

Abstract
The estrogenic properties of bisphenol A (BPA), a ubiquitous synthetic monomer that can leach into the food and water supply, have prompted considerable research into exposure-associated health risks in humans. Endocrine-disrupting properties of BPA suggest it may impact developmental plasticity during early life, predisposing individuals to disease at doses below the oral reference dose (RfD) established by the Environmental Protection Agency in 1982. Herein, we review the current in vivo literature evaluating the carcinogenic properties of BPA. We conclude that there is substantial evidence from rodent studies indicating that early-life BPA exposures below the RfD lead to increased susceptibility to mammary and prostate cancer. Based on the definitions of “carcinogen” put forth by the International Agency for Research on Cancer and the National Toxicology Program, we propose that BPA may be reasonably anticipated to be a human carcinogen in the breast and prostate due to its tumor promoting properties.
Keywords: Bisphenol A, Cancer, Mammary, Prostate, Uterus, Ovary, Estrogen Receptor, Testes
1. Introduction
Incidence and prevalence of cancers of endocrine target organs, including prostate, breast, and testis, as well as other diseases such as infertility and obesity began steadily increasing in the 1970's and reached an elevated plateau in 2002 [1, 2]. The incidence of breast cancer increased by 26% during this time period , while prostrate and testicular cancers increased by 94% and 56%, respectively. Increased exposure to environmental synthetic estrogens, such as BPA, has been postulated to contribute, in part, to this increasing incidence [3, 4]. BPA is a synthetic monomer used in the production of polycarbonate plastics, epoxy resin linings of canned foods and beverage containers, dental sealants, and thermal receipt paper. In 2003, more than 6 billion pounds of BPA were produced worldwide [5], and production is expected to exceed 5.4 million tons this year (for detailed international market analysis, see Bisphenol A (BPA): 2015 World Market Outlook and Forecast up to 2019 from mcgroup.co.uk). The United States Environmental Protection Agency (EPA) estimates that over 1 million pounds of BPA leaches into the environment each year and over 90% of tested humans have detectable BPA in their systems with the highest levels found in infants and children [6-11].
BPA is an estrogenic compound [12]. It has a similar structure as the highly potent estrogen receptor (ER) agonist, diethylstilbestrol (DES), and binds classical nuclear ER alpha and beta, as well as membrane-associated GPR30, (Figure 1) albeit with lower affinity [13]. Thus, BPA is expected to have effects on ER function in addition to other nuclear hormone receptors and most of the studies on BPA action have focused on hormone sensitive tissues. The ubiquitous presence of BPA in the environment, concomitant with the increased prevalence of endocrine-related cancers, has led to numerous studies evaluating the role of BPA in carcinogenesis. In 1982, the National Toxicology Program (NTP) conducted a toxicology analysis and concluded that while pharmacological doses of BPA induced some cancers in both male and female adult rodents, it was not a robust carcinogen at doses relevant to human exposure [14, 15]. Hence, the EPA and the U.S. Food and Drug Administration established a safe reference dose (RfD) for humans at 50 μg/kg/day, based on a 1,000-fold reduction of the dose used in the NTP study (www.epa.gov/iris/subst/0356.htm). While dose scaling is valid for agents that follow linear dose-response relationships, many endocrine-disruptors, like their endogenous hormonal counterparts, demonstrate a non-monotonic dose-response curve. In this case, lower doses are as relevant as higher doses [16-18]. In addition, cancer susceptibility may be established during fetal and postnatal organ development and exposure during these times was not assessed by the NTP study. During various developmental windows, tissues are finely attuned to endocrine input for establishing tissue architecture. Altering this milieu can predispose individuals to diseases manifested later in life (reviewed in [19]). BPA has been measured in maternal serum and ovarian follicular fluid, as well as in amniotic fluid and fetal plasma, indicating passage across the placenta during pregnancy [20, 21]. Furthermore, studies in primate and rodent fetuses and newborns suggest that the liver has a limited ability to metabolize BPA, creating the potential for BPA to be detrimental during critical developmental stages [22, 23]. Given these limitations, the impact of BPA on sex-steroid responsive organs required additional study beyond the NTP analysis.
Figure 1.
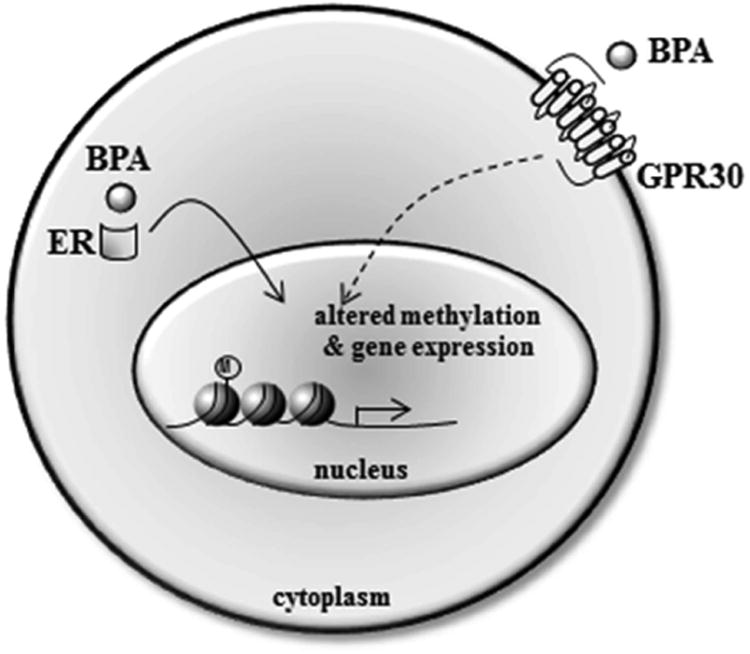
While the mechanism of action of BPA in humans is largely unknown, in vitro BPA effects are primarily mediated through its binding to classical nuclear ERs or through membrane-initiated non-genomic signaling by GRP30. In vivo, early life exposure to BPA rewrites the epigenetic code to stably alter gene expression in the breast, uterus and prostate. M=methylation.
RTI International conducted a study sponsored by the Polycarbonate/BPA Global Group, an organization that promotes the interests and welfare of the major manufacturers of polycarbonate plastic and BPA, to evaluate the effects of early life BPA exposures on multiple reproductive parameters utilizing the rat strain, Crl:CD(SD), derived from Sprague-Dawley by Charles River Laboratories. [24]. The three-generation study revealed no increase in cancer of any organ system examined with chronic BPA exposure from gestation through adulthood at both low (0.001-5 mg/kg/day) and high doses (50-500 mg/kg/day) [24]. While these data suggest that fetal BPA exposure does not increase cancer incidence, limitations of the study included use of an animal model that is resistant to endocrine disruption and a narrow selection of organs analyzed. Additionally, animals were examined in young adulthood (3-4 months of age) prior to when cancer endpoints are typically observed without a carcinogenic challenge [17, 25]. In an attempt to address some of these concerns, RTI conducted a subsequent two-generation study utilizing CD-1 mice, estrogen treatment as a positive control, and low-doses of BPA (0.003-600 mg/kg/day) from gestation through adulthood [26]. While histopathological analysis revealed no cancer in any tissue examined, animals were only aged ∼15-20 weeks, a time point too early for most cancers to become evident. Further, the mice displayed a relative insensitivity to estrogen compared to other studies as well as other strains, diminishing the ability to make strong conclusions regarding the effects of low doses of BPA [26, 27]. Indeed, many studies on the reproductive effects of BPA exposure continue to report dichotomous results due to disparity in the use of animal models, dose, timing, and route of exposure [28-30].
The debate over the health risk of BPA exposure spurred the National Institutes of Health (NIEHS, NIDCR) and the EPA to assemble a panel of experts in endocrine-disruption to review the literature and compile a consensus report evaluating the association between BPA exposure and human health risk. As a result of this meeting, we extensively reviewed the weight of evidence for the carcinogenicity of BPA in 2007. Based on the scientific evidence at that time, we were confident that BPA displayed estrogenic properties and acted as an endocrine-disruptor [28]. We also concluded that BPA was likely to be associated with increased malignancies of the testes and hematopoietic system and increased susceptibility to neoplastic lesions in mammary and prostate glands following early-life exposures. However, insufficient data on tumor formation in response to BPA in vivo, coupled with vastly varying experimental designs, precluded conclusions specifically on the carcinogenic impact of BPA. The review panel then established guidelines to address these discrepancies with the purpose of firmly addressing BPA-induced health risks using environmentally relevant doses, often referred to as ‘low doses’ or doses below the LOAEL (50mg/kg/day). These guidelines have since been followed by numerous groups evaluating the carcinogenic impact of BPA, resulting in an expansion of our understanding of the effects of this endocrine disruptor.
Herein, we present an updated analysis of the weight of evidence for the carcinogenicity of BPA. Studies using BPA doses at or below the RfD (50 μg/kg/day) are given greater weight and are considered here as ‘low dose’ because they more closely model the most conservative estimates of environmentally relevant BPA exposures and the RfD is the dose considered “safe” by the EPA and the U.S. Food and Drug Administration. While we refer to all other BPA doses as “high doses”, the actual extent of human exposure to BPA, as well as the route of exposure, is under debate as bio-monitoring studies suggest that human-relevant exposures are indeed higher than the RfD [31, 32]. While critical, accurate approaches for measuring the BPA exposome (i.e. the total human life-time exposure of BPA) have not yet been developed and validated; hence we consider examining doses below the RfD as the most conservative approach for assessing the potential impact of BPA on establishing cancer risk.
While the NTP study conducted in 1982 concluded that BPA exposure could increase the incidence of hematopoietic malignancies in Fischer 344 rats, no further analyses of this malignancy have been reported. Due to the estrogenic action of BPA, this review primarily focuses on in vivo assessments of carcinogenesis in the male and female reproductive tracts as well as other proposed estrogen target tissues (a summary of studies is presented in Table 1). However, we do not exclude any non-reproductive tissues from this analysis as the list of estrogen target tissues continues to expand [33-35]. Similar to the NTP and the International Agency on Research on Cancer (IARC), we evaluate the strength of the evidence to gauge the carcinogenicity of BPA and not the potency of its carcinogenic activity. Further, we note that negative studies do not necessarily indicate that BPA is not a carcinogen, as these experiments are conducted under a specific set of conditions; standards also exercised by the NTP. Lastly, we include a series of conclusions regarding the current state of this field and areas requiring further study.
Table 1. Summary of In Vivo Studies Since 2007 on the Carcinogenicity of BPA.
