

Abstract
Eleven new lignans and neolignans, named acortatarinowins G–N (1−8), including three pairs of enantiomers (1a/1b−3a/3b) and five optically pure lignans and neolignans (4−8), along with five known analogs (9−14), were isolated from the rhizomes of Acorus tatarinowii Schott. Compounds 1−3 were successfully separated by chiral HPLC to afford 1a/1b−3a/3b. The planar structures of 1−8 were elucidated by extensive spectroscopic analyses including HRESIMS and NMR. Their absolute configurations were determined by analyses of single-crystal X-ray diffraction and a modified Mosher’s method, assisted by experimental and calculated electronic circular dichroism (ECD) data. Compounds 1a and 1b were rare 7,8′-epoxy-8,7′-oxyneolignane. Compounds 1−8 were evaluated for their antioxidant activities using 2,2-diphenyl-1-picrylhydrazyl (DPPH) reducing antioxidant power assay. Compound 6, exhibiting strong DPPH radical scavenging capacity with IC50 value of 16.4 ± 0.22 μg/mL, could interpret the herbal traditional usage.
Reactive oxygen free radical, produced in normal or pathological cell metabolism, is oxygenated to provide energy to fuel biological processes for many living organisms1. However, many diseases, such as cirrhosis and arteriosclerosis, rheumatoid arthritis, cancer as well as neurodegenerative diseases associated with aging, are due to the accumulation of oxygen derived free radicals1,2,3.
Acorus tatarinowii Schott (family Araceae) is a well-known traditional Chinese medical plant, whose rhizomes are historically used to treat neurodegenerative diseases, such as apoplexy and dementia, amnesia, epilepsy, improvement of learning and memory, especially Alzheimer’s disease (AD)4,5. AD is a devastating neurodegenerative disease, and no effective treatment is available, affecting more than 35 million people worldwide6. Therefore, it is increasingly urgent to search for bioactive compounds on the cure of AD from traditional Chinese medicine.
As part of our continuing efforts to discover bioactive natural products in the medical plant A. tatarinowii, a series of enantiomeric lignans and norlignans were previously reported7. In our current study, further investigation on chemical constituents of the rhizomes of A. tatarinowii in different regions has led to the isolation of eleven new lignans and neolignans (1a/1b−3a/3b and 4−8), and six known analogs (9−14). All of these new isolates were evaluated for their antioxidant activities to interpret the herbal traditional usage and to discover new potential agent for AD. Herein, we report the isolation, structural elucidation, and the absolute configuration determination of all new compounds.
Results and Discussion
Structure Elucidation
The EtOAc extract of the air-dried rhizomes of A. tatarinowii was subjected to a series of chromatographic separations, to yield eleven new (1a/1b−3a/3b and 4−8) and six known (9−14) neolignans and lignans (Fig. 1). The structures of the known compounds (9−14) were identified as tatarinoid C (9)8, (+)-veraguensin (10)9, (−)-galgravin (11)10, and (+)-saucernetindiol (12)11, (−)-machilin-I (13)12 and (+)-verrucosn (14)11, respectively, by comparing their NMR data with those reported in literatures.
Figure 1. Structures of isolated compounds.

(±)-Acortatarinowin G (1a/1b), obtained as block crystals in MeOH (mp 186–187 °C), had a molecular formula of C24H32O8 based on 13C NMR data and a sodiated molecular ion in positive HRESIMS at m/z 471.1977 [M + Na]+ (calcd for C24H32NaO8, 471.1995). The 1H NMR (Table 1) data of 1 showed signals for two 1,2,4,5-tetrasubstituted phenyl groups (δH 7.08, 1H, s; 7.07, 1H, s; 6.51, 1H, s; and 6.49, 1H, s), four oxygenated methines (δH 5.25, 1H, d, J = 3.2 Hz; δH 5.02, 1H, d, J = 9.3 Hz; δH 4.34, 1H, m; and δH 3.76, 1H, d, m), six methoxy groups (δH 3.90, 2*3.89, 3.88, 3.83, and 3.81), and two methyl signals (δH 1.12, 3H, J = 6.8 Hz and δH 1.08, 3H, J = 6.3 Hz). The 13C NMR and DEPT (Table 2) of 1 revealed 24 carbons, which were resolved into 12 aromatic carbons, four oxygenated methine carbons, six methoxy groups, and two methyl carbons. The spectroscopic data suggested that 1 was a 7,8-dioxane neolignan which was very similar to verimol H except that two 1,4-dimethoxyphenyl moieties in verimol H were replaced by two 1,2,4,5-tetramethoxyphenyl groups in 113. This conclusion was supported by the HMBC correlations from H-7 (δH 5.02) to C-1 (δC 119.8), C-2 (δC 151.8), C-6 (δC 111.7), C-8 (δC 78.8), C-9 (δC 17.1), and C-8′ (δC 70.8), and from H-7′ (δH 5.25) to C-1′ (δC 119.5), C-2′ (δC 150.0), C-6′ (δC 111.2), C-8′ (δC 70.8), C-9′ (δC 12.3), and C-8 (δC 78.8). In addition, the HMBC correlations of OMe-2/C-2, OMe-4/C-4, OMe-5/C-5, OMe-2′/C-2′, OMe-4′/C-4′ and OMe-5′/C-5′, verified that compound 1 was 2,4,5, 2′,4′,5′-hexamethoxy-7,8′-epoxy-8,7′-oxyneolignane.
Table 1. H NMR Data for Compounds 1−8 (400 MHz, J in Hz).
| No. | 1b | 2b | 3a | 4b | 5b | 6a | 7a | 8a |
|---|---|---|---|---|---|---|---|---|
| 2 | 6.82 s | 6.62 s | 7.04 s | 6.63 s | 6.76 s | 6.77 s | ||
| 3 | 6.51 s | |||||||
| 4 | 6.93 s | |||||||
| 5 | 5.88 s | |||||||
| 6 | 7.07 s | 6.82 s | 6.62 s | 6.93 s | 6.63 s | 6.76 s | 6.77 s | |
| 7 | 5.02 d (9.3) | 6.45 s | 4.41 d (9.3) | 4.38 d (9.5) | 4.41 d (9.0) | 4.52 d (7.4) | 4.71 d (5.9) | 4.73 d (4.9) |
| 8 | 3.76 m | 1.77 m | 2.62 m | 2.45 m | 2.24 m | 4.42 m | 4.65 m | |
| 9 | 1.09 d (6.3) | 1.86 s | 1.08 d (6.6) | 1.07 d (6.6) | 1.03 d (6.8) | 1.12 d (7.0) | 1.13 d (6.3) | 1.31 d (6.1) |
| 2′ | 6.956 d (2.0) | 7.56 d (2.0) | 7.58 d (2.0) | 6.78 d (1.8) | 7.33 s | 7.53 d (2.0) | ||
| 3′ | 6.50 s | 6.53 s | ||||||
| 5′ | 6.97 d (8.2) | 6.92 d (8.8) | 7.08 d (8.5) | 6.72 d (8.0) | 6.97d (8.5) | |||
| 6′ | 7.08 s | 6.45 s | 6.97 dd (8.2, 2.0) | 7.55 dd (8.8, 2.0) | 7.71 dd (8.5, 2.0) | 6.64 dd (8.0, 1.8) | 7.33 s | 7.57 dd (8.5, 2.0) |
| 7′ | 5.25 d (3.2) | 4.44 s | 5.15 d (8.8) | 2.83 dd (13.4, 5.0), 2.45 dd (13.4, 11.1) | ||||
| 8′ | 4.34 m | 2.31 q (7.3) | 2.28 m | 3.83 m | 4.03 m | 2.61 m | ||
| 9′a | 1.12 d (6.8) | 1.11 d (7.2) | 0.66 d (7.0) | 4.30 dd (9.3, 8.8) | 4.28 dd (9.0, 8.5) | 4.02 dd (8.4, 6.3) | ||
| 9′b | 4.15 dd (9.3, 7.5) | 4.11 dd (9.0, 6.8) | 3.73 dd (8.4, 5.1) | |||||
| OMe-2 | 3.82 s | |||||||
| OMe-3 | 3.87 s | 3.87 s | 3.86 s | 3.83 s | 3.83 s | 3.82 s | ||
| OMe-4 | 3.89 s | 3.79 s | 3.78 s | 3.83 s | 3.75 s | 3.74 s | 3.70 s | |
| OMe-5 | 3.88 s | 3.87 s | 3.87 s | 3.86 s | 3.83 s | 3.83 s | 3.82 s | |
| OMe-2′ | 3.83 s | 3.90 s | ||||||
| OMe-3′ | 3.83 s | 3.94 s | 3.90 | 3.89 s | 3.85 s | |||
| OMe-4′ | 3.89 s | 3.84 s | 3.83 s | 3.95 s | 3.93 | 3.83 s | ||
| OMe-5′ | 3.90 s | 3.64 s | 3.89 s | |||||
| OMe-7′ | 3.90 s | 3.86 s |
ain methanol-d4.
