

Abstract
The gastrointestinal (GI) and cutaneous systems are closely linked in origin. Skin manifestations are frequently seen as a part of different GI syndromes. Gastroenterologists play an important role in recognizing the symptoms, patient workup and arriving at appropriate diagnoses, often in consultation with dermatologists. This review discusses the diseases with both cutaneous and intestinal involvement. Hereditary polyposis GI cancers, hereditary nonpolyposis colorectal cancers (CRCs), hamartomatous disorders, and inflammatory bowel disease (IBD) are reviewed with emphasis on the genetic basis, diagnostic, histologic findings, screening modalities, and therapeutic options.
Keywords: Skin, intestine, hereditary polyposis gastrointestinal (GI) cancers, hereditary nonpolyposis colorectal cancers (CRCs), hamartomatous disorders, inflammatory bowel disease (IBD)
Introduction
Dermatological findings are commonly associated with underlying gastrointestinal (GI) disease. In fact, in a lot of circumstances the cutaneous presentations herald the clinical symptoms of the existing GI disease. The concomitant finding of GI and skin manifestations can also be a part genetic syndromes and hereditary GI malignancies. Recognizing these symptoms can guide the clinicians to prompt diagnosis of occult GI disease and start the proper treatment. Besides, the other family members will benefit from genetic counselling and appropriate screening tests. In this review we will describe some of the most common disorders involving both the integumentary and alimentary systems.
Hereditary gastrointestinal (GI) cancers associated with cutaneous manifestation
Colorectal cancer (CRC) is a very common malignancy worldwide. About 140,000 people are diagnosed with CRC every year in United States and 900,000 cases worldwide. Hereditary conditions account about 15% of CRCs (1). This group includes familial adenomatous polyposis (FAP), hereditary nonpolyposis colon cancer (Muir-Torre; Lynch syndrome), Peutz-Jeghers syndrome (PJS), juvenile polyposis syndrome (JPS), Cowden syndrome, Bannayan Riley-Ruvulcaba syndrome and Cronkhite-Canada syndrome (CCS) (2). Typically a single gene, a combination of genes, or a combination of genetic and environmental factors can cause familial CRC. Early diagnosis and screening is crucial for managing and timely administration of treatment of the patients. Distinct skin manifestations of these syndromes may hint at the underlying disease and therefore have an important value in prompt diagnosis. Although these syndromes are uncommon, it is essential for the clinician to consider them due to the strong association of these syndromes with morbidity and mortality secondary to both malignant and nonmalignant conditions. The availability of clinical genetic testing for these disorders makes accurate diagnosis and timely managing of the patient possible (3) (Table 1).
Table 1. Hereditoty gastriintstinal conditions associated with cutaneous manifestations.
| Syndromes | Inheritance pattern | Affected gene | Involved chromosome | Cutaneous manifestations |
|---|---|---|---|---|
| Familial adenomatous polyposis/Gardener syndrome | Autosomal dominant | APC | 5q21-q22 | Epidermoid cyst, lipomas, desmoids tumors |
| Peutz-Jegheres syndrome | Autosomal dominant | STK11 | 19p13.3 | Hyperpigmented macules on face and fingers |
| Hereditary nonpolyposis Colorectal syndrome (Lynch syndrome, Muir-Torre syndrome) | Autosomal dominant | MLH1, MSH2, MSH6, PS2 | 2p16, 3p21b, 2q32, 7p22 | Sebaceous adenoma, epithelioma, sebaceous adenomas, sebaceous carcinoma and multiple keratoacanthoma |
| Cowden syndrome | Autosomal dominant | PTEN | 10q23.31 | Tricholemomas, oral papillomatosis, facial papules, acral keratosis |
| Bannayan-Riley-Ruvalcaba syndrome | Autosomal dominant | PTEN | 10q23.31 | Hyperpigmented macules on genitalia, acrochordons, lipomas |
| Cronkhite-Canada syndrome | – | – | – | Nail dystrophy, alopecia, onycholysis, onychomadesis, hyperpigmemtation |
| Juvenile polyposis syndrome | Autosomal dominant | MADH4, BMPR1A | 18q21.1,10q22.3, 17q11.2 | – |
Familial adenomatous polyposis (FAP)
FAP is an autosomal dominant disorder caused by a germline mutation in the APC gene located on 5q21-q22. This mutation cause widespread polyposis in colon and rectum. In most number of these cases the number of polyps is in thousands. Therefore there is a potential risk of developing adenocarcinoma if patient left untreated. Duodenal cancer is the second most common malignancy in FAP. These patients are also predisposed to other malignancies, including thyroid, brain (most often medulloblastomas), adrenal, and liver cancers in addition to CRC. Prophylactic colectomy should be performed before age 25 years old. Annual upper GI endoscopy and thyroid examination is also recommended (4).
Gardner syndrome (GS)
GS is a variant of FAP characterized by numerous GI adenomatous polyps. Extracolonic manifestations may also occur, such as multiple osteomas of skull, mandible, skin tumors, dental abnormalities, and congenital hypertrophy of the retinal pigment epithelium. Cutaneous manifestations include epidermoid cysts, lipomas and multiple soft tissue neoplasms such as desmoid tumors (DTs). DTs are locally aggressive mesenchymal neoplasms originated from proliferating mature fibroblasts (5). Thirty seven to 50% of DTs occur in the abdominal region. The most common extra-abdominal sites include the shoulder girdle, chest wall, and inguinal regions. A skin examination may reveal a nontender, well-circumscribed, firm, flesh-colored tumor (6). Epidermoid cysts are usually multiple in number and present on the face or extremities. They are frequently seen before the appearance of intestinal polyps. Skin manifestations can be treated through a variety of excisions and therapy depending on location, size, and number of malignancies (7)
One early sign of this syndrome is congenital hypertrophy of retinal pigmented epithelium (CHRPE). On ophthalmologic examination CHRPE is a patch of darkly pigmented retinal epithelium. Although it is not specific for FAP and GS, presence of the bilateral, large or multiple CHRPE is considered a phenotypic marker for FAP or GS (8). Dental abnormalities include odontomas, and supernumerary or unerupted teeth (5).
Peutz-Jeghers syndrome (PJS)
This autosomal-dominant disorder is caused by mutations in the STK11 (a tumor suppressor) gene located on chromosome 19p13.3 (9). PJS is characterized by hamartomatous polyposis, mucocutaneous pigmentation, and an increased risk of visceral malignancy (1). The GI manifestations include the presence of multiple hyperplastic polyps/serrated adenomas (4). Several studies have proven the increased lifetime risk for both CRC and gastric cancer. Polyps are found throughout the GI tract, but mostly within the jejunum and colon. They could also be found in the gallbladder, bronchi, bladder, and ureter. These polyps can cause bleeding, obstruction and also intussusception. So far, there is no universal consensus for treating the patients. Endoscopic polypectomy appears to be sufficient in some patients whereas in patients with multiple polyps, severe symptoms or family history of CRC, subtotal colectomy and gastrectomy should be considered. Microscopically these polyps have a “Christmas tree” appearance at low power with broad bands of muscularis mucosa smooth muscle especially in the center (Figure 1).
