

Abstract
Over the last few years, incretin‐based therapies have emerged as important agents in the treatment of type 2 diabetes (T2D). These agents exert their effect via the incretin system, specifically targeting the receptor for the incretin hormone glucagon‐like peptide 1 (GLP‐1), which is partly responsible for augmenting glucose‐dependent insulin secretion in response to nutrient intake (the ‘incretin effect’). In patients with T2D, pharmacological doses/concentrations of GLP‐1 can compensate for the inability of diabetic β cells to respond to the main incretin hormone glucose‐dependent insulinotropic polypeptide, and this is therefore a suitable parent compound for incretin‐based glucose‐lowering medications. Two classes of incretin‐based therapies are available: GLP‐1 receptor agonists (GLP‐1RAs) and dipeptidyl peptidase‐4 (DPP‐4) inhibitors. GLP‐1RAs promote GLP‐1 receptor (GLP‐1R) signalling by providing GLP‐1R stimulation through ‘incretin mimetics’ circulating at pharmacological concentrations, whereas DPP‐4 inhibitors prevent the degradation of endogenously released GLP‐1. Both agents produce reductions in plasma glucose and, as a result of their glucose‐dependent mode of action, this is associated with low rates of hypoglycaemia; however, there are distinct modes of action resulting in differing efficacy and tolerability profiles. Furthermore, as their actions are not restricted to stimulating insulin secretion, these agents have also been associated with additional non‐glycaemic benefits such as weight loss, improvements in β‐cell function and cardiovascular risk markers. These attributes have made incretin therapies attractive treatments for the management of T2D and have presented physicians with an opportunity to tailor treatment plans. This review endeavours to outline the commonalities and differences among incretin‐based therapies and to provide guidance regarding agents most suitable for treating T2D in individual patients.
Keywords: DPP‐4 inhibitor, GLP‐1, GLP‐1 receptor agonist, glucagon‐like peptide‐1, incretin enhancer, incretin mimetics, mode of action, type 2 diabetes mellitus
Introduction
Type 2 diabetes (T2D) is a chronic, progressive disease characterized by defective gluco‐regulation caused by a combination of insulin resistance, impaired β‐cell function progressively declining over several years, hyperglucagonaemia and inappropriate hepatic glucose production 1. The progressive nature makes pharmacological intervention necessary eventually in most patients. Typical comorbidities are obesity and hypertension, which contribute to a high cardiovascular disease burden and mortality 2. Unfortunately, many existing glucose‐lowering agents cause unwanted consequences, such as hypoglycaemia and weight gain, which may reduce adherence to treatment 3. In contrast, ideal diabetes medications would control glycaemia without a risk of hypoglycaemia, while providing additional beneficial effects on β‐cell function, body weight, lipoprotein profiles, hypertension and cardiovascular risk in more general terms.
Incretin‐based therapies have emerged that make use of the incretin system, which comprises the incretin hormones glucagon‐like peptide‐1 (GLP‐1) and glucose‐dependent insulinotropic polypeptide (GIP) that stimulate the release of insulin from pancreatic β cells at elevated glucose concentrations 4. Glucose administered orally elicits a higher insulin secretory response than glucose infused intravenously, even at isoglycaemic concentrations, a phenomenon referred to as the ‘incretin effect’ (Figure 1) 5. In T2D, the incretin effect is reduced 6, but therapeutically, incretin activity can be provided by supraphysiological dosages of GLP‐1 or related agents stimulating the GLP‐1 receptor (GLP‐1R) 7. There are two classes of incretin therapies: dipeptidyl peptidase‐4 (DPP‐4) inhibitors, which prevent the proteolytic breakdown and inactivation of GLP‐1 and GLP‐1 receptor agonists (GLP‐1RAs), which provide supraphysiological concentrations of ligands that stimulate the GLP‐1R 8. Incretin hormones may have effects beyond the stimulation of insulin secretion 8, and the proteolytic activity of DPP‐4 is not restricted to the degradation and inactivation of the incretin hormones 9. Consequently, GLP‐1RAs and DPP‐4 inhibitors exhibit differences in efficacy, safety and tolerability, and may have additional benefits, such as promoting weight loss and improving (markers of) cardiovascular risk, which add to their attractive panel of clinical effects. The aim of the present review was to highlight the common features in the modes of action of GLP‐1RAs and DPP‐4 inhibitors, and also the differences between them.
Figure 1.
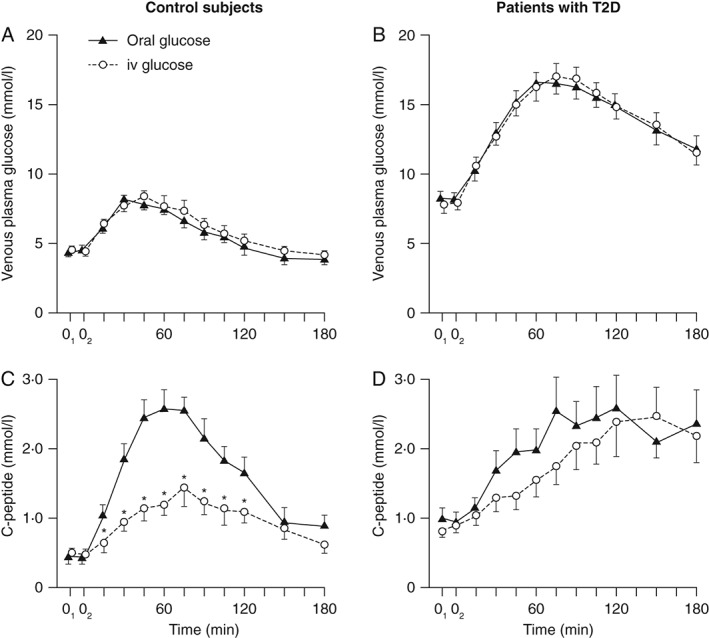
The incretin effect in control subjects and patients with type 2 diabetes (T2D). Venous plasma glucose and integrated incremental β‐cell secretory responses to oral glucose loads (black triangles) or ‘isoglycaemic’ intravenous glucose infusion (open circles). After an overnight fast, oral glucose (50 g glucose/400 ml) was ingested (time 0) and blood samples taken every 15–120 min and then every 30 min for the final two samples. Isoglycaemic intravenous glucose infusions were designed to mimic glucose concentration profiles after glucose ingestion. Asterisks denote significance (p < 0.05) to the respective value after oral load. © Springer‐Verlag 1986, reproduced with permission from Nauck et al. Diabetologia 1986; 29: 46–52 5. iv, intravenous.
Incretin System in Healthy Individuals and Those with Type 2 Diabetes
In healthy individuals, oral ingestion of nutrients stimulates secretion of multiple gut hormones involved in regulating digestion, motility and metabolism, including insulin 8. The gut hormones GIP and GLP‐1, which are secreted from the intestinal K‐ and L‐cells, respectively, within minutes of food ingestion 8, 10, mediate major and minor proportions of the incretin effect, respectively, and together account for up to 70% of insulin secretory responses after nutrient ingestion 6, 11.
In T2D, the incretin effect is severely reduced, and the defective incretin‐mediated stimulation is partially responsible for the generally defective insulin secretion in these patients (Figure 1) 5. Despite reports that GLP‐1 concentrations may be reduced in patients with T2D, incretin hormone loss is not a universal characteristic of T2D and several studies actually report minor increases in GIP secretion in T2D 10. Recent meta‐analyses did not reveal systematic differences in GLP‐1 or GIP secretion between healthy subjects and patients with T2D 10, 12. A major focus of incretin‐based research has therefore been on understanding the differences in insulinotropic and glucagonostatic responses to the incretin hormones in people with T2D and healthy subjects 10.
Insulinotropic Effects of the Incretin Hormones
In T2D, the insulinotropic effects of GIP are virtually absent (Figure 2A) 7, 14, 15. Even pharmacological doses of GIP only have marginal effects on insulin secretion 16. By contrast, the insulinotropic effects of GLP‐1 are at least partially preserved in T2D, although endogenous GLP‐1‐mediated insulin secretion does not compensate for the loss of the insulinotropic activity of GIP 7. At nearly all stages of T2D, pharmacological dosages of GLP‐1 significantly restore insulin secretion to levels approaching normal (Figure 2B, C) 7, 14 and can normalize fasting plasma glucose (FPG) levels in the short term 13. This indicates that the insulin secretory mechanism responds better to GLP‐1 than to other secretagogues, albeit with reduced potency compared with healthy subjects. Furthermore, as for healthy subjects, the insulinotropic actions of exogenously administered GLP‐1 are glucose‐dependent (with identical glucose thresholds) in patients with T2D 17, 18. It is of interest that in T2D the effects of exogenously administered GLP‐1 cannot be augmented by GIP (Figure 2D, E) 15. This may reflect a decreased responsiveness of β cells to GIP in individuals with T2D 10.
Figure 2.
