

Abstract
Chronic viral infections represent a unique challenge to the infected host. Persistently replicating viruses outcompete or subvert the initial antiviral response, allowing the establishment of chronic infections that result in continuous stimulation of both the innate and adaptive immune compartments. This causes a profound reprogramming of the host immune system, including attenuation and persistent low levels of type I interferons, progressive loss (or exhaustion) of CD8+ T cell functions, and specialization of CD4+ T cells to produce interleukin-21 and promote antibody-mediated immunity and immune regulation. Epigenetic, transcriptional, posttranscriptional, and metabolic changes underlie this adaptation or recalibration of immune cells to the emerging new environment in order to strike an often imperfect balance between the host and the infectious pathogen. In this review we discuss the common immunological hallmarks observed across a range of different persistently replicating viruses and host species, the underlying molecular mechanisms, and the biological and clinical implications.
Keywords: chronic viral infection, interferon, T cells, HIV, HCV, LCMV
INTRODUCTION
Chronic viral infections are a significant global health burden. For example, human immunodeficiency virus types 1 and 2 (HIV-1 and HIV-2) are the causative agents of acquired immune deficiency syndrome (AIDS), which results in an estimated 1.5 million deaths worldwide annually. Hepatitis C virus (HCV) and hepatitis B virus (HBV) are major causes of viral hepatitis. Moreover, HCV is the major risk factor for the development of hepatocellular carcinoma, the fifth most common cancer in the world.
Viruses that cause chronic infections have evolved a range of distinct mechanisms that allow them to be retained within their host. Endogenous retroviruses, for instance, exist in the genome of every member of a host species, are present for the lifetime of the host, and are transmitted vertically from parent to offspring (1). Other strategies utilized by viruses during chronic infections include viral latency and persistent active replication. Herpes simplex virus 1 and 2 (HSV-1 and HSV-2, which cause oral and genital herpes, respectively), Epstein-Barr virus (EBV, which causes mononucleosis and a number of lymphomas), and human and mouse cytomegaloviruses are all examples of latent viruses. Initial infection with these viruses is associated with acute symptoms before the virus enters a period of latency with sporadic reactivation. During these quiescent periods, the viruses enter a transcriptionally silent state in which new infectious virions are not generated, allowing the virus to avoid immune surveillance (reviewed in 2). In contrast, other viruses—such as HIV-1, HCV, and HBV in humans; simian immunodeficiency virus (SIV) in primates; and certain variants of lymphocytic choriomeningitis virus (LCMV) in mice [e.g., Clone 13 (Cl13) and Docile]—display the capacity to continuously replicate and package new viral particles despite the presence of an antiviral immune response. Viral integration, latency, and persistent replication are all effective strategies to establish chronic infection (and indeed some viruses, such as HIV-1, utilize more than one strategy at a time).
Notably, the presence of persistent viral products, a common feature of infections with continuously replicating viruses, causes drastic and sustained alterations in the host immune system. This often results in altered susceptibility to secondary infections, cancers, and inflammatory disorders. Thus, at the risk of neglecting the unique effects of individual viruses on their respective hosts, in this review we discuss selected examples of the overlapping immune hallmarks found in infections with persistently replicating viruses, including LCMV infection in mice; SIV infection in primates; and HIV, HBV, and HCV infections in humans.
INNATE IMMUNITY: DYNAMICS AND OPPOSING EFFECTS OF TYPE I INTERFERONS DURING CHRONIC VIRAL INFECTION
The host innate immune response is a highly potent barrier of defense comprising many innate immune cells, such as dendritic cells (DCs), macrophages, and natural killer (NK) cells. These cells are capable of recognizing a wide range of viral products through a complex and overlapping molecular network of pattern-recognition receptors (PRRs), including Toll-like receptors (TLRs) as well as cytosolic sensors such as the helicases retinoic acid–inducible gene 1 (RIG-I) and MDA5 (3). Activation of innate cells through these PRRs promotes rapid viral sensing and the release of key antiviral cytokines with the potential to rapidly restrain viral replication and drive further innate and adaptive immune responses (3). In particular, the type I interferons (IFN-I) are a pleiotropic cytokine family consisting of thirteen IFNα isoforms and one IFNβ isoform that signal through a shared ubiquitously expressed receptor (IFNαβR) (4). Signaling through IFNαβR leads to both autocrine and paracrine activation, resulting in phosphorylation of signal transducer and activator of transcription 1 and 2 (STAT1 and STAT2). This induces hundreds of IFN-I-stimulated genes (ISGs) that promote an antiviral state and activate multiple immune cells (4). Despite this sophisticated innate response, many viruses, including those that achieve chronic infection, are able to suppress and/or evade these early innate host responses to favor their own replication and transmission. In this section, we focus on IFN-I to illustrate the dynamic changes that the innate immune system undergoes and the significant and broad consequences that such changes can have during chronic viral infections.
Type I Interferon Induction by Persisting Viruses
Within days after HIV detection in plasma from infected patients, and within hours after infection with LCMV Cl13 in mice, elevated levels of systemic IFN-I are detected (5, 6). Furthermore, enhanced ISG expression is evident in tissues and/or T cells after LCMV Cl13, HCV, and SIV infections (Figure 1) (7–10). In contrast, genes with innate immune function are undetectable early after in vivo HBV infection in chimpanzees (11).
Figure 1.
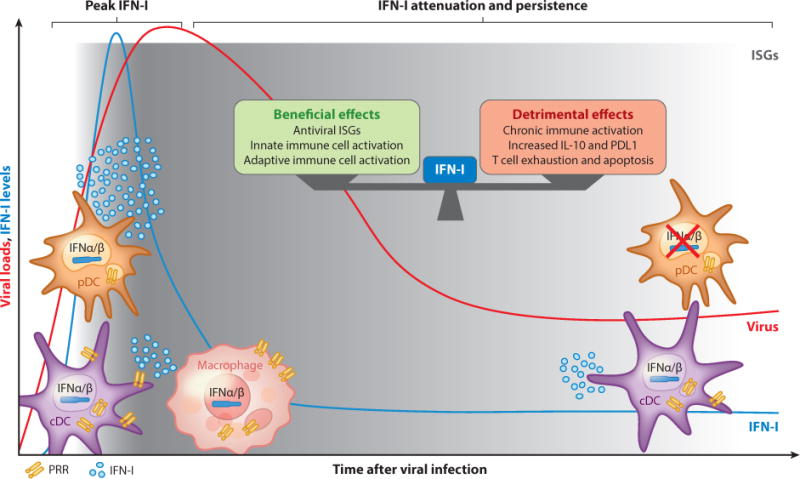
Type I interferon (IFN-I) dynamics and opposing effects during chronic viral infections. Upon infection with most persistent viruses, IFN-I are powerfully and systemically induced; this initial response is rapidly attenuated, although low IFN-I and IFN-I-stimulated gene (ISG) levels still persist in multiple cells and tissues. Several innate cells, including macrophages, conventional dendritic cells (cDCs), and plasmacytoid dendritic cells (pDCs) (and possibly nonhematopoietic cells, which are not depicted), sense viral products via pattern-recognition receptors (PRRs) to contribute to IFN-I levels. Although they are the most powerful IFN-I producers early after challenge with persistent viruses, pDCs show an impaired capacity to produce IFN-I during chronic stages of lymphocytic choriomeningitis virus (LCMV), human immunodeficiency virus (HIV), hepatitis C virus (HCV), and hepatitis B virus (HBV) infections. IFN-I exert both beneficial and detrimental effects on immune responses and viral control, which appear conserved among different persistent infections. The relative degree and impact on viral replication of such opposing IFN-I effects may vary for individual cases.
Several nonhematopoietic and hematopoietic cells, including macrophages, conventional DCs (cDCs), and plasmacytoid DCs (pDCs), can contribute to the initially robust IFN-I response by recognizing and responding to viral infection through PRR-mediated signaling pathways. For example, in LCMV Cl13 infection, cDCs and macrophages produce IFN-I through MDA5/MAVS-dependent pathways (8, 12–14). Although the cellular source and the specific type of IFN (e.g., IFN-I and/or IFN-III) that induces elevated ISGs in the liver upon HCV infection are still unclear, studies indicate that hepatocytes are capable of producing IFN-I via RIG-I signaling in response to HCV infection (15). Finally, monocyte-derived DCs can sense HIV-2 (and HIV-1 when its replication is not restricted by the host enzyme SAMHD1) via cyclic guanosine monophosphate–adenosine monophosphate synthase (cGAS) pathways, inducing proinflammatory cytokines and IFN-I production (reviewed in 16).
Despite the fact that almost all cell types can produce IFN-I upon viral encounter, pDCs are highly specialized for secreting these cytokines, producing up to 1,000 times more IFN-I than any other blood cell type and synthesizing a broader range of IFN-I isoforms upon TLR stimulation (17). pDCs are able to directly recognize several persistent viruses, including HIV and HCV in humans and LCMV in mice, through TLR7 (18–20). pDCs have been shown to be essential for T cell responses and viral control during chronic LCMV infection (21), although another study using a different LCMV variant and an inducible pDC deletion system observed only minimal effects (14). Notably, in contrast to IFN-I production in most cell types, pDC IFN-I production does not require viral replication, and the majority of DC IFN-I comes from uninfected pDCs early after LCMV Cl13 infection (19). Similarly, studies with HCV found that uninfected pDCs are able to sense neighboring infected hepatocytes by recognizing HCV RNA–containing exosomes (20, 22), and neither viral fusion nor replication is required for pDC recognition of HIV (16).
In conclusion, these studies indicate that the majority of persistently replicating viruses can be sensed by innate cells via PRRs, resulting in a robust IFN-I response in the early stages of in vivo chronic infection.
Rapid Attenuation and Persistence of Type I Interferon Responses
Following the initial IFN-I response after infection, systemic IFN-I becomes undetectable at later stages, despite maintained viral loads (Figure 1) (5, 6). This is accompanied by a profound reduction in pDC IFN-I production capacity during LCMV, HIV, and HCV infections that often coincides with altered innate responses to secondary, unrelated pathogens (6, 18, 23, 24). However, it is important to note that in LCMV, HCV, HIV, and SIV infections, IFN-I or ISG transcripts are persistently detected in total DCs, CD4+ and CD8+ T cells, and/or tissues, albeit at lower amounts than those detected during the peak IFN-I response (7, 10, 12, 25, 26). Several mechanisms may contribute to the attenuation of IFN-I in the postacute stages of chronic viral infections, including (a) direct suppression of innate pathways by viral products and (b) host immunomodulatory mechanisms. Examples of direct mechanisms of IFN-I inhibition include HCV NS3–4A protein inhibition of TRIF (the TLR3 adaptor), RIG-I, and MAVS viral sensing pathways upstream of IFN production (27–29) as well as LCMV nucleoprotein (NP) blockade of IRF3 activation (30). Similarly, the HIV proteins Vpr and Vif induce IRF3 degradation (31), and direct HIV gp120 interaction with pDCs inhibits TLR9 (but not TLR7) responses, including pDC activation and IFNα secretion (32). The HIV capsid also interacts with selected host cofactors to evade PRR sensing in macrophages (33). The aforementioned undetectable levels of innate genes early after in vivo HBV infection (11) may be partly explained by HBV polymerase interference with IRF3 phosphorylation and/or by inhibition of MAVS signaling by the viral HBx protein (34–37). Moreover, in line with the reduced TLR2 expression in hepatocytes, monocytes, and Kupffer cells (liver-resident macrophages) from chronic HBV patients, HBV soluble antigen can reduce expression of TLR2 in hepatic cell lines (38). Similarly, HBV soluble antigen also suppresses TLR9 (but not TLR7) signaling in pDCs, and TLR9 expression is reduced in blood pDCs from chronically infected patients (39, 40). In addition to the direct IFN-I inhibitory activities of viral proteins, viruses that establish chronic infections also promote host immunomodulatory responses that may cause aberrant functioning of innate cells. For example, during chronic HCV infection, pDC IFN-I production capacity is impaired, at least in part, by monocyte-derived tumor necrosis factor α (TNFα) and interleukin (IL)-10 (24).
