

Abstract
The generation of natural product libraries containing column fractions, each with only a few small molecules, using a high-throughput, automated fractionation system, has made it possible to implement an improved dereplication strategy for selection and prioritization of leads in a natural product discovery program. Analysis of databased UPLC-MS-ELSD-PDA information of three leads from a biological screen employing the ependymoma cell line EphB2-EPD generated details on the possible structures of active compounds present. The procedure allows the rapid identification of known compounds and guides the isolation of unknown compounds of interest. Three previously known flavanone-type compounds homoeriodictyol (1), hesperetin (2), and sterubin (3) were identified in a selected fraction derived from the leaves of Eriodictyon angustifolium. The lignan compound deoxypodophyllotoxin (8) was confirmed to be an active constituent in two lead fractions derived from the bark and leaves of Thuja occidentalis. In addition, two new but inactive labdane-type diterpenoids with an uncommon triol side chain were also identified as coexisting with deoxypodophyllotoxin in a lead fraction from the bark of T. occidentalis. Both diterpenoids were isolated in acetylated form, and their structures were determined as 14S,15-diacetoxy-13R-hydroxy-labd-8(17)-en-19-oic acid (9) and 14R,15-diacetoxy-13S-hydroxy-labd-8(17)-en-19-oic acid (10), respectively, by spectroscopic data interpretation and X-ray crystallography. This work demonstrates that a UPLC-MS-ELSD-PDA database produced during fractionation may be used a powerful dereplication tool to facilitate compound identification from chromatographically tractable small molecule natural product libraries.
Graphic abstract
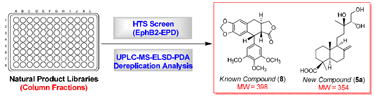
Natural products historically have played a vital role in drug discovery by serving as both prototype drugs and leads for the synthesis of improved drugs. They have also played important roles as probes for elucidating new medically important biological targets, especially in the therapeutic areas of cancer and infectious diseases.1-4 However, the past two decades have witnessed a decrease in natural product drug discovery efforts in the U.S. pharmaceutical industry due to technological challenges in matching the historical discovery paradigm with modern drug discovery strategies. Significant challenges in a natural product drug discovery program include: (1) the complexities of interpreting the observed activities for crude extracts due to low concentrations of active compounds and the potential antagonism/synergism of multiple active compounds; (2) interference of nuisance compounds, making it difficult to prioritize leads for bioassay-guided isolation; (3) increased costs for the time-consuming isolation and structure elucidation processes; and (4) the re-isolation of already known active compounds that hinders the discovery effort. In recent years, several studies have attempted to address these issues,5-16 including a disclosure of an automated high-throughput system to fractionate crude natural product extracts into column fractions plated in microtiter format for high-throughput screening (HTS) driven drug discovery.14 Prior work has demonstrated the removal of potentially interfering compounds such as polyphenols, sugars, and amino acids, and enrichment of the active compounds by this process. The resulting column fractions contain only small organic molecules, and these are generally present in a quantity (>0.5 mg) that is sufficient for contemporary bioassays. Analytical data for the column fractions was acquired routinely during fractionation by ultra-performance liquid chromatography (UPLC) in standard 3-minute runs coupled with multiple channel detection including positive and negative electrospray ionization (ESI) mass spectrometry (MS), evaporative light scanning detection (ELSD), and UV photodiode array spectroscopy (PDA). As column fractions generally contain only a few compounds with similar polarities, these relatively “clean” samples are ideal for biological screening and the available database of analytical data facilitates the rapid characterization of active compounds.
In the current work, a biological screen has been conducted to identify compounds that block proliferation of the ependymoma EphB2-EPD cell model on 16,000 column fractions derived from plants. EphB2-EPD was generated from primary mouse radial glial cells transformed with EphB2, which plays a role in regulating the Ras-MAPK pathway associated with cytoskeletal reorganization and adhesion responses in neuronal growth cones.17 The tumors produced in mice are similar histologically and genetically to human ependymoma. Thus, compounds active against this cell line may serve as leads to develop anticancer drugs for the treatment of childhood brain cancers, which lack effective drugs in the current clinical setting.
After primary screening at a fixed concentration, active column fractions were subjected to dose-response experiments to establish potency. This produced seven prioritized leads that showed EC50 values ranging from 0.02 to 4.1 μg/mL. As described in the referenced paper,17 compounds active against this model were tested to establish potency against untransformed BJ fibroblasts (a normal human foreskin fibroblast cell line available from ATCC) and only compounds with differential activity were followed up. This serves to remove grossly cytotoxic compounds from the workflow. In addition to this immediate control, the fractions have been screened against multiple primary cell types and cell lines and attention focused to those fractions, such as the ones reported herein, that are fairly selective for individual tumor models. In the current study, data from the three most potent fractions from this screen are presented to illustrate how UPLC-MS-ELSD-PDA data can be utilized to guide rapid and efficient dereplication and subsequent structure determination for natural product discovery efforts.
RESULTS AND DISCUSSION
The lead coded as 78821-c4, derived from the leaves of Eriodictyon angustifolium Nutt. (Hydrophyllaceae), was active against EphB2-EPD with an EC50 of 1.5 μg/mL. The UPLC-MS-ELSD-PDA profiles are shown in Figure 1. The minor shifts for the retention times (Rt) of the three major compounds (1–3) in this lead in the respective chromatograms (Figures 1a-1d) are due to the sequential four-channel detection system. The universal ELSD detection method is accurate in assessing the relative content of individual compounds in a mixture that may not be UV active; compound 3 with Rt 1.02 min was present in the highest concentration and represented 50.01% of the total mass. The positive- and negative-ion ESIMS detection procedures showed different sensitivities (Figures 1c and 1d), with the negative mode producing a strong total ion chromatogram (TIC). Thus, the negative ESIMS and UV data were utilized to determine structural information of the compounds present.
Figure 1.