bin CDCl3.
Table 2. 13C NMR Data for Compounds 1−8 (100 MHz).
| No. | 1b | 2b | 3a | 4b | 5b | 6a | 7a | 8a |
|---|---|---|---|---|---|---|---|---|
| 1 | 119.8 | 137.3 | 138.7 | 136.0 | 134.3 | 140.0 | 138.4 | 138.7 |
| 2 | 151.8 | 132.5 | 104.9 | 103.7 | 111.4 | 104.2 | 105.5 | 105.6 |
| 3 | 97.7 | 181.5 | 154.6 | 153.4 | 150.7 | 154.5 | 154.2 | 154.1 |
| 4 | 149.4 | 159.6 | 138.1 | 137.8 | 112.7 | 138.4 | 138.4 | 138.3 |
| 5 | 143.3 | 106.2 | 154.6 | 153.4 | 150.7 | 154.5 | 154.2 | 154.1 |
| 6 | 111.7 | 186.6 | 104.9 | 103.7 | 120.8 | 104.2 | 105.5 | 105.6 |
| 7 | 67.9 | 113.3 | 88.9 | 89.2 | 90.2 | 88.6 | 78.5 | 77.1 |
| 8 | 78.8 | 153.3 | 49.8 | 45.9 | 47.7 | 46.6 | 84.9 | 80.0 |
| 9 | 17.1 | 23.4 | 15.1 | 15.4 | 15.1 | 12.8 | 17.1 | 15.6 |
| 1′ | 119.5 | 121.4 | 135.1 | 130.3 | 131.4 | 133.5 | 126.4 | 123.9 |
| 2′ | 150.0 | 151.1 | 112.4 | 110.2 | 111.8 | 113.5 | 107.9 | 114.0 |
| 3′ | 97.0 | 99.1 | 149.9 | 149.4 | 150.5 | 145.8 | 154.4 | 150.1 |
| 4′ | 148.8 | 148.9 | 150.2 | 153.9 | 155.6 | 149.0 | 142.2 | 153.1 |
| 5′ | 143.6 | 143.0 | 112.7 | 110.5 | 111.9 | 116.2 | 154.4 | 115.4 |
| 6′ | 111.2 | 112.2 | 120.9 | 123.2 | 124.9 | 122.2 | 107.9 | 124.6 |
| 7′ | 75.1 | 35.0 | 84.6 | 197.6 | 200.2 | 34.3 | 168.1 | 168.4 |
| 8′ | 70.8 | 41.5 | 47.1 | 54.4 | 55.1 | 45.4 | ||
| 9′ | 12.3 | 19.5 | 15.3 | 70.8 | 71.5 | 73.4 | ||
| OMe-2 | 56.8 | |||||||
| OMe-3 | 56.7 | 56.3 | 56.5 | 56.6 | 56.7 | 56.5 | ||
| OMe-4 | 56.3 | 56.3 | 61.1 | 60.9 | 61.1 | 61.1 | 61.1 | |
| OMe-5 | 56.3 | 56.7 | 56.3 | 56.5 | 56.6 | 56.7 | 56.5 | |
| OMe-2′ | 56.8 | 57.2 | ||||||
| OMe-3′ | 56.5 | 56.1 | 56.5 | 56.6 | 56.5 | |||
| OMe-4′ | 56.3 | 56.3 | 56.5 | 56.2 | 56.6 | 56.4 | ||
| OMe-5′ | 56.7 | 57.2 | 56.6 | |||||
| OMe-7′ | 52.8 | 52.5 |
ain methanol-d4.
bin CDCl3.
The relative configuration of compound 1 was determined through a NOESY experiment and the coupling constants, and further confirmed by X-ray crystallographic analyses. The large coupling constant of H-7/H-8 (J7,8 = 9.3 Hz) suggested a trans-relationship between H-7 and H-8, and the small coupling constant of H-7′/H-8′ (J7′,8′ = 3.2 Hz) indicated H-7′ and H-8′ were in a cis position. The key cross-peak of H-7′/H-8 in the NOESY spectrum indicated the relative configuration of acortatarinowin G was (7R*, 8R*, 7′S*, 8′R*). Fortunately, crystals of 1 were obtained from MeOH, and a single X-ray diffraction experiment was performed using Cu Kα radiation (Fig. 2), which supported the above conclusion. Surprisingly, the crystal of 1 had the centrosymmetric space group P-2yc, indicating its racemic nature13,14, which was supported by the absence of optical rotation and any Cotton effects in the ECD spectrum. Subsequent chiral resolution led to the isolation of 1a and 1b (Fig. 3, 1a:1b = 1:1) with opposite specific rotation and mirror imaged Cotton effects in their ECD spectrum (Fig. 4). An 8S configuration for 1a was established by a positive Cotton effect around 230 nm in the ECD spectrum (Fig. 4)15,16,17. Furthermore, the experiment ECD spectrum of 1a/1b matched well with calculated ECD spectrum of (7S, 8S, 7′R, 8′S)-1 and (7R, 8R, 7′S, 8′R)-1 (Fig. 4), respectively. Thus, the absolute configurations of 1a and 1b were established as shown (Fig. 1).
Figure 2. ORTEP drawing of compounds 1 and 4.

Figure 3. Chiral HPLC separation profiles of compounds 1a/1b−3a/3b.

Figure 4. The experimental ECD spectra of 1a/1b−3a/3b, and the calculated ECD spectra for 1a/1b.

(±)-Acortatarinowin H (2a/2b), obtained as brown-red amorphous powder, had a molecular formula of C22H24O6 as revealed by HRESIMS spectrum (m/z 407.1493 [M + Na]+) and 13C NMR data. The 1H NMR data showed signals (Table 1) attributable to two methyl groups, two methine protons, four methoxy protons, two olefinic protons, and two aromatic protons. The 13C-NMR spectrum showed the presence of 22 carbon signals, corresponding to two methyl carbons, two methine carbons, four methoxy groups, 12 olefinic carbons and two conjugated carbonyl carbons, which resembled those of 7′-(2′,4′,5′-trimethoxyphenyl)-4-methoxy-8,8′-dimethyl-2,5 quinone18. A side-by-side comparison of the 1H and 13C NMR data of 1 with those of 7′-(2′,4′,5′-trimethoxyphenyl)-4-methoxy-8,8′-dimethyl-2,5-quinone revealed that the small differences were the absence of two olefinic carbons and the appearance of two methine carbons in 2. Based on the aforementioned evidence, 2 was determined to be 4,2′,4′,5′-tetramethoxy-7-ene-3,6-dione-2,7′-cyclolignane, which was further supported by the 1H−1H COSY correlations of H-7′/H-8′ and H-8′/H-9′, and the key HMBC correlations from H-7′ (δH 4.44) to C-1′ (δC 121.4), C-2′ (δC 151.1), C-6′ (δC 112.2), C-8′ (δC 41.5), C-9′ (δC 19.5), C-1 (δC 137.3), C-3 (δC 181.5), C-2 (δC 132.5), and C-8 (δC 153.3).