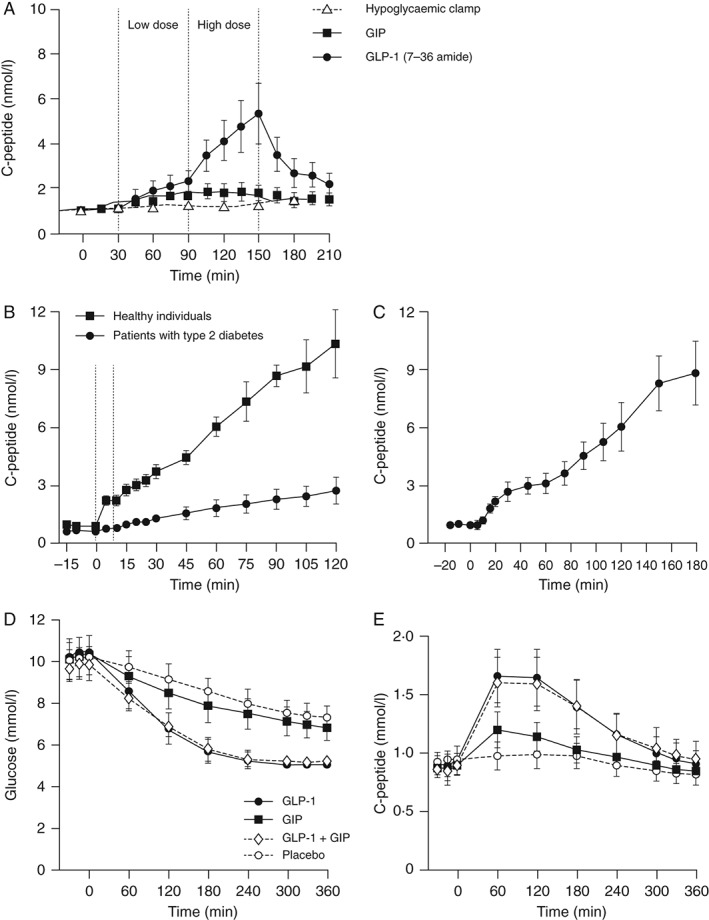
Insulinotropic activities of the incretin hormones glucagon‐like peptide 1 (GLP‐1) and glucose‐dependent insulinotropic polypeptide (GIP). (A) GLP‐1, but not GIP, can increase both early and late stage insulin secretion. Nine patients with type 2 diabetes (T2D) and nine subjects with normal glucose tolerance were included. All antidiabetic drugs were withheld until experimental completion and each experiment was performed following an overnight fast. At time 0, patients received intravenous glucose to a plasma glucose concentration of 8.75 mmol/l, at which point a hyperglycaemic glucose clamp was initiated. Either exogenous synthetic human GIP or GLP‐1 (7‐36 amide) was then infused to concentrations equivalent to physiological levels (0.8 and 0.4 pmol/kg/min, respectively) between 30 and 90 min (dotted lines). Between 90 and 150 min, infusion rates were increased threefold to supraphysiological concentrations (2.4 and 1.2 pmol/kg/min, respectively). C‐peptide level was determined from blood samples by radioimmunoassay. Data are mean ± standard error of the mean. © The American Society for Clinical Investigation 1993, reproduced with permission from Nauck et al. J Clin Invest 1993; 91: 301–7 7. (B, C): Insulin responses to physiological levels of GLP‐1 are severely impaired in T2D but can be restored by pharmacological doses of GLP‐1. Sixteen obese patients with T2D [eight patients per experiment (B, C)] underwent hyperglycaemic clamp (15 mmol/l) and infusion of physiological [0.5 pmol/kg/min (B)] or pharmacological [1 pmol/kg/min (C)] levels of GLP‐1 or GLP‐ 1 (7‐36 amide), respectively. C‐peptide concentrations were measured by Autodelphia automatic fluoroimmunoassay. © Springer‐Verlag 2009 and 2002, reproduced with permission from Højberg et al. Diabetologia 2009; 52: 199–207 13 (B) and Vilsbøll et al. Diabetologia 2002; 45: 1111–9 14 (C). (D, E) GIP does not augment the insulinotropic activity of GLP‐1. Twelve patients with T2D were included. Antidiabetic drugs were discontinued 1 day before each experiment and each experiment was performed after an overnight fast. Placebo (0.9% NaCl with 1% human serum albumin), GIP (4 pmol/kg/min), GLP‐1 (7‐36 amide; 1.2 pmol/kg/min), or a combination of GIP and GLP‐1 (7‐36 amide) was infused over a period of 360 min. Plasma glucose concentrations and C‐peptide secretion rates were determined from blood drawn in the basal state and during infusions. Glucose was measured using a glucose oxidase assay and C‐peptide determined by immunoassay. © American Diabetes Association 2011, reproduced with permission from Mentis et al. Diabetes 2011; 60: 1270–6 15.
Glucagonostatic Effects of GLP‐1
Glucagon is secreted from pancreatic α cells and opposes the action of insulin by regulating hepatic glucose production 19. In healthy subjects, oral or intravenous glucose induces suppression of glucagon secretion 20. In T2D, defective glucagon suppression produces hyperglucagonaemia, both in the fasting and post‐nutrient state 20. Paradoxically, a rise in glucagon is often observed in subjects with T2D after nutrient ingestion, and it has been postulated that reduced sensitivity of α cells to glucose (and insulin) may be responsible for this hyperglucagonemia 21. Incretin hormones also have significant roles in the regulation of glucagon release, and GLP‐1 appears to be a physiological inhibitor of glucagon secretion 7. By contrast, GIP can (under certain circumstances) stimulate glucagon secretion and antagonize GLP‐1‐mediated suppression of glucagon release 22, 23. In T2D, supraphysiological doses of GLP‐1 can fully restore α‐cell glucose sensitivity, facilitating normal glucose‐induced inhibition of glucagon secretion 7, 17. The mechanisms underlying these glucagonostatic effects are not entirely clear, but may involve direct α‐cell inhibition 24 or indirect inhibition via stimulation of somatostatin release from neighbouring δ cells 25. Clinical studies have indicated that GLP‐1‐induced inhibition of glucagon secretion contributes significantly to the glucose‐lowering capacity of GLP‐1‐based therapies and may be as important as their insulinotropic effects 26.
Development of Incretin‐based Therapies
As the insulinotropic effects of GLP‐1 are (partially) preserved in T2D, most pharmacological efforts have focused on making use of the preserved ability of GLP‐1 to stimulate the GLP‐1R (rather than the functionally ineffective GIP receptor) to re‐introduce ‘incretin’ activity 8. Native GLP‐1 has a half‐life of ∼2 min as a result of degradation by DPP‐4 and rapid renal clearance of both the intact and degraded GLP‐1 molecules 8. Continuous intravenous GLP‐1 infusion is therefore required to provide glycaemic benefits in subjects with T2D 17, 18. As this is not clinically feasible, alternative strategies have been pursued in the development of therapeutically viable incretin‐based therapies. Two strategies were adopted: (i) DPP‐4 enzyme inhibition, preventing proteolytic degradation and inactivation of GLP‐1 8, 27, 28, 29; thus, making use of glucose‐lowering properties of endogenously secreted GLP‐1 from the gut and (ii) administration of GLP‐1RA (GLP‐1 analogues with slower degradation by DPP‐4 and prolonged half‐life 8, 30, either found in nature, or designed for the purpose).
Structure, Formulation and Administration of Incretin Therapies
DPP‐4 Inhibitors
Sitagliptin was the first selective inhibitor of DPP‐4, followed by vildagliptin, saxagliptin, linagliptin and, most recently, alogliptin (Figure 3 and Table S1). Sitagliptin is a non‐peptide heterocyclic compound with rapid onset and long duration of action, whereas vildagliptin and saxagliptin are both cyanopyrrolidines with slow onset and prolonged action upon binding to the DPP‐4 enzyme. Linagliptin (a methylxanthine) and alogliptin (a heterocyclic aminopiperidine) differ still further 27, 29. These distinct chemical structures affect pharmacokinetic properties, formulation and daily dosing (Table S1) and may determine specificity, tolerability and safety profiles. DPP‐4 inhibitors are administered orally and formulated either as a single agent or in combination with other antihyperglycaemic compounds, typically metformin 27, 29, 31.
Figure 3.
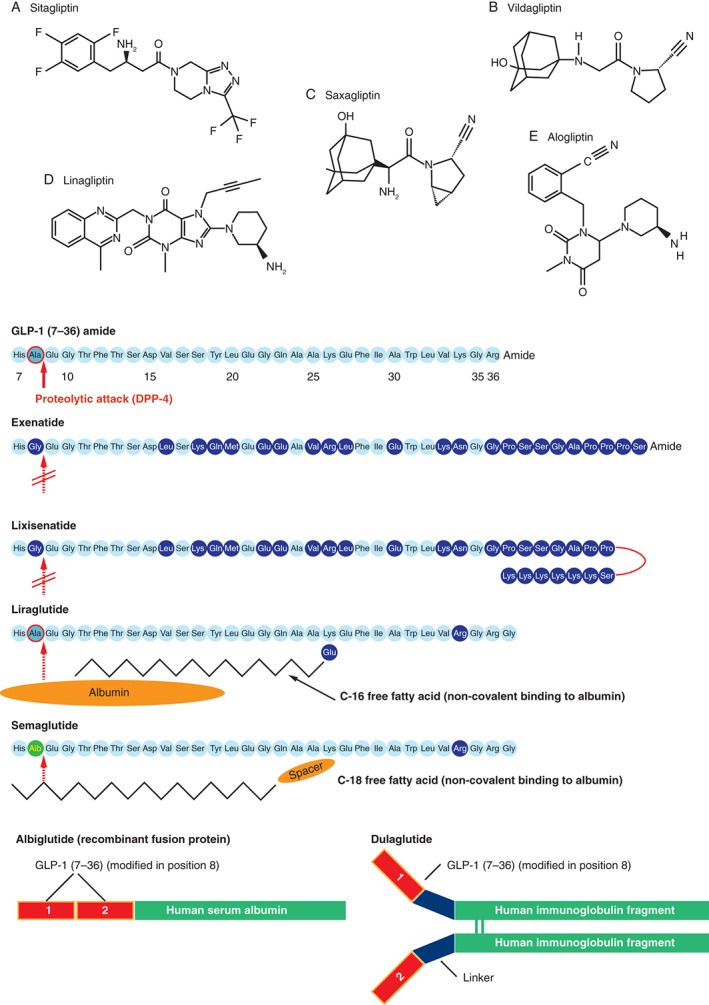
The chemically distinct structures of dipeptidyl peptidase 4 (DPP‐4) inhibitors [(A–E) for DPP‐4 inhibitors indicates order of approval] and GLP‐1RAs. Images (A–E) © of Wiley and reproduced with permission from Deacon et al., Diabetes Obes Metab 2011; 13: 7–18; Drucker and Nauck. Lancet 2006; 368: 1696–705 [glucagon‐like peptide‐1 (GLP‐1), exenatide, liraglutide] 8, 27.
