrix. “Cell type and composition” refers to the fact that neural tissues are composed of many subtypes of neurons and also different types of glial cells. The latter comprise the large, and sometimes heterogeneous, groups of astrocytes, oligodendrocytes, Schwann cells and microglial cells. Multicellular units are fundamental for the performance of the nervous system and for many underlying biological processes such as synapse formation and function. The presence of glial cells is of utmost importance since they can be both targets and mediators of chemically induced brain injury. Moreover, the presence of glia may affect or modulate the toxicity of chemicals for neurons (e.g., Eskes et al., 2003; Giordano et al., 2009; Kuegler et al., 2012, 2010; Schildknecht et al., 2009; Viviani et al., 1998; Zurich et al., 2004, 2002, 1998), or they may be drivers of neuro-inflammation (e.g., Boillee et al., 2006a,b; Falsig et al., 2004, 2008; Henn et al., 2011, 2009; Hoing et al., 2012; Monnet-Tschudi et al., 2007). The proportion of the different types of glial cells is also an important factor since it modulates the release of bioactive molecules upon toxic exposure (Eskes et al., 2002; Simon et al., 2002). Glial cells can also determine the reaction of the overall tissue to injury, as they take over the role of phagocytosis of dying neurons and neurite debris (Hirt et al., 2000; Hirt and Leist, 2003). Therefore, the inclusion of the different types of glial cells is an important consideration for the setup of a neural 3D model. In the same vein, it may be considered to combine different types of neurons, or different types of glia, to cover all processes affected by toxicants. For modelling of neuroinflammation even inclusion of, e.g., lymphocytes may be required, as they modulate many brain processes (Kipnis and Schwartz, 2005) and extreme synergy can be generated between macrophages and lymphocytes (Gantner et al., 1996).
The “structural organization” affects the connectivity of the cells, the networks they may form, and the signals they can exchange. This has immediate secondary effects, e.g., on the activity of cells, on their metabolism or on their control of specific differentiation processes (e.g., myelin formation by oligodendrocytes or Schwann cells wrapping around neurites). Several metabolic pathways in the brain run across different cell types, e.g., astrocytes take up glutamate, transform it to glutamine and feed this to neurons, or they provide neurons with specific energy substrates or essential thiols. A particular example of a 3D structure formed by several cell types is the blood-brain barrier, and an understanding of its function has a profound importance for the prediction of neurotoxicity. In an adult organism, the blood-brain barrier has a large influence on the exposure of brain cells. Information on the concentration of compounds reaching the brain (or the target cell in 3D organotypic cultures) has to be derived from in vitro or in silico models for the blood-brain barrier. Different 3D models have been established in this area (Vandenhaute et al., 2012). The exposure of the developing brain to chemicals would be modified by the placental barrier, which also requires modeling for predictions from in vitro models to man (Giordano et al., 2009). Figure 5 shows schematic representation of 3D in vitro models of the nervous system.
Fig. 5. Schematic representation of 3D in vitro models of the nervous system.

The first 3D in vitro models used rodent acute or organotypic brain slices. These are still a major model for electrophysiological studies (A). Alternative approaches use co-culture models consisting of, e.g., a layer of neurons directly on top of a glial cell layer (B). Transwell culture models are used, e.g., for in vitro blood-brain barrier models by growing endothelial cells on a perforated membrane above a glial cell layer. Neurons may be attached to the bottom compartment (C). Re-aggregating brain cell culture models are formed by spontaneous re-aggregation of dissociated rodent brain cells under continuous shaking (D). Floating neurospheres can be generated from neural stem cells/neural progenitor cells, kept in low adherence plastic plates (E). Secondary 3D cultures are produced by plating neurospheres onto a planar adhesive substrate, which facilitates outgrowth of radial glia and migrating neurons (F). Engineered neural tissue (ENT) is generated by growing highly concentrated suspensions of stem cells on a membrane floating at the air-liquid interphase. This tissue is polarized (e.g., astrocytes at the bottom), consists of several subtypes of neurons and shows rosettes as neural tube-like structures (G).
At the intersection of structural organization and cell composition, there is another important aspect of 3D models: the formation of highly specific cell biological niches in the developing and developed brain. Such niches are formed by specific cell compositions and gradients of signalling molecules, and they provide a highly defined environment (e.g., enriched or deprived of certain signalling molecules) for the development and function of cells. Specialized glial cells are often important for such niches and are therefore required for modelling them. Examples are, e.g., the neurogenic niche of the adult brain, different complex niches in the retina, or the niche formed by satellite glial cells and pain neuron somata in trigeminal ganglia (Jasmin et al., 2010). Gradients of signalling molecules and niches are particularly important during development for the guidance of cell migration, for cell differentiation and thus for the overall patterning of the brain. 3D test systems have obvious advantages over 2D cultures when the modeling of niches is a major objective of the experiments. For instance, the combination of the intrinsic characteristics of stem cells and their microenvironment facilitates self-renewal and symmetric division, while differentiation occurs by asymmetric cell division (Fuchs et al., 2004; Morrison and Spradling, 2008).
The final fundamental aspect to be considered is cell maturity. There is an important difference whether a model is established to predict toxicity to the adult, mature and relatively static nervous system, or whether the center of interest is disturbances of brain development. The first types of models are pertinent to neurotoxicity (NT) and the second to developmental neurotoxicity (DNT). DNT is a sub-discipline of toxicology that belongs to the larger field of developmental/reproductive toxicity, while NT is usually considered in the context of systemic acute or chronic organ-specific toxicity. Thus, the models, and the questions to them, are largely different. Concerning maturity, the first question to be answered of any model is whether it addresses changing states of maturity (DNT models) or a stable state (NT models). The second issue to be addressed is the definition of the initial maturity state of the cells, e.g., at the time when toxicant exposure starts. For the definition of maturity and for the effects of toxicants, modern omics methods may deliver richer information than traditional single endpoints (Balmer et al., 2012; Ramirez et al., 2013; Waldmann et al., 2014). This would also include information on the epigenetic state and changes of the cells (Balmer and Leist, 2014).
3.2 Issues and future challenges to be addressed by 3D in vitro nervous system models
NT and DNT are currently assessed for regulatory purposes by conventional in vivo methods that are regulated e.g., for chemicals by OECD guidelines (OECD, 1997, 2007). These experiments are time consuming and costly, and, for this reason, the tests are often not performed at all. Therefore, many chemicals lack NT/DNT information altogether. This has spurred the demand for predictive toxicological in vitro methods to assess chemicals quicker and in a more economically feasible manner (Bal-Price et al., 2010a). Several promising in vitro models have been developed for examination of either NT or DNT (Bal-Price et al., 2010a,b, 2012; Coecke et al., 2007; Honegger et al., 1979, 2011; Kadereit et al., 2012; Krause et al., 2013; Krug et al., 2013a,b; Kuegler et al., 2010; Stiegler et al., 2011; van Thriel et al., 2012).
Many of them are based on 2D cell cultures. Such models have proven to be useful, e.g., for studying the molecular mechanisms of neuronal growth and survival (Gebhardt and Jonitza, 1991; Stiegler et al., 2011; Volbracht et al., 2001, 1999). However, modeling the complexity of neurite networks and brain cell architecture in vitro is limited in 2D neural cell cultures. For example, glial cells have an important role during the cell migration process of brain development: guiding the neurons to the right position. Such processes cannot be mimicked in monocultures. They require contribution of at least two cell types and they require a defined, often 3D, organization. Thus, it appears that 3D models have advantages for reproducing the complexity of directional growth and connections and of tissue structuring (Limongi et al., 2013), and most likely many other processes specific for the nervous system.
The transition from 2D to 3D does not necessarily solve all predictivity issues of in vitro models and important challenges remain for the future: The brain has a complex, non-repetitive 3D structure that determines its connectivity. The specific connectivity may be more important than the three-dimensionality as such. This situation is different from several other organs. For instance, the liver has a highly repetitive structural organization, in which one given unit (liver lobule or liver acinus) has the same function as any other of these basic units. The situation is more complex in lungs and kidneys but, also there, a multitude of exchangeable functional units can be found across the entire organ (terminal broncheoli in lung, nephrons in kidney). This is not the case in the brain. It may rather be compared to a complex integrated circuit. The exact connections between the elements are important, not their three-dimensionality as such. In fact, examples from microelectronics show that two dimensions with a minimal extent of a third dimension are perfectly sufficient to model most complex neuronal networks. The generation of such structured neuronal networks from stem cells using, e.g., cell 3D printing, sophisticated 3D matrixes or optimized differentiation protocols is just about to start, and such technologies may revolutionize the future.
Similar to neurons as a whole, also their neurite structures are organized in highly specific ways. For instance, they are organized within a nerve, and there are pronounced differences between dendrites and axons (Kim et al., 2007; Lein et al., 2007). Specific 3D models will eventually need to take this into account. The organization of the peripheral nervous system requires long neurites that emerge, e.g., from ganglia situated close to the spinal cord. The somata of the respective neurons are closely associated with glia in specific 3D structures. Myelin formation in the peripheral nervous system is a typical process that requires defined cell-cell interactions, for example, receptors expressed on neuronal and Schwann cells membranes (e.g., neuregulin-1 and Neecl4) are essential for forming mature myelin.
The ratio between different cell types is an important issue but, in the brain, it is not only the average ratio but the many different local ratios that make the biological difference. Reconstruction of such locally specialized cell units and of the associated niches is a formidable task.
The complexity of structure will need most likely to be reflected by specific functional endpoints. Optical or electronic measurements with simultaneously high temporal and spatial resolution will be required for many applications. Tissue that is not organized in a point symmetric way will also need to be manipulated and fixed with a defined spatial orientation for such readouts.
3.3 Different types of technologies used to generate 3D nervous system models
Several technologies have been established to generate 3D cultures and co-cultures employing different cell populations. The approaches may be categorized according to complexity, cell types involved, source of cells, or their purpose, e.g., for DNT vs. NT testing. One approach is layering, e.g., neurons on top of glia, to allow formation of neuronal-glial functional units (Schildknecht et al., 2012; Viviani, 2006). Such models have only a small extension in one (z) of the dimensions, but the layers may be analyzed separately, e.g., by the use of confocal microscopy. There are several variations of such models with only 2–5 cell layers in one dimension. For instance, DNT models operating at the stage of neural tube induction often use very high cell densities that may form more than one cell layer. This partial extension into the third dimension is important for the generation of defined cell biological niches (Chambers et al., 2012; Zimmer et al., 2012). Cells can also be layered on a perforated membrane. For instance, combinations of layered glial cells and endothelial cells may be used for blood-brain barrier models to measure drug transport. Also, strongly 3-dimensional complex blood-brain barrier models have been generated. These do not allow the study of drug transport, but may allow the examination of toxicant effects on the blood-brain barrier structure (Al Ahmad et al., 2011). A variant of the membrane technique is the formation of 3D tissue at an air liquid interphase (Dubois-Dauphin et al., 2010; Preynat-Seauve et al., 2009). Alternatively, cells may be grown in artificial scaffolds to promote three-dimensionality (Lu et al., 2012a).
Novel 3D in vitro models are continuously emerging in parallel areas (e.g., regenerative medicine). Therefore, not all technologies required for drug discovery 3D models need to be developed by toxicologists and pharmacologists. It is rather important to remain open to progress in other fields. For instance, research into the development of surgical biodegradable nerve guides for the repair of traumatic injury requires in vitro testing. Such test models have generated 3D scaffolds consisting of parallel aligned polycaprolactone microfilaments upon which neuronal cells, Schwann cells or dorsal root ganglia can be cultured. This approach results in highly aligned neuronal-glial co-cultures resembling “mini peripheral nerves” and, importantly, permits an organized 3D structure whereby intercellular contact between the Schwann cells and axons arises (Daud et al., 2012).
A classical approach to generate highly complex 3D in vitro models is the preparation of tissue slices. Acutely prepared slices or blocks remain viable for a few hours only, but they represent the models with highest resemblance to the in vivo situation. They have been used a lot for electrophysiological approaches to neurotoxicity research or for neuroprotection studies (Volbracht et al., 2006). The so-called organotypic slices usually dedifferentiate for a while after preparation, but they can then be used for weeks or up to months, e.g., to study prion replication (Falsig et al., 2012) or excitotoxicity (Montero et al., 2007). Variations of the technology frequently use nerve explants, ganglion explants (especially dorsal root ganglia) or retina explants. All of the above technologies mainly make use of rodent tissue. Obtaining human tissue is not feasible for routine testing, but the field of stem cell research promises interesting future approaches. Layered 3D cortical tissue has already been generated (Lancaster et al., 2013), and approaches to form 3D retinal tissue or adrenal tissue have also been described (Jin et al., 2011; Nistor et al., 2010; Tsukada et al., 2013).
Another large group of 3D cultures are spheroid cultures formed by spontaneous aggregation of dissociated cells. Aggregation may, for instance, be caused by gravity in so-called hanging drops (Leist et al., 2012b). An alternative approach is the so-called re-aggregating brain cell culture. These are formed from dissociated rodent brain cells that undergo spontaneous aggregation into spheroids of a narrowly defined diameter when they are stirred (Honegger et al., 2011, 1979; Honegger and Richelson, 1976; Moscona, 1960, 1961). Such cultures allow for formation of histotypic structures, such as natural extracellular matrix, mature synapses, functional neuronal networks and myelinated axons (Honegger et al., 2011). Re-aggregating brain cell cultures proved to be suitable for the detection of organ-specific toxicity (Zurich et al., 2013) and have been used successfully for NT and DNT investigations.
Spheroids may also be formed by proliferation of neural precursor cells under conditions allowing all progeny of initially few cells to stick together. For instance, culturing coarse human or rodent brain cell homogenates (containing individual cells) in the presence of growth factors (Chiasson et al., 1999; Kuhn and Svendsen, 1999) eliminates most cell types originally present in the tissue and allows the few stem cells to proliferate and form neurospheres. NSCs and neural progenitor cells grown in suspension are responsive to mitogenic factors such as FGF-2 and EGF. These maintain cell proliferation and promote formation of sphere-like aggregates (neurospheres) (Reynolds et al., 1992; Singec et al., 2006). Neurospheres have the ability to produce their own extracellular matrix molecules (fibronectins, laminins), β-integrins and growth factors (Campos et al., 2004; Lobo et al., 2003). The cellular milieu of the neurosphere has been suggested to exhibit an in vitro microenvironment similar to the in vivo compartment (Conti and Cattaneo, 2010). This may allow generation of physiologically relevant cellular systems in drug discovery and toxicology screenings. In addition, since neurospheres provide a microenvironment for key events during neurodevelopment such as NSC maintenance, proliferation and differentiation, the neurosphere system enables the assessment of neurodevelopmental toxicants (Fritsche et al., 2011; Moors et al., 2009).
Secondary 3D cultures may be generated from neurospheres by plating them onto a planar substrate. They may then, e.g., be used for electrophysiological studies or to follow differentiation during radial migration on a laminin-coated surface (Moors et al., 2007, 2009; van Vliet et al., 2007). It is noteworthy that despite differentiation onto a surface, this manner of differentiation differs from simple 2D single cell plating because there is glial cell guidance with neuronal growth on top in a process-organized manner out of an organoid cell system (Ahlenius and Kokaia, 2010; Bal-Price et al., 2012; Liu and Sun, 2011; Schreiber et al., 2010). This phenomenon can be seen in the organized process of cell migration, which can also be quantified in such a system (Moors et al., 2007, 2009).
Generation of neural stem cells (NSC) from pluripotent stem cells is an alternative to the isolation of NSC from fetal brains. However, there are still some differences: so far only few groups could successfully expand NSC cultures for long periods of time (Koch et al., 2009; Palmer et al., 1997). When such cells are kept in 2D cultures, the gene expression level and cellular functionality often differs from original NSC or from cells growing in 3D (Birgersdotter et al., 2005; Kanebratt and Andersson, 2008; Zahir and Weaver, 2004). The expression profile in 3D cell cultures appeared to be more similar to the gene expression levels in vivo than that of their 2D counterpart (Abbott, 2003). Thus, toxicological and pharmacological screenings performed in monolayer cell culture do not necessary identify in vivo toxicity (Griffith and Swartz, 2006). Thus, toxicological and pharmacological screenings performed in monolayer cell culture do not necessary identify in vivo toxicity (Griffith and Swartz, 2006).
3.4 Sources of cells for 3D cultures
To provide human cellular material, which is currently seen as the new gold standard in toxicology of the 21st century (Leist et al., 2008b; NRC, 2007), new technologies are emerging that allow differentiation of human embryonic stem cells (hESC), human induced pluripotent stem cells (hiPSC) or fetal neural progenitor cells (NPCs) into neurons and glial cells (Bal-Price et al., 2012; Fritsche et al., 2011; Leist et al., 2008a). NSC may also be generated (from scratch) from human pluripotent stem cells that are driven towards neural differentiation. This allows the formation of neurospheres with highly defined characteristics (Hoelting et al., 2013). Rodent neurons, glia, neural stem cells and tissues can be isolated from either fetal, newborn or adult brain.
Although human cell systems are in the end desired for making predictions of human health, rodent systems can also be of high value for risk assessment. For instance, rodent models may be used in parallel to a human cell-based model. By using both cultures, neurotoxicity observed in human cells in vitro can be extrapolated to the large database on neurotoxicity in rodents in vivo. Such species extrapolations that use comparison of in vitro and in vivo data are called the “parallelogram approach of toxicology”. For instance, species-specific differences in sensitivity observed between human and rat organoid cultures are most likely to reflect, and to predict, disparities in cellular toxicodynamics. These experimentally-derived toxicodynamic factors can then be employed for hazard and risk assessment. However, degrees of maturation between human and rodent systems have to be carefully assessed, as speed of maturation differs between species, not only in vivo but also in vitro (Baumann et al., 2014).
3.5 Benefits and advantages of neuronal 3D models
Often, 3D models result in improved structure and cell connectivity. Moreover three-dimensional cultures often show enhanced survival and neuronal differentiation compared to traditional monolayer cultures (Brannvall et al., 2007; Peretz et al., 2007). The enhanced survival could be related to the improved cell-cell and cell-matrix contact, as it improves cell signaling and gap junction connections, but these factors may need further investigation, as also in vivo survival after, e.g., injection of cells in the brain is still not clarified (Hansson et al., 2000; Schierle et al., 1999). Cell-cell signaling can also promote proliferation and differentiation of glial and neural stem cells, which in turn contributes to improved culture longevity and functional relevance. Local secretion and paracrine signaling of neurotrophic factors, such as nerve growth factor (NGF) and brain-derived neurotrophic factor (BDNF), together with intercellular communication between neuregulin-1 and Necl4 are important in this regard (Maurel and Salzer, 2000; Michailov et al., 2004). Designing in vitro models to enable this is therefore an important consideration. The ability to keep cultures for prolonged times is an important prerequisite for experiments addressing chronic forms of toxicity.
Improved cell-cell interactions in 3D structures, especially between astrocytes and neurons, may enhance neurogenesis and synapse formation. Functional synapses have been observed in three-dimensional neuronal cultures at similar or earlier time points compared to 2D models (O’Shaughnessy et al., 2003; van Vliet et al., 2007). Furthermore, neural stem/progenitor three-dimensional cell cultures showed an increased differentiation into neurons compared with 2D cultures (Brannvall et al., 2007). The phenotype of microglial cells is highly dependent on the culture conditions. Often microglial cells show an amoeboid morphology in 2D cultures, even in control cultures, in the absence of any toxicant. This is characteristic for phagocytic cells that have been pre-activated. However, when cultured in an inert 3D matrix, microglial BV-2 (Henn et al., 2009) cells shifted from an amoeboid form to a population dominated by multipolar (resting) phenotypes (Pottler et al., 2006). Furthermore, in untreated 3D aggregating brain cell cultures devoid of any type of inert matrix, microglial cells exhibit a ramified morphology typical of quiescent cells. Only activated (directly by a toxicant or indirectly by injured neurons or glial cells) microglia exhibit amoeboid morphology (Monnet-Tschudi et al., 2007). The presence of quiescent microglial cells in control cultures is due to the intense cell-cell interactions occurring in 3D between all brain cell types in the aggregating brain cell cultures. The dampening effect of brain tissue on microglia has been shown also by transcriptomics approaches (Higgins et al., 2010).
3.6 Issues/disadvantages of neuronal 3D models
Some issues are specific for 3D models while others apply equally to 2D models. For instance, the models use mostly complex culture media that do not necessarily resemble the cerebrospinal fluid (CSF) in brain. Also, the models usually do not have a barrier function that restricts access of toxicants similar to the blood-brain or the placental barrier.
A specific problem of 3D models may be limited compound diffusion to the cells. In many models and for many compounds this may not be an issue at all, while, e.g., for larger models and high molecular mass “compounds” (e.g., nanoparticles), limited accessibility of the cells far from the surface needs to be considered.
4 3D models of the respiratory tract
The respiratory tract has a large internal surface area (>150 m2) and a very thin air-blood tissue barrier (<1 μm) (Gehr et al., 1978; Ochs and Weibel, 2008), both of which are essential for an optimal gas exchange between the air and blood by diffusion. The respiratory tract can be sub-divided into functionally and structurally distinct regions (Ochs and Weibel, 2008; Weibel, 1979). Most proximally is the extrathoracic region, consisting of the nasal cavity, mouth, pharynx and larynx, followed by the tracheobronchiolar region, consisting of the trachea, main bronchi, bronchi and bronchioles including the terminal bronchioles. The main tasks of this region are air conditioning and air conduction. Ambient air is efficiently modified and cleansed of much of the larger particulate material by mucociliary activity (fast particle clearance) before being conducted deep into the lungs. The tracheobronchiolar region is followed by the proximal part of the alveolar-interstitial region, consisting of the respiratory bronchioli with only a few alveoli, the task of which is air conduction, a small amount of gas exchange and slow clearance of particulate material. The distal part of the alveolar-interstitial region consists of the most peripheral airways, the alveolar ducts, with their “walls” completely covered with alveoli (i.e., alveolar entrances), and the alveolar sacs (i.e., alveolar ducts with alveoli closing the end of the terminal ducts), including the interstitial connective tissue. The main task of this region is gas exchange of oxygen and carbon dioxide with the blood (Gehr, 1994).
A series of structural and functional barriers protect the respiratory system against both harmful and innocuous particulate material (Nicod, 2005). The barriers comprise the surfactant film (Gil and Weibel, 1971; Schurch et al., 1990), the aqueous surface lining layer including the mucociliary escalator (Kilburn, 1968), macrophages (professional phagocytes) (Brain, 1988; Lehnert, 1992), the epithelial cellular layer endowed with tight junctions and adherens junctions between the cells (Godfrey, 1997; Schneeberger and Lynch, 1984), and a network of dendritic cells (DCs) inside and underneath the epithelium (Holt and Schon-Hegrad, 1987; McWilliams, 2000). In the respiratory tract, dendritic cells (DCs) crucially maintain the fragile equilibrium between induction of tolerance against innocuous substances and mounting an active immune response against a potentially harmful pathogen. Besides the lung epithelium, the basement membrane (Maina and West, 2005), the connective tissue (Dunsmore and Rannels, 1996), and the capillary endothelium (Dudek and Garcia, 2001; Schneeberger, 1977) serve as structural barriers.
4.1 In vitro respiratory tract models
Since the morphology and function of the respiratory tract changes completely from the upper to the lower airways, many cell culture models have been established using primary cells or cell lines to represent the area of interest for a certain study.