The relative configurations at C-7′ and C-8′ of the core skeleton of 2 were established by analyses of coupling constants aided by molecular models generated by Chem3D. According to those models, H-7′ and H-8′ in a trans position matched well with the coupling constant (J7′,8′ = 0 Hz), which was supported by a single peak at δH 4.44 (H-7′) and a quartet peak at δH 2.31 (H-8′). The absence of any Cotton effects in the ECD spectrum and optical rotation revealed compound 2 was a pair of racemic mixture as well. These findings were supported by chiral HPLC analyses of 2 (Fig. 3). Compounds 2a and 2b were successfully obtained using a Daicel IC column, showing opposite specific rotations (2a: [α +14; 2b: [α
+14; 2b: [α −14) and opposite Cotton effects (Fig. 4) [2a: 213 (Δε +26.05), 243 (Δε−12.82), 315 (Δε−9.33); 2b: 213 (Δε−27.01), 243 (Δε +13.02), 315 (Δε +10.31)].A negative Cotton effect around 315 nm indicated an 8′S for 2a19,20. Therefore, the absolute configurations of 2a and 2b were determined as 7′R, 8′S and 7′S, 8′R, respectively.
−14) and opposite Cotton effects (Fig. 4) [2a: 213 (Δε +26.05), 243 (Δε−12.82), 315 (Δε−9.33); 2b: 213 (Δε−27.01), 243 (Δε +13.02), 315 (Δε +10.31)].A negative Cotton effect around 315 nm indicated an 8′S for 2a19,20. Therefore, the absolute configurations of 2a and 2b were determined as 7′R, 8′S and 7′S, 8′R, respectively.
(±)-Acortatarinowin I (3a/3b), obtained as a light yellow gum, its NMR data and positive optical rotation were the same as those of 3,4,5,3′,4′-pentamethoxy-7,7′-epoxylignan reported in the literature with absolute configuration not determined21. Based on our previous research7, compound 3 was subjected to a Daicel IC column to check its partial racemic nature, and (−)-3a and (+)-3b (Fig. 3, 3a:3b ≈1:3) were successfully obtained, showing typical antipodal ECD curves (Fig. 4) and specific rotations of opposite sign. The absolute configuration for 3b was determined as 7S,8S,7′R,8′S by comparing the ECD curve of 3b with that of closely related compound hydroxyvera22. Hence, that of 3a was determined as 7R,8R,7′S,8′R (Fig. 1).
Acortatarinowin J (4), was isolated as colourless crystals (MeOH) with mp 158–159 °C. The molecular formula of 4 was determined as C23H28O7 according to 13C NMR data and HREIMS data m/z 439.1718 [M + Na]+ (calcd for C23H28NaO7, 439.1733). The IR spectrum indicated the presence of conjugated carbonyl group (1674 cm–1) and aromatic rings (1594 and 1515 cm–1). The NMR data (Tables 1 and 2) showed signals attributable to a 1,3,4,5-tetrasubstituted and a 1,3,4-trisubstituted aromatic moiety, five methoxy groups, three methines including an oxygenated methine, an oxygenated methylene unit, a methyl group and a conjugated carbonyl group. These findings indicated that compound 4 is a 7,9′-epoxylignan similar to magnone B23. Comparison of the NMR data indicated that the only small difference between 4 and magnone B was that the hydroxymethyl group (C-8) in magnone B was reduced to a methyl group. This deduction was supported by the HMBC correlations from H-7 (δH 4.38) to C-1 (δC 136.0), C-2,6 (δC 103.7), C-8 (δC 45.9), C-9 (δC 15.4), C-8′ (δC 54.4), and C-9′ (δC 70.8), from H-8 (δH 2.62) to C-1(δC 136.0), C-7(δC 89.2), C-8′ (δC 54.4), and C-7′ (δC 197.6), and from H-2′/6′ (δH 7.55/7.56) to C-7′ (δC 197.6). In addition, the HMBC correlations for OMe-3/C-3, OMe-4/C-4, OMe-5/C-5, OMe-3′/C-3′, and OMe-4′/C-4′ verified the location of these substituents. Thus, the structure of 4 was determined as 3,3′,4,4′, 5-pentamethoxy-7,9′-epoxylignan-7′-one.
The relative configuration of 4 was established through a NOESY experiment and coupling constant. The big coupling constant (J = 9.5 Hz) between H-7 and H-8 indicated the trans-orientation, and the strong NOESY correlation of H-7 and H-8′ verified they were in a cis-orientation. Therefore, the 2D structure of 4 was determined as (7R*, 8R*, 8′R*)-3,3′,4,4′,5-pentamethoxy-7,9′-epoxylignan-7′-one. Fortunately, the crystals of 4 were obtained from MeOH, and the absolute configuration of acortatarinowin J was finally elucidated as 7R,8R,8′R on the basis of single crystal X-ray diffraction (Fig. 2).
Acortatarinowin K (5), obtained as white amorphous powder, possesses the molecular formula C22H26O6, as determined from a sodiated molecular ion in the positive HRESIMS at m/z 387.1797 [M + H]+ (calcd for C22H27O6, 387.1808) and its NMR data. A side-by-side comparison of the 13C NMR data of 5 with those of 4 revealed that 5 was closely related to 4 except the absence of a methoxy group. Correspondingly, the signals attributable to a symmetrical 1,3,4,5-tetrasubstituted aromatic moiety in 4 was replaced by those of a 1,3,5-trisubstituted aromatic moiety in the 1H NMR spectrum. Therefore, compound 5 was surmised to be 3,3′,4′, 5-tetramethoxy-7,9′-epoxylignan-7′-one. This deduction was confirmed by 2D NMR data analyses. The big coupling constant (J7,8 = 9.5 Hz) and key NOESY correlation from H-7 to H-8′ indicated the relative configuration of 5 was the same as 4. Furthermore, the experiment ECD spectrum of 5 matched well with that of 4 (Supporting Information, SI, Figs S37 and S47). Thus, the absolute configuration of 5 was determined as 7R,8R,8′R.
Acortatarinowin L (6), had the molecular formula C22H28O6, determined on the basis of a sodiated molecular ion in the positive HRESIMS at m/z 411.1774 [M + Na]+ (calcd for C22H28NaO6, 411.1784) and its NMR data. A comparison of the NMR data (Tables 1 and 2) between 6 and 4 indicated that they differed in the presence of a methylene unit and the absence of a conjugated carbonyl and a methoxy group in 6. Thus, compound 6 was determined as 4′-hydroxy-3,3′,4,5-tetramethoxy-7,9′-epoxylignan. The NOESY correlations from H-9 to H-7′ and H-7 revealed H-7 was oriented opposite to H-8 and H-8′. Thus, the relative configuration of 6 was determined as 7R*, 8R*, 8′S*. Compound 6 displayed a coupled Cotton effect, negative at 235 nm and positive at 215 nm (SI, Fig. S57), indicating exciton coupling in the π→π* transition of the phenyl chromophores24. The positive chirality ([α +18) revealed the 7R, 8R, 8′S configuration for 6 on the basis of the ECD exciton chirality rule17,24,25. Therefore, the absolute configuration of 6 was assigned as 7R, 8R, 8′S.