GLP‐1RAs
There are five GLP‐1RAs currently approved for use in the treatment of T2D: exenatide, liraglutide, lixisenatide, dulaglutide and albiglutide. These molecules were developed based on human GLP‐1 or exendin‐4, a 39‐amino acid peptide from the saliva of the lizard Heloderma suspectum, and there are key structural differences between the GLP‐1RAs. These differences affect product half‐life and dosing interval and have resulted in GLP‐1RAs being classified as short‐ or long‐acting. All of the currently approved products are administered as subcutaneous injections.
Short‐acting GLP‐1RAs
Exenatide was the first therapeutic GLP‐1RA. It is a synthetic version of exendin‐4, having ∼50% sequence identity to native GLP‐1 32. It is a potent agonist of GLP‐1Rs and is resistant to degradation by DPP‐4 because of the lack of alanine or proline at position 2 (Figure 3) 8, 33. Exenatide was first made available as a twice‐daily (>6 h apart) formulation for subcutaneous injection within 60 min of the two main meals of the day (Table S1).
Apart from exenatide twice daily, lixisenatide is the only other short‐acting GLP‐1RA and is also a derivative of exendin‐4 34. Lixisenatide differs from exenatide by the deletion of a proline residue and the addition of six lysine residues at the C‐terminal end 35. While lixisentide has a half‐life of ∼3 h , which is similar to that for exenatide twice daily (2.4 h), lixisenatide is administered once daily (Table S1). Lixisentide is currently approved in Europe but not in the USA.
Long‐acting GLP‐1RAs
Liraglutide is a GLP‐1 analogue with 97% amino acid sequence identity to native human GLP‐1 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36 (Figure 3) and was the first long‐acting compound in this class. The addition of a C16 fatty acid side chain to lysine (Lys26) and replacement of lysine with arginine at position 34 facilitates non‐covalent binding to albumin, and also promotes self‐association into heptamers 36. Heptamer formation retards absorption from the subcutis 37 and albumin binding provides stability, resistance to DPP‐4 and limits renal clearance. These modifications prolong the half‐life and facilitate once‐daily dosing at dosages of 1.2 or 1.8 mg, with the timing of injection being independent from meals (Table S1).
A second (long‐acting) formulation of exenatide, designed for once‐weekly administration at any time, irrespective of meals (exenatide once weekly; Table S1) has also been developed. Exenatide once weekly is formulated as exenatide and poly‐(d, l lactide‐co‐glycolide) microspheres that slowly degrade in situ, releasing exenatide in a sustained fashion 38. Unlike exenatide twice daily, exenatide once weekly requires patients to reconstitute the powdered agent before administration.
Albiglutide and dulaglutide consist of a dimer of human GLP‐1 molecules that is fused to recombinant human albumin and a modified human immunoglobulin G4 heavy chain, respectively. Amino acid substitutions render both molecules resistant to DPP‐4 and their extended half‐life (Table S1) allows weekly dosing 34, 39.
Semaglutide is a novel GLP‐1RA that is currently in phase III trials and is being developed for once‐weekly subcutaneous administration 40 and once‐daily oral administration. Addionally, a GLP‐1RA/basal insulin combination has recently been approved and other combination products are in development 41.
Clinical Utility of Incretin Therapies
The differing characteristics of DPP‐4 inhibitors and GLP‐1RAs are responsible for differing clinical performance, tolerability and pharmacokinetic profiles, and such differences are also observed within each drug class (Tables S2 and S3). Appreciating these differences is therefore important so the best and most suitable agent can be chosen for each patient.
DPP‐4 Inhibitors
Pharmacokinetics
The main clinically relevant pharmacokinetic characteristics of the DPP‐4 inhibitors are outlined in Table S1. Typically, DPP‐4 inhibitors reduce serum DPP‐4 activity by >80%, resulting in a doubling of intact, biologically active GLP‐1 concentrations 42. This is associated with increased insulin and suppressed glucagon secretion, resulting in reductions in postprandial glucose (PPG) levels 42, 43. The relatively long half‐lives of sitagliptin, linagliptin and alogliptin facilitate once‐daily dosing. Saxagliptin is also suitable for once‐daily dosing as a result of the presence of the active metabolite, BMS‐510849, which inhibits DPP‐4 27. The shorter half‐life of vildagliptin requires twice‐daily dosing 27.
As vildagliptin and saxagliptin are partially eliminated via the liver, dose reductions are also recommended in patients with hepatic functional impairment 27, 30.
For sitagliptin, vildagliptin, linagliptin and alogliptin, there are no known clinically significant drug–drug interactions; however, saxagliptin is metabolized via hepatic cytochrome P450 (CYP450) and may be exposed to drug–drug interactions. Hence, patients co‐administering saxagliptin and CYP3A4/5 inhibitors should reduce saxagliptin dosages 27, 30.
In general, the pharmacokinetic profiles of DPP‐4 inhibitors are not directly influenced by gender, age, race or BMI and are similar in patients with T2D and healthy subjects 27, 30.
An important differentiating characteristic among DPP‐4 inhibitors is their route of elimination. Sitagliptin and alogliptin are primarily excreted renally, whereas saxagliptin undergoes both renal and hepatic clearance (Table S1). Linagliptin is predominately (∼90%) secreted unchanged in the faeces 44. Metabolism therefore plays a minor role in the elimination of these DPP‐4 inhibitors. By contrast, vildagliptin is metabolized via at least four pathways before excretion 27, 29; however, the renal clearance of sitagliptin, vildagliptin, saxagliptin and alogliptin means that patients with renal impairment are at risk of increased drug exposure, and dose reductions are therefore recommended 27, 29. Linagliptin may provide a suitable alternative for such patients, with no reason to adjust doses in the case of chronic renal functional impairment.
Glycaemic Efficacy
All DPP‐4 inhibitors achieve glycaemic efficacy in a similar fashion and elevate GLP‐1 levels by 1.5‐ to 3‐fold (i.e. near the physiological range) 27, 29. Once DPP‐4 is maximally inhibited, glycated haemoglobin (HbA1c) reductions plateau and, consequently, HbA1c reductions are similar across this class (weighted mean differences of <0.8% as monotherapy) and there is no obvious basis for differentiation regarding efficacy 45. DPP‐4 inhibitors may be preferred in patients with modest elevations in HbA1c and FPG as, in these circumstances, there is a realistic chance of achieving common HbA1c targets 33.
Safety and Tolerability
In general, the tolerability of DPP‐4 inhibitors is excellent and, as a result of their glucose‐dependent mode of action, they are associated with a low risk of hypoglycaemia (unless used in combination with sulphonylureas or insulin) 45, 46. Overall, the number of adverse events (AEs) reported in clinical trials with DPP‐4 inhibitors were similar in patients receiving the drug compared with those receiving placebo 46. Initial surveys recorded a slight increase in infections, predominantly upper respiratory tract infections, in patients treated with a DPP‐4 inhibitor 47. An immune system imbalance has been suspected, but more recent, larger analyses have not confirmed these trends 27, 46, 48, 49.
Some post‐market reporting and analysis of large AE databases saw sitagliptin associated with an increased risk of pancreatitis 50; however, the validity of the conclusions based on these data have been widely criticized as data may often be incomplete, over‐reported and biased 51. Detailed discussions of pancreatitis potentially induced by DPP‐4 inhibitors have been published 51, 52. Importantly, the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) agreed in 2014 that a causal association between incretin‐based drugs and pancreatitis or pancreatic cancer, as expressed in the scientific literature, was inconsistent with the data available at the time 53. These data included two large cardiovascular outcomes trials, one with saxagliptin and one with alogliptin. A further, more recently reported, cardiovascular outcomes trial in which sitagliptin or placebo was added to usual care showed no significant between‐group differences in the rates of acute pancreatitis or pancreatic cancer 54.
GLP‐1RAs
Pharmacokinetics
The clinically relevant pharmacokinetic characteristics of the six currently available GLP‐1RAs are outlined in Table S1.
Short‐acting GLP‐1RAs
The short half‐life (2.4 h) of exenatide requires twice‐daily dosing (with meals). Whereas the majority of novel developments within the class of GLP‐1RAs aim at prolonged action and once‐weekly administration, lixisenatide has recently been introduced as a novel short‐acting GLP‐1RA, similar to exenatide, but recommended for once‐daily administration (Table S1). Peak plasma concentrations (Cmax) are achieved within 1.5–3.5 h of lixisenatide administration 55.
Long‐acting GLP‐1RAs
The longer half‐life of liraglutide (13 h) permits once‐daily dosing (independent of meal times). Peak plasma concentrations are greater for liraglutide compared with either formulation of exenatide (once daily or once weekly), most likely as a result of substantial albumin binding. It is estimated that only 1–2% of liraglutide is ‘free’ and able to diffuse into target tissues and interact with receptors 36. Exenatide once weekly has a prolonged duration of action with a biphasic release pattern, peaking initially, then being followed by a sustained release. Therapeutic drug levels are achieved after 2 weeks, and steady state plasma concentrations after ≥6 weeks 56.
The half‐life of albiglutide is ∼5 days, with steady‐state concentrations reached within 3–5 weeks 57. Dulaglutide's half‐life is ∼4.5 days, with steady‐state concentrations occurring within 2–4 weeks 58.
A DPP‐4 inhibitor increases endogenous GLP‐1 to physiological levels (10–25 pmol/l), whereas GLP‐1RAs reach higher pharmacological levels (e.g. free active liraglutide levels are in the range 60–90 pmol/l) 43, 59, 60. It is not known, however, whether tissue distribution is similar for all GLP‐1 receptors and it would be interest to understand what level of GLP‐1 exposure occurs on β cells as well as in the autonomic and central nervous system.
The predominant elimination route for exenatide is via glomerular filtration with subsequent proteolytic degradation. Exposure to exenatide increases in patients with end‐stage renal disease (ESRD) and moderate renal impairment, compared with subjects with normal renal function (Table S1). Exenatide twice daily and once weekly are therefore not recommended in patients with ESRD or severe renal impairment, and caution is advised in patients with moderate renal impairment.