Figure 1.
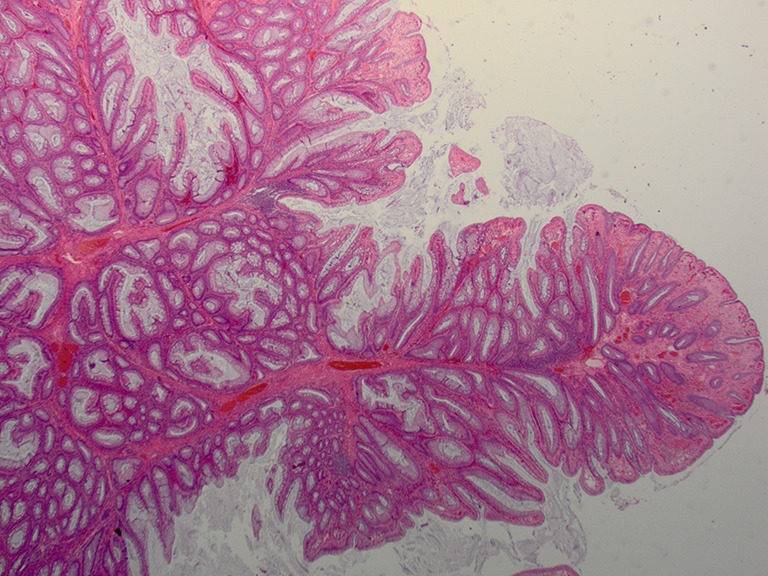
Hyperplastic polyp in Peutz-Jeghers syndrome (Hematoxyline and Eosin stain; original magnification: ×2).
The cutaneous manifestations are characterized by presence of 1-5 mm hyperpigmented macules on the lips, buccal mucosa, periorbital area, nose, genitalia, and on the fingers (Figure 2). These macules often precede GI symptoms. The cutaneous lesions often fade after puberty while oral-buccal mucosal pigmentation is usually permanent (9,10).
Figure 2.
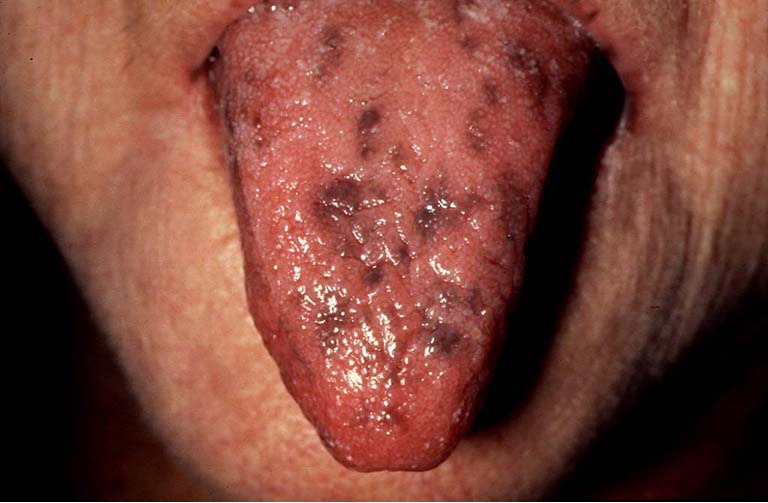
Hyperpigmentation of the tongue in Peutz-Jeghers syndrome.
Hereditary nonpolyposis colorectal cancer (HNPCC)
Lynch syndrome is the most common form of hereditary CRC, accounting for about 2-4% of all CRCs. LS is an autosomal dominant disease resulting from germ line mutations in DNA mismatch repair (MMR) genes, including MSH2, MLH1, MSH6 and PMS2. The MMR system is necessary for correcting single-base mismatches and insertion-deletion loops that form during DNA replication (11,12). They are located on chromosomes 2p16, 3p21, 2q32, and 7p22. This syndrome was first described by Warthin in 1913. Later in 1967, Lynch defined it comprehensively by in-depth analysis of families with this syndrome (13-15). Different malignancies including colon, endometrial, urologic, small intestine, ovarian, hepatobiliary and brain tumors have been reported in these patients (3).
Muir-Torre syndrome (MTS)
MTS is a variant of LS described in the 1960s by Muir et al. (16) and Torre (17). It is observed in 1% up to 9% of patients with HNPCC (3,4). In 1967, Muir et al. presented a case of a Maltese man with cancer of the colon, duodenum, and larynx presenting with a keratoacanthoma of the face. In 1968, Torre noted the involvement of sebaceous tumors in a patient with carcinoma of the ampulla of Vater. The disease appears to have a slight male predominance, with a male to female ratio of 3:2 (18).
The majority of cutaneous manifestations of LS are associated with this variant. Skin manifestations include sebaceous tumors, epitheliomas, carcinomas, or multiple keratoacanthomas. The sebaceous tumors are sometimes difficult to classify, but they most resemble sebaceous adenoma, sebaceoma, or occasionally sebaceous carcinoma. The cutaneous tumors may precede or follow the visceral malignancies, and may be observed in sporadically in other family members (19).
The most common skin lesion is the sebaceous adenoma, presenting as yellow papules or nodules predominantly on the face (20) (Figure 3). Histologically sebaceous adenoma has a nodular lobulated growth with dark and light areas corresponding to generative cells (dark) and sebaceous cells (light) with cytoplasmic lipid vacuoles (Figure 4). Sebaceous epitheliomas differ in the degree of differentiation to adenomas and microscopically may appear similar to a basal cell carcinoma, but with focal sebaceous differentiation. Sebaceous carcinomas are typically found on the eyelids as yellow nodules with tendency to ulceration and are locally aggressive in nature. Excision and cryotherapy are helpful techniques for removal of the skin lesions. Isotetinoin and interferon-α2A are been suggested in treating early lesions and also preventing tumor development (19). Keratoacanthomas may appear in 20% of patients with typical histologic features except on the rare occasion when they may show sebaceous differentiation (1).
Figure 3.