+18) revealed the 7R, 8R, 8′S configuration for 6 on the basis of the ECD exciton chirality rule17,24,25. Therefore, the absolute configuration of 6 was assigned as 7R, 8R, 8′S.
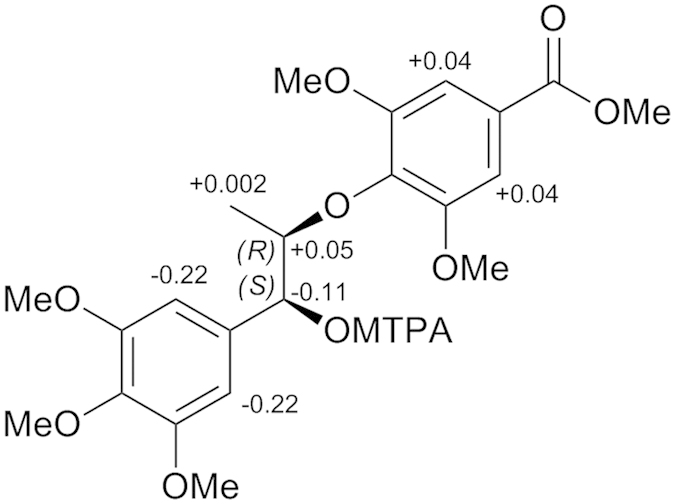
Acortatarinowin M (7) was obtained as a light yellow gum. The HRESIMS ion at m/z 459.1619 [M + Na]+ (calcd for C22H28NaO9, 459.1631) and the NMR data established its molecular formula as C22H28O9. The NMR data (Tables 1 and 2) displayed signals for two symmetric tetrasubstituted aromatic moieties, six methoxy groups, two oxygenated methines, a methyl, and a conjugated carbonyl. These findings indicated that compound 7 was an 8-O-4′ type neolignan and was similar to acortatarinowin C with the difference in the presence of an methoxy group in 77. The key HMBC correlation from H-2′/H-6′ to C-7′ revealed compound 7 was 5′-methoxy derivative of acortatarinowin C. The absolute configuration at C-7 was determined as 7S by modified Mosher’s method (Fig. 5), and C-8 was R due to a negative Cotton effect around 237 nm15,16,17.
Figure 5. ΔδH(S−R) values (in ppm) for the MTPA esters of 7.
Acortatarinowin N (8) was obtained as a light yellow gum. The NMR data and an HRESIMS [M + Na]+ ion at m/z 429.1464 (calcd for C21H26NaO8, 429.1525) allowed the assignment of the molecular formula of C21H26O8 the same as acortatarinowin C7. A side-by-side comparison of the 1H and 13C NMR data of 8 with those of acortatarinowin C indicated there were only small changes in chemical shifts from H-7/C-7 to H-9/C-9 [(δH 4.73 (1H, d, J = 4.9 Hz, H-7), δC 77.1 (C-7); 4.65 (1H, dq, J = 6.1, 4.9 Hz, H-8), 80.0 (C-8); and 1.31 (3H, d, J = 6.1 Hz, H-9,15.6 (C-9 in 8); (δH 4.75 (1H, d, J = 5.4 Hz, H-7), δC 77.1 (C-7); 4.69 (1H, dq, J = 6.2, 5.4 Hz, H-8), 80.0 (C-8); 1.17 (3H, d, J = 6.2 Hz, H-9,16.1 (C-9) in acortatarinowin C)]. On the basis of these observations, along with 2D NMR analyses, compound 8 was deduced to be the stereoisomer of acortatarinowin C with an erythro configuration (7R*, 8S*) due to its small coupling constant (J7,8 = 4.9 Hz). The positive ECD Cotton effect near 238 nm of 8 agreed well with that of (−)-acortatarinowin B7 and its structurally related neolignan compound (7R, 8S)-balanophonin25,26. Thus, the absolute configuration of acortatarinowin N was determined as 7R,8S.
Antioxidant Activity Evaluation
The abilities of the compounds 1−8 to scavenge DPPH radical were evaluated from 6.25 to 100 μg/mL. Trolox (vitamin E) was used as positive control for antioxidant activity (Table 3). Among the compounds tested, compound 6 exhibited the most potent antioxidant activity with IC50 value of 16.437 ± 0.22 μg/mL, whereas the other compounds did not exhibit any activity with the maximum concentration 100 μg/mL. By comparing the structures of all the compounds tested, it was concluded that the presence of phenolic hydroxyl functionality was required for the antioxidant activity of these lignans and neolignans.
Table 3. DPPH Radical Scavenging Activity of New Compounds in vitro.
| Compounds | Concentrations (μg/mL) | Scavenging ratio (%) | IC50 (μg/mL) |
|---|---|---|---|
| 1a | 100 | −2.292 ± 0.263 | |
| 1b | 100 | −2.893 ± 0.278 | |
| 2a | 100 | −2.477 ± 0.351 | |
| 2b | 100 | −4.892 ± 0.088 | |
| 3a | 100 | −1.487 ± 0.351 | |
| 3b | 100 | −1.363 ± 0 | |
| 4 | 100 | −1.301 ± 0.263 | |
| 5 | 100 | −1.796 ± 0.088 | |
| 6 | 100 | 84.784 ± 0.756 | 16.437 ± 0.22 |
| 50 | 71.794 ± 0.611 | ||
| 25 | 59.629 ± 0.328 | ||
| 12.5 | 43.712 ± 0.435 | ||
| 6.25 | 29.609 ± 0.993 | ||
| 7 | 100 | −1.796 ± 0.088 | |
| 8 | 100 | −2.145 ± 0.078 | |
| Trolox | 50 | 95.629 ± 0.372 | 3.895 ± 0.38 |
| 25 | 94.805 ± 0.611 | ||
| 12.5 | 94.433 ± 0.682 | ||
| 6.25 | 69.238 ± 3.335 | ||
| 3.125 | 41.114 ± 4.279 |
The data are expressed as the means ± SEM. Three independent experiments were performed. Trolox (vitamin E) was used as the positive control.
Conclusion
In this study, eleven new lignans and neolignans, including three pairs of enantiomers 1a/1b–3a/3b, were isolated from the rhizomes of A. tatarinowii. Compounds 1a and 1b were rare 7,8′-epoxy-8,7′-oxyneolignanes. The absolute configurations of the two 7,8′-epoxy-8,7′-oxyneolignanes (1a/1b) were determined for the first time by comparison of their experimental and calculated ECD spectrum. Biologically, the antioxidant activities of the new isolates were evaluated. Compound 6 possessing a phenolic hydroxyl moiety exhibited the most potent antioxidant activity, whereas the other compounds, containing no phenolic hydroxyl groups in their structures, did not show activity in DPPH assay. Currently, pathogenetic and biochemical studies have consistently suggested oxidative stress have played an important role in AD pathogenesis27, and many studies have focused on searching therapeutic agents for AD through inhibiting reactive oxygen species production28. Thus, the results of the study could elucidate the bioactivity of A. tatarinowii in some extent. Further investigations on in vivo animal experiments and structure-function relationship for developing more excellent agent for AD are performing.