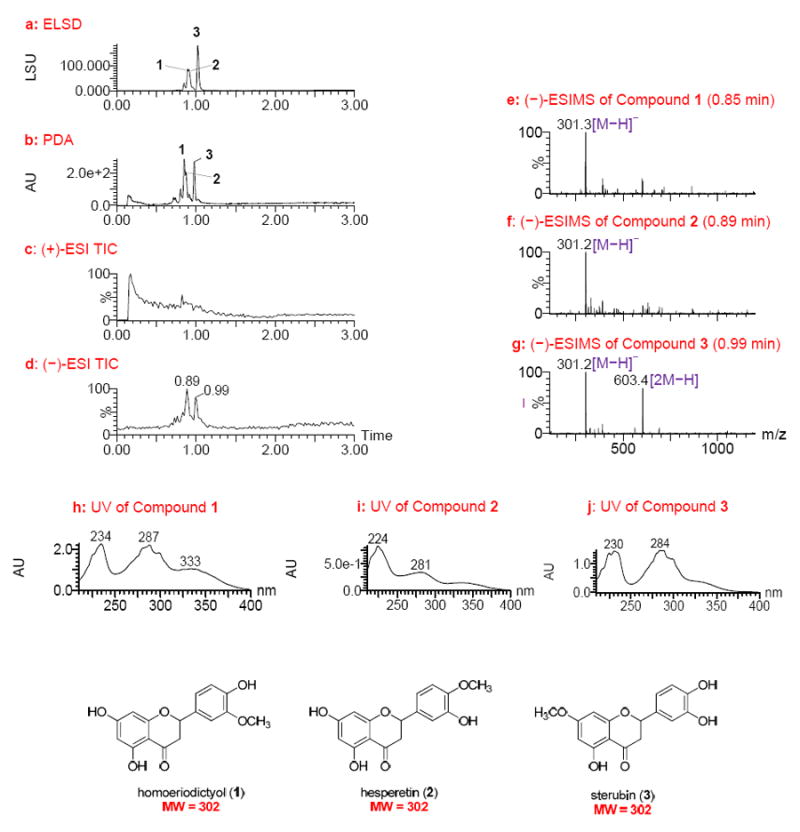
UPLC-MS-ELSD-PDA analysis of lead 78821-c4 (obtained from the leaves of Eriodictyon angustifolia). a: ELSD chromatogram showing compounds 1 – 3 with retention times of 0.89, 0.91, and 1.02 min, respectively; b: PDA chromatogram showing retention times of 0.85, 0.87, and 0.98 min, respectively; c and d: positive and negative ESIMS total-ion chromatograms (TIC), respectively; e-g: negative ESIMS of compounds 1 – 3 with retention times of 0.85, 0.89, and 0.98 min, respectively; and h-j: UV spectra of compounds 1 – 3. UPLC conditions: Acquity UPLC BEH C18 column (2.1 × 50 mm, 1.7 μm); gradient elution starting at 15%, ramping to 20% in 0.2 min, then to 95% CH3CN in water with 0.1% HCOOH in 2.65 min at a flow rate of 1.0 mL/min.
Compound 1 showed a quasimolecular ion base peak at m/z 301.3 [M − H]− in the ESIMS, indicating a molecular weight (MW) of 302 (Figure 1e). Compounds 2 and 3 were determined to have the same MW of 302 based on analysis of their ESIMS (Figures 1f and 1g). In addition, compound 3 generated a strong dimeric quasimolecular ion peak at m/z 603.4 [2M − H]− in the ESIMS (Figure 1g). The UV spectra of the three compounds were found to be similar (Figures 1h-1j), displaying absorptions around 230, 280, and 330 (sh) nm that are characteristic of the flavanone chemotype.18,19 The three compounds were thus identified as homoeriodictyol, hesperetin, and sterubin (1–3, Figure 1), all of which are known constituents of E. californicum, a closely related species within the same genus taxonomically.20,21 Authentic samples of the three compounds were then analyzed using the same UPLC-MS-ELSD-PDA method, confirming compounds 1–3 as homoeriodictyol, hesperetin, and sterubin, respectively, on the basis of analysis of their retention times and ESIMS data (Supporting Information). The strong dimeric quasimolecular ion of sterubin (3) present in the ESIMS (Figure 1g) is likely associated with the catechol structural nature of its B-ring.
Sterubin (3) was reported to possess in vivo antitumor activity in mice and rats against melanoma B16.22 Homoeriodictyol (1) showed weak antimicrobial activity,23 and hesperetin (2) exhibited antioxidative, cardiovascular, neuroprotective, anti-allergic, and antimicrobial activities.24 It was assumed that one or more of the three compounds is responsible for the observed activity in the 78821-c4 sample. Considering the known skeleton, the previously reported biological activities of these compounds, and the well-known issues with the development of flavanones, this lead fraction was deprioritized and further isolation and structure elucidation steps were deemed unnecessary. However, this analysis demonstrates the power of the UPLC-MS-ELSD-PDA technique for the rapid dereplication of known compounds in fractions, thus eliminating time-consuming isolation work.
From the bark of Thuja occidentalis L. (Cuppressaceae), lead 79865-c7 exhibited potent activity against EphB2-EPD with an EC50 of 0.03 μg/mL. The UPLC-MS-ELSD-PDA profiles (Figure 2) indicated that this sample fraction contained five major compounds (4–8), as assessed by ELSD detection with Rt values at 0.90, 0.92, 0.99, 1.02, and 1.19 min (Figure 2a). Among these, compounds 4 and 8 had strong UV absorptions (Figures 2b, 2j, and 2k). All five compounds were well ionized in the positive-ion ESIMS detection mode (Figure 2c), while compound 8 was poorly ionized in the negative-ion ESIMS detection mode (Figure 2d). This again demonstrates that all four detection methods in the system used are complementary, providing comprehensive information including relative concentrations, UV characteristics, and molecular weights of all compounds belonging to different chemotypes.
Figure 2.
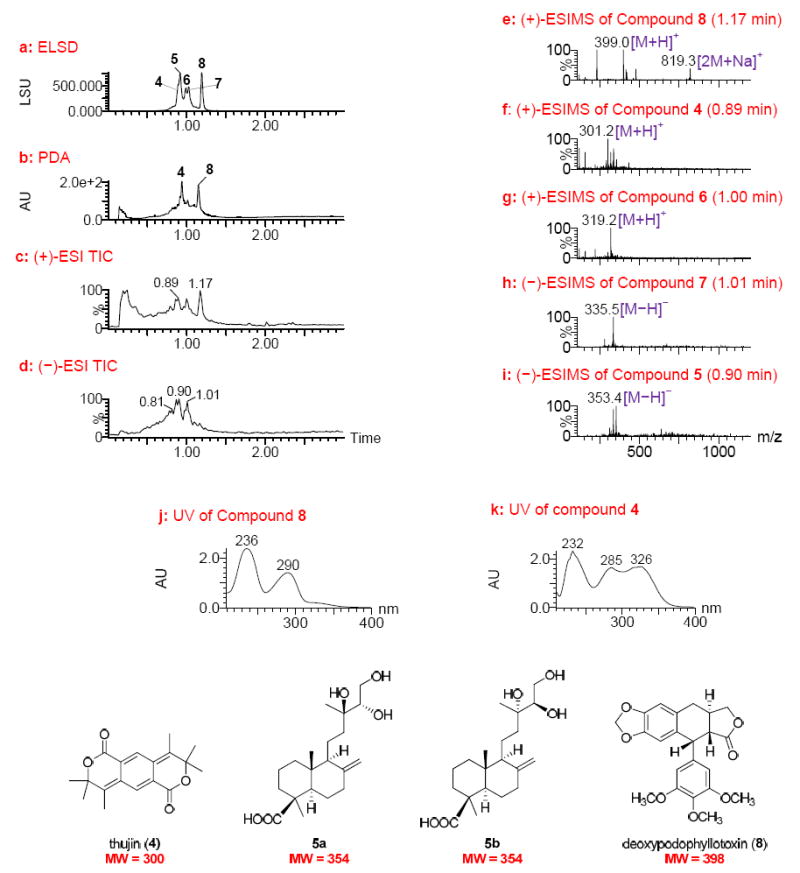
UPLC-MS-ELSD-PDA analysis of lead 79865-c7 (obtained from the bark of Thuja occidentalis). a: ELSD chromatogram showing compounds 4 – 8 with retention times of 0.90, 0.92, 0.99, 1.02, and 1.19 min, respectively; b: PDA chromatogram; c and d: positive and negative ESIMS total-ion chromatograms (TIC), respectively; e-g: positive ESIMS of compounds 8, 4 and 6 with retention times of 1.17, 0.89, and 1.00 min, respectively; h and i: negative ESIMS of compounds with retention times of 1.01 and 0.90 min, respectively; and j and k: UV spectra of compounds 8 and 4. UPLC conditions: Acquity UPLC BEH C18 column (2.1 × 50 mm, 1.7 μm); gradient elution starting at 15%, ramping to 20% in 0.2 min, then to 95% CH3CN in water with 0.1% HCOOH in 2.65 min at a flow rate of 1.0 ml/min.