| Organ | Species/Strain | Dose | Route | Timing | Tumor Initiator | Outcome | Reference |
|---|---|---|---|---|---|---|---|
| Females | |||||||
| Mammary | Mice/Balb/c | 25μg/kg/day | Oral | Puberty-3 weeks | DMBA | Increased lateral branches and hyperplasia; neoplastic lesions in MaSC regenerated mammary glands | (Wang et al. 2014) |
| Mammary | Mice/C57BL/6-ERα+/- | 250ng/kg/day | Osmotic Pump | E8 to parturition | -- | Global changes in the stromal and epithelial transcriptome, altered stromal and epithelial compartments in the mammary gland | (Wadia et al, 2013) |
| Mammary | Rats/SD | 50-2 50μg/kg/day | Oral | Birth to PND21 | DMBA | Increased number of mammary tumors | (Betancourt et al. 2012) |
| Mammary | Mice/MMTV-erbB2 | 2.5 -2,500μg/L drinking water | Oral | 56 to 252 days of age | activated erbB2 | Decreased tumor latency, increased tumor multiplicity, tumor burden and | (Jenkins et al. 2011) |
| Mammary | Mice/FVB/N | 2 -250μg/kg/day | Oral | E8 to parturition | DMBA | Decreased tumor latency and increased tumor susceptibility | (Weber Lozada et al. 2011) |
| Mammary | Mice/C57BL/6 | 0.6μg - 1.2mg/kg/day | Oral | E1 to PND24 | -- | Increased number of TEB and PR expressing mammary epithelial cells | (Ayyanan et al. 2011) |
| Mammary | Rats/SD | 25-250μg/kg/day | Oral | PND2 to PND21 and E2 to E20 | DMBA | Decreased tumor latency, and increased number of mammary tumors. | (Lamartiniere et al.2011) |
| Mammary | Mice/CD-1 | 5mg/kg/day | IP | E9 to parturition | -- | No effect on EZH2 mRNA expression, increased EZH2 protein expression and | (Doherty et al. 2010) |
| Mammary | Rats/SD | 25-250 μg/kg/day | Oral | E10 to E21 | DMBA | Decreased tumor latency and increased tumor susceptibility | (Betancourt et al. 2010) |
| Mammary | Rats/SD | 25- 250 μg/kg day | Oral | PND2 - PND20 | DMBA | Decreased tumor latency and increase in mammary tumor formation | (Jenkins et al. 2009) |
| Mammary | Rats/SD | 25-250μg/kg/day | Oral | E10 to parturition | -- | Increased number of TEBs, TDs, and lobular structures during development | (Moral et al. 2008) |
| Mammary | Rats/Wistar | 25μg/kg/day | Osmotic pump | E8 to E23 | NMU | Increase in ductal hyperplasia formation | (Durando et al. 2007) |
| Mammary Mammary | Rats/Wistar/furth Rats/SD | 2.5 -1000μg/kg/day; 0.25 to 250μg/kg/day | Osmotic Pump; Osmotic pump | E9 to PND1;E9 to PND 1 and E9 to PND21 | --- | Increase in ductal hyperplasia formation, presence of DCISPalpable tumors (carcinomas) | (Murray et al. 2007) (Acevedo et al 2013) |
| Mammary | Rats/W/F | 250μg/kg/day | Osmuotic pump | E9 to PND1 | - | Global epigenome alterations (DNA methylation at PND4, 21, 50 Altered gene expression pattern at PND50 | (Dhimolea et al, 2014) |
| Ovary | Rats/Eker | 50mg/kg/day | s.c. | PND10-12 | Tsc Ek/+ mutation | normal ovary morphology: presence of corpora lutea, normal estrous cyclicity | (Greathouse et al. 2012) |
| Ovary | Mice/CD-1 | 10mg/kg/day | s.c. | PND15-18 | -- | No morphological abnormalities | (Nikaido et al. 2005) |
| Ovary | Mice/CD-1 | 10-1000μg/kg/day | s.c. | PND1-5 | -- | Increased ovarian cysts (100μg/kg) and progressive proliferative lesions | (Newbold et al. 2007) |
| Ovary | Mice/CD-1 | 0.1-1000 μg/kg/day | s.c. | E9-16 | -- | Increased ovarian cysts (1μg/kg only ) and cystadenomas; increased progressive proliferative lesions | (Newbold et al. 2009) |
| Ovary | Rats/SD | 5-500μg/kg/day | s.c. | PND1-10 | -- | Increased polycystic ovaries and infertility (500μg/kg/day only) | (Fernandez et al. 2010) |
| Uterus | Mice/CD-1 | 10-1000μg/kg/day | s.c. | PND1-5 | -- | Increased incidence of cystic endometrial hyperplasia, increased trend in adenomyosis, increased leiomyomas | (Newbold et al. 2007) |
| Uterus | Mice/CD-1 | 0.1-1000 μg/kg/day | s.c. | E9-16 | -- | No instances of leiomyoma, some atypical hyperplasia and stromal polyps, sarcoma of uterine cervix | (Newbold et al. 2009) |
| Uterus | Rats/Wistar | 0.5-50mg/kg/day | s.c. | ∼PND91-95 | -- | No change in uterine morphology | (Okuda et al. 2010) |
| Uterus | Rats/Eker | 50mg/kg/day | s.c. | PND10-12 | -- | No increased frequency of multiplicity of leiomyoma | (Greathouse et al. 2012) |
| Vagina | Rats/Eker | 50mg/kg/day | s.c. | PND10-12 | Tsc Ek/+ mutation | Normal vaginal morphology | (Greathouse et al. 2012) |
| Males | |||||||
| Prostate | Rat/SD | 10μg/kg/day | s.c. | PND1, 3, 5 | E | Hypomethylation of PDE4D4 with increased expression | (Prins et al. 2008) |
| Prostate | Rats/SD | 10-90μg/kg/day | i.g. | adult- 4 week treatment | T | Increased prostate weight, volume, epithelial cell height, decreased testosterone and increased PSA | (Wu et al. 2011) |
| Prostate | Rats/SD | 10μg/kg/day | s.c and oral | PND1,3&5 | T+E | Increased incidence of PINs | (Prins et al. 2011) |
| Prostate | Rat/SD | 10μg/kg/day | s.c. | PND1,3,5 | T+E | Altered methylation/expression of Nsbp1, Hpcal1 and genes involved in methylation | (Tang et al. 2011) |
| Prostate | Mice/C57BL/6 | 20μg/kg/day | Oral | E13-E16 | - | Elevated CYP19A1 activity and increased E2 levels in the urogenital | (Arase et al. 2011) |
| Prostate | Rats/Wistar | 25-600μg/kg/day | s.c. | adult-4day treatment | -- | Increased 5α-reductase and plasma E:T ratio | (Castro et al. 2013) |
| Prostate | Rats/SD | 25-250μg/kg/day | Oral gavage | E10-E21 | -- | Increased incidence of multifocal hyperplasia/dysplasia; PINs | (Brandt et al. 2014) |
| Prostate | Mice/Nude | 100-250μg/kg/day | Oral | Daily treatment for 2 weeks post xenograft | T+E | Increased PINS and prostate adenocarcinoma of human prostate epithelium in renal capsule xenograft model | (Prins et al. 2014) |
| Prostate | Rats/SD | 2-50μg/kg/day | s.c./oral | PND1,3,5 | T+E | Increased lateral prostatic inflammation and PIN lesions | (Wong et al. 2015) |
| Testes | Rats/Long-Evans | 2.5-25μg/kg/day | Oral | E12-PND21 | -- | Increased Leydig cell number | (Nanjappa et al. 2012) |
| Testes | Mice/C57BL/6 | 50-1000μg/kg/day | Oral | E10-16 | -- | No changes in sperm production, germ cell apoptosis, serminiferous tubule histology, serum testosterone | (LaRocca et al. 2011) |
| Testes | Rats/SD | 285.4ppm (food) | Oral | adult-3month treatment | -- | No histological abnormalities | (Zhang et al. 2013) |
| Females and Males | |||||||
| Liver | Mice/Avy C3HeJ/C57BL/6 | 0.5ng-50mg/kg/day | Oral | E0-PND22 | -- | Increased hepatic neoplastic and preneoplastic lesions | (Weinhouse et al. 2014) |
2. Methods
A search for published manuscripts analyzing the effects of BPA treatment in in vivo mammalian models was performed utilizing Pubmed and Google Scholar search engines, among others. Principle searches were conducted using “cancer” and “bisphenol” as key words and subsequently refined with additional key words such as testes, brain, mammary, etc.
3. Epidemiological Data
In 2008, Lang, et al. reported the levels of urinary BPA in samples collected by the National Health and Nutrition Examination Survey (NHANES) of U.S. adults that had also completed a health survey [36]. No association between elevated BPA concentrations and cancer was evident. However, “cancer” was listed as a single category, potentially masking associations of BPA with specific malignancies. Indeed, the cross-sectional design of this study provides limited data regarding extent, timing and duration of BPA exposure as only a single time point was investigated. Further, patient cancer history was dependent on self-reporting and it is not known whether the subjects had occult tumors at the time of urine collection. While small studies have reported BPA concentrations in cancer patients with selected tumor types, large-scale epidemiological data is lacking. Such studies are necessary to obtain sufficient statistical power to identify concurrent low-dose exposures to BPA and associated cancers as well as patient outcomes from specific disease subtypes. Most importantly, large birth cohorts documenting fetal/early life BPA exposure with lifetime follow-up are needed to fully evaluate the impact of environmental BPA exposure during early life on cancer risk. While informative, data from such studies would not be available for decades due to the nature of the study design, the current inability to measure the timing and extent of exposure, and the lack of a funding mechanism to collect, analyze and house the data generated by a life-time cohort. Thus, it is critical to focus on other data streams to assess the potential impact of BPA exposure on cancer risk.
4. Cancers of the female breast and reproductive tract
4.1 BPA and Breast Cancer Incidence
Other than skin cancer, breast cancer is the most common malignancy among American women, and the second leading cause of cancer death (ACS Statistics, 2012). Breast cancer incidence steadily increased from 1970-2000 (National Cancer Institute's Surveillance, Epidemiology and End Results Program), correlating with increased production of BPA [37]. The potential impact of BPA specifically on breast cancer in women has been evaluated in two studies. Yang, et al analyzed 167 blood samples collected between 1994 and 1997 from Korean women, and measured the level of BPA in blood by HPLC/FD. Blood BPA levels were not associated with increased breast cancer risk [38]. However, given the large variability in BPA levels across the sample set, this study was likely to be underpowered to identify an association. It also did not assess exposures during breast development. Recently, a study was conducted analyzing the urinary BPA metabolite, BPA-glucuronide (BPA-G), in postmenopausal Polish breast cancer patients at the time of diagnosis [39]. BPA-G measurement is reflective of BPA metabolism, thus reducing the potential of sample contamination with BPA during collection and analysis. Data on 575 invasive breast cancer cases recruited from 2 sites (Warsaw or Lodz, Poland), including 384 ER+ and 191 ER- cases, was collected as part of a previously described Polish Breast cancer study and compared to age and study-site matched controls. [40]. Overnight urine samples were collected in propylene tubes and creatinine-adjusted unconjugated and BPA-G measured by HPLC/MS/MS in the same laboratory by the same technician. No association between urinary BPA-G and breast cancer risk was noted, nor were there significant differences in urinary BPA-G between patients with ER+ versus ER- cancer. However, BPA-G levels were significantly elevated in women from Warsaw (n=420) compared to those from Lodz (n=155), possibly confounding risk estimates. Like the Korean breast cancer study, one-time, adult assessment of BPA levels is inadequate to accurately discern BPA exposure levels during the critical windows of mammary gland development or a cumulative life-time of BPA exposure.
Rat Models of BPA-induced mammary carcinogenesis
Throughout its development, the mammary gland undergoes hormonally-induced morphogenetic changes in the stromal and epithelial compartments. At the cellular level, mammotropic hormones regulate cell proliferation, apoptosis and the expression of specific genes and proteins. At the tissue level, hormones regulate ductal elongation, branching, lobuloalveolar development and post-weaning remodeling. All these phenomena involve stromal-epithelial interactions [41]. During these morphogenetic processes mammary tissue is exquisitely sensitive to neoplastic insults, as observed in women exposed to the synthetic estrogen diethylstilbestrol during pregnancy (mothers) and gestation (daughters), and may be highly susceptible to the presence of endocrine disrupting chemicals such as BPA [42-44]. It is noteworthy that dysregulated cell signaling, manifested as hyperplasia, is considered to be one of the first steps toward transformation and neoplasia [45].
The majority of in vivo studies examining BPA effects on mammary gland carcinogenesis have used rat models. While multiple exposure time points have been examined, a consistent impact of BPA on mammary gland architecture has been revealed particularly when exposure occurs during embryogenesis. Fetal exposure of Wistar-Furth rats significantly increases intraductal hyperplasia at postnatal day (PND) 50 and PND95 when dams received 2.5 μg/kg/day BPA by implanted osmotic pump [46]. Both Ki67 and ERα expression were significantly increased in the hyperplastic lesions compared to normal ducts in controls, indicating increased proliferation and estrogenic activity. During puberty, fetal exposure to BPA also results in an increase in the proliferation/apoptosis ratio in both the epithelial and stromal compartments. These changes translated to an increase in the number of hyperplastic ducts by post-natal day 110; the ducts were also surrounded by a more fibroblastic and dense stroma in the BPA-treated cohort, suggestive of a desmoplastic reaction.
When a range of doses (2.5-1,000 μg/kg/day) were used in an in utero exposure paradigm with Wistar-Furth rats that involved BPA delivery by mini-osmotic pumps implanted in the mothers on embryonic day 9, BPA caused comparable changes in the mammary gland, including a 3-4 fold increase in hyperplastic ducts and an increase in estrogen receptor (ER) positive cells. Of note, this study also revealed a BPA-associated incidence of mammary intraepithelial neoplasias (MIN), described as cribriform carcinoma in situ in 25-33% of adult rats following fetal exposure [46]. Further supporting the conclusion that BPA increases mammary tumor incidence in rats, Sprague Dawley rats exposed to a range of BPA doses (0.25, 2.5, 25, 250 μg BPA/kg/day) via osmotic pumps during gestation or during both gestation and post-natally through the mother's milk developed macroscopic tumors by PND90, at a rate of 3.5%. Although free BPA could only be measured in 100% of the dams and fetuses exposed to 250 μg BPA/kg/day, carcinomas appeared at doses as low as 0.25 μg/kg/day. These results suggest that BPA may have carcinogenic properties when administered perinatally [47].
An increase in the number of terminal end buds (TEB) and lobular structures of the mammary gland were also observed in 21 day old SD female rats exposed in utero to a high dose of BPA (250 μg/kg/day) but not to a low dose (25 μg/kg/day) delivered by oral gavage to the mother. Although early morphological changes were consistent between all studies using different rat strains, no change in proliferation was observed at any dose or stage of development in SD rats [48]. At 100 days, in utero exposure (250 μg/kg/day) also resulted in variations in gene expression of differentiation genes that may modify gland development in SD rats [48]. However, the specific role of these expression changes on BPA response and cancer susceptibility is not yet known.
Using similar dosing protocols, the impact of BPA on susceptibility to mammary tumor formation in rats has been assessed following carcinogenic insults with N-nitroso-N-methylurea (NMU) or 7,12-Dimethylbenz(a)anthracene (DMBA). During postnatal development, the mammary gland contains a large number of TEBs. As indicated above, these structures are highly sensitive to carcinogens due to their high rate of proliferation and undifferentiated state [49, 50]. The surrounding stroma is also susceptible to insult and can contribute to the subsequent formation of cancers [51, 52]. As indicated above, fetal BPA exposure (25 μg/kg/day, osmotic pump beginning E8 to E23) to Wistar rats increased the number of hyperplastic ducts at PND110 and PND180 and these were associated with desmoplastic stroma and increased mast cells [53]. To further investigate the likelihood of a susceptibility effect on mammary tumorigenesis, another cohort was treated in utero from E8 to E23, with low dose BPA (25 μg/kg/day), and additionally administered a subcarcinogenic dose of NMU (25 mg/kg) at 50 days of age. BPA-exposed animals had an increase of 2.8 and 2.3 fold in the number of preneoplastic lesions in response to NMU at PND 110 and PND180; respectively. In addition, 2 of 15 mice had cribriform ductal carcinoma in situ [53]. This study complements those demonstrating an increase in neoplastic lesions with BPA exposure, suggesting that BPA can also increase susceptibility to an NMU-induced carcinogenic event later in life.
Several studies utilizing DMBA as the carcinogenic insult in Sprague-Dawley CD rats offer similar evidence that BPA can increase mammary cancer susceptibility. Jenkins, et al presented the first evidence that delivery of high dose (250 μg/kg/day) BPA by oral gavage to lactating SD dams from PND2 through PND20, can significantly increase susceptibility of the offspring to DMBA-induced mammary tumors. A similar trend was observed for low dose (25 μg/kg/day) exposure [54]. The higher dose cohort also had a shorter tumor latency and increased multiplicity. Similarly, in utero exposure to BPA beginning on embryonic day 10 (E10) through embryonic day 21 (E21), also increased mammary tumor susceptibility to DMBA in SD-CD rats, but this was dependent upon the timing of carcinogenic insult [55]. When DMBA was given at PND50, there was no difference in tumor latency or in tumor multiplicity in either the low (25 μg/kg/day) or high (250 μg/kg/day) dose cohorts compared to controls. However, when DMBA was administered at PND100, a significant increase in tumor incidence was observed in the high dose BPA cohort (250 μg/kg/day). In addition to tumors, rats exposed in utero to high dose BPA and PND100 DMBA treatment also displayed an increase in proliferation and significant upregulation of ERa as well as its downstream targets, progesterone receptor (PR) and Bcl-2 in the mammary gland, suggesting that BPA may change the gland's sensitivity to circulating estrogens. A similar shift in the window of DMBA susceptibility was observed with in utero exposure (E2 through PND21) of SD-CD rats to BPA at low (25 μg/kg/day) or high (250 μg/kg/day) doses. DMBA failed to induce tumor formation when given at PND50, while increased tumor formation was observed when DMBA exposure occurred at PND100 following high dose (250 μg/kg/day) fetal administration of BPA [56]. Together, these studies indicated that the timing of a carcinogenic event is critical to observe an effect of BPA exposure on mammary tumor susceptibility (Table 1). Importantly, windows of breast cancer susceptibility also exist in women. Exposure of pubertal girls to radiation from the atomic bombs in Japan increased their risk for developing breast cancer later in life whereas exposure of older women did not alter their breast cancer incidence [57].