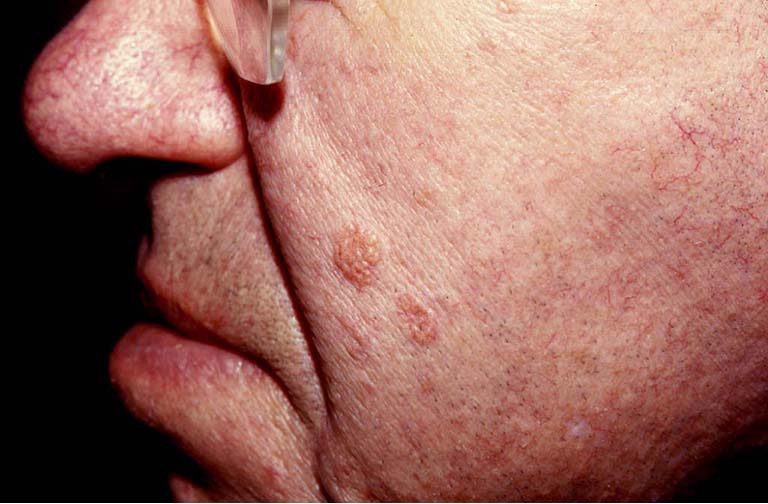
Sebaceous adenoma.
Figure 4.
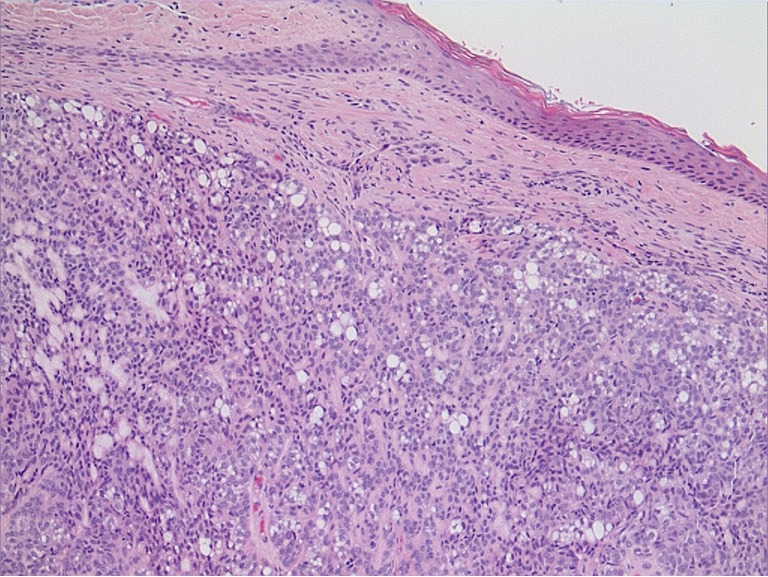
Sebaceous adenoma (Hematoxyline and Eosin stain; original magnification: ×20).
The diagnosis requires a detailed family history, immunohistochemistry of the visceral tumors, and possible microsatellite instability analysis. Vasen et al. study showed reduction risk from 10% to 6% after increasing the frequency of surveillance colonoscopy examinations from 3 to 1-2 years. In patients with this syndrome, the recommended age of starting colorectal surveillance is 20-25 years, based on the finding that the risk of developing CRC before this age is low. Regular dermatologic follow-up is recommended annually (21).
Cowden’s syndrome (CS)
CS or multiple hamartoma syndromes (MHSs) is an autosomal dominant disorder with multiple hamartomas and increased risk of malignancies. The malignancies can develop from increased cellular proliferation of all 3 embryogenic germ cell lines. CS was first described by Lioyd and Dennis in 1963 (22). The syndrome is named after patient “Rachel Cowden” who presented with multiple hamartomas, unusual skin and facial findings, abnormal CNS findings, and fibrocystic breast disease. Later, in 1972, Weary et al. described similar symptoms in case series of five patients.
CS is inherited in an autosomal dominant fashion with incomplete penetrance and variable expressivity. The responsible gene is located on chromosome 10q22-23 and is known as PTEN (phosphatase and tensin homologue). Inheriting a mutation in one allele of PTEN, and subsequent “second hit” mutation of the other PTEN allele causes the loss or reduction of PTEN activity. Malfunctioning of this gene results in increased phosphorylation of many key cellular proteins, which in turn can affect processes such as cell cycle progression, metabolism, translation, growth, migration, invasion, angiogenesis, and apoptosis through various signaling pathways.
The most recent diagnostic criteria of CS, published by United States the national comprehensive cancer network (NCCN) is based on pathogenomic criteria with major and minor diagnostic criteria. CS-like syndrome is referred to subset of patients who do not meet the full diagnostic criteria for CS (23,24) (Table 2). This group of patients may have germline mutations in the succinate dehydrogenase subunits B or D or mutations in the Killin gene (a tumor suppressor gene, an inhibitor of DNA synthesis regulated by p53) (28,29).
Table 2. International Cowden Syndrome Consortium diagnostic criteria. Cowden syndrome can be diagnosed by the following.
| Any pathognomonic lesion |
| Macrocephaly and one other major criterion |
| One major and 3 minor criteria |
| Four minor criteria |
| Pathognomonic criteria |
| Mucocutaneous lesions |
| Facial trichilemmomas |
| Acral keratosis |
| Papillomatous papules |
| Major criteria |
| Breast carcinoma |
| Nonmedullary (follicular) thyroid carcinoma |
| Macrocephaly (>97th percentile) |
| Endometrial carcinoma |
| Lhermitte-Duclos syndrome |
| Minor criteria |
| Other thyroid lesions (e.g., adenoma, multinodular goiter) |
| Mental retardation |
| Gastrointestinal hamartomas |
| Fibrocystic disease of the breast |
| Lipomas |
| Fibromas |
| Genitourinary tumors (renal cell carcinoma and uterine fibroids) |
| Mucocutaneous lesions |
| Pathognomonic lesion then the following subcriteria must be fulfilled: |
| ≥6 facial papules (>3 trichilemmomas) |
| Cutaneous facial papules and oral mucosal papillomatosis |
| Acral keratoses and oral mucosal papillomatosis |
| ≥6 palmoplantar keratoses (National Comprehensive) |
GI involvement is seen in 70-80% 0f patients. Polyps can involve the entire the entire GI tract. The rectosigmoid region is almost always involved and small bowel is rarely involved (3). The polyps range in size from 1 mm to several centimeters in diameter. The histological features of the polyps are not specific. The most common type is hamartomatous, however other subtypes including adenomatous, lipomatous, inflammatory, fibromatous, and hyperplastic lesions are reported (23).