There are numerous different in vitro models that can be used to evaluate the human pulmonary epithelial tissue barrier, such as epithelial airway or alveolar cells. They range from simple 2D monocultures (Fuchs et al., 2002; Steimer et al., 2005) to highly sophisticated 3D models involving different cell types (Rothen-Rutishauser et al., 2008). The 3D models represent a more realistic physiological situation (Carterson et al., 2005). Studies have shown that when cells are removed from their host tissue and are grown as monolayers on impermeable surfaces, they undergo dedifferentiation and lose specialized functions, which is thought to be, in part, due to the disassociation of cells from their native three-dimensional tissue structure in vivo (Freshney, 2000). Co-cultures of different cell types have been shown to have an influence on the outcome of the results, since cells continuously cross-talk in vivo through intercellular signaling to maintain homeostasis and to coordinate immune responses (Roggen et al., 2006). However, these models fail to provide organ-level functionalities (molecular transport across tissue-tissue interfaces, contributions of vascular and air flow, etc.) that are required for the development of meaningful physiological models (Huh et al., 2011, 2012b). A potential solution to this problem is the development of human “organs-on-chips” in which microscale engineering technologies are combined with cultured living human cells to create microfluidic devices that recapitulate the physiological and mechanical microenvironment of whole living organs (Huh et al., 2011, 2012b). These organomimetic microdevices enable the study of complex human physiology in an organ-specific context and, more importantly, they offer the potential opportunity to develop specialized in vitro human disease models that could revolutionize drug development.
In vitro co-cultures to mimic the alveolar epithelial barrier with two cell types have been described in the literature. Two cell co-culture models with epithelial (A549 cells) and endothelial cells were established to examine events in the pathogenesis of bacteria (Bermudez et al., 2002; Birkness et al., 1995). Recently, a primary co-culture system to simulate the human alveolar-capillary barrier by using primary cells was developed. Human pulmonary microvascular endothelial cells were co-cultivated with primary human type II alveolar epithelial cells on opposite sides of a permeable filter support to study the impact of nanocarriers (Hermanns et al., 2009 2010).
Recently, a triple cell culture in vitro model of the human airway wall to study the cellular interplay and the cellular response of epithelial cells, human blood monocyte-derived macrophages and dendritic cells to particles (Rothen-Rutishauser et al., 2008) has been developed. In this model, monolayers of two different epithelial cell lines representing alveolar as well as bronchial regions, A549 (Rothen-Rutishauser et al., 2005) and 16HBE14o-epithelia (Blank et al., 2007), as well as primary epithelial type I cells (Lehmann et al., 2011), can be grown on a microporous membrane in a two chamber system. In addition, a quadruple-culture containing epithelial and endothelial cells, macrophages, as well as mast cells, has been established (Alfaro-Moreno et al., 2008). Studies using such co-culture cell systems have reported that they observe different reactions compared to monoculture analysis when the cells are exposed to nanomaterials (Lehmann et al., 2009; Muller et al., 2010); however, such reactions observed from a culture containing two, three or four different types of cells merely cultured in the same dish, are not as specific as those that would occur in the human body. Thus, the architecture of the in vitro cell co-culture model, in regard to the specific organ they represent, is essential when nanomaterials’ effects are studied. Figure 6 shows the schematic representation of 3D in vitro models of lung for transwell co-culture technique and the currently most advanced in vitro lung model.
Fig. 6. Schematic representation of 3D in vitro models of lung.
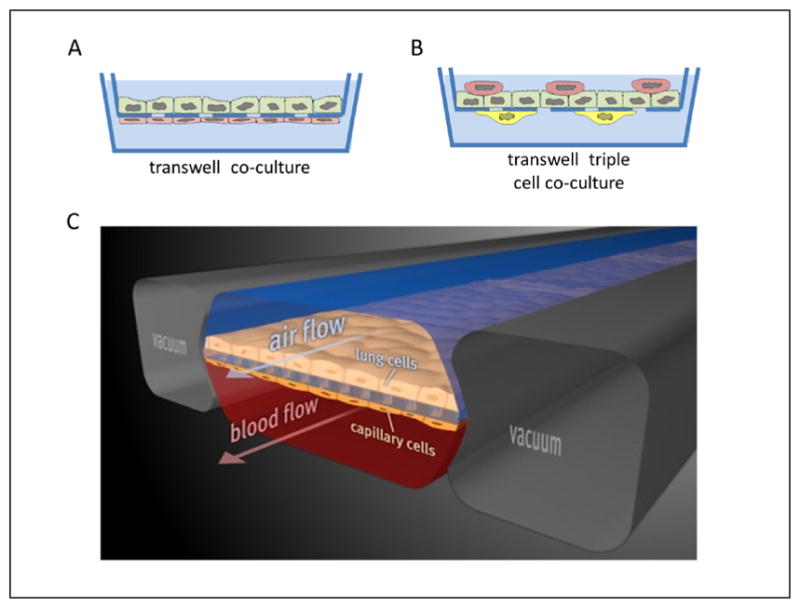
Many co-cultures mimicking the alveolar barrier are described in the literature (A). The most common approach is to use permeable membrane inserts in a two chamber system with epithelial cells grown on the upper side and endothelial cells on the opposite (B). Design of the triple cell co-culture composed of epithelial cells with macrophages on top and dendritic cells on the opposite of the insert membrane. (C) The microfabricated lung device uses compartmentalized polydimethylsiloxane (PDMS) microchannels to form an alveolar-capillary barrier on a thin, porous, flexible PDMS membrane coated with extracellular matrix. The device recreates physiological breathing movements by applying vacuum to the side chambers and causing mechanical stretching of the PDMS membrane forming the alveolar-capillary barrier (reprinted with permission from Huh et al., 2010).
4.2 Benefits and uses of 3D organotypic respiratory system models
The recently (micro)engineered “lung-on-a-chip” reproduces key structural, functional and mechanical properties of the living lung by microfabricating a microfluidic system containing two closely apposed microchannels separated by a thin (10 μm) porous flexible membrane made of polydimethylsiloxane (PDMS) (Huh et al., 2010). The intervening membrane was coated with extracellular matrix (ECM), and human alveolar epithelial cells and human pulmonary microvascular endothelial cells were cultured on opposite sides of the membrane. Once the cells were grown to confluence, air was introduced into the epithelial compartment to create an air-liquid interface and more precisely mimic the lining of the alveolar air space. In addition, this microsystem reproduces breathing movements and the associated cyclic strain experienced by cells at the alveolar-capillary interface by applying cyclic suction to neighboring hollow microchambers that deforms the central porous membrane and attached cell layers.
Most importantly, this lung-on-a-chip microdevice replicates the complex organ-level responses of living human lung to physiological inflammatory stimuli, such as bacteria or cytokines introduced into the air space. The underlying endothelial cells rapidly become activated in response to these cues, which induces adhesion of primary human neutrophils flowing in the capillary channel, as well as their transmigration across the capillary-alveolar interface and into the alveolar space where they then engulf the bacteria. Because of the optical clarity of the PDMS, the entire human inflammatory response can be visualized in real-time within this microdevice (Huh et al., 2011).
The lung-on-a-chip has been used for nanotoxicology studies by introducing various types of nanoparticles into the air channel. These studies revealed that physiological breathing movements accentuate toxic and inflammatory responses of the lung to silica nanoparticles (12 nm) used to mimic airborne particulates. Mechanical strain also enhanced epithelial and endothelial uptake of nanoparticulates and stimulated their transport into the underlying microvascular channel. Importantly, similar effects of physiological breathing on nanoparticle absorption are observed in whole mouse lung (Huh et al., 2012b).
Most recently, the same group has microengineered a disease model of pulmonary edema in human lungs (Huh et al., 2012a). Specifically, they established a model of pulmonary edema induced by a life-threatening side effect of chemotherapy based on interleukin-2 (IL-2), known as a vascular leakage syndrome (VLS). They successfully reconstituted the essential features of this disease including fluid accumulation and fibrin deposition in the lung. This disease model revealed previously unknown effects of physiological mechanical force on the development and progression of VLS and that circulating immune cells are not required for the development of this disease. These studies also led to identification of potential new therapeutics that might prevent this life-threatening toxicity of IL-2 in the future. These data demonstrate the ability to leverage the unique capabilities of the human lung-on-a-chip microdevice to create a clinically relevant human disease model in vitro, and ultimately to reliably predict drug toxicity and efficacy in humans. Importantly, these findings suggest that a microengineered in vitro human disease model can potentially replace preclinical animal models of pulmonary edema currently used by the pharmaceutical industry to evaluate the effects of drugs on VLS in the lung.
4.3 Issues/disadvantages of 3D lung models
One of the main problems for 2D as well as 3D lung models is that no cell line – neither for the upper nor the lower airways – exists that represents the optimal function-structure relationship. On the other side, the isolation and handling of primary cells is much more time-consuming and the variability between the different isolations is high. Therefore, the limitation of each model which has been described so far needs to be carefully considered for the interpretation of observed effects. In addition, since the lung surface is exposed to air, the cell model(s) also need to be cultured at the air-liquid interface to mimic the lung more realistically. This is however difficult to perform over longer exposure durations due to the limited life time of the cells (i.e., mainly a problem for cell lines) and/or due to an overgrowth of cells forming multiple cells instead of a monolayer (Paur et al., 2011). In addition, the vast majority of lung models are still lacking in mimicking the specific mechanics of the lung surface. In particular, they typically present a static air-liquid interface, which is clearly unrealistic given the cyclical motion of the alveolar surface during breathing. A recent exception is provided by Huh and colleagues (Huh et al., 2011) whose lung on a chip model features mechanical stretching of an interface combining alveolar epithelial and microvascular endothelial cells to simulate the dynamics of breathing. This microenvironment with the mechanical activation can maintain cells in culture for several weeks. These cells also exhibit more highly differentiated phenotypes when presented with physiologically relevant microenvironmental cues than when maintained under conventional 2D or 3D culture condition.
5 3D models of the skin
The skin is the largest organ of the human body and also the first “line of defense” against many external insults. All manner of chemicals, cosmetics and medicines must be tested for skin irritation and/or sensitization. This has been a major source of animal use in testing, particularly for cosmetics. Even before the recent outlawing of animal testing in cosmetics in the European Union, in vitro skin testing was perhaps the furthest along of all alternative testing methods, regarding acceptance by regulatory authorities and use in toxicological testing.
5.1 In vitro skin models
Human keratinocyte and fibroblast monolayer cultures are well established (Fig. 7), particularly ones using murine fibroblasts (3T3 cells) and the spontaneously transformed human keratinocyte cell line HaCaT. However, the latter does not reflect the primary keratinocyte in situ, which can be easily visualized, and has been shown, for example, by the spontaneous chromosomal aberrations in immortalized keratinocytes. This raises the question of whether monolayer-based (2D) assays are suitable for drug development.
Fig. 7. In monolayer culture, cellular morphology differs from in vivo morphology.
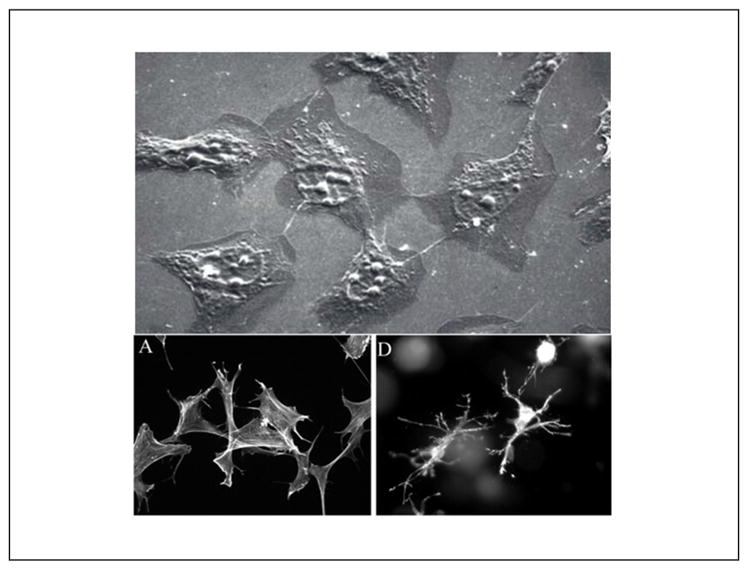
Upper picture: Fibroblasts in monolayer culture on lamella observed by scanning electron microscopy (SEM): population sub-confluence. Cells have rather starry shapes. Length of the line 10 μm (Image: A. Minondo, L’Oréal); lower pictures: human fibroblasts project a dendritic network of extensions in collagen matrices but not on collagen-coated coverslips. Fibroblasts were incubated for 5 h on collagen-coated surfaces (A) or in collagen matrices (D). At the end of the incubations, samples were fixed and stained for actin. Reprinted with permission from Grinnell et al. (2003).
Although skin metabolism takes place mostly in epidermal keratinocytes, only 3D constructs mimic xenobiotic biotransformation in human skin properly.
In the 1980s it first became feasible to predict cytotoxicity (OECD, 2013a), phototoxicity (in 3T3 cells; (OECD, 2004a)) or induction of cell migration (scratch assay) using 2D assays. In fact, OECD guideline 432, adopted in 2004, is the first in vitro test replacing hazard analysis in the animal almost entirely (with drug development being the only exception). But monolayer cultures cannot be used to investigate percutaneous absorption or biotransformation of compounds, and processes involving these (e.g., systemic toxicity, skin sensitizing and genotoxicity) therefore require more complex approaches. In contrast, multilayer (3D) reconstructed human epidermis has proved to be suitable for in vitro skin absorption testing. A schematic set up for reconstructing human skin is shown in Figure 8.
Fig. 8. Reconstructing human skin.
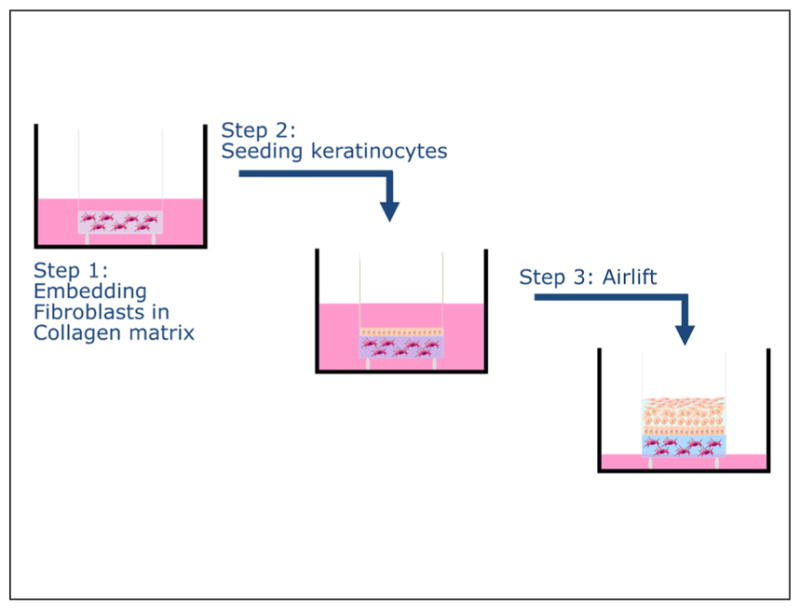
Normal fibroblasts and keratinocytes are isolated from, e.g., human foreskin and expanded in monolayer culture. Fibroblasts are embedded into a collagen matrix (step 1) and the collagen is allowed to contract (growth medium). Keratinocytes are seeded on top and medium is changed to differentiation medium (step 2). Finally the culture is lifted to the air-medium interface (step 3). Total culture duration is about 14 days. For the reconstruction of human epidermis step 1 is omitted and keratinocytes are seeded on a supporting membrane.
In addition, monolayer cultures do not mimic cell-cell interactions of a multilayer tissue consisting of different cell types. Several types of cells contribute to skin function in order to prevent damage by UV irradiation, xenobiotics or microorganisms. Skin morphology varies during lifetime and according to the body site. Disturbed barrier function and/or immunosuppression lead to severe or chronic/recurrent diseases (e.g., cancer, septicemia or atopic dermatitis).
5.2 Benefits and uses of 3D organotypic skin models
Multilayer skin constructs represent only epidermis or only epidermis plus dermis. Reconstructed human epidermis, built up by proliferating and differentiating human keratinocytes, or reconstructed human skin (full-thickness skin construct), in which the epidermis equivalent covers a fibroblast embedding collagen layer, are state-of-the-art in dermal tissue engineering. Constructs of high quality are commercially available (Fig. 8); these are close to native human skin in terms of morphology, lipid composition and differentiation. These approaches are intended to address issues in which 2D assays fail due to the lack of the extensive barrier of intact skin. Examples can be found in OECD TG 432 (tiered test for phototoxicity, if a substance is suspected to penetrate skin only poorly), OECD TG 431 (test for skin corrosion) or OECD TG 439 (skin irritation) (OECD, 2004a, 2013a).
In most 3D-models, a scaffold provides the geometrical structure for tissue growth. Additionally, scaffolds can present relevant markers and proteins to foster cell adhesion and to guide tissue development. Furthermore, a scaffold can support the development of sophisticated co-culture systems. The third dimension allows the culture of different cell types in a tissue-specific formation. For instance, full-thickness skin equivalents are comprised of a dermal and an epidermal layer (Fig. 3A). In each layer, specific cells are cultured. The dermal layer is usually generated by seeding fibroblasts into a collagen hydrogel scaffold. To mimic the basement membrane of the skin, the surface of the dermal compartment can be coated with fibronectin. This artificial basement membrane allows for the culture of keratinocytes. To include an additional barrier, the bottom surface can be populated with endothelial cells.
One approach to generate 2D co-culture models is to use porous membranes in order to establish different culture compartments based on the insert technology. A specific cell type is seeded in each compartment. Cellular cross talk between the two cell types is restricted to diffusion in the liquid phase and across the pores of the membrane. Therefore, concentration gradients have to be established that are strongly determined by the pore parameters such as pore diameter, porosity, pore length, and the diffusion coefficients of the messenger substances in the cell culture media. In more complex liquid systems, such as blood plasma, the diffusion coefficients of proteins, e.g., urea, creatinine, and uric acid, are approximately 30% lower compared to water. Therefore, mass transport is biased when compared to the in vivo situation, which has a significant impact on patterning that is based on the spatial and temporal distribution of messenger proteins. Patterning guides tissue or cell development, and thus, the 2D model may have different structural or functional properties in comparison to native tissue due to the parameters of the experimental setting such as pore diameter or membrane thickness.
In contrast, the 3D environment in a scaffold fosters a higher complexity of cellular cross talk. Messenger proteins diffuse in an extracellular matrix (ECM) and, hence, the diffusion is affected by the ECM components. Additionally, further mechanisms such as protein binding to specific binding sites of the ECM are possible. Moreover, the communication distance in a 3D scaffold is determined by the cell distribution and not by the artificial gap between well plate and porous membrane. In conclusion, culture of cells in 2D is far too artificial and lacks many parameters known to be important for an accurately reproduced tissue structure and physiology.
Recently, several more advanced states of tissue culturing have been realized. Co-culture of reconstructed human skin with malignant cells has been used for nonclinical assessment of photodynamic therapy. Candida albicans seeding allows building mycosis models for the analysis of host-pathogen interactions at the molecular level. Moreover, gene silencing in normal human epidermal keratinocytes can be used to generate diseased skin models, such as for congenital ichthyosis by corneodesmosin knock down or diseases linked to a barrier defect by filaggrin knock-down.
Development of 3D models for non-melanoma skin cancer, candidiasis and loss of barrier function was a priority in 3D model development due to the prevalence of these diseases. Mycoses have become a major health problem: the frequencies of mucosal and cutaneous fungal infections are increasing worldwide and host immunosuppression facilitates invasive candidiasis, resulting in septicemia. Oral and vaginal mucosa models of epithelial candida infections based on commercially available reconstructed tissues offer a faster approach for the development of new drugs. The value of these models cannot be over-estimated, since pathological processes in the current in vivo guinea pig model do not reflect mucosal fungal infections and it is difficult to compare guinea pig cutaneous fungal infections to fungal infections in humans. Non-melanoma skin cancer is also a major health problem: increased incidences of non-melanoma skin cancer and invasive and life-threatening diseases due to immunosuppression as treatment, in particular in organ transplant receivers, are a major cause of morbidity and mortality. Field cancerization, in which dozens of lesions occur simultaneously, requires efficient field-directed therapy. Recently, the efficacy of one approach, photodynamic therapy, was assessed by reconstructed human skin integrating malignant cells. Atopic dermatitis affects currently about 10–20% of children and 1–3% of adults. Although atopic dermatitis models do not reflect the multifactorial disease pattern in full, a well-characterized model can be used for testing of glucocorticoids or calcineurin inhibitors and the respective formulations for topical use.
5.3 Disadvantages of 3D skin models
The general procedure of building, maintaining and characterizing 3D skin models is published; these well-known techniques are then further adapted to disease models. But the current 3D constructs need much more detailed characterization, going beyond morphology and lipid analysis. To be used for regulatory purposes the construct itself has to reflect the in vivo situation and the methodology for proving efficacy and/or identifying hazards has to be validated, and then achieve regulatory acceptance. Successfully navigating this process will allow 3D constructs and disease models to be used as alternatives to animal or 2D approaches.
Though quite advanced, current 3D constructs still do not yet contain all essential cell types – in particular dendritic cells are most difficult to (co-)culture. However, they are needed for realistic models of a number of important diseases, such as atopic dermatitis and psoriasis. Some initial approaches of embedding macrophages into dermal equivalents are described in the literature, but they still need improvement regarding epidermal structure. An epidermis close to the in vivo situation is imperative, particularly to recapitulate complex pharmacokinetics and pharmacodynamics. Another important step is to integrate blood vessels or, at least, an endothelial layer, below the dermal equivalent. This type of integrated skin would be a major part of the “body-on-a-chip”, mentioned previously.
Finally, from a supply-perspective, the broad spectrum of potential 3D construct applications as well as a major increase in REACH-based testing will likely lead to primary cell shortages. To ensure sufficient supply of primary cells, inducible stem cells offer a viable option. Besides the general approach of cell-differentiation, there is a more specific one with respect to keratinocytes: Progenitor cells are found in hair bulbs, which are removed by hair plugs and can be expanded for reconstruction of human epidermis.
Research in the field of alternative testing has been reinforced in order to avoid the testing of cosmetics and ingredients of cosmetic formulations in animals (Leist et al., 2008b). In vivo, the toxicity of a test substance is the result of complex events involving multifactorial and dynamic physiological processes. These mechanisms are dependent on the interaction between the substance and the cell as well as skin penetration, metabolism or inactivation. Although an in vitro model cannot reproduce all the events observed in vivo, in vitro assays based on 3D models constructed using human cells are very powerful tools. They are a simplified representation of reality that allows studying a compound under standardized conditions with controlled parameters. This given, 3D models may even allow to overcome the high failure rates in clinical testing because of the poor transferability of animal data to man (Hartung, 2007; Hartung et al., 2013), both with respect to rodents (Seok et al., 2013) and the dog (Bailey et al., 2013). Today, reconstructed human epidermis (RHE) is commercially available in high quality (e.g., EpiDerm™, SkinEthic™, EpiSkin™) for hazard analysis (Fig. 9). RHE from other origin (in-house constructs, Open-Source Reconstructed Epidermismodell, OS-REp) also can be used if quality is proven.
Fig. 9.
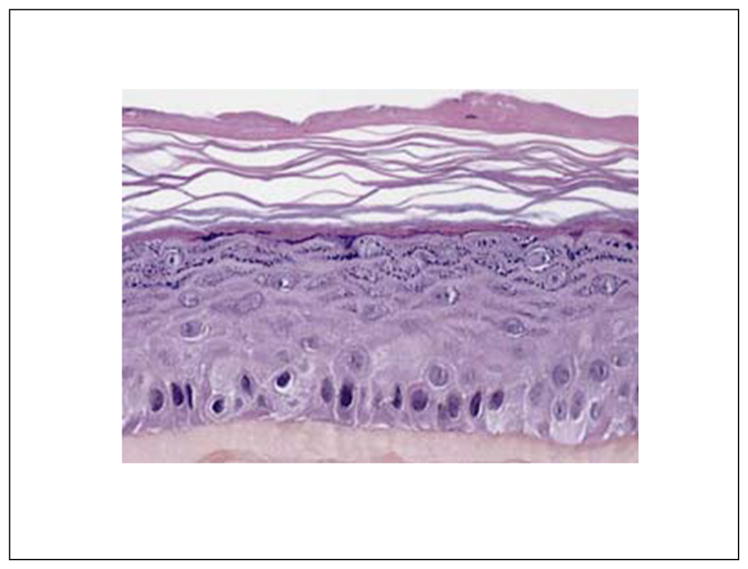
Histology of a reconstructed epidermis, EpiSkin™
Keratinocytes cannot reproduce all the physiological processes present in human skin. This underlines the need for more complex constructs to address complex toxicological endpoints. Micro-environmental factors, such as the dynamic crosstalk between epithelium and connective tissue, are known to regulate epidermal morphogenesis and homeostasis. Thus, reconstructed human full-thickness skin (RHS) presenting an additional dermal layer (commercial products: EpiDerm FT™, Phenion-FT™) may be helpful. Because of differences in enzyme expression in keratinocytes and fibroblasts RHS may be superior to RHE when investigating dermal biotransformation of xenobiotics and related endpoints (sensitization, genotoxicity). Further advancements of RHS, such as long-term stable constructs and models with co-cultured dendritic cells, are highly desirable for several applications.