Elimination of lixisenatide is also through the kidneys. Caution is advised with moderate renal impairment, and lixisenatide is not recommended in patients with ESRD or severe renal impairment 55.
Liraglutide is endogenously metabolized, with no single organ providing the major route of elimination 61. Liraglutide exposure is not elevated in patients with renal impairment but is not currently recommended in patients with severe renal impairment, including ESRD, because of limited clinical experience in this population (Table S1). Ongoing and recently completed clinical trials are investigating liraglutide use in patients with moderate and severe renal impairment (NCT01620489, NCT01394341 and NCT01179048).
Albiglutide and dulaglutide are metabolized and do not require dose adjustment with moderate renal impairment, but are not recommended for patients with severe renal impairment, including ESRD 57, 58.
Limited data exist regarding the use of GLP‐1RAs in patients with hepatic functional impairment 62, 63. Although hepatic functional impairment is not anticipated to adversely affect exposure levels, GLP‐1RAs are not recommended in patients with mild, moderate or severe hepatic impairment because of limited clinical experience.
Glycaemic Efficacy
Compared with placebo, GLP‐1RAs reduce HbA1c levels by ∼1% 64; however, reductions are dependent on the choice of agent, dose, baseline HbA1c and ongoing treatment. The short half‐life of exenatide twice daily and its requirement for twice‐daily dosing at meal times means it predominantly reduces PPG levels. By contrast, the prolonged actions of exenatide once weekly and liraglutide produce reductions in both FPG and PPG as a result of suppression of fasting glucagon (exenatide once weekly) 56 or increase in fasting insulin levels (liraglutide) 65. Significantly greater HbA1c changes are therefore observed when compared with exenatide twice daily in head‐to‐head trials [−1.9% (exenatide once weekly) vs −1.5% (exenatide twice daily); p = 0.0023 56 and −1.12% (liraglutide) vs −0.79 (exenatide twice daily); p < 0.0001] 65. In a 26‐week head‐to‐head trial between liraglutide and exenatide once weekly, treatment with liraglutide produced greater reductions in mean HbA1c compared with patients receiving exenatide (−1.48% vs −1.28%, respectively) 66. Similarly, in a 32‐week trial, treatment with liraglutide led to greater reductions in HbA1c compared with albiglutide (–0.99% vs –0.78%, respectively) 67.
Dulaglutide treatment was superior in lowering HbA1c compared with exenatide twice daily in patients who were on maximum tolerated doses of metformin and pioglitazone 68. Another head‐to‐head trial reported that dulaglutide was non‐inferior to liraglutide in lowering HbA1c (least‐squares mean reduction in HbA1c was −1.42% with dulaglutide vs −1.36% for liraglutide) 69. Similar reductions in HbA1c were reported for lixisenatide vs exenatide in the GetGoal‐X study 68, 70.
Exenatide twice daily and once weekly are associated with the formation of anti‐exenatide antibodies, and glycaemic efficacy can be reduced in patients with high‐titre, compared with absent or low‐titres of anti‐exenatide antibodies 56; however, the clinical significance of antibody generation is not yet clear. The percentage of patients developing antibodies against liraglutide is much lower 71.
Safety and Tolerability
As a result of their glucose‐dependent mode of action, GLP‐1RAs are associated with a very low risk of hypoglycaemia; however, when used in conjunction with other medications able to provoke episodes of hypoglycaemia (e.g. sulphonylureas), the glucose‐lowering effect of GLP‐1RAs may increase the risk of hypoglycaemia 64. The most commonly observed AE is transient nausea, which can be minimized by initiating treatment with low doses and gradually increasing the dose 38. If nausea, which typically diminishes after a few weeks with liraglutide and exenatide 65, persists, symptoms can be mitigated by transient dose reductions. Nausea is also the most frequently reported AE with exenatide once weekly; however, because of the small initial release of this formulation, large weekly doses (2 mg) are tolerated 38. The incidence of nausea and vomiting was also significantly lower with once‐weekly albiglutide compared with liraglutide 67. Despite GLP‐1RA‐related or insulin ‘gastrointestinal’ AEs occurring early in a treatment course, some patients find these events intolerable and need to cease treatment 64.
A possible association of GLP‐1RAs with acute pancreatitis was raised as a result of some post‐marketing case reports from the use of exenatide 72. Although few cases of pancreatitis were reported in clinical trials with liraglutide, the incidence among patients receiving liraglutide was greater than those among patients receiving placebo or comparator medications 73. Establishing or excluding a cause and effect link between GLP‐1RAs and pancreatitis in T2D is difficult as patients have a threefold greater pancreatitis risk compared with healthy subjects 74. Retrospective analysis of AE reporting databases also correlated GLP‐1RAs with an increased risk of pancreatitis 50; however, as mentioned previously, an extensive review by the FDA and EMA in 2014 concluded that, studies so far do not show a causal link between incretin‐based drugs and either pancreatitis or pancreatic cancer 53.
Preclinical studies indicated liraglutide could increase the risk of C‐cell proliferation and medullary thyroid adenomas and carcinomas (MTCs) in rodents via GLP‐1R activation and calcitonin release 36; however, compared with rodents, human and non‐human primates have far fewer C‐cells that express lower levels of GLP‐1Rs and do not release calcitonin (a biomarker for human MTC) in response to GLP‐1 36. In accordance with these observations, a 2‐year clinical study could not demonstrate plasma calcitonin increases in patients treated with liraglutide 36, even in high doses used to treat obesity 75. Despite the FDA concluding that there is a low risk of carcinoma development in humans, liraglutide remains contraindicated in patients with a history of MTC. Epidemiological and animal studies are currently underway to explore possible associations between liraglutide and MTC and liraglutide clinical trials [NCT01179048 (LEADER)] are monitoring calcitonin levels and analysing the incidence of MTC. Patients at elevated risk of MTC (familial MTC, multiple endocrine neoplasia syndrome 2) should not be treated with incretin‐based drugs. The same reasoning applies to other GLP‐1RAs 76.
Head‐to‐head Comparisons Between GLP‐1RAs and DPP‐4 Inhibitors
Comparative clinical trials have highlighted important differences between DPP‐4 inhibitors and GLP‐1RAs in patients with T2D.
Glycaemic Effects
Head‐to‐head studies between sitagliptin and GLP‐1RAs (liraglutide, exenatide twice daily and once weekly) have been performed in patients with T2D as an add‐on to metformin 77, 78, 79. Compared with sitagliptin, liraglutide treatment (1.2 or 1.8 mg) produces significantly greater reductions from baseline in HbA1c [−1.29% (1.2 mg) and −1.51% (1.8 mg) vs −0.88% (sitagliptin)] and FPG [−1.71 mmol/l (1.2 mg) and −2.04 mmol/l (1.8 mg) vs −0.59 mmol/l (sitagliptin); p < 0.0001 all comparisons] 79. Significant PPG reductions from baseline were noted for exenatide twice daily [6.2 ± 0.3 mmol/l vs 2.1 ± 0.3 mmol/l (sitagliptin); p < 0.0001] 78.
Exenatide once weekly significantly reduced HbA1c (−1.5% vs −0.9%; p < 0.0001) and FPG (−1.8% vs −0.9 mmol/l; p < 0.0001) values from baseline compared with sitagliptin, and significantly greater numbers of patients receiving exenatide once weekly achieved an FPG target of 7 mmol/l (60% vs 35%; p < 0.0001) 77. Dulaglutide treatment was also found to be super ior in lowering HbA1c compared with sitagliptin (–1.10 and –0.87% with dulaglutide 1.5 and 0.75 mg, respectively, vs –0.39% with sitagliptin 100 mg; p < 0.001 for both doses vs sitagliptin) 80. Furthermore, albiglutide produced superior reductions in HbA1c versus sitagliptin: (model‐adjusted mean difference −0.4%; p = 0.0001) 81. These trials clearly indicate that GLP‐1RAs are more effective at controlling glycaemia than are DPP‐4 inhibitors, potentially because of the differences in GLP‐1/GLP‐1RA concentrations. Nevertheless, DPP‐4 inhibitors do promote meaningful reductions in plasma glucose.
This relatively strong clinical effectiveness of DPP‐4 inhibitors relative to the GLP‐1 plasma concentrations achieved may be related to the differential distribution patterns of activated GLP‐1Rs by DPP‐4 inhibitors compared with that induced by GLP‐1RAs. Exogenously administered GLP‐1RAs injected into subcutaneous adipose tissue interact mainly with GLP‐1Rs in the systemic circulation; however, native GLP‐1 released from the gut can stimulate GLP‐1Rs in the gut 82 and furthermore, GLP‐1 not degraded by local DPP‐4 can interact with hepato‐portal GLP‐1Rs 82, 83. Recent studies show that activation of hepato‐portal GLP‐1Rs and, consequently, vagal afferents of the parasympathetic nervous system can lead to increases in glucose‐stimulated insulin secretion 83.
Glucose‐lowering by DPP‐4 inhibitors may therefore be mediated via portal vein GLP‐1R signalling and GLP‐1 sensing targeting neural pathways, and may explain the relatively prominent effects relative to the GLP‐1 plasma levels that DPP‐4 inhibitors induce in terms of reductions in plasma glucose concentrations compared with those achieved when employing GLP‐1RAs. This is shown in Figure 4.
Figure 4.