The mucocutaneous lesions are found in 100% of patients. These lesions include tricholemomas, oral papillomatosis, facial papules, and acral keratosis. Tricholemmomas are benign hamartomatous lesions of the hair follicle outer root sheath (trichilemma). They are slow growing, flesh colored, usually measuring 1-5 mm in diameter and most often found close to the hairline of the face and neck (29). Microscopically, trichilemma is characterized by vertically oriented bulbous hyperplasia of infandibular epithelial cells confined by peripherally palisading clear cells (Figure 5). Papillomatous papules are benign mucocutaneous lesions that occur most often on the oral mucosa, face, and pressure points such as the palmar and plantar surfaces. They produce a cobblestone pattern on the buccal and gingival mucosa (30). Lipomas are seen in 30% of the patients and café au lait spots in 9% of patients with CS. Other mucocutaneous lesions include hemangiomas, neuromas, xanthomas, vitiligo, acanthosis nigricans, peritoneal and acral lentigines, keratosis punctata and speckled pigmentation of penis (1,31,32).
Figure 5.
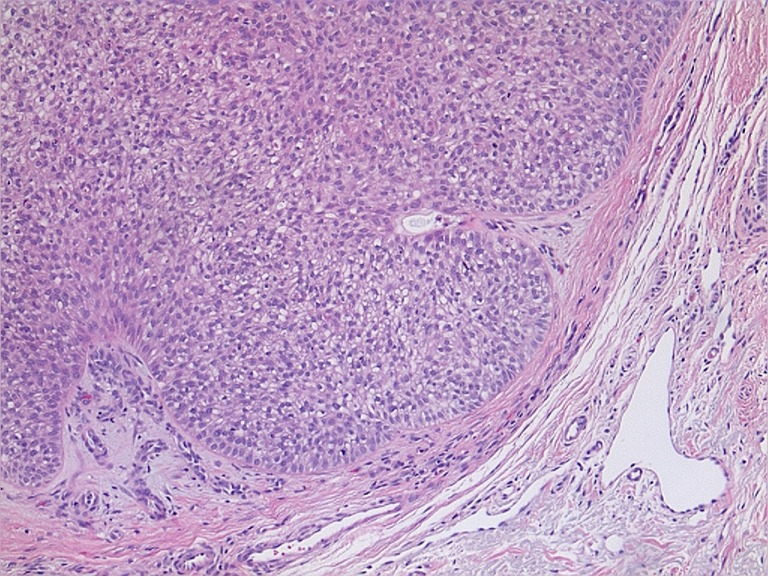
Tricholemmoma (Hematoxyline and Eosin stain; original magnification: ×20).
The most common type of visceral malignancy in CS is breast cancer, which is 50% more common than in the general population. The second and third most common cancers are thyroid and colon, respectively. The most common histological subtype for thyroid cancer is papillary and follicular and the most common type of breast cancer is invasive ductal carcinoma, reflecting similar distribution seen in the general population. The other reported malignancies include endometrial, testicular seminoma and mixed germ cell tumors. Therefore, CS patients must undergo surveillance and screening particularly for breast, thyroid and CRCs. Biannual breast examinations starting at 25 years of age, followed by mammography after 35 years old are recommended. Thyroid examination with ultrasound exam should be started at 18 years old. The treatment of mucocutaneous lesions includes topical 5-fluorouracil, CO2 laser ablation, cryotherapy, dermabrasion, interferon-2 alfa, bleomycin, and surgery (23,33).
Bannayan-Riley-Ruvalcaba syndrome (BRRS)
BRRS is an autosomal dominant genodermatosis characterized by GI hamartomatous polyps, macrocephaly, hyperpigmentation of the glans penis, developmental delay and hemangiomas. Germline mutation of the PTEN gene can be detected in 60% of individuals. This syndrome originally described as the triad of macrocephaly, lipomatosis and pigmentation of glans penis. Although BRRS has overlapping features with CS, it is usually diagnosed at younger ages with 68% male predominance. In contrast, CS often occurs later in life and more frequently seen in women. GI manifestations include hamartomatous polyps found in 50% of patients, diarrhea, intussusceptions and anemia. Polyps can be seen in the entire GI tract, however they are more common in distal ileum and colon. BRRS is not associated with CRC or other GI malignancies however these patients are at risk of malignancies of PTEN mutations including breast, thyroid, kidney, and endometrium (34,35).
The most specific related cutaneous manifestation of BRRS is Hyperpigmented macules involving the glans penis or vulva. Other skin findings include genital lentigines, facial verrucae-like or acanthosis nigricans-like lesions, multiple acrochordons of the neck, axilla, and groin, vascular malformations, and lipomas (3,36). Histologically the hyperpigmented lesions appear as lentiginous epidermal hyperplasia, with an increased number of melanosomes and a slight increase in melanocytes (37).
Other reported findings include central nervous system symptoms like hypotonia, delayed psychomotor development, seizures and ocular abnormalities involving retina and cornea (3).
All the patients with BRRS regardless of phenotypic expression are at risk of malignancies. Therefore, comprehensive management focused on early diagnosis by frequent screening of the organs at risk of cancer is required. The current guidelines are similar to CS (38).
Cronkhite-Canada syndrome (CCS)
CCS was first described by Cronkhite and Canada in 1995 (39). So far, about 400 cases mainly from European or Asian descent with mean age of 59 years old have been reported. CCS is characterized by diffuse GI tract polyposis and unique ectodermal manifestations. The disease has never been reported as having occurred in a relative of an affected patient (40). The etiology of CCS is not well understood but some studies have shown association with elevated antinuclear antibody (ANA) and IgG4 levels (41,42). There is also an association between CCS with hypothyroidism and various autoimmune diseases such as systemic lupus erythematous, rheumatoid arthritis, and scleroderma, all of which point toward an autoimmune etiology (40,43). The mean age of onset is estimated to be in the fifth to sixth decade, with a slight male predominance of 3:2 ratio (44).
The most common presenting symptoms include diarrhea, weight loss, nausea, vomiting, hypogeusia and anorexia (45). Endoscopic examination reveals diffuse polyposis of the entire GI tract. Histopathologically, polyps in CCS are most frequently described as being of the hamartomatous or juvenile type. In microscopic examination intestinal mucosa may also reveal edema of the lamina propria, cystic dilation of glands and inflammatory cell infiltration with mononuclear cells and eosinophils. These polyps can cause different complications like enteropathy, GI bleeding, intussusception, and prolapse. The malignant potential of polyps in CCS is controversial (44). However, it appears that these clinical manifestations more commonly occur in patients with the larger “giant” polyps (46).
The cutaneous manifestation of disease might be partly due to malabsorption and nutritional deficiency (3). Nail dystrophy has been described in 98% of patients and can include thinning, splitting, onycholysis, and onychomadesis (44). Alopecia and hyperpigmentation also occur in these patients. Alopecia starts as patchy lesion and can progress to alopecia totalis, involving both the scalp and body hair (3,45). Hyperpigmentation ranges from light to dark macular lesions predominantly involving the face, palms, soles, and neck (3,44). Microscopic examination of biopsied skin reveals abnormally increased melanin deposition with or without increased melanocyte proliferation (47,48).