Methods
General experimental procedures
Melting points were measured using a Beijing Tech X-5 micro-melting point apparatus and were uncorrected. UV, FT-IR, and ECD spectra were obtained from a Varian Cary 50, a Bruker Vertex 70, and a JASCO-810 spectrometer, respectively. Optical rotations were measured in MeOH/CH2Cl2 on a Perkin-Elmer 341 polarimeter equipped with a 1 dm 75 microcell and a sodium lamp (589 nm). 1D and 2D NMR spectra were acquired on a Bruker AM-400 spectrometer and the 1H and 13C NMR chemical shifts were referenced to the solvent or solvent impurity peaks for methanol-d4 (δH 3.31 and δC 49.0) and CDCl3 (δH 7.26 and δC 77.0). High-resolution electrospray ionization mass spectra (HRESIMS) were acquired in the positive ion mode with a Thermo Fisher LC-LTQ-Orbitrap XL spectrometer. Column chromatography was performed using silica gel (100–200 and 200–300 mesh; Qingdao Marine Chemical Inc., China), octadecylsilyl (ODS, 50 μm, YMC Co. Ltd., Japan), and Sephadex LH-20 (40–70 μm, Amersham Pharmacia Biotech AB, Uppsala, Sweden, Sweden). The crystallographic data were acquired on a Bruker SMART APEX-II CCD diffractometer equipped with graphite-monochromatised Cu Kα radiation (λ = 1.54178 Å). Semi-preparative HPLC was carried out on an Agilent 1200 quaternary system with a UV detector or on a Dionex HPLC system equipped with an Ultimate 3000 pump, an Ultimate 3000 autosampler injector, and an Ultimate 3000 diode array detector (DAD) using a reversed-phased C18 column (5 μm, 10 × 250 mm) at a flow rate of 2.5 mL/min. Thin-layer chromatography (TLC) was performed with silica gel 60 F254 (Yantai Chemical Industry Research Institute) and RP-C18 F254 plates (Merck, Germany).
Plant material
The rhizomes of A. tatarinowii were collected at Jiujiang, Jiangxi Province, P. R. China in September 2013, and were identified by one of the authors, Prof. J. P. Wang. A voucher specimen (No. 2013–0916A) was deposited with the Herbarium of Materia Medica, Faculty of Pharmacy, Tongji Medical College of Huazhong University of Science and Technology, China.
Extraction and isolation
The air-dried rhizomes of A. tatarinowii (40 kg) were extracted with 95% EtOH three times at room temperature, and the extract was partitioned with petroleum ether, EtOAc, and n-BuOH against water. The EtOAc fraction (400.0 g) was separated by chromatography to obtain six fractions (Fr.1–6) using silica gel CC eluting with a gradient of petroleum ether–acetone (20:1–1:1). Fr. 2 was further purified by column chromatography (silica gel CC, 10 × 100 cm), eluting with a gradient of petroleum ether–EtOAc (15:1–1:1), to yield five fractions (Fr.2.1–2.5). Fr. 2.3 were subjected to an ODS column (MeOH–H2O, 30:70–70:30) and a Sephadex LH-20 (CH2Cl2−MeOH, 1:1) column to yield three mixtures (A–C). Mixture A was then purified by semi-preparative HPLC (CH3CN-H2O, 45:55) to obtain compound 1 (tR 56.0 min, 10.0 mg). Compounds 9 (19.5 mg) and 10 (12.6 mg) were got from mixture B using an RP C-18 column eluting with MeOH–H2O (65:35), and compound 11 (5 mg) was obtained from Mixture C. Fr.2.4 was separated by repeated silica gel CC eluted with a petroleum ether–EtOAc gradient and Sephadex LH-20 (CH2Cl2−MeOH, 1:1) to yield two mixtures (D and F). Compounds 2 (tR 60.0 min, 8.2 mg, CH3CN-H2O, 50:50) and 3 (tR 90.0 min, 6.0 mg, CH3CN-H2O, 45:55) were purified from mixtures D and F by semi-preparative HPLC, respectively. Compound 1 (1a/1b) was further purified by chiral HPLC eluting with EtOH to give 1a (tR 7.0 min, 2.0 mg) and 1b (tR 11.5 min, 2.0 mg). Compounds 2a (tR 10.0 min, 2.8 mg), 2b (tR 17.0 min, 2.8 mg), 3a (tR 20.0 min, 1.5 mg), and 3b (tR 30.0 min, 4.0 mg) were successfully separated using the same IC column as 1 eluting with EtOH–n-hexane 15:85 and 25:75, respectively. Fr. 4 was subjected to a silica gel again eluting with petroleum ether–EtOAc (10:1–3:1) to yield four subfractions, named Fr.4.1–4.4. Fr.4.2 and Fr.4.3 were further partitioned with ODS column (MeOH–H2O, 30:70–70:30) and Sephadex LH-20 (CH2Cl2−MeOH, 1:1) to obtain four subfractions, named Fr.4.2.1–4.2.2 and Fr.4.3.1–4.3.2, respectively. Compounds 7 (tR 49.0 min, 4.0 mg) and a mixture of 4 and 5 (tR 44.0 min, 27.0 mg) were separated by semi-preparative HPLC (MeOH–H2O, 55:45) equipped with an RP-C18 column from Fr.4.2.1, and compounds 4 and 5 were finally purified by semi-preparative HPLC eluting with CH3CN-H2O (45:55). Compound 8 (tR 63.5 min, 6.0 mg) was obtained from Fr.4.2.2 by semi-preparative HPLC (MeOH–H2O, 50:50). Fr.4.3.1 was subjected to an RP-C18 column to obtain compound 6 (tR 87.4 min, 4.0 mg) and a mixture of 12–14 (tR 74.0 min, 15.0 mg) eluting with CH3CN-H2O (40:60). Compounds 12–14 were purified by repeated semi-preparative HPLC equipped with an RP C18 column and a SiO2 column, respectively.
(±)-Acortatarinowin G (1)
Colorless block crystals (MeOH), mp 186–187 °C; 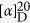 0 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 205 (4.91), 230 (4.41), 291 (4.13) nm; IR (KBr) νmax 1612, 1511, 1465, 1399, 1317, 1206, 1130, 1086, 1034, 862, 781 cm–1; 1H NMR (CDCl3, 400 MHz) data, see Table 1; 13C NMR (CDCl3, 100 MHz) data, see Table 2; (+)-HREIMS m/z 471.1977 [M + Na]+ (calcd for C24H32NaO8, 471.1995).
0 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 205 (4.91), 230 (4.41), 291 (4.13) nm; IR (KBr) νmax 1612, 1511, 1465, 1399, 1317, 1206, 1130, 1086, 1034, 862, 781 cm–1; 1H NMR (CDCl3, 400 MHz) data, see Table 1; 13C NMR (CDCl3, 100 MHz) data, see Table 2; (+)-HREIMS m/z 471.1977 [M + Na]+ (calcd for C24H32NaO8, 471.1995).
(+)-Acortatarinowin G (1a)
White amporous powder; 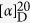 +28 (c 0.1, MeOH); ECD (MeOH) 210 (Δε +13.03), 230 (Δε +3.33), 260 (Δε +0.52).
+28 (c 0.1, MeOH); ECD (MeOH) 210 (Δε +13.03), 230 (Δε +3.33), 260 (Δε +0.52).
(−)-Acortatarinowin G (1b)
White amporous powder; 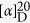 –28 (c 0.1, MeOH); ECD (MeOH) 210 (Δε−13.51), 230 (Δε−3.53), 260 (Δε−0.54).
–28 (c 0.1, MeOH); ECD (MeOH) 210 (Δε−13.51), 230 (Δε−3.53), 260 (Δε−0.54).
(±)-Acortatarinowin H (2)
Brown-red amorphous powder, 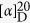 0 (c = 0.2, CH2Cl2); UV (MeOH) λmax (log ε) = 208 (4.21), 243 (3.83), 290 (3.43), and 315 (3.42) nm; IR νmax =1650, 1573, 1509, 1456, 1299, 1243, 1218, 1036, 843, 810 cm−1; 1H NMR (CDCl3, 400 MHz) data, see Table 1; 13C NMR (CDCl3, 100 MHz) data, see Table 2; HRESIMS m/z 407.1493 [M + Na]+ (calcd for C22H24NaO6, 407.1471).