It should be noted that not all aspects of innate responses are attenuated during chronic viral infections; indeed, select pathways are enhanced. For instance, whereas LCMV Cl13–infected mice respond poorly to TLR9 ligand (CpG) challenge, they produce abnormally high IFN-I levels upon TLR4 ligand (LPS) stimulation, leading to rapid death (6). In line with these observations, chronic HCV infection also promotes hyperresponsive macrophages, including Kupffer cells, contributing to the chronic liver inflammation that is characteristic of chronic HCV infection (41, 42). Finally, macrophages treated with HIV and antigen-presenting cells isolated from HIV-infected patients show hyperresponsiveness to unrelated TLR ligands and commensal bacteria, respectively (43, 44).
Together, these studies highlight that although innate immune cells are capable of a robust IFN-I response in the early stages of most chronic viral infections, blockade of innate pathways by viral proteins together with host immunomodulatory molecules leads to a profound (albeit incomplete) attenuation of IFN-I at later stages of infection.
Beneficial and Deleterious Type I Interferon Effects on Antiviral Immunity
IFN-I antiviral effects are mediated by induction of several ISGs that function to impair viral replication through multiple mechanisms [e.g., protein translation inhibition, viral RNA degradation (reviewed in 4)]. IFN-I also orchestrate both innate and adaptive immune cells such as DCs, macrophages, NK cells, and T cells following viral infections, providing activation and survival signals (4, 45). Consistently, studies using persistent LCMV infection, in which the virus is cleared from the blood and most tissues by 2–3 months postinfection, demonstrate that mice lacking IFNαβR establish lifelong viremia (46). Furthermore, Sandler et al. (47) demonstrated that treatment of rhesus macaques with IFNαβR neutralizing antibody during acute SIV infection resulted in enhanced CD4+ T cell depletion, decreased antiviral gene expression, reduced proportions of cytotoxic NK cells, and increased SIV reservoirs, all correlating with faster progression to simian AIDS. In addition, treatment with recombinant IFN-I can often boost the host defense and enhance viral control. Wang et al. (14) showed that treatment with recombinant IFNα5 and IFNβ between days 2 and 5 after LCMV Cl13 infection (perhaps equivalent to delaying the IFN-I attenuation described above) resulted in early viral containment accompanied by enhanced virus-specific CD8+ T cell responses, although the same treatment in the chronic phase of infection had no effect. Similarly, prophylactic treatment with pegylated IFNα2a (pIFNα) prior to SIV mucosal challenge led to increased resistance to infection mediated in part by an increase in CD56+ NK cells; however, continuous pIFNα treatment resulted in an IFN-I desensitization state with reduced ISG levels and increased cell-associated viral loads (47). Notably, pIFNα also enhances expression of several ISGs known to have direct anti-HIV activity (reviewed in 48). However, treatment with pIFNα in HIV-infected patients has yielded mixed results; some studies have shown improved viral control as a result of increased ISG levels, whereas other studies have reported only modest or null effects (49–52). Finally, current therapeutic strategies for HCV infection include pIFNα administration, resulting in a success rate that varies from 40% to 80% depending on time of treatment, HCV genotype, basal IFN-I signature, and polymorphisms associated with the IFN-III cytokines (reviewed in 53).
In addition to the beneficial roles of IFN-I, several studies in HCV and HIV/SIV demonstrate that persisting IFN-I and ISGs correlate with poor disease prognosis (25, 54, 55). Moreover, comparative studies of SIV-infected sooty mangabeys and African green monkeys (which remain healthy despite high viral replication) versus rhesus macaques (which progress to simian AIDS) have shown that a reduced IFN-I signature in both tissues and T cells correlates with better clinical outcome (7, 9, 56). Further studies have also demonstrated that sustained IFN-I signatures correlate with reduced viral control in chronic stages of HIV infection (57, 58). Recently, two reports using LCMV Cl13–infected mice demonstrated that treatment with neutralizing antibody to IFNαβR prior to or during infection led to accelerated viral control despite inducing early enhancement in viremia (likely due to the attenuation of the IFN-I-mediated antiviral effects described above). This transient blockade of IFNαβR resulted in reduced expression of IL-10 and PDL1 (both of which contribute to CD8+ T cell exhaustion, as described below) as well as improved splenic architecture and virus-specific CD4+ T cell responses (10, 59). Previous studies have shown that IFN-I can also impair the development of cDCs in both LCMV and HIV (60, 61) and can mediate HIV-induced apoptosis of CD4+ T cells (62). These deleterious IFN-I effects may contribute to the improvement in immune responses upon IFNαβR blockade.
In conclusion, whereas IFN-I have easily demonstrable effects in controlling replication of persistent viruses in many different chronic infections, they can also limit antiviral immune responses and viral control. Although the relative contributions of positive and negative IFN-I effects may vary depending on the nature of the viral infection, gaining a better understanding of how IFN-I production and silencing are regulated in the context of persistent viral replication may allow manipulation of endogenous IFN-I levels and selected cellular sources to improve viral containment.
CD8+ T CELLS: KEY FEATURES AND DRIVERS OF FUNCTIONAL EXHAUSTION DURING CHRONIC VIRAL INFECTION
CD8+ T cells are specialized in the elimination of intracellular pathogens. Their cytotoxic function includes the secretion of cytolytic proteins such as granzyme B and perforin and of cytokines such as IFNγ and TNFα. Consistently, CD8+ T cell cytolytic responses are required for direct control of persistently replicating viruses such as LCMV in mice and SIV or HBV in macaques (63–67). Furthermore, robust CD8+ T cell responses correlate with disease protection in HIV-, HCV-, and HBV-infected patients (68–71), and human leukocyte antigen class I haplotypes correlate with disease protection in HIV (72). In addition, vigorous CD8+ T cell responses can rapidly drive viral escape mutations in both persistent LCMV and HIV infections (73, 74). However, CD8+ T cell functions are typically inferior during chronic viral infections in comparison with the potent effector and memory T cells generated during acute infections. This state of T cell hyporesponsiveness has been termed exhaustion and is common to numerous chronic viral infections in mice and humans as well as cancer (75). In this section we give an overview of the general features of CD8+ T cell exhaustion during chronic viral infection and highlight the most recent advances in understanding the underlying transcriptional, epigenetic, and metabolic mechanisms as well as potential therapies designed to restore CD8+ T cell functions (Figure 2).
Figure 2.
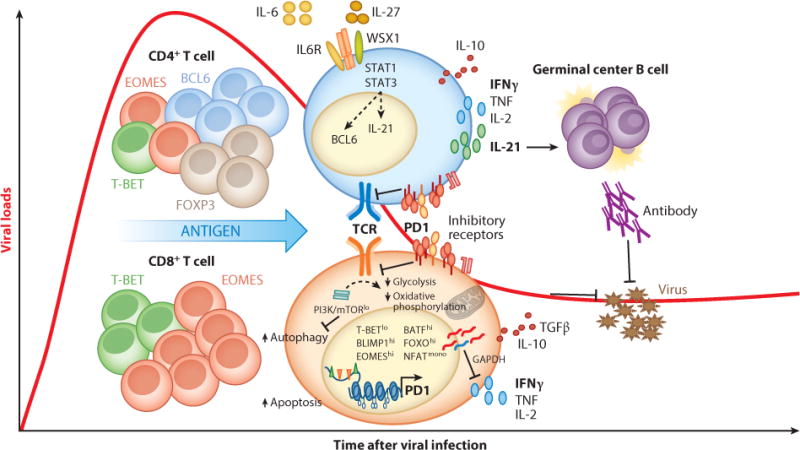
CD4+ and CD8+ T cell adaptation during chronic viral infections. Persistent antigen drives recalibration of T cell responsiveness by inducing transcriptional (e.g., reduced TBET and increased EOMES), epigenetic (e.g., DNA methylation of the PD1 gene locus), and metabolic (e.g., reduced glycolysis and spare respiratory capacity) changes that result in limited functional properties [e.g., CD8+ T cell cytotoxicity, T helper 1 (Th1) cytokine production] and/or functional specialization [e.g., IL-21 elevation and T follicular helper (TFH) cell differentiation]. Recent studies have begun uncovering the heterogeneity in the CD4+ and CD8+ T cell compartment during chronic viral infection. Importantly, although they are partially hyporesponsive, CD4+ and CD8+ T cells are essential to control persistently replicating viruses in mice and humans.
Cardinal Features of Exhausted CD8+ T Cells
Exhaustion in CD8+ T cells from LCMV Cl13–infected mice was first characterized as a progressive loss of antiviral functions, including the antigen-specific secretion of IL-2, followed by secretion of TNFα, and finally by IFNγ secretion upon cognate peptide recognition (75–77). Although exhausted T cells retain expression of the cytolytic molecules granzyme A and B, their cytotoxic capacity is progressively reduced (77–79). Exhausted CD8+ T cells also fail to differentiate into potent memory cells (80, 81), although a memory-like CD8+ T cell precursor during chronic LCMV infection has recently been proposed (82).
The cellular phenotype of exhaustion is characterized by the surface expression of inhibitory receptors—most prominently PD1, but also TIM-3, LAG3, 2B4, and CD160—which directly contribute to CD8+ T cell dysfunction in several chronic viral infections (83–89). Exhaustion also encompasses a reduced response to cytokines including IL-7 (81, 90), IL-2 (91), and IL-15 (92) that normally promote proliferation and survival in effector and/or memory CD8+ T cells. In addition, CD8+ T cell responsiveness to IL-12, IL-18, and IL-21 has recently been shown to be reduced during chronic LCMV infection (93). In contrast, virus-specific CD8+ T cells show enhanced production of the suppressive cytokines IL-10 and/or TGFβ in LCMV-, HCV-, and HIV-infected hosts (94–98).
Importantly, chronic antigen stimulation maintains the exhausted state of CD8+ T cells in LCMV Cl13 infection (80, 92, 99); the amount of antigen correlates with the degree of exhaustion (100, 101). Similarly, levels of exhaustion mirror viremia in HIV- and HCV-infected patients (102–104). Consistently, CD8+ T cells specific for LCMV nucleoprotein (the most abundant viral protein) are more susceptible to deletion, an extreme form of exhaustion (77). This is similar to the loss of high-avidity T cells that occurs early after HIV infection (105).
Thus, a combination of diminished effector functions, expression of inhibitory receptors, and cytokine hyporesponsiveness characterizes a unique exhausted state in CD8+ T cells during chronic viral infections.
Transcriptional Regulation of Exhausted CD8+ T Cells
CD8+ T cell exhaustion has also been defined at the transcriptional level (106, 107). It is now well established that CD8+ T cell exhaustion is transcriptionally distinct from other forms of T cell hyporesponsiveness, such as senescence and anergy as well as memory, even in the presence of continuous antigen stimulation (106, 108, 109). Importantly, these findings in mice translate to human chronic infection, as global gene patterns in CD8+ T cells of HIV-positive elite controllers and progressors were found to resemble CD8+ T cells from mice infected with acute and persistent LCMV variants, respectively (110).
Exhausted CD8+ T cells are characterized by many alterations in transcription factors that normally drive CD8+ T cell cytolytic function, including reduced T-BET expression and NFATc1 nuclear translocation and upregulation of EOMES, BLIMP1, and BATF (66, 111–115). Reduction in T-BET expression correlates with CD8+ T cell dysfunction during chronic HIV, HCV, HBV, and LCMV infections (114–118), and its presence is necessary to limit the expression of multiple inhibitory receptors as well as to retain partial CD8+ T cell functionality during chronic LCMV infection (114). In contrast, the T-BET homologous T-box transcription factor EOMES promotes CD8+ T cell exhaustion. However, EOMES also mediates the antigen-induced conversion (and proliferation) of T-BEThiEOMESloPD1int CD8+ T cells into T-BETloEOMEShiPD1hi terminally differentiated cells and is critical to maintaining a pool of partially effective CD8+ T cells during chronic LCMV infection (66). Notably, the aforementioned low T-BET and high EOMES expression is predominant in CD8+ T cells from chronic (versus resolved) HCV infection (66), and HIV-specific CD8+ T cells maintain this phenotype for years after initiation of antiretroviral therapy (115).