Compound 8 showed a quasimolecular ion peak at m/z 399.2 [M + H]+ and a dimeric quasimolecular ion peak at m/z 819.3 [2M + Na]+ in the positive ESIMS, indicating a MW of 398 (Figure 2e). This compound was judged most likely to be deoxypodophyllotoxin, a substance previously reported to occur in T. occidentalis.25 The UV spectrum for this compound showed maximum absorptions at λmax 236 and 290 nm (Figure 2j) consistent with those reported for deoxypodophyllotoxin.26 The reported cytotoxicity of deoxypodophyllotoxin against several cancer cell lines25,27,28 reinforced the prediction that this compound was present in lead 79865-c7 and contributed to the observed activity against EphB2-EPD.
The UPLC-MS-ELSD-PDA data of the remaining compounds in this sample afforded additional interesting structural information. Compound 4 with a MW of 300 as indicated by its positive-ion ESIMS (Figure 2f) and UV absorptions at λmax 232, 285, and 326 nm was likely to be the aromatic lactone, thujin, known to occur in Thuja plicata.29 Compounds 6 and 7 with MWs of 318 and 336, respectively, deduced from their ESIMS (Figures 2g and 2h) were inferred as being diterpenoids, since these compounds are common in T. occidentalis.25,30-33 In particular, compound 5 with a MW of 354 (Figure 2i) was of interest because this MW did not match any of the previously reported natural lignans or diterpenoids found in a search of the Dictionary of Natural Products online database (Chapman Hall/CRC) and SciFinder® (Chemical Abstracts Service).
To confirm the above deductions indicating deoxypodophyllotoxin (8) as the active compound and compound 5 as potential new (and possibly active) natural product in lead 79865-c7, a scale-up of the extraction and isolation from the bark of T. occidentalis was performed in order to isolate these two compounds. A methanol extract of the dried plant material was fractionated into hexanes- and chloroform-methanol-water soluble portions. The later was chromatographed on silica gel to afford column fractions, which were subjected to UPLC-MS analysis and biological testing against EphB2-EPD cells. The compound with a MW of 398 was present in the active column fraction and subsequent separation by reversed-phase silica gel chromatography afforded (−)-deoxypodophyllotoxin (8), which was confirmed by comparison of its optical rotation and NMR spectroscopic data with those reported in the literature.25,34 All other column fractions showed negligible activities, confirming that deoxypodophyllotoxin was the primary active compound in the lead fraction.
Compound 5 was present in a relatively polar column fraction that was detected by UPLC-MS. This fraction contained a mixture of two compounds (5a and 5b) with the same MW of 354 and close retention times that both semi-preparative and preparative HPLC failed to separate. Initial 13C NMR spectroscopic analyses of the mixture indicated the presence of labdane-type diterpenes.35-37 Acetylation of the mixture and subsequent separation by reversed-phase HPLC yielded compounds 9 and 10 (Figure 3), which were diacetates of 5a and 5b, respectively, based on the analysis of their ESIMS and NMR spectroscopic data.
Figure 3.
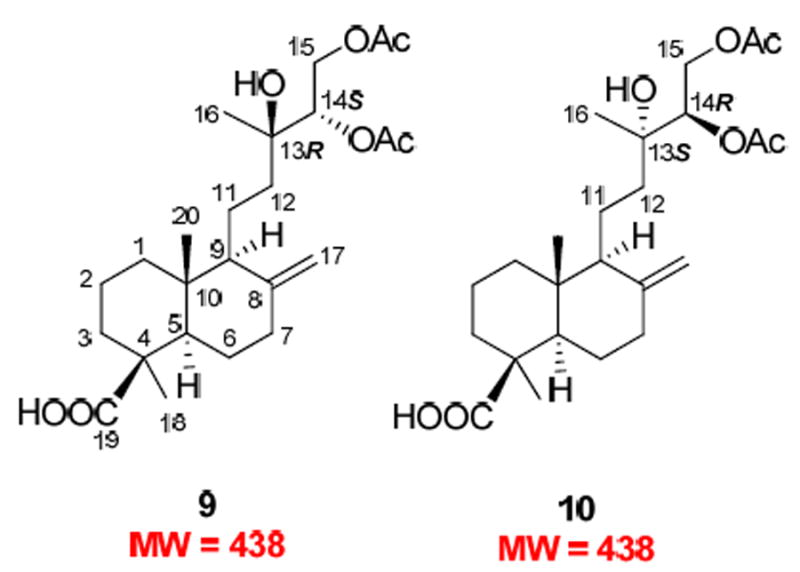
Structures of new acetylated diterpenes 9 and 10 from lead 79865-c7 (obtained from the bark of Thuja occidentalis).