In addition to prenatal BPA exposure, the impact of early postnatal exposures on mammary cancer susceptibility has been evaluated. SD-CD female rats were exposed to low (25 μg/kg/day) and high (250 μg/kg/day) doses of BPA by oral gavage of the mother, delivering BPA to the pups through the milk from PND2-21. This was followed by treatment with DMBA at PND50. BPA had a dose-dependent effect on mammary tumor formation with a significant increase in the number of tumors observed in the high dose cohort (250 μg/kg/day). Tumor latency was also significantly decreased for BPA-treated rats compared to controls for both doses. BPA exposure led to an age-dependent increase in mammary epithelial cell proliferation, decreased apoptosis, up-regulation of several proteins involved in cell growth, and an increase in AKT signaling [56]. Similar outcomes were observed with pre-pubertal exposure to BPA followed by DMBA where administration of high dose BPA (250 μg/kg/day) to pre-pubertal SD-CD rats, and subsequent exposure to DMBA at PND50, significantly increased the number of mammary tumors compared to controls [58]. In addition to the increase in tumor formation, Betancourt, et al. also observed differential expression of several proteins crucial to extracellular matrix composition, indicating a potential effect of BPA on both stromal and epithelial cells, both of which may contribute to the increased susceptibility to carcinogenic insults.
As a whole, these studies using distinct rat strains indicate that low dose exposures to BPA (25 μg/kg/day) in utero or pre-pubertally cause a range of mammary gland changes including increased proliferation, decreased apoptosis, hyperplasia, and increased proliferative structures such as TEBs. These results were obtained in the majority of reports for BPA exposure of female rats at a low dose. When BPA exposure is coupled with either NMU or DMBA treatment, several studies reported an increase in mammary tumor formation, a decrease in latency, and an increase in multiplicity. The most reported effects occurred upon exposure to high doses of BPA (250 μg/kg/day) either in utero or pre-pubertally. In addition to the high dose results, two different labs also reported a statistically significant increase in tumor susceptibility with low dose BPA (25 μg/kg/day). Differences in the dosing/timing of susceptibility may be explained by the presence or absence of tumor-susceptible terminal end buds and terminal ducts during the time of treatment with carcinogen, or an effect of BPA on stromal susceptibility. Based on these findings we conclude that BPA increases the susceptibility of the adult mammary gland to subsequent exposures to chemical carcinogens. Importantly, BPA exposure resulted in some neoplasias in multiple studies in the absence of a NMU or DMBA challenge, suggesting that BPA can act as a carcinogen.
Mouse models of BPA-induced mammary carcinogenesis
The effects of BPA exposure on the mammary gland have also been investigated in various murine models using several genetic strains and transgenic mice. Studies in CD-1 mice revealed that in utero or perinatal exposure to BPA caused intraductal mammary epithelial hyperplasias [59]. Vandenberg, et al., reported that in utero exposure from E8 to E18 to 0.25 μg/kg/day BPA by osmotic pumps in outbred CD-1 mice significantly increased adipose differentiation of the stroma, altered the organization of the extracellular matrix (ECM), increased ductal area and length and inhibited lumen formation in mammary glands at E18 indicating that BPA exposure during fetal development can alter normal mammary gland morphogenesis [60]. Subsequently, Wadia, et al. examined the transcriptome of the stroma and epithelium of C57BL/6 female mice at E19, showing that 0.025 μg BPA/kg/day, osmotic pump beginning at E8, induced comparable changes to those obtained with ethinylestradiol [61]. Moreover, the transcriptome patterns were consistent with the morphological changes reported by Vandenberg, et al. [60], supporting the postulate that BPA acts through ERs located in the stroma and affects stromal-epithelial interactions. Using the same protocol in CD-1 mice, a series of publications have shown that 0.025μg and 25μg BPA/kg/day administered gestationally (osmotic pumps) results in morphological alterations at puberty, increased sensitivity to estradiol, earlier expression of progesterone receptor (PR) positive epithelial cells, increased lateral branching, increased lobuloalveolar development and intraductal hyperplasias [59-62].
Addition of BPA (0.6 μg-1.2 mg/kg/day) to the drinking water of C57BL/6 breeding pairs exposes the pregnant dam as well offspring to BPA from gestation through lactation [63]. The mammary glands of female offspring displayed a significantly increased number of terminal end buds at puberty when their mothers were given a 3 μg/kg/day dose. Adult female offspring also had an elevated number of mammary epithelial cells following a 6 μg/kg/day or 12 μg/kg/day dose compared to controls. These increases were comparable to those seen in mice treated with diethylstilbestrol (DES), a positive-control estrogenic compound with established pro-tumorigenic effects in the mammary gland [63]. There were also an increased number of progesterone receptor positive cells in the luminal epithelium, indicating elevated estrogenic activity. The increase in PR expression provides a potential mechanism for the findings of Munoz de Toro, et al., 2005 where early exposure to BPA enhanced sensitivity of the mammary gland to progesterone [43].
To determine if prenatal exposure to BPA could induce precocious puberty or alter mammary gland development of inbred FVB/N mice, pregnant dams were treated beginning on E8 through parturition with vehicle or low dose BPA (25 μg/kg/day, oral gavage). Female pups exposed to BPA in utero exhibited early puberty as demonstrated by accelerated vaginal opening. There were no changes in ductal length or notable qualitative morphological differences in mammary gland development at any of the stages examined including prepuberty, puberty, postpuberty, pregnancy, lactation, or involution [64]. These results contrast with previous studies and may be due to difference in strain, BPA delivery, dose or end point measured [43, 63]. Indeed, Soto and colleagues reported a non-monotonic effect when examining the impact of BPA on TEB number [65].
As in rats, the DMBA-induced mammary gland carcinogenesis model has been used to assess the impact of BPA on tumor susceptibility. Exposure of pregnant FVB/N dams to BPA (25 μg/kg/day or 250 μg/kg/day, oral gavage), followed by treatment of female offspring with DMBA during puberty at 5 and 6 weeks of age, revealed a dose-dependent increase in mammary tumor susceptibility compared to controls [64]. These results further support the conclusion that prenatal BPA exposure increases mammary tumor susceptibility later in life. Recently, Wang et al. examined the effect of postnatal exposure to low dose (25 μg/kg/day) BPA with a subsequent DMBA challenge on the ability of mammary stem cells to recapitulate the mammary gland in a BALB/c xenograft model. Prepubertal (21 days old) females were orally gavaged with BPA for 3 weeks, followed by one oral dose of DMBA at 8 weeks of age. Mammary stem cells were isolated at various ages and transplanted. Lateral branching, hyperplasia, and luminal progenitors were all increased by exposure to the combination of BPA and DMBA. In addition, glands reconstituted from mammary stem cells that were previously exposed to BPA during puberty formed neoplastic lesions similar to glands exposed to DMBA. This is the first report that exposure to BPA alters the function of mammary stem cells [66]. Given the proposed roles of stem cells in mammary tumorigenesis, these studies provide a plausible mechanism for the ability of BPA to increase tumor susceptibility [67].
Low dose BPA also accelerated mammary tumorigenesis and increased metastasis in a transgenic mouse model of erbB2/HER2-induced breast cancer [68]. Beginning after puberty at 56 days of age, female MMTV-neuT mice were exposed to BPA via the drinking water until PND 252. Lower doses of BPA (2.5 μg/kg/day and 25 μg/kg/day) significantly reduced the time to initial tumor detection, increased tumor multiplicity and volume, and lung metastases. Interestingly, none of these effects were manifested with higher doses of BPA, indicating again that the effects of BPA can follow an inverted-U dose–response, with larger effects at lower doses. These data further indicate that exposure to BPA during adulthood can also increase susceptibility to mammary tumorigenesis, in this case induced by erbB2/HER2 overexpression.
In summary, several studies reported a significant impact of BPA on mouse mammary architecture and suggest that perinatal exposure to BPA increases the sensitivity of the gland to estradiol and progesterone, hormones that control cancer risk in this tissue. However, it is difficult to derive comprehensive and consistent conclusions on the effects that BPA has on the morphology of the mouse mammary gland due to lack of consistent use of strain, delivery methods, and end points studied (Table 1). Reports using distinct mouse breast cancer models and dosing paradigms have repeatedly found that BPA increases tumor susceptibility when using a variety of tumor models and the available mouse data support findings derived from rats. Thus, these data strongly support a carcinogenic role of BPA in the mammary gland.
Epigenetic effects of BPA on the mammary gland
The principal mechanism of action of BPA appears to be its estrogenic activity [61] (Figure 1), yet many of its effects are observed long past the duration of exposure. Thus, the impact of BPA must extend beyond immediate transcriptional regulation in both stromal and epithelial cells. Indeed, alterations of the transcriptomes of both tissue compartments observed during the period of exposure may explain the morphological and functional effects observed later in life ([61] and reviewed in [65]). Microarray analyses of rat mammary glands exposed to BPA in utero revealed a host of changes in gene expression in low (25μg/kg/day) and high (250μg/kg/day, oral gavage) dose cohorts [48]. While changes in gene expression were modest at PND21 and 35; the number of differentially-regulated genes increased at both PND50 and PND100. Expression changes included genes involved in cell death, immune function, response to stress, and differentiation, among others [48]. Although not directly tested, it is likely that at least some of these changes impact mammary cancer susceptibility. The fact that many gene expression changes were delayed also supports the possibility that BPA may have an impact on the mammary epigenome or the systemic hormonal milieu. Supporting an epigenetic impact, Dhimolea, et al. examined the methylation profile of the DNA of mammary glands of WF rats exposed in utero (ED9 to PND1) to BPA at 250 μg/kg/day using osmotic pumps. Of the 58,207 genomic loci probed, 812 were hyper-methylated and 675 were hypo-methylated at PND 4, 1,904 were hyper-methylated and 1,787 hypo-methylated at PND 21, and 1,072 were hyper-methylated and 1,162 hypo-methylated at PND 50 in the BPA-exposed animals compared with vehicle controls. The methylation patterns induced by BPA showed dynamic changes over time. Relatively few genomic DNA methylation differences between BPA- and vehicle-treated groups were maintained throughout the three time-points. This may suggest a time-dependent methylation change in the vehicle-treated group that did not occur in the BPA-treated group [69]. Of note, transcriptome changes were only observed at PND 50, i.e., the time at which increased incidence of intraductal hyperplasias and DCIS are observed in this rat strain. Genes whose expression was elevated in the BPA-treated group included those associated with cell cycle regulation (eg. JUN and CDKN1c). These changes appear to be a consequence of the developmental alterations caused by BPA rather than causal events that result in neoplastic development later in adulthood. Given their timing, they may reflect alterations in stromal-epithelial communication during gland morphogenesis rather than direct carcinogenic pathways within the epithelial cells and may reflect sustained effects on epithelial-stromal communication within the gland.
Together, animal modeling studies suggest that BPA increases breast cancer susceptibility. While no human data are currently available to confirm this postulate, one primate study has been reported. Female neonates born to rhesus monkeys exposed during pregnancy to orally-delivered BPA (400 μg/kg/day BW, ED100 to ED165) exhibit a significantly increased density of mammary buds [70]. These data are consistent with those at E18 in CD-1 mice [60]. As a whole, studies using multiple animal models indicate that early life exposure to BPA increases mammary cancer risk. A new study in SD rats reported that low doses of BPA (0.25, 2.5, and 25 μg/kg BW from ED 9-birth and from ED 9-PND 21, osmotic pump) induce mammary carcinomas on their own, supporting the notion that BPA is a direct breast carcinogen [47]. With the addition of this study, the weight of evidence in rodent models reported by independent laboratories supports the conclusion that BPA at doses below the RfD increases the susceptibility of the mammary gland to develop cancer [47, 60, 64]. These findings and conclusions are summarized in the Overall Conclusions of this report.
4.2 Ovarian cancer
Low doses of BPA alter oocyte meiotic events [71-73], yet data demonstrating an association between BPA exposure and ovarian cancer is lacking. The study conducted by RTI discussed above concluded that there was no increase in cancer risk in many reproductive tissues, although detailed histological analysis of the ovary was not reported [24]. In separate studies, Eker rats and CD-1 mice treated with BPA pre-pubertally [50 mg/kg/day subcutaneously (s.c.) PND10-12 and 10mg/kg/day s.c. PND15-18, respectively] also showed no histological ovarian abnormalities [74, 75]. In contrast to these analyses that utilized very high doses, Newbold et al treated CD-1 mice with lower doses of BPA (10-1000 μg/kg/day s.c.) during PND 1-5. By 18 months, mice receiving 100 μg/kg/day BPA had more ovarian cysts compared to controls, although cystic ovaries and progressive proliferative lesions of the oviduct were common in both treated and control groups [76]. The high incidence of lesions in the control group is likely due to exposure to environmental estrogens from housing materials. Most importantly, when in utero exposure (ED 9-16, s.c.) was assessed under BPA-free housing conditions, a significant increase in ovarian cysts occurred at a much lower BPA dose (1 μg/kg/day) compared to controls [77]. Other ovarian abnormalities included benign cystadenomas, although this was not statistically significant. Polycystic ovaries have also been noted in SD rats treated from PND 0-9 with 4 mg/kg/day BPA s.c. [78] and PND 1-10 with ∼25mg/kg/day BPA s.c. [79] at ∼2.5 and 4-5months, respectively. While the studies using SD rats independently confirm that BPA induces cystic ovarian lesions, BPA was administered at levels well above the RfD (currently set at 50 μg/kg/day) and higher than estimated human exposure levels. Given the non-monotonic effects observed in other tissues, additional studies using lower doses are necessary to determine if BPA exposure may impact ovarian cancer susceptibility in rodents.