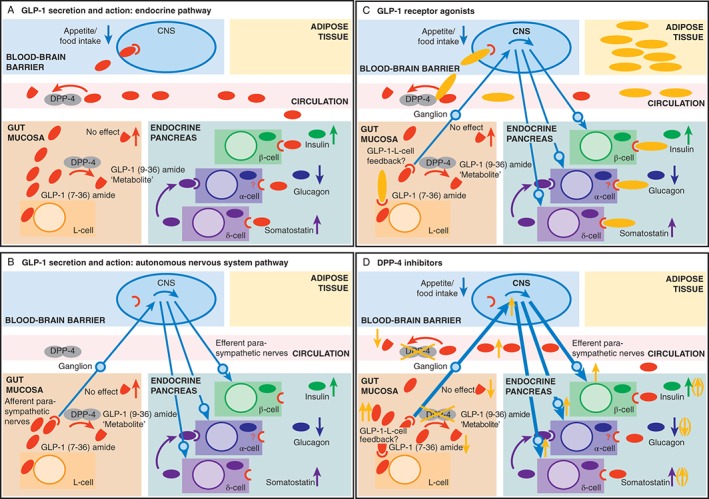
Illustrations of the endocrine (A) and nerve‐stimulating (B) elements of the mode of action of glucagon‐like peptide‐1 (GLP‐1) and of the predominant modes of action of GLP‐1 receptor agonists (GLP‐1RAs) (C) and DPP‐4 inhibitors (D). (A) GLP‐1 (depicted by red ovals) is released from L‐cells in the gut mucosa, and is partially degraded and inactivated by DPP‐4 in the vicinity of L‐cells in the gut mucosa and other compartments (circulatory system and other tissues). GLP‐1 surviving in its intact, biologically active form reaches target cells expressing the GLP‐1 receptor (GLP‐1R) via the bloodstream. (B) Afferent vagal nerve endings with GLP‐1Rs respond to GLP‐1 immediately after its release from L‐cells. The signal reaches the brain via ganglia belonging to the parasympathetic nervous system. The brain then sends efferent impulses to target organs for GLP‐1 activity, such as the endocrine pancreas, where insulin secretion is stimulated and glucagon secretion is suppressed. The endocrine and nerve‐stimulating elements of the mode of action of GLP‐1 co‐exist and may vary in their relative importance. (C) GLP‐1RAs (yellow ovals) are injected into the adipose tissue compartment and, from there, mainly reach target cells via the general circulation. (D) For DPP‐4 inhibitors, a substantial proportion of the effects may be mediated through enhanced interactions of GLP‐1 maintained in its intact, biologically active form, with receptors on afferent vagal fibres (i.e. the neural pathway).
Non‐glycaemic Effects
Both GLP‐1RAs and DPP‐4 inhibitors also lead to various non‐glycaemic effects, including weight decreases (GLP‐1RAs only), improvements in β‐cell function, and improvements in systolic blood pressure and cardiovascular risk markers, such as triglycerides and total cholesterol.
In all direct comparisons with sitagliptin, GLP‐1RAs resulted in significantly greater reductions in body weight 77, 78, 84, 85. Exenatide twice daily reduced weight by −0.8 kg [vs −0.3 kg (sitagliptin); p = 0.0056] 78, whereas greater reductions were observed with exenatide once weekly, in drug‐naive patients [−2.0 kg vs −0.8 kg (sitagliptin); p < 0.001] 85 and those treated with metformin (−2.3 kg vs −1.5 kg [sitagliptin]; p = 0.0002) 77. In a 52‐week extension trial, weight reductions of 2.78 and 3.68 kg were observed in patients receiving liraglutide (1.2 and 1.8 mg, respectively), compared with a reduction of 1.16 kg in the sitagliptin group (p < 0.0001 vs both liraglutide doses) 84.
Exenatide once weekly and liraglutide were associated with improvements in β‐cell function as determined by homeostatic model assessment, the insulinogenic index or insulin secretion rate 84, 85, 86. These measurements were made while the patients were still exposed to the GLP‐1RAs, and thus were most likely attributable to an acute stimulation of insulin secretion by the drug. In separate studies, exenatide once weekly, liraglutide and dulaglutide produced more favourable improvements in β‐cell function versus sitagliptin 79, 84, 85.
With regard to cardiovascular risk markers, exenatide twice daily produced greater reductions in postprandial triglycerides compared with sitagliptin (mean ratio exenatide to sitagliptin, 0.90 ± 0.04; p = 0.0118) 78. In patients switching from sitagliptin to exenatide once weekly, significant (p < 0.05) systolic blood pressure (−2.7 mmHg) and total cholesterol (−0.26 mmol/l) reductions were observed 87. No significant improvements in cardiovascular risk markers (systolic blood pressure and lipid profiles) were reported in a 52‐week sitagliptin and liraglutide trial extension 84. Treatment satisfaction (potentially driven by weight reductions) was greater for liraglutide, which is noteworthy given that liraglutide is injected whereas sitagliptin is taken orally 88. This observation may prove important given that treatment satisfaction correlates with adherence, which, in turn, can lead to improved clinical outcomes 84.
Safety and Tolerability
Mild‐to‐moderate gastrointestinal AEs were common in these head‐to‐head trials, and their incidence (particularly nausea) was higher with GLP‐1RAs compared with sitagliptin 77, 78, 79, 84. This is most likely attributable to the pharmacological vs relatively physiological levels of GLP‐1 activity. Nausea was again transient and declined with treatment duration, such that after 26 weeks of liraglutide vs sitagliptin, the proportions of subjects experiencing nausea were similar 79.
Implications for Treatment Choice
GLP‐1RAs and DPP‐4 inhibitors are becoming established therapies for T2D, used alone or in combination with other glucose‐lowering agents, and are recognized as a vital component of ongoing therapy by the American and European Diabetes Associations 89, 90. Improvements in β‐cell function, weight and markers of cardiovascular risk, not generally seen with other glucose‐lowering treatments, provide hope that incretin‐based therapies may slow down disease progression and address some of the common comorbidities of T2D.
The differing modes of actions of these therapies produce varied efficacy and tolerability profiles and additional treatment benefits beyond glycaemic control. This provides an opportunity to tailor treatment plans to the requirements of each patient, thereby facilitating treatment individualization. Table 1 provides an overview of situations where incretin therapies may find clinical utility 8, 27, 29, 42, 89, 90, 91, 92, 93. The table outlines situations, in the author's estimation, where incretin therapies may be the preferred option ahead of other glucose‐lowering agents, as well as under what circumstances the choice of agent should be a GLP‐1RA or a DPP‐4 inhibitor. Furthermore instances have been included where specific incretin therapies are contraindicated.
Table 1.
Patient characteristics influencing preferred use of dipeptidyl peptidase‐4 inhibitors or glucagon‐like peptide‐1 receptor agonists.
| Patient characteristics | DPP‐4 inhibitor therapy | GLP‐1RA therapy | Therapy with other antidiabetic drugs (as specified) |
|---|---|---|---|
| Glycaemic‐related | |||
| HbA1c | 0.5–1.0% above target* | 1.0–1.5% above target | >1.5% above target |
| Unsuitable: DPP‐4 inhibitor | |||
| Possible: GLP‐1RA | |||
| Preferred: insulin | |||
| FPG | 0–1.7 mmol/l (0–30 mg/dl) above target 42 | 1.1–3.9 mmol/l (20–70 mg/dl) above target | >3.9 mmol/l (70 mg/dl) above target |
| Liraglutide > exenatide 91 | Preferred: insulin 92 | ||
| Unsuitable: DPP‐4 inhibitor or GLP‐1RA 8 | |||
| PPG | Δ above preprandial >3.3 mmol/l (60 mg/dl) (±elevated FBG) 42 | Δ above preprandial >3.3–5.6 (60–100 mg/dl) | Δ above preprandial >5.6 mmol/l (100 mg/dl) insulin – prandial or mealtime 90, 92 |
| Exenatide > liraglutide 91 | |||
| Necessity to avoid hypoglycaemia* | Preferred | Preferred | Unsuitable: insulin or SUs |
| Non‐glycaemic‐related | |||
| Necessity to reduce body mass index | Preferred to insulin | Preferred to DPP‐4 inhibitor | Possible: metformin, pramlintide |
| Unsuitable: insulin, SUs and glitazones | |||
| Preference for oral treatment/injection phobia | Preferred over GLP‐1RAs and insulin | Not suitable | Suitable: SUs, if DPP‐4 inhibitors contraindicated |
| Inability or unwillingness for blood glucose self‐monitoring* | — | Preferred over insulin | Unsuitable: insulin |
| Sensitivity to gastrointestinal events | Suitable | Not suitable | Unsuitable: metformin, acarbose |
| Poor compliance* | Possible: (neutral) | Preferred: long‐acting GLP‐1RA | Unsuitable: insulin |
| Comorbidities | |||
| Renal insufficiency | Preferred: linagliptin | Preferred: liraglutide | Unsuitable: metformin (lactic acidosis) 90; sulphonylureas (hypoglycaemia) |
| Dose adjustment required for other DPP‐4 inhibitors 27, 29 | Possible: exenatide (mild/moderate renal impairment); exenatide once weekly (mild renal impairment) | ||
| Liver disease† | Suitable (except saxagliptin) | Suitable: (unlimited data) | Preferred: insulin |
| Unsuitable: secretagogues (severe hepatic disease) 90 | |||
| Cardiovascular disease | Preferred to insulin | Preferred | Preferred: metformin, acarbose |
| Unsuitable: intensive insulin therapy, SUs90 | |||
| Economics | |||
| Cost | Preferred over GLP‐1RA | More costly than DPP‐4 inhibitors, similar to insulin treatment (including blood glucose self‐monitoring) | Preferred: metformin, SUs |
Treatment choice should be in line with primary treatment goal of achieving glycaemic control and as an adjunct to lifestyle interventions, but patients' preferences and various patient, disease and drug characteristics 90 (such as susceptibility to side effects, potential for weight gain and hypoglycaemia) should be considered, where no preference for DPP‐4 inhibitor/GLP‐1RA is apparent. DPP‐4, dipeptidyl peptidase‐4; FBG, fasting blood glucose; FPG, fasting plasma glucose; GLP‐1RAs, glucagon‐like peptide‐1 receptor agonists; OADs, oral antidiabetic drugs; PPG, postprandial glucose; SU, sulphonylurea.