The 1H NMR spectrum of 9 showed resonances of an exocyclic double bond at δ 4.85 (br s) and 4.51 (br s) and three methyl singlets at δ 1.24, 1.20, and 0.61. Resonances appearing at δ 5.06 (1H, dd, J = 8.6, 2.7 Hz), 4.49 (1H, dd, J = 12.1, 2.4 Hz), and 4.10 (1H, dd, J = 12.1, 8.7 Hz) suggested a CH(OH)-CH2OH structural moiety. A carboxyl functionality (δC 183.5) and three oxygen-bearing carbons [δC 73.6 (s), 76.0 (d), and 63.0 (t)] were evident in the 13C NMR spectrum. Further comparison of the 1H and 13C NMR data of 9 with those of isocupressic acid35 suggested that it possesses an 8(17)-labden-19-oic acid skeleton with a 13,14,15-triol side chain, which was confirmed by 2D-NMR experiments as follows. In the HMBC spectrum, H-14 at δH 5.06 correlated with C-12, 13, 15, 16, and the carbonyl carbon of the C-14 acetoxy group at δC 37.5, 73.6, 63.0, 23.5, and 170.6, respectively. The H-15 signal at δH 4.49 and 4.10 showed cross peaks with signals due to C-13, -14 and the carbonyl carbon of the C-15 acetoxy group, at δC 73.6, 76.0, and 171.0, respectively. In the NOESY spectrum, the key NOE correlations between Me-20 (δH 0.61) and H-11a and H-11b (δH 1.45 and 1.62) and between Me-20 and H-17b (δH 4.51) as well as the absence of an NOE correlation between Me-20 and Me-18 supported the presence of a labdane skeleton. The absolute configuration of the triol system, however, could not be resolved by NMR spectroscopy, although an NOE correlation between Me-16 and H-14 was observed. Finally, a single crystal from 9 was successfully obtained and analysis of its X-ray crystallographic data defined a 13R,14S absolute configuration (Figure 4).
Figure 4.
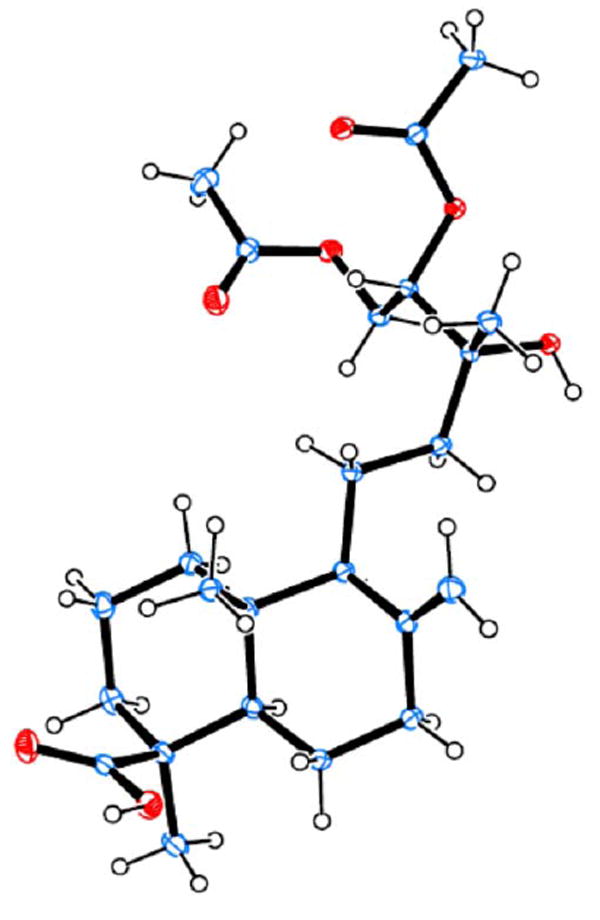
ORTEP drawing of compound 9.
The 1H and 13C NMR spectra of 10 were almost superimposable on those of 9, with only minor differences evident for the chemical shifts of H-12 at δH 1.77 and 1.16 for 9 and δH 1.67 and 1.31 for 10 (see the full 1H NMR spectra of 9 and 10 in the Supporting Information). In addition, the coupling constants between H-14 and H-15 for both compounds were exactly the same. These data indicated that 10 should possess an opposite configuration at C-13 and C-14, imposing minor effects on the 1H NMR of the achiral C2 unit (C-11 and C-12) that separates the chiral labdane skeleton from the chiral C-13-C-15 side chain. Such different absolute configurations in 9 and 10 presumably result from stereoselective oxidations of the side chain in their downstream biosynthetic pathways.
It has been a challenge to determine the absolute configuration of the hydroxy-substituted carbons on the side chains of labdane-type diterpenes, especially the absolute configuration at C-13.38,39 For example, the absolute configuration of C-13 in labda-8(17),14-diene-2α,13-diol-19-oic acid,40 excoecarins G1 and G2,41 and botryosphaerin E42 remain undefined. It may be noted that biotransformation of cupressic acid [13-hydroxy-8(17),14-labdadien-19-oic acid] using Fusarium gramiearum produced 13,14,15-trihydroxy-8(17)-labden-19-oic acid, which is an inseparable mixture of C-13 and/or C-14 diastereomers.43 The present work represents the first report of the absolute configuration determination of this type of triol system among the labdane diterpenes.
The purified deoxypodophyllotoxin (8) was tested for activity against EphB2-EPD cells and gave an EC50 of 1.93 nM. The mixture of 5a and 5b (in approximately 1:1 ratio) was also tested and was confirmed inactive (EC50 > 100 μg/mL). Thus, deoxypodophyllotoxin was detected as the active compound responsible for the potent activity of lead 79865-c7.
This study shows that the potent activity of fraction 79863-c9 (EC50, 0.02 μg/mL) from the leaves of T. occidentalis against EphB2-EPD cells was also due to the presence of deoxypodophyllotoxin (8), as indicated by its UPLC-MS-ELSD-PDA profiles (Figure 5). Deoxypodophyllotoxin was identified as the major compound with Rt values of 1.18 and 1.15 min in the ELSD and PDA chromatograms (Figures 5a and 5b), respectively, showing a quasimolecular ion peak at m/z 399.0 in the positive-ion ESIMS (Figure 5e). Thujin (4), with Rt value of 0.92 min in the PDA chromatogram (Figure 5b) was also present in this fraction. In addition, compound 11 with Rt value of 1.20 min in the ELSD chromatogram (Figure 5a) gave a MW of 400 in the positive-ion ESIMS (Figure 5f) and UV absorptions at λmax 232 and 285 nm (Figure 5g). This compound was predicted to be deoxypodorhizone,26 a biosynthetic precursor of deoxypodophyllotoxin (8) that is much less cytotoxic to cancer cell lines.25 Coincidently, this compound was found to be the major compound in sample 79864-c9, a column fraction derived from the stems of T. occidentalis (Supporting Information, Figure S14). The 1H NMR spectrum of this fraction confirmed the identity of this compound as deoxypodorhizone (Supporting Information, Figure S15).34 Although a trace amount of deoxypodophyllotoxin appears to be in fraction 79864-c9 (<5% mass), the mixture did not produce sufficient activity and thus was not identified as a lead. Other column fractions from the stems of T. occidentalis also lacked deoxypodophyllotoxin. The above analysis indicates that deoxypodophyllotoxin (8) was the active compound present in the bark and leaves of T. occidentalis.
Figure 5.