5.4 Key issues of normal reconstructed human epidermis (RHE)
Reconstructed from human non-transformed epidermal keratinocytes, RHE closely mimics the morphological (well-stratified epithelium with basal, spinous, granulous and corneal layers), biochemical and physiological properties of the human epidermis. Quality assurance according to OECD TG 431 (OECD, 2013a) includes the declaration of cell types, cell handling, type of the construct, histology, immunohistology, lipid analysis, permeability for standard substances as well as resistance to irritants. The barrier function is highly relevant for the use of RHE in hazard analysis.
Constructs are built from keratinocytes isolated from juvenile foreskin or adult abdominal/breast skin and expanded in culture media. The cells are seeded into transwells, and after a short submersed cultivation an airlift is performed in order to induce keratinocyte differentiation, which is a continuous process. After about two weeks keratinocytes will be fully differentiated and all the epidermal layers of normal human skin are presented. Transcriptome analysis proved human skin and RHE to be similar to each other, expressing epidermal differentiation markers – e.g., filaggrin, loricrin, involucrin, receptors and transcription factors. In contrast, the expression of many cell cycle and DNA replication genes and the lack of differentiation markers prove that keratinocytes in monolayers fail to differentiate (Gazel et al., 2003).
Lipid analysis demonstrated great similarities of stratum corneum lipids between normal human skin and RHE. Differences in specific lipids (e.g., less essential long-chain free fatty acids in RHE) and in particular the lipid order (native human stratum corneum presents two lamellar phases with repeat distances of 13.4 and 6.4 nm, RHE presents long periodicity only), however, can explain the poorer barrier function. This might be even worse with RHE grown from the HaCaT cell line which showed an impaired capacity for lipid synthesis (Ponec et al., 2001). Currently, a construct with a barrier as good as human skin is not available (Thakoersing et al., 2012). The investigation of specific effects of defined stratum corneum lipids on the barrier function becomes possible by, e.g., spraying lipid mixtures dissolved in organic solvents onto an appropriate support material (Groen et al., 2011).
5.5 Key issues of normal reconstructed human skin (RHS)
Full-thickness human skin (RHS) can be reconstructed from human keratinocytes seeded on collagen-embedded fibroblasts (Fig. 10). Quality assurance is as described for RHE. Morphology of the constructs – including fibroblast morphology and the organization of cell-derived matrix components (Cukierman et al., 2001) – and lipid formation is close to normal human skin. The order of stratum corneum lipids is not perfect but improves over time whereas the skin surface pH 5.5 is well in accordance with human skin already in the early stage of development (Vávrová et al., in press). This holds true also with a construct generated from the HaCaT cell line replacing primary keratinocytes. Acidic pH at the skin surface increased up to 7.0 in deeper layers. In the intercellular matrix pH is ≤ 6.6 (Hanson et al., 2002; Niesner et al., 2005).
Fig. 10. Matrix effects on 3D RHS.

Both 3D organization and cell-derived matrix components are required for formation of 3D matrix adhesions (triple label localization of α5 integrin (green, A), paxillin (red, B), and fibronectin blue, (B)) in RHS. The yellow color (A) represents the merged labels of α5 in green and paxillin in red. Triple merged images for the indicated matrices are shown in (C) to (F). Note that complete triple co-localization (white) is observed only with in vivo-like, cell-derived 3D matrix (C) or mouse tissue-derived 3D matrix (F) in 3D matrix adhesions (arrows). Reprinted with permission from Cukierman et al. (2001).
3D constructs, which are stable for up to 6 months, appear to make it possible to avoid collagen as a component of the dermal equivalent (Stark et al., 2006). Other major aims are the construction of vascularized and immune-competent skin, which may become possible by integration of, e.g., a vascular endothelial cell line and dermal-type macrophages (Bechetoille et al., 2011) or CD34+ cells after keratinocyte seeding. Epidermal Langerhans cells and dermal dendritic cells can be formed depending on the culture condition (Dezutter-Dambuyant et al., 2006; Auxenfans et al., 2009).
5.6 Benefits of 3D skin constructs and special requirements
In monolayers, the cells are immersed in an aqueous culture medium which limits the testing to water soluble substances. RHE overcomes this limitation and various substances and various vehicles (liquid, powder, gel …) can be applied directly to the skin surface. 3D constructs enable the investigation of dermal penetration and various biological effects in the skin can be derived by quantitation of several biological endpoints, e.g., by biomarkers or two-photon microscopy
Primary cells are preferred for 3D models due to their wild-type nature, which translates to in vitro cultures with cells preserving better the structural and biochemical complexity found in vivo. Yet, primary cells are most sensitive to their surroundings, handling and use require sophisticated techniques and advanced tissue culture training in order to obtain the high level of organization of 3D constructs. Cell migration and differentiation are very sensitive to any modification of the culture environment. This multiplies sources of variability in the resulting 3D models and RHE/RHS require distinct quality control during their life cycle. Test protocols must always include assay acceptance criteria (negative and positive controls, comparison to historical data, replicates, etc.).
5.7 RHE and RHS for studies of (per-)cutaneous absorption and biotransformation
(Per-)cutaneous absorption in human skin ex vivo closely approximates data obtained in humans given the protocols are appropriately matched (Lehman et al., 2011), and as with human skin, intercorneocyte penetration is the predominant uptake pathway of low molecular weight xenobiotics in RHE. The still imperfect structural organization of stratum corneum lipids explains the over-predictive power for (per-)cutaneous absorption of RHE. Nevertheless, using a strictly controlled protocol conforming to OECD TG 428 (OECD, 2004b) a validation study indicated the suitability of EpiDerm™, EpiSkin™, and SkinEthic™ for in vitro absorption studies. These constructs were appropriate alternatives to human skin ex vivo when testing nine compounds widely varying in molecular mass and partition coefficient. The permeation of RHE was strongly related to the permeation of human epidermis sheet and the absolute cut-off level of penetration (800 g Mol-1) of skin and RHE was in accordance, too. Both infinite and finite dose experiments were feasible (Schäfer-Korting et al., 2008a). The protocol was transferable to RHS and discriminated vehicle effects (Schäfer-Korting et al., 2008b; Ackermann et al., 2010). Thus RHE/RHS may find a major place in (per-)cutaneous absorption testing.
In addition to the barrier function, metabolic competence of RHE and RHS is of relevance. Cutaneous biotransformation (Hewitt et al., 2013) can inactivate toxic agents, yet biotransformation may also be the cause of sensitization (Hagvall et al., 2008; Ott et al., 2009; Samuelsson et al., 2011) and genotoxicity (Hu et al., 2009; Brinkmann et al., 2012). This is often linked to the activation of a compound by biotransformation in keratinocytes and dendritic cells. Comprehensive investigations on the expression and functionality of enzymes involved in cutaneous biotransformation of xenobiotics in human skin ex vivo, RHE and RHS (Oesch et al., 2007; Harris et al., 2002; Luu-The et al., 2009; Götz et al., 2012; Jäckh et al., 2012; Mitriaikina and Müller-Goymann, 2009; Bätz et al., 2013) are ongoing and the knowledge is currently improving. Biotransformation-competent RHE or RHS may be used for the study of penetration and biotransformation in a single step and RHE/RHS may be suitable to study genotoxicity (Aardema et al., 2010) and sensitization (Mehling et al., 2012) of compounds in the skin. Moreover, the skin is also important for synthesis and metabolism of steroids, and EpiSkin™ expresses the enzymes for steroid metabolism (van Eijl et al., 2012; Luu-The et al., 2007).
5.8 Choice of endpoints for hazard analysis based on reconstructed human skin models
The use of RHE for the study of corrosivity and irritancy is well established. OECD has accepted the respective TG 431 (OECD, 2013a) and 439 (OECD, 2010b). OECD TG 431 was updated in 2013 to include a set of performance standards for the assessment of similar and modified RHE-based test methods (OECD, 2013a). Importantly, the update also comprises the possibility for sub-categorization among corrosive chemicals, classifying substances in the Globally Harmonised System of Hazard Classification and Labeling (GHS). In fact, Transportation of Dangerous Goods Regulations request a classification of corrosives into 3 subcategories (1A – severe, 1B moderately severe, and 1C minor danger) and GHS 1A classification of a chemical has huge impact on transport and storage conditions. OECD TG 431 now allows a classification according to the categories 1A and 1B/1C. Adoption of a revision of OECD TG 439 (OECD, 2010b) is expected by 2014, once more in order to allow classifying substances in GHS categories. Category 3 (mild irritants) is optional in Europe but required in some countries and by some authorities (Kolle et al., 2012).
Corrosivity and irritancy testing is based on the MTT reduction test. A limitation of this read-out is the non-specific interaction of MTT with some test substances (colored substances, test substances that directly reduce MTT). Then, additional steps have to be taken and the test substance may even be incompatible with the test system. In order to extend the applicability domain, dead tissue may be used for control. Alternatively, the quantification of ATP levels can solve the case of direct MTT reducers. For colored substances, use of HPLC as complement to photometry is currently assessed to address this limitation.
Another widely accepted test for hazard analysis is phototoxicity testing with RHE as an adjunct test to the 3T3 Neutral Red Uptake Phototoxicity Test (3T3 NRU PT; OECD TG 432 (OECD, 2004c)). Despite the fact that the 3T3 NRU PT is over-sensitive (Lynch and Wilcox, 2011), it is still considered to be the first test in a tiered testing phototoxicity strategy as the assay identifies the phototoxic hazard with 100% sensitivity (Ceridono et al., 2012). Other than monolayers, the barrier function of RHE limits access of substances to viable cells. Yet, until today a formal validation of phototoxicity tests using 3D models is lacking.
The complex cascade of events underlying skin sensitization asks for an integrated testing strategy for the replacement of animal testing (Local Lymph Node Assay, LLNA (OECD, 2010a)). The compound’s penetration into viable skin, the potential biotransformation, the covalent binding to proteins and the activation of keratinocytes and dendritic cells and subsequent processing resulting in the expression of surface markers of activated dendritic cells have to be tested stepwise (Mehling et al., 2012). Among the most advanced in the validation process at ECVAM are the direct peptide reactivity assay (DPRA), the KeratinoSens™ assay and the human cell line activation test (h-CLAT). These assays cover three key steps: protein reactivity, keratinocyte activation and dendritic cell activation. Most recently, two reports have demonstrated that the combination of these methods yields a high accuracy to predict skin sensitizers (Bauch et al., 2012; Natsch et al., 2013). However, these approaches, similarly to other traditional submerged cell culture systems, have the potential limitation that testing of substances presenting little or no water solubility could be a major challenge. The same is true for compounds that are instable in water. In order to overcome these potential problems, some reports have suggested the use of RHE (Saito et al., 2013; Teunis et al., 2013). Using 16 reference chemicals, the expression profile of five genes was in complete agreement with the results from LLNA (Saito et al., 2013). In the future, an approach based on RHS co-cultured with dendritic cells may allow development of an alternative strategy to address this endpoint.
As with sensitization, a complex cascade of events also underlies genotoxicity. The assessment of the genotoxic potential of compounds is primarily based on a battery of bacterial and mammalian in vitro cell assays allowing the detection of mutagenicity at the gene level, structural chromosome damage (clastogenicity) or numerical chromosome aberration (aneugenicity). However, the specificity of the mammalian cell tests is limited and irrelevant positive results are reported. There is strong evidence that the low predictivity is caused by the use of cell lines of less relevance with regard to metabolic capacity, p53 expression and/or DNA repair. Based on an appropriate cytotoxicity measurement the highest applied dose can be adjusted and thereby the experimental outcome of a study is improved (Fowler et al., 2012a,b). For substances in contact with the skin surface, cutaneous uptake and biotransformation are once more of pivotal relevance and RHE/RHS seems to be a suitable test matrix as species- and organ-specific metabolism, normal cell cycle control and DNA repair capacity can be assumed.
Two in vitro genotoxicity approaches using 3D skin models have been proposed. The human reconstructed skin micronucleus (RSMN) assay combines the EpiDerm® skin model with the established endpoint of micronuclei analysis (Curren et al., 2006) and allows identification of direct-acting mutagens as well as pro-mutagens (Aardema et al., 2013). The method has been transferred to other laboratories (Aardema et al., 2010) and a global validation study is underway. The 3D Skin Comet assay detects complementary DNA damage, including damage that arises from gene mutations. The initial approach in RHE used EpiDerm® (Reus et al., 2013), moreover, EpiDerm FT and Phenion® FT were studied. Based on results generated by five independent laboratories (Pfuhler et al., 2010), the Phenion® FT and EpiDerm™ FT were selected among the three tissues to enter the following validation phase, in which a total of 30 chemicals will be investigated. Once validated, the RSMN and the 3D Skin Comet assay may be used as follow-ups for positive results from the current in vitro genotoxicity test battery in a weight-of-evidence approach.
5.9 Recombinant human skin models (RHS) for drug development
Other than with hazard analysis, the use of RHS for the evaluation of beneficial and adverse drug effects is in its infancy. Glucocorticoid induced anti-inflammatory effects (decline of interleukin-6 and interleukin-8 secretion, histological analysis) were seen following the induction of inflammation by UVB irradiation or TNFα exposure. Unwanted effects were observable by a decline in collagen amounts (Weindl et al., 2011; Zöller et al., 2008), which reflects the most relevant adverse effect of long-term topical glucocorticoid therapy, i.e., skin atrophy.
Various disease models have been developed based on the techniques for the generation of normal RHS. These include barrier-deficient human skin, which becomes possible by knock-down of the filaggrin gene (Küchler et al., 2011; Mildner et al., 2010), which is often defective in patients suffering from atopic dermatitis, ichthyosis vulgaris and – possibly also – psoriasis (Brown et al., 2012). Other than with normal RHS, the order of stratum corneum lipids in the knock-down models does not improve with an extended culture period. Whereas filaggrin deficiency thus results in major disorder of the lipid arrangement, skin surface pH is normal due to compensatory enhanced phospholipase activity and activation of a proton transporter (Vávrová et al., in press). The disturbed barrier induced by filaggrin silencing enhances permeability of the construct for the lipophilic marker substance testosterone and the resilience against the irritant sodium dodecylsulfate declines (Küchler et al., 2011). This is in accordance with the enhanced transepidermal water loss in atopic patients presenting filaggrin abnormalities (Winge et al., 2011). Moreover, the model of peeling skin disease is also based on gene silencing, i.e., of transglutaminase (Oji et al., 2010).
In vitro skin tumor models include non-melanoma skin cancer (NMSC) and melanoma. For reconstruction, normal keratinocytes and a transformed cell line are seeded on a dermal equivalent. The protocol (time of addition of tumor cell line, ratio of normal and transformed cells) determines the tissue architecture. The models can be used for investigation of the impact of the interaction between tumor cells and their microenvironment on cancer progression (Carlson et al., 2008). IL-4 stimulation induced degradation of the basal membrane and enhanced collagenolysis favored tumor cell invasion (Linde et al., 2012). Non-clinical studies of anticancer drugs is also possible (Ali-von Laue et al., in press; Hoeller Obrigkeit et al., 2009). Other coculture based disease models include candidiasis of epidermal, oral and vaginal epithelia which unraveled TLR-4 mediated defense mechanisms induced by leukocyte co-cultures (Weindl et al., 2007; Schaller et al., 2006) and allowed demonstration of the efficacy of antifungal treatment (Schaller et al., 2000).
3D wound models can be generated from normal RHS, inducing defined wounding, e.g., by incision of a scalpel (Carlson et al., 2008) or CO2 laser irradiation (Küchler et al., 2010). The constructs allow the histological study of key response parameters of wounded epithelium, such as cell proliferation, migration differentiation and growth factor responses.
5.10 Shortcomings of 3D skin models and future challenges
Several studies have shown that RHE is able to reproduce the differentiation process and exhibit all the different layers of a normal epidermis. The expression of differentiation markers (filaggrin, transglutaminase, etc.) in the lower epidermis reflects a hyperproliferative state of the constructs close to that seen in healing processes (Smiley et al., 2005) and may be overcome by avoiding the embedding of fibroblasts into a collagen matrix (Stark et al., 2006). Because of much lower barrier properties of current RHE/RHS, skin permeation is overestimated when compared to the flux across human skin. This weakness of the barrier function impacts other toxicological endpoints by increasing the access of topically applied xenobiotics to viable skin. Although the predictive model of the validated protocol for skin absorption (Schäfer-Korting et al., 2008a) may corrects this, it would be useful to have a more physiological construct. Another limit of RHE and RHS is that they do not desquamate. Increasing the culture period causes a thickening of the stratum corneum and a modification of its penetrability. The lack of desquamation excludes the use of the constructs for the assessment of toxicity induced by repetitive application of test agents.
Nevertheless, thanks to their ability to mimic skin penetration and biotransformation of chemicals RHE/RHS is essential to the future of toxicology. However, the road is still long to achieve the ultimate goal of complete replacement of animals in toxicology. Different ways can be explored to improve the capacity of these models to predict some toxicological endpoints and to facilitate their regulatory recognition.
5.11 Improvement of regulatory implementation
Integrated testing strategy (ITS)
The one-for-one replacement vision of alternative methods (one in vitro test method to replace one animal study) is only possible for a limited number of toxicity endpoints. The way forward is in integrated testing strategies, in particular when addressing complex toxicological endpoints. ITS are stepwise processes which offer a framework for analyzing in a weight of evidence approach all the existent data and the new data generated in the context of the ITS. This given, ITS will allow to evaluate the risk of a chemical for human health.
The REACH system encourages registrants of substances to develop and implement ITS that make use of information from pre-existing classical in vivo toxicological experimentation, information from structurally-related substances, and data from in vitro methodologies and non-testing methods (including computational techniques), thereby minimizing the need for the generation of new toxicological data in vivo. Results from an in vitro test method may fully or partly replace an in vivo test in an ITS if the assay has been validated according to internationally agreed principles.
Replacement of cell viability by other biological endpoints
This point can benefit from the adverse outcome pathway (AOP) strategy as described in Toxicity Testing in the 21st Century: a Vision and a Strategy (Krewski et al., 2010). AOPs are cellular response pathways that, when sufficiently perturbed, are expected to result in adverse health effects. This approach is based on the rapidly evolving understanding of how genes, proteins and small molecules interact to form molecular pathways that maintain cell function. The goal is to determine how some chemicals perturb these pathways causing a cascade of subsequent key events leading to a toxic response. Mapping AOPs will permit to identify some key biomarkers specific of their activation. Considering the advantages discussed above, RHE/RHS should be powerful tools to study these pathways of toxicology and to assess toxic effects on a molecular level. This given, RHE/RHS must be comprehensively characterized (proteomics, transcriptomics, metabolomics …) to improve their predictability.
Combining skin models with other organoids
Combining the advantages of RHE/RHS with the high potential of microfluidic technologies (organs-on-a-chip; Marx et al., 2012) offers the chance to build up donor pools with defined genetics.
For 3D constructs, keratinocytes and fibroblasts are most often isolated from foreskin. To build healthy constructs, interfering (skin) diseases of the donors must be excluded, as they affect tissue features. For lab safety, samples containing HIV, hepatitis B and C, and cytomegalovirus must be excluded. Cell pooling of at least three donors is a way to reduce donor related variability in routine testing, but single donor constructs are needed for more specific purposes. Co-culture with malignant cells raises the question whether to use cell lines or patient biopsies – at the moment there is a tendency to use well-defined cell lines. When integrating immune cells, the effect of activation caused by primary cells is not yet elucidated. However, the more cell types are integrated into a construct the more complex the timing, medium composition and histology, and further characterization may be another important step forward. The system consists of microchannels that reproduce a circulation system between several organ constructs. Microfluidics provide a very fine control of nutrition, waste disposal and establishment of concentration gradients of cytokines or other biochemical substances which may be even closer to the situation in vivo compared to 3D constructs cultivated in transwells.
6 Other 3D models, such as intestine and tumor tissues
Models (3D organoids) for several further organs and tissues have been developed. These include, for instance, lymphoid tissue to address immunological niches, or they involve the coupling of several organoids in multi-organoid systems. Only two examples are briefly described below to give a perspective on this wide field (Atac et al., 2013; Materne et al., 2013; Schimek et al., 2013; Wagner et al., 2013).
6.1 3D models of the intestine
Besides the lung-on-a-chip mentioned above, the same approach has been used to develop a human gut-on-a-chip that features peristalsis and trickling flow. This system uses a variation of the lung chip platform technology to develop a gut chip that is lined by human intestinal epithelial cells and cultured under flow conditions to produce physiological shear stress (~0.02 dyne/cm2) while simultaneously exerting cyclic mechanical strain (10% elongation, 0.15 Hz). Human CaCO2 cells cultured under these conditions differentiate by changing their entire transcriptome (measured using gene microarrays) and form 3D villus structures that match the height of the microfluidic interstitial channel (Kim et al., 2012). The physiologically-relevant conditions recreated in the gut-on-a-chip allow the culture of living gut bacteria (Lactobacillus) directly on top of the living human gut epithelium and hence open the possibility to investigate the influence of the gut microbiome on drug and nutrient absorption and metabolism.
6.2 In vitro multicellular tumor spheroids (MCTS)
MCTS have been used intensively in the academic world for several decades. The first publications demonstrating biological differences of tumor cells in 2D and 3D go back to the early 70s: Folkman and Hochberg described differences in proliferation of tumor cells in monolayer versus spheroids (Folkman and Hochberg, 1973). Later, highly increased drug resistance was observed if cells were cultured as MCTS rather than as monolayers (Sutherland et al., 1979). Even though the biological similarities of tumor spheroids and native tumors have been described over many years, this model has not yet found its way into routine drug discovery processes. However, recently the spheroid model is attracting increased attention as novel cell culture technologies allow its implementation in automated workflows, thus enabling its use in drug candidate screening.
Proliferating cells, such as those from tumors, show higher expression and activity of multidrug resistance transporters (e.g., MRP2). This was recently reported in HepG2 spheroids which showed highest EC50 values for tamoxifen when compared with collagen sandwich and monolayer cultures of HepG2 cells. The monolayer cultures showed no MRP2 activity (Mueller et al., 2011a). The study also showed that the spheroids of the proliferating HepG2 cells were increasing in size but the diameters decreased after reaching a critical size.
Growth profiling
In 1977 Yuhas and coworkers had already compared the growth profile of three mouse cell lines in monolayer versus spheroid cultures. MCTS grew at a rate that was proportional to the in vivo growth rate, while the growth rate of monolayers did not correlate. Yuhas concluded that the spheroid allows for greater expression of inherent growth characteristics and depends less on the particulars of the media used. Furthermore the growth profile of the spheroids depends not only on cellular doubling times but also on the size of the microtissue (Yuhas et al., 1977).
Diffusion resistance
The efficacy and toxicity of a molecule depends strongly on its tissue penetration capabilities (Minchinton and Tannock, 2006). In contrast to cells in a monolayer format, cells in a 3-dimensional spheroid format develop cell-cell contacts and secrete high amounts of extracellular matrix (Kelm et al., 2012). Consequently, this leads to increased diffusion barriers that block the penetration of drug molecules and biological agents. Similar diffusion barriers have to be overcome in vivo, whereas in monolayer cultures all cells are exposed in the same way to the test substance. Therefore, a concentration profile in a spheroid model more closely reflects the in vivo situation than a homogenously exposed cell population. This is of particular concern to companies developing large molecules such as antibodies, as targeted antibodies could become stuck within the first cell layers depending on their physical-chemical properties and the amount of target expressed on the cells.