0 (c = 0.2, CH2Cl2); UV (MeOH) λmax (log ε) = 208 (4.21), 243 (3.83), 290 (3.43), and 315 (3.42) nm; IR νmax =1650, 1573, 1509, 1456, 1299, 1243, 1218, 1036, 843, 810 cm−1; 1H NMR (CDCl3, 400 MHz) data, see Table 1; 13C NMR (CDCl3, 100 MHz) data, see Table 2; HRESIMS m/z 407.1493 [M + Na]+ (calcd for C22H24NaO6, 407.1471).
(+)-Acortatarinowin H (2a)
Brown-red amorphous powder; 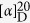 +14 (c 0.1, CH2Cl2); ECD (MeOH) 213(Δε+26.05), 243 (Δε−12.82), 315 (Δε−9.33).
+14 (c 0.1, CH2Cl2); ECD (MeOH) 213(Δε+26.05), 243 (Δε−12.82), 315 (Δε−9.33).
(−)-Acortatarinowin H (2b)
Brown-red amorphous powder; 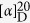 –14 (c 0.1, CH2Cl2); ECD (MeOH) 213 (Δε−27.01), 243 (Δε +13.02), 315 (Δε +10.31).
–14 (c 0.1, CH2Cl2); ECD (MeOH) 213 (Δε−27.01), 243 (Δε +13.02), 315 (Δε +10.31).
(±)-Acortatarinowin I (3)
Light yellow gum, 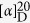 +13 (c = 0.4, MeOH); UV (MeOH) λmax (log ε) = 207 (4.72), 232 (4.51), and 278 (4.04) nm; IR νmax = 927, 1590, 1509, 1454, 1417, 1381, 1328, 1233, 1125, 1027, 812, 766, and 713 cm−1; 1H NMR (methanol-d4, 400 MHz) data, see Table 1; 13C NMR (methanol-d4, 100 MHz) data, see Table 2; HRESIMS m/z 425.1958 [M + Na]+ (calcd for C23H30NaO6, 425.1940).
+13 (c = 0.4, MeOH); UV (MeOH) λmax (log ε) = 207 (4.72), 232 (4.51), and 278 (4.04) nm; IR νmax = 927, 1590, 1509, 1454, 1417, 1381, 1328, 1233, 1125, 1027, 812, 766, and 713 cm−1; 1H NMR (methanol-d4, 400 MHz) data, see Table 1; 13C NMR (methanol-d4, 100 MHz) data, see Table 2; HRESIMS m/z 425.1958 [M + Na]+ (calcd for C23H30NaO6, 425.1940).
(−)-Acortatarinowin I (3a)
Light yellow gum; 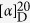 –32 (c 0.1, MeOH); ECD (MeOH) 210 (Δε−9.71), 233 (Δε−3.10);
–32 (c 0.1, MeOH); ECD (MeOH) 210 (Δε−9.71), 233 (Δε−3.10);
(+)-Acortatarinowin I (3b)
light yellow gum; 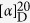 +32 (c 0.3, MeOH); ECD (MeOH) 210 (Δε +10.32), 230 (Δε +2.55).
+32 (c 0.3, MeOH); ECD (MeOH) 210 (Δε +10.32), 230 (Δε +2.55).
Acortatarinowin J (4)
Colorless block crystals, mp 158–159 °C; 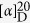 −1.4 (c = 0.4, CH2Cl2); UV (MeOH) λmax (log ε) = 207 (5.19), 231(4.60), 278 (4.11), and 303 (3.95) nm; ECD (MeOH) λmax (Δε) 210 (−3.51), 237 (+1.32), 275 (−0.80), 305 (−0.92) nm; IR νmax = 2934, 2030, 1674, 1594, 1514, 1486, 1419, 1266, 1127, 1023, 829, and 766 cm−1; 1H NMR (CDCl3, 400 MHz) data, see Table 1; 13C NMR (CDCl3, 100 MHz) data, see Table 2; HRESIMS m/z 439.1718 [M + Na]+ (calcd for C23H28NaO7, 439.1733).
−1.4 (c = 0.4, CH2Cl2); UV (MeOH) λmax (log ε) = 207 (5.19), 231(4.60), 278 (4.11), and 303 (3.95) nm; ECD (MeOH) λmax (Δε) 210 (−3.51), 237 (+1.32), 275 (−0.80), 305 (−0.92) nm; IR νmax = 2934, 2030, 1674, 1594, 1514, 1486, 1419, 1266, 1127, 1023, 829, and 766 cm−1; 1H NMR (CDCl3, 400 MHz) data, see Table 1; 13C NMR (CDCl3, 100 MHz) data, see Table 2; HRESIMS m/z 439.1718 [M + Na]+ (calcd for C23H28NaO7, 439.1733).
Acortatarinowin K (5)
White amorphous powder, 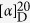 −1.2 (c = 0.1, MeOH); UV (MeOH) λmax (log ε) = 205 (4.45), 231(4.24), 278 (3.93), and 308 (3.77) nm; ECD (MeOH) λmax (Δε) 210 (−1.05), 233 (+0.71), 266 (−0.20), 303 (−0.48) nm; IR νmax = 2934, 2031, 1673, 1633, 1518, 1446, 1419, 1299, 1166, 1022, 813, and 765 cm−1; 1H NMR (methanol-d4, 400 MHz) data, see Table 1; 13C NMR (methanol-d4, 100 MHz) data, see Table 2; HRESIMS m/z 387.1797 [M + H]+ (calcd for C22H27O6, 387.1808).
−1.2 (c = 0.1, MeOH); UV (MeOH) λmax (log ε) = 205 (4.45), 231(4.24), 278 (3.93), and 308 (3.77) nm; ECD (MeOH) λmax (Δε) 210 (−1.05), 233 (+0.71), 266 (−0.20), 303 (−0.48) nm; IR νmax = 2934, 2031, 1673, 1633, 1518, 1446, 1419, 1299, 1166, 1022, 813, and 765 cm−1; 1H NMR (methanol-d4, 400 MHz) data, see Table 1; 13C NMR (methanol-d4, 100 MHz) data, see Table 2; HRESIMS m/z 387.1797 [M + H]+ (calcd for C22H27O6, 387.1808).
Acortatarinowin L (6)
Light yellow gum, 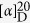 +18 (c = 0.1, MeOH); UV (MeOH) λmax (log ε) = 205 (4.81), 230 (4.15), and 280 (3.53) nm; ECD (MeOH) λmax (Δε) 210 (+3.92), 228 (+0.81), 275 (+0.44) nm; IR νmax = 3428, 2934, 1590, 1513, 1461, 1418, 1370, 1327, 1271, 1234, 1125, 1032, 1005, 799, and 707 cm−1; 1H NMR (methanol-d4, 400 MHz) data, see Table 1; 13C NMR (methanol-d4, 100 MHz) data, see Table 2; HRESIMS m/z 411.1774 [M + Na]+ (calcd for C22H28NaO6, 411.1784).
+18 (c = 0.1, MeOH); UV (MeOH) λmax (log ε) = 205 (4.81), 230 (4.15), and 280 (3.53) nm; ECD (MeOH) λmax (Δε) 210 (+3.92), 228 (+0.81), 275 (+0.44) nm; IR νmax = 3428, 2934, 1590, 1513, 1461, 1418, 1370, 1327, 1271, 1234, 1125, 1032, 1005, 799, and 707 cm−1; 1H NMR (methanol-d4, 400 MHz) data, see Table 1; 13C NMR (methanol-d4, 100 MHz) data, see Table 2; HRESIMS m/z 411.1774 [M + Na]+ (calcd for C22H28NaO6, 411.1784).