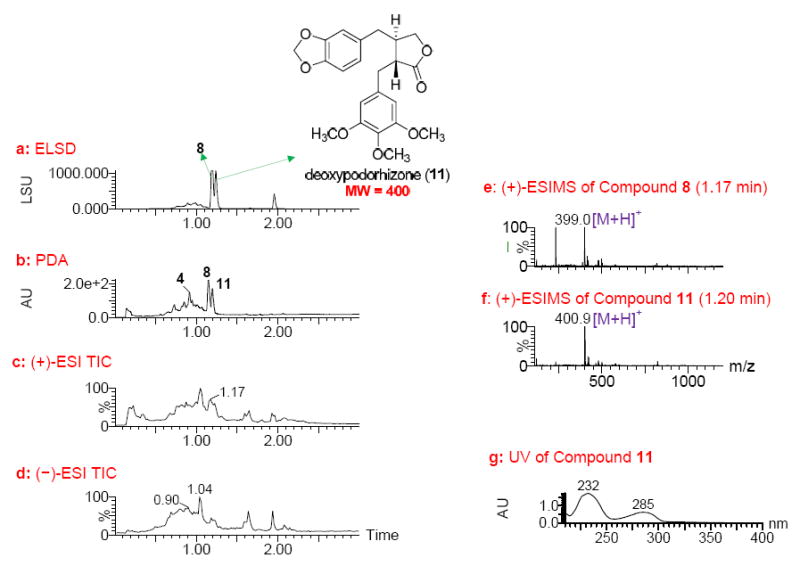
UPLC-MS-ELSD-PDA analysis of lead 79863-c9 (obtained from the leaves of Thuja occidentalis). a: ELSD chromatogram showing compounds 8 and 11 with retention times of 1.18 and 1.20 min, respectively; b: PDA chromatogram showing compounds 4, 8 and 11 with retention times of 0.92, 1.15, and 1.17 min, respectively; c and d: positive and negative ESIMS total-ion chromatograms (TIC), respectively; e and f: positive ESIMS spectra of 8 and 11 with retention times of 1.17 and 1.20 min, respectively; and g: UV spectrum of 11. UPLC conditions: Acquity UPLC BEH C18 column (2.1 × 50 mm, 1.7 μm); gradient elution starting at 10%, ramping to 45% in 0.2 min, then to 100% MeOH in water with 0.1% HCOOH in 1.1 min at a flow rate of 1. 0 mL/min, holding for 1.65 min.
In conclusion, it has been shown that UPLC-MS-ELSD-PDA analytical data collected during the process of the automated fractionation of plant extracts provides a powerful resource for rapid dereplication to identify known compounds from biologically active column fractions. The relative simplicity of compound composition in such fractions makes it possible to interpret the UPLC-MS-ELSD-PDA data effectively, facilitating lead selection and prioritization at an early stage of a natural product drug discovery program. Clearly, this dereplication strategy is superior to one that might be performed at the level of a crude extract with a complex chromatographic profile. As shown in the examples discussed above, current UPLC conditions may not produce optimal compound separations. However, the high-throughput analytical system, combined with the biological screening of fractions, allows a rapid focus on any novel bioactive compounds present in the extracts. This allows the resource- and effort-intensive isolation and structure determination work to focus only on the highest priority leads. It is believed that more structural information will be obtained with improved separations, which, if necessary, can be accomplished readily by a different UPLC solvent eluting system in follow-up studies. For example, a better separation for compounds 1–3 has been achieved by a slightly modified condition, as shown in the Supporting Information (Figure S1). It has been shown also that UPLC-MS-ELSD-PDA data can predict novel structural information and serve as a guide to isolate specific unknown compounds of interest, as demonstrated in the isolation of the new labdane diterpenes (compounds 5a and 5b) from the lead 79865-c7 (obtained from the bark of T. occidentalis). Utilizing this approach, novel active compounds can be isolated so long as such compounds do indeed occur. In the case of dealing with potentially novel compounds in a particular lead sample, the UPLC-MS-ELSD-PDA data should be carefully examined for every single compound, including those occurring only in minute quantities, to ensure the isolation of all active compounds.
EXPERIMENTAL SECTION
General Experimental Procedures
Specific rotations were measured on an Autopol IV polarimeter. 1H and 13C NMR spectra were recorded on a Bruker DRX NMR spectrometer operating at 400 (1H) and 100 (13C) MHz. Chemical shifts are expressed in ppm relative to the solvent residue signals. High-resolution ESIMS data were obtained on an Agilent Series 1100 SL mass spectrometer. Column chromatography (CC) was performed on silica gel (40 μm, J. T. Baker) and reversed-phase silica gel (C18, 40 μm, J. T. Baker). Semi-preparative HPLC separation was carried out using a Waters LC Module 1 system. The column was a Supelco Discovery® C18 column (250 × 10 mm, 5 μm). Silica gel 60 F254 TLC plates (Merck, Darmstadt, Germany) and reversed-phase TLC plates (C18, Merck, Darmstadt, Germany) were used for analytical TLC. A UPLC-MS was used for the analysis of column fractions and run on an Agilent 1290 Infinity series chromatograph with a dual pump, autosampler, thermostated column compartment, and a diode array detector. The chromatograph was coupled with an Agilent 6120 single quadruple mass spectrometer with a dual APCI/ESI source operated in both the positive and negative modes. The system was controlled by ChemStation software. A Waters Acquity UPLC BEH C18 column (2.1 × 150 mm, 1.7 μm) was used. The experiments were carried out at a gradient elution from 5% to 95% CH3CN in H2O containing 0.1% HCOOH in 15 min, and then held for 5 min. The quadruple mass analyzer was operated in the scan mode with the mass range from 100 to 1000. The drying gas was 10 L/min at 250 °C, the nebulizer pressure 30 psi, and the vaporizer temperature was 350 °C. The capillary voltage used was 3 kV, the corona current 10 μA, and the charging voltage was 2 kV. The fragmentor was set to 120 V.
UPLC-MS-ELSD-PDA Analysis
Detailed procedures for generation of natural product libraries by an automated high-throughput system have been described in a previous paper.14 UPLC-MS-ELSD-PDA data were obtained with a Waters Acquity UPLCMS system (Waters Corp., Milford, MA, USA). An Acquity UPLC BEH C18 column (2.1 × 50 mm, 1.7 μm) was used. The mobile phase consisted of H2O containing 0.1% HCOOH and CH3CN or MeOH. The total run time for each analysis was 3.0 min. Ionization and detection of natural products were carried out on a Waters SQ mass spectrometer using both the positive and negative ESI modes. The capillary voltage was set at 3.4 kV. The extractor voltage was 2 V. Nitrogen was used as the nebulizing gas, and the source temperature was set at 130 °C. The scan range was m/z 130-1400. Data processing was performed automatically with OpenLynx by extracting all graphic information, such as retention times and UV and ELSD peak areas, and converted to text to allow transfer to a database for storage and analysis. Each 384-well plate could be analyzed in 20 h.