BLIMP1, a transcriptional repressor, is known to promote CD8+ T cell terminal differentiation and effector functions (112). However, high levels of BLIMP1 promote expression of inhibitory molecules, including PD1 and likely IL-10 in CD8+ T cells from LCMV Cl13–infected mice (112, 119). Interestingly, the reduced amounts of BLIMP1 found in hemizygous mice appear sufficient to promote partial effector functions but insufficient to maximize inhibitory receptor expression in exhausted CD8+ T cells. Such mice exhibit enhanced control of LCMV Cl13 (112). Furthermore, monomeric NFAT transcription factors (devoid of AP-1 binding) directly induce BLIMP1 and inhibitory receptors in exhausted CD8+ T cells (120). In addition, while suppressing CD8+ T cell responses by attenuating T cell receptor (TCR) signaling (121) and decreasing cell motility (122), PD1 also induces BATF transcriptional activity, which decreases proliferation as well as IFNγ and IL-2 production in CD8+ T cells from HIV-infected patients (111).
In conclusion, the state of CD8+ T cell exhaustion does not depend on a master transcriptional regulator but appears to be sustained by a unique cohort of transcription factors that regulate different genes during acute versus chronic viral infection (66, 106, 123).
Metabolic Changes in Exhausted CD8+ T Cells
There has been increasing interest in the metabolic state of exhausted CD8+ T cells, and recent microarray analysis suggested a metabolic adaptation conserved among different persistent viral infections, including HIV and LCMV (124, 125). LCMV-specific CD8+ T cells exhibit decreased PI3K-AKT-mTOR signaling upon antigen stimulation, likely due to desensitization of TCR signaling by PD1 (121), which underlies defects in both glycolysis and spare respiratory capacity (123). Reduced glycolysis may increase the availability of glycolytic enzymes, such as GAPDH. When not engaged in glycolysis, this dual-functional enzyme diminishes translation of IFNγ by binding to its mRNA 3′ untranslated region, which may explain the posttranscriptional reduction in IFNγ levels previously described in exhausted CD8+ T cells (126, 127). Decreased AKT activity also causes reduced phosphorylation (and enhanced nuclear retention) of the transcription factor FOXO1, which represses antiviral molecules such as granzyme B and T-BET while inducing expression of PD1, EOMES, and the survival molecule BCL2 (123). Of note, FOXO3, which is also inactivated downstream of mTOR signaling, promotes apoptosis (128), thereby supporting a contrasting role for FOXO1 and FOXO3 in exhausted CD8+ T cells.
In addition, mTOR signaling uses hypoxia-inducible factors (HIFs) to drive glycolysis-related genes (129). During chronic LCMV infection, enforcement of HIF accumulation by deleting the von Hippel–Lindau (VHL) tumor suppressor (a negative regulator of HIFs) promotes glycolysis and the expression of effector molecules (e.g., granzyme B), which leads to pathology and death in VHL-null mice (130). mTOR signaling also reduces autophagy (131), which is required by LCMV-specific T cells to form memory, and T cells lacking autophagy proteins during chronic LCMV infection expand normally but do not persist, causing delayed viral control (132). These results suggest that defects in a variety of metabolic processes (perhaps triggered by nutrient and/or oxygen restrictions) might underlie T cell exhaustion but may be necessary to prevent lethal immunopathology.
Epigenetic Regulation of CD8+ T Cell Exhaustion
Interestingly, when transgenic virus-specific CD8+ T cells isolated early after LCMV Cl13 infection are transferred into a similarly chronic infectious environment in the absence of their cognate antigen, their function and memory formation are restored (74, 99, 100). In contrast, transfer of exhausted CD8+ T cells later during infection into naive animals allows only partial restoration of function, and these cells maintain PD1 expression (82, 133). These results suggest antigen-driven, progressive, heritable genomic changes in exhausted CD8+ T cells. This is consistent with decreased DNA methylation of the PD1 locus in CD8+ T cells from both chronic LCMV and HIV infections (134, 135). Furthermore, Zhang et al. (136) recently showed reduced diacetylated histone 3 in exhausted LCMV-specific CD8+ T cells, and treatment with a histone deacetylase inhibitor increased IFNγ production and memory differentiation during chronic infection. Interestingly, many DNA/histone methylation marks promote HIV latency via long terminal repeat silencing; consequently, the histone deacetylase inhibitor SAHA (vorinostat) is proceeding to clinical trials as potential treatment to reduce the latent HIV reservoir (137). It will be interesting to evaluate whether this inhibitor also restores T cell function.
Although global epigenetic maps in exhausted CD8+ T cells have not yet been published, the aforementioned studies indicate that epigenetic modifications help maintain this cellular state. Future studies might allow therapeutic reprogramming of T cell responsiveness.
CD8+ T Cell Exhaustion Can Be Therapeutically Reversed
Characterization of exhausted CD8+ T cells has opened numerous therapeutic windows, many of which are related to modulation of γC cytokine pathways, namely the IL-2, IL-7, IL-15, and IL-21 pathways (138). Treatment with IL-7, IL-2, or IL-21 in established chronic LCMV infection is successful in amplifying the T cell response and reducing the viral titer (91, 139–142). In contrast, multiple phase III clinical trials of IL-2 treatment in HIV-positive patients concluded with no difference in outcome (143). IL-7 is currently in phase II trials, and IL-15 and IL-21 did not lower viremia in SIV-infected macaques despite increasing CD8+ T cell numbers (144–146).
In recent studies, therapeutic targeting of inhibitory surface receptors or cytokines induced strong restoration of T cell function, in particular the PD1/PDL1 and IL-10/IL-10R pathways (96, 147–150). Targeting other pathways [CTLA-4, TIM-3, 4-1BB, or TGFβ (151, 152)] alone is not effective to reduce viral loads. However, coblockade of α-PDL1 with α-TIM-3 (153) or α-IL-10 (147); α-PDL1 combined with costimulation with 4-1BB (154) or OX40 (155); or administration of low-dose IL-2 (156) results in synergistic enhancement of T cell numbers and function during LCMV Cl13 infection to reduce viral titer. Anti-PD1 clinical trials in HIV-infected patients are now in progress (157). These results support the idea that further understanding of the complex mechanisms underlying CD8+ T cell exhaustion may illuminate and refine effective therapeutic interventions that simultaneously enhance immune function, diminish viral load, and minimize immunopathology.
CD4+ T CELLS: DISTINCT FUNCTIONAL SPECIALIZATION DURING CHRONIC VIRAL INFECTIONS
Following vaccination and acute viral infections, CD4+ T cells have the capacity to differentiate into a number of subsets that sustain and enhance both CD8+ T cell– and antibody-mediated immunity. In HIV-1, HBV, and HCV infections, enhanced virus-specific CD4+ T cell responses are associated with improved viral control (158–161). Consistently, major histocompatibility complex class II human leukocyte antigen haplotypes correlate with disease prognosis in HIV-positive individuals (162, 163). Furthermore, CD4+ T cell–depleted chimpanzees fail to clear HBV or HCV and develop severe liver disease (164, 165). Similarly, CD4+ T cell–depleted mice fail to control persistent LCMV infection (63, 166), whereas the adoptive transfer of virus-specific CD4+ T cells into LCMV Cl13–infected mice helps restore CD8+ T cell exhaustion (167) and can provide B cell help (168).
Altogether, these studies indicate that CD4+ T cells are essential in the immune responses to chronic viral infections. However, excessive numbers of CD4+ T cells, caused by NK cell depletion or by vaccines that solely elicit an antiviral CD4+ T cell response, can result in pronounced inflammation and mortality during chronic LCMV infection, suggesting that enhanced CD4+ T cell responses should be sought carefully (169, 170). In the following sections we discuss key features of CD4+ T cell functional specialization and heterogeneity during chronic viral infections (Figure 2).
Reduced T Helper 1 Features and Enhanced Interleukin-21 Production
The prototypic antiviral CD4+ T cell response is characterized by the differentiation of T helper 1 (Th1) cells and their secretion of IFNγ, TNFα, and IL-2. Like CD8+ T cells (discussed above), CD4+ T cells also exhibit altered function during chronic infection that has historically been considered a form of functional exhaustion. Although less is known about the precise mechanisms behind this process, this CD4+ T cell state is characterized by reduced IL-2, TNFα, and IFNγ production (168, 171, 172). Similar to CD8+ T cells, virus-specific CD4+ T cells from LCMV-, HCV-, and HIV-infected individuals also express higher levels of inhibitory receptors (173–177), and blockade of PD1/PDL1 is sufficient to restore the function of HIV- and HCV-specific CD4+ T cells in vitro (174, 176). Chronic viral infections also increase the expression of the inhibitory cytokine IL-10 by CD4+ T cells (reviewed in 178), and loss of IL-10 signaling during chronic LCMV infection improves CD4+ T cell functionality and viral control (95, 96, 150, 179).
Despite reduced expression of prototypic Th1 cytokines, CD4+ T cells show enhanced production of IL-21 during chronic versus acute LCMV infection (142, 180–182). IL-21 is critical for the proliferation and survival of germinal center B cells (183). However, IL-21 not only promotes higher antiviral antibody levels but also is essential for the maintenance of virus-specific CD8+ T cells, suppression of regulatory T cell (Treg) expansion, and subsequent viral control during chronic LCMV infection (142, 180–182). Elevated IL-21 levels and increased frequencies of IL-21+CD4+ T cells are also associated with improved viral control in HIV, HBV, and HCV infections (184–190). Although the signals that promote IL-21 production in CD4+ T cells during human chronic infections have yet to be investigated, we demonstrated that, during chronic LCMV infection, cell-intrinsic signaling via the shared IL-6 family cytokine coreceptor, gp130, is essential for IL-21 production by virus-specific CD4+ T cells (191). Overall, the aforementioned studies are consistent with a model in which, rather than being exhausted, virus-specific CD4+ T cells favor distinct functional specialization (e.g., IL-21 production) during chronic viral infections.
Regulatory T Cells Suppress Antiviral Responses and Limit Pathology
Foxp3+ Tregs maintain immune system quiescence and prevent reactivity that would otherwise promote immunopathology (reviewed in 192). Chronic HIV, HCV, LCMV, and Friend virus (FV) infections all result in the expansion of Tregs (182, 193–197). In vitro, these Tregs have a potent capacity to suppress virus-specific CD8+ T cell function (194, 197–199), and depletion of Tregs during murine LCMV and FV infections has established the same role in vivo (200, 201). Of note, Treg depletion enhances CD8+ T cell responses and improves viral control during FV infection, but it has no effect on viral control during LCMV Cl13 infection [despite increased CD8+ T cell responses (182, 200, 201)]. This failure in viral control was associated with increased expression of PDL1, and a combination of Treg ablation and PDL1 blockade resulted in a greater reduction in viral titers than did PDL1 blockade alone (201). Tregs therefore appear to negatively impact the ability of the host to resolve an actively replicating chronic viral infection. There is evidence, however, that Tregs may play an important role in limiting pathology and morbidity during chronic LCMV infection (182). During HIV infection, reduced frequency and functionality of Tregs are associated with the onset of immune activation, increased viral load, and overall HIV disease progression (202, 203). Similar observations of higher Treg frequency associated with reduced pathology have also been made in HCV and SIV infections (204, 205). Thus, Tregs may be part of a disease tolerance mechanism (206) that minimizes the tissue damage caused by antiviral immune responses and thereby reduces the host’s fitness costs associated with a chronic viral infection.
T Follicular Helper Cell Escalation and Antibody-Mediated Immunity
T follicular helper (TFH) cells are a CD4+ T cell subset specialized to provide B cell help and promote antibody-mediated immunity (reviewed in 207, 208). Among other molecular features, they are primarily characterized by high expression of the chemokine receptor CXCR5 (which promotes their migration to the B cell zone of secondary lymphoid organs), the transcriptional repressor BCL6 (which is necessary for TFH differentiation), and the cytokine IL-21.