Targets refer to those established for individual patients. The American Association of Clinical Endocrinologists and American College of Endocrinology 93, and Amercian Diabetes Association/European Association for the Study of Diabetes 89, 90 provide standard recommendations on glycaemic targets. In some patients, the optimum treatment may be the result of a compromise between what is desirable and what can be realistically achieved. In these cases, the suggested glycaemic ranges may not fully apply.
Incretin therapy suitable unless history of pancreatitis 91.
Conclusions
The GLP‐1RAs and DPP‐4 inhibitors add vital new tools to the physician's armoury in the fight against T2D. Generally, their benefit‐to‐risk profile is favourable and their additional benefits distinguish these two classes of glucose‐lowering treatments apart from traditional agents.
The varied pharmacological action and kinetic profiles across the incretin therapies (between classes and among agents of the same class) offer physicians and patients alike a wide variety of treatment options. These options may simply improve treatment flexibility and/or convenience (oral vs injection), whereas in other patient populations incretin therapies may provide vital treatment solutions (patients with elevated BMI or renal impairment). Physicians can therefore use incretin therapies to manage patient care effectively and provide tailored treatment regimens that may promote increased adherence to therapy, improve outcomes and potentially slow down disease progression. The full potential of the incretin therapies, however, may not yet have been appreciated. GLP‐1Rs are widely distributed and DPP‐4 participates in numerous different biological processes. Consequently, the actions of GLP‐1RAs and DPP‐4 inhibitors extend beyond glycaemic control (Tables S2 and S3) and can result in additional beneficial effects, such as weight reductions and improvements in β‐cell function and cardiovascular risk markers. Similarly, incretin therapies also have the potential to affect many crucial biological pathways adversely, potentially resulting in unwanted extra‐glycaemic effects. It is therefore important to continue characterization of the incretin system and incretin‐based therapies so that the full potential of these agents can be realized and their involvement in biological processes unrelated to glycaemic control can be fully understood. Pertinent cardiovascular outcome studies are underway with all agents.
Conflict of Interest
M. N. has served on the advisory boards for Berlin Chemie AG, Amylin Pharmaceuticals Inc, Boehringer Ingelheim, Eli Lilly & Co, GlaxoSmithKline, Hoffmann‐La Roche, MSD Sharp & Dohme GmbH, Merck Sharp & Dohme Ltd, Novo Nordisk Pharma GmbH, Sanofi‐Aventis Deutschland GmbH, Versatis Inc and Intarcia Therapeutics Inc., has provided consultancy for Amylin Pharmaceuticals, AstraZeneca, Berlin Chemie, Boehringer Ingelheim, Diartis Pharmaceuticals Inc, Eli Lilly & Co, GlaxoSmithKline, Hoffmann‐La Roche, Intarcia Therapeutics Inc, MSD Sharp & Dohme GmbH, Novartis Pharmaceuticals Corp., Janssen Global Services, Novo Nordisk A/S, Sanofi‐Aventis Pharma, Versatis; grant/research support received from Boehringer Ingelheim, Novartis Pharma, MSD Sharp & Dohme GmbH, Metacure, AstraZeneca, GlaxoSmithKline, Roche Pharma AG, Novo Nordisk Pharma GmbH and ToleRx, and has served on the speaker's bureau for AstraZeneca, Berlin Chemie AG, Boehringer Ingelheim, Lilly Deutschland GmbH, MSD Sharp & Dohme GmbH, Novartis Pharma, Novo Nordisk A/S, Roche Pharma. Other relationships with Diabate/Boehringer Ingelheim, MSD Sharp & Dohme GmbH, Incretin Expert Program/Lilly Deutschland GmbH and Medscape LLC are also disclosed.
Supporting information
Table S1. The main pharmacokinetic parameters of the incretin therapies.
Table S2. Clinical effects of incretin therapies compared with non‐pancreatic physiological effects of native glucagon‐like peptide‐1.
Table S3. Clinical effects of incretin therapies compared with pancreatic physiological effects of native glucagon‐like peptide‐1.
Acknowledgements
The author is grateful Watermeadow Medical for writing and editing assistance in the development of this manuscript. This assistance was funded by Novo Nordisk, which also had a role in the review of the manuscript for scientific accuracy.
References
- 1. Defronzo RA. Banting Lecture. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus. Diabetes 2009; 58: 773–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lorber D. Importance of cardiovascular disease risk management in patients with type 2 diabetes mellitus. Diabetes Metab Syndr Obes 2014; 7: 169–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ross SA. Breaking down patient and physician barriers to optimize glycemic control in type 2 diabetes. Am J Med 2013; 126: S38–S48. [DOI] [PubMed] [Google Scholar]
- 4. Nauck MA. Unraveling the science of incretin biology. Eur J Intern Med 2009; 20(Suppl. 2): S303–S308. [DOI] [PubMed] [Google Scholar]
- 5. Nauck M, Stockmann F, Ebert R, Creutzfeldt W. Reduced incretin effect in type 2 (non‐insulin‐dependent) diabetes. Diabetologia 1986; 29: 46–52. [DOI] [PubMed] [Google Scholar]
- 6. Nauck MA, Homberger E, Siegel EG et al. Incretin effects of increasing glucose loads in man calculated from venous insulin and C‐peptide responses. J Clin Endocrinol Metab 1986; 63: 492–498. [DOI] [PubMed] [Google Scholar]
- 7. Nauck MA, Heimesaat MM, Orskov C, Holst JJ, Ebert R, Creutzfeldt W. Preserved incretin activity of glucagon‐like peptide 1 [7‐36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type‐2 diabetes mellitus. J Clin Invest 1993; 91: 301–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Drucker DJ, Nauck MA. The incretin system: glucagon‐like peptide‐1 receptor agonists and dipeptidyl peptidase‐4 inhibitors in type 2 diabetes. Lancet 2006; 368: 1696–1705. [DOI] [PubMed] [Google Scholar]
- 9. Kim NH, Yu T, Lee DH. The nonglycemic actions of dipeptidyl peptidase‐4 inhibitors. Biomed Res Int 2014; 2014: 368703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nauck MA. Incretin‐based therapies for type 2 diabetes mellitus: properties, functions, and clinical implications. Am J Med 2011; 124: S3–S18. [DOI] [PubMed] [Google Scholar]
- 11. Holst JJ, Vilsboll T, Deacon CF. The incretin system and its role in type 2 diabetes mellitus. Mol Cell Endocrinol 2009; 297: 127–136. [DOI] [PubMed] [Google Scholar]
- 12. Calanna S, Christensen M, Holst JJ et al. Secretion of glucose‐dependent insulinotropic polypeptide in patients with type 2 diabetes: systematic review and meta‐analysis of clinical studies. Diabetes Care 2013; 36: 3346–3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hojberg PV, Vilsboll T, Rabol R et al. Four weeks of near‐normalisation of blood glucose improves the insulin response to glucagon‐like peptide‐1 and glucose‐dependent insulinotropic polypeptide in patients with type 2 diabetes. Diabetologia 2009; 52: 199–207. [DOI] [PubMed] [Google Scholar]
- 14. Vilsboll T, Krarup T, Madsbad S, Holst JJ. Defective amplification of the late phase insulin response to glucose by GIP in obese Type II diabetic patients. Diabetologia 2002; 45: 1111–1119. [DOI] [PubMed] [Google Scholar]
- 15. Mentis N, Vardarli I, Kothe LD et al. GIP does not potentiate the antidiabetic effects of GLP‐1 in hyperglycemic patients with type 2 diabetes. Diabetes 2011; 60: 1270–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kim W, Egan JM. The role of incretins in glucose homeostasis and diabetes treatment. Pharmacol Rev 2008; 60: 470–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nauck MA, Kleine N, Orskov C, Holst JJ, Willms B, Creutzfeldt W. Normalization of fasting hyperglycaemia by exogenous glucagon‐like peptide 1 (7‐36 amide) in type 2 (non‐insulin‐dependent) diabetic patients. Diabetologia 1993; 36: 741–744. [DOI] [PubMed] [Google Scholar]
- 18. Nauck MA, Meier JJ. Glucagon‐like peptide 1 and its derivatives in the treatment of diabetes. Regul Pept 2005; 128: 135–148. [DOI] [PubMed] [Google Scholar]
- 19. Taborsky GJ Jr. The physiology of glucagon. J Diabetes Sci Technol 2010; 4: 1338–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gerich JE. Abnormal glucagon secretion in type 2 (noninsulin‐dependent) diabetes mellitus: causes and consequences In: Creutzfeldt W, Lefèbvre P, eds. Diabetes Mellitus: Pathophysiology and Therapy. Berlin, Heidelberg: Springer‐Verlag, 1989; 127–133. [Google Scholar]
- 21. Menge BA, Gruber L, Jorgensen SM et al. Loss of inverse relationship between pulsatile insulin and glucagon secretion in patients with type 2 diabetes. Diabetes 2011; 60: 2160–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Meier JJ, Deacon CF, Schmidt WE, Holst JJ, Nauck MA. Suppression of glucagon secretion is lower after oral glucose administration than during intravenous glucose administration in human subjects. Diabetologia 2007; 50: 806–813. [DOI] [PubMed] [Google Scholar]