EphB2-EPD Assay
A high-throughput screening approach has been previously reported.17 Briefly, cells were seeded in 30 μL of neurobasal medium in each well of 384-well plates (Corning) using an automated plate filler (Wellmate, Matrix). After 24 h, 25 nL of solution containing appropriate compounds were pin transferred into the 384-well plates resulting in approximately 8.3 mM final drug concentration. Each plate included DMSO and cycloheximide as controls. The cell number was determined in each well using the Cell Titer Glo reagent (Promega) and read in an automated Envision plate reader (Perkin-Elmer) after 96 h incubation. Luminescence data were normalized by log10 transformation and the percentage inhibition was calculated. Secondary screens were conducted in a similar manner although compounds were applied in a dilution series (8.3 mM to 0.5 nM final concentration) and repeated in triplicate. All data processing and visualization was performed using custom programs written in the Pipeline Pilot platform (Accelrys, v.7.0.1) and the R program.
Plant Material
The leaves of Eriodictyon angustifolium were collected in Gila, AZ, USA with coordinates of 33°21′40″N 110°48′40″W by Zachary Scott Rogers in May, 2005 and identified by Greg Gust. The bark, leaves, and stems of Thuja occidentalis were collected in Sheboygan, WI, USA with coordinates of 43°52′22″N 087°56′33″W by Andrew Townesmith and G. Gust in August, 2005 and identified by A. Townesmith. The voucher specimen of E. angustifolium (No. 2793053) and T. occidentalis (No. 2909789) are deposited in the Herbarium of the Missouri Botanical Garden, St Louis, MO, USA.
Extraction and Isolation
The air-dried powdered bark of T. occidentalis (181.5 g) was extracted at room temperature with MeOH. The solvent was evaporated under reduced pressure at 40 °C to yield 40.8 g extract. The MeOH extract (40.3 g) was dissolved in MeOH-H2O (9:1) and extracted with hexanes (200 mL × 3) and then CHCl3 (200 mL). The CHCl3-MeOH-H2O layer was concentrated to give a residue (16.2 g), which was subjected to silica gel CC using CHCl3 first and then a gradient of CHCl3-MeOH-H2O (30:20:1), to afford 24 fractions. UPLC-MS analysis indicated that deoxypodophyllotoxin (8) was in fraction 6 (367.2 mg), which was chromatographed by reversed-phase silica gel using 70-90% MeOH in H2O to afford deoxypodophyllotoxin (8) (10.5 mg), [α]D25 −112 (c 0.18, CHCl3). Fraction 19 (350.7 mg), containing the compound(s) with a MW of 354 as indicated by UPLC-MS, was subjected to reversed-phase silica gel CC and eluted with 50-80% MeOH in H2O to afford seven sub-fractions. Sub-fraction 4 (52.0 mg) was acetylated with Ac2O-pyridine (1:1, 1 mL) and the resultant products were purified by semi-preparative HPLC (50% CH3CN in H2O with a flow rate at 1.5 mL/min and detected with UV at 205 nm) to yield compounds 9 (8.5 mg, Rt 52.93 min) and 10 (5.4 mg, Rt 51.34 min).
14S,15-Diacetoxy-13R-hydroxy-labd-8(17)-en-19-oic acid (9): white powder; [α]25D +7.7 (c 0.26, MeOH); 1H NMR (CDCl3, 400 MHz) δ5.06 (1H, dd, J = 8.6, 2.7 Hz, H-14), 4.85 (1H, br s, H-17a), 4.51 (1H, br s, H-17b), 4.49 (1H, dd, J = 12.1, 2.4 Hz, H-15a), 4.10 (1H, dd, J = 12.1, 8.7 Hz, H-15b), 2.40 (1H, dd, J = 9.5, 3.2 Hz, H-7a), 2.17 (1H, m, H-3a), 2.13 (3H, s, - COOCH3) , 2.04 (3H, s, -COOCH3), 1.98 (1H, br d, J = 6.8 Hz, H-6a), 1.93 (1H, m, H-6b), 1.87 (1H, m, H-2a), 1.86 (1H, m, H-1a), 1.84 (1H, m, H-7b), 1.77 (1H, m, H-12a), 1.62 (1H, m, H-11a), 1.55 (1H, m, H-9), 1.52 (1H, m, H-2b), 1.45 (1H, m, H-11b), 1.32 (1H, m, H-5), 1.24 (3H, s, H-18), 1.20 (3H, s, H-16), 1.16 (1H, m, H-12b), 1.09 (1H, m, H-3b), 1.05 (1H, m, H-1b), 0.61 (3H, s, H-20); 13C NMR (CDCl3, 100 MHz) δ 39.1 (CH2, C-1), 19.9 (CH2, C-2), 37.9 (CH2, C-3), 44.2 (C, C-4), 56.3 (CH, C-5), 26.0 (CH2, C-6), 38.7 (CH2, C-7), 147.7 (C, C-8), 56.5 (CH, C-9), 40.7 (C, C-10), 17.3 (CH2, C-11), 37.5 (CH2, C-12), 73.6 (C, C-13), 76.0 (CH, C-14), 63.0 (CH2, C-15), 23.5 (CH3, C-16), 106.7 (CH2, C-17), 29.0 (CH3, C-18), 183.5 (C, C-19), 12.8 (CH3, C-20), 170.6 (C, COOCH3), 171.0 (C, COOCH3), 20.9 (CH3, COOCH3), and 21.0 (CH3, COOCH3); HRESIMS m/z 899.5151 (calcd for [2M(C24H38O7) + Na]+, 899.5128), 461.2542 (calcd for [C24H38O7 + Na]+, 461.2510), 439.2702 (calcd for [C24H38O7 + H]+, 439.2690), 421.2604 (calcd for [C24H38O7 – H2O + H]+, 421.2585.