Multiple different treatment approaches have been tried in these patients. However, the most common approach is aggressive nutritional support, including electrolyte replacement and parenteral nutrition when necessary. There have been reports of patients achieving remission for more than 5 years with complete symptomatic improvement and resolution of ectodermal changes. But, unfortunately, most of the patients do not respond to nutritional therapy alone, and other treatments are required. Antibiotics, corticosteroids, histamine receptor antagonists, have been used with anecdotal success in few patients (40,49-51). The CCS carries a poor long-term prognosis, with a 55% mortality rate documented at least in one study (40).
Juvenile polyposis syndrome (JPS)
JPS is an autosomal dominant disease characterized by predisposition to multiple juvenile polyps in the GI tract and is associated with an increased risk of colorectal and ventricular cancer. Diagnosis is by the presence of any of the following three criteria: 3 to 5 juvenile polyps in the colorectum, juvenile polyps throughout the entire GI tract, or any number of juvenile polyps in an individual with a family history of JPS. If the polyps are left untreated, they may cause bleeding and anemia. Most juvenile polyps are benign; however, malignant transformation can occur. Risk for GI cancers in families with JPS ranges from 9% to 50%. Most of this increased risk is attributed to colon cancer, but cancers of the stomach, upper GI tract, and pancreas have also been reported.
20% of patients have germline mutations in the SMAD4 and BMPR1A genes that encode proteins involved in the transforming growth factor-beta signaling pathway. Also PTEN mutations have been reported in 5% of patients (3,35).
Although JPS is not associated with skin manifestations, occasionally it presents with hereditary hemorrhagic telangiectasia (HHT) or Osler-Weber-Rendu disease (especially in SMAD4 pathogenic variant) (35,52). Patients with combined HHT and JPS will present with cutaneous features of HHT including multiple punctate telangiectasiaes in the skin and mucous membranes (52).
Inflammatory bowel disease (IBD)
IBD is a disease that affects the intestinal tract via an inflammatory process. Ulcerative colitis (UC) and Crohn’s disease (CD) are two major types of IBD. The incidence of UC and CD are similar, at 7.3 cases per 100,000 and 5.8 cases per 100,000 people per year, respectively (53,54). Multiple extraintestinal manifestations in various organ systems have been reported to be associated with IBD. Skin is one of the organs that commonly get involved through different mechanisms. Cutaneous manifestations may precede, occur with or postdate the onset of the intestinal disease and can significantly contribute to the morbidity and impairment of the affected patients. Although there are many overlaps between UC and CD, but they are unique in many different ways. UC mainly involves colon and rectum, whereas CD can affect any part of GI tract from mouth to anus. Histologically inflammation is limited to mucosal and submucosal surface in UC and full thickness involvement is seen in CD. Human leukocyte antigen B27 (HLA-B27) is found in most patients with UC, whereas the IBD1 gene is associated with susceptibility to CD (55).
Skin manifestations of IBD are divided into three discrete categories based on the pathogenesis.
Specific lesions
These lesions are caused by direct involvement of the skin by the same pathological mechanism seen in GI tract involvement. Therefore, it is only seen in CD because UC usually does not involve mucosal membranes (55,56).
Fissures and fistulae: one study has shown that perianal fissures and fistulas are seen in 36% of patients with CD. The majority of these people have active perianal disease compared to patients with disease confined to small bowel. Histologically these lesions are similar to GI tract lesions with transmural inflammation and granulomatous process. Perianal abscess, skin tags, acrochordons, and chronic edematous inflammation are often seen in association of these lesions (55).
Oral Crohn’s disease: oral lesions occur in 5-20% of patients with CD. The presentation and severity of these lesions do not always correlate with the activity of the underlying bowel disease. Clinically they manifest as aphthous stomatitis, pyostomatitis vegetans, angular cheilitis, ulceration, mucosal nodularity (cobblestoning of the buccal mucosa), nodules of gingival and alveolar mucosa, and indurated fissuring of lower lips. These lesions are often painful and disruptive to eating (56).
Metastatic Crohn’s disease (MCD): similar to other specific lesions MCD are specific granulomatous cutaneous lesions with the same histopathology (non-caseating granulomas with multinucleated giant cells in the dermis) as the intestinal lesions. MCP is predominantly found on the extremities or in intertriginous areas, with facial and genital lesions rarely reported (56). Treatment of underlying CD is the treatment of choice for MCD, however surgical resection of involved segment of bowel does not guarantee resolving of the cutaneous lesions (55).
Reactive lesions
These lesions are seen in both UC and CD. Reactive lesions are caused by the underlying IBD without exhibiting similar pathologic features of the GI tract lesions. Cross antigenicity between the skin and intestine mucosa might be responsible for reactive lesions (57).
Erythema nodosum (EN): EN is the most common cutaneous manifestation of IBD, presenting in 4-6% of the patients. It has a higher incidence in CD than in UC. EN is often associated with systemic symptoms of fevers, chills, arthralgias, or arthritis (58-60). Typical EN lesions present as painful, tender, warm nodules (1-5 cm in diameter), and raised bluish-red subcutaneous lesions or plaques located on the extensor surfaces of extremities, predominantly on the anterior surface of the lower extremities (56-61). Unusual cases with atypically located case on the trunk, neck, and even on the face have been reported in literature (59,60,62). Biopsy is generally not necessary as the diagnosis of EN may be confirmed based on the clinical presentation. The classic histological presentation is lympho-histocytic infiltrate of lower dermis and focal panniculitis (56). Direct immunofluorescence of EN lesions reveal perivascular deposits of immunoglobulins and complement, suggesting that the pathogenesis of reactive cutaneous manifestation of IBD is due to the abnormal immunological response of common antigens between bowel bacteria and skin (63). EM lesions usually mirror IBD activity in colon and appropriate treatment of underlying disease results in resolution of the lesions (56).