While the animal studies remain incomplete, studies in women also suggest that BPA exposure may be associated with increased incidence of ovarian cysts. Circulating BPA levels were ∼46% higher in a cohort of 71 women with polycystic ovary syndrome (1.05 ± 0.56 ng/ml) compared to 100 controls (0.72 ± .37 ng/ml) [80]. While intriguing, long term follow-up studies with large cohorts are necessary to determine if women with elevated BPA concomitant with PCOS have an increased risk of developing ovarian cancer. Ovarian cystadenomas have a significant impact on health, however these lesions have no association with increased risk of ovarian carcinoma [81, 82]. In contrast, women with polycystic ovary disease have a 2-3 fold increased risk of endometrial and ovarian cancers [83, 84]. Together, the human and rodent studies reveal that BPA exposure consistently induces ovarian pathologies that may ultimately be associated with increased cancer risk. However, the lack of overt tumor formation in the currently available reports suggests that BPA may not have carcinogenic activity in the ovary. Future studies focusing on the role of BPA in cancer progression utilizing environmentally relevant dosing paradigms in accurate animal models of ovarian cancer are necessary to more fully elucidate its impact on ovarian cancer risk.
4.3 Uterine cancer
The uterus is exquisitely regulated by estrogens and progestins, which control growth of the endometrium. Like the ovary and breast, prolonged or elevated estrogenic exposure is associated with increased risk of endometrial cancer. Indeed, early life exposure to another endocrine disruptor, DES, induces leiomyoma and uterine cancer in mice [85]. In humans, it was reported that lower serum BPA levels were associated with endometrial hyperplasia with malignant potential as well as endometrial cancer in a small cohort of premenopausal Japanese women [86]. However, this study was limited to nine premenopausal women with hyperplasia and seven postmenopausal women with cancer. Furthermore, serum BPA was measured by enzyme-linked immunosorbent assay (ELISA), the use of which has been questioned due to the potential for cross reactivity with other compounds [87, 88]. A larger, more recent study found no association with urinary BPA and the odds of leiomyoma diagnosis in 495 women across 14 clinical sites in the United States [89]. Notably, only a single BPA measurement was performed at the time of examination or study enrollment in either study, a typical limitation of epidemiological studies examining the association of BPA with human disease. To fully discern the longitudinal impact of BPA on uterine physiology, animal models have been necessary.
It was expected that adult or gestational exposure to BPA, due to its endocrine disruptor properties, would increase the incidence of uterine anomalies in animal models. However, treatment of SD rats with high doses of BPA (0.1-50 mg/kg/day) in utero (ED 6-21) until weaning failed to increase the incidence of benign leiomyomas or uterine carcinomas when examined at 4 months of age [90]. Spontaneous development of leiomyomas and uterine carcinomas is rare in SD rats and this strain is generally resistant to endocrine disruption in the vagina and uterus by estrogens [91-93]. In fact, in utero exposure to the powerful synthetic estrogen, DES, only induces benign uterine lesions at the extended age of 14 months in SD rats, suggesting evaluation after only 4 months would be insufficient to observe any BPA-induced pathology [94]. Nevertheless, this study did find that BPA decreased uterine epithelial wall thickness and caused a two-fold increase in the accumulation of pronounced nuclei with condensed chromatin [90].
A similar lack of impact on the uterus was observed when adult female ovariectomized Wistar rats were treated with high BPA doses (0.5-50 mg/kg/day, s.c.) for 5 days [95]. However, studies in rodents have shown that the uterus is less sensitive to the proliferative effects of estrogen during adulthood when compared to neonatal exposure, with PND 3-12 being the most sensitive window to induce developmental reprogramming [74, 96]. Consistently, no evidence of uterine pathologies was found in outbred CD-1 mice after being treated at 14-18 days of age with high-dose BPA (10 mg/kg/day, s.c.) and then analyzed at 24 weeks [75]. Again, this is an earlier time point than the observation of uterine pathologies induced by DES in this strain (∼13months of age) and doses well above the RfD were used [97].
In contrast to the above studies, when using a neonatal exposure protocol (PND 1-5) of 100 μg/kg/day of BPA administered s.c. to CD-1 mice, a significant seven-fold increase in cystic endometrial hyperplasia (CEH) was observed when the mice reached 18 months of age. Leiomyoma was observed at each BPA dose (10, 100 and 1000 μg/kg/day) and not in controls, although the increased incidence of leiomyoma did not reach statistical significance [76]. In the same mouse strain, gestational exposures (0.1-1000 μg/kg/day) on embryonic days 9-16 yielded no increase in incidence of CEH in the offspring at 18 months of age in exposed versus control animals and evidence of leiomyoma was not reported [77]. This may be due to BPA exposure occurring prior to the developmental window when the uterus is most sensitive (PND 3-12) to reprogramming by xenoestrogens [74, 96].
BPA effects have also been studied using rodent models that are susceptible to adverse effects of endocrine disruption in the uterus, including the genetically-predisposed Eker rats as well as Donryu rats treated with the carcinogen N-ethyl-N′-nitro-N-nitrosoguanidine (ENNG) [98-100]. A 2-day treatment of neonatal Eker rats with high doses of BPA s.c. (PND10-12 at 50 mg/kg/day) failed to increase frequency or multiplicity of leiomyomas by 16 months of age despite observing increased pathologies with DES treatment, a key positive control [74]. Early life exposure to low doses of BPA (6 μg/kg/day, ED2-PND20) in Donryu rats with the additional carcinogenic insult of ENNG also did not increase the incidence of uterine malignancies over controls at 15 months of age [101]. These data suggest that early life exposure to BPA does not induce neoplastic changes in the uterus. However, each of these studies has significant shortcomings that preclude definitive conclusions. Neonatal BPA treatment for two days at the end of the developmental window, as in the 2012 Greathouse study [74], may be insufficient to induce uterine pathologies with age and, as mentioned previously, higher BPA doses may be less effective and are less environmentally relevant. While the study in Donyru rats utilized conservative estimates of environmentally relevant doses of BPA, serum and tissue analyses revealed detectable BPA levels in all study animals, including the controls. This is likely due to the BPA contamination reported by the authors in the drinking water and pellet diet [101].
Given the limitations of the current studies, it remains unclear whether BPA can increase the risk of developing uterine cancer or benign leiomyomas. Rodent studies examining BPA are confounded by strain-specific differences in uterine sensitivity to estrogenic input, including the dose and timing of BPA exposure. Additionally, while morphological changes have been reported in the vagina with BPA treatment, studies are limited and no evidence of overt tumors has been reported [74, 75, 77, 102].
5. Cancers of the Male Reproductive Tract
5.1 Prostate Cancer
The first clinical evidence that BPA exposure may be associated with prostate cancer has recently been reported. In a cohort of 60 urologic patients, urinary BPA levels were normalized to creatinine content to adjust for variability of urine dilution. This study revealed that there were significantly higher urinary BPA levels in biopsy-confirmed prostate cancer patients than those without prostate cancer and this difference was even more profound in prostate cancer patients less than 65 years of age [103]. In these younger patients, serum prostate specific antigen (PSA) levels were negatively associated with urine BPA measurements, suggesting that urinary BPA may be a better diagnostic biomarker of prostate cancer than the traditional PSA analysis. Although a single urine sample with elevated BPA represents only a snap-shot in the patient's lifetime, it may be indicative of a lifestyle that sustains higher levels of BPA exposure. The preliminary association between elevated BPA levels and prostate cancer is remarkable particularly in light of a growing body of evidence supporting a cancer promoting potential of BPA in animal models as described below.
Dysregulation of the temporal pattern of estrogen exposure impairs prostate morphogenesis in rodents [104]. While estrogens are not required for prostate budding [105], sustained estrogen or estrogen mimetics, such as DES, predispose the gland to prominent dysplasia analogous to prostatic intraepithelial neoplasms (PINs) in humans. In adult rodents, prolonged 17β-estradiol and testosterone treatment (E+T) also induces PINs as well as invasive prostate adenocarcinomas [106]. Moreover, adult men with chronically elevated estrogens are at an increased risk of prostate cancer [107]. A novel mouse model has been developed to assess the effects of estrogenic compounds on human prostate morphogenesis [108]. This model uses human prostate stem-progenitor cells from young, disease-free human organ donors mixed with rat embryonic mesenchyme to generate chimeric prostate-like humanized tissues when grown under the renal capsule of host mice. Exposure of the PSA-expressing, human prostate epithelium to elevated estradiol in an androgen-supported milieu for 2-4 months in vivo transforms the epithelium and induces prostate cancer albeit at a low incidence of 11% [108]. Nonetheless, this model provided direct evidence that estradiol is a carcinogen for human prostatic epithelium. This leads to the question of whether estrogenic compounds such as BPA may also be carcinogenic in the prostate.
Prior to 2007, studies examining the impact of early-life BPA exposures on prostate cancer risk yielded conflicting results, likely due to differences in experimental design and dosing regimens. Fisher 344 rats exposed to BPA (0.05-120 mg/kg/day from E1 through lactation, orally to dams) and subsequently treated at 5 weeks of age with DMBA had the same incidence of PIN or prostate cancer as controls at 65 weeks of age [109]. The equivalent incidence of pathologies between the treatment groups may be explained by the failure to control for exposures to environmental estrogens leached from the caging, water bottles and diet, thus exposing all animals in this study to BPA. No serum BPA measurements were reported, making it difficult to reach definitive conclusions from these data. In a separate evaluation, exposures to 0.001-500 mg/kg/day BPA from ED 0 throughout adulthood also failed to induce prostate cancer in SD rats when examined at 15 weeks of age [24]. Analysis at 15 weeks is likely too early to observe pathological changes associated with BPA treatment, as epithelial hyperplasia and dysplasia is not even evident until at least 6 months of age with neonatal DES treatment [110]. It is also noteworthy that circulating BPA measurements were also not reported and no positive controls were utilized in this study.
Since these studies were reported, evidence has been accumulating that supports a cancer-promoting effect of BPA on the prostatic epithelium. BPA treatment of adult SD rats (10 μg/kg/day; i.p. for 4 weeks) potentiates testosterone-induced benign prostate hyperplasia and increases prostate weight in SD rats. These effects decreased with increasing doses of BPA, indicative of a non-monotonic dose response [111]. Recently, a study evaluating low and high doses of BPA (25 or 250 μg/kg/day) administered in utero (ED 10-21) to SD rats (orally to the dams) was conducted to determine whether gestational exposure to indole-3-carbinol (I3C), a bioactive compound found in cruciferous vegetables, could reverse the harmful effects of BPA on the prostate. At the lower doses, BPA alone was sufficient to induce a significant increase in hyperplasia/dysplasia of the ventral prostate at PND180 (0% incidence in controls vs 62% incidence with BPA treatment) which was reversed with I3C [112]. While this study did not report overt cancers induced by BPA, it did reveal that doses of BPA below the RfD initiate precancerous events in the prostate epithelium. Moreover, when a carcinogenic regimen that mimics the rising estrogen:testosterone ratio observed in aging men was used in another report, BPA (10 μg/kg/day, s.c. on PND 1, 3 and 5) treated neonatal rats developed significantly more PIN lesions at 28 weeks of age than animals that did not receive BPA [113]. To address concerns regarding subcutaneous administration of BPA, Prins and colleagues have dosed neonatal rats with 10 μg/kg/day BPA s.c. or orally to compare susceptibilities to (E+T)-induced prostate cancer [114]. While free plasma BPA was modestly higher within the 30 min. immediately following s.c injection versus oral ingestion, values of free BPA for both routes of exposure were within the range observed in humans (0.2-20 ng/ml serum) [115]. Notably, BPA exposure by either route (10 μg/kg/day on PND 1,3 and 5), followed by the E+T regimen, caused a similar increase in PIN lesions, prostatic epithelial hyperplasia and chronic inflammation when compared to controls receiving E+T alone. Thus, despite higher circulating BPA levels during the first hour of exposure, subcutaneous delivery of BPA in neonatal rodents results in environmentally relevant doses of free BPA with physiological outcomes that phenocopy those induced with oral administration. Most recently, a dose-response analysis examined neonatal (PND 1, 3 and 5) exposure to BPA at 0.1, 1, 10, 100 or 5,000 μg/kg/day delivered s.c. in oil (Prins and Ho, 2013, unpublished data). Serum levels of free and glucuronidated BPA were measured by HPLC-MS-MS 1 hour post-exposure in PND 3 pups and although free BPA was undetectable in oil controls and the 0.1 and 1.0 μg BPA dose, the 10 μg/kg/day dose resulted in 0.72 ng/ml serum levels. Levels then rose linearly with dose thereafter. Neonatal BPA exposure alone did not produce PIN lesions in any prostate lobe at any dose when assessed at 7 months. However, with adult T+E exposure, all doses of neonatal BPA, including those with undetectable free BPA, resulted in a significant increase in PIN incidence in the lateral prostate lobe compared to controls given T+E alone. Further, an inverted U-shaped dose-response in PIN incidence was noted in the dorsal and ventral lobes given adult T+E with a peak in PIN lesions occurring with 100 μg BPA/kg/day.
Recently, this approach was repeated by an independent group [116]). In that study, SD rats were treated neonatally with oil or BPA (s.c. at 10 μg/kg/day or oral at 2, 10 or 50 μg/kg/day), then given T+E as adults and assessed for pathology at 1 year. Pharmacokinetic analysis by HPLC-MS-MS showed similar free BPA levels in serum between the s.c. 10 μg/kg/day and oral 50 μg/kg/day BPA exposures at day 3. A low incidence of well-differentiated adenocarcinoma was observed in the periurethral prostatic ducts as a function of prolonged adult T+E treatment, however, the incidence and severity of these lesions were not altered by neonatal BPA exposures at any dose. The principal prostatic lesions observed in 12 month-old rats exposed to BPA were lateral prostatic inflammation and PIN lesions. Compared to controls, overall PIN lesions were significantly increased in rats treated subcutaneously with 10 μg/kg/day of BPA. Further, greater than minimal PIN incidence was significantly higher in both the subcutaneous (10 μg/kg/day) and orally (50 μg/kg/day) exposed lateral prostates as compared to controls. This provides additional support that early-life BPA exposures increase susceptibility to estrogen-driven carcinogenesis. PIN lesions have been classified by the Mouse Models of Human Cancer Consortium as focal, atypical and progressive lesions, and are frequently, but not always, associated with progression to cancer with age [117]. Whether PINs induced by early life BPA exposure will progress to invasive carcinoma is unknown and remains to be established. It is notable that s.c. BPA delivery (25-600μg/kg/day) for four days in adult Wistar rats increases the plasma E:T ratios in a dose dependent manner [118]. Correspondingly, levels of 5α-reductase type 3, a proposed biomarker for malignant prostate epithelial cells in humans, were also increased [119]. Additionally, fetal BPA exposure at the onset of prostatic budding (20 μg/kg/day orally from ED13-16) elevated CYP19A1 activity and estradiol levels in the urogenital sinuses of PND1 C57BL/6 mice [120].