14R,15-Diacetoxy-13S-hydroxy-labd-8(17)-en-19-oic acid (10): white powder; [α]25D +46.4 (c 0.45, MeOH); 1H NMR (CDCl3, 400 MHz) δ 5.06 (1H, dd, J = 8.4, 2.6 Hz, H-14), 4.85 (1H, br s, H-17a), 4.50 (1H, br s, H-17b), 4.48 (1H, dd, J = 12.5, 2.6 Hz, H-15a), 4.11 (1H, dd, J = 12.1, 8.6 Hz, H-15b), 2.41 (1H, dd, J = 9.2, 3.4 Hz, H-7a), 2.16 (1H, m, H-3a), 2.12 (3H, s, COOCH3) , 2.04 (3H, s, COOCH3), 1.98 (1H, br d, J = 6.8 Hz, H-6a), 1.90 (1H, m, H-6b), 1.86 (1H, m, H-2a), 1.84 (1H, m, H-1a), 1.67 (1H, m, H-12a), 1.65 (1H, m, H-11a), 1.56 (1H, m, H-9), 1.50 (1H, m, H-2b), 1.38 (1H, m, H-11b), 1.34 (1H, m, H-5), 1.31 (1H, m, H-12b), 1.24 (3H, s, H-18), 1.21 (3H, s, H-16), 1.08 (1H, m, H-7b), 1.02 (1H, m, H-1b), 0.61 (3H, s, H-20); 13C NMR (CDCl3, 100 MHz) δ 39.1 (CH2, C-1), 19.9 (CH2, C-2), 37.9 (CH2, C-3), 44.2 (C, C-4), 56.4 (CH, C-5), 26.1 (CH2, C-6), 38.7 (CH2, C-7), 148.0 (C, C-8), 56.7 (CH, C-9), 40.7 (C, C-10), 17.1 (CH2, C-11), 37.5 (CH2, C-12), 73.6 (C, C-13), 76.2 (CH, C-14), 63.1 (CH2, C-15), 23.4 (CH3, C-16), 106.5 (CH2, C-17), 29.1 (CH3, C-18), 183.4 (C, C-19), 12.8 (CH3, C-20), 170.6 (C, COOCH3), 171.0 (C, COOCH3), 20.9 (CH3, COOCH3), and 21.0 (CH3, COOCH3); HRESIMS m/z 899.5097 (calcd for [2M(C24H38O7) + Na]+, 899.5128), 461.2501 (calcd for [C24H38O7 +Na]+), 461.2510), 439.2687 (calcd for [C24H38O7 + H]+, 439.2690), 421.2586 (calcd for [C24H38O7 – H2O + H]+, 421.2585).
X-Ray Crystallographic Analysis of Compound 9
A translucent colorless block-like specimen of the compound from CH3CN-H2O (9:1) with approximate dimensions 0.19 × 0.20 × 0.40 mm was used for the X-ray crystallographic analysis. The X-ray intensity data were measured. A total of 6401 frames were collected. The total exposure time was 17.78 h. The frames were integrated with the Bruker SAINT software package using a narrow-frame algorithm. The integration of the data using a monoclinic unit cell yielded a total of 9654 reflections to a maximum θ angle of 64.93° (0.85 Å resolution), of which 3756 were independent (average redundancy 2.570, completeness = 99.2%, Rint = 2.09%, Rsig = 2.22%) and 3750 (99.84%) were greater than 2σ(F2). The final cell constants of a = 7.0889(2) Å, b = 23.3669(5) Å, c = 7.3183(2) Å, β = 91.7930(10)°, volume = 1211.65(5) Å3, are based upon the refinement of the XYZ-centroids of 8645 reflections above 20 σ(I) with 7.566° < 2θ < 137.7°. Data were corrected for absorption effects using the multi-scan method (SADABS). The ratio of minimum to maximum apparent transmission was 0.903. The calculated minimum and maximum transmission coefficients (based on crystal size) are 0.7639 and 0.8767.
The structure was solved and refined using the Bruker SHELXTL-2013 Software Package, using the space group P21, with Z = 2 for the formula unit, C24H38O7. The final anisotropic full-matrix least-squares refinement on F2 with 288 variables converged at R1 = 2.53%, for the observed data and wR2 = 6.75% for all data. The goodness-of-fit was 1.039. The largest peak in the final difference electron density synthesis was 0.183 e-/Å3 and the largest hole was -0.140 e-/Å3 with an RMS deviation of 0.030 e-/Å3. The refined flack parameter χ = 0.09(3) for the structure and χ = 0.91(3) for the inverted structure indicated that the right absolute configuration was assigned. On the basis of the final model, the calculated density was 1.202 g/cm3 and F(000), 476 e-. The supplementary crystallographic data can be obtained free of charge from The Cambridge Crystallographic Data Centre, reference number CCDC 973386, via www.ccdc.cam.ac.uk/data_request/cif.
Supplementary Material
Acknowledgments
The authors thank Dr. Ilias Muhammad for providing an authentic sample deoxypodophyllotoxin for TLC comparison purposes, Dr. Bharathi Avula for recording high-resolution ESIMS, and Dr. Jon Parcher for reading through the manuscript. This work was supported by the USDA Agricultural Research Service Specific Cooperative Agreement No. 58-6408-1-603, the American Lebanese Syrian Associated Charities, St Jude Children’s Research Hospital, and the China Scholarship Council.
Footnotes
Supporting Information Available: UPLC-MS-ELSD-PDA data of homoeriodictyol (1), hesperetin (2), sterubin (3), lead 78821-c4, and column fraction 79864-c9; NMR spectra of acetylated 5a/5b; NMR spectroscopic and HRESIMS data of compounds 9 and 10, and X-ray data of compound 9. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
References
- 1.Newman DJ. J Med Chem. 2008;51:2589–2599. doi: 10.1021/jm0704090. [DOI] [PubMed] [Google Scholar]
- 2.Cragg MG, Grothaus GP, Newman DJ. Chem Rev. 2009109:3012–3043. doi: 10.1021/cr900019j. [DOI] [PubMed] [Google Scholar]
- 3.Newman DJ, Cragg GM. J Nat Prod. 2012;75:311–315. doi: 10.1021/np200906s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Butler MS, Cooper MA. J Antibiot. 2011;64:413–425. doi: 10.1038/ja.2011.44. [DOI] [PubMed] [Google Scholar]
- 5.Cruz PG, Auld DS, Schultz PJ, Lovell S, Battaile KP, MacArthur R, Shen M, Tamayo-Castillo G, Inglese J, Sherman DH. Chem Biol. 2011;18:1442–1452. doi: 10.1016/j.chembiol.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ekdrudge GR, Vervoort HC, Lee CM, Cremin PA, Williams CT, Hart SM, Goering MG, O’Neil-Johnson M, Zeng L. Anal Chem. 2002;74:3963–3971. doi: 10.1021/ac025534s. [DOI] [PubMed] [Google Scholar]
- 7.Cremin PA, Zeng L. Anal Chem. 2002;74:5492–5500. doi: 10.1021/ac025516a. [DOI] [PubMed] [Google Scholar]