Pyoderma gangrenosum (PG): PG is the second most common skin manifestation of IBD, documented in 3% of patients (64,65). PG is more common in UC (5-12%) than CD (1-2%) and like EN some studies have shown a female predilection (56,66,67). PG was first described by Brocq in 1916 and was initially considered to represent an infectious process (68). PG initially presents as pustules, and nodules. These pustules and nodules eventually expand outward and develop into painful deep ulcers with undermined wound edges. The ulcer has sharply circumscribed and demarcated borders with a necrotic, granulated yellowish base. The ulcers can be single or multiple, unilateral or bilateral, and can range in size from several centimeters to the surface of an entire limb (56,65,68). PG usually occurs on the extensor surface of the legs, but can appear anywhere on the skin (69). A unique PG presentation found frequently in the setting of IBD is peristomal PG at the stoma site created after bowel resection, most likely reflecting pathergy (70). PG does not necessarily follow the course of the underlying IBD (70,71). Localized lesions with IBD might response to topical therapy with potent corticosteroids, tacrolimus, or intralesional corticosteroids, especially if treatment starts early while lesions are still small and superficial. Widespread or refractory lesions need systemic therapy. Tumor necrosis factor alfa inhibitors are considered first-line therapy in these situations, with infliximab showing the most rapid response (72,73).
Pyostomatitis vegetans (PSV): PSV is a rare skin manifestation of IBD characterized by presents of pustules and ulcerations in skin folds such as axillary or inguinal area, but can also be present on the trunk or extremities (65). The pustules can quickly rupture and form erosions with hemorrhagic ground and large raised (vegetating) well-demarcated plaques. PSV shares some similarities with PG of the skin (74). PSV, is the oral equivalent of PSV on skin, and is relatively specific for IBD, particularly UC (61,75). Lesions are erythematous, thickened, and hyperplastic folds of the buccal and labial mucosa with multiple gray to yellow pustules, which progress to erosions forming shallow, folded, and fissured “snail tract” ulcers, due to degeneration, ulceration, and suppuration of vegetating pustules (75). Although the pathophysiology of this condition is not fully understood, but it is widely believed that immune system dysregulation is the cause (61,76). PDV was first reported by Hallopeau in 1898 (77). He described two patients with unusual pustular dermatosis and oral lesions for which he named “pyodermite vegetans” (56,77). In 1949 McCarthy observed similar lesions isolated in the oral cavity and proposed the term “pyostomatitis vegetans” (56,78). Recently these two entities are considered to be variants of the same disease termed pyodermatitis–pyostomatitis vegetans (PPV) (56,75). Local treatment of lesions including hydrogen peroxide, topical steroids, and antibacterial mouth-washes are usually not effective in controlling disease activity (56). Treatment of the underlying IBD is essential for ultimate controlling these lesions (55).
Associated conditions
Associated conditions are likely related to HLA linkage and the chronic inflammatory nature of IBD. These include psoriasis, vitiligo, reactive arthritis, eczema, clubbing of the nails, and acrodermatitis enteropathica (55).
Psoriasis is the most common associated cutaneous diseased, occurs in about 7-10% of patients with IBD compared to 1-2% of general population (56,64). The association between IBD and psoriasis is both genetically and immunologically. Certain gene loci on chromosomes 3, 4, 6, and 16 are found to be associated with both CD and psoriasis (65).
Conclusions
The accompanying involvement of GI and cutaneous systems is frequently seen in different disorders. We reviewed the cutaneous manifestations of GI disease with emphasize of collaborative role of dermatologist, gastroenterologists and other specialties in early diagnosis and treatment of these patients. The comprehensive history taking and detailed physical examination is essential for accurate diagnosis in concurrent involvement of cutis and intestinal tract. The new advances in genetic testing have enabled the precise diagnosis of most of these syndromes. Moreover, many patients will now benefit from new pharmacological agents, and surgical methods.
Acknowledgements
None.
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Cancer facts and figures 2014. Available online: http://www.cancer.org/acs/groups/content/@research/documents/webcontent/acspc-042151.pdf
- 2.Genetics of Colorectal Cancer (PDQ®) Major Genetic Syndromes. National Cancer Institute, 2014. Available online: http://www.cancer.gov/cancertopics/pdq/genetics/colorectal/HealthProfessional/page3#top
- 3.Shah KR, Boland CR, Patel M, et al. Cutaneous manifestations of gastrointestinal disease: part I. J Am Acad Dermatol 2013;68:189.e1-21; quiz 210. [DOI] [PubMed]
- 4.Rosai J, editor. Rosai and Ackerman's Surgical Pathology, 9th ed. New York: Mosby, 2004:755-61. [Google Scholar]
- 5.Pinheiro LV, Fagundes JJ, Coy CS, et al. Multiple desmoid tumors in a patient with Gardner's syndrome - Report of a case. Int J Surg Case Rep 2014;5:370-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shields CJ, Winter DC, Kirwan WO, et al. Desmoid tumours. Eur J Surg Oncol 2001;27:701-6. [DOI] [PubMed] [Google Scholar]
- 7.Juhn E, Khachemoune A. Gardner syndrome: skin manifestations, differential diagnosis and management. Am J Clin Dermatol 2010;11:117-22. [DOI] [PubMed] [Google Scholar]
- 8.Baker RH, Heinemann MH, Miller HH, et al. Hyperpigmented lesions of the retinal pigment epithelium in familial adenomatous polyposis. Am J Med Genet 1988;31:427-35. [DOI] [PubMed] [Google Scholar]
- 9.Ghevariya V, Singhal S, Anand S. The skin: a mirror to the gut. Int J Colorectal Dis 2013;28:889-913. [DOI] [PubMed] [Google Scholar]