Recent studies indicate that TFH cell differentiation is favored during chronic viral infections. Chronic LCMV infection leads to the pronounced accumulation of TFH cells that migrate into the B cell zone, enhance the quantity and quality of germinal center B cell and virus-specific antibodies, and promote LCMV Cl13 control (209, 210). Increased frequencies of circulating TFH-like or lymph node TFH cells are also observed in patients infected with HIV, HBV, or HCV and in macaques infected with SIV and correlate with the levels of germinal centers, plasma cells, and/or virus-specific antibodies (211–214). Moreover, higher frequencies of circulating TFH cells correlate with the appearance of HIV-specific broadly neutralizing antibodies, and improved TFH function is associated with better vaccination outcomes in HIV-positive patients (215, 216). However, circulating TFH-like and lymph node TFH cells from HIV-infected patients exhibit reduced B cell helper capacity (217, 218), which can be restored by blockade of PD1/PDL1 on HIV-specific lymph node TFH cells (217). Importantly, TFH cell frequencies do not correlate with viral control during SIV or HIV infections (213–215). This is different from the role of TFH cells in the control of persistent LCMV discussed above and could be related to the efficacy of antibody-mediated immunity in containing distinct persistent viruses. Indeed, permanent control of LCMV requires the presence of B cells (219, 220) and the production of virus-specific antibodies (221, 222). However, the presence of antibodies does not prevent reinfection with similar strains of HCV in either humans or chimpanzees (223, 224), nor does it prevent progression of HIV to AIDS (225), possibly due to antibody escape mutants (226). The late appearance of neutralizing or broadly neutralizing antibodies during chronic infection or vaccination could also contribute to their limited efficacy (reviewed in 227). This view is consistent with the observation that, when administered before HIV infection or even during HCV infection, broadly neutralizing antibodies are capable of promoting viral containment (228, 229). One confounding factor that may influence the capacity of TFH cells to aid in the control of HIV-1 or SIV infections is that they are preferentially infected by both HIV-1 and SIV (213, 214, 230–233). This could result in the presence of a viral reservoir, the localization of which provides the virus with protection from clearance by CD8+ T cells (230, 231). Indeed, it was recently shown that TFH cells act as a viral sanctuary in SIV-infected rhesus macaques (234).
The accumulation of TFH cells during the late stages of LCMV infection is, in part, driven by increased IL-6 signaling, which also promotes optimal levels of functional molecules (e.g., ICOS) in virus-specific TFH cells (210). Consistently, increased IL-6 signaling in TFH cells is also observed during the chronic stage of SIV infection (210, 214). Another IL-6 family member, IL-27, promotes survival of virus-specific CD4+ T cells during chronic LCMV infection and is essential for viral control (191). Another factor promoting TFH cell accumulation is the presence of chronic TCR stimulation; mice receiving either a chronic viral infection or recurrent antigen administration have increased TFH cell frequencies compared with mice receiving acute inflammatory stimuli (209, 235, 236). Moreover, whereas TFH cell development during acute infection or vaccination requires cognate B cell interactions (207, 208), B cells are not required in the context of chronic antigen (209, 236). Finally, the miR-17~92 family of microRNAs is essential for TFH cell formation and viral control during LCMV Cl13 infection (237).
Thus, the escalation of TFH cells during chronic viral infections is induced by a combination of signals, including cytokines and continuous TCR stimulation. Concomitant with attenuated Th1 functions and expanded numbers of Tregs, biased TFH cell differentiation may be part of a common host mechanism to drive a less pathogenic response mediated by antiviral antibodies.
Distinctive CD4+ T Cell Transcriptional Signature and Heterogeneity
A recent microarray analysis of virus-specific CD4+ T cells from LCMV Cl13–infected mice revealed a transcriptional signature that is distinct from the prototypic CD4+ T cell fates (e.g., Th1, Th2, Th17, induced Treg) (238). This study highlighted the expression of several key transcription factors in CD4+ T cells from chronically infected mice, including low expression of Tbx21 (T-BET) and increased expression of Prdm1 (BLIMP1), Ikzf2 (HELIOS), and Eomes (EOMES). As mentioned above, in virus-specific CD8+ T cells, T-BET is critical for maintaining function and high BLIMP1 expression is associated with increased inhibitory receptor expression and exhaustion, and it is conceivable that these transcription factors would play similar roles in CD4+ T cells (112, 114, 238). Neither HELIOS nor EOMES has been previously implicated in T cell dysfunction during chronic viral infection; however, the Ikaros family of transcription factors, which includes HELIOS, is associated with cytokine production by CD4+ T cells (239). By contrast, CD4+ T cell expression of EOMES has recently been found to drive a distinct subset of cytotoxic CD4+ T cells in melanomas (240). Interestingly, Crawford et al. (238) found that high expression of BLIMP1 and EOMES was restricted to distinct populations of CD4+ T cells during chronic LCMV infection.
These studies highlight the presence of CD4+ T cell heterogeneity during chronic viral infection and the potentially distinct differentiation and/or abundance of CD4+ T cell subsets with respect to vaccinations or acute infections.
CONCLUDING REMARKS
Given the hyporesponsiveness of innate and adaptive immune cells during chronic viral infections, the term exhaustion could be applied to almost all aspects of immunity discussed in this review (e.g., pDCs and T cells). In all cases, however, an argument could be made to switch this terminology to adaptation or recalibration of immune cells, as has recently been proposed for CD8+ T cells (241). Adaptation or recalibration (rather than exhaustion) emphasizes reprogramming of innate and adaptive immune cells to establish an equilibrium with the new environment while remaining partially effective during chronic viral infections. This involves multiple layers of cell-intrinsic transcriptional, epigenetic, and posttranscriptional processes that respond to cell-extrinsic changes, including sustained stimulation via TCRs, B cell receptors, and/or PRRs; a distinct inflammatory milieu; altered nutrient and oxygen levels; and, likely, increased damage-associated molecular patterns and tissue repair factors. Notably, the molecular mechanisms underlying immune adaptation appear to be conserved in great part during chronic infections with distinct viruses in a range of host species. It is important to emphasize, however, that the ultimate effectiveness of particular immune mediators (e.g., IFN-I, TFH cells, antibodies) in promoting viral control depends on the specific life cycle and immune-evasion strategies of each infectious agent (e.g., tropism, mutation rate, susceptibility to ISGs, etc.).
Technological advances will continue to allow greater understanding of innate and adaptive immune regulation during chronic viral infections. For instance, advances in single-cell sequencing in combination with multiparameter flow cytometry, including mass cytometry, should provide clarification on the extent of heterogeneity in different immune cell compartments during chronic versus acute viral infections. Similarly, high-throughput approaches to epigenetic, posttranscriptional, and metabolomic processes should provide greater clarity about their roles in immunity to chronic viral infections. Additionally, the increasing evidence for cross talk between the host’s immune system and microbiome should also prove an intriguing avenue of discovery.
In conclusion, the molecular networks underlying immune cell adaptation likely evolved as a safety rheostat to counteract immune responses that, although well tolerated for a limited time in an acute infection, have the capacity to cause considerable pathology in the presence of persistent pathogens. Sterilizing therapeutics will likely benefit from a combination of drugs or gene therapies that boost different arms of the immune system and target key steps in the virus life cycle. Further understanding of the unique and sophisticated adaptation of immune cells to a chronic infectious environment will move us closer to this goal.
Acknowledgments
We apologize for being unable due to space constraints to cite all studies that have improved our understanding of immune regulation during chronic viral infections. We thank Zuniga lab members and Stephen Hedrick for critically reading this review. Some of the findings described were supported in part by National Institutes of Health (NIH) grants AI081923 and AI0113923 (to E.I.Z.). E.I.Z. is a Leukemia and Lymphoma Society Scholar. G.L. is supported by NIH grant AI081923, and M.M. is supported by an American Cancer Society Scholar Research Grant (to E.I.Z.) J.A.H. is supported by a Sir Henry Dale Fellowship (101372/Z/13/Z) from the Royal Society and Wellcome Trust.
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Kassiotis G. Endogenous retroviruses and the development of cancer. J Immunol. 2014;192:1343–49. doi: 10.4049/jimmunol.1302972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Virgin HW, Wherry EJ, Ahmed R. Redefining chronic viral infection. Cell. 2009;138:30–50. doi: 10.1016/j.cell.2009.06.036. [DOI] [PubMed] [Google Scholar]
- 3.Aoshi T, Koyama S, Kobiyama K, Akira S, Ishii KJ. Innate and adaptive immune responses to viral infection and vaccination. Curr Opin Virol. 2011;1:226–32. doi: 10.1016/j.coviro.2011.07.002. [DOI] [PubMed] [Google Scholar]
- 4.Garcia-Sastre A, Biron CA. Type 1 interferons and the virus-host relationship: a lesson in détente. Science. 2006;312:879–82. doi: 10.1126/science.1125676. [DOI] [PubMed] [Google Scholar]
- 5.Stacey AR, Norris PJ, Qin L, Haygreen EA, Taylor E, et al. Induction of a striking systemic cytokine cascade prior to peak viremia in acute human immunodeficiency virus type 1 infection, in contrast to more modest and delayed responses in acute hepatitis B and C virus infections. J Virol. 2009;83:3719–33. doi: 10.1128/JVI.01844-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zuniga EI, Liou LY, Mack L, Mendoza M, Oldstone MB. Persistent virus infection inhibits type I interferon production by plasmacytoid dendritic cells to facilitate opportunistic infections. Cell Host Microbe. 2008;4:374–86. doi: 10.1016/j.chom.2008.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bosinger SE, Li Q, Gordon SN, Klatt NR, Duan L, et al. Global genomic analysis reveals rapid control of a robust innate response in SIV-infected sooty mangabeys. J Clin Investig. 2009;119:3556–72. doi: 10.1172/JCI40115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clingan JM, Ostrow K, Hosiawa KA, Chen ZJ, Matloubian M. Differential roles for RIG-I-like receptors and nucleic acid-sensing TLR pathways in controlling a chronic viral infection. J Immunol. 2012;188:4432–40. doi: 10.4049/jimmunol.1103656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jacquelin B, Mayau V, Targat B, Liovat AS, Kunkel D, et al. Nonpathogenic SIV infection of African green monkeys induces a strong but rapidly controlled type I IFN response. J Clin Investig. 2009;119:3544–55. doi: 10.1172/JCI40093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wilson EB, Yamada DH, Elsaesser H, Herskovitz J, Deng J, et al. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science. 2013;340:202–7. doi: 10.1126/science.1235208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wieland S, Thimme R, Purcell RH, Chisari FV. Genomic analysis of the host response to hepatitis B virus infection. PNAS. 2004;101:6669–74. doi: 10.1073/pnas.0401771101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hahm B, Trifilo MJ, Zuniga EI, Oldstone MB. Viruses evade the immune system through type I interferon-mediated STAT2-dependent, but STAT1-independent, signaling. Immunity. 2005;22:247–57. doi: 10.1016/j.immuni.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 13.Diebold SS, Montoya M, Unger H, Alexopoulou L, Roy P, et al. Viral infection switches non-plasmacytoid dendritic cells into high interferon producers. Nature. 2003;424:324–28. doi: 10.1038/nature01783. [DOI] [PubMed] [Google Scholar]