Cancer cell metabolism
The aforementioned diffusion resistance in tumor spheroids also results in oxygen and metabolite gradients. In cancer, it is assumed that glycolysis is the main pathway to generate energy, especially in tumors with hypoxic regions. This physiological phenomenon is reflected in tumor spheroids. Depending on the size and metabolic activity of the cancer cells, hypoxia starts to develop at a diameter of 400 μm. Rodríguez-Enríquez and coworkers (2008) evaluated the differences in glycolysis in monolayer versus spheroid cultures and measured a two- to four-fold increased glycolytic flux in the 3D culture providing 80% of total ATP consumption. Throughout the spheroid, different metabolic regions can be distinguished, e.g., lactate concentration increases towards the core of the spheroid, which leads to acidification of the extracellular environment, adding an additional pH gradient within the spheroid.
Cell population
The spheroid format leads to heterogeneous cell populations within the tissue due to glucose and oxygen gradients, which are critical in the development of non-proliferating and non-clonogenic cell subpopulations (Freyer and Sutherland, 1986). In the outer layers, the cells are proliferating; in the underlying regions, the cells are non-proliferative; and in the core of the tissue, cells become necrotic (layering strongly depends on spheroid size). Moreover, tumors do not only consist of one cell type: Tumor spheroids allow the implementation of stromal components such as fibroblasts and/or endothelial cells (Kelm et al., 2006).
The 3D format can alter drug sensitivity significantly, as shown for vinblastine: Multicellular drug resistance is attributed to the contacts between cells or between cells and the extracellular matrix. The mechanisms underlying the multicellular drug resistance are still being evaluated. However, it is thought that hypoxia, non-proliferative cell populations, cell-cell contacts and extracellular matrix environment impact drug sensitivity. The spheroid is the ideal model to study this kind of resistance since it so closely recapitulates patient tumor resistance (Desoize et al., 1998). Importantly, however, the 3D format can also lead to increased drug sensitivity, for example for compounds which are only active in a low oxygen/hypoxic environment or whose targets are not expressed in 2D. In the early 1970s, Durand and co-workers described spheroids composed of Chinese hamster V79-171b cells, containing about 10–25 cells, which were more resistant to killing by ionizing radiation than their 2D counterparts. Subsequent studies revealed that spheroids showed fewer radiation-induced mutations than monolayers exposed to the same dose, implicating differences in cellular capacity to sustain and repair damage (Olive and Durand, 1994).
Data analysis
Finally, the spheroid approach allows for histological analysis, which is not possible with monolayer cultures. This enables direct morphological comparison of the in vitro model with preclinical and clinical pathological assessment.
6.3 Benefits and uses of MCTS
Recently, Fayad and co-workers (2011) demonstrated the use of a tumor spheroid model to prioritize the hits of a screening campaign to identify apoptosis-inducing compounds. They started with a standard 10,000 compound screen, which resulted in about 400 hits. These hits were further assessed on a 3D model of the same cell type. Using the same concentration as in the monolayer screen, the number of hits was reduced by a factor of 10. Further decreasing the concentration in a third screening round, the number of hits was reduced to 11. Interestingly, if only the 100 most potent 2D hits had been investigated, 10 of the 11 final 3D hits would have been lost.
6.4 Issues/disadvantages of MCTS
Current approaches to produce spheroids are highly manual, low-throughput processes, which are not industry-compatible. However, new technologies are emerging that address robust automation-compatible production processes, allowing reproducible production of tumor spheroids. The spheroid approach to coax cells into the third dimension does not require the addition of foreign material, but even so biochemical assays may require adaptation. Tissue lysis is more complex due to high quantities of extracellular matrix and high content analysis becomes more challenging as the z-dimension is greatly increased and the signal from fluorescent dyes can only be detected to a certain depth in the tissue. Thus, more complex analytics, such as high content analysis, have to be refined, since the added z-dimension to the culture leads to lower resolution on a single cell level. Novel approaches based on in vivo imaging technologies are currently being developed to address this issue.
In general, the field of 3D modeling is lacking consistent publicly available data sets, from in vitro to in vivo evaluations, demonstrating that by introducing a 3D-based screen the number of false-positives but also false-negatives can be reduced, for example by performing a retrospective screen. To move the field forward, and increase the likelihood of regulatory acceptance, this kind of data set would be required not only to highlight the biological similarities to in vivo tumors but also to verify the added value of a more complex tumor model over simple cell culture in the drug discovery process.
7 Overall discussion
7.1 Benefits and uses of on-a-chip models
Use of this microengineering approach allows one to develop the simplest model possible that retains physiological relevance, and then to add complexity to the system as necessary, which is not possible in animal models. The in vitro organ chip platform is being applied to model diseases in other organs and to predict different drug efficacies and toxicities in humans. It is also being leveraged to engineer a “human body-on-a-chip” with the goal of recapitulating multi-organ physiology by linking multiple organ chips through microfluidic channels. Human organ-on-chip disease models could provide a new approach to enhance our fundamental understanding of complex disease processes and to enable more rapid, accurate, cost-effective and clinically relevant testing of drugs, as well as cosmetics, chemicals and environmental toxins. In addition, it might be possible one day to inhabit these devices with cells obtained from different human genetic populations (e.g., HLA subgroups or people who express particular molecular transporters or metabolic enzymes) so that drugs can be developed for specific human subpopulations and, eventually, so that virtual human clinical trials may be carried out using these microengineered models. In this manner, on-chip human organ models have the potential to accelerate the translation of basic discoveries into effective new treatment strategies (Huh et al., 2011, 2012b).
7.2 Issues/disadvantages of 3D organ-on-a-chip models
3D organ-on-a-chip models represent a great improvement over planar 2D models in recapitulating organ-level functionality. Reconstituting the appropriate tissue microarchitecture, complex biochemical milieu and dynamic mechanical microenvironment (e.g., cyclic mechanical strain, fluid shear) have been key in these improvements (Huh et al., 2011, 2012b). As discussed previously, a key challenge will be identifying the optimal source of human cells that best mimic in vivo responses. Recent organ-on-a-chip studies suggest that established human cell lines can be used to model complex lung functions (e.g., inflammatory response) in vitro and that these cells exhibit more highly differentiated phenotypes when presented with physiologically relevant microenvironmental cues than when maintained under conventional 2D or 3D culture conditions (Li et al., 2012). Inducible pluripotent stem (iPS) cells or embryonic stem (ES) cells may provide a solution, however, additional development is required in order to drive these cells from a neonate-like state to the more mature adult phenotype required for use in organ-on-a-chip microsystems.
While organ-on-a-chip systems can be designed and developed by leveraging engineering principles to provide the required function/physiology, several issues will need to be addressed to allow for broader utility. These include overcoming technical issues and complexity from a manufacturing perspective, implementation and ease of use in the laboratory, automation, as well as developing intuitive and easy user interfaces. Technical challenges include integrating these organ chips with sensors that can detect and measure optical, chemical, electrical and mechanical signals from the cells, development of automated flow and pressure control within the microfluidic devices and multiplexing these microsystems for higher-throughput analysis and screening. Also, in order to move towards large scale commercial manufacturing, novel materials will need to be developed that are more amenable to mass production. Polydimethylsiloxane (PDMS) is most commonly used in microfluidic systems because of its optical transparency, flexibility, biocompatibility and fabrication by casting. However, small hydrophobic molecules can partition into the bulk of PDMS, which limits its utility for drug development applications. Recently, alternative materials that are resistant to absorption of small hydrophobic molecules but are similar to PDMS in terms of optical transparency, flexibility and castability, have been identified (Domansky et al., 2013).
Future in silico modeling paradigms will need to be developed to utilize endpoints generated within these systems and to predict human translation. This will also entail the development of appropriate in vitro biomarkers and assays so that results can be modeled and extrapolated for human translation.
7.3 Quality control
When developing organotypic models, quality control can be thought of in terms of model predictivity (i.e., how well does it reflect the in vivo situation), as well as technical validity (i.e., standardization and reproducibility of the model as a whole) (Leist et al., 2010). The model should adequately and stably reflect the physical and functional parameters of the organ in question. Use of 3D modules seems only justified if data can be provided that show an improvement over existing 2D models for the same organ system or systems.
During initial stages of developing a test using an organotypic model, requirements for validation and fitness for purpose are low, but once the test progresses to a stage at which data are related to regulatory decisions, standardization, reproducibility, transferability, predictive relevance, applicability domain, performance and fitness for purpose become more formalized and important. Due to the mass transport difficulties discussed above (due to increased complexity) and due to limited throughput (due to costs, complexity and training, or all of the above), validation is challenging. This is worth the effort if applicability (due to improved reflection of the in vivo situation), predictivity and relevance are clearly demonstrated.
8 Recommendations to improve the quality of organotypic models and increase utility for toxicological risk assessment and regulatory acceptance
Organotypic models have great potential for utility in toxicological investigations and regulatory risk assessment. Perhaps the greatest difficulty for organotypic systems is also their greatest advantage: organotypic systems are created to mimic the in vivo situation as closely as possible. The complexity of the system reduces the chance that key aspects of biology are missing, but increases the difficulties for reproducibility, quality control and interpretation of results. Therefore it is necessary to create, choose and use models which are as simple as possible, yet as complex as necessary, to address a certain question (Hartung, 2007; Hartung et al., 2004, 2013; Leist et al., 2010, 2012a). To that end, a call for data-sharing (Breeze et al., 2012; Hanson et al., 2011; Murray-Rust et al., 2010) and incorporation of bioinformatics approaches and other new and useful technologies for data mining and interpretation (Blaauboer et al., 2012; Leist et al., 2012b) is deemed vital to the future of organotypic models in toxicological investigations. The participants in the 3D modelling symposium included these and a number of other recommendations as important for improving the quality and utility of organotypic models and increasing the probability of regulatory acceptance as a replacement for in vivo testing. The short form of the recommendations is summarized in Box 1.
Box 1. Recommendations for improving the quality and utility of organotypic models, and increasing the probability of regulatory acceptance as a replacement for in vivo testing.
Design the model “fit for purpose”, i.e., to be applicable to the specific task at hand (as complex as necessary, as simple as possible).
Recruit robust and sustainable cell sources.
Reflect human variability (genotypes, transgene expression) in cell sources.
Utilize and justify appropriate dose ranges (drug concentrations).
Create a web portal or similar open access database for reference data on organotypic models.
Openly share detailed protocols for model production and use.
Couple funding and support for model development to adherence to minimal quality control measures.
Follow good cell culture practice (GCCP) protocols (Coeke et al., 2005), and pay special attention to spatial effects and longer-term effects.
Strive to apply non-invasive online monitoring.
Consider adding omics endpoints, high throughput capability and/or modern reporter readouts.
Combine 3D model use with in silico technology and bioinformatics.
Promote 3D models as alternatives to mouse experiments in all biomedical communities.
Bring together users and developers of 3D models for multiple purposes (drug development, toxicological investigation, regenerative medicine).
Aim high, but always be aware of current downsides and shortcomings.
9 Conclusions
The routine use of 3D models is not necessarily cheaper than animal experiments, but human-derived tissues used under defined test conditions offer several advantages. Issues of species extrapolation and ethical questions arising from the use of experimental animals are avoided. Integrated approaches using 2D and 3D in vitro methods, as well as bioinformatics, can reduce time to market and costs in drug development. They also have the potential to improve the accuracy of toxicological risk assessment in addition to increasing our understanding of disease mechanisms and drug actions. This provides the most important incentive for the further development of 3D models: the true cost of a test should be measured by the probability that a wrong decision in drug discovery or toxicology is made based on the test results – and 3D models are expected to reduce such errors.
Acknowledgments
We are grateful to many participants of the CAAT-Europe workshop and information day on 3D models for sharing their ideas and for giving input. The valuable help of many members of the groups and laboratories represented by the authors that wrote this review is acknowledged.
Footnotes
a report of t4 – the transatlantic think tank for toxicology, a collaboration of the toxicologically oriented chairs in Baltimore, Konstanz and Utrecht sponsored by the Doerenkamp-Zbinden Foundation; participants do not represent their institutions and do not necessarily endorse all recommendations made.
References
- Aardema MJ, Barnett BC, Khambatta Z, et al. International prevalidation studies of the EpiDerm 3D human reconstructed skin micronucleus (RSMN) assay: Transferability and reproducibility. Mutat Res. 2010;701:123–131. doi: 10.1016/j.mrgentox.2010.05.017. http://dx.doi.org/10.1016/j.mrgentox.2010.05.017. [DOI] [PubMed] [Google Scholar]
- Aardema MJ, Barnett BB, Mun GC, et al. Evaluation of chemicals requiring metabolic activation in the EpiDerm 3D human reconstructed skin micronucleus (RSMN) assay. Mutat Res. 2013;750:40–49. doi: 10.1016/j.mrgentox.2012.08.009. http://dx.doi.org/10.1016/j.mrgentox.2012.08.009. [DOI] [PubMed] [Google Scholar]
- Abbott A. Cell culture: Biology’s new dimension. Nature. 2003;424:870–872. doi: 10.1038/424870a. http://dx.doi.org/10.1038/424870a. [DOI] [PubMed] [Google Scholar]
- Ackermann K, Borgia SL, Korting HC, et al. The Phenion full-thickness skin model for percutaneous absorption testing. Skin Pharmacol Physiol. 2010;23:105–112. doi: 10.1159/000265681. http://dx.doi.org/10.1159/000265681. [DOI] [PubMed] [Google Scholar]
- Ahlenius H, Kokaia Z. Isolation and generation of neurosphere cultures from embryonic and adult mouse brain. Methods Mol Biol. 2010;633:241–252. doi: 10.1007/978-1-59745-019-5_18. http://dx.doi.org/10.1007/978-1-59745-019-5_18. [DOI] [PubMed] [Google Scholar]
- Aird WC. Endothelial cell heterogeneity. Cold Spring Harb Perspect Med. 2012;2:a006429. doi: 10.1101/cshperspect.a006429. http://dx.doi.org/10.1101/cshperspect.a006429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Ahmad A, Taboada CB, Gassmann M, et al. Astrocytes and pericytes differentially modulate blood-brain barrier characteristics during development and hypoxic insult. J Cereb Blood Flow Metab. 2011;31:693–705. doi: 10.1038/jcbfm.2010.148. http://dx.doi.org/10.1038/jcbfm.2010.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfaro-Moreno E, Nawrot TS, Vanaudenaerde BM, et al. Co-cultures of multiple cell types mimic pulmonary cell communication in response to urban PM10. Eur Respir J. 2008;32:1184–1194. doi: 10.1183/09031936.00044008. http://dx.doi.org/10.1183/09031936.00044008. [DOI] [PubMed] [Google Scholar]
- Ali-von Laue C, Zoschke C, Do N, et al. Improving topical non-melanoma skin cancer treatment – in vitro efficacy of a novel guanosine phosphonate analogue. Skin Pharmacol Physiol. doi: 10.1159/000354118. in press. [DOI] [PubMed] [Google Scholar]
- Andersson TB, Kanebratt KP, Kenna JG. The HepaRG cell line: A unique in vitro tool for understanding drug metabolism and toxicology in human. Expert Opin Drug Metab Toxicol. 2012;8:909–920. doi: 10.1517/17425255.2012.685159. http://dx.doi.org/10.1517/17425255.2012.685159. [DOI] [PubMed] [Google Scholar]
- Atac B, Wagner I, Horland R, et al. Skin and hair on-a-chip: In vitro skin models versus ex vivo tissue maintenance with dynamic perfusion. Lab Chip. 2013;13:3555–3561. doi: 10.1039/c3lc50227a. http://dx.doi.org/10.1039/c3lc50227a. [DOI] [PubMed] [Google Scholar]
- Auxenfans C, Fradette J, Lequeux C, et al. Evolution of three dimensional skin equivalent models reconstructed in vitro by tissue engineering. Eur J Dermatol. 2009;19:107–113. doi: 10.1684/ejd.2008.0573. [DOI] [PubMed] [Google Scholar]
- Bailey J, Thew M, Balls M. An analysis of the use of dogs in predicting human toxicology and drug safety. Altern Lab Anim. 2013;41:335–350. doi: 10.1177/026119291304100504. [DOI] [PubMed] [Google Scholar]
- Bal-Price AK, Hogberg HT, Buzanska L, et al. Relevance of in vitro neurotoxicity testing for regulatory requirements: Challenges to be considered. Neurotoxicol Teratol. 2010a;32:36–41. doi: 10.1016/j.ntt.2008.12.003. http://dx.doi.org/10.1016/j.ntt.2008.12.003. [DOI] [PubMed] [Google Scholar]
- Bal-Price AK, Hogberg HT, Buzanska L, et al. In vitro developmental neurotoxicity (DNT) testing: Relevant models and endpoints. Neurotoxicology. 2010b;31:545–554. doi: 10.1016/j.neuro.2009.11.006. http://dx.doi.org/10.1016/j.neuro.2009.11.006. [DOI] [PubMed] [Google Scholar]
- Bal-Price AK, Coecke S, Costa L, et al. Advancing the science of developmental neurotoxicity (DNT): Testing for better safety evaluation. ALTEX. 2012;29:202–215. doi: 10.14573/altex.2012.2.202. http://dx.doi.org/10.14573/altex.2012.2.202. [DOI] [PubMed] [Google Scholar]
- Balmer NV, Weng MK, Zimmer B, et al. Epigenetic changes and disturbed neural development in a human embryonic stem cell-based model relating to the fetal valproate syndrome. Hum Mol Genet. 2012;21:4104–4114. doi: 10.1093/hmg/dds239. http://dx.doi.org/10.1093/hmg/dds239. [DOI] [PubMed] [Google Scholar]
- Balmer NV, Leist M. Epigenetics and transcriptomics to detect adverse drug effects in model systems of human development. Basic Clin Pharmacol Toxicol. 2014;115:59–68. doi: 10.1111/bcpt.12203. http://dx.doi.org/10.1111/bcpt.12203. [DOI] [PubMed] [Google Scholar]
- Bätz FM, Klipper W, Korting HC, et al. Esterase activity in excised and reconstructed human skin – biotransformation of prednicarbate and the model dye fluorescein diacetate. Eur J Pharm Biopharm. 2013;84:374–385. doi: 10.1016/j.ejpb.2012.11.008. http://dx.doi.org/10.1016/j.ejpb.2012.11.008. [DOI] [PubMed] [Google Scholar]
- Bauch C, Kolle SN, Ramirez T, et al. Putting the parts together: Combining in vitro methods to test for skin sensitizing potentials. Regul Toxicol Pharmacol. 2012;63:489–504. doi: 10.1016/j.yrtph.2012.05.013. http://dx.doi.org/10.1016/j.yrtph.2012.05.013. [DOI] [PubMed] [Google Scholar]
- Baumann J, Barenys M, Gassmann K, et al. Comparative human and rat ‘Neurosphere Assay’ for developmental neurotoxicity testing. Current Protocols in Toxicology. 2014;59:12.21.11–12.21.24. doi: 10.1002/0471140856.tx1221s59. http://dx.doi.org/10.1002/0471140856.tx1221s59. [DOI] [PubMed] [Google Scholar]
- Bechetoille N, Vachon H, Gaydon A, et al. A new organotypic model containing dermal-type macrophages. Exp Dermatol. 2011;20:1035–1037. doi: 10.1111/j.1600-0625.2011.01383.x. http://dx.doi.org/10.1111/j.1600-0625.2011.01383.x. [DOI] [PubMed] [Google Scholar]