Several studies have identified plausible mechanisms of action of BPA on prostate cancer risk. On a molecular level, evidence for epigenetic alterations in the prostate with early life BPA exposure provides a potential mechanism of action for low dose BPA [121]. With normal aging, hypermethylation of the Pde4d4 and nucleosome-binding protein 1 (Nsbp1) genes is associated with decreased gene expression. However, subcutaneous BPA treatment (10 μg/kg/day) on neonatal days 1, 3 and 5 in SD rats reverses this trend, resulting in hypomethylation and aberrant expression of Pde4d4 and Nsbp1 later in life [122, 123]. Expression of both PDE4D4 and NSBP1 is associated with cancer cell proliferation [124, 125]. Indeed, NSBP1 is required for growth of xenografted human prostate cancer cells and its expression is upregulated in human prostate cancers compared to normal tissue [126, 127]. Similarly, secretoglobin family 2A member 1 (Scgb2a1) expression is upregulated over 100-fold in the adult prostates of rats following neonatal BPA treatment [116]. Significant CpG hypomethylation was identified within 10kb upstream of the Scgb2a1 promoter and associated with acetylation at H3K9, suggesting neonatal BPA exposure results in sustained epigenetic reprogramming that is maintained in the adult prostate. SCGB2A1 overexpression is also a marker of disseminated breast tumor cells and is predictive of resistance to chemo- and radiotherapy in colorectal cancer. It increases proliferation and cancer stem cell survival [128, 129]. Thus, these epigenetic changes are consistent with early life BPA increasing susceptibility to prostate cancer as well as response to chemotherapeutics later in life.
To determine whether the human prostate is similarly sensitive to BPA, a recent study utilized human prostate epithelial stem-like cells cultured from prostates of young, disease-free human donors [130]. These cells express estrogen receptors ERα, ERβ and GPR30. Similar to 17β-estradiol (E2), BPA increased human prostate stem/progenitor cell self-renewal and expression of stem-related genes in a dose-dependent manner, in vitro. Further, 10 nM BPA or E2 possessed equimolar membrane-initiated signaling with robust induction of phosphorylated Akt and Erk at 15 min. Subsequent studies determined that 7 days of 10, 200 or 1000 nM BPA exposure altered the transcriptome of the human prostate progenitor cells with an overrepresentation for multiple SNORDS, a class of non-coding RNAs [131]. Furthermore, BPA-induced repression of several SNORDs was associated with specific histone modifications (H3K4me3 and H3K27me3 loss and gain of H3K9me3) at their promoter regions, thus identifying an epigenetic basis for prostate progenitor cell reprogramming.
To assess in vivo carcinogenicity, human prostate stem/progenitor cells combined with rat mesenchyme were grown as renal grafts in nude mice, forming histologically normal human prostate epithelium at 1 month. Developmental BPA exposure was achieved through oral administration of 100 μg or 250 μg/kg/day to hosts for 2 weeks post-grafting, producing free-BPA levels of 0.39 and 1.35 ng/ml serum, respectively. Carcinogenesis was driven by E+T treatment for 2-4 months to model rising E2 levels in aging men. The incidence of prostate intraepithelial neoplasia (HG-PIN) and adenocarcinoma markedly increased from 13% in oil-fed controls to 33-36% in grafts exposed in vivo to either BPA dose. Continuous developmental BPA exposure to prostate stem/progenitors in vitro (200 nM) plus in vivo BPA treatment to engrafted mice (250 μg/kg/day) significantly increased HG-PIN/cancer incidence to 45%. These findings demonstrate that human prostate stem/progenitor cells are direct BPA targets and that developmental exposure to BPA at low-doses increases steroid hormone-dependent cancer risk in the human prostatic epithelium. In total, these new findings suggest that the effects of BPA in rodent prostates are translatable to the human gland and that developmental exposures may increase risk of adult-onset prostate cancers in humans.
Together, these data indicate that early life BPA exposure induces/increases prostate lesions in rodent prostate cancer models as well as in human xenografts. Moreover, elevated urinary BPA levels are associated with prostate cancer in humans [103]. While mounting evidence indicates that BPA likely has carcinogenic activity in the prostate, further studies from independent investigators replicating some of the above findings are necessary to determine if BPA is a bona fide carcinogen in human prostate epithelial cells.
5.2 Testicular cancer
The NTP study of 1982 reported that pharmacological doses of BPA increased the incidence of testicular interstitial cancers in mice and rats, although the controls also had an elevated baseline of tumor incidence compared to historical controls [14]. In contrast, an analysis sponsored by the plastics industry determined that BPA exposure [0.001-500 mg/kg/day BPA (in food from gestation throughout adulthood)] alone was insufficient to induce testicular cancer in SD rats when examined at 15weeks of age [24]. While no effects were observed in the second study, circulating BPA levels were not described and no positive controls were included in the report. An independent analysis also failed to detect histological anomalies of the testes in adult SD rats fed BPA (285.4 ppm) for 3 months [132]. Additionally, treatment of C57BL/6 mice with BPA (50-1000μg/kg/day, oral gavage) in utero yielded no significant changes in sperm production, seminiferous tubule histology, germ cell apoptosis, or serum testosterone [133]. These results are tempered by a recent study examining BPA-induced alterations in testicular meiotic events in 3 mouse strains. While CD-1 and C3H/HeJ mice that were orally exposed to 20 or 500 μg/kg/day BPA (PND1-12) had reduced meiotic recombination events in the testicular meiocytes, no such changes were observed in the BPA-exposed C57BL/6 strain. These data indicate that the C57BL/6 strain may be resistant to BPA-induced changes in the testes [134]. Recent studies have also shown that low doses of BPA given postnatally (2.5-25 μg/kg/day, E12-weaning) may increase Leydig cell numbers in adult Long-Evans rats [135]; however, the development of testicular neoplasias is unknown. While these studies suggest that BPA is not carcinogenic in the testes, they are limited by the lack of measurements of serum BPA levels; failure to include an estrogenic positive control, such as DES; use of resistant animal models; and a lack of long-term follow-up with assessment of testis pathology. Furthermore, these studies primarily focused on the potential for BPA to induce nascent tumors. As with other cancers of the reproductive tract, low doses of BPA may promote testicular cancer progression, but not act as initiators of tumors. This possibility remains to be examined.
6. Cancers in Non-reproductive Estrogen Target Tissues
6.1 Hepatic Cancer
While free BPA levels range from undetectable to 0.38ng/g tissue in the adult human liver, greater than 70% of human fetal livers contain detectable levels of BPA (0.1-50.5 ng/g of free BPA and 0.1-49.5 ng/g conjugated BPA) and this is associated with increases in methylation of genes encoding xenobiotic metabolizing enzymes [136, 137]. Recently, increased incidence of hepatic tumors or preneoplastic lesions was reported for mice treated in utero with low-dose BPA [138]. Dams from a mixed genetic background of C3H/HeJ (6.25-25%) and C57BL/6J (75-93.75%) that were heterozygous for the viable yellow Agouti (Avy) allele were exposed to one of 4 doses of BPA (0μg, 0.05μg, 50μg or 50mg/kg/day in diet) two weeks prior to mating. Dams were maintained on a BPA-containing diet throughout pregnancy and weaning of litters at PND22. Pre-neoplastic or neoplastic lesions (hepatocellular carcinomas or hepatic adenomas) were evident in ∼23% of combined male and female offspring receiving 50mg/kg/day BPA in the diet. This represented a statistically significant increase compared to controls, even when preneoplastic lesions were excluded from the analysis. It is also noteworthy that hepatic tumors occurred in the absence of additional carcinogenic insult even though this study used the C56BL/6 mouse strain, a model that is considered “relatively resistant” to hepatocellular carcinoma [139]. It should be noted, however, that the doses of BPA used to achieve significant induction of liver pathologies were above the RfD, yet female offspring exhibited a significant linear dose-response for combined neoplastic and preneoplastic lesions [138]. In humans, females are at a lower risk of developing liver cancer than males, in part due to the protective effects of estrogen [140]. The analysis of BPA impact on murine livers revealed that early-life BPA exposure may eliminate that protective effect. While it is possible that BPA acts as a partial/competitive agonist for estrogen during a critical window of fetal liver development, the specific mechanisms by which BPA exerts its actions in the liver requires further study.
7. Overall Conclusions
The pervasiveness of BPA exposure early in life combined with the complexities of adult exposure and BPA pharmacodynamics precludes the use of cross-sectional, short-term epidemiological studies to draw definitive conclusions on cancer risk. Instead, large-scale human studies examining cumulative life-time BPA exposure with long-term follow-up are needed. Unfortunately, such studies may not be feasible because most cancer cases occur in individuals over 50 years of age. In addition, the BPA exposome has not yet been elucidated. Defining accurate surrogate markers of life-long BPA exposure will be essential to facilitate completion of such studies. In contrast, monitoring of urine BPA levels in patients with newly diagnosed hormonally-responsive cancers followed by long-term follow-up should shed light onto the tumor promoting potential of environmental BPA exposures. Until such studies are possible, we must examine the weight of evidence for the carcinogenicity of conservative estimates of human-relevant BPA exposures (below the RfD) using animal models of disease.
There is substantial evidence in mice and rats generated by independent investigators indicating an increased susceptibility to cancer of the mammary gland following exposure to BPA at doses below the RfD [47, 55, 64]. Specifically, early life BPA exposure within the ranges observed in humans increases the number of mammary tumors with carcinogen treatment in 2 separate studies in rats [53]. Similarly, early life BPA exposure decreases tumor latency and increases tumor susceptibility in mice using similar treatment paradigms [64]. Another report demonstrated that BPA enhances tumor multiplicity, burden, and metastasis and decreases tumor latency in a mouse model of HER2 positive mammary cancer [68]. Lastly, two studies assessed the carcinogenicity of BPA alone (i.e, without secondary insult by a chemical carcinogen); one study showed that BPA-induced carcinomas in situ and the other macroscopic mammary tumors, both in rats [46, 47]. While the evidence is not as extensive for BPA-induced prostate cancer, three studies have found that cancer risk is enhanced with early-life BPA exposure in a rat model mimicking rising estrogen levels in aging men and one study found that BPA can induce dysplastic lesions without additional carcinogenic insults in rats [112-114]. Human prostatic epithelium exhibits a similarly increased susceptibility to cancer with BPA exposure when evaluated in a xenografted tissue reconstitution model [130]. Moreover, elevated urinary BPA levels are associated with prostate cancer in humans [103].
The International Agency for Research on Cancer (IARC) states that chemical carcinogenesis is “…the induction by chemicals of neoplasms that are not usually observed, the earlier induction by chemicals of neoplasms that are usually observed, and/or the induction by chemicals of more neoplasms than are usually found…” [141]. Further, the IARC states that if sufficient evidence exists supporting carcinogenicity in experimental animals in the absence of conclusive epidemiological studies, then it is “biologically plausible that agents for which there is sufficient evidence of carcinogenicity in experimental animals also present a carcinogenic hazard to humans” as was the case for DES [142, 143] (http://monographs.iarc.fr/ENG/Preamble/currentb3studiesanimals0706.php). Finally, the IARC categorizes agents into one of 5 groups, all defined by the evidence for carcinogenicity in humans. With these guidelines in mind and considering the enhanced susceptibility to cancer with low-dose BPA exposure in multiple studies in mammary and prostate combined, we propose that BPA be classified as a chemical carcinogen in “Group 2A: The agent is probably carcinogenic to humans as there is inadequate evidence of carcinogenicity in humans, yet sufficient evidence of the carcinogenicity in experimental animals and strong evidence that the carcinogenesis is mediated by a mechanism that also operates in humans.” (http://monographs.iarc.fr/ENG/Preamble/currentb6evalrationale0706.php)
The guidelines established by the NTP for defining a substance as “reasonably anticipated to be a human carcinogen” states that a substance can be listed as a carcinogen if “there is sufficient evidence of carcinogenicity from studies in experimental animals, which indicates there is an increased incidence of malignant and/or a combination of malignant and benign tumors (1) in multiple species or at multiple tissue sites, or (2) by multiple routes of exposure, or (3) to an unusual degree with regard to incidence, site, or type of tumor, or age at onset” (http://ntp.niehs.nih.gov/?objectid=47B37760-F1F6-975E-7C15022B9C93B5A6). By these standards, we submit that BPA may be presumed to be a human carcinogen due to its ability to enhance tumor susceptibility and promote tumorigenic properties in the breast and prostate glands. This notion is further strengthened by the fact that BPA is estrogenic [12] and that data gathered in humans have shown that the synthetic estrogen DES, a compound structurally related to BPA, induces clear cell adenocarcinoma of the vagina and increases the risk of breast cancer following fetal exposure [144, 145].
Based on the current weight of existing evidence for doses of BPA below the RfD, we are confident that:
BPA acts as an endocrine disruptor with estrogenic effects in the rodent mammary gland.
Prenatal exposure to low doses of BPA alters mammary gland morphogenesis, including proliferation and apoptotic pathways in the mammary glands of rodents, and increases risk of mammary cancer later in life at doses below the RfD.
Early life BPA exposure alters methylation patterning of several genes expressed in the prostate and mammary glands of rodents.
BPA may be reasonably anticipated to be a human carcinogen in the breast independent of route of exposure.
Based on the weight of existing evidence, we believe the following to be likely, but requiring confirmation:
Pharmacological doses of BPA may be associated with increased malignancies of the hematopoietic system and testicular interstitial-cell tumors in rodents.
The human prostate epithelium may be significantly influenced by developmental BPA exposures resulting in increased susceptibility towards neoplasia and cancer with aging.
Elevated urinary BPA levels are associated with prostate cancer in humans and may be an independent diagnostic marker in prostate cancer patients.
Early life BPA exposure may elevate the incidence of simple ovarian cysts and polycystic ovaries in rodents at environmentally relevant doses.
BPA alone does not induce testicular malignancies at doses below the RfD.
Areas requiring further investigation:
Continued analyses of uterine, ovarian, prostate and hepatic pathologies in rodents and higher mammals with early life BPA exposure at environmentally relevant doses are necessary.
Evaluation of the cancer promoting potential of early-life BPA exposures when coupled with additional carcinogenic insults in prostate, uterus, ovary and testis is needed.