- 10.Jelsig AM, Qvist N, Brusgaard K, et al. Hamartomatous polyposis syndromes: a review. Orphanet J Rare Dis 2014;9:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Samadder NJ, Jasperson K, Burt RW. Hereditary and common familial colorectal cancer: evidence for colorectal screening. Dig Dis Sci 2015;60:734-47. [DOI] [PubMed] [Google Scholar]
- 12.Rustgi AK. The genetics of hereditary colon cancer. Genes Dev 2007;21:2525-38. [DOI] [PubMed] [Google Scholar]
- 13.Classics in oncology. Heredity with reference to carcinoma as shown by the study of the cases examined in the pathological laboratory of the University of Michigan, 1895-1913. By Aldred Scott Warthin. 1913. CA Cancer J Clin 1985;35:348-59. [DOI] [PubMed] [Google Scholar]
- 14.Lynch HT, Lynch PM, Lanspa SJ, et al. Review of the Lynch syndrome: history, molecular genetics, screening, differential diagnosis, and medicolegal ramifications. Clin Genet 2009;76:1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lynch HT, Shaw MW, Magnuson CW, et al. Hereditary factors in cancer. Study of two large midwestern kindreds. Arch Intern Med 1966;117:206-12. [PubMed] [Google Scholar]
- 16.Muir EG, Bell AJ, Barlow KA. Multiple primary carcinomata of the colon, duodenum, and larynx associated with kerato-acanthomata of the face. Br J Surg 1967;54:191-5. [DOI] [PubMed] [Google Scholar]
- 17.Torre D. Multiple sebaceous tumors. Arch Dermatol 1968;98:549-51. [DOI] [PubMed] [Google Scholar]
- 18.Svec J, Schwarzová L, Janošíková B, et al. Synchronous gastric and sebaceous cancers, a rare manifestation of MLH1-related Muir-Torre syndrome. Int J Clin Exp Pathol 2014;7:5196-202. [PMC free article] [PubMed] [Google Scholar]
- 19.Weedon D, Geoffrey S. Tumors of cutaneous appendages. In: Weedon D. editors. Skin pathology. London: Churchill Livingstone, 2004:874-5. [Google Scholar]
- 20.Banse-Kupin L, Morales A, Barlow M. Torre's syndrome: report of two cases and review of the literature. J Am Acad Dermatol 1984;10:803-17. [DOI] [PubMed] [Google Scholar]
- 21.Vasen HF, Abdirahman M, Brohet R, et al. One to 2-year surveillance intervals reduce risk of colorectal cancer in families with Lynch syndrome. Gastroenterology 2010;138:2300-6. [DOI] [PubMed] [Google Scholar]
- 22.Lloyd KM, 2nd, Dennis M. Cowden's disease. A possible new symptom complex with multiple system involvement. Ann Intern Med 1963;58:136-42. [DOI] [PubMed] [Google Scholar]
- 23.Farooq A, Walker LJ, Bowling J, et al. Cowden syndrome. Cancer Treat Rev 2010;36:577-83. [DOI] [PubMed] [Google Scholar]
- 24.Weary PE, Gorlin RJ, Gentry WC, Jr, et al. Multiple hamartoma syndrome (Cowden's disease). Arch Dermatol 1972;106:682-90. [PubMed] [Google Scholar]
- 25.National Comprehensive Cancer Network web site. Clinical practice guidelines in oncology. Cowden syndrome. Accessed on September 5, 2011. Available online: http://www.nccn.org/professionals/physician_gls/f_guidelines.asp
- 26.Eng C. Will the real Cowden syndrome please stand up: revised diagnostic criteria. J Med Genet 2000;37:828-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pilarski R, Burt R, Kohlman W, et al. Cowden syndrome and the PTEN hamartoma tumor syndrome: systematic review and revised diagnostic criteria. J Natl Cancer Inst 2013;105:1607-16. [DOI] [PubMed] [Google Scholar]
- 28.Ni Y, Zbuk KM, Sadler T, et al. Germline mutations and variants in the succinate dehydrogenase genes in Cowden and Cowden-like syndromes. Am J Hum Genet 2008;83:261-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brownstein MH, Mehregan AH, Bilowski JB. Trichilemmomas in Cowden's disease. JAMA 1977;238:26. [PubMed] [Google Scholar]
- 30.Jornayvaz FR, Philippe J. Mucocutaneous papillomatous papules in Cowden's syndrome. Clin Exp Dermatol 2008;33:151-3. [DOI] [PubMed] [Google Scholar]
- 31.Ferran M, Bussaglia E, Lazaro C, et al. Acral papular neuromatosis: an early manifestation of Cowden syndrome. Br J Dermatol 2008;158:174-6. [DOI] [PubMed] [Google Scholar]
- 32.Schaffer JV, Kamino H, Witkiewicz A, et al. Mucocutaneous neuromas: an underrecognized manifestation of PTEN hamartoma-tumor syndrome. Arch Dermatol 2006;142:625-32. [DOI] [PubMed] [Google Scholar]
- 33.Wheeland RG, McGillis ST. Cowden's disease--treatment of cutaneous lesions using carbon dioxide laser vaporization: a comparison of conventional and superpulsed techniques. J Dermatol Surg Oncol 1989;15:1055-9. [DOI] [PubMed] [Google Scholar]
- 34.Schreibman IR, Baker M, Amos C, et al. The hamartomatous polyposis syndromes: a clinical and molecular review. Am J Gastroenterol 2005;100:476-90. [DOI] [PubMed] [Google Scholar]
- 35.Zbuk KM, Eng C. Hamartomatous polyposis syndromes. Nat Clin Pract Gastroenterol Hepatol 2007;4:492-502. [DOI] [PubMed] [Google Scholar]
- 36.Teresi RE, Zbuk KM, Pezzolesi MG, et al. Cowden syndrome-affected patients with PTEN promoter mutations demonstrate abnormal protein translation. Am J Hum Genet 2007;81:756-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Erkek E, Hizel S, Sanlý C, et al. Clinical and histopathological findings in Bannayan-Riley-Ruvalcaba syndrome. J Am Acad Dermatol 2005;53:639-43. [DOI] [PubMed] [Google Scholar]
- 38.Fargnoli MC, Orlow SJ, Semel-Concepcion J, et al. Clinicopathologic findings in the Bannayan-Riley-Ruvalcaba syndrome. Arch Dermatol 1996;132:1214-8. [PubMed] [Google Scholar]
- 39.Cronkhite LW, Jr, Canada WJ. Generalized gastrointestinal polyposis; an unusual syndrome of polyposis, pigmentation, alopecia and onychotrophia. N Engl J Med 1955;252:1011-5. [DOI] [PubMed] [Google Scholar]
- 40.Daniel ES, Ludwig SL, Lewin KJ, et al. The Cronkhite-Canada Syndrome. An analysis of clinical and pathologic features and therapy in 55 patients. Medicine (Baltimore) 1982;61:293-309. [PubMed] [Google Scholar]
- 41.Chan HL, Ho KT, Khoo OT. Cronkhite-Canada syndrome in a Malay. Arch Dermatol 1979;115:98-9. [PubMed] [Google Scholar]
- 42.Takeuchi Y, Yoshikawa M, Tsukamoto N, et al. Cronkhite-Canada syndrome with colon cancer, portal thrombosis, high titer of antinuclear antibodies, and membranous glomerulonephritis. J Gastroenterol 2003;38:791-5. [DOI] [PubMed] [Google Scholar]