- 23. Meier JJ, Gallwitz B, Siepmann N et al. Gastric inhibitory polypeptide (GIP) dose‐dependently stimulates glucagon secretion in healthy human subjects at euglycaemia. Diabetologia 2003; 46: 798–801. [DOI] [PubMed] [Google Scholar]
- 24. Kiec‐Klimczak ME, Pach DM, Pogwizd ME, Hubalewska‐Dydejczyk AB. Incretins yesterday, pleiotropic gastrointestinal hormones today:glucagon‐like peptide‐1 (GLP‐1) and glucose‐dependent insulinotropic polypeptide (GIP). Recent Patents Endocr Metab Immune Drug Discov 2011; 5: 176–182. [DOI] [PubMed] [Google Scholar]
- 25. de Heer J, Rasmussen C, Coy DH, Holst JJ. Glucagon‐like peptide‐1, but not glucose‐dependent insulinotropic peptide, inhibits glucagon secretion via somatostatin (receptor subtype 2) in the perfused rat pancreas. Diabetologia 2008; 51: 2263–2270. [DOI] [PubMed] [Google Scholar]
- 26. Hare KJ, Vilsboll T, Asmar M, Deacon CF, Knop FK, Holst JJ. The glucagonostatic and insulinotropic effects of glucagon‐like peptide 1 contribute equally to its glucose‐lowering action. Diabetes 2010; 59: 1765–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Deacon CF. Dipeptidyl peptidase‐4 inhibitors in the treatment of type 2 diabetes: a comparative review. Diabetes Obes Metab 2011; 13: 7–18. [DOI] [PubMed] [Google Scholar]
- 28. Deacon CF, Hughes TE, Holst JJ. Dipeptidyl peptidase IV inhibition potentiates the insulinotropic effect of glucagon‐like peptide 1 in the anesthetized pig. Diabetes 1998; 47: 764–769. [DOI] [PubMed] [Google Scholar]
- 29. Scheen AJ. Pharmacokinetics of dipeptidylpeptidase‐4 inhibitors. Diabetes Obes Metab 2010; 12: 648–658. [DOI] [PubMed] [Google Scholar]
- 30. Deacon CF, Knudsen LB, Madsen K, Wiberg FC, Jacobsen O, Holst JJ. Dipeptidyl peptidase IV resistant analogues of glucagon‐like peptide‐1 which have extended metabolic stability and improved biological activity. Diabetologia 1998; 41: 271–278. [DOI] [PubMed] [Google Scholar]
- 31. Russell S. Incretin‐based therapies for type 2 diabetes mellitus: a review of direct comparisons of efficacy, safety and patient satisfaction. Int J Clin Pharm 2013; 35: 159–172. [DOI] [PubMed] [Google Scholar]
- 32. Eng J, Kleinman WA, Singh L, Singh G, Raufman JP. Isolation and characterization of exendin‐4, an exendin‐3 analogue, from Heloderma suspectum venom. Further evidence for an exendin receptor on dispersed acini from guinea pig pancreas. J Biol Chem 1992; 267: 7402–7405. [PubMed] [Google Scholar]
- 33. Nauck M, Smith U. Incretin‐based therapy: how do incretin mimetics and DPP‐4 inhibitors fit into treatment algorithms for type 2 diabetic patients? Best Pract Res Clin Endocrinol Metab 2009; 23: 513–523. [DOI] [PubMed] [Google Scholar]
- 34. Tella SH, Rendell MS. Glucagon‐like polypeptide agonists in type 2 diabetes mellitus: efficacy and tolerability, a balance. Ther Adv Endocrinol Metab 2015; 6: 109–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Horowitz M, Rayner CK, Jones KL. Mechanisms and clinical efficacy of lixisenatide for the management of type 2 diabetes. Adv Ther 2013; 30: 81–101. [DOI] [PubMed] [Google Scholar]
- 36. Bjerre Knudsen L, Madsen LW, Andersen S et al. Glucagon‐like Peptide‐1 receptor agonists activate rodent thyroid C‐cells causing calcitonin release and C‐cell proliferation. Endocrinology 2010; 151: 1473–1486. [DOI] [PubMed] [Google Scholar]
- 37. Garber AJ. Long‐acting glucagon‐like peptide 1 receptor agonists: a review of their efficacy and tolerability. Diabetes Care 2011; 34(Suppl. 2): S279–S284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fineman M, Flanagan S, Taylor K et al. Pharmacokinetics and pharmacodynamics of exenatide extended‐release after single and multiple dosing. Clin Pharmacokinet 2011; 50: 65–74. [DOI] [PubMed] [Google Scholar]
- 39. Rosenstock J, Reusch J, Bush M, Yang F, Stewart M, Albiglutide SG. Potential of albiglutide, a long‐acting GLP‐1 receptor agonist, in type 2 diabetes: a randomized controlled trial exploring weekly, biweekly, and monthly dosing. Diabetes Care 2009; 32: 1880–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nauck MA, Petrie JR, Sesti G et al. A phase 2, randomized, dose‐finding study of the novel once‐weekly human GLP‐1 analog, semaglutide, compared with placebo and open‐label liraglutide in patients with type 2 diabetes. Diabetes Care 2015: dc150165. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 41. Novo Nordisk A/S. Xultophy (insulin degludec/liraglutide) summary of product characteristics. 2015. Available from URL: https://www.medicines.org.uk/emc/medicine/29493. Accessed 21 July 2015.
- 42. Ahren B, Landin‐Olsson M, Jansson PA, Svensson M, Holmes D, Schweizer A. Inhibition of dipeptidyl peptidase‐4 reduces glycemia, sustains insulin levels, and reduces glucagon levels in type 2 diabetes. J Clin Endocrinol Metab 2004; 89: 2078–2084. [DOI] [PubMed] [Google Scholar]
- 43. Herman GA, Bergman A, Stevens C et al. Effect of single oral doses of sitagliptin, a dipeptidyl peptidase‐4 inhibitor, on incretin and plasma glucose levels after an oral glucose tolerance test in patients with type 2 diabetes. J Clin Endocrinol Metab 2006; 91: 4612–4619. [DOI] [PubMed] [Google Scholar]
- 44. Forst T, Pfutzner A. Linagliptin, a dipeptidyl peptidase‐4 inhibitor with a unique pharmacological profile, and efficacy in a broad range of patients with type 2 diabetes. Expert Opin Pharmacother 2012; 13: 101–110. [DOI] [PubMed] [Google Scholar]
- 45. Craddy P, Palin HJ, Johnson KI. Comparative effectiveness of dipeptidylpeptidase‐4 inhibitors in type 2 diabetes: a systematic review and mixed treatment comparison. Diabetes Ther 2014; 5: 1–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Goossen K, Graber S. Longer term safety of dipeptidyl peptidase‐4 inhibitors in patients with type 2 diabetes mellitus: systematic review and meta‐analysis. Diabetes Obes Metab 2012; 14: 1061–1072. [DOI] [PubMed] [Google Scholar]
- 47. Willemen MJ, Mantel‐Teeuwisse AK, Straus SM, Meyboom RH, Egberts TC, Leufkens HG. Use of dipeptidyl peptidase‐4 inhibitors and the reporting of infections: a disproportionality analysis in the World Health Organization VigiBase. Diabetes Care 2011; 34: 369–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ligueros‐Saylan M, Foley JE, Schweizer A, Couturier A, Kothny W. An assessment of adverse effects of vildagliptin versus comparators on the liver, the pancreas, the immune system, the skin and in patients with impaired renal function from a large pooled database of Phase II and III clinical trials. Diabetes Obes Metab 2010; 12: 495–509. [DOI] [PubMed] [Google Scholar]
- 49. Williams‐Herman D, Engel SS, Round E et al. Safety and tolerability of sitagliptin in clinical studies: a pooled analysis of data from 10,246 patients with type 2 diabetes. BMC Endocr Disord 2010; 10: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Elashoff M, Matveyenko AV, Gier B, Elashoff R, Butler PC. Pancreatitis, pancreatic, and thyroid cancer with glucagon‐like peptide‐1‐based therapies. Gastroenterology 2011; 141: 150–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nauck MA, Friedrich N. Do GLP‐1‐based therapies increase cancer risk? Diabetes Care 2013; 36: S245–252; DOI: 10.2337/dcS13–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nauck MA. A critical analysis of the clinical use of incretin‐based therapies: the benefits by far outweigh the potential risks. Diabetes Care 2013; 36: 2126–2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Egan AG, Blind E, Dunder K et al. Pancreatic safety of incretin‐based drugs–FDA and EMA assessment. N Engl J Med 2014; 370: 794–797. [DOI] [PubMed] [Google Scholar]
- 54. Green JB, Bethel MA, Armstrong PW et al. Effect of sitagliptin on cardiovascular outcomes in type 2 diabetes. N Engl J Med 2015; 373: 232–242. [DOI] [PubMed] [Google Scholar]
- 55. Sanofi. Lyxumia (lixisenatide) summary of product characteristics. 2014. Available from URL: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Product_Information/human/002445/WC500140401.pdf. Accessed 25 June 2015.
- 56. Drucker DJ, Buse JB, Taylor K et al. Exenatide once weekly versus twice daily for the treatment of type 2 diabetes: a randomised, open‐label, non‐inferiority study. Lancet 2008; 372: 1240–1250. [DOI] [PubMed] [Google Scholar]
- 57. GlaxoSmithKline. Eperzan (albiglutide) summary of product characteristics. 2015. Available from URL: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Product_Information/human/002735/WC500165117.pdf. Accessed 25 June 2015.
- 58. Lilly. Trulicity (dulaglutide) summary of product characteristics. 2014. Available from URL: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Product_Information/human/002825/WC500179470.pdf. Accessed 25 June 2015.