- 14.Wang Y, Swiecki M, Cella M, Alber G, Schreiber RD, et al. Timing and magnitude of type I interferon responses by distinct sensors impact CD8 T cell exhaustion and chronic viral infection. Cell Host Microbe. 2012;11:631–42. doi: 10.1016/j.chom.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schoggins JW, Rice CM. Innate immune responses to hepatitis C virus. Curr Top Microbiol Immunol. 2013;369:219–42. doi: 10.1007/978-3-642-27340-7_9. [DOI] [PubMed] [Google Scholar]
- 16.Silvin A, Manel N. Innate immune sensing of HIV infection. Curr Opin Immunol. 2015;32:54–60. doi: 10.1016/j.coi.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 17.Reizis B, Bunin A, Ghosh HS, Lewis KL, Sisirak V. Plasmacytoid dendritic cells: recent progress and open questions. Annu Rev Immunol. 2011;29:163–83. doi: 10.1146/annurev-immunol-031210-101345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fitzgerald-Bocarsly P, Jacobs ES. Plasmacytoid dendritic cells in HIV infection: striking a delicate balance. J Leukoc Biol. 2010;87:609–20. doi: 10.1189/jlb.0909635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Macal M, Lewis GM, Kunz S, Flavell R, Harker JA, Zuniga EI. Plasmacytoid dendritic cells are productively infected and activated through TLR-7 early after arenavirus infection. Cell Host Microbe. 2012;11:617–30. doi: 10.1016/j.chom.2012.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takahashi K, Asabe S, Wieland S, Garaigorta U, Gastaminza P, et al. Plasmacytoid dendritic cells sense hepatitis C virus-infected cells, produce interferon, and inhibit infection. PNAS. 2010;107:7431–36. doi: 10.1073/pnas.1002301107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cervantes-Barragan L, Lewis KL, Firner S, Thiel V, Hugues S, et al. Plasmacytoid dendritic cells control T-cell response to chronic viral infection. PNAS. 2012;109:3012–17. doi: 10.1073/pnas.1117359109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dreux M, Garaigorta U, Boyd B, Decembre E, Chung J, et al. Short-range exosomal transfer of viral RNA from infected cells to plasmacytoid dendritic cells triggers innate immunity. Cell Host Microbe. 2012;12:558–70. doi: 10.1016/j.chom.2012.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee LN, Burke S, Montoya M, Borrow P. Multiple mechanisms contribute to impairment of type 1 interferon production during chronic lymphocytic choriomeningitis virus infection of mice. J Immunol. 2009;182:7178–89. doi: 10.4049/jimmunol.0802526. [DOI] [PubMed] [Google Scholar]
- 24.Dolganiuc A, Chang S, Kodys K, Mandrekar P, Bakis G, et al. Hepatitis C virus (HCV) core protein-induced, monocyte-mediated mechanisms of reduced IFN-α and plasmacytoid dendritic cell loss in chronic HCV infection. J Immunol. 2006;177:6758–68. doi: 10.4049/jimmunol.177.10.6758. [DOI] [PubMed] [Google Scholar]
- 25.Rotger M, Dalmau J, Rauch A, McLaren P, Bosinger SE, et al. Comparative transcriptomics of extreme phenotypes of human HIV-1 infection and SIV infection in sooty mangabey and rhesus macaque. J Clin Investig. 2011;121:2391–400. doi: 10.1172/JCI45235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Su AI, Pezacki JP, Wodicka L, Brideau AD, Supekova L, et al. Genomic analysis of the host response to hepatitis C virus infection. PNAS. 2002;99:15669–74. doi: 10.1073/pnas.202608199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Foy E, Li K, Sumpter R, Jr, Loo YM, Johnson CL, et al. Control of antiviral defenses through hepatitis C virus disruption of retinoic acid-inducible gene-I signaling. PNAS. 2005;102:2986–91. doi: 10.1073/pnas.0408707102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li K, Foy E, Ferreon JC, Nakamura M, Ferreon AC, et al. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. PNAS. 2005;102:2992–97. doi: 10.1073/pnas.0408824102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, et al. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437:1167–72. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- 30.Martinez-Sobrido L, Zuniga EI, Rosario D, Garcia-Sastre A, de la Torre JC. Inhibition of the type I interferon response by the nucleoprotein of the prototypic arenavirus lymphocytic choriomeningitis virus. J Virol. 2006;80:9192–99. doi: 10.1128/JVI.00555-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Okumura A, Alce T, Lubyova B, Ezelle H, Strebel K, Pitha PM. HIV-1 accessory proteins VPR and Vif modulate antiviral response by targeting IRF-3 for degradation. Virology. 2008;373:85–97. doi: 10.1016/j.virol.2007.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martinelli E, Cicala C, Van Ryk D, Goode DJ, Macleod K, et al. HIV-1 gp120 inhibits TLR9-mediated activation and IFN-α secretion in plasmacytoid dendritic cells. PNAS. 2007;104:3396–401. doi: 10.1073/pnas.0611353104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rasaiyaah J, Tan CP, Fletcher AJ, Price AJ, Blondeau C, et al. HIV-1 evades innate immune recognition through specific cofactor recruitment. Nature. 2013;503:402–5. doi: 10.1038/nature12769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kumar M, Jung SY, Hodgson AJ, Madden CR, Qin J, Slagle BL. Hepatitis B virus regulatory HBx protein binds to adaptor protein IPS-1 and inhibits the activation of beta interferon. J Virol. 2011;85:987–95. doi: 10.1128/JVI.01825-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang X, Li Y, Mao A, Li C, Tien P. Hepatitis B virus X protein suppresses virus-triggered IRF3 activation and IFN-β induction by disrupting the VISA-associated complex. Cell Mol Immunol. 2010;7:341–48. doi: 10.1038/cmi.2010.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wei C, Ni C, Song T, Liu Y, Yang X, et al. The hepatitis B virus X protein disrupts innate immunity by downregulating mitochondrial antiviral signaling protein. J Immunol. 2010;185:1158–68. doi: 10.4049/jimmunol.0903874. [DOI] [PubMed] [Google Scholar]
- 37.Yu S, Chen J, Wu M, Chen H, Kato N, Yuan Z. Hepatitis B virus polymerase inhibits RIG-I-and Toll-like receptor 3-mediated beta interferon induction in human hepatocytes through interference with interferon regulatory factor 3 activation and dampening of the interaction between TBK1/IKKε and DDX3. J Gen Virol. 2010;91:2080–90. doi: 10.1099/vir.0.020552-0. [DOI] [PubMed] [Google Scholar]
- 38.Visvanathan K, Skinner NA, Thompson AJ, Riordan SM, Sozzi V, et al. Regulation of Toll-like receptor-2 expression in chronic hepatitis B by the precore protein. Hepatology. 2007;45:102–10. doi: 10.1002/hep.21482. [DOI] [PubMed] [Google Scholar]
- 39.Xie Q, Shen HC, Jia NN, Wang H, Lin LY, et al. Patients with chronic hepatitis B infection display deficiency of plasmacytoid dendritic cells with reduced expression of TLR9. Microbes Infect. 2009;11:515–23. doi: 10.1016/j.micinf.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 40.Xu Y, Hu Y, Shi B, Zhang X, Wang J, et al. HBsAg inhibits TLR9-mediated activation and IFN-α production in plasmacytoid dendritic cells. Mol Immunol. 2009;46:2640–46. doi: 10.1016/j.molimm.2009.04.031. [DOI] [PubMed] [Google Scholar]
- 41.Dolganiuc A, Norkina O, Kodys K, Catalano D, Bakis G, et al. Viral and host factors induce macrophage activation and loss of toll-like receptor tolerance in chronic HCV infection. Gastroenterology. 2007;133:1627–36. doi: 10.1053/j.gastro.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Negash AA, Ramos HJ, Crochet N, Lau DT, Doehle B, et al. IL-1β production through the NLRP3 inflammasome by hepatic macrophages links hepatitis C virus infection with liver inflammation and disease. PLOS Pathog. 2013;9:e1003330. doi: 10.1371/journal.ppat.1003330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brown JN, Kohler JJ, Coberley CR, Sleasman JW, Goodenow MM. HIV-1 activates macrophages independent of Toll-like receptors. PLOS ONE. 2008;3:e3664. doi: 10.1371/journal.pone.0003664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nagy LH, Grishina I, Macal M, Hirao LA, Hu WK, et al. Chronic HIV infection enhances the responsiveness of antigen presenting cells to commensal Lactobacillus. PLOS ONE. 2013;8:e72789. doi: 10.1371/journal.pone.0072789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Crouse J, Bedenikovic G, Wiesel M, Ibberson M, Xenarios I, et al. Type I interferons protect T cells against NK cell attack mediated by the activating receptor NCR1. Immunity. 2014;40:961–73. doi: 10.1016/j.immuni.2014.05.003. [DOI] [PubMed] [Google Scholar]
- 46.Ou R, Zhou S, Huang L, Moskophidis D. Critical role for alpha/beta and gamma interferons in persistence of lymphocytic choriomeningitis virus by clonal exhaustion of cytotoxic T cells. J Virol. 2001;75:8407–23. doi: 10.1128/JVI.75.18.8407-8423.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sandler NG, Bosinger SE, Estes JD, Zhu RT, Tharp GK, et al. Type I interferon responses in rhesus macaques prevent SIV infection and slow disease progression. Nature. 2014;511:601–5. doi: 10.1038/nature13554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Borrow P. Innate immunity in acute HIV-1 infection. Curr Opin HIV AIDS. 2011;6:353–63. doi: 10.1097/COH.0b013e3283495996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Asmuth DM, Murphy RL, Rosenkranz SL, Lertora JJ, Kottilil S, et al. Safety, tolerability, and mechanisms of antiretroviral activity of pegylated interferon alfa-2a in HIV-1-monoinfected participants: a phase II clinical trial. J Infect Dis. 2010;201:1686–96. doi: 10.1086/652420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Azzoni L, Foulkes AS, Papasavvas E, Mexas AM, Lynn KM, et al. Pegylated interferon alfa-2a monotherapy results in suppression of HIV type 1 replication and decreased cell-associated HIV DNA integration. J Infect Dis. 2013;207:213–22. doi: 10.1093/infdis/jis663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Boue F, Reynes J, Rouzioux C, Emilie D, Souala F, et al. Alpha interferon administration during structured interruptions of combination antiretroviral therapy in patients with chronic HIV-1 infection: INTERVAC ANRS 105 trial. AIDS. 2011;25:115–18. doi: 10.1097/QAD.0b013e328340a1e7. [DOI] [PubMed] [Google Scholar]
- 52.Hatzakis A, Gargalianos P, Kiosses V, Lazanas M, Sypsa V, et al. Low-dose IFN-α monotherapy in treatment-naive individuals with HIV-1 infection: evidence of potent suppression of viral replication. J Interferon Cytokine Res. 2001;21:861–69. doi: 10.1089/107999001753238114. [DOI] [PubMed] [Google Scholar]
- 53.Laidlaw SM, Dustin LB. Interferon lambda: opportunities, risks, and uncertainties in the fight against HCV. Front Immunol. 2014;5:545. doi: 10.3389/fimmu.2014.00545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sarasin-Filipowicz M, Oakeley EJ, Duong FH, Christen V, Terracciano L, et al. Interferon signaling and treatment outcome in chronic hepatitis C. PNAS. 2008;105:7034–39. doi: 10.1073/pnas.0707882105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Harris LD, Tabb B, Sodora DL, Paiardini M, Klatt NR, et al. Downregulation of robust acute type I interferon responses distinguishes nonpathogenic simian immunodeficiency virus (SIV) infection of natural hosts from pathogenic SIV infection of rhesus macaques. J Virol. 2010;84:7886–91. doi: 10.1128/JVI.02612-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Durudas A, Milush JM, Chen HL, Engram JC, Silvestri G, Sodora DL. Elevated levels of innate immune modulators in lymph nodes and blood are associated with more-rapid disease progression in simian immunodeficiency virus-infected monkeys. J Virol. 2009;83:12229–40. doi: 10.1128/JVI.01311-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rotger M, Dang KK, Fellay J, Heinzen EL, Feng S, et al. Genome-wide mRNA expression correlates of viral control in CD4+ T-cells from HIV-1-infected individuals. PLOS Pathog. 2010;6:e1000781. doi: 10.1371/journal.ppat.1000781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Herbeuval JP, Nilsson J, Boasso A, Hardy AW, Kruhlak MJ, et al. Differential expression of IFN-α and TRAIL/DR5 in lymphoid tissue of progressor versus nonprogressor HIV-1-infected patients. PNAS. 2006;103:7000–5. doi: 10.1073/pnas.0600363103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Teijaro JR, Ng C, Lee AM, Sullivan BM, Sheehan KC, et al. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science. 2013;340:207–11. doi: 10.1126/science.1235214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kodama A, Tanaka R, Zhang LF, Adachi T, Saito M, et al. Impairment of in vitro generation of monocyte-derived human dendritic cells by inactivated human immunodeficiency virus-1: involvement of type I interferon produced from plasmacytoid dendritc cells. Hum Immunol. 2010;71:541–50. doi: 10.1016/j.humimm.2010.02.020. [DOI] [PubMed] [Google Scholar]