While difficult to accomplish, large, well-controlled human studies beginning during fetal development and including lifetime follow-up may be necessary to determine if early life BPA exposure correlates with any increase in tumor incidence.
As more estrogen target organs are identified, a focus on the effects of BPA_on cancer susceptibility in those tissues is necessary.
Evaluation of the effects of early-life BPA exposure on genomic stability, epigenome alterations and gene expression changes, particularly in non-reproductive estrogen-target tissues such as the liver are warranted.
Acknowledgments
This work was supported by NIH grants CA090398 and CA154384 (RAK), CA172220 (GSP), ES018758 (GSP, SMH), ES015584 (GSP, SMH), ES018822, ES08314 and ES020888 (AMS). We thank Dr. Jerry Heindel for his constructive input to this review.
Abbreviations
- BPA
bisphenol A
- ER
estrogen receptor-α
- PND
postnatal day
- GD
gestational day
- ED
embryonic day
- DES
diethylstilbestrol
Footnotes
Declaration of Financial Interests: None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.De Coster S, van Larebeke N. Endocrine-disrupting chemicals: associated disorders and mechanisms of action. J Environ Public Health. 2012;2012:713696. doi: 10.1155/2012/713696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Howlander N, Noone AM, Krapcho M, Garshell J, Miller D, Altekruse SF, et al. SEER Cancer Statistics Review, 1975-2012. National Cancer Institute; p based on November 2014 SEER data submission. [Google Scholar]
- 3.Maffini MV, Rubin BS, Sonnenschein C, Soto AM. Endocrine disruptors and reproductive health: the case of bisphenol-A. Mol Cell Endocrinol. 2006:254–255. 179–86. doi: 10.1016/j.mce.2006.04.033. [DOI] [PubMed] [Google Scholar]
- 4.Brody JG, Rudel RA. Environmental pollutants and breast cancer. Environ Health Perspect. 2003;111:1007–19. doi: 10.1289/ehp.6310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burridge E. Bisphenol A: product profile. European Chemical News. 2003:14–20. [Google Scholar]
- 6.Kuroda N, Kinoshita Y, Sun Y, Wada M, Kishikawa N, Nakashima K, et al. Measurement of bisphenol A levels in human blood serum and ascitic fluid by HPLC using a fluorescent labeling reagent. J Pharm Biomed Anal. 2003;30:1743–9. doi: 10.1016/s0731-7085(02)00516-2. [DOI] [PubMed] [Google Scholar]
- 7.Lee YJ, Ryu HY, Kim HK, Min CS, Lee JH, Kim E, et al. Maternal and fetal exposure to bisphenol A in Korea. Reprod Toxicol. 2008;25:413–9. doi: 10.1016/j.reprotox.2008.05.058. [DOI] [PubMed] [Google Scholar]
- 8.Calafat AM, Weuve J, Ye X, Jia LT, Hu H, Ringer S, et al. Exposure to bisphenol A and other phenols in neonatal intensive care unit premature infants. Environ Health Perspect. 2009;117:639–44. doi: 10.1289/ehp.0800265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Edginton AN, Ritter L. Predicting plasma concentrations of bisphenol A in children younger than 2 years of age after typical feeding schedules, using a physiologically based toxicokinetic model. Environ Health Perspect. 2009;117:645–52. doi: 10.1289/ehp.0800073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nepomnaschy PA, Baird DD, Weinberg CR, Hoppin JA, Longnecker MP, Wilcox AJ. Within-person variability in urinary bisphenol A concentrations: measurements from specimens after long-term frozen storage. Environ Res. 2009;109:734–7. doi: 10.1016/j.envres.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Calafat AM, Kuklenyik Z, Reidy JA, Caudill SP, Ekong J, Needham LL. Urinary concentrations of bisphenol A and 4-nonylphenol in a human reference population. Environ Health Perspect. 2005;113:391–5. doi: 10.1289/ehp.7534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dodds EC. The Pharmacological Action and Clinical Use of Drugs with a Camphor- and Coramine-like Action: (Section of Therapeutics and Pharmacology) Proc R Soc Med. 1936;29:655–7. [PMC free article] [PubMed] [Google Scholar]
- 13.Thomas P, Dong J. Binding and activation of the seven-transmembrane estrogen receptor GPR30 by environmental estrogens: a potential novel mechanism of endocrine disruption. J Steroid Biochem Mol Biol. 2006;102:175–9. doi: 10.1016/j.jsbmb.2006.09.017. [DOI] [PubMed] [Google Scholar]
- 14.NTP. Carcinogenesis Bioassay of Bisphenol A (CAS No. 80-05-7) in F344 Rats and B6C3F1 Mice (Feed Study) Natl Toxicol Program Tech Rep Ser. 1982;215:1–116. [PubMed] [Google Scholar]
- 15.Huff J. Does exposure to bisphenol A represent a human health risk? Regul Toxicol Pharmacol. 2003;37:407–8. doi: 10.1016/s0273-2300(03)00009-6. [DOI] [PubMed] [Google Scholar]
- 16.Welshons WV, Thayer KA, Judy BM, Taylor JA, Curran EM, vom Saal FS. Large effects from small exposures. I. Mechanisms for endocrine-disrupting chemicals with estrogenic activity. Environ Health Perspect. 2003;111:994–1006. doi: 10.1289/ehp.5494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.vom Saal FS, Hughes C. An extensive new literature concerning low-dose effects of bisphenol A shows the need for a new risk assessment. Environ Health Perspect. 2005;113:926–33. doi: 10.1289/ehp.7713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vandenberg LN, Colborn T, Hayes TB, Heindel JJ, Jacobs DR, Jr, Lee DH, et al. Hormones and endocrine-disrupting chemicals: low-dose effects and nonmonotonic dose responses. Endocr Rev. 2012;33:378–455. doi: 10.1210/er.2011-1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vandenberg LN, Ehrlich S, Belcher SM, Ben-Jonathan N, Dolinoy DC, Hugo ER, et al. Low dose effects of bisphenol A: An integrated review of in vitro, laboratory animal, and epidemiology studies. Endocrine Disruptors. 2013;1:0–1. [Google Scholar]
- 20.Schonfelder G, Wittfoht W, Hopp H, Talsness CE, Paul M, Chahoud I. Parent bisphenol A accumulation in the human maternal-fetal-placental unit. Environ Health Perspect. 2002;110:A703–7. doi: 10.1289/ehp.110-1241091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ikezuki Y, Tsutsumi O, Takai Y, Kamei Y, Taketani Y. Determination of bisphenol A concentrations in human biological fluids reveals significant early prenatal exposure. Hum Reprod. 2002;17:2839–41. doi: 10.1093/humrep/17.11.2839. [DOI] [PubMed] [Google Scholar]
- 22.Taylor JA, Vom Saal FS, Welshons WV, Drury B, Rottinghaus G, Hunt PA, et al. Similarity of bisphenol A pharmacokinetics in rhesus monkeys and mice: relevance for human exposure. Environ Health Perspect. 2011;119:422–30. doi: 10.1289/ehp.1002514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Taylor JA, Welshons WV, vom Saal FS. No effect of route of exposure (oral; subcutaneous injection) on plasma bisphenol A throughout 24h after administration in neonatal female mice. Reprod Toxicol. 2008;25:169–76. doi: 10.1016/j.reprotox.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tyl RW, Myers CB, Marr MC, Thomas BF, Keimowitz AR, Brine DR, et al. Three-generation reproductive toxicity study of dietary bisphenol A in CD Sprague-Dawley rats. Toxicol Sci. 2002;68:121–46. doi: 10.1093/toxsci/68.1.121. [DOI] [PubMed] [Google Scholar]
- 25.Welshons WV, Nagel SC, vom Saal FS. Large effects from small exposures. III. Endocrine mechanisms mediating effects of bisphenol A at levels of human exposure. Endocrinology. 2006;147:S56–69. doi: 10.1210/en.2005-1159. [DOI] [PubMed] [Google Scholar]
- 26.Tyl RW, Myers CB, Marr MC, Sloan CS, Castillo NP, Veselica MM, et al. Two-generation reproductive toxicity study of dietary bisphenol A in CD-1 (Swiss) mice. Toxicol Sci. 2008;104:362–84. doi: 10.1093/toxsci/kfn084. [DOI] [PubMed] [Google Scholar]
- 27.Spearow JL, Doemeny P, Sera R, Leffler R, Barkley M. Genetic variation in susceptibility to endocrine disruption by estrogen in mice. Science. 1999;285:1259–61. doi: 10.1126/science.285.5431.1259. [DOI] [PubMed] [Google Scholar]
- 28.Keri RA, Ho SM, Hunt PA, Knudsen KE, Soto AM, Prins GS. An evaluation of evidence for the carcinogenic activity of bisphenol A. Reprod Toxicol. 2007;24:240–52. doi: 10.1016/j.reprotox.2007.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goodman JE, Witorsch RJ, McConnell EE, Sipes IG, Slayton TM, Yu CJ, et al. Weight-of-evidence evaluation of reproductive and developmental effects of low doses of bisphenol A. Crit Rev Toxicol. 2009;39:1–75. doi: 10.3109/10408440903279946. [DOI] [PubMed] [Google Scholar]
- 30.Melnick R, Lucier G, Wolfe M, Hall R, Stancel G, Prins G, et al. Summary of the National Toxicology Program's report of the endocrine disruptors low-dose peer review. Environ Health Perspect. 2002;110:427–31. doi: 10.1289/ehp.02110427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vandenberg LN, Chahoud I, Padmanabhan V, Paumgartten FJ, Schoenfelder G. Biomonitoring studies should be used by regulatory agencies to assess human exposure levels and safety of bisphenol A. Environ Health Perspect. 2010;118:1051–4. doi: 10.1289/ehp.0901717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.vom Saal FS, Welshons WV. Evidence that bisphenol A (BPA) can be accurately measured without contamination in human serum and urine, and that BPA causes numerous hazards from multiple routes of exposure. Mol and Cell Endocrinol. 2014;398:101–13. doi: 10.1016/j.mce.2014.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dave N, Chow LML, Gudelsky GA, LaSance K, Qi X, Desai PB. Preclinical Pharmacological Evaluation of Letrozole as a Novel Treatment for Gliomas. Mol Cancer Ther. 2015;14:857–64. doi: 10.1158/1535-7163.MCT-14-0743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Siegfried JM, Hershberger PA, Stabile LP. Estrogen Receptor Signaling in Lung Cancer. Semin Oncol. 2009;36:524. doi: 10.1053/j.seminoncol.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Caiazza F, Ryan EJ, Doherty G, Winter DC, Sheahan K. Estrogen receptors and their implications in colorectal carcinogenesis. Front Oncol. 2015;5 doi: 10.3389/fonc.2015.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lang IA, Galloway TS, Scarlett A, Henley WE, Depledge M, Wallace RB, et al. Association of urinary bisphenol A concentration with medical disorders and laboratory abnormalities in adults. JAMA. 2008;300:1303–10. doi: 10.1001/jama.300.11.1303. [DOI] [PubMed] [Google Scholar]
- 37.Tsai WT. Human health risk on environmental exposure to Bisphenol-A: a review. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev. 2006;24:225–55. doi: 10.1080/10590500600936482. [DOI] [PubMed] [Google Scholar]
- 38.Yang M, Ryu JH, Jeon R, Kang D, Yoo KY. Effects of bisphenol A on breast cancer and its risk factors. Arch Toxicol. 2009;83:281–5. doi: 10.1007/s00204-008-0364-0. [DOI] [PubMed] [Google Scholar]
- 39.Trabert B, Falk R, Figueroa J, Graubard B, Garcia-Closas M, Lissowska J, et al. Urinary bisphenol A-glucuronide and postmenopausal breast cancer in Poland. Cancer Causes Control. 2014;25:1587–93. doi: 10.1007/s10552-014-0461-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garcia-Closas M, Brinton LA, Lissowska J, Chatterjee N, Peplonska B, Anderson WF, et al. Established breast cancer risk factors by clinically important tumour characteristics. Br J Cancer. 2006;95:123–9. doi: 10.1038/sj.bjc.6603207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Neville MC, McFadden TB, Forsyth I. Hormonal regulation of mammary differentiation and milk secretion. Journal of Mammary Gland Biology and Neoplasia. 2002;7:49–66. doi: 10.1023/a:1015770423167. [DOI] [PubMed] [Google Scholar]
- 42.Russo IH, Russo J. Mammary gland neoplasia in long-term rodent studies. Environ Health Perspect. 1996;104:938–67. doi: 10.1289/ehp.96104938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Munoz-de-Toro M, Markey CM, Wadia PR, Luque EH, Rubin BS, Sonnenschein C, et al. Perinatal exposure to bisphenol-A alters peripubertal mammary gland development in mice. Endocrinology. 2005;146:4138–47. doi: 10.1210/en.2005-0340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reed CE, Fenton SE. Exposure to diethylstilbestrol during sensitive life stages: a legacy of heritable health effects. Birth Defects Res C Embryo Today. 2013;99:134–46. doi: 10.1002/bdrc.21035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Agius LM. Theory in the Pathophysiology of Carcinogenesis. Bentham Science Publishers; 2011. [Google Scholar]
- 46.Murray TJ, Maffini MV, Ucci AA, Sonnenschein C, Soto AM. Induction of mammary gland ductal hyperplasias and carcinoma in situ following fetal bisphenol A exposure. Reprod Toxicol. 2007;23:383–90. doi: 10.1016/j.reprotox.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Acevedo N, Davis B, Schaeberle CM, Sonnenschein C, Soto AM. Perinatally administered bisphenol a as a potential mammary gland carcinogen in rats. Environ Health Perspect. 2013;121:1040–6. doi: 10.1289/ehp.1306734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moral R, Wang R, Russo IH, Lamartiniere CA, Pereira J, Russo J. Effect of prenatal exposure to the endocrine disruptor bisphenol A on mammary gland morphology and gene expression signature. J Endocrinol. 2008;196:101–12. doi: 10.1677/JOE-07-0056. [DOI] [PubMed] [Google Scholar]