- 59. Degn KB, Juhl CB, Sturis J et al. One week's treatment with the long‐acting glucagon‐like peptide 1 derivative liraglutide (NN2211) markedly improves 24‐h glycemia and alpha‐ and beta‐cell function and reduces endogenous glucose release in patients with type 2 diabetes. Diabetes 2004; 53: 1187–1194. [DOI] [PubMed] [Google Scholar]
- 60. Mari A, Sallas WM, He YL et al. Vildagliptin, a dipeptidyl peptidase‐IV inhibitor, improves model‐assessed beta‐cell function in patients with type 2 diabetes. J Clin Endocrinol Metab 2005; 90: 4888–4894. [DOI] [PubMed] [Google Scholar]
- 61. Neumiller JJ. Differential chemistry (structure), mechanism of action, and pharmacology of GLP‐1 receptor agonists and DPP‐4 inhibitors. J Am Pharm Assoc (2003) 2009; 49(Suppl. 1): S16–29. [DOI] [PubMed] [Google Scholar]
- 62. Flint A, Nazzal K, Jagielski P, Hindsberger C, Zdravkovic M. Influence of hepatic impairment on pharmacokinetics of the human GLP‐1 analogue, liraglutide. Br J Clin Pharmacol 2010; 70: 807–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Giorda C, Nada E, Tartaglino B. Pharmacokinetics, safety, and efficacy of DPP‐4 inhibitors and GLP‐1 receptor agonists in patients with type 2 diabetes mellitus and renal or hepatic impairment. A systematic review of the literature. Endocrine 2014; 46: 406–419. [DOI] [PubMed] [Google Scholar]
- 64. Shyangdan DS, Royle PL, Clar C, Sharma P, Waugh NR, Snaith A. Glucagon‐like peptide analogues for type 2 diabetes mellitus. Cochrane Database Syst Rev 2011; 5: CD006423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Buse JB, Rosenstock J, Sesti G et al. Liraglutide once a day versus exenatide twice a day for type 2 diabetes: a 26‐week randomised, parallel‐group, multinational, open‐label trial (LEAD‐6). Lancet 2009; 374: 39–47. [DOI] [PubMed] [Google Scholar]
- 66. Buse JB, Nauck M, Forst T et al. Exenatide once weekly versus liraglutide once daily in patients with type 2 diabetes (DURATION‐6): a randomised, open‐label study. Lancet 2013; 381: 117–124. [DOI] [PubMed] [Google Scholar]
- 67. Pratley RE, Nauck MA, Barnett AH et al. Once‐weekly albiglutide versus once‐daily liraglutide in patients with type 2 diabetes inadequately controlled on oral drugs (HARMONY 7): a randomised, open‐label, multicentre, non‐inferiority phase 3 study. Lancet Diabetes Endocrinol 2014; 2: 289–297. [DOI] [PubMed] [Google Scholar]
- 68. Wysham C, Blevins T, Arakaki R et al. Efficacy and safety of dulaglutide added onto pioglitazone and metformin versus exenatide in type 2 diabetes in a randomized controlled trial (AWARD‐1). Erratum in Diabetes Care 2014; 37: 2895. Diabetes Care 2014; 37: 2159–2167. [DOI] [PubMed] [Google Scholar]
- 69. Dungan KM, Povedano ST, Forst T et al. Once‐weekly dulaglutide versus once‐daily liraglutide in metformin‐treated patients with type 2 diabetes (AWARD‐6): a randomised, open‐label, phase 3, non‐inferiority trial. Lancet 2014; 384: 1349–1357. [DOI] [PubMed] [Google Scholar]
- 70. Rosenstock J, Raccah D, Koranyi L et al. Efficacy and safety of lixisenatide once daily versus exenatide twice daily in type 2 diabetes inadequately controlled on metformin: a 24‐week, randomized, open‐label, active‐controlled study (GetGoal‐X). Diabetes Care 2013; 36: 2945–2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Buse JB, Garber A, Rosenstock J et al. Liraglutide treatment is associated with a low frequency and magnitude of antibody formation with no apparent impact on glycemic response or increased frequency of adverse events: results from the Liraglutide Effect and Action in Diabetes (LEAD) trials. J Clin Endocrinol Metab 2011; 96: 1695–1702. [DOI] [PubMed] [Google Scholar]
- 72. FDA . Exenatide (marketed as BYETTA): acute pancreatitis. FDA Drug Safety Newsletter: Postmarketing Reviews – Volume 1, Number 2, Winter 2008. Available from URL: http://www.fda.gov/Drugs/DrugSafety/DrugSafetyNewsletter/ucm119034.htm. Accessed 25 June 2015.
- 73. Jensen TM, Saha K, Steinberg WM. Is there a link between liraglutide and pancreatitis? A post hoc review of pooled and patient‐level data from completed liraglutide type 2 diabetes clinical trials. Diabetes Care 2015; 38: 1058–1066. [DOI] [PubMed] [Google Scholar]
- 74. Noel RA, Braun DK, Patterson RE, Bloomgren GL. Increased risk of acute pancreatitis and biliary disease observed in patients with type 2 diabetes: a retrospective cohort study. Diabetes Care 2009; 32: 834–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hegedus L, Moses AC, Zdravkovic M, Le Thi T, Daniels GH. GLP‐1 and calcitonin concentration in humans: lack of evidence of calcitonin release from sequential screening in over 5000 subjects with type 2 diabetes or nondiabetic obese subjects treated with the human GLP‐1 analog, liraglutide. J Clin Endocrinol Metab 2011; 96: 853–860. [DOI] [PubMed] [Google Scholar]
- 76. Reid T. Choosing GLP‐1 receptor agonists and DPP‐4 inhibitors: weighing the clinical trial evidence. Clin Diabetes 2012; 30: 3–12. [Google Scholar]
- 77. Bergenstal RM, Wysham C, Macconell L et al. Efficacy and safety of exenatide once weekly versus sitagliptin or pioglitazone as an adjunct to metformin for treatment of type 2 diabetes (DURATION‐2): a randomised trial. Lancet 2010; 376: 431–439. [DOI] [PubMed] [Google Scholar]
- 78. DeFronzo RA, Okerson T, Viswanathan P, Guan X, Holcombe JH, MacConell L. Effects of exenatide versus sitagliptin on postprandial glucose, insulin and glucagon secretion, gastric emptying, and caloric intake: a randomized, cross‐over study. Curr Med Res Opin 2008; 24: 2943–2952. [DOI] [PubMed] [Google Scholar]
- 79. Pratley RE, Nauck M, Bailey T et al. Liraglutide versus sitagliptin for patients with type 2 diabetes who did not have adequate glycaemic control with metformin: a 26‐week, randomised, parallel‐group, open‐label trial. Lancet 2010; 375: 1447–1456. [DOI] [PubMed] [Google Scholar]
- 80. Nauck M, Weinstock RS, Umpierrez GE, Guerci B, Skrivanek Z, Milicevic Z. Efficacy and safety of dulaglutide versus sitagliptin after 52 weeks in type 2 diabetes in a randomized controlled trial (AWARD‐5). Erratum in Diabetes Care 2015; 38: 538. Diabetes Care 2014; 37: 2149–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ahren B, Johnson SL, Stewart M et al. HARMONY 3: 104‐week randomized, double‐blind, placebo‐ and active‐controlled trial assessing the efficacy and safety of albiglutide compared with placebo, sitagliptin, and glimepiride in patients with type 2 diabetes taking metformin. Diabetes Care 2014; 37: 2141–2148. [DOI] [PubMed] [Google Scholar]
- 82. Hansen L, Deacon CF, Orskov C, Holst JJ. Glucagon‐like peptide‐1‐(7‐36)amide is transformed to glucagon‐like peptide‐1‐(9‐36)amide by dipeptidyl peptidase IV in the capillaries supplying the L cells of the porcine intestine. Endocrinology 1999; 140: 5356–5363. [DOI] [PubMed] [Google Scholar]
- 83. Vahl TP, Tauchi M, Durler TS et al. Glucagon‐like peptide‐1 (GLP‐1) receptors expressed on nerve terminals in the portal vein mediate the effects of endogenous GLP‐1 on glucose tolerance in rats. Endocrinology 2007; 148: 4965–4973. [DOI] [PubMed] [Google Scholar]
- 84. Pratley R, Nauck M, Bailey T et al. One year of liraglutide treatment offers sustained and more effective glycaemic control and weight reduction compared with sitagliptin, both in combination with metformin, in patients with type 2 diabetes: a randomised, parallel‐group, open‐label trial. Int J Clin Pract 2011; 65: 397–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Russell‐Jones D, Cuddihy RM, Hanefeld M et al. Efficacy and safety of exenatide once weekly versus metformin, pioglitazone, and sitagliptin used as monotherapy in drug‐naive patients with type 2 diabetes (DURATION‐4): a 26‐week double‐blind study. Diabetes Care 2012; 35: 252–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Pratley RE, Nauck MA, Bailey T et al. Efficacy and safety of switching from the DPP‐4 inhibitor sitagliptin to the human GLP‐1 analog liraglutide after 52 weeks in metformin‐treated patients with type 2 diabetes: a randomized, open‐label trial. Diabetes Care 2012; 35: 1986–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Wysham C, Bergenstal R, Malloy J et al. DURATION‐2: efficacy and safety of switching from maximum daily sitagliptin or pioglitazone to once‐weekly exenatide. Diabet Med 2011; 28: 705–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Davies M, Pratley R, Hammer M, Thomsen AB, Cuddihy R. Liraglutide improves treatment satisfaction in people with Type 2 diabetes compared with sitagliptin, each as an add on to metformin. Diabet Med 2011; 28: 333–337. [DOI] [PubMed] [Google Scholar]
- 89. Association AD. Standards of care in diabetes – 2015. J Clin Appl Res Educ 2015; 38: s1–s99. [Google Scholar]
- 90. Inzucchi SE, Bergenstal RM, Buse JB et al. Management of hyperglycemia in type 2 diabetes, 2015: a patient‐centered approach: update to a position statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 2015; 38: 140–149. [DOI] [PubMed] [Google Scholar]
- 91. Diamant M, Van Gaal L, Stranks S et al. Once weekly exenatide compared with insulin glargine titrated to target in patients with type 2 diabetes (DURATION‐3): an open‐label randomised trial. Lancet 2010; 375: 2234–2243. [DOI] [PubMed] [Google Scholar]
- 92. Yki‐Jarvinen H, Ryysy L, Nikkila K, Tulokas T, Vanamo R, Heikkila M. Comparison of bedtime insulin regimens in patients with type 2 diabetes mellitus. A randomized, controlled trial. Ann Intern Med 1999; 130: 389–396. [DOI] [PubMed] [Google Scholar]
- 93. Endocrinology AAoCEaACo . Clinical practice guidelines for developing a diabetes mellitus comprehensive care plan – 2015. Endocr Pract 2015; 21: s1–s87. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. The main pharmacokinetic parameters of the incretin therapies.
Table S2. Clinical effects of incretin therapies compared with non‐pancreatic physiological effects of native glucagon‐like peptide‐1.
Table S3. Clinical effects of incretin therapies compared with pancreatic physiological effects of native glucagon‐like peptide‐1.