- Bermudez LE, Sangari FJ, Kolonoski P, et al. The efficiency of the translocation of Mycobacterium tuberculosis across a bilayer of epithelial and endothelial cells as a model of the alveolar wall is a consequence of transport within mononuclear phagocytes and invasion of alveolar epithelial cells. Infect Immun. 2002;70:140–146. doi: 10.1128/IAI.70.1.140-146.2002. http://dx.doi.org/10.1128/IAI.70.1.140-146.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birgersdotter A, Sandberg R, Ernberg I. Gene expression perturbation in vitro – a growing case for three-dimensional (3D) culture systems. Semin Cancer Biol. 2005;15:405–412. doi: 10.1016/j.semcancer.2005.06.009. http://dx.doi.org/10.1016/j.semcancer.2005.06.009. [DOI] [PubMed] [Google Scholar]
- Birkness KA, Swisher BL, White EH, et al. A tissue culture bilayer model to study the passage of Neisseria meningitidis. Infect Immun. 1995;63:402–409. doi: 10.1128/iai.63.2.402-409.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaauboer BJ, Boekelheide K, Clewell HJ, et al. The use of biomarkers of toxicity for integrating in vitro hazard estimates into risk assessment for humans. ALTEX. 2012;29:411–425. doi: 10.14573/altex.2012.4.411. http://dx.doi.org/10.14573/altex.2012.4.411. [DOI] [PubMed] [Google Scholar]
- Blank F, Rothen-Rutishauser B, Gehr P. Dendritic cells and macrophages form a transepithelial network against foreign particulate antigens. Am J Respir Cell Mol Biol. 2007;36:669–677. doi: 10.1165/rcmb.2006-0234OC. http://dx.doi.org/10.1165/rcmb.2006-0234OC. [DOI] [PubMed] [Google Scholar]
- Boillee S, Vande Velde C, Cleveland DW. ALS: A disease of motor neurons and their nonneuronal neighbors. Neuron. 2006a;52:39–59. doi: 10.1016/j.neuron.2006.09.018. http://dx.doi.org/10.1016/j.neuron.2006.09.018. [DOI] [PubMed] [Google Scholar]
- Boillee S, Yamanaka K, Lobsiger CS, et al. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006b;312:1389–1392. doi: 10.1126/science.1123511. http://dx.doi.org/10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- Bokhari M, Carnachan RJ, Cameron NR, et al. Culture of HepG2 liver cells on three dimensional polystyrene scaffolds enhances cell structure and function during toxicological challenge. J Anat. 2007;211:567–576. doi: 10.1111/j.1469-7580.2007.00778.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brain JD. Lung macrophages: How many kinds are there? What do they do? Am Rev Respir Dis. 1988;137:507–509. doi: 10.1164/ajrccm/137.3.507. http://dx.doi.org/10.1164/ajrccm/137.3.507. [DOI] [PubMed] [Google Scholar]
- Brandon EF, Raap CD, Meijerman I, et al. An update on in vitro test methods in human hepatic drug biotransformation research: Pros and cons. Toxicol Appl Pharmacol. 2003;189:233–246. doi: 10.1016/s0041-008x(03)00128-5. http://dx.doi.org/10.1016/S0041-008X(03)00128-5. [DOI] [PubMed] [Google Scholar]
- Brannvall K, Bergman K, Wallenquist U, et al. Enhanced neuronal differentiation in a three-dimensional collagen-hyaluronan matrix. J Neurosci Res. 2007;85:2138–2146. doi: 10.1002/jnr.21358. http://dx.doi.org/10.1002/jnr.21358. [DOI] [PubMed] [Google Scholar]
- Breeze JL, Poline JB, Kennedy DN. Data sharing and publishing in the field of neuroimaging. Gigascience. 2012;1:9. doi: 10.1186/2047-217X-1-9. http://dx.doi.org/10.1186/2047-217X-1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkmann J, Stolpmann K, Trappe S, et al. Metabolically competent human skin models: Activation and genotoxicity of benzo[a]pyrene. Toxicol Sci. 2012;131:351–359. doi: 10.1093/toxsci/kfs316. http://dx.doi.org/10.1093/toxsci/kfs316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brophy CM, Luebke-Wheeler JL, Amiot BP, et al. Rat hepatocyte spheroids formed by rocked technique maintain differentiated hepatocyte gene expression and function. Hepatology. 2009;49:578–586. doi: 10.1002/hep.22674. http://dx.doi.org/10.1002/hep.22674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown SJ, Kroboth K, Sandilands A, et al. Intragenic copy number variation within filaggrin contributes to the risk of atopic dermatitis with a dose-dependent effect. J Invest Dermatol. 2012;132:98–104. doi: 10.1038/jid.2011.342. http://dx.doi.org/10.1038/jid.2011.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkard A, Dahn C, Heinz S, et al. Generation of proliferating human hepatocytes using Upcyte(R) technology: Characterisation and applications in induction and cytotoxicity assays. Xenobiotica. 2012;42:939–956. doi: 10.3109/00498254.2012.675093. http://dx.doi.org/10.3109/00498254.2012.675093. [DOI] [PubMed] [Google Scholar]
- Butler P, Fox K, Shaw J. Methods for improving in vitro prediction of intrinsic clearance utilising 3D cell culture models (by Cyprotex using InSphero technology). 10th International ISSXMeeting; Toronto, Ontario, Canada. 2013. [Google Scholar]
- Campos LS, Leone DP, Relvas JB, et al. Beta1 integrins activate a MAPK signalling pathway in neural stem cells that contributes to their maintenance. Development. 2004;131:3433–3444. doi: 10.1242/dev.01199. http://dx.doi.org/10.1242/dev.01199. [DOI] [PubMed] [Google Scholar]
- Carlson MW, Alt-Holland A, Egles C, et al. Three-dimensional tissue models of normal and diseased skin. Curr Protoc Cell Biol. 2008;Chapter 19(Unit 19.9) doi: 10.1002/0471143030.cb1909s41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438:932–936. doi: 10.1038/nature04478. http://dx.doi.org/10.1038/nature04478. [DOI] [PubMed] [Google Scholar]
- Carterson AJ, Honer zu Bentrup K, Ott CM, et al. A549 lung epithelial cells grown as three-dimensional aggregates: Alternative tissue culture model for Pseudomonas aeruginosa pathogenesis. Infect Immun. 2005;73:1129–1140. doi: 10.1128/IAI.73.2.1129-1140.2005. http://dx.doi.org/10.1128/IAI.73.2.1129-1140.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceridono M, Tellner P, Bauer D, et al. The 3T3 neutral red uptake phototoxicity test: Practical experience and implications for phototoxicity testing – the report of an ECVAM-EFPIA workshop. Regul Toxicol Pharmacol. 2012;63:480–488. doi: 10.1016/j.yrtph.2012.06.001. http://dx.doi.org/10.1016/j.yrtph.2012.06.001. [DOI] [PubMed] [Google Scholar]
- Chambers SM, Qi Y, Mica Y, et al. Combined small-molecule inhibition accelerates developmental timing and converts human pluripotent stem cells into nociceptors. Nat Biotechnol. 2012;30:715–720. doi: 10.1038/nbt.2249. http://dx.doi.org/10.1038/nbt.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiasson BJ, Tropepe V, Morshead CM, et al. Adult mammalian forebrain ependymal and subependymal cells demonstrate proliferative potential, but only subependymal cells have neural stem cell characteristics. J Neurosci. 1999;19:4462–4471. doi: 10.1523/JNEUROSCI.19-11-04462.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien S. Mechanotransduction and endothelial cell homeostasis: The wisdom of the cell. Am J Physiol Heart Circ Physiol. 2007;292:H1209–1224. doi: 10.1152/ajpheart.01047.2006. http://dx.doi.org/10.1152/ajpheart.01047.2006. [DOI] [PubMed] [Google Scholar]
- Chrobak KM, Potter DR, Tien J. Formation of perfused, functional microvascular tubes in vitro. Microvasc Res. 2006;71:185–196. doi: 10.1016/j.mvr.2006.02.005. http://dx.doi.org/10.1016/j.mvr.2006.02.005. [DOI] [PubMed] [Google Scholar]
- Coecke S, Balls M, Bowe G, et al. Guidance on good cell culture practice. a report of the second ECVAM task force on good cell culture practice. Altern Lab Anim. 2005;33:261–287. doi: 10.1177/026119290503300313. [DOI] [PubMed] [Google Scholar]
- Coecke S, Goldberg AM, Allen S, et al. Workgroup report: Incorporating in vitro alternative methods for developmental neurotoxicity into international hazard and risk assessment strategies. Environ Health Perspect. 2007;115:924–931. doi: 10.1289/ehp.9427. http://dx.doi.org/10.1289/ehp.9427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colton CK, Smith KA, Merrill EW, et al. Diffusion of organic solutes in stagnant plasma and red cell suspensions. Chemical Engineering Progress, Symposium Series. 1970;66:85–100. [Google Scholar]
- Conti L, Cattaneo E. Neural stem cell systems: Physiological players or in vitro entities? Nat Rev Neurosci. 2010;11:176–187. doi: 10.1038/nrn2761. http://dx.doi.org/10.1038/nrn2938. [DOI] [PubMed] [Google Scholar]
- Coon MJ, Ding XX, Pernecky SJ, et al. Cytochrome P450: Progress and predictions. FASEB J. 1992;6:669–673. doi: 10.1096/fasebj.6.2.1537454. [DOI] [PubMed] [Google Scholar]
- Cukierman E, Pankov R, Stevens DR, et al. Taking cell-matrix adhesions to the third dimension. Science. 2001;294:1708–1712. doi: 10.1126/science.1064829. http://dx.doi.org/10.1126/science.1064829. [DOI] [PubMed] [Google Scholar]
- Curren RD, Mun GC, Gibson DP, et al. Development of a method for assessing micronucleus induction in a 3D human skin model (EpiDerm) Mutat Res. 2006;607:192–204. doi: 10.1016/j.mrgentox.2006.04.016. http://dx.doi.org/10.1016/j.mrgentox.2006.04.016. [DOI] [PubMed] [Google Scholar]
- Darnell M, Schreiter T, Zeilinger K, et al. Cytochrome P450-dependent metabolism in HepaRG cells cultured in a dynamic three-dimensional bioreactor. Drug Metab Dispos. 2011;39:1131–1138. doi: 10.1124/dmd.110.037721. http://dx.doi.org/10.1124/dmd.110.037721. [DOI] [PubMed] [Google Scholar]
- Datta AK. Biological and Bioenvironmental Heat and Mass Transfer. CRC Press LLC; 2002. http://dx.doi.org/10.1201/9780203910184. [Google Scholar]
- Daud MF, Pawar KC, Claeyssens F, et al. An aligned 3D neuronal-glial co-culture model for peripheral nerve studies. Biomaterials. 2012;33:5901–5913. doi: 10.1016/j.biomaterials.2012.05.008. http://dx.doi.org/10.1016/j.biomaterials.2012.05.008. [DOI] [PubMed] [Google Scholar]
- Desoize B, Gimonet D, Jardiller JC. Cell culture as spheroids: An approach to multicellular resistance. Anticancer Res. 1998;18:4147–4158. [PubMed] [Google Scholar]
- Dezutter-Dambuyant C, Black A, Bechetoille N, et al. Evolutive skin reconstructions: From the dermal collagen-glycosaminoglycan-chitosane substrate to an immunocompetent reconstructed skin. Biomed Mater Eng. 2006;16:S85–94. [PubMed] [Google Scholar]
- Di L, Trapa P, Obach RS, et al. A novel relay method for determining low-clearance values. Drug Metab Dispos. 2012;40:1860–1865. doi: 10.1124/dmd.112.046425. http://dx.doi.org/10.1124/dmd.112.046425. [DOI] [PubMed] [Google Scholar]
- Domansky K, Inman W, Serdy J, et al. Perfused multiwell plate for 3D liver tissue engineering. Lab Chip. 2010;10:51–58. doi: 10.1039/b913221j. http://dx.doi.org/10.1039/b913221j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domansky K, Leslie DC, McKinney J, et al. Clear castable polyurethane elastomer for fabrication of microfluidic devices. Lab Chip. 2013;13:3956–3964. doi: 10.1039/c3lc50558h. http://dx.doi.org/10.1039/c3lc50558h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan D, Brown NJ, Bishop ET, et al. Comparison of three in vitro human ‘angiogenesis’ assays with capillaries formed in vivo. Angiogenesis. 2001;4:113–121. doi: 10.1023/a:1012218401036. http://dx.doi.org/10.1023/A:1012218401036. [DOI] [PubMed] [Google Scholar]
- Dubois-Dauphin ML, Toni N, Julien SD, et al. The long-term survival of in vitro engineered nervous tissue derived from the specific neural differentiation of mouse embryonic stem cells. Biomaterials. 2010;31:7032–7042. doi: 10.1016/j.biomaterials.2010.06.017. http://dx.doi.org/10.1016/j.biomaterials.2010.06.017. [DOI] [PubMed] [Google Scholar]
- Dudek SM, Garcia JG. Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol (1985) 2001;91:1487–1500. doi: 10.1152/jappl.2001.91.4.1487. [DOI] [PubMed] [Google Scholar]
- Dunn JC, Yarmush ML, Koebe HG, et al. Hepatocyte function and extracellular matrix geometry: Long-term culture in a sandwich configuration. FASEB J. 1989;3:174–177. doi: 10.1096/fasebj.3.2.2914628. [DOI] [PubMed] [Google Scholar]
- Dunn JC, Tompkins RG, Yarmush ML. Long-term in vitro function of adult hepatocytes in a collagen sandwich configuration. Biotechnol Prog. 1991;7:237–245. doi: 10.1021/bp00009a007. http://dx.doi.org/10.1021/bp00009a007. [DOI] [PubMed] [Google Scholar]
- Dunsmore SE, Rannels DE. Extracellular matrix biology in the lung. Am J Physiol. 1996;270:L3–27. doi: 10.1152/ajplung.1996.270.1.L3. [DOI] [PubMed] [Google Scholar]
- Eskes C, Honegger P, Juillerat-Jeanneret L, et al. Microglial reaction induced by noncytotoxic methylmercury treatment leads to neuroprotection via interactions with astrocytes and IL-6 release. Glia. 2002;37:43–52. doi: 10.1002/glia.10019. http://dx.doi.org/10.1002/glia.10019. [DOI] [PubMed] [Google Scholar]
- Eskes C, Juillerat-Jeanneret L, Leuba G, et al. Involvement of microglia-neuron interactions in the tumor necrosis factor-alpha release, microglial activation, and neurodegeneration induced by trimethyltin. J Neurosci Res. 2003;71:583–590. doi: 10.1002/jnr.10508. http://dx.doi.org/10.1002/jnr.10508. [DOI] [PubMed] [Google Scholar]
- Falsig J, Latta M, Leist M. Defined inflammatory states in astrocyte cultures: Correlation with susceptibility towards CD95-driven apoptosis. J Neurochem. 2004;88:181–193. doi: 10.1111/j.1471-4159.2004.02144.x. http://dx.doi.org/10.1111/j.1471-4159.2004.02144.x. [DOI] [PubMed] [Google Scholar]
- Falsig J, van Beek J, Hermann C, et al. Molecular basis for detection of invading pathogens in the brain. J Neurosci Res. 2008;86:1434–1447. doi: 10.1002/jnr.21590. http://dx.doi.org/10.1002/jnr.21590. [DOI] [PubMed] [Google Scholar]
- Falsig J, Sonati T, Herrmann US, et al. Prion pathogenesis is faithfully reproduced in cerebellar organotypic slice cultures. PLoS Pathog. 2012;8:e1002985. doi: 10.1371/journal.ppat.1002985. http://dx.doi.org/10.1371/journal.ppat.1002985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fayad W, Rickardson L, Haglund C, et al. Identification of agents that induce apoptosis of multicellular tumour spheroids: Enrichment for mitotic inhibitors with hydrophobic properties. Chem Biol Drug Des. 2011;78:547–557. doi: 10.1111/j.1747-0285.2011.01170.x. http://dx.doi.org/10.1111/j.1747-0285.2011.01170.x. [DOI] [PubMed] [Google Scholar]
- Folkman J, Hochberg M. Self-regulation of growth in three dimensions. J Exp Med. 1973;138:745–753. doi: 10.1084/jem.138.4.745. http://dx.doi.org/10.1084/jem.138.4.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler P, Smith K, Young J, et al. Reduction of misleading (“false”) positive results in mammalian cell genotoxicity assays. I. Choice of cell type. Mutat Res. 2012a;742:11–25. doi: 10.1016/j.mrgentox.2011.10.014. http://dx.doi.org/10.1016/j.mrgentox.2011.10.014. [DOI] [PubMed] [Google Scholar]
- Fowler P, Smith R, Smith K, et al. Reduction of misleading (“false”) positive results in mammalian cell genotoxicity assays. II. Importance of accurate toxicity measurement. Mutat Res. 2012b;747:104–117. doi: 10.1016/j.mrgentox.2012.04.013. http://dx.doi.org/10.1016/j.mrgentox.2012.04.013. [DOI] [PubMed] [Google Scholar]
- Freshney RI. Culture of Animal Cells: A Manual of Basic Technique. Vol. 4. New York: Wiley-Liss Publication; 2000. [Google Scholar]
- Freyer JP, Sutherland RM. Regulation of growth saturation and development of necrosis in EMT6/Ro multicellular spheroids by the glucose and oxygen supply. Cancer Res. 1986;46:3504–3512. [PubMed] [Google Scholar]
- Fritsche E, Gassmann K, Schreiber T. Neurospheres as a model for developmental neurotoxicity testing. Methods Mol Biol. 2011;758:99–114. doi: 10.1007/978-1-61779-170-3_7. http://dx.doi.org/10.1007/978-1-61779-170-3_7. [DOI] [PubMed] [Google Scholar]
- Fuchs E, Tumbar T, Guasch G. Socializing with the neighbors: Stem cells and their niche. Cell. 2004;116:769–778. doi: 10.1016/s0092-8674(04)00255-7. http://dx.doi.org/10.1016/S0092-8674(04)00255-7. [DOI] [PubMed] [Google Scholar]
- Fuchs S, Gumbleton MUSF, et al. Models of alveolar epithelium. In: Lehr CM, editor. Cell Culture Models of Biological Barriers. In vitro Test Systems for Drug Absorption and Delivery. London, New York: CRC Press; 2002. [Google Scholar]
- Gantner F, Leist M, Kusters S, et al. T-cell stimulus-induced crosstalk between lymphocytes and liver macrophages results in augmented cytokine release. Exp Cell Res. 1996;229:137–146. doi: 10.1006/excr.1996.0351. http://dx.doi.org/10.1006/excr.1996.0351. [DOI] [PubMed] [Google Scholar]
- Gazel A, Ramphal P, Rosdy M, et al. Transcriptional profiling of epidermal keratinocytes: Comparison of genes expressed in skin, cultured keratinocytes, and recon stituted epidermis, using large DNA microarrays. J Invest Dermatol. 2003;121:1459–1468. doi: 10.1111/j.1523-1747.2003.12611.x. http://dx.doi.org/10.1111/j.1523-1747.2003.12611.x. [DOI] [PubMed] [Google Scholar]
- Gebhardt R, Jonitza D. Different proliferative responses of periportal and perivenous hepatocytes to EGF. Biochem Biophys Res Commun. 1991;181:1201–1207. doi: 10.1016/0006-291x(91)92066-s. http://dx.doi.org/10.1016/0006-291X(91)92066-S. [DOI] [PubMed] [Google Scholar]
- Gehr P, Bachofen M, Weibel ER. The normal human lung: Ultrastructure and morphometric estimation of diffusion capacity. Respir Physiol. 1978;32:121–140. doi: 10.1016/0034-5687(78)90104-4. http://dx.doi.org/10.1016/0034-5687(78)90104-4. [DOI] [PubMed] [Google Scholar]
- Gehr P. In Human respiratory tract model for radiological protection: A report of the International Commission on Radiological Protection. Oxford: Elsevier Science, Ltd; 1994. Anatomy and morphology of the respiratory tract: Annexe A. [Google Scholar]
- Gerets HH, Tilmant K, Gerin B, et al. Characterization of primary human hepatocytes, HepG2 cells, and HepaRG cells at the mRNA level and CYP activity in response to inducers and their predictivity for the detection of human hepatotoxins. Cell Biol Toxicol. 2012;28:69–87. doi: 10.1007/s10565-011-9208-4. http://dx.doi.org/10.1007/s10565-011-9208-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlach JC, Mutig K, Sauer IM, et al. Use of primary human liver cells originating from discarded grafts in a bioreactor for liver support therapy and the prospects of culturing adult liver stem cells in bioreactors: A morphologic study. Transplantation. 2003;76:781–786. doi: 10.1097/01.TP.0000083319.36931.32. http://dx.doi.org/10.1097/01.TP.0000083319.36931.32. [DOI] [PubMed] [Google Scholar]
- Gil J, Weibel ER. Extracellular lining of bronchioles after perfusion-fixation of rat lungs for electron microscopy. Anat Rec. 1971;169:185–199. doi: 10.1002/ar.1091690205. http://dx.doi.org/10.1002/ar.1091690205. [DOI] [PubMed] [Google Scholar]
- Giordano G, Kavanagh TJ, Costa LG. Mouse cerebellar astrocytes protect cerebellar granule neurons against toxicity of the polybrominated diphenyl ether (PBDE) mixture DE-71. Neurotoxicology. 2009;30:326–329. doi: 10.1016/j.neuro.2008.12.009. http://dx.doi.org/10.1016/j.neuro.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godfrey RW. Human airway epithelial tight junctions. Microsc Res Tech. 1997;38:488–499. doi: 10.1002/(SICI)1097-0029(19970901)38:5<488::AID-JEMT5>3.0.CO;2-E. http://dx.doi.org/10.1002/(SICI)1097-0029(19970901)38:5<488::AID-JEMT5>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Godoy P, Hengstler JG, Ilkavets I, et al. Extracellular matrix modulates sensitivity of hepatocytes to fibroblastoid dedifferentiation and transforming growth factor beta-induced apoptosis. Hepatology. 2009;49:2031–2043. doi: 10.1002/hep.22880. http://dx.doi.org/10.1002/hep.22880. [DOI] [PubMed] [Google Scholar]
- Godoy P, Hewitt NJ, Albrecht U, et al. Recent advances in 2D and 3D in vitro systems using primary hepatocytes, alternative hepatocyte sources and non-parenchymal liver cells and their use in investigating mechanisms of hepatotoxicity, cell signaling and ADME. Arch Toxicol. 2013;87:1315–1530. doi: 10.1007/s00204-013-1078-5. http://dx.doi.org/10.1007/s00204-013-1078-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Lechon MJ, Jover R, Donato T, et al. Long-term expression of differentiated functions in hepatocytes cultured in three-dimensional collagen matrix. J Cell Physiol. 1998;177:553–562. doi: 10.1002/(SICI)1097-4652(199812)177:4<553::AID-JCP6>3.0.CO;2-F. http://dx.doi.org/10.1002/(SICI)1097-4652(199812)177:4<553::AID-JCP6>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Götz C, Pfeiffer R, Tigges J, et al. Xenobiotic metabolism capacities of human skin in comparison with a 3D-epidermis model and keratinocyte-based cell culture as in vitro alternatives for chemical testing: Phase II enzymes. Exp Dermatol. 2012;21:364–369. doi: 10.1111/j.1600-0625.2012.01478.x. http://dx.doi.org/10.1111/j.1600-0625.2012.01478.x. [DOI] [PubMed] [Google Scholar]
- Griffith LG, Swartz MA. Capturing complex 3D tissue physiology in vitro. Nat Rev Mol Cell Biol. 2006;7:211–224. doi: 10.1038/nrm1858. http://dx.doi.org/10.1038/nrm1858. [DOI] [PubMed] [Google Scholar]
- Grinnell F, Ho CH, Tamariz E, et al. Dendritic fibroblasts in three-dimensional collagen matrices. Mol Biol Cell. 2003;14:384–395. doi: 10.1091/mbc.E02-08-0493. http://dx.doi.org/10.1091/mbc.E02-08-0493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groen D, Poole DS, Gooris GS, et al. Investigating the barrier function of skin lipid models with varying compositions. Eur J Pharm Biopharm. 2011;79:334–342. doi: 10.1016/j.ejpb.2011.05.007. http://dx.doi.org/10.1016/j.ejpb.2011.05.007. [DOI] [PubMed] [Google Scholar]