- 49.Gullino PM, Pettigrew HM, Grantham FH. N-Nitrosomethylurea as Mammary-Gland Carcinogen in Rats. J Natl Cancer Inst. 1975;54:401–14. [PubMed] [Google Scholar]
- 50.Russo J, Wilgus G, Russo IH. Susceptibility of the mammary gland to carcinogenesis: I Differentiation of the mammary gland as determinant of tumor incidence and type of lesion. Am J Pathol. 1979;96:721–36. [PMC free article] [PubMed] [Google Scholar]
- 51.Maffini MV, Soto AM, Calabro JM, Ucci AA, Sonnenschein C. The stroma as a crucial target in rat mammary gland carcinogenesis. J Cell Sci. 2004;117:1495–502. doi: 10.1242/jcs.01000. [DOI] [PubMed] [Google Scholar]
- 52.Barcellos-Hoff MH, Ravani SA. Irradiated mammary gland stroma promotes the expression of tumorigenic potential by unirradiated epithelial cells. Cancer Res. 2000;60:1254–60. [PubMed] [Google Scholar]
- 53.Durando M, Kass L, Piva J, Sonnenschein C, Soto AM, Luque EH, et al. Prenatal bisphenol A exposure induces preneoplastic lesions in the mammary gland in Wistar rats. Environ Health Perspect. 2007;115:80–6. doi: 10.1289/ehp.9282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jenkins S, Raghuraman N, Eltoum I, Carpenter M, Russo J, Lamartiniere CA. Oral exposure to bisphenol a increases dimethylbenzanthracene-induced mammary cancer in rats. Environ Health Perspect. 2009;117:910–5. doi: 10.1289/ehp.11751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Betancourt AM, Eltoum IA, Desmond RA, Russo J, Lamartiniere CA. In Utero Exposure to Bisphenol A Shifts the Window of Susceptibility for Mammary Carcinogenesis in the Rat. Environ Health Perspect. 2010;118:1614–9. doi: 10.1289/ehp.1002148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lamartiniere CA, Jenkins S, Betancourt AM, Wang J, Russo J. Exposure to the Endocrine Disruptor Bisphenol A Alters Susceptibility for Mammary Cancer. Horm Mol Biol Clin Investig. 2011;5:45–52. doi: 10.1515/HMBCI.2010.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tokunaga M, Land CE, Yamamoto T, Asano M, Tokuoka S, Ezaki H, et al. Incidence of female breast cancer among atomic bomb survivors, Hiroshima and Nagasaki, 1950-1980. Radiat Res. 1987;112:243–72. [PubMed] [Google Scholar]
- 58.Betancourt AM, Wang J, Jenkins S, Mobley J, Russo J, Lamartiniere CA. Altered carcinogenesis and proteome in mammary glands of rats after prepubertal exposures to the hormonally active chemicals bisphenol a and genistein. J Nutr. 2012;142:1382S–8S. doi: 10.3945/jn.111.152058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vandenberg LN, Maffini MV, Schaeberle CM, Ucci AA, Sonnenschein C, Rubin BS, et al. Perinatal exposure to the xenoestrogen bisphenol-A induces mammary intraductal hyperplasias in adult CD-1 mice. Reprod Toxicol. 2008;26:210–9. doi: 10.1016/j.reprotox.2008.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vandenberg LN, Maffini MV, Wadia PR, Sonnenschein C, Rubin BS, Soto AM. Exposure to environmentally relevant doses of the xenoestrogen bisphenol-A alters development of the fetal mouse mammary gland. Endocrinology. 2007;148:116–27. doi: 10.1210/en.2006-0561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wadia PR, Cabaton NJ, Borrero MD, Rubin BS, Sonnenschein C, Shioda T, et al. Low-Dose BPA Exposure Alters the Mesenchymal and Epithelial Transcriptomes of the Mouse Fetal Mammary Gland. PLoS ONE. 2013;8:e63902. doi: 10.1371/journal.pone.0063902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wadia PR, Vandenberg LN, Schaeberle CM, Rubin BS, Sonnenschein C, Soto AM. Perinatal bisphenol A exposure increases estrogen sensitivity of the mammary gland in diverse mouse strains. Environ Health Perspect. 2007;115:592–8. doi: 10.1289/ehp.9640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ayyanan A, Laribi O, Schuepbach-Mallepell S, Schrick C, Gutierrez M, Tanos T, et al. Perinatal Exposure to Bisphenol A Increases Adult Mammary Gland Progesterone Response and Cell Number. Molecular Endocrinology. 2011;25:1915–23. doi: 10.1210/me.2011-1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Weber Lozada K, Keri RA. Bisphenol A increases mammary cancer risk in two distinct mouse models of breast cancer. Biol Reprod. 2011;85:490–7. doi: 10.1095/biolreprod.110.090431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Soto A, Brisken C, Schaeberle C, Sonnenschein C. Does Cancer Start in the Womb? Altered Mammary Gland Development and Predisposition to Breast Cancer due to in Utero Exposure to Endocrine Disruptors. J Mammary Gland Biol Neoplasia. 2013;18:199–208. doi: 10.1007/s10911-013-9293-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang D, Gao H, Bandyopadhyay A, Wu A, Yeh IT, Chen Y, et al. Pubertal Bisphenol A Exposure Alters Murine Mammary Stem Cell Function Leading to Early Neoplasia in Regenerated Glands. Cancer Prev Res (Phila) 2014;7:445–55. doi: 10.1158/1940-6207.CAPR-13-0260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tiede B, Kang Y. From milk to malignancy: the role of mammary stem cells in development, pregnancy and breast cancer. Cell Res. 2011;21:245–57. doi: 10.1038/cr.2011.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jenkins S, Wang J, Eltoum I, Desmond R, Lamartiniere CA. Chronic oral exposure to bisphenol A results in a nonmonotonic dose response in mammary carcinogenesis and metastasis in MMTV-erbB2 mice. Environ Health Perspect. 2011;119:1604–9. doi: 10.1289/ehp.1103850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dhimolea E, Wadia PR, Murray TJ, Settles ML, Treitman JD, Sonnenschein C, et al. Prenatal Exposure to BPA Alters the Epigenome of the Rat Mammary Gland and Increases the Propensity to Neoplastic Development. PLoS ONE. 2014;9:e99800. doi: 10.1371/journal.pone.0099800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tharp AP, Maffini MV, Hunt PA, VandeVoort CA, Sonnenschein C, Soto AM. Bisphenol A alters the development of the rhesus monkey mammary gland. Proc Natl Acad Sci U S A. 2012;109:8190–5. doi: 10.1073/pnas.1120488109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Susiarjo M, Hassold TJ, Freeman E, Hunt PA. Bisphenol A exposure in utero disrupts early oogenesis in the mouse. PLoS Genet. 2007;3:e5. doi: 10.1371/journal.pgen.0030005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hunt PA, Koehler KE, Susiarjo M, Hodges CA, Ilagan A, Voigt RC, et al. Bisphenol a exposure causes meiotic aneuploidy in the female mouse. Curr Biol. 2003;13:546–53. doi: 10.1016/s0960-9822(03)00189-1. [DOI] [PubMed] [Google Scholar]
- 73.Hunt PA, Lawson C, Gieske M, Murdoch B, Smith H, Marre A, et al. Bisphenol A alters early oogenesis and follicle formation in the fetal ovary of the rhesus monkey. Proc Natl Acad Sci U S A. 2012;109:17525–30. doi: 10.1073/pnas.1207854109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Greathouse KL, Bredfeldt T, Everitt JI, Lin K, Berry T, Kannan K, et al. Environmental Estrogens Differentially Engage the Histone Methyltransferase EZH2 to Increase Risk of Uterine Tumorigenesis. Mol Cancer Res. 2012;10:546–57. doi: 10.1158/1541-7786.MCR-11-0605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nikaido Y, Danbara N, Tsujita-Kyutoku M, Yuri T, Uehara N, Tsubura A. Effects of prepubertal exposure to xenoestrogen on development of estrogen target organs in female CD-1 mice. In Vivo. 2005;19:487–94. [PubMed] [Google Scholar]
- 76.Newbold RR, Jefferson WN, Padilla-Banks E. Long-term adverse effects of neonatal exposure to bisphenol A on the murine female reproductive tract. Reprod Toxicol. 2007;24:253–8. doi: 10.1016/j.reprotox.2007.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Newbold RR, Jefferson WN, Padilla-Banks E. Prenatal exposure to bisphenol a at environmentally relevant doses adversely affects the murine female reproductive tract later in life. Environ Health Perspect. 2009;117:879–85. doi: 10.1289/ehp.0800045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kato H, Ota T, Furuhashi T, Ohta Y, Iguchi T. Changes in reproductive organs of female rats treated with bisphenol A during the neonatal period. Reprod Toxicol. 2003;17:283–8. doi: 10.1016/s0890-6238(03)00002-9. [DOI] [PubMed] [Google Scholar]
- 79.Fernandez M, Bourguignon N, Lux-Lantos V, Libertun C. Neonatal exposure to bisphenol a and reproductive and endocrine alterations resembling the polycystic ovarian syndrome in adult rats. Environ Health Perspect. 2010;118:1217–22. doi: 10.1289/ehp.0901257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kandaraki E, Chatzigeorgiou A, Livadas S, Palioura E, Economou F, Koutsilieris M, et al. Endocrine Disruptors and Polycystic Ovary Syndrome (PCOS): Elevated Serum Levels of Bisphenol A in Women with PCOS. J Clin Endocrinol & Metab. 2011;96:E480–E4. doi: 10.1210/jc.2010-1658. [DOI] [PubMed] [Google Scholar]
- 81.Bailey CL, Ueland FR, Land GL, DePriest PD, Gallion HH, Kryscio RJ, et al. The malignant potential of small cystic ovarian tumors in women over 50 years of age. Gynecol Oncol. 1998;69:3–7. doi: 10.1006/gyno.1998.4965. [DOI] [PubMed] [Google Scholar]
- 82.Modesitt SC, Pavlik EJ, Ueland FR, DePriest PD, Kryscio RJ, van Nagell JR., Jr Risk of malignancy in unilocular ovarian cystic tumors less than 10 centimeters in diameter. Obstet Gynecol. 2003;102:594–9. doi: 10.1016/s0029-7844(03)00670-7. [DOI] [PubMed] [Google Scholar]
- 83.Schildkraut JM, Schwingl PJ, Bastos E, Evanoff A, Hughes C. Epithelial ovarian cancer risk among women with polycystic ovary syndrome. Obstet Gynecol. 1996;88:554–9. doi: 10.1016/0029-7844(96)00226-8. [DOI] [PubMed] [Google Scholar]
- 84.Dumesic DA, Lobo RA. Cancer risk and PCOS. Steroids. 2013 doi: 10.1016/j.steroids.2013.04.004. [DOI] [PubMed] [Google Scholar]
- 85.Newbold RR, Bullock BC, McLachlan JA. Uterine adenocarcinoma in mice following developmental treatment with estrogens: a model for hormonal carcinogenesis. Cancer Res. 1990;50:7677–81. [PubMed] [Google Scholar]
- 86.Hiroi H, Tsutsumi O, Takeuchi T, Momoeda M, Ikezuki Y, Okamura A, et al. Differences in serum bisphenol a concentrations in premenopausal normal women and women with endometrial hyperplasia. Endocr J. 2004;51:595–600. doi: 10.1507/endocrj.51.595. [DOI] [PubMed] [Google Scholar]
- 87.Fukata H, Miyagawa H, Yamazaki N, Mori C. Comparison of Elisa- and LC-MS-Based Methodologies for the Exposure Assessment of Bisphenol A. Toxicol Mech Methods. 2006;16:427–30. doi: 10.1080/15376520600697404. [DOI] [PubMed] [Google Scholar]
- 88.Dekant W, Volkel W. Human exposure to bisphenol A by biomonitoring: methods, results and assessment of environmental exposures. Toxicol Appl Pharmacol. 2008;228:114–34. doi: 10.1016/j.taap.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 89.Pollack AZ, Buck Louis GM, Chen Z, Sun L, Trabert B, Guo Y, et al. Bisphenol A, benzophenone-type ultraviolet filters, and phthalates in relation to uterine leiomyoma. Environ Res. 2015;137:101–7. doi: 10.1016/j.envres.2014.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Schonfelder G, Friedrich K, Paul M, Chahoud I. Developmental effects of prenatal exposure to bisphenol a on the uterus of rat offspring. Neoplasia. 2004;6:584–94. doi: 10.1593/neo.04217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Prejean JD, Peckham JC, Casey AE, Griswold DP, Weisburger EK, Weisburger JH. Spontaneous tumors in Sprague-Dawley rats and Swiss mice. Cancer Res. 1973;33:2768–73. [PubMed] [Google Scholar]
- 92.Kitamura T, Nishimura S, Sasahara K, Yoshida M, Ando J, Takahashi M, et al. Transplacental administration of diethylstilbestrol (DES) causes lesions in female reproductive organs of Donryu rats, including endometrial neoplasia. Cancer Lett. 1999;141:219–28. doi: 10.1016/s0304-3835(99)00108-1. [DOI] [PubMed] [Google Scholar]
- 93.Long X, Steinmetz R, Ben-Jonathan N, Caperell-Grant A, Young PC, Nephew KP, et al. Strain differences in vaginal responses to the xenoestrogen bisphenol A. Environ Health Perspect. 2000;108:243–7. doi: 10.1289/ehp.00108243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rothschild TC, Calhoon RE, Boylan ES. Genital tract abnormalities in female rats exposed to diethylstilbestrol in utero. Reprod Toxicol. 1987;1:193–202. doi: 10.1016/s0890-6238(87)80033-3. [DOI] [PubMed] [Google Scholar]
- 95.Okuda K, Takiguchi M, Yoshihara S. In vivo estrogenic potential of 4-methyl-2,4-bis(4-hydroxyphenyl)pent-1-ene, an active metabolite of bisphenol A, in uterus of ovariectomized rat. Toxicol Lett. 2010;197:7–11. doi: 10.1016/j.toxlet.2010.04.017. [DOI] [PubMed] [Google Scholar]
- 96.Greathouse KL, Cook JD, Lin K, Davis BJ, Berry TD, Bredfeldt TG, et al. Identification of Uterine Leiomyoma Genes Developmentally Reprogrammed by Neonatal Exposure to Diethylstilbestrol. Reprod Sci. 2008;15:765–78. doi: 10.1177/1933719108322440. [DOI] [PubMed] [Google Scholar]
- 97.Newbold RR, Moore AB, Dixon D. Characterization of Uterine Leiomyomas in CD-1 Mice Following Developmental Exposure to Diethylstilbestrol (DES) Toxicol Pathol. 2002;30:611–6. doi: 10.1080/01926230290105839. [DOI] [PubMed] [Google Scholar]