Acortatarinowin M (7)
Light yellow gum, 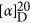 +12 (c = 0.1, MeOH); UV (MeOH) λmax (log ε) = 208 (4.73) and 272 (4.01) nm; ECD (MeOH) λmax (Δε) 210 (+0.02), 221 (−1.41), 241 (−0.93), and 296 (−0.41) nm; IR νmax = 3479, 2937, 1717, 1590, 1500, 1461, 1416, 1335, 1228, 1126, 1033, 1000, 831, and 763 cm−1; 1H NMR (methanol-d4, 400 MHz) data, see Table 1; 13C NMR (methanol-d4, 100 MHz) data, see Table 2; HRESIMS m/z 459.1619 [M + Na]+ (calcd for C22H28NaO9, 459.1631).
+12 (c = 0.1, MeOH); UV (MeOH) λmax (log ε) = 208 (4.73) and 272 (4.01) nm; ECD (MeOH) λmax (Δε) 210 (+0.02), 221 (−1.41), 241 (−0.93), and 296 (−0.41) nm; IR νmax = 3479, 2937, 1717, 1590, 1500, 1461, 1416, 1335, 1228, 1126, 1033, 1000, 831, and 763 cm−1; 1H NMR (methanol-d4, 400 MHz) data, see Table 1; 13C NMR (methanol-d4, 100 MHz) data, see Table 2; HRESIMS m/z 459.1619 [M + Na]+ (calcd for C22H28NaO9, 459.1631).
Acortatarinowin N (8)
Light yellow gum, 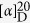 −23 (c = 0.1, MeOH); UV (MeOH) λmax (log ε) = 207 (4.77), 263 (4.22), and 293 (3.93) nm; ECD (MeOH) 215 (Δε −0.70), 224 (Δε −0.78), 238 (Δε +2.13), 296 (−1.68) nm; IR (KBr) νmax 3456, 2939, 2714, 1714, 1593, 1508, 1463, 1416, 1328, 1270, 1219, 1126, 1057, 1004, 762 cm–1; 1H NMR (methanol-d4, 400 MHz) data, see Table 1; 13C NMR (methanol-d4, 100 MHz) data, see Table 2; (+)-HREIMS m/z 429.1464 [M + Na]+ (calcd for C21H26NaO8, 429.1525).
−23 (c = 0.1, MeOH); UV (MeOH) λmax (log ε) = 207 (4.77), 263 (4.22), and 293 (3.93) nm; ECD (MeOH) 215 (Δε −0.70), 224 (Δε −0.78), 238 (Δε +2.13), 296 (−1.68) nm; IR (KBr) νmax 3456, 2939, 2714, 1714, 1593, 1508, 1463, 1416, 1328, 1270, 1219, 1126, 1057, 1004, 762 cm–1; 1H NMR (methanol-d4, 400 MHz) data, see Table 1; 13C NMR (methanol-d4, 100 MHz) data, see Table 2; (+)-HREIMS m/z 429.1464 [M + Na]+ (calcd for C21H26NaO8, 429.1525).
(S)-MTPA Derivative of 7
1H NMR (methanol-d4, 400 MHz) δH 7.50−7.15 (5H, overlap, aromatic protons), 6.59 (2H, s, H-2,6), 6.10 (1H, d, J = 7.2 Hz, H-7), 4.85 (m, H-8), 1.016 (1H, d, J = 6.4 Hz, H-9), 7.35 (2H, s, H-2′,6′), HRESIMS m/z 675.2020 [M + Na]+ (calcd for C32H35F3NaO11, 675.2029).
(R)-MTPA Derivative of 7
1H NMR (methanol-d4, 400 MHz) δH 7.50−7.15 (5H, overlap, aromatic protons), 6.81 (2H, s, H-2,6), 6.21 (1H, d, J = 7.2 Hz, H-7), 4.80 (m, H-8), 1.014 (1H, d, J = 6.4 Hz, H-9), 7.31 (2H, s, H-2′,6′), HRESIMS m/z 675.2019 [M + Na]+ (calcd for C32H35F3NaO11, 675.2029).
Single Crystal X-ray Diffraction Analyses and Crystallographic Data of Compounds (±)-1 and 4
The crystallographic data of (±)−1 CCDC 1434906) and 4 (CCDC 1028833) have been deposited in the Cambridge Crystallographic Data Centre.
Crystallographic Data of Compound (±)-1
C24H32O8, M = 448.5, monoclinic, a = 14.8444(2) Å, b = 10.0477(2) Å, c = 8.05590(10) Å, α = 90.00°, β = 91.0230(10)°, γ = 90.00°, V = 1201.36(3) Å3, T = 296 (2) K, space group P-2yc, Z = 2, μ(CuKα) = 1.54178 mm−1, 9895 reflections measured. The final R1 values were 0.0226 (I > 2σ (I)). The final wR(F2) values were 0.0911 (I > 2σ(I)). The final R1 values were 0.0338 (all data). The final wR(F2) values were 0.0906 (all data). The goodness of fit on F2 was 1.048. Flack parameter = 0.19 (12).
Crystallographic Data of Compound 4
C23H28O7, M = 416.45, monoclinic, a = 9.8071(4) Å, b = 7.9422(3) Å, c = 13.6711(5) Å, α = 90.00°, β = 99.0390(10)°, γ = 90.00°, V = 1051.62(7) Å3, T = 100(2) K, space group P21, Z = 2, μ(CuKα) = 0.801 mm−1, 8314 reflections measured, 3314 independent reflections (Rint = 0.0370). The final R1 values were 0.0338 (I > 2σ(I)). The final wR(F2) values were 0.0983 (I > 2σ(I)). The final R1 values were 0.0339 (all data). The final wR(F2) values were 0.0983 (all data). The goodness of fit on F2 was 1.094. Flack parameter = 0.17(15). The Hooft parameter is 0.09(5) for 1354 Bijvoet pairs.
Preparation of the (R) and (S)-MTPA Ester of 729
Compound 7 (0.6 mg) was dissolved in 2.0 mL anhydrous CH2Cl2. Dimethylaminopyridine (30.0 mg), 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide hydrochloride (20 μL), and (R)-MTPA chloride (25.0 μL) were added in sequentially. The reaction mixture was stirred for 3 h at room temperature under N2. The solution was evaporated under reduced pressure. The residue was passed to a small silica gel CC eluting with petroleum ether–acetone (5:1) to afford the (S)-MTPA ester of 7. The (R)-MTPA ester of 7 was prepared with (S)-MTPA chloride in the same manner.
DPPH Radical Scavenging Activity
The free radical scavenging activity of each isolated new neolignans and lignans was tested using a DPPH assay previously described30. Briefly, the tested compounds (6.25, 12.5, 25, 50, 75, and 100μg/mL) were reacted with DPPH (100 μg/mL) in EtOH (with 5% DMSO). The reaction was placed in the wells of 96-well plates at room temperature in the dark. After 30 min of incubation, the optical density of the reaction mixture at 515 nm was read using a microplate reader. All the tests were performed three times, and the results were averaged. Vitamin E was used as the positive control. An EtOH solution (with 5% DMSO) was used as a control.
Additional Information
How to cite this article: Lu, Y. et al. Antioxidant Lignans and Neolignans from Acorus tatarinowii. Sci. Rep. 6, 22909; doi: 10.1038/srep22909 (2016).