- Guillouzo A, Corlu A, Aninat C, et al. The human hepatoma HepaRG cells: A highly differentiated model for studies of liver metabolism and toxicity of xenobiotics. Chem Biol Interact. 2007;168:66–73. doi: 10.1016/j.cbi.2006.12.003. http://dx.doi.org/10.1016/j.cbi.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Gunness P, Mueller D, Shevchenko V, et al. 3D organotypic cultures of human HepaRG cells: A tool for in vitro toxicity studies. Toxicol Sci. 2013;133:67–78. doi: 10.1093/toxsci/kft021. http://dx.doi.org/10.1093/toxsci/kft021. [DOI] [PubMed] [Google Scholar]
- Hagvall L, Baron JM, Borje A, et al. Cytochrome P450-mediated activation of the fragrance compound geraniol forms potent contact allergens. Toxicol Appl Pharmacol. 2008;233:308–313. doi: 10.1016/j.taap.2008.08.014. http://dx.doi.org/10.1016/j.taap.2008.08.014. [DOI] [PubMed] [Google Scholar]
- Hansmann J, Groeber F, Kahlig A, et al. Bioreactors in tissue engineering – principles, applications and commercial constraints. Biotechnol J. 2013;8:298–307. doi: 10.1002/biot.201200162. http://dx.doi.org/10.1002/biot.201200162. [DOI] [PubMed] [Google Scholar]
- Hanson B, Sugden A, Alberts B. Making data maximally available. Science. 2011;331:649. doi: 10.1126/science.1203354. http://dx.doi.org/10.1126/science.1203354. [DOI] [PubMed] [Google Scholar]
- Hanson KM, Behne MJ, Barry NP, et al. Two-photon fluorescence lifetime imaging of the skin stratum corneum pH gradient. Biophys J. 2002;83:1682–1690. doi: 10.1016/S0006-3495(02)73936-2. http://dx.doi.org/10.1016/S0006-3495(02)73936-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson O, Castilho RF, Kaminski Schierle GS, et al. Additive effects of caspase inhibitor and lazaroid on the survival of transplanted rat and human embryonic dopamine neurons. Exp Neurol. 2000;164:102–111. doi: 10.1006/exnr.2000.7406. http://dx.doi.org/10.1006/exnr.2000.7406. [DOI] [PubMed] [Google Scholar]
- Harris IR, Siefken W, Beck-Oldach K, et al. Comparison of activities dependent on glutathione S-transferase and cytochrome P-450 IA1 in cultured keratinocytes and reconstructed epidermal models. Skin Pharmacol Appl Skin Physiol. 2002;15(Suppl 1):59–67. doi: 10.1159/000066676. http://dx.doi.org/10.1159/000066676. [DOI] [PubMed] [Google Scholar]
- Hartung T, Bremer S, Casati S, et al. A modular approach to the ECVAM principles on test validity. Altern Lab Anim. 2004;32:467–472. doi: 10.1177/026119290403200503. [DOI] [PubMed] [Google Scholar]
- Hartung T. Food for thought … on validation. ALTEX. 2007;24:67–80. doi: 10.14573/altex.2007.2.67. http://www.altex.ch/All-issues/Issue.50.html?iid=86&aid=3. [DOI] [PubMed] [Google Scholar]
- Hartung T, Hoffmann S, Stephens M. Mechanistic validation. ALTEX. 2013;30:119–130. doi: 10.14573/altex.2013.2.119. http://dx.doi.org/10.14573/altex.2013.2.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haycock JW. 3D cell culture: A review of current approaches and techniques. Methods Mol Biol. 2011;695:1–15. doi: 10.1007/978-1-60761-984-0_1. http://dx.doi.org/10.1007/978-1-60761-984-0_1. [DOI] [PubMed] [Google Scholar]
- Henn A, Lund S, Hedtjarn M, et al. The suitability of BV2 cells as alternative model system for primary microglia cultures or for animal experiments examining brain inflammation. ALTEX. 2009;26:83–94. doi: 10.14573/altex.2009.2.83. http://www.altex.ch/All-issues/Issue.50.html?iid=105&aid=2. [DOI] [PubMed] [Google Scholar]
- Henn A, Kirner S, Leist M. TLR2 hypersensitivity of astrocytes as functional consequence of previous inflammatory episodes. J Immunol. 2011;186:3237–3247. doi: 10.4049/jimmunol.1002787. http://dx.doi.org/10.4049/jimmunol.1002787. [DOI] [PubMed] [Google Scholar]
- Hermanns MI, Fuchs S, Bock M, et al. Primary human coculture model of alveolo-capillary unit to study mechanisms of injury to peripheral lung. Cell Tissue Res. 2009;336:91–105. doi: 10.1007/s00441-008-0750-1. http://dx.doi.org/10.1007/s00441-008-0750-1. [DOI] [PubMed] [Google Scholar]
- Hermanns MI, Kasper J, Dubruel P, et al. An impaired alveolar-capillary barrier in vitro: Effect of proinflammatory cytokines and consequences on nanocarrier interaction. J R Soc Interface. 2010;7(Suppl 1):S41–54. doi: 10.1098/rsif.2009.0288.focus. http://dx.doi.org/10.1098/rsif.2009.0288.focus. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewitt NJ, Buhring KU, Dasenbrock J, et al. Studies comparing in vivo:in vitro metabolism of three pharmaceutical compounds in rat, dog, monkey, and human using cryopreserved hepatocytes, microsomes, and collagen gel immobilized hepatocyte cultures. Drug Metab Dispos. 2001;29:1042–1050. [PubMed] [Google Scholar]
- Hewitt NJ, Edwards RJ, Fritsche E, et al. Use of human in vitro skin models for accurate and ethical risk assessment: Metabolic considerations. Toxicol Sci. 2013;133:209–217. doi: 10.1093/toxsci/kft080. http://dx.doi.org/10.1093/toxsci/kft080. [DOI] [PubMed] [Google Scholar]
- Higgins PG, Dammhayn C, Hackel M, et al. Global spread of carbapenem-resistant Acinetobacter baumannii. J Antimicrob Chemother. 2010;65:233–238. doi: 10.1093/jac/dkp428. http://dx.doi.org/10.1093/jac/dkp428. [DOI] [PubMed] [Google Scholar]
- Hirt UA, Gantner F, Leist M. Phagocytosis of nonapoptotic cells dying by caspase-independent mechanisms. J Immunol. 2000;164:6520–6529. doi: 10.4049/jimmunol.164.12.6520. http://dx.doi.org/10.4049/jimmunol.164.12.6520. [DOI] [PubMed] [Google Scholar]
- Hirt UA, Leist M. Rapid, noninflammatory and PS-dependent phagocytic clearance of necrotic cells. Cell Death Differ. 2003;10:1156–1164. doi: 10.1038/sj.cdd.4401286. http://dx.doi.org/10.1038/sj.cdd.4401286. [DOI] [PubMed] [Google Scholar]
- Hoekstra D, Zegers MM, van Ijzendoorn SC. Membrane flow, lipid sorting and cell polarity in HepG2 cells: Role of a subapical compartment. Biochem Soc Trans. 1999;27:422–428. doi: 10.1042/bst0270422. [DOI] [PubMed] [Google Scholar]
- Hoekstra R, Nibourg GA, van der Hoeven TV, et al. The HepaRG cell line is suitable for bioartificial liver application. Int J Biochem Cell Biol. 2011;43:1483–1489. doi: 10.1016/j.biocel.2011.06.011. http://dx.doi.org/10.1016/j.biocel.2011.06.011. [DOI] [PubMed] [Google Scholar]
- Hoeller Obrigkeit D, Jugert FK, Beermann T, et al. Effects of photodynamic therapy evaluated in a novel three-dimensional squamous cell carcinoma organ construct of the skin. Photochem Photobiol. 2009;85:272–278. doi: 10.1111/j.1751-1097.2008.00432.x. http://dx.doi.org/10.1111/j.1751-1097.2008.00432.x. [DOI] [PubMed] [Google Scholar]
- Hoelting L, Scheinhardt B, Bondarenko O, et al. A 3-dimensional human embryonic stem cell (hESC)-derived model to detect developmental neurotoxicity of nanoparticles. Arch Toxicol. 2013;87:721–733. doi: 10.1007/s00204-012-0984-2. http://dx.doi.org/10.1007/s00204-012-0984-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann SA, Muller-Vieira U, Biemel K, et al. Analysis of drug metabolism activities in a miniaturized liver cell bioreactor for use in pharmacological studies. Biotechnol Bioeng. 2012;109:3172–3181. doi: 10.1002/bit.24573. http://dx.doi.org/10.1002/bit.24573. [DOI] [PubMed] [Google Scholar]
- Hoing S, Rudhard Y, Reinhardt P, et al. Discovery of inhibitors of microglial neurotoxicity acting through multiple mechanisms using a stem-cell-based phenotypic assay. Cell Stem Cell. 2012;11:620–632. doi: 10.1016/j.stem.2012.07.005. http://dx.doi.org/10.1016/j.stem.2012.07.005. [DOI] [PubMed] [Google Scholar]
- Holt PG, Schon-Hegrad MA. Localization of Tcells, macrophages and dendritic cells in rat respiratory tract tissue: Implications for immune function studies. Immunology. 1987;62:349–356. [PMC free article] [PubMed] [Google Scholar]
- Honegger P, Richelson E. Biochemical differentiation of mechanically dissociated mammalian brain in aggregating cell culture. Brain Res. 1976;109:335–354. doi: 10.1016/0006-8993(76)90534-5. http://dx.doi.org/10.1016/0006-8993(76)90534-5. [DOI] [PubMed] [Google Scholar]
- Honegger P, Lenoir D, Favrod P. Growth and differentiation of aggregating fetal brain cells in a serum-free defined medium. Nature. 1979;282:305–308. doi: 10.1038/282305a0. http://dx.doi.org/10.1038/282305a0. [DOI] [PubMed] [Google Scholar]
- Honegger P, Defaux A, Monnet-Tschudi F, et al. Preparation, maintenance, and use of serum-free aggregating brain cell cultures. Methods Mol Biol. 2011;758:81–97. doi: 10.1007/978-1-61779-170-3_6. http://dx.doi.org/10.1007/978-1-61779-170-3_6. [DOI] [PubMed] [Google Scholar]
- Hu T, Bailey RE, Morrall SW, et al. Dermal penetration and metabolism of p-aminophenol and p-phenylenediamine: Application of the EpiDerm human reconstructed epidermis model. Toxicol Lett. 2009;188:119–129. doi: 10.1016/j.toxlet.2009.03.019. http://dx.doi.org/10.1016/j.toxlet.2009.03.019. [DOI] [PubMed] [Google Scholar]
- Huh D, Matthews BD, Mammoto A, et al. Reconstituting organ-level lung functions on a chip. Science. 2010;328:1662–1668. doi: 10.1126/science.1188302. http://dx.doi.org/10.1126/science.1188302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh D, Hamilton GA, Ingber DE. From 3D cell culture to organs-on-chips. Trends Cell Biol. 2011;21:745–754. doi: 10.1016/j.tcb.2011.09.005. http://dx.doi.org/10.1016/j.tcb.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh D, Leslie DC, Matthews BD, et al. A human disease model of drug toxicity-induced pulmonary edema in a lung-on-a-chip microdevice. Sci Transl Med. 2012a;4:159ra147. doi: 10.1126/scitranslmed.3004249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh D, Torisawa YS, Hamilton GA, et al. Micro-engineered physiological biomimicry: Organs-on-chips. Lab Chip. 2012b;12:2156–2164. doi: 10.1039/c2lc40089h. http://dx.doi.org/10.1039/c2lc40089h. [DOI] [PubMed] [Google Scholar]
- Inamori M, Mizumoto H, Kajiwara T. An approach for formation of vascularized liver tissue by endothelial cell-covered hepatocyte spheroid integration. Tissue Eng Part A. 2009;15:2029–2037. doi: 10.1089/ten.tea.2008.0403. http://dx.doi.org/10.1089/ten.tea.2008.0403. [DOI] [PubMed] [Google Scholar]
- Jäckh C, Fabian E, van Ravenzwaay B, et al. Relevance of xenobiotic enzymes in human skin in vitro models to activate pro-sensitizers. J Immunotoxicol. 2012;9:426–438. doi: 10.3109/1547691X.2012.664578. http://dx.doi.org/10.3109/1547691X.2012.664578. [DOI] [PubMed] [Google Scholar]
- James LP, Mayeux PR, Hinson JA. Acetaminophen-induced hepatotoxicity. Drug Metab Dispos. 2003;31:1499–1506. doi: 10.1124/dmd.31.12.1499. http://dx.doi.org/10.1124/dmd.31.12.1499. [DOI] [PubMed] [Google Scholar]
- Jasmin L, Vit JP, Bhargava A, et al. Can satellite glial cells be therapeutic targets for pain control? Neuron Glia Biol. 2010;6:63–71. doi: 10.1017/S1740925X10000098. http://dx.doi.org/10.1017/S1740925X10000098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin ZB, Okamoto S, Osakada F, et al. Modeling retinal degeneration using patient-specific induced pluripotent stem cells. PLoS One. 2011;6:e17084. doi: 10.1371/journal.pone.0017084. http://dx.doi.org/10.1371/journal.pone.0017084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungermann K, Kietzmann T. Zonation of parenchymal and nonparenchymal metabolism in liver. Annu Rev Nutr. 1996;16:179–203. doi: 10.1146/annurev.nu.16.070196.001143. http://dx.doi.org/10.1146/annurev.nu.16.070196.001143. [DOI] [PubMed] [Google Scholar]
- Kadereit S, Zimmer B, van Thriel C, et al. Compound selection for in vitro modeling of developmental neurotoxicity. Front Biosci (Landmark Ed) 2012;17:2442–2460. doi: 10.2741/4064. http://dx.doi.org/10.2741/4064. [DOI] [PubMed] [Google Scholar]
- Kalig A, Hansmann J, Groeber F, et al. In silico approaches for the identification of optimal culture condition for tissue engineered bone substitutes. Curr Anal Chem. 2013;9:16–28. http://dx.doi.org/10.2174/157341113804486563. [Google Scholar]
- Kanebratt KP, Andersson TB. Evaluation of HepaRG cells as an in vitro model for human drug metabolism studies. Drug Metab Dispos. 2008;36:1444–1452. doi: 10.1124/dmd.107.020016. http://dx.doi.org/10.1124/dmd.107.020016. [DOI] [PubMed] [Google Scholar]
- Kelm JM, Timmins NE, Brown CJ, et al. Method for generation of homogeneous multicellular tumor spheroids applicable to a wide variety of cell types. Biotechnol Bioeng. 2003;83:173–180. doi: 10.1002/bit.10655. http://dx.doi.org/10.1002/bit.10655. [DOI] [PubMed] [Google Scholar]
- Kelm JM, Fussenegger M. Microscale tissue engineering using gravity-enforced cell assembly. Trends Biotechnol. 2004;22:195–202. doi: 10.1016/j.tibtech.2004.02.002. http://dx.doi.org/10.1016/j.tibtech.2004.02.002. [DOI] [PubMed] [Google Scholar]
- Kelm JM, Djonov V, Ittner LM, et al. Design of custom-shaped vascularized tissues using microtissue spheroids as minimal building units. Tissue Eng. 2006;12:2151–2160. doi: 10.1089/ten.2006.12.2151. http://dx.doi.org/10.1089/ten.2006.12.2151. [DOI] [PubMed] [Google Scholar]
- Kelm JM, Breitbach M, Fischer G, et al. 3D microtissue formation of undifferentiated bone marrow mesenchymal stem cells leads to elevated apoptosis. Tissue Eng Part A. 2012;18:692–702. doi: 10.1089/ten.TEA.2011.0281. http://dx.doi.org/10.1089/ten.tea.2011.0281. [DOI] [PubMed] [Google Scholar]
- Kilburn KH. A hypothesis for pulmonary clearance and its implications. Am Rev Respir Dis. 1968;98:449–463. doi: 10.1164/arrd.1968.98.3.449. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Huh D, Hamilton G, et al. Human gut-on-a-chip inhabited by microbial flora that experiences intestinal peristalsis-like motions and flow. Lab Chip. 2012;12:2165–2174. doi: 10.1039/c2lc40074j. http://dx.doi.org/10.1039/c2lc40074j. [DOI] [PubMed] [Google Scholar]
- Kim JH, Ha HC, Lee MS, et al. Effect of Tremella fuciformis on the neurite outgrowth of PC12h cells and the improvement of memory in rats. Biol Pharm Bull. 2007;30:708–714. doi: 10.1248/bpb.30.708. http://dx.doi.org/10.1248/bpb.30.708. [DOI] [PubMed] [Google Scholar]
- Kipnis J, Schwartz M. Controlled autoimmunity in CNS maintenance and repair: Naturally occurring CD4+CD25+ regulatory T-Cells at the crossroads of health and disease. Neuromolecular Med. 2005;7:197–206. doi: 10.1385/NMM:7:3:197. http://dx.doi.org/10.1385/NMM:7:3:197. [DOI] [PubMed] [Google Scholar]
- Kirouac DC, Ito C, Csaszar E, et al. Dynamic interaction networks in a hierarchically organized tissue. Mol Syst Biol. 2010;6:417. doi: 10.1038/msb.2010.71. http://dx.doi.org/10.1038/msb.2010.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein S, Mueller D, Schevchenko V, et al. Long-term maintenance of HepaRG cells in serum-free conditions and application in a repeated dose study. J Appl Toxicol. 2013 doi: 10.1002/jat.2929. Epub ahead of print. http://dx.doi.org/10.1002/jat.2929. [DOI] [PubMed]
- Koch P, Opitz T, Steinbeck JA, et al. A rosette-type, self-renewing human ES cell-derived neural stem cell with potential for in vitro instruction and synaptic integration. Proceedings of the National Academy of Sciences. 2009;106:3225–3230. doi: 10.1073/pnas.0808387106. http://dx.doi.org/10.1073/pnas.0808387106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolle SN, Basketter DA, Casati S, et al. Performance standards and alternative assays: Practical insights from skin sensitization. Regul Toxicol Pharmacol. 2012;65:278–285. doi: 10.1016/j.yrtph.2012.12.006. http://dx.doi.org/10.1016/j.yrtph.2012.12.006. [DOI] [PubMed] [Google Scholar]
- Krause KH, van Thriel C, De Sousa PA, et al. Monocrotophos in Gandaman village: India school lunch deaths and need for improved toxicity testing. Arch Toxicol. 2013;87:1877–1881. doi: 10.1007/s00204-013-1113-6. http://dx.doi.org/10.1007/s00204-013-1113-6. [DOI] [PubMed] [Google Scholar]
- Krewski D, Acosta D, Jr, Andersen M, et al. Toxicity testing in the 21st century: A vision and a strategy. J Toxicol Environ Health B Crit Rev. 2010;13:51–138. doi: 10.1080/10937404.2010.483176. http://dx.doi.org/10.1080/10937404.2010.483176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug AK, Balmer NV, Matt F, et al. Evaluation of a human neurite growth assay as specific screen for developmental neurotoxicants. Arch Toxicol. 2013a;87:2215–2231. doi: 10.1007/s00204-013-1072-y. http://dx.doi.org/10.1007/s00204-013-1072-y. [DOI] [PubMed] [Google Scholar]
- Krug AK, Kolde R, Gaspar JA, et al. Human embryonic stem cell-derived test systems for developmental neurotoxicity: A transcriptomics approach. Arch Toxicol. 2013b;87:123–143. doi: 10.1007/s00204-012-0967-3. http://dx.doi.org/10.1007/s00204-012-0967-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Küchler S, Wolf NB, Heilmann S, et al. 3D-wound healing model: Influence of morphine and solid lipid nanoparticles. J Biotechnol. 2010;148:24–30. doi: 10.1016/j.jbiotec.2010.01.001. http://dx.doi.org/10.1016/j.jbiotec.2010.01.001. [DOI] [PubMed] [Google Scholar]
- Küchler S, Henkes D, Eckl KM, et al. Hallmarks of atopic skin mimicked in vitro by means of a skin disease model based on FLG knock-down. Altern Lab Anim. 2011;39:471–480. doi: 10.1177/026119291103900508. [DOI] [PubMed] [Google Scholar]
- Kuegler PB, Zimmer B, Waldmann T, et al. Markers of murine embryonic and neural stem cells, neurons and astrocytes: Reference points for developmental neurotoxicity testing. ALTEX. 2010;27:17–42. doi: 10.14573/altex.2010.1.16. http://www.altex.ch/All-issues/Issue.50.html?iid=112&aid=2. [DOI] [PubMed] [Google Scholar]
- Kuegler PB, Baumann BA, Zimmer B, et al. GFAP-independent inflammatory competence and trophic functions of astrocytes generated from murine embryonic stem cells. Glia. 2012;60:218–228. doi: 10.1002/glia.21257. http://dx.doi.org/10.1002/glia.21257. [DOI] [PubMed] [Google Scholar]
- Kuhn HG, Svendsen CN. Origins, functions, and potential of adult neural stem cells. Bioessays. 1999;21:625–630. doi: 10.1002/(SICI)1521-1878(199908)21:8<625::AID-BIES1>3.0.CO;2-6. http://dx.doi.org/10.1002/(SICI)1521-1878(199908)21:8<625::AID-BIES1>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Lancaster MA, Renner M, Martin CA, et al. Cerebral organoids model human brain development and microcephaly. Nature. 2013;501:373–379. doi: 10.1038/nature12517. http://dx.doi.org/10.1038/nature12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer O, Muller M. Methods to assess tissue-specific distribution and metabolism of drugs. Curr Drug Metab. 2004;5:463–481. doi: 10.2174/1389200043335379. http://dx.doi.org/10.2174/1389200043335379. [DOI] [PubMed] [Google Scholar]
- Latta M, Kunstle G, Leist M, et al. Metabolic depletion of ATP by fructose inversely controls CD95- and tumor necrosis factor receptor 1-mediated hepatic apoptosis. J Exp Med. 2000;191:1975–1985. doi: 10.1084/jem.191.11.1975. http://dx.doi.org/10.1084/jem.191.11.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauer B, Tuschl G, Kling M, et al. Species-specific toxicity of diclofenac and troglitazone in primary human and rat hepatocytes. Chem Biol Interact. 2009;179:17–24. doi: 10.1016/j.cbi.2008.10.031. http://dx.doi.org/10.1016/j.cbi.2008.10.031. [DOI] [PubMed] [Google Scholar]
- Lehman PA, Raney SG, Franz TJ. Percutaneous absorption in man: In vitro-in vivo correlation. Skin Pharmacol Physiol. 2011;24:224–230. doi: 10.1159/000324884. http://dx.doi.org/10.1159/000324884. [DOI] [PubMed] [Google Scholar]
- Lehmann AD, Blank F, Baum O, et al. Diesel exhaust particles modulate the tight junction protein occludin in lung cells in vitro. Part Fibre Toxicol. 2009;6:26. doi: 10.1186/1743-8977-6-26. http://dx.doi.org/10.1186/1743-8977-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann AD, Daum N, Bur M, et al. An in vitro triple cell co-culture model with primary cells mimicking the human alveolar epithelial barrier. Eur J Pharm Biopharm. 2011;77:398–406. doi: 10.1016/j.ejpb.2010.10.014. http://dx.doi.org/10.1016/j.ejpb.2010.10.014. [DOI] [PubMed] [Google Scholar]
- Lehnert BE. Pulmonary and thoracic macrophage subpopulations and clearance of particles from the lung. Environ Health Perspect. 1992;97:17–46. doi: 10.1289/ehp.929717. http://dx.doi.org/10.1289/ehp.929717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein PJ, Guo X, Shi GX, et al. The novel GTPase Rit differentially regulates axonal and dendritic growth. J Neurosci. 2007;27:4725–4736. doi: 10.1523/JNEUROSCI.5633-06.2007. http://dx.doi.org/10.1523/JNEUROSCI.5633-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leist M, Bremer S, Brundin P, et al. The biological and ethical basis of the use of human embryonic stem cells for in vitro test systems or cell therapy. ALTEX. 2008a;25:163–190. http://www.altex.ch/All-issues/Issue.50.html?iid=101&aid=3. [PubMed] [Google Scholar]
- Leist M, Hartung T, Nicotera P. The dawning of a new age of toxicology. ALTEX. 2008b;25:103–114. http://www.altex.ch/All-issues/Issue.50.html?iid=99&aid=3. [PubMed] [Google Scholar]
- Leist M, Efremova L, Karreman C. Food for thought … considerations and guidelines for basic test method descriptions in toxicology. ALTEX. 2010;27:309–317. doi: 10.14573/altex.2010.4.309. http://www.altex.ch/All-issues/Issue.50.html?iid=121&aid=7. [DOI] [PubMed] [Google Scholar]
- Leist M, Hasiwa N, Daneshian M, et al. Validation and quality control of replacement alternatives – current status and future challenges. Toxicol Res. 2012a;1:8–22. [Google Scholar]
- Leist M, Lidbury BA, Yang C, et al. Novel technologies and an overall strategy to allow hazard assessment and risk prediction of chemicals, cosmetics, and drugs with animal-free methods. ALTEX. 2012b;29:373–388. doi: 10.14573/altex.2012.4.373. http://dx.doi.org/10.14573/altex.2012.4.373. [DOI] [PubMed] [Google Scholar]
- Leite SB, Wilk-Zasadna I, Zaldivar JM, et al. Three-dimensional HepaRG model as an attractive tool for toxicity testing. Toxicol Sci. 2012;130:106–116. doi: 10.1093/toxsci/kfs232. http://dx.doi.org/10.1093/toxsci/kfs232. [DOI] [PubMed] [Google Scholar]