- 43.Qiao M, Lei Z, Nai-Zhong H, et al. Cronkhite-Canada syndrome with hypothyroidism. South Med J 2005;98:575-6. [DOI] [PubMed] [Google Scholar]
- 44.Kao KT, Patel JK, Pampati V. Cronkhite-Canada syndrome: a case report and review of literature. Gastroenterol Res Pract 2009;2009:619378. [DOI] [PMC free article] [PubMed]
- 45.Ward EM, Wolfsen HC. Review article: the non-inherited gastrointestinal polyposis syndromes. Aliment Pharmacol Ther 2002;16:333-42. [DOI] [PubMed] [Google Scholar]
- 46.Riegert-Johnson DL, Osborn N, Smyrk T, et al. Cronkhite-Canada syndrome hamartomatous polyps are infiltrated with IgG4 plasma cells. Digestion 2007;75:96-7. [DOI] [PubMed] [Google Scholar]
- 47.Jørgensen H, Mogensen AM, Svendsen LB. Hyperplastic polyposis of the large bowel. Three cases and a review of the literature. Scand J Gastroenterol 1996;31:825-30. [DOI] [PubMed] [Google Scholar]
- 48.Herzberg AJ, Kaplan DL. Cronkhite-Canada syndrome. Light and electron microscopy of the cutaneous pigmentary abnormalities. Int J Dermatol 1990;29:121-5. [DOI] [PubMed] [Google Scholar]
- 49.Ortonne JP, Bazex J, Berbis P. Cronkhite-Canada disease. Discussion apropos of a case and study of the pigmentation. Ann Dermatol Venereol 1985;112:951-8. [PubMed] [Google Scholar]
- 50.Russell DM, Bhathal PS, St John DJ. Complete remission in Cronkhite-Canada syndrome. Gastroenterology 1983;85:180-5. [PubMed] [Google Scholar]
- 51.Russell DM, Bhathal PS, St John DJ. Sustained remission in Cronkhite-Canada syndrome. Gastroenterology 1986;91:1580. [DOI] [PubMed] [Google Scholar]
- 52.Pagon RA, Adam MP, Ardinger HH, et al, editors. Juvenile Polyposis Syndrome. Seattle (WA): University of Washington, Seattle, 1993-2015. [PubMed] [Google Scholar]
- 53.Lashner B. Inflammatory bowel disease. In: Carey WD, editor. Cleveland Clinic: current clinical medicine 2009. Philadelphia: Saunders, 2009. [Google Scholar]
- 54.Loftus EV, Jr, Silverstein MD, Sandborn WJ, et al. Ulcerative colitis in Olmsted County, Minnesota, 1940-1993: incidence, prevalence, and survival. Gut 2000;46:336-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thrash B, Patel M, Shah KR, et al. Cutaneous manifestations of gastrointestinal disease: part II. J Am Acad Dermatol 2013;68:211.e1-33; quiz 244-6. [DOI] [PubMed]
- 56.Huang BL, Chandra S, Shih DQ. Skin manifestations of inflammatory bowel disease. Front Physiol 2012;3:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rachmilewitz D, Stamler JS, Karmeli F, et al. Peroxynitrite-induced rat colitis--a new model of colonic inflammation. Gastroenterology 1993;105:1681-8. [DOI] [PubMed] [Google Scholar]
- 58.Freeman HJ. Erythema nodosum and pyoderma gangrenosum in 50 patients with Crohn's disease. Can J Gastroenterol 2005;19:603-6. [DOI] [PubMed] [Google Scholar]
- 59.Greenstein AJ, Janowitz HD, Sachar DB. The extra-intestinal complications of Crohn's disease and ulcerative colitis: a study of 700 patients. Medicine (Baltimore) 1976;55:401-12. [DOI] [PubMed] [Google Scholar]
- 60.Farhi D, Cosnes J, Zizi N, et al. Significance of erythema nodosum and pyoderma gangrenosum in inflammatory bowel diseases: a cohort study of 2402 patients. Medicine (Baltimore) 2008;87:281-93. [DOI] [PubMed] [Google Scholar]
- 61.Timani S, Mutasim DF. Skin manifestations of inflammatory bowel disease. Clin Dermatol 2008;26:265-73. [DOI] [PubMed] [Google Scholar]
- 62.Basler RS, Dubin HV. Ulcerative colitis and the skin. Arch Dermatol 1976;112:531-4. [PubMed] [Google Scholar]
- 63.Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature 2011;474:307-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Danese S, Semeraro S, Papa A, et al. Extraintestinal manifestations in inflammatory bowel disease. World J Gastroenterol 2005;11:7227-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Georgiou S, Pasmatzi E, Monastirli A, et al. Cutaneous manifestations of inflammatory bowel disease. Hospital Chronicles 2006;1:158-68. [Google Scholar]
- 66.Trost LB, McDonnell JK. Important cutaneous manifestations of inflammatory bowel disease. Postgrad Med J 2005;81:580-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bernstein CN, Blanchard JF, Rawsthorne P, et al. The prevalence of extraintestinal diseases in inflammatory bowel disease: a population-based study. Am J Gastroenterol 2001;96:1116-22. [DOI] [PubMed] [Google Scholar]
- 68.Brocq L. New contribution to the study of geometric phagedaenism. Ann Dermatol Syphiligr 1916;6:1-39. [Google Scholar]
- 69.Lebwohl M, Lebwohl O. Cutaneous manifestations of inflammatory bowel disease. Inflamm Bowel Dis 1998;4:142-8. [DOI] [PubMed] [Google Scholar]
- 70.Keltz M, Lebwohl M, Bishop S. Peristomal pyoderma gangrenosum. J Am Acad Dermatol 1992;27:360-4. [DOI] [PubMed] [Google Scholar]
- 71.Hughes AP, Jackson JM, Callen JP. Clinical features and treatment of peristomal pyoderma gangrenosum. JAMA 2000;284:1546-8. [DOI] [PubMed] [Google Scholar]
- 72.Reichrath J, Bens G, Bonowitz A, et al. Treatment recommendations for pyoderma gangrenosum: an evidence-based review of the literature based on more than 350 patients. J Am Acad Dermatol 2005;53:273-83. [DOI] [PubMed] [Google Scholar]
- 73.Chow RK, Ho VC. Treatment of pyoderma gangrenosum. J Am Acad Dermatol 1996;34:1047-60. [DOI] [PubMed] [Google Scholar]
- 74.Canpolat F, Cemil BÇ, Yimazer D, et al. Pyoderma vegetans associated with ulcerative colitis: a case with good response to steroids. Case Rep Dermatol 2011;3:80-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Femiano F, Lanza A, Buonaiuto C, et al. Pyostomatitis vegetans: a review of the literature. Med Oral Patol Oral Cir Bucal 2009;14:E114-7. [PubMed] [Google Scholar]
- 76.Damm DD, Fantasia JE. Chronic diffuse yellow eruption. Pyostomatitis vegetans. Gen Dent 2002;50:561, 564. [PubMed]
- 77.Hallopeau H. "Pyodermite vegetante". ihre Beziehungen zur dermatitis herpetiformis und dem pemphigus vegetans. Arch Dermatol Syphilol 1898;43:289-306. [Google Scholar]
- 78.McCARTHY FP . Pyostomatitis vegetans; report of three cases. Arch Derm Syphilol 1949;60:750-64. [PubMed] [Google Scholar]