- 61.Sevilla N, McGavern DB, Teng C, Kunz S, Oldstone MB. Viral targeting of hematopoietic progenitors and inhibition of DC maturation as a dual strategy for immune subversion. J Clin Investig. 2004;113:737–45. doi: 10.1172/JCI20243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Herbeuval JP, Grivel JC, Boasso A, Hardy AW, Chougnet C, et al. CD4+ T-cell death induced by infectious and noninfectious HIV-1: role of type 1 interferon-dependent, TRAIL/DR5-mediated apoptosis. Blood. 2005;106:3524–31. doi: 10.1182/blood-2005-03-1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Matloubian M, Concepcion RJ, Ahmed R. CD4+ T cells are required to sustain CD8+ cytotoxic T-cell responses during chronic viral infection. J Virol. 1994;68:8056–63. doi: 10.1128/jvi.68.12.8056-8063.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schmitz JE, Kuroda MJ, Santra S, Sasseville VG, Simon MA, et al. Control of viremia in simian immunodeficiency virus infection by CD8+ lymphocytes. Science. 1999;283:857–60. doi: 10.1126/science.283.5403.857. [DOI] [PubMed] [Google Scholar]
- 65.Jin X, Bauer DE, Tuttleton SE, Lewin S, Gettie A, et al. Dramatic rise in plasma viremia after CD8+ T cell depletion in simian immunodeficiency virus-infected macaques. J Exp Med. 1999;189:991–98. doi: 10.1084/jem.189.6.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Paley MA, Kroy DC, Odorizzi PM, Johnnidis JB, Dolfi DV, et al. Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science. 2012;338:1220–25. doi: 10.1126/science.1229620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Thimme R, Wieland S, Steiger C, Ghrayeb J, Reimann KA, et al. CD8+ T cells mediate viral clearance and disease pathogenesis during acute hepatitis B virus infection. J Virol. 2003;77:68–76. doi: 10.1128/JVI.77.1.68-76.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Maini MK, Boni C, Ogg GS, King AS, Reignat S, et al. Direct ex vivo analysis of hepatitis B virus-specific CD8+ T cells associated with the control of infection. Gastroenterology. 1999;117:1386–96. doi: 10.1016/s0016-5085(99)70289-1. [DOI] [PubMed] [Google Scholar]
- 69.Borrow P, Lewicki H, Hahn BH, Shaw GM, Oldstone MB. Virus-specific CD8+ cytotoxic T-lymphocyte activity associated with control of viremia in primary human immunodeficiency virus type 1 infection. J Virol. 1994;68:6103–10. doi: 10.1128/jvi.68.9.6103-6110.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Edwards BH, Bansal A, Sabbaj S, Bakari J, Mulligan MJ, Goepfert PA. Magnitude of functional CD8+ T-cell responses to the Gag protein of human immunodeficiency virus type 1 correlates inversely with viral load in plasma. J Virol. 2002;76:2298–305. doi: 10.1128/jvi.76.5.2298-2305.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jagannathan P, Osborne CM, Royce C, Manion MM, Tilton JC, et al. Comparisons of CD8+ T cells specific for human immunodeficiency virus, hepatitis C virus, and cytomegalovirus reveal differences in frequency, immunodominance, phenotype, and interleukin-2 responsiveness. J Virol. 2009;83:2728–42. doi: 10.1128/JVI.02128-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Horton H, Frank I, Baydo R, Jalbert E, Penn J, et al. Preservation of T cell proliferation restricted by protective HLA alleles is critical for immune control of HIV-1 infection. J Immunol. 2006;177:7406–15. doi: 10.4049/jimmunol.177.10.7406. [DOI] [PubMed] [Google Scholar]
- 73.Price DA, Goulder PJ, Klenerman P, Sewell AK, Easterbrook PJ, et al. Positive selection of HIV-1 cytotoxic T lymphocyte escape variants during primary infection. PNAS. 1997;94:1890–95. doi: 10.1073/pnas.94.5.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Blattman JN, Wherry EJ, Ha SJ, van der Most RG, Ahmed R. Impact of epitope escape on PD-1 expression and CD8 T-cell exhaustion during chronic infection. J Virol. 2009;83:4386–94. doi: 10.1128/JVI.02524-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wherry EJ. T cell exhaustion. Nat Immunol. 2011;12:492–99. doi: 10.1038/ni.2035. [DOI] [PubMed] [Google Scholar]
- 76.Zajac AJ, Blattman JN, Murali-Krishna K, Sourdive DJ, Suresh M, et al. Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med. 1998;188:2205–13. doi: 10.1084/jem.188.12.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J Virol. 2003;77:4911–27. doi: 10.1128/JVI.77.8.4911-4927.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jo J, Bengsch B, Seigel B, Rau SJ, Schmidt J, et al. Low perforin expression of early differentiated HCV-specific CD8+ T cells limits their hepatotoxic potential. J Hepatol. 2012;57:9–16. doi: 10.1016/j.jhep.2012.02.030. [DOI] [PubMed] [Google Scholar]
- 79.Zhang D, Shankar P, Xu Z, Harnisch B, Chen G, et al. Most antiviral CD8 T cells during chronic viral infection do not express high levels of perforin and are not directly cytotoxic. Blood. 2003;101:226–35. doi: 10.1182/blood-2002-03-0791. [DOI] [PubMed] [Google Scholar]
- 80.Wherry EJ, Barber DL, Kaech SM, Blattman JN, Ahmed R. Antigen-independent memory CD8 T cells do not develop during chronic viral infection. PNAS. 2004;101:16004–9. doi: 10.1073/pnas.0407192101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wherry EJ, Day CL, Draenert R, Miller JD, Kiepiela P, et al. HIV-specific CD8 T cells express low levels of IL-7Rα: implications for HIV-specific T cell memory. Virology. 2006;353:366–73. doi: 10.1016/j.virol.2006.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Utzschneider DT, Legat A, Fuertes Marraco SA, Carrie L, Luescher I, et al. T cells maintain an exhausted phenotype after antigen withdrawal and population reexpansion. Nat Immunol. 2013;14:603–10. doi: 10.1038/ni.2606. [DOI] [PubMed] [Google Scholar]
- 83.Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol. 2009;10:29–37. doi: 10.1038/ni.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bengsch B, Seigel B, Ruhl M, Timm J, Kuntz M, et al. Coexpression of PD-1, 2B4, CD160 and KLRG1 on exhausted HCV-specific CD8+ T cells is linked to antigen recognition and T cell differentiation. PLOS Pathog. 2010;6:e1000947. doi: 10.1371/journal.ppat.1000947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schlaphoff V, Lunemann S, Suneetha PV, Jaroszewicz J, Grabowski J, et al. Dual function of the NK cell receptor 2B4 (CD244) in the regulation of HCV-specific CD8+ T cells. PLOS Pathog. 2011;7:e1002045. doi: 10.1371/journal.ppat.1002045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yamamoto T, Price DA, Casazza JP, Ferrari G, Nason M, et al. Surface expression patterns of negative regulatory molecules identify determinants of virus-specific CD8+ T-cell exhaustion in HIV infection. Blood. 2011;117:4805–15. doi: 10.1182/blood-2010-11-317297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nakamoto N, Kaplan DE, Coleclough J, Li Y, Valiga ME, et al. Functional restoration of HCV-specific CD8 T cells by PD-1 blockade is defined by PD-1 expression and compartmentalization. Gastroenterology. 2008;134:1927–37. doi: 10.1053/j.gastro.2008.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Trautmann L, Janbazian L, Chomont N, Said EA, Gimmig S, et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat Med. 2006;12:1198–202. doi: 10.1038/nm1482. [DOI] [PubMed] [Google Scholar]
- 89.Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. 2006;443:350–54. doi: 10.1038/nature05115. [DOI] [PubMed] [Google Scholar]
- 90.Fuller MJ, Hildeman DA, Sabbaj S, Gaddis DE, Tebo AE, et al. Cutting edge: emergence of CD127high functionally competent memory T cells is compromised by high viral loads and inadequate T cell help. J Immunol. 2005;174:5926–30. doi: 10.4049/jimmunol.174.10.5926. [DOI] [PubMed] [Google Scholar]
- 91.Blattman JN, Grayson JM, Wherry EJ, Kaech SM, Smith KA, Ahmed R. Therapeutic use of IL-2 to enhance antiviral T-cell responses in vivo. Nat Med. 2003;9:540–47. doi: 10.1038/nm866. [DOI] [PubMed] [Google Scholar]
- 92.Shin H, Blackburn SD, Blattman JN, Wherry EJ. Viral antigen and extensive division maintain virus-specific CD8 T cells during chronic infection. J Exp Med. 2007;204:941–49. doi: 10.1084/jem.20061937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ingram JT, Yi JS, Zajac AJ. Exhausted CD8 T cells downregulate the IL-18 receptor and become unresponsive to inflammatory cytokines and bacterial co-infections. PLOS Pathog. 2011;7:e1002273. doi: 10.1371/journal.ppat.1002273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Elrefaei M, Burke CM, Baker CA, Jones NG, Bousheri S, et al. TGF-β and IL-10 production by HIV-specific CD8+ T cells is regulated by CTLA-4 signaling on CD4+ T cells. PLOS ONE. 2009;4:e8194. doi: 10.1371/journal.pone.0008194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Brockman MA, Kwon DS, Tighe DP, Pavlik DF, Rosato PC, et al. IL-10 is up-regulated in multiple cell types during viremic HIV infection and reversibly inhibits virus-specific T cells. Blood. 2009;114:346–56. doi: 10.1182/blood-2008-12-191296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Brooks DG, Trifilo MJ, Edelmann KH, Teyton L, McGavern DB, Oldstone MB. Interleukin-10 determines viral clearance or persistence in vivo. Nat Med. 2006;12:1301–9. doi: 10.1038/nm1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tinoco R, Alcalde V, Yang Y, Sauer K, Zuniga EI. Cell-intrinsic transforming growth factor-β signaling mediates virus-specific CD8+ T cell deletion and viral persistence in vivo. Immunity. 2009;31:145–57. doi: 10.1016/j.immuni.2009.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Alatrakchi N, Graham CS, van der Vliet HJ, Sherman KE, Exley MA, Koziel MJ. Hepatitis C virus (HCV)-specific CD8+ cells produce transforming growth factor βthat can suppress HCV-specific T-cell responses. J Virol. 2007;81:5882–92. doi: 10.1128/JVI.02202-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Brooks DG, McGavern DB, Oldstone MB. Reprogramming of antiviral T cells prevents inactivation and restores T cell activity during persistent viral infection. J Clin Investig. 2006;116:1675–85. doi: 10.1172/JCI26856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mueller SN, Ahmed R. High antigen levels are the cause of T cell exhaustion during chronic viral infection. PNAS. 2009;106:8623–28. doi: 10.1073/pnas.0809818106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Richter K, Brocker T, Oxenius A. Antigen amount dictates CD8+ T-cell exhaustion during chronic viral infection irrespective of the type of antigen presenting cell. Eur J Immunol. 2012;42:2290–304. doi: 10.1002/eji.201142275. [DOI] [PubMed] [Google Scholar]
- 102.Oxenius A, Sewell AK, Dawson SJ, Gunthard HF, Fischer M, et al. Functional discrepancies in HIV-specific CD8+ T-lymphocyte populations are related to plasma virus load. J Clin Immunol. 2002;22:363–74. doi: 10.1023/a:1020656300027. [DOI] [PubMed] [Google Scholar]