- Li XJ, Valadez AV, Zuo P, et al. Microfluidic 3D cell culture: Potential application for tissue-based bioassays. Bioanalysis. 2012;4:1509–1525. doi: 10.4155/bio.12.133. http://dx.doi.org/10.4155/bio.12.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limongi T, Cesca F, Gentile F, et al. Nanostructured superhydrophobic substrates trigger the development of 3D neuronal networks. Small. 2013;9:402–412. doi: 10.1002/smll.201201377. http://dx.doi.org/10.1002/smll.201201377. [DOI] [PubMed] [Google Scholar]
- Lin RZ, Chang HY. Recent advances in three-dimensional multicellular spheroid culture for biomedical research. Biotechnol J. 2008;3:1172–1184. doi: 10.1002/biot.200700228. http://dx.doi.org/10.1002/biot.200700228. [DOI] [PubMed] [Google Scholar]
- Linde N, Gutschalk CM, Hoffmann C, et al. Integrating macrophages into organotypic co-cultures: A 3D in vitro model to study tumor-associated macrophages. PLoS One. 2012;7:e40058. doi: 10.1371/journal.pone.0040058. http://dx.doi.org/10.1371/journal.pone.0040058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linke K, Schanz J, Hansmann J, et al. Engineered liver-like tissue on a capillarized matrix for applied research. Tissue Eng. 2007;13:2699–2707. doi: 10.1089/ten.2006.0388. http://dx.doi.org/10.1089/ten.2006.0388. [DOI] [PubMed] [Google Scholar]
- Liu XW, Sun JW. Primary culture of neurospheres obtained from fetal mouse central nervous system and generation of inner ear hair cell immunophenotypes in vitro. J Laryngol Otol. 2011;125:686–691. doi: 10.1017/S0022215110002914. http://dx.doi.org/10.1017/S0022215110002914. [DOI] [PubMed] [Google Scholar]
- Lobo MV, Alonso FJ, Redondo C, et al. Cellular characterization of epidermal growth factor-expanded free-floating neurospheres. J Histochem Cytochem. 2003;51:89–103. doi: 10.1177/002215540305100111. http://dx.doi.org/10.1177/002215540305100111. [DOI] [PubMed] [Google Scholar]
- Lu HF, Lim SX, Leong MF, et al. Efficient neuronal differentiation and maturation of human pluripotent stem cells encapsulated in 3D microfibrous scaffolds. Biomaterials. 2012a;33:9179–9187. doi: 10.1016/j.biomaterials.2012.09.006. http://dx.doi.org/10.1016/j.biomaterials.2012.09.006. [DOI] [PubMed] [Google Scholar]
- Lu Y, Zhang G, Shen C, et al. A novel 3D liver organoid system for elucidation of hepatic glucose metabolism. Biotechnol Bioeng. 2012b;109:595–604. doi: 10.1002/bit.23349. http://dx.doi.org/10.1002/bit.23349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubberstedt M, Muller-Vieira U, Mayer M, et al. HepaRG human hepatic cell line utility as a surrogate for primary human hepatocytes in drug metabolism assessment in vitro. J Pharmacol Toxicol Methods. 2011;63:59–68. doi: 10.1016/j.vascn.2010.04.013. http://dx.doi.org/10.1016/j.vascn.2010.04.013. [DOI] [PubMed] [Google Scholar]
- Lubberstedt M, Muller-Vieira U, Biemel KM, et al. Serum-free culture of primary human hepatocytes in a miniaturized hollow-fibre membrane bioreactor for pharmacological in vitro studies. J Tissue Eng Regen Med. 2012 doi: 10.1002/term.1652. in print. http://dx.doi.org/10.1002/term.1652. [DOI] [PubMed]
- Lum H, Malik AB. Regulation of vascular endothelial barrier function. Am J Physiol. 1994;267:L223–241. doi: 10.1152/ajplung.1994.267.3.L223. [DOI] [PubMed] [Google Scholar]
- Luu-The V, Ferraris C, Duche D, et al. Steroid metabolism and profile of steroidogenic gene expression in Episkin: High similarity with human epidermis. J Steroid Biochem Mol Biol. 2007;107:30–36. doi: 10.1016/j.jsbmb.2007.03.036. http://dx.doi.org/10.1016/j.jsbmb.2007.03.036. [DOI] [PubMed] [Google Scholar]
- Luu-The V, Duche D, Ferraris C, et al. Expression profiles of phases 1 and 2 metabolizing enzymes in human skin and the reconstructed skin models Episkin and full thickness model from Episkin. J Steroid Biochem Mol Biol. 2009;116:178–186. doi: 10.1016/j.jsbmb.2009.05.011. http://dx.doi.org/10.1016/j.jsbmb.2009.05.011. [DOI] [PubMed] [Google Scholar]
- Lynch AM, Wilcox P. Review of the performance of the 3T3 NRU in vitro phototoxicity assay in the pharmaceutical industry. Exp Toxicol Pathol. 2011;63:209–214. doi: 10.1016/j.etp.2009.12.001. http://dx.doi.org/10.1016/j.etp.2009.12.001. [DOI] [PubMed] [Google Scholar]
- Maina JN, West JB. Thin and strong! The bioengineering dilemma in the structural and functional design of the blood-gas barrier. Physiol Rev. 2005;85:811–844. doi: 10.1152/physrev.00022.2004. http://dx.doi.org/10.1152/physrev.00022.2004. [DOI] [PubMed] [Google Scholar]
- Mandenius CF, Andersson TB, Alves PM, et al. Toward preclinical predictive drug testing for metabolism and hepatotoxicity by using in vitro models derived from human embryonic stem cells and human cell lines – a report on the Vitrocellomics EU-project. Altern Lab Anim. 2011;39:147–171. doi: 10.1177/026119291103900210. [DOI] [PubMed] [Google Scholar]
- Marion TL, Leslie EM, Brouwer KL. Use of sandwich-cultured hepatocytes to evaluate impaired bile acid transport as a mechanism of drug-induced hepatotoxicity. Mol Pharm. 2007;4:911–918. doi: 10.1021/mp0700357. http://dx.doi.org/10.1021/mp0700357. [DOI] [PubMed] [Google Scholar]
- Marx U, Walles H, Hoffmann S, et al. ‘Human-on-a-chip’ developments: A translational cutting-edge alternative to systemic safety assessment and efficiency evaluation of substances in laboratory animals and man? Altern Lab Anim. 2012;40:235–257. doi: 10.1177/026119291204000504. [DOI] [PubMed] [Google Scholar]
- Materne EM, Tonevitsky AG, Marx U. Chip-based liver equivalents for toxicity testing – organotypicalness versus cost-efficient high throughput. Lab Chip. 2013;13:3481–3495. doi: 10.1039/c3lc50240f. http://dx.doi.org/10.1039/c3lc50240f. [DOI] [PubMed] [Google Scholar]
- Maurel P, Salzer JL. Axonal regulation of Schwann cell proliferation and survival and the initial events of myelination requires PI 3-kinase activity. J Neurosci. 2000;20:4635–4645. doi: 10.1523/JNEUROSCI.20-12-04635.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWilliams AH. Dendritic cells as sentinals of immune surveillance in the airways. In: Gehr PH, editor. Particle-lung interations. New York, Basel: Marcel Dekker; 2000. http://dx.doi.org/10.1201/b14423-16. [Google Scholar]
- Mehling A, Eriksson T, Eltze T, et al. Non-animal test methods for predicting skin sensitization potentials. Arch Toxicol. 2012;86:1273–1295. doi: 10.1007/s00204-012-0867-6. http://dx.doi.org/10.1007/s00204-012-0867-6. [DOI] [PubMed] [Google Scholar]
- Messner S, Agarkova I, Moritz W, et al. Multi-cell type human liver microtissues for hepatotoxicity testing. Arch Toxicol. 2013;87:209–213. doi: 10.1007/s00204-012-0968-2. http://dx.doi.org/10.1007/s00204-012-0968-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michailov GV, Sereda MW, Brinkmann BG, et al. Axonal neuregulin-1 regulates myelin sheath thickness. Science. 2004;304:700–703. doi: 10.1126/science.1095862. http://dx.doi.org/10.1126/science.1095862. [DOI] [PubMed] [Google Scholar]
- Miki T, Ring A, Gerlach J. Hepatic differentiation of human embryonic stem cells is promoted by three-dimensional dynamic perfusion culture conditions. Tissue Eng Part C Methods. 2011;17:557–568. doi: 10.1089/ten.TEC.2010.0437. http://dx.doi.org/10.1089/ten.tec.2010.0437. [DOI] [PubMed] [Google Scholar]
- Mildner M, Jin J, Eckhart L, et al. Knockdown of filaggrin impairs diffusion barrier function and increases UV sensitivity in a human skin model. J Invest Dermatol. 2010;130:2286–2294. doi: 10.1038/jid.2010.115. http://dx.doi.org/10.1038/jid.2010.115. [DOI] [PubMed] [Google Scholar]
- Miller JS, Stevens KR, Yang MT, et al. Rapid casting of patterned vascular networks for perfusable engineered three-dimensional tissues. Nat Mater. 2012;11:768–774. doi: 10.1038/nmat3357. http://dx.doi.org/10.1038/nmat3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nat Rev Cancer. 2006;6:583–592. doi: 10.1038/nrc1893. http://dx.doi.org/10.1038/nrc1893. [DOI] [PubMed] [Google Scholar]
- Miranda JP, Leite SB, Muller-Vieira U, et al. Towards an extended functional hepatocyte in vitro culture. Tissue Eng Part C Methods. 2009;15:157–167. doi: 10.1089/ten.tec.2008.0352. http://dx.doi.org/10.1089/ten.tec.2008.0352. [DOI] [PubMed] [Google Scholar]
- Miranda JP, Rodrigues A, Tostoes RM, et al. Extending hepatocyte functionality for drug-testing applications using high-viscosity alginate-encapsulated three-dimensional cultures in bioreactors. Tissue Eng Part C Methods. 2010;16:1223–1232. doi: 10.1089/ten.TEC.2009.0784. http://dx.doi.org/10.1089/ten.tec.2009.0784. [DOI] [PubMed] [Google Scholar]
- Mitriaikina S, Müller-Goymann CC. Comparative permeation studies of nondiluted and diluted betamethasone-17-valerate semisolid formulations through isolated human stratum corneum and artificial skin construct. Skin Pharmacol Physiol. 2009;22:142–150. doi: 10.1159/000203368. http://dx.doi.org/10.1159/000203368. [DOI] [PubMed] [Google Scholar]
- Monnet-Tschudi F, Zurich MG, Honegger P. Neurotoxicant-induced inflammatory response in three-dimensional brain cell cultures. Hum Exp Toxicol. 2007;26:339–346. doi: 10.1177/0960327107074589. http://dx.doi.org/10.1177/0960327107074589. [DOI] [PubMed] [Google Scholar]
- Montero M, Poulsen FR, Noraberg J, et al. Comparison of neuroprotective effects of erythropoietin (EPO) and carbamylerythropoietin (CEPO) against ischemia-like oxygen-glucose deprivation (OGD) and NMDA excitotoxicity in mouse hippocampal slice cultures. Exp Neurol. 2007;204:106–117. doi: 10.1016/j.expneurol.2006.09.026. http://dx.doi.org/10.1016/j.expneurol.2006.09.026. [DOI] [PubMed] [Google Scholar]
- Moors M, Cline JE, Abel J, et al. ERK-dependent and -independent pathways trigger human neural progenitor cell migration. Toxicol Appl Pharmacol. 2007;221:57–67. doi: 10.1016/j.taap.2007.02.018. http://dx.doi.org/10.1016/j.taap.2007.02.018. [DOI] [PubMed] [Google Scholar]
- Moors M, Rockel TD, Abel J, et al. Human neurospheres as three-dimensional cellular systems for developmental neurotoxicity testing. Environ Health Perspect. 2009;117:1131–1138. doi: 10.1289/ehp.0800207. http://dx.doi.org/10.1289/ehp.0800207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison SJ, Spradling AC. Stem cells and niches: Mechanisms that promote stem cell maintenance throughout life. Cell. 2008;132:598–611. doi: 10.1016/j.cell.2008.01.038. http://dx.doi.org/10.1016/j.cell.2008.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscona A. Metaplastic changes in the chorioallantoic membrane. Transplant Bull. 1960;26:120–124. doi: 10.1097/00006534-196007000-00043. http://dx.doi.org/10.1097/00006534-196007000-00043. [DOI] [PubMed] [Google Scholar]
- Moscona A. Rotation-mediated histogenetic aggregation of dissociated cells. A quantifiable approach to cell interactions in vitro. Exp Cell Res. 1961;22:455–475. doi: 10.1016/0014-4827(61)90122-7. http://dx.doi.org/10.1016/0014-4827(61)90122-7. [DOI] [PubMed] [Google Scholar]
- Mueller D, Koetemann A, Noor F. Organotypic cultures of Hepg2 cells for in vitro toxicity studies. J Bioengineer Biomedical Sci. 2011a;S2-002 [Google Scholar]
- Mueller D, Tascher G, Muller-Vieira U, et al. In-depth physiological characterization of primary human hepatocytes in a 3D hollow-fiber bioreactor. J Tissue Eng Regen Med. 2011b;5:e207–218. doi: 10.1002/term.418. http://dx.doi.org/10.1002/term.418. [DOI] [PubMed] [Google Scholar]
- Mueller D, Muller-Vieira U, Biemel KM, et al. Biotransformation of diclofenac and effects on the metabolome of primary human hepatocytes upon repeated dose exposure. Eur J Pharm Sci. 2012;45:716–724. doi: 10.1016/j.ejps.2012.01.014. http://dx.doi.org/10.1016/j.ejps.2012.01.014. [DOI] [PubMed] [Google Scholar]
- Mueller D, Heinzle E, Noor F. 3D hepatic in vitro models as tools for toxicity studies. Current Tissue Engineering. 2013a;2:78–89. http://dx.doi.org/10.2174/2211542011302010007. [Google Scholar]
- Mueller D, Tascher G, Damm G, et al. Real-time in situ viability assessment in a 3D bioreactor with liver cells using resazurin assay. Cytotechnology. 2013b;65:297–305. doi: 10.1007/s10616-012-9486-6. http://dx.doi.org/10.1007/s10616-012-9486-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller L, Riediker M, Wick P, et al. Oxidative stress and inflammation response after nanoparticle exposure: Differences between human lung cell monocultures and an advanced three-dimensional model of the human epithelial airways. J R Soc Interface. 2010;7(Suppl 1):S27–40. doi: 10.1098/rsif.2009.0161.focus. http://dx.doi.org/10.1098/rsif.2009.0161.focus. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray-Rust P, Neylon C, Pollock R, Wilbanks J. Panton principles, principles for open data in science. 2010 Retrieved from http://pantonprinciples.org/
- Natsch A, Ryan CA, Foertsch L, et al. A dataset on 145 chemicals tested in alternative assays for skin sensitization undergoing prevalidation. J Appl Toxicol. 2013 doi: 10.1002/jat.2868. Epub ahead of print. http://dx.doi.org/10.1002/jat.2868. [DOI] [PubMed]
- Nicod LP. Lung defences: An overview. Eur Respir Rev. 2005;14:45–50. http://dx.doi.org/10.1183/09059180.05.00009501. [Google Scholar]
- Niesner R, Peker B, Schlusche P, et al. 3D-resolved investigation of the pH gradient in artificial skin constructs by means of fluorescence lifetime imaging. Pharm Res. 2005;22:1079–1087. doi: 10.1007/s11095-005-5304-6. http://dx.doi.org/10.1007/s11095-005-5304-6. [DOI] [PubMed] [Google Scholar]
- Niklas J, Noor F, Heinzle E. Effects of drugs in subtoxic concentrations on the metabolic fluxes in human hepatoma cell line Hep G2. Toxicol Appl Pharmacol. 2009;240:327–336. doi: 10.1016/j.taap.2009.07.005. http://dx.doi.org/10.1016/j.taap.2009.07.005. [DOI] [PubMed] [Google Scholar]
- Nistor G, Seiler MJ, Yan F, et al. Three-dimensional early retinal progenitor 3D tissue constructs derived from human embryonic stem cells. J Neurosci Methods. 2010;190:63–70. doi: 10.1016/j.jneumeth.2010.04.025. http://dx.doi.org/10.1016/j.jneumeth.2010.04.025. [DOI] [PubMed] [Google Scholar]
- Noor F, Niklas J, Muller-Vieira U, et al. An integrated approach to improved toxicity prediction for the safety assessment during preclinical drug development using Hep G2 cells. Toxicol Appl Pharmacol. 2009;237:221–231. doi: 10.1016/j.taap.2009.03.011. http://dx.doi.org/10.1016/j.taap.2009.03.011. [DOI] [PubMed] [Google Scholar]
- Novosel EC, Kleinhans C, Kluger PJ. Vascularization is the key challenge in tissue engineering. Adv Drug Deliv Rev. 2011;63:300–311. doi: 10.1016/j.addr.2011.03.004. http://dx.doi.org/10.1016/j.addr.2011.03.004. [DOI] [PubMed] [Google Scholar]
- NRC. Toxicity Testing in the 21st Century: A Vision and a Strategy. Washington, DC, USA: The National Academies Press; 2007. http://www.nap.edu/openbook.php?record_id=11970. [Google Scholar]
- Nyberg SL, Remmel RP, Mann HJ, et al. Primary hepatocytes outperform Hep G2 cells as the source of biotransformation functions in a bioartificial liver. Ann Surg. 1994;220:59–67. [PMC free article] [PubMed] [Google Scholar]
- O’Brien PJ, Irwin W, Diaz D, et al. High concordance of drug-induced human hepatotoxicity with in vitro cytotoxicity measured in a novel cell-based model using high content screening. Arch Toxicol. 2006;80:580–604. doi: 10.1007/s00204-006-0091-3. http://dx.doi.org/10.1007/s00204-006-0091-3. [DOI] [PubMed] [Google Scholar]
- O’Shaughnessy TJ, Lin HJ, Ma W. Functional synapse formation among rat cortical neurons grown on three-dimensional collagen gels. Neurosci Lett. 2003;340:169–172. doi: 10.1016/s0304-3940(03)00083-1. http://dx.doi.org/10.1016/S0304-3940(03)00083-1. [DOI] [PubMed] [Google Scholar]
- Obach RS. Predicting clearance in humans from in vitro data. Curr Top Med Chem. 2011;11:334–339. doi: 10.2174/156802611794480873. http://dx.doi.org/10.2174/156802611794480873. [DOI] [PubMed] [Google Scholar]
- Ochs M, Weibel ER. Functional design of the human lung for gas exchange. In: Fishman A, Elias J, Fishman J, et al., editors. Fishman’s Pulmonary Diseases and Disorders. New York, USA: McGraw-Hill; 2008. [Google Scholar]
- OECD. Test No. 424: Neurotoxicity Study in Rodents. OECD Publishing; 1997. http://dx.doi.org/10.1787/9789264071025-en. [Google Scholar]
- OECD. Test No. 432: In vitro 3T3 NRU Phototoxicity Test. OECD Publishing; 2004a. http://dx.doi.org/10.1787/9789264071162-en. [Google Scholar]
- OECD. Test No. 428: Skin Absorption: In vitro Method. OECD Guidelines for the Testing of Chemicals, Section 4. OECD Publishing; 2004b. http://dx.doi.org/10.1787/9789264071087-en. [Google Scholar]
- OECD. Test No. 432: In vitro 3T3 NRU Phototoxicity Test. OECD Guidelines for the Testing of Chemicals, Section 4. OECD Publishing; 2004c. http://dx.doi.org/10.1787/9789264071162-en. [Google Scholar]
- OECD. Test No. 426: Developmental Neurotoxicity Study. OECD Publishing; 2007. http://dx.doi.org/10.1787/9789264067394-en. [Google Scholar]
- OECD. Test No. 429: Skin Sensitisation. OECD Publishing; 2010a. http://dx.doi.org/10.1787/9789264071100-en. [Google Scholar]
- OECD. Test Guideline 439: In vitro skin irritation: Reconstructed human epidermis test method. OECD Guidelines for the Testing of Chemicals, Section 4. OECD Publishing; 2010b. [Google Scholar]
- OECD. Test No. 431: In vitro Skin Corrosion: Reconstructed Human Epidermis (RHE) Test Method. OECD Publishing; 2013a. http://dx.doi.org/10.1787/9789264203822-en. [Google Scholar]
- OECD. Test No. 439: In vitro Skin Irritation – Reconstructed Human Epidermis Test Method. OECD Publishing; 2013b. http://dx.doi.org/10.1787/9789264203884-en. [Google Scholar]
- Oesch F, Fabian E, Oesch-Bartlomowicz B, et al. Drug-metabolizing enzymes in the skin of man, rat, and pig. Drug Metab Rev. 2007;39:659–698. doi: 10.1080/03602530701690366. http://dx.doi.org/10.1080/03602530701690366. [DOI] [PubMed] [Google Scholar]
- Ogimura E, Sekine S, Horie T. Bile salt export pump inhibitors are associated with bile acid-dependent drug-induced toxicity in sandwich-cultured hepatocytes. Biochem Biophys Res Commun. 2011;416:313–317. doi: 10.1016/j.bbrc.2011.11.032. http://dx.doi.org/10.1016/j.bbrc.2011.11.032. [DOI] [PubMed] [Google Scholar]
- Oji V, Eckl KM, Aufenvenne K, et al. Loss of corneodesmosin leads to severe skin barrier defect, pruritus, and atopy: unraveling the peeling skin disease. Am J Hum Genet. 2010;87:274–281. doi: 10.1016/j.ajhg.2010.07.005. http://dx.doi.org/10.1016/j.ajhg.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olive PL, Durand RE. Drug and radiation resistance in spheroids: cell contact and kinetics. Cancer Metastasis Rev. 1994;13:121–138. doi: 10.1007/BF00689632. http://dx.doi.org/10.1007/BF00689632. [DOI] [PubMed] [Google Scholar]
- Ott H, Bergstrom MA, Heise R, et al. Cutaneous metabolic activation of carvoxime, a self-activating, skin-sensitizing prohapten. Chem Res Toxicol. 2009;22:399–405. doi: 10.1021/tx8003642. http://dx.doi.org/10.1021/tx8003642. [DOI] [PubMed] [Google Scholar]
- Palmer TD, Takahashi J, Gage FH. The adult rat hippocampus contains primordial neural stem cells. Mol Cell Neurosci. 1997;8:389–404. doi: 10.1006/mcne.1996.0595. http://dx.doi.org/10.1006/mcne.1996.0595. [DOI] [PubMed] [Google Scholar]
- Paur I, Carlsen MH, Halvorsen BL, Blomhoff R. Antioxidants in Herbs and Spices: Roles in Oxidative Stress and Redox Signaling. In: Benzie IFF, Wachtel-Galor S, editors. Herbal Medicine: Biomolecular and Clinical Aspects. 2. Boca Raton, FL, USA: CRC Press; 2011. http://dx.doi.org/10.1201/b10787-3. [PubMed] [Google Scholar]
- Pavelka M, Roth J. Functional Ultrastructure: Atlas of Tissue Biology and Pathology. Vienna, New York: Springer Verlag; 2005. [Google Scholar]
- Peretz H, Talpalar AE, Vago R, et al. Superior survival and durability of neurons and astrocytes on 3-dimensional aragonite biomatrices. Tissue Eng. 2007;13:461–472. doi: 10.1089/ten.2005.0522. http://dx.doi.org/10.1089/ten.2005.0522. [DOI] [PubMed] [Google Scholar]
- Pfuhler S, Kirst A, Aardema M, et al. A tiered approach to the use of alternatives to animal testing for the safety assessment of cosmetics: genotoxicity. A COLIPA analysis. Regul Toxicol Pharmacol. 2010;57:315–324. doi: 10.1016/j.yrtph.2010.03.012. http://dx.doi.org/10.1016/j.yrtph.2010.03.012. [DOI] [PubMed] [Google Scholar]
- Ponec M, Gibbs S, Pilgram G, et al. Barrier function in reconstructed epidermis and its resemblance to native human skin. Skin Pharmacol Appl Skin Physiol. 2001:63–71. doi: 10.1159/000056392. http://dx.doi.org/10.1159/000056392. [DOI] [PubMed]
- Pottler M, Zierler S, Kerschbaum HH. An artificial three-dimensional matrix promotes ramification in the microglial cell-line, BV-2. Neurosci Lett. 2006;410:137–140. doi: 10.1016/j.neulet.2006.09.082. http://dx.doi.org/10.1016/j.neulet.2006.09.082. [DOI] [PubMed] [Google Scholar]
- Powers MJ, Domansky K, Kaazempur-Mofrad MR, et al. A microfabricated array bioreactor for perfused 3D liver culture. Biotechnol Bioeng. 2002;78:257–269. doi: 10.1002/bit.10143. http://dx.doi.org/10.1002/bit.10143. [DOI] [PubMed] [Google Scholar]
- Preynat-Seauve O, Suter DM, Tirefort D, et al. Development of human nervous tissue upon differentiation of embryonic stem cells in three-dimensional culture. Stem Cells. 2009;27:509–520. doi: 10.1634/stemcells.2008-0600. http://dx.doi.org/10.1634/stemcells.2008-0600. [DOI] [PubMed] [Google Scholar]