Supplementary Material
Acknowledgments
The authors would like to thank the Analytical and Testing Center at Huazhong University of Science and Technology for assistance in conducting ECD and IR analyses. This work was financially supported by the Program for New Century Excellent Talents in University, State Education Ministry of China (NCET-2008-0224), the National Natural Science Foundation of China (Nos. 31370372, 81573316, 31570361, and 31200258).
Footnotes
Author Contributions G.D. and Y.Z. designed the experiments and commented the manuscript. Y.L. conducted the main experiments with the help of C.Q. analyzed the data, and wrote the manuscript; X.L. conducted the single crystal X-ray diffraction analyses; J.L. did the ECD calculations; S.C. performed the Mosher’s experiment; Y.X. and J.W. authenticated the plant material; H.Z., J.Z. and Y.X. polished this manuscript; All authors reviewed the manuscript.
References
- Mau J.-L., Chao G.-R. & Wu K.-T. Antioxidant Properties of Methanolic Extracts from Several Ear Mushrooms. J Agr Food Chem 49, 5461–5467 (2001). [DOI] [PubMed] [Google Scholar]
- Saleh M. A., Clark S., Woodard B. & Deolu-Sobogun S. A. Antioxidant and free radical scavenging activities of essential oils. Ethn Dis 20, S1-78–82 (2010). [PubMed] [Google Scholar]
- Hazra B., Biswas S. & Mandal N. Antioxidant and free radical scavenging activity of Spondias pinnata. BMC Complem Altern M 8, 63 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P., Kong M., Yuan S., Liu J. & Wang P. History and Experience: A Survey of Traditional Chinese Medicine Treatment for Alzheimer’s Disease. Evid-Based Compl Alt 2014, 5 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang C., Ye Y., Feng Y. & Quinn R. J. TCM, brain function and drug space. Nat. Prod. Rep. 33, 6–25 (2016) [DOI] [PubMed] [Google Scholar]
- Prince M., Prina M. & Guerchet M. World Alzheimer Report. Alzheimer’s Disease International: London, UK; pp 1–92 (2013). [Google Scholar]
- Lu Y. et al. (±)-Acortatarinowins A–F, Norlignan, Neolignan, and Lignan Enantiomers from Acorus tatarinowii. J Nat Prod 78, 2205–2214 (2015). [DOI] [PubMed] [Google Scholar]
- Tong X.-G. et al. Compounds from Acorus tatarinowii: Determination of Absolute Configuration by Quantum Computations and cAMP Regulation Activity. J. Nat. Prod. 73, 1160–1163 (2010). [DOI] [PubMed] [Google Scholar]
- Prasad A. K. et al. Neolignans and a lignan from Piper clarkii. Phytochemistry 39, 655–658 (1995). [Google Scholar]
- Li X.-N. et al. Lignans with Anti-HIV Activity from Schisandra propinqua var. sinensis. J. Nat. Prod. 72, 1133–1141 (2009). [DOI] [PubMed] [Google Scholar]
- Nishiwaki H., Nakayama K., Shuto Y. & Yamauchi S. Synthesis of All Stereoisomers of 3,3′-Dimethoxy-7,7′-epoxylignane-4,4′-diol and Their Plant Growth Inhibitory Activity. J. Ag. Food Chem. 62, 651–659 (2014). [DOI] [PubMed] [Google Scholar]
- Shimomura H., Sashida Y. & Oohara M. Lignans from Machilus thunbergii. Phytochemistry 27, 634–636 (1988). [Google Scholar]
- Sy L.-K. & Brown G. D. Novel Phenylpropanoids and Lignans from Illicium verum. J. Nat. Prod. 61, 987–992 (1998). [DOI] [PubMed] [Google Scholar]
- Zhu H. et al. A pair of unprecedented cyclohexylethanoid enantiomers containing unusual trioxabicyclo[4.2.1]nonane ring from Clerodendrum bungei. Tetrahedron Lett. 55, 2277–2279 (2014). [Google Scholar]
- Harada N. & Nakanishi K. A Method for Determining the Chirality of Two Aromatic Chromophores and the Absolute Configurations of Chromomycin A3 and Related Antibiotics. J. Am. Chem. Soc. 91, 5896 (1969). [DOI] [PubMed] [Google Scholar]
- Huo C., Liang H., Zhao Y., Wang B. & Zhang Q. Neolignan glycosides from Symplocos caudata. Phytochemistry 69, 788–795 (2008). [DOI] [PubMed] [Google Scholar]
- Li Y. et al. Bioactive Neolignans and Lignans from the Bark of Machilus robusta. J. Nat. Prod. 74, 1444–1452 (2011). [DOI] [PubMed] [Google Scholar]
- Lee S. Y., Moon E., Kim S. Y., Choi S. U. & Lee K. R. Quinone Derivatives from the Rhizomes of Acorus gramineus and Their Biological Activities. Biosci., Biotech., and Bioch. 77, 276–280 (2013). [DOI] [PubMed]
- da Silva T. & Lopes L. M. X. Aryltetralone lignans and 7,8-seco-lignans from Holostylis reniformis. Phytochemistry 65, 751–759 (2004). [DOI] [PubMed] [Google Scholar]
- da Silva T. & Lopes L. M. X. Aryltetralol and aryltetralone lignans from Holostylis reniformis. Phytochemistry 67, 929–937 (2006). [DOI] [PubMed] [Google Scholar]
- Lopes N. P., de Almeida Blumenthal E. E., Cavalheiro A. J., Kato M. J. & Yoshida M. Lignans, γ-lactones and propiophenones of Virola surinamensis. Phytochemistry 43, 1089–1092 (1996). [Google Scholar]
- Sawasdee K. et al. New neolignans and a lignan from Miliusa fragrans, and their anti-herpetic and cytotoxic activities. Tetrahedron Lett. 54, 4259–4263 (2013). [Google Scholar]
- Jung K. Y. et al. Magnone A and B, Novel Anti-PAF Tetrahydrofuran Lignans from the Flower Buds of Magnolia fargesii. J. Nat. Prod. 61, 808–811 (1998). [DOI] [PubMed] [Google Scholar]
- Harada N. & Nakanishi K. Circular Dichroic Spectroscopy-Exciton Coupling in Organic Stereochemistry; University Science Books, Mill Valley, CA, and Oxford University Press: Oxford (1983).
- León A., Cogordán J. A., Sterner O. & Delgado G. Enantiomeric Derivatives of Tokinolide B: Absolute Configuration and Biological Properties. J. Nat. Prod. 75, 859–864 (2012). [DOI] [PubMed] [Google Scholar]
- Sólyomváry A. et al. Identification and isolation of new neolignan and sesquineolignan species: Their acid-catalyzed ring closure and specific accumulation in the fruit wall of Cirsium eriophorum (L.) Scop. Process Biochem. 50, 853–858 (2015). [Google Scholar]
- Pratico D. & Sung S. Lipid peroxidation and oxidative imbalance: early functional events in Alzheimer’s disease. J. Alzheimers Dis. 6, 171–175 (2004). [DOI] [PubMed] [Google Scholar]
- Pontikia E., Kontogiorgis C., Xu Y., Hadjipavlou-Litina D. & Luo Y. New Lipoxygenase Inhibitors of Reactive Oxygen Species Production in Cellular Models of Amyloid (A2) Toxicities. J. Alzheimers Dis. 34, 215–230 (2013). [DOI] [PubMed] [Google Scholar]
- Ohtani I., Kusumi T., Kashman Y. & Kakisawa H. High-field FT NMR application of Mosher’s method. The absolute configurations of marine terpenoids. J. Am. Chem. Soc. 113, 4092–4096 (1991). [Google Scholar]
- Krishnaiah D., Sarbatly R. & Nithyanandam R. A review of the antioxidant potential of medicinal plant species. Food Bioprod. Process 89, 217–233 (2011). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.