- 103.Rehr M, Cahenzli J, Haas A, Price DA, Gostick E, et al. Emergence of polyfunctional CD8+ T cells after prolonged suppression of human immunodeficiency virus replication by antiretroviral therapy. J Virol. 2008;82:3391–404. doi: 10.1128/JVI.02383-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lechner F, Gruener NH, Urbani S, Uggeri J, Santantonio T, et al. CD8+ T lymphocyte responses are induced during acute hepatitis C virus infection but are not sustained. Eur J Immunol. 2000;30:2479–87. doi: 10.1002/1521-4141(200009)30:9<2479::AID-IMMU2479>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 105.Lichterfeld M, Yu XG, Mui SK, Williams KL, Trocha A, et al. Selective depletion of high-avidity human immunodeficiency virus type 1 (HIV-1)-specific CD8+ T cells after early HIV-1 infection. J Virol. 2007;81:4199–214. doi: 10.1128/JVI.01388-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Doering TA, Crawford A, Angelosanto JM, Paley MA, Ziegler CG, Wherry EJ. Network analysis reveals centrally connected genes and pathways involved in CD8+ T cell exhaustion versus memory. Immunity. 2012;37:1130–44. doi: 10.1016/j.immuni.2012.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Larsson M, Shankar EM, Che KF, Saeidi A, Ellegard R, et al. Molecular signatures of T-cell inhibition in HIV-1 infection. Retrovirology. 2013;10:31. doi: 10.1186/1742-4690-10-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wirth TC, Xue HH, Rai D, Sabel JT, Bair T, et al. Repetitive antigen stimulation induces stepwise transcriptome diversification but preserves a core signature of memory CD8+ T cell differentiation. Immunity. 2010;33:128–40. doi: 10.1016/j.immuni.2010.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. 2007;27:670–84. doi: 10.1016/j.immuni.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 110.Gaiha GD, McKim KJ, Woods M, Pertel T, Rohrbach J, et al. Dysfunctional HIV-specific CD8+ T cell proliferation is associated with increased caspase-8 activity and mediated by necroptosis. Immunity. 2014;41:1001–12. doi: 10.1016/j.immuni.2014.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Quigley M, Pereyra F, Nilsson B, Porichis F, Fonseca C, et al. Transcriptional analysis of HIV-specific CD8+ T cells shows that PD-1 inhibits T cell function by upregulating BATF. Nat Med. 2010;16:1147–51. doi: 10.1038/nm.2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Shin H, Blackburn SD, Intlekofer AM, Kao C, Angelosanto JM, et al. A role for the transcriptional repressor Blimp-1 in CD8+ T cell exhaustion during chronic viral infection. Immunity. 2009;31:309–20. doi: 10.1016/j.immuni.2009.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Agnellini P, Wolint P, Rehr M, Cahenzli J, Karrer U, Oxenius A. Impaired NFAT nuclear translocation results in split exhaustion of virus-specific CD8+ T cell functions during chronic viral infection. PNAS. 2007;104:4565–70. doi: 10.1073/pnas.0610335104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kao C, Oestreich KJ, Paley MA, Crawford A, Angelosanto JM, et al. Transcription factor T-bet represses expression of the inhibitory receptor PD-1 and sustains virus-specific CD8+ T cell responses during chronic infection. Nat Immunol. 2011;12:663–71. doi: 10.1038/ni.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Buggert M, Tauriainen J, Yamamoto T, Frederiksen J, Ivarsson MA, et al. T-bet and Eomes are differentially linked to the exhausted phenotype of CD8+ T cells in HIV infection. PLOS Pathog. 2014;10:e1004251. doi: 10.1371/journal.ppat.1004251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kurktschiev PD, Raziorrouh B, Schraut W, Backmund M, Wachtler M, et al. Dysfunctional CD8+ T cells in hepatitis B and C are characterized by a lack of antigen-specific T-bet induction. J Exp Med. 2014;211:2047–59. doi: 10.1084/jem.20131333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ribeiro-dos-Santos P, Turnbull EL, Monteiro M, Legrand A, Conrod K, et al. Chronic HIV infection affects the expression of the 2 transcription factors required for CD8 T-cell differentiation into cytolytic effectors. Blood. 2012;119:4928–38. doi: 10.1182/blood-2011-12-395186. [DOI] [PubMed] [Google Scholar]
- 118.Hersperger AR, Martin JN, Shin LY, Sheth PM, Kovacs CM, et al. Increased HIV-specific CD8+ T-cell cytotoxic potential in HIV elite controllers is associated with T-bet expression. Blood. 2011;117:3799–808. doi: 10.1182/blood-2010-12-322727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Parish IA, Marshall HD, Staron MM, Lang PA, Brustle A, et al. Chronic viral infection promotes sustained Th1-derived immunoregulatory IL-10 via BLIMP-1. J Clin Investig. 2014;124:3455–68. doi: 10.1172/JCI66108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Martinez GJ, Pereira RM, Aijo T, Kim EY, Marangoni F, et al. The transcription factor NFAT promotes exhaustion of activated CD8+ T cells. Immunity. 2015;42:265–78. doi: 10.1016/j.immuni.2015.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sheppard KA, Fitz LJ, Lee JM, Benander C, George JA, et al. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3ζ signalosome and downstream signaling to PKCθ. FEBS Lett. 2004;574:37–41. doi: 10.1016/j.febslet.2004.07.083. [DOI] [PubMed] [Google Scholar]
- 122.Zinselmeyer BH, Heydari S, Sacristan C, Nayak D, Cammer M, et al. PD-1 promotes immune exhaustion by inducing antiviral T cell motility paralysis. J Exp Med. 2013;210:757–74. doi: 10.1084/jem.20121416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Staron MM, Gray SM, Marshall HD, Parish IA, Chen JH, et al. The transcription factor FoxO1 sustains expression of the inhibitory receptor PD-1 and survival of antiviral CD8 T cells during chronic infection. Immunity. 2014;41:802–14. doi: 10.1016/j.immuni.2014.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Trautmann L, Mbitikon-Kobo FM, Goulet JP, Peretz Y, Shi Y, et al. Profound metabolic, functional, and cytolytic differences characterize HIV-specific CD8 T cells in primary and chronic HIV infection. Blood. 2012;120:3466–77. doi: 10.1182/blood-2012-04-422550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Schurich A, Henson SM. The many unknowns concerning the bioenergetics of exhaustion and senescence during chronic viral infection. Front Immunol. 2014;5:468. doi: 10.3389/fimmu.2014.00468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Chang CH, Curtis JD, Maggi LB, Jr, Faubert B, Villarino AV, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. 2013;153:1239–51. doi: 10.1016/j.cell.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Mackerness KJ, Cox MA, Lilly LM, Weaver CT, Harrington LE, Zajac AJ. Pronounced virus-dependent activation drives exhaustion but sustains IFN-γ transcript levels. J Immunol. 2010;185:3643–51. doi: 10.4049/jimmunol.1000841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sullivan JA, Kim EH, Plisch EH, Suresh M. FOXO3 regulates the CD8 T cell response to a chronic viral infection. J Virol. 2012;86:9025–34. doi: 10.1128/JVI.00942-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Finlay DK, Rosenzweig E, Sinclair LV, Feijoo-Carnero C, Hukelmann JL, et al. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J Exp Med. 2012;209:2441–53. doi: 10.1084/jem.20112607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Doedens AL, Phan AT, Stradner MH, Fujimoto JK, Nguyen JV, et al. Hypoxia-inducible factors enhance the effector responses of CD8+ T cells to persistent antigen. Nat Immunol. 2013;14:1173–82. doi: 10.1038/ni.2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–41. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Xu X, Araki K, Li S, Han JH, Ye L, et al. Autophagy is essential for effector CD8+ T cell survival and memory formation. Nat Immunol. 2014;15:1152–61. doi: 10.1038/ni.3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Angelosanto JM, Blackburn SD, Crawford A, Wherry EJ. Progressive loss of memory T cell potential and commitment to exhaustion during chronic viral infection. J Virol. 2012;86:8161–70. doi: 10.1128/JVI.00889-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Youngblood B, Noto A, Porichis F, Akondy RS, Ndhlovu ZM, et al. Cutting edge: Prolonged exposure to HIV reinforces a poised epigenetic program for PD-1 expression in virus-specific CD8 T cells. J Immunol. 2013;191:540–44. doi: 10.4049/jimmunol.1203161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Youngblood B, Oestreich KJ, Ha SJ, Duraiswamy J, Akondy RS, et al. Chronic virus infection enforces demethylation of the locus that encodes PD-1 in antigen-specific CD8+ T cells. Immunity. 2011;35:400–12. doi: 10.1016/j.immuni.2011.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zhang F, Zhou X, DiSpirito JR, Wang C, Wang Y, Shen H. Epigenetic manipulation restores functions of defective CD8+ T cells from chronic viral infection. Mol Ther. 2014;22:1698–706. doi: 10.1038/mt.2014.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Rasmussen TA, Tolstrup M, Winckelmann A, Ostergaard L, Sogaard OS. Eliminating the latent HIV reservoir by reactivation strategies: advancing to clinical trials. Hum Vaccines Immunother. 2013;9:790–99. doi: 10.4161/hv.23202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Cox MA, Kahan SM, Zajac AJ. Anti-viral CD8 T cells and the cytokines that they love. Virology. 2013;435:157–69. doi: 10.1016/j.virol.2012.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Nanjappa SG, Kim EH, Suresh M. Immunotherapeutic effects of IL-7 during a chronic viral infection in mice. Blood. 2011;117:5123–32. doi: 10.1182/blood-2010-12-323154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Pellegrini M, Calzascia T, Toe JG, Preston SP, Lin AE, et al. IL-7 engages multiple mechanisms to overcome chronic viral infection and limit organ pathology. Cell. 2011;144:601–13. doi: 10.1016/j.cell.2011.01.011. [DOI] [PubMed] [Google Scholar]
- 141.Pellegrini M, Calzascia T, Elford AR, Shahinian A, Lin AE, et al. Adjuvant IL-7 antagonizes multiple cellular and molecular inhibitory networks to enhance immunotherapies. Nat Med. 2009;15:528–36. doi: 10.1038/nm.1953. [DOI] [PubMed] [Google Scholar]
- 142.Yi JS, Du M, Zajac AJ. A vital role for interleukin-21 in the control of a chronic viral infection. Science. 2009;324:1572–76. doi: 10.1126/science.1175194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Keating SM, Jacobs ES, Norris PJ. Soluble mediators of inflammation in HIV and their implications for therapeutics and vaccine development. Cytokine Growth Factor Rev. 2012;23:193–206. doi: 10.1016/j.cytogfr.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Lugli E, Mueller YM, Lewis MG, Villinger F, Katsikis PD, Roederer M. IL-15 delays suppression and fails to promote immune reconstitution in virally suppressed chronically SIV-infected macaques. Blood. 2011;118:2520–29. doi: 10.1182/blood-2011-05-351155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Mueller YM, Do DH, Altork SR, Artlett CM, Gracely EJ, et al. IL-15 treatment during acute simian immunodeficiency virus (SIV) infection increases viral set point and accelerates disease progression despite the induction of stronger SIV-specific CD8+ T cell responses. J Immunol. 2008;180:350–60. doi: 10.4049/jimmunol.180.1.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Pallikkuth S, Rogers K, Villinger F, Dosterii M, Vaccari M, et al. Interleukin-21 administration to rhesus macaques chronically infected with simian immunodeficiency virus increases cytotoxic effector molecules in T cells and NK cells and enhances B cell function without increasing immune activation or viral replication. Vaccine. 2011;29:9229–38. doi: 10.1016/j.vaccine.2011.09.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Brooks DG, Ha SJ, Elsaesser H, Sharpe AH, Freeman GJ, Oldstone MB. IL-10 and PD-L1 operate through distinct pathways to suppress T-cell activity during persistent viral infection. PNAS. 2008;105:20428–33. doi: 10.1073/pnas.0811139106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Blackburn SD, Shin H, Freeman GJ, Wherry EJ. Selective expansion of a subset of exhausted CD8 T cells by αPD-L1 blockade. PNAS. 2008;105:15016–21. doi: 10.1073/pnas.0801497105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–87. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 150.Ejrnaes M, Filippi CM, Martinic MM, Ling EM, Togher LM, et al. Resolution of a chronic viral infection after interleukin-10 receptor blockade. J Exp Med. 2006;203:2461–72. doi: 10.1084/jem.20061462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Garidou L, Heydari S, Gossa S, McGavern DB. Therapeutic blockade of transforming growth factor beta fails to promote clearance of a persistent viral infection. J Virol. 