- Pusch J, Votteler M, Gohler S, et al. The physiological performance of a three-dimensional model that mimics the microenvironment of the small intestine. Biomaterials. 2011;32:7469–7478. doi: 10.1016/j.biomaterials.2011.06.035. http://dx.doi.org/10.1016/j.biomaterials.2011.06.035. [DOI] [PubMed] [Google Scholar]
- Ramirez T, Daneshian M, Kamp H, et al. Metabolomics in toxicology and preclinical research. ALTEX. 2013;30:209–225. doi: 10.14573/altex.2013.2.209. http://dx.doi.org/10.14573/altex.2013.2.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reus AA, Reisinger K, Downs TR, et al. Comet assay in reconstructed 3D human epidermal skin models – investigation of intra- and inter-laboratory reproducibility with coded chemicals. Mutagenesis. 2013;28:709–720. doi: 10.1093/mutage/get051. http://dx.doi.org/10.1093/mutage/get051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds BA, Tetzlaff W, Weiss S. A multipotent EGF-responsive striatal embryonic progenitor cell produces neurons and astrocytes. J Neurosci. 1992;12:4565–4574. doi: 10.1523/JNEUROSCI.12-11-04565.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringel M, von Mach MA, Santos R, et al. Hepatocytes cultured in alginate microspheres: an optimized technique to study enzyme induction. Toxicology. 2005;206:153–167. doi: 10.1016/j.tox.2004.07.017. http://dx.doi.org/10.1016/j.tox.2004.07.017. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Enriquez S, Gallardo-Perez JC, Aviles-Salas A, et al. Energy metabolism transition in multi-cellular human tumor spheroids. J Cell Physiol. 2008;216:189–197. doi: 10.1002/jcp.21392. http://dx.doi.org/10.1002/jcp.21392. [DOI] [PubMed] [Google Scholar]
- Roggen EL, Soni NK, Verheyen GR. Respiratory immunotoxicity: an in vitro assessment. Toxicol In Vitro. 2006;20:1249–1264. doi: 10.1016/j.tiv.2006.03.009. http://dx.doi.org/10.1016/j.tiv.2006.03.009. [DOI] [PubMed] [Google Scholar]
- Rothen-Rutishauser BM, Kiama SG, Gehr P. A three-dimensional cellular model of the human respiratory tract to study the interaction with particles. Am J Respir Cell Mol Biol. 2005;32:281–289. doi: 10.1165/rcmb.2004-0187OC. http://dx.doi.org/10.1165/rcmb.2004-0187OC. [DOI] [PubMed] [Google Scholar]
- Rothen-Rutishauser B, Blank F, Muhlfeld C, et al. In vitro models of the human epithelial airway barrier to study the toxic potential of particulate matter. Expert Opin Drug Metab Toxicol. 2008;4:1075–1089. doi: 10.1517/17425255.4.8.1075. http://dx.doi.org/10.1517/17425255.4.8.1075. [DOI] [PubMed] [Google Scholar]
- Ruhrberg C, Gerhardt H, Golding M, et al. Spatially restricted patterning cues provided by heparin-binding VEGF-A control blood vessel branching morphogenesis. Genes Dev. 2002;16:2684–2698. doi: 10.1101/gad.242002. http://dx.doi.org/10.1101/gad.242002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K, Nukada Y, Takenouchi O, et al. Development of a new in vitro skin sensitization assay (Epidermal Sensitization Assay; EpiSensA) using reconstructed human epidermis. Toxicol In Vitro. 2013;27:2213–2224. doi: 10.1016/j.tiv.2013.08.007. http://dx.doi.org/10.1016/j.tiv.2013.08.007. [DOI] [PubMed] [Google Scholar]
- Saltzman WM. Tissues Engineering: Engineering Principles for the Design of Replacement Organs and Tissuess. Oxford, New York: Oxford University Press; 2004. [Google Scholar]
- Samuelsson K, Bergstrom MA, Jonsson CA, et al. Diphenylthiourea, a common rubber chemical, is bioactivated to potent skin sensitizers. Chem Res Toxicol. 2011;24:35–44. doi: 10.1021/tx100241z. http://dx.doi.org/10.1021/tx100241z. [DOI] [PubMed] [Google Scholar]
- Schaefer O, Ohtsuki S, Kawakami H, et al. Absolute quantification and differential expression of drug transporters, cytochrome P450 enzymes, and UDP-glucuronosyltransferases in cultured primary human hepatocytes. Drug Metab Dispos. 2012;40:93–103. doi: 10.1124/dmd.111.042275. http://dx.doi.org/10.1124/dmd.111.042275. [DOI] [PubMed] [Google Scholar]
- Schäfer-Korting M, Bock U, Diembeck W, et al. The use of reconstructed human epidermis for skin absorption testing: Results of the validation study. Altern Lab Anim. 2008a;36:161–187. doi: 10.1177/026119290803600207. [DOI] [PubMed] [Google Scholar]
- Schäfer-Korting M, Mahmoud A, Lombardi Borgia S, et al. Reconstructed epidermis and full-thickness skin for absorption testing: influence of the vehicles used on steroid permeation. Altern Lab Anim. 2008b;36:441–452. doi: 10.1177/026119290803600405. [DOI] [PubMed] [Google Scholar]
- Schaller M, Schackert C, Korting HC, et al. Invasion of Candida albicans correlates with expression of secreted aspartic proteinases during experimental infection of human epidermis. J Invest Dermatol. 2000;114:712–717. doi: 10.1046/j.1523-1747.2000.00935.x. http://dx.doi.org/10.1046/j.1523-1747.2000.00935.x. [DOI] [PubMed] [Google Scholar]
- Schaller M, Zakikhany K, Naglik JR, et al. Models of oral and vaginal candidiasis based on in vitro reconstituted human epithelia. Nat Protoc. 2006;1:2767–2773. doi: 10.1038/nprot.2006.474. http://dx.doi.org/10.1038/nprot.2006.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheller K, Dally I, Hartmann N, et al. Upcyte(R) microvascular endothelial cells repopulate decellularized scaffold. Tissue Eng Part C Methods. 2013;19:57–67. doi: 10.1089/ten.tec.2011.0723. http://dx.doi.org/10.1089/ten.tec.2011.0723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schierle GS, Hansson O, Leist M, et al. Caspase inhibition reduces apoptosis and increases survival of nigral transplants. Nat Med. 1999;5:97–100. doi: 10.1038/4785. http://dx.doi.org/10.1038/4785. [DOI] [PubMed] [Google Scholar]
- Schildknecht S, Poltl D, Nagel DM, et al. Requirement of a dopaminergic neuronal phenotype for toxicity of low concentrations of 1-methyl-4-phenylpyridinium to human cells. Toxicol Appl Pharmacol. 2009;241:23–35. doi: 10.1016/j.taap.2009.07.027. http://dx.doi.org/10.1016/j.taap.2009.07.027. [DOI] [PubMed] [Google Scholar]
- Schildknecht S, Kirner S, Henn A, et al. Characterization of mouse cell line IMA 2.1 as a potential model system to study astrocyte functions. ALTEX. 2012;29:261–274. doi: 10.14573/altex.2012.3.261. http://dx.doi.org/10.14573/altex.2012.3.261. [DOI] [PubMed] [Google Scholar]
- Schimek K, Busek M, Brincker S, et al. Integrating biological vasculature into a multi-organ-chip microsystem. Lab Chip. 2013;13:3588–3598. doi: 10.1039/c3lc50217a. http://dx.doi.org/10.1039/c3lc50217a. [DOI] [PubMed] [Google Scholar]
- Schneeberger EE. Ultrastructure of intercellular junctions in the freeze fractured alveolar-capillary membrane of mouse lung. Chest. 1977;71:299–300. doi: 10.1378/chest.71.2_supplement.299. http://dx.doi.org/10.1378/chest.71.2_Supplement.299. [DOI] [PubMed] [Google Scholar]
- Schneeberger EE, Lynch RD. Tight junctions. Their structure, composition, and function. Circ Res. 1984;55:723–733. doi: 10.1161/01.res.55.6.723. http://dx.doi.org/10.1161/01.RES.55.6.723. [DOI] [PubMed] [Google Scholar]
- Schreiber T, Gassmann K, Gotz C, et al. Polybrominated diphenyl ethers induce developmental neurotoxicity in a human in vitro model: evidence for endocrine disruption. Environ Health Perspect. 2010;118:572–578. doi: 10.1289/ehp.0901435. http://dx.doi.org/10.1289/ehp.0901435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schurch S, Gehr P, Im Hof V, et al. Surfactant displaces particles toward the epithelium in airways and alveoli. Respir Physiol. 1990;80:17–32. doi: 10.1016/0034-5687(90)90003-h. http://dx.doi.org/10.1016/0034-5687(90)90003-H. [DOI] [PubMed] [Google Scholar]
- Schutte J, Hagmeyer B, Holzner F, et al. “Artificial micro organs” – a microfluidic device for dielectrophoretic assembly of liver sinusoids. Biomed Microdevices. 2011a;13:493–501. doi: 10.1007/s10544-011-9517-7. http://dx.doi.org/10.1007/s10544-011-9517-7. [DOI] [PubMed] [Google Scholar]
- Schutte M, Fox B, Baradez MO, et al. Rat primary hepatocytes show enhanced performance and sensitivity to acetaminophen during three-dimensional culture on a polystyrene scaffold designed for routine use. Assay Drug Dev Technol. 2011b;9:475–486. doi: 10.1089/adt.2011.0371. http://dx.doi.org/10.1089/adt.2011.0371. [DOI] [PubMed] [Google Scholar]
- Seok J, Warren HS, Cuenca AG, et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci U S A. 2013;110:3507–3512. doi: 10.1073/pnas.1222878110. http://dx.doi.org/10.1073/pnas.1222878110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shtrichman R, Germanguz I, Itskovitz-Eldor J. Induced pluripotent stem cells (iPSCs) derived from different cell sources and their potential for regenerative and personalized medicine. Curr Mol Med. 2013;13:792–805. doi: 10.2174/1566524011313050010. http://dx.doi.org/10.2174/1566524011313050010. [DOI] [PubMed] [Google Scholar]
- Simon BM, Malisan F, Testi R, et al. Disialoganglioside GD3 is released by microglia and induces oligodendrocyte apoptosis. Cell Death Differ. 2002;9:758–767. doi: 10.1038/sj.cdd.4401027. http://dx.doi.org/10.1038/sj.cdd.4401027. [DOI] [PubMed] [Google Scholar]
- Singec I, Knoth R, Meyer RP, et al. Defining the actual sensitivity and specificity of the neurosphere assay in stem cell biology. Nat Methods. 2006;3:801–806. doi: 10.1038/nmeth926. http://dx.doi.org/10.1038/nmeth926. [DOI] [PubMed] [Google Scholar]
- Sivertsson L, Synnergren J, Jensen J, et al. Hepatic differentiation and maturation of human embryonic stem cells cultured in a perfused three-dimensional bioreactor. Stem Cells Dev. 2013;22:581–594. doi: 10.1089/scd.2012.0202. http://dx.doi.org/10.1089/scd.2012.0202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smiley AK, Klingenberg JM, Aronow BJ, et al. Microarray analysis of gene expression in cultured skin substitutes compared with native human skin. J Invest Dermatol. 2005;125:1286–1301. doi: 10.1111/j.0022-202X.2005.23971.x. http://dx.doi.org/10.1111/j.0022-202X.2005.23971.x. [DOI] [PubMed] [Google Scholar]
- Stark HJ, Boehnke K, Mirancea N, et al. Epidermal homeostasis in long-term scaffold-enforced skin equivalents. J Investig Dermatol Symp Proc. 2006:93–105. doi: 10.1038/sj.jidsymp.5650015. http://dx.doi.org/10.1038/sj.jidsymp.5650015. [DOI] [PubMed]
- Steimer A, Haltner E, Lehr CM. Cell culture models of the respiratory tract relevant to pulmonary drug delivery. J Aerosol Med. 2005;18:137–182. doi: 10.1089/jam.2005.18.137. http://dx.doi.org/10.1089/jam.2005.18.137. [DOI] [PubMed] [Google Scholar]
- Stiegler NV, Krug AK, Matt F, et al. Assessment of chemical-induced impairment of human neurite outgrowth by multiparametric live cell imaging in high-density cultures. Toxicol Sci. 2011;121:73–87. doi: 10.1093/toxsci/kfr034. http://dx.doi.org/10.1093/toxsci/kfr034. [DOI] [PubMed] [Google Scholar]
- Strigun A, Noor F, Pironti A, et al. Metabolic flux analysis gives an insight on verapamil induced changes in central metabolism of HL-1 cells. J Biotechnol. 2011a;155:299–307. doi: 10.1016/j.jbiotec.2011.07.028. http://dx.doi.org/10.1016/j.jbiotec.2011.07.028. [DOI] [PubMed] [Google Scholar]
- Strigun A, Wahrheit J, Beckers S, et al. Metabolic profiling using HPLC allows classification of drugs according to their mechanisms of action in HL-1 cardiomyocytes. Toxicol Appl Pharmacol. 2011b;252:183–191. doi: 10.1016/j.taap.2011.02.008. http://dx.doi.org/10.1016/j.taap.2011.02.008. [DOI] [PubMed] [Google Scholar]
- Strigun A, Wahrheit J, Niklas J, et al. Doxorubicin increases oxidative metabolism in HL-1 cardiomyocytes as shown by 13C metabolic flux analysis. Toxicol Sci. 2012;125:595–606. doi: 10.1093/toxsci/kfr298. http://dx.doi.org/10.1093/toxsci/kfr298. [DOI] [PubMed] [Google Scholar]
- Sutherland RM, Eddy HA, Bareham B, et al. Resist ance to adriamycin in multicellular spheroids. Int J Radiat Oncol Biol Phys. 1979;5:1225–1230. doi: 10.1016/0360-3016(79)90643-6. http://dx.doi.org/10.1016/0360-3016(79)90643-6. [DOI] [PubMed] [Google Scholar]
- Teunis M, Corsini E, Smits M, et al. Transfer of a two-tiered keratinocyte assay: IL-18 production by NCTC2544 to determine the skin sensitizing capacity and epidermal equivalent assay to determine sensitizer potency. Toxicol In Vitro. 2013;27:1135–1150. doi: 10.1016/j.tiv.2012.06.004. http://dx.doi.org/10.1016/j.tiv.2012.06.004. [DOI] [PubMed] [Google Scholar]
- Thakoersing VS, Gooris GS, Mulder A, et al. Unraveling barrier properties of three different in-house human skin equivalents. Tissue Eng Part C Methods. 2012;18:1–11. doi: 10.1089/ten.TEC.2011.0175. http://dx.doi.org/10.1089/ten.tec.2011.0175. [DOI] [PubMed] [Google Scholar]
- Tostoes RM, Leite SB, Miranda JP, et al. Perfusion of 3D encapsulated hepatocytes – a synergistic effect enhancing long-term functionality in bioreactors. Biotechnol Bioeng. 2011;108:41–49. doi: 10.1002/bit.22920. http://dx.doi.org/10.1002/bit.22920. [DOI] [PubMed] [Google Scholar]
- Tostoes RM, Leite SB, Serra M, et al. Human liver cell spheroids in extended perfusion bioreactor culture for repeated-dose drug testing. Hepatology. 2012;55:1227–1236. doi: 10.1002/hep.24760. http://dx.doi.org/10.1002/hep.24760. [DOI] [PubMed] [Google Scholar]
- Tsukada T, Kouki T, Fujiwara K, et al. Reassembly of anterior pituitary organization by hanging drop three-dimensional cell culture. Acta Histochem Cytochem. 2013;46:121–127. doi: 10.1267/ahc.13015. http://dx.doi.org/10.1267/ahc.13015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuschl G, Hrach J, Walter Y, et al. Serum-free collagen sandwich cultures of adult rat hepatocytes maintain liver-like properties long term: a valuable model for in vitro toxicity and drug-drug interaction studies. Chem Biol Interact. 2009;181:124–137. doi: 10.1016/j.cbi.2009.05.015. http://dx.doi.org/10.1016/j.cbi.2009.05.015. [DOI] [PubMed] [Google Scholar]
- Uehara T, Kiyosawa N, Shimizu T, et al. Species-specific differences in coumarin-induced hepatotoxicity as an example toxicogenomics-based approach to assessing risk of toxicity to humans. Hum Exp Toxicol. 2008;27:23–35. doi: 10.1177/0960327107087910. http://dx.doi.org/10.1177/0960327107087910. [DOI] [PubMed] [Google Scholar]
- van Eijl S, Zhu Z, Cupitt J, et al. Elucidation of xenobiotic metabolism pathways in human skin and human skin models by proteomic profiling. PLoS One. 2012;7:e41721. doi: 10.1371/journal.pone.0041721. http://dx.doi.org/10.1371/journal.pone.0041721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Thriel C, Westerink RH, Beste C, et al. Translating neurobehavioural endpoints of developmental neurotoxicity tests into in vitro assays and readouts. Neurotoxicology. 2012;33:911–924. doi: 10.1016/j.neuro.2011.10.002. http://dx.doi.org/10.1016/j.neuro.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Vliet E, Stoppini L, Balestrino M, et al. Electrophysiological recording of re-aggregating brain cell cultures on multi-electrode arrays to detect acute neurotoxic effects. Neurotoxicology. 2007;28:1136–1146. doi: 10.1016/j.neuro.2007.06.004. http://dx.doi.org/10.1016/j.neuro.2007.06.004. [DOI] [PubMed] [Google Scholar]
- Vandenhaute E, Sevin E, Hallier-Vanuxeem D, et al. Case study: Adapting in vitro blood-brain barrier models for use in early-stage drug discovery. Drug Discov Today. 2012;17:285–290. doi: 10.1016/j.drudis.2011.10.006. http://dx.doi.org/10.1016/j.drudis.2011.10.006. [DOI] [PubMed] [Google Scholar]
- Vávrová K, Henkes D, Strüver K, et al. Filaggrin deficiency leads to impaired lipid profile and altered acidification pathways in a 3D skin construct. J Invest Dermatol. 134:746–53. doi: 10.1038/jid.2013.402. in press. http://dx.doi.org/10.1038/jid.2013.402. [DOI] [PubMed] [Google Scholar]
- Viviani B, Corsini E, Galli CL, et al. Glia increase degeneration of hippocampal neurons through release of tumor necrosis factor-alpha. Toxicol Appl Pharmacol. 1998;150:271–276. doi: 10.1006/taap.1998.8406. http://dx.doi.org/10.1006/taap.1998.8406. [DOI] [PubMed] [Google Scholar]
- Viviani B. Preparation and coculture of neurons and glial cells. Curr Protoc Cell Biol. 2006;Chapter 2(Unit 27) doi: 10.1002/0471143030.cb0207s32. http://dx.doi.org/10.1002/0471143030.cb0207s32. [DOI] [PubMed] [Google Scholar]
- Volbracht C, Leist M, Nicotera P. ATP controls neuronal apoptosis triggered by microtubule breakdown or potassium deprivation. Mol Med. 1999;5:477–489. [PMC free article] [PubMed] [Google Scholar]
- Volbracht C, Leist M, Kolb SA, et al. Apoptosis in caspase-inhibited neurons. Mol Med. 2001;7:36–48. [PMC free article] [PubMed] [Google Scholar]
- Volbracht C, van Beek J, Zhu C, et al. Neuroprotective properties of memantine in different in vitro and in vivo models of excitotoxicity. Eur J Neurosci. 2006;23:2611–2622. doi: 10.1111/j.1460-9568.2006.04787.x. http://dx.doi.org/10.1111/j.1460-9568.2006.04787.x. [DOI] [PubMed] [Google Scholar]
- Votteler M, Kluger PJ, Walles H, et al. Stem cell microenvironments – unveiling the secret of how stem cell fate is defined. Macromol Biosci. 2010;10:1302–1315. doi: 10.1002/mabi.201000102. http://dx.doi.org/10.1002/mabi.201000102. [DOI] [PubMed] [Google Scholar]
- Wagner I, Materne EM, Brincker S, et al. A dynamic multi-organ-chip for long-term cultivation and substance testing proven by 3D human liver and skin tissue co-culture. Lab Chip. 2013;13:3538–3547. doi: 10.1039/c3lc50234a. http://dx.doi.org/10.1039/c3lc50234a. [DOI] [PubMed] [Google Scholar]
- Waldmann T, Rempel E, Balmer NV, et al. Design principles of concentration-dependent transcriptome deviations in drug-exposed differentiating stem cells. Chem Res Toxicol. 2014;27:408–420. doi: 10.1021/tx400402j. http://dx.doi.org/10.1021/tx400402j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weibel ER. Stereological Methods. Vol I: Practical Methods for Biological Morphometry. London-New York-Toronto: Academic Press; 1979. [Google Scholar]
- Weindl G, Naglik JR, Kaesler S, et al. Human epithelial cells establish direct antifungal defense through TLR4-mediated signaling. J Clin Invest. 2007;117:3664–3672. doi: 10.1172/JCI28115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weindl G, Castello F, Schafer-Korting M. Evaluation of anti-inflammatory and atrophogenic effects of glucocorticoids on reconstructed human skin. Altern Lab Anim. 2011;39:173–187. doi: 10.1177/026119291103900212. [DOI] [PubMed] [Google Scholar]
- Winge MC, Hoppe T, Berne B, et al. Filaggrin genotype determines functional and molecular alterations in skin of patients with atopic dermatitis and ichthyosis vulgaris. PLoS One. 2011;6:e28254. doi: 10.1371/journal.pone.0028254. http://dx.doi.org/10.1371/journal.pone.0028254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada KM, Cukierman E. Modeling tissue morphogenesis and cancer in 3D. Cell. 2007;130:601–610. doi: 10.1016/j.cell.2007.08.006. http://dx.doi.org/10.1016/j.cell.2007.08.006. [DOI] [PubMed] [Google Scholar]
- Yuhas JM, Li AP, Martinez AO, et al. A simplified method for production and growth of multicellular tumor spheroids. Cancer Res. 1977;37:3639–3643. [PubMed] [Google Scholar]
- Zahir N, Weaver VM. Death in the third dimension: apoptosis regulation and tissue architecture. Curr Opin Genet Dev. 2004;14:71–80. doi: 10.1016/j.gde.2003.12.005. http://dx.doi.org/10.1016/j.gde.2003.12.005. [DOI] [PubMed] [Google Scholar]
- Zeilinger K, Sauer IM, Pless G, et al. Three-dimensional co-culture of primary human liver cells in bioreactors for in vitro drug studies: effects of the initial cell quality on the long-term maintenance of hepatocyte-specific functions. Altern Lab Anim. 2002;30:525–538. doi: 10.1177/026119290203000506. [DOI] [PubMed] [Google Scholar]
- Zeilinger K, Schreiter T, Darnell M, et al. Scaling down of a clinical three-dimensional perfusion multicompartment hollow fiber liver bioreactor developed for extracorporeal liver support to an analytical scale device useful for hepatic pharmacological in vitro studies. Tissue Eng Part C Methods. 2011;17:549–556. doi: 10.1089/ten.TEC.2010.0580. http://dx.doi.org/10.1089/ten.tec.2010.0580. [DOI] [PubMed] [Google Scholar]
- Zimmer B, Lee G, Balmer NV, et al. Evaluation of developmental toxicants and signaling pathways in a functional test based on the migration of human neural crest cells. Environ Health Perspect. 2012;120:1116–1122. doi: 10.1289/ehp.1104489. http://dx.doi.org/10.1289/ehp.1104489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zöller NN, Kippenberger S, Thaci D, et al. Evaluation of beneficial and adverse effects of glucocorticoids on a newly developed full-thickness skin model. Toxicol In Vitro. 2008;22:747–759. doi: 10.1016/j.tiv.2007.11.022. http://dx.doi.org/10.1016/j.tiv.2007.11.022. [DOI] [PubMed] [Google Scholar]
- Zurich MG, Monnet-Tschudi F, Berode M, et al. Lead acetate toxicity in vitro: Dependence on the cell composition of the cultures. Toxicol In Vitro. 1998;12:191–196. doi: 10.1016/s0887-2333(97)00089-1. http://dx.doi.org/10.1016/S0887-2333(97)00089-1. [DOI] [PubMed] [Google Scholar]
- Zurich MG, Eskes C, Honegger P, et al. Maturation-dependent neurotoxicity of lead acetate in vitro: implication of glial reactions. J Neurosci Res. 2002;70:108–116. doi: 10.1002/jnr.10367. http://dx.doi.org/10.1002/jnr.10367. [DOI] [PubMed] [Google Scholar]
- Zurich MG, Honegger P, Schilter B, et al. Involvement of glial cells in the neurotoxicity of parathion and chlorpyrifos. Toxicol Appl Pharmacol. 2004;201:97–104. doi: 10.1016/j.taap.2004.05.003. http://dx.doi.org/10.1016/j.taap.2004.05.003. [DOI] [PubMed] [Google Scholar]
- Zurich MG, Stanzel S, Kopp-Schneider A, et al. Evaluation of aggregating brain cell cultures for the detection of acute organ-specific toxicity. Toxicol In Vitro. 2013;27:1416–1424. doi: 10.1016/j.tiv.2012.06.018. http://dx.doi.org/10.1016/j.tiv.2012.06.018. [DOI] [PubMed] [Google Scholar]