- 98.Everitt JI, Wolf DC, Howe SR, Goldsworthy TL, Walker C. Rodent model of reproductive tract leiomyomata. Clinical and pathological features. Am J Pathol. 1995;146:1556–67. [PMC free article] [PubMed] [Google Scholar]
- 99.Walker CL, Hunter D, Everitt JI. Uterine leiomyoma in the Eker rat: a unique model for important diseases of women. Genes Chromosomes Cancer. 2003;38:349–56. doi: 10.1002/gcc.10281. [DOI] [PubMed] [Google Scholar]
- 100.Maekawa A, Onodera H, Tanigawa H, Furuta K, Matsuoka C, Kanno J, et al. Spontaneous neoplastic and non-neoplastic lesions in aging Donryu rats. Jpn J Cancer Res. 1986;77:882–90. [PubMed] [Google Scholar]
- 101.Yoshida M, Shimomoto T, Katashima S, Watanabe G, Taya K, Maekawa A. Maternal exposure to low doses of bisphenol a has no effects on development of female reproductive tract and uterine carcinogenesis in Donryu rats. J Reprod Dev. 2004;50:349–60. doi: 10.1262/jrd.50.349. [DOI] [PubMed] [Google Scholar]
- 102.Schonfelder G, Flick B, Mayr E, Talsness C, Paul M, Chahoud I. In utero exposure to low doses of bisphenol A lead to long-term deleterious effects in the vagina. Neoplasia. 2002;4:98–102. doi: 10.1038/sj.neo.7900212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tarapore P, Ying J, Ouyang B, Burke B, Bracken B, Ho SM. Exposure to bisphenol A correlates with early-onset prostate cancer and promotes centrosome amplification and anchorage-independent growth in vitro. PLoS ONE. 2014;9:e90332. doi: 10.1371/journal.pone.0090332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Greene RR, Burrill MW, Ivy AC. Experimental intersexuality. The effects of estrogens on the antenatal sexual development of the rat. Am J Anat. 1940;67:305–45. [Google Scholar]
- 105.Allgeier SH, Vezina CM, Lin TM, Moore RW, Silverstone AE, Mukai M, et al. Estrogen signaling is not required for prostatic bud patterning or for its disruption by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol Appl Pharmacol. 2009;239:80–6. doi: 10.1016/j.taap.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Leav I, Ho SM, Ofner P, Merk FB, Kwan PW, Damassa D. Biochemical alterations in sex hormone-induced hyperplasia and dysplasia of the dorsolateral prostates of Noble rats. J Natl Cancer Inst. 1988;80:1045–53. doi: 10.1093/jnci/80.13.1045. [DOI] [PubMed] [Google Scholar]
- 107.Modugno F, Weissfeld JL, Trump DL, Zmuda JM, Shea P, Cauley JA, et al. Allelic variants of aromatase and the androgen and estrogen receptors: toward a multigenic model of prostate cancer risk. Clin Cancer Res. 2001;7:3092–6. [PubMed] [Google Scholar]
- 108.Hu WY, Shi GB, Lam HM, Hu DP, Ho SM, Madueke IC, et al. Estrogen-Initiated Transformation of Prostate Epithelium Derived from Normal Human Prostate Stem-Progenitor Cells. Endocrinology. 2011;152:2150–63. doi: 10.1210/en.2010-1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ichihara T, Yoshino H, Imai N, Tsutsumi T, Kawabe M, Tamano S, et al. Lack of carcinogenic risk in the prostate with transplacental and lactational exposure to bisphenol A in rats. J Toxicol Sci. 2003;28:165–71. doi: 10.2131/jts.28.165. [DOI] [PubMed] [Google Scholar]
- 110.Prins GS, Birch L, Couse JF, Choi I, Katzenellenbogen B, Korach KS. Estrogen imprinting of the developing prostate gland is mediated through stromal estrogen receptor alpha: studies with alpha-ERKO and beta-ERKO mice. Cancer Res. 2001;61:6089–97. [PubMed] [Google Scholar]
- 111.Wu JH, Jiang XR, Liu GM, Liu XY, He GL, Sun ZY. Oral exposure to low-dose bisphenol A aggravates testosterone-induced benign hyperplasia prostate in rats. Toxicol Ind Health. 2011;27:810–9. doi: 10.1177/0748233711399310. [DOI] [PubMed] [Google Scholar]
- 112.Brandt JZ, Silveira LTR, Grassi TF, Anselmo-Franci JA, Fávaro WJ, Felisbino SL, et al. Indole-3-carbinol attenuates the deleterious gestational effects of bisphenol A exposure on the prostate gland of male F1 rats. Reprod Toxicol. 2014;43:56–66. doi: 10.1016/j.reprotox.2013.11.001. [DOI] [PubMed] [Google Scholar]
- 113.Ho SM, Tang WY, Belmonte de Frausto J, Prins GS. Developmental Exposure to Estradiol and Bisphenol A Increases Susceptibility to Prostate Carcinogenesis and Epigenetically Regulates Phosphodiesterase Type 4 Variant 4. Cancer Res. 2006;66:5624–32. doi: 10.1158/0008-5472.CAN-06-0516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Prins GS, Ye SH, Birch L, Ho Sm, Kannan K. Serum bisphenol A pharmacokinetics and prostate neoplastic responses following oral and subcutaneous exposures in neonatal Sprague–Dawley rats. Reprod Toxicol. 2011;31:1–9. doi: 10.1016/j.reprotox.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Vandenberg LN, Hauser R, Marcus M, Olea N, Welshons WV. Human exposure to bisphenol A (BPA) Reprod Toxicol. 2007;24:139–77. doi: 10.1016/j.reprotox.2007.07.010. [DOI] [PubMed] [Google Scholar]
- 116.Wong RL, Wang Q, Trevino LS, Bosland MC, Chen J, Medvedovic M, et al. Identification of secretaglobin Scgb2a1 as a target for developmental reprogramming by BPA in the rat prostate. Epigenetics. 2015;10:127–34. doi: 10.1080/15592294.2015.1009768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ittmann M, Huang J, Radaelli E, Martin P, Signoretti S, Sullivan R, et al. Animal models of human prostate cancer: the consensus report of the New York meeting of the Mouse Models of Human Cancers Consortium Prostate Pathology Committee. Cancer Res. 2013;73:2718–36. doi: 10.1158/0008-5472.CAN-12-4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Castro B, Sánchez P, Torres JM, Preda O, del Moral RG, Ortega E. Bisphenol A Exposure during Adulthood Alters Expression of Aromatase and 5α-Reductase Isozymes in Rat Prostate. PLoS ONE. 2013;8:e55905. doi: 10.1371/journal.pone.0055905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Godoy A, Kawinski E, Li Y, Oka D, Alexiev B, Azzouni F, et al. 5alpha-reductase type 3 expression in human benign and malignant tissues: a comparative analysis during prostate cancer progression. Prostate. 2011;71:1033–46. doi: 10.1002/pros.21318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Arase S, Ishii K, Igarashi K, Aisaki K, Yoshio Y, Matsushima A, et al. Endocrine disrupter bisphenol A increases in situ estrogen production in the mouse urogenital sinus. Biol Reprod. 2011;84:734–42. doi: 10.1095/biolreprod.110.087502. [DOI] [PubMed] [Google Scholar]
- 121.Ho SM, Johnson A, Tarapore P, Janakiram V, Zhang X, Leung YK. Environmental epigenetics and its implication on disease risk and health outcomes. ILAR J. 2012;53:289–305. doi: 10.1093/ilar.53.3-4.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Prins GS, Tang WY, Belmonte J, Ho SM. Perinatal Exposure to Oestradiol and Bisphenol A Alters the Prostate Epigenome and Increases Susceptibility to Carcinogenesis. Basic Clin Pharmacol Toxicol. 2008;102:134–8. doi: 10.1111/j.1742-7843.2007.00166.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Tang Wy, Morey LM, Cheung YY, Birch L, Prins GS, Ho Sm. Neonatal Exposure to Estradiol/Bisphenol A Alters Promoter Methylation and Expression of Nsbp1 and Hpcal1 Genes and Transcriptional Programs of Dnmt3a/b and Mbd2/4 in the RatProstate Gland Throughout Life. Endocrinology. 2012;153:42–55. doi: 10.1210/en.2011-1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Chen TC, Wadsten P, Su S, Rawlinson N, Hofman FM, Hill CK, et al. The type IV phosphodiesterase inhibitor rolipram induces expression of the cell cycle inhibitors p21(Cip1) and p27(Kip1), resulting in growth inhibition, increased differentiation, and subsequent apoptosis of malignant A-172 glioma cells. Cancer Biol Ther. 2002;1:268–76. doi: 10.4161/cbt.80. [DOI] [PubMed] [Google Scholar]
- 125.Narita M, Murata T, Shimizu K, Sugiyama T, Nakagawa T, Manganiello VC, et al. Phosphodiesterase 4 in osteoblastic osteosarcoma cells as a potential target for growth inhibition. Anticancer Drugs. 2003;14:377–81. doi: 10.1097/00001813-200306000-00009. [DOI] [PubMed] [Google Scholar]
- 126.Jiang N, Zhou LQ, Zhang XY. Downregulation of the nucleosome-binding protein 1 (NSBP1) gene can inhibit the in vitro and in vivo proliferation of prostate cancer cells. Asian J Androl. 2010;12:709–17. doi: 10.1038/aja.2010.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Song G, Zhou LQ, Weng M, He Q, He ZS, Hao JR, et al. Expression of nucleosomal binding protein 1 in normal prostate benign prostate hyperplasia, and prostate cancer and significance thereof. Zhonghua Yi Xue Za Zhi. 2006;86:1962–5. [PubMed] [Google Scholar]
- 128.Lacroix M. Significance, detection and markers of disseminated breast cancer cells. Endocr Relat Cancer. 2006;13:1033–67. doi: 10.1677/ERC-06-0001. [DOI] [PubMed] [Google Scholar]
- 129.Munakata K, Uemura M, Takemasa I, Ozaki M, Konno M, Nishimura J, et al. SCGB2A1 is a novel prognostic marker for colorectal cancer associated with chemoresistance and radioresistance. Int J Oncol. 2014;44:1521–8. doi: 10.3892/ijo.2014.2316. [DOI] [PubMed] [Google Scholar]
- 130.Prins GS, Hu WY, Shi GB, Hu DP, Majumdar S, Li G, et al. Bisphenol A Promotes Human Prostate Stem-Progenitor Cell Self-Renewal and Increases in vivo Carcinogenesis in Human Prostate Epithelium. Endocrinology. 2014;155 doi: 10.1210/en.2013-1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ho SM, Cheong A, Lam HM, Hu WY, Shi GB, Zhu X, et al. Exposure of human prostaspheres to Bisphenol A epigenetically regulates SNORD family non-coding RNAs via histone modification. Endocrinology. 2015:en20151067. doi: 10.1210/en.2015-1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zhang WZ, Yong L, Jia XD, Li N, Fan YX. Combined subchronic toxicity of bisphenol A and dibutyl phthalate on male rats. Biomed Environ Sci. 2013;26:63–9. doi: 10.3967/0895-3988.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 133.LaRocca J, Boyajian A, Brown C, Smith SD, Hixon M. Effects of in utero exposure to Bisphenol A or diethylstilbestrol on the adult male reproductive system. Birth Defects Res B Dev Reprod Toxicol. 2011;92:526–33. doi: 10.1002/bdrb.20336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Vrooman LA, Oatley JM, Griswold JE, Hassold TJ, Hunt PA. Estrogenic exposure alters the spermatogonial stem cells in the developing testis, permanently reducing crossover levels in the adult. PLoS Genet. 2015;11:e1004949. doi: 10.1371/journal.pgen.1004949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Nanjappa MK, Simon L, Akingbemi BT. The industrial chemical bisphenol A (BPA) interferes with proliferative activity and development of steroidogenic capacity in rat Leydig cells. Biol Reprod. 2012;86:135, 1–12. doi: 10.1095/biolreprod.111.095349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Nahar MS, Liao C, Kannan K, Dolinoy DC. Fetal liver bisphenol A concentrations and biotransformation gene expression reveal variable exposure and altered capacity for metabolism in humans. J Biochem Mol Toxicol. 2013;27:116–23. doi: 10.1002/jbt.21459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Nahar MS, Kim JH, Sartor MA, Dolinoy DC. Bisphenol A-associated alterations in the expression and epigenetic regulation of genes encoding xenobiotic metabolizing enzymes in human fetal liver. Environ Mol Mutagen. 2013;55:184–95. doi: 10.1002/em.21823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Weinhouse C, Anderson OS, Bergin IL, Vandenbergh DJ, Gyekis JP, Dingman MA, et al. Dose-Dependent Incidence of Hepatic Tumors in Adult Mice following Perinatal Exposure to Bisphenol A. Environ Health Perspect. 2014;122:485–91. doi: 10.1289/ehp.1307449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Maronpot RR. Biological Basis of Differential Susceptibility to Hepatocarcinogenesis among Mouse Strains. J Toxicol Pathol. 2009;22:11–33. doi: 10.1293/tox.22.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.IARC. Cancer incidence in five continents. VIII. IARC Sci Publ; 2002. pp. 1–781. [PubMed] [Google Scholar]
- 141.IARC. International Agency for Research on Cancer Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Humans. Lyons, France: 1982. [Google Scholar]
- 142.Dunning WF, Curtis MR, Maun ME. The Effect of Dietary Fat and Carbohydrate on Diethylstilbestrol-induced Mammary Cancer in Rats. Cancer Res. 1949;9:354–61. [PubMed] [Google Scholar]
- 143.Kirkman H, Bacon RL. Estrogen-induced tumors of the kidney. I. Incidence of renal tumors in intact and gonadectomized male golden hamsters treated with diethylstilbestrol. J Natl Cancer Inst. 1952;13:745–55. [PubMed] [Google Scholar]
- 144.Herbst AL, Ulfelder H, Poskanzer DC. Adenocarcinoma of the vagina. Association of maternal stilbestrol therapy with tumor appearance in young women. N Engl J Med. 1971;284:878–81. doi: 10.1056/NEJM197104222841604. [DOI] [PubMed] [Google Scholar]
- 145.Hoover RN, Hyer M, Pfeiffer RM, Adam E, Bond B, Cheville AL, et al. Adverse health outcomes in women exposed in utero to diethylstilbestrol. N Engl J Med. 2011;365:1304–14. doi: 10.1056/NEJMoa1013961. [DOI] [PubMed] [Google Scholar]