2012;86:7060–71. doi: 10.1128/JVI.00164-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Boettler T, Cheng Y, Ehrhardt K, von Herrath M. TGF-β blockade does not improve control of an established persistent viral infection. Viral Immunol. 2012;25:232–38. doi: 10.1089/vim.2011.0079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Jin HT, Anderson AC, Tan WG, West EE, Ha SJ, et al. Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. PNAS. 2010;107:14733–38. doi: 10.1073/pnas.1009731107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Vezys V, Penaloza-MacMaster P, Barber DL, Ha SJ, Konieczny B, et al. 4-1BB signaling synergizes with programmed death ligand 1 blockade to augment CD8 T cell responses during chronic viral infection. J Immunol. 2011;187:1634–42. doi: 10.4049/jimmunol.1100077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Boettler T, Choi YS, Salek-Ardakani S, Cheng Y, Moeckel F, et al. Exogenous OX40 stimulation during lymphocytic choriomeningitis virus infection impairs follicular Th cell differentiation and diverts CD4 T cells into the effector lineage by upregulating Blimp-1. J Immunol. 2013;191:5026–35. doi: 10.4049/jimmunol.1300013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.West EE, Jin HT, Rasheed AU, Penaloza-MacMaster P, Ha SJ, et al. PD-L1 blockade synergizes with IL-2 therapy in reinvigorating exhausted T cells. J Clin Investig. 2013;123:2604–15. doi: 10.1172/JCI67008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Gardiner D, Lalezari J, Lawitz E, DiMicco M, Ghalib R, et al. A randomized, double-blind, placebo-controlled assessment of BMS-936558, a fully human monoclonal antibody to programmed death-1 (PD-1), in patients with chronic hepatitis C virus infection. PLOS ONE. 2013;8:e63818. doi: 10.1371/journal.pone.0063818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Porichis F, Kaufmann DE. HIV-specific CD4 T cells and immune control of viral replication. Curr Opin HIV AIDS. 2011;6:174–80. doi: 10.1097/COH.0b013e3283454058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Thimme R, Oldach D, Chang KM, Steiger C, Ray SC, Chisari FV. Determinants of viral clearance and persistence during acute hepatitis C virus infection. J Exp Med. 2001;194:1395–406. doi: 10.1084/jem.194.10.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Diepolder HM, Zachoval R, Hoffmann RM, Wierenga EA, Santantonio T, et al. Possible mechanism involving T-lymphocyte response to non-structural protein 3 in viral clearance in acute hepatitis C virus infection. Lancet. 1995;346:1006–7. doi: 10.1016/s0140-6736(95)91691-1. [DOI] [PubMed] [Google Scholar]
- 161.Loggi E, Gamal N, Bihl F, Bernardi M, Andreone P. Adaptive response in hepatitis B virus infection. J Viral Hepatol. 2014;21:305–13. doi: 10.1111/jvh.12255. [DOI] [PubMed] [Google Scholar]
- 162.Julg B, Moodley ES, Qi Y, Ramduth D, Reddy S, et al. Possession of HLA class II DRB1*1303 associates with reduced viral loads in chronic HIV-1 clade C and B infection. J Infect Dis. 2011;203:803–9. doi: 10.1093/infdis/jiq122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Ranasinghe S, Cutler S, Davis I, Lu R, Soghoian DZ, et al. Association of HLA-DRB1-restricted CD4+ T cell responses with HIV immune control. Nat Med. 2013;19:930–33. doi: 10.1038/nm.3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Grakoui A, Shoukry NH, Woollard DJ, Han JH, Hanson HL, et al. HCV persistence and immune evasion in the absence of memory T cell help. Science. 2003;302:659–62. doi: 10.1126/science.1088774. [DOI] [PubMed] [Google Scholar]
- 165.Asabe S, Wieland SF, Chattopadhyay PK, Roederer M, Engle RE, et al. The size of the viral inoculum contributes to the outcome of hepatitis B virus infection. J Virol. 2009;83:9652–62. doi: 10.1128/JVI.00867-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Battegay M, Moskophidis D, Rahemtulla A, Hengartner H, Mak TW, Zinkernagel RM. Enhanced establishment of a virus carrier state in adult CD4+ T-cell-deficient mice. J Virol. 1994;68:4700–4. doi: 10.1128/jvi.68.7.4700-4704.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Aubert RD, Kamphorst AO, Sarkar S, Vezys V, Ha SJ, et al. Antigen-specific CD4 T-cell help rescues exhausted CD8 T cells during chronic viral infection. PNAS. 2011;108:21182–87. doi: 10.1073/pnas.1118450109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Oxenius A, Zinkernagel RM, Hengartner H. Comparison of activation versus induction of unresponsiveness of virus-specific CD4+ and CD8+ T cells upon acute versus persistent viral infection. Immunity. 1998;9:449–57. doi: 10.1016/s1074-7613(00)80628-7. [DOI] [PubMed] [Google Scholar]
- 169.Penaloza-MacMaster P, Barber DL, Wherry EJ, Provine NM, Teigler JE, et al. Vaccine-elicited CD4 T cells induce immunopathology after chronic LCMV infection. Science. 2015;347:278–82. doi: 10.1126/science.aaa2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Waggoner SN, Cornberg M, Selin LK, Welsh RM. Natural killer cells act as rheostats modulating antiviral T cells. Nature. 2012;481:394–98. doi: 10.1038/nature10624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Fuller MJ, Zajac AJ. Ablation of CD8 and CD4 T cell responses by high viral loads. J Immunol. 2003;170:477–86. doi: 10.4049/jimmunol.170.1.477. [DOI] [PubMed] [Google Scholar]
- 172.Brooks DG, Teyton L, Oldstone MB, McGavern DB. Intrinsic functional dysregulation of CD4 T cells occurs rapidly following persistent viral infection. J Virol. 2005;79:10514–27. doi: 10.1128/JVI.79.16.10514-10527.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Kasprowicz V, Schulze Zur Wiesch J, Kuntzen T, Nolan BE, Longworth S, et al. High level of PD-1 expression on hepatitis C virus (HCV)-specific CD8+ and CD4+ T cells during acute HCV infection, irrespective of clinical outcome. J Virol. 2008;82:3154–60. doi: 10.1128/JVI.02474-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Raziorrouh B, Ulsenheimer A, Schraut W, Heeg M, Kurktschiev P, et al. Inhibitory molecules that regulate expansion and restoration of HCV-specific CD4+ T cells in patients with chronic infection. Gastroenterology. 2011;141:1422–31. doi: 10.1053/j.gastro.2011.07.004. [DOI] [PubMed] [Google Scholar]
- 175.Kaufmann DE, Walker BD. PD-1 and CTLA-4 inhibitory cosignaling pathways in HIV infection and the potential for therapeutic intervention. J Immunol. 2009;182:5891–97. doi: 10.4049/jimmunol.0803771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Kassu A, Marcus RA, D’Souza MB, Kelly-McKnight EA, Golden-Mason L, et al. Regulation of virus-specific CD4+ T cell function by multiple costimulatory receptors during chronic HIV infection. J Immunol. 2010;185:3007–18. doi: 10.4049/jimmunol.1000156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Kaufmann DE, Kavanagh DG, Pereyra F, Zaunders JJ, Mackey EW, et al. Upregulation of CTLA-4 by HIV-specific CD4+ T cells correlates with disease progression and defines a reversible immune dysfunction. Nat Immunol. 2007;8:1246–54. doi: 10.1038/ni1515. [DOI] [PubMed] [Google Scholar]
- 178.Ng CT, Oldstone MB. IL-10: achieving balance during persistent viral infection. Curr Top Microbiol Immunol. 2014;380:129–44. doi: 10.1007/978-3-662-43492-5_6. [DOI] [PubMed] [Google Scholar]
- 179.Kaplan DE, Ikeda F, Li Y, Nakamoto N, Ganesan S, et al. Peripheral virus-specific T-cell interleukin-10 responses develop early in acute hepatitis C infection and become dominant in chronic hepatitis. J Hepatol. 2008;48:903–13. doi: 10.1016/j.jhep.2008.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Elsaesser H, Sauer K, Brooks DG. IL-21 is required to control chronic viral infection. Science. 2009;324:1569–72. doi: 10.1126/science.1174182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Frohlich A, Kisielow J, Schmitz I, Freigang S, Shamshiev AT, et al. IL-21R on T cells is critical for sustained functionality and control of chronic viral infection. Science. 2009;324:1576–80. doi: 10.1126/science.1172815. [DOI] [PubMed] [Google Scholar]
- 182.Schmitz I, Schneider C, Frohlich A, Frebel H, Christ D, et al. IL-21 restricts virus-driven Treg cell expansion in chronic LCMV infection. PLOS Pathog. 2013;9:e1003362. doi: 10.1371/journal.ppat.1003362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Ozaki K, Spolski R, Feng CG, Qi CF, Cheng J, et al. A critical role for IL-21 in regulating immunoglobulin production. Science. 2002;298:1630–34. doi: 10.1126/science.1077002. [DOI] [PubMed] [Google Scholar]
- 184.Chevalier MF, Julg B, Pyo A, Flanders M, Ranasinghe S, et al. HIV-1-specific interleukin-21+ CD4+ T cell responses contribute to durable viral control through the modulation of HIV-specific CD8+ T cell function. J Virol. 2011;85:733–41. doi: 10.1128/JVI.02030-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Feng G, Zhang JY, Zeng QL, Jin L, Fu J, et al. HCV-specific interleukin-21+CD4+ T cells responses associated with viral control through the modulation of HCV-specific CD8+ T cells function in chronic hepatitis C patients. Mol Cells. 2013;36:362–67. doi: 10.1007/s10059-013-0181-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Iannello A, Boulassel MR, Samarani S, Debbeche O, Tremblay C, et al. Dynamics and consequences of IL-21 production in HIV-infected individuals: a longitudinal and cross-sectional study. J Immunol. 2010;184:114–26. doi: 10.4049/jimmunol.0901967. [DOI] [PubMed] [Google Scholar]
- 187.Publicover J, Goodsell A, Nishimura S, Vilarinho S, Wang ZE, et al. IL-21 is pivotal in determining age-dependent effectiveness of immune responses in a mouse model of human hepatitis B. J Clin Investig. 2011;121:1154–62. doi: 10.1172/JCI44198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Ren G, Esser S, Jochum C, Schlaak JF, Gerken G, et al. Interleukin 21 augments the hepatitis B virus-specific CD8+ T-cell response in vitro in patients coinfected with HIV-1. AIDS. 2012;26:2145–53. doi: 10.1097/QAD.0b013e328359b7ae. [DOI] [PubMed] [Google Scholar]
- 189.Williams LD, Bansal A, Sabbaj S, Heath SL, Song W, et al. Interleukin-21-producing HIV-1-specific CD8 T cells are preferentially seen in elite controllers. J Virol. 2011;85:2316–24. doi: 10.1128/JVI.01476-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Yue FY, Lo C, Sakhdari A, Lee EY, Kovacs CM, et al. HIV-specific IL-21 producing CD4+ T cells are induced in acute and chronic progressive HIV infection and are associated with relative viral control. J Immunol. 2010;185:498–506. doi: 10.4049/jimmunol.0903915. [DOI] [PubMed] [Google Scholar]
- 191.Harker JA, Dolgoter A, Zuniga EI. Cell-intrinsic IL-27 and gp130 cytokine receptor signaling regulates virus-specific CD4+ T cell responses and viral control during chronic infection. Immunity. 2013;39:548–59. doi: 10.1016/j.immuni.2013.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol. 2012;30:531–64. doi: 10.1146/annurev.immunol.25.022106.141623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Iwashiro M, Messer RJ, Peterson KE, Stromnes IM, Sugie T, Hasenkrug KJ. Immunosuppression by CD4+ regulatory T cells induced by chronic retroviral infection. PNAS. 2001;98:9226–30. doi: 10.1073/pnas.151174198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Kinter AL, Hennessey M, Bell A, Kern S, Lin Y, et al. CD25+CD4+ regulatory T cells from the peripheral blood of asymptomatic HIV-infected individuals regulate CD4+ and CD8+ HIV-specific T cell immune responses in vitro and are associated with favorable clinical markers of disease status. J Exp Med. 2004;200:331–43. doi: 10.1084/jem.20032069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Weiss L, Donkova-Petrini V, Caccavelli L, Balbo M, Carbonneil C, Levy Y. Human immunodeficiency virus-driven expansion of CD4+CD25+ regulatory T cells, which suppress HIV-specific CD4 T-cell responses in HIV-infected patients. Blood. 2004;104:3249–56. doi: 10.1182/blood-2004-01-0365. [DOI] [PubMed] [Google Scholar]
- 196.Boettler T, Spangenberg HC, Neumann-Haefelin C, Panther E, Urbani S, et al. T cells with a CD4+CD25+ regulatory phenotype suppress in vitro proliferation of virus-specific CD8+ T cells during chronic hepatitis C virus infection. J Virol. 2005;79:7860–67. doi: 10.1128/JVI.79.12.7860-7867.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Punkosdy GA