- 8.Lang G, Mitova MI, Ellis G, Van der Sar S, Phipps RK, Blunt JW, Cummings NJ, Cole ALJ, Munro MHG. J Nat Prod. 2006;69:621–624. doi: 10.1021/np0504917. [DOI] [PubMed] [Google Scholar]
- 9.Bugni TS, Richards B, Bhoite L, Cimbora D, Harper MK, Ireland CM. J Nat Prod. 2008;71:1095–1098. doi: 10.1021/np800184g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lang G, Mayhudin NA, Mitova MI, Sun L, van der Sar S, Blunt JW, Cole ALJ, Ellis G, Laatsch H, Munro MHG. J Nat Prod. 2008;71:1595–1599. doi: 10.1021/np8002222. [DOI] [PubMed] [Google Scholar]
- 11.Mitova MI, Murphy AC, Lang G, Blunt JW, Cole ALJ, Ellis G, Munro MHG. J Nat Prod. 2008;71:1600–1603. doi: 10.1021/np800221b. [DOI] [PubMed] [Google Scholar]
- 12.Johnson AT, Morgan VCM, Aratow AN, Estee AS, Sashidhara VK, Loveridge TS, Segraves LN, Crews P. J Nat Prod. 2010;73:359–364. doi: 10.1021/np900565a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson AT, Sohn J, Inman DW, Estee AS, Loveridge TS, Vervoort CH, Tenney K, Liu J, Ang KK, Ratnam J, Bray MW, Gassner CN, Shen YY, Lokey SR, McKerrow HJ, Boundy-Mills K, Nukanto A, kanti A, Julistiono H, Kardono LBS, Bjeldanes FL, Crews P. J Nat Prod. 2011;74:2545–2555. doi: 10.1021/np200673b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tu Y, Jeffries C, Ruan H, Nelson C, Smithson D, Shelat AA, Brown MK, Li XC, Hester PJ, Smillie T, Khan AI, Walker L, Guy K, Yan B. J Nat Prod. 2010;73:751–754. doi: 10.1021/np9007359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nielsen KF, Maansson M, Rank C, Frisvad JC, Larsen TO. J Nat Prod. 2011;74:2338–2348. doi: 10.1021/np200254t. [DOI] [PubMed] [Google Scholar]
- 16.El-Elimat T, Figueroa M, Ehrmann BM, Cech NB, Pearce CJ, Oberlies NH. J Nat Prod. 2013;76:1709–1716. doi: 10.1021/np4004307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Atkinson JM, Shelat AA, Carcaboso AM, Kranenburg TA, Arnold LA, Boulos N, Wright K, Johnson RA, Poppleton H, Mohankumar KM, Féau C, Phoenix T, Gibson P, Zhu L, Tong Y, Eden C, Ellison DW, Priebe W, Koul D, Yung WK, Gajjar A, Stewart CF, Guy RK, Gilbertson RJ. Cancer Cell. 2011;13:384–399. doi: 10.1016/j.ccr.2011.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Horowitz MR, Gentili B. J Am Chem Soc. 1960;82:2803–2806. [Google Scholar]
- 19.Horowitz MR, Jurd L. J Org Chem. 1961;26:2446–2449. [Google Scholar]
- 20.Ley J, Reiss I, Blings M, Hoffmann-Luecke P, Herzog J. PCT Int Appl. 2004 WO 2004041804 A2. [Google Scholar]
- 21.Ley JP, Krammer G, Reinders G, Gatfield IL, Bertram HJ. J Agrc Food Chem. 2005;15:6061–6066. doi: 10.1021/jf0505170. [DOI] [PubMed] [Google Scholar]
- 22.Chemesova II, Belenovskaya LM, Stukov AN. Rastitel’nye Resursy. 1987;23:100–103. [Google Scholar]
- 23.Mossa JS, Sattar EA, Abou-Shoer M, Galal AM. Int J Pharm. 1996;34:198–201. [Google Scholar]
- 24.Liu XR, Zhang Y, Lin ZQ. Zhongguo Xinyao Zazhi. 2011;20:329–333. [Google Scholar]
- 25.Chang LC, Song LL, Park JE, Luyengi L, Lee KJ, Farnsworth RN, Pezzuto JM, Kinghorn AD. J Nat Prod. 2000;63:1235–1238. doi: 10.1021/np0001575. [DOI] [PubMed] [Google Scholar]
- 26.Jeong GS, Kwon OK, Park BY, Oh SR, Ahn KS, Chang MJ, Oh WK, Kim JC, Min BS, Kim YC, Lee HK. Biol Pharm Bull. 2007;30:1340–1343. doi: 10.1248/bpb.30.1340. [DOI] [PubMed] [Google Scholar]
- 27.Castro MA, Miguel Del Corral JM, Gordaliza M, Gomez-Zurita MA, Garcia PA, San Feliciano A. Phytochem Rev. 2003;2:219–233. [Google Scholar]
- 28.Gordaliza M, Garcia AP, Miguel del Corral MJ, Gomez-Zurita AM. Toxicon. 2004;44:441–459. doi: 10.1016/j.toxicon.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 29.Jin L, Wilson JW. Can J Chem. 1988;66:51–53. [Google Scholar]
- 30.Witte L, Berlin J, Wray V, Schubert W, Kohl W, Hofle G, Hammer J. Planta Med. 1983;49:216–221. doi: 10.1055/s-2007-969854. [DOI] [PubMed] [Google Scholar]
- 31.Berlin J, Witte L. Phytochemistry. 1988;27:127–132. [Google Scholar]
- 32.Kamdem PD, Hanover JW, Gage DA. J Essent Oil Res. 1993;5:117–122. [Google Scholar]
- 33.Hafez SS. Mansoura J Pharm Sci. 2004;20:34–47. [Google Scholar]
- 34.Iketa R, Nagao T, Okabe H, Nakano Y, Matsunaga H, Katano M, Mori M. Chem Pharm Bull. 1998;46:871–874. doi: 10.1248/cpb.46.871. [DOI] [PubMed] [Google Scholar]
- 35.Muhammad I, Mossa SJ, Al-Yahya AM, Ramadan FA, El-Feraly FS. Phytother Res. 1995;9:584–588. [Google Scholar]
- 36.Fang JM, Sou YC, Chiu YH, Cheng YS. Phytochemistry. 1993;34:1581–1584. [Google Scholar]
- 37.Popova PM, Chinou BI, Marekov NI, Bankova SV. Phytochemistry. 2009;70:1262–1271. doi: 10.1016/j.phytochem.2009.07.025. [DOI] [PubMed] [Google Scholar]
- 38.Wang YZ, Tang CP, Ke CQ, Weiss HC, Gesing ER, Ye Y. Phytochemistry. 2008;69:518–526. doi: 10.1016/j.phytochem.2007.07.023. [DOI] [PubMed] [Google Scholar]
- 39.Bolster GM, Jansen JMB, Groot DA. Tetrahedron. 2001;57:5663–5679. [Google Scholar]
- 40.Lin SJ, Rosazza JPN. J Nat Prod. 1998;61:922–966. doi: 10.1021/np980064b. [DOI] [PubMed] [Google Scholar]
- 41.Konishi T, Konoshima T, Fujiwara Y, Kiyousawa S, Miyahara K, Nish M. Chem Pharm Bull. 1999;47:456–458. doi: 10.1248/cpb.49.318. [DOI] [PubMed] [Google Scholar]
- 42.Yuan L, Zhao PJ, Ma J, Lu CH, Shen YM. Helv Chem Acta. 2009;92:1118–1125. [Google Scholar]
- 43.Rodrigues-Filho E, Magnani RF, Xie W, Mirocha CJ, Pathre SV. J Braz Chem Soc. 2002;13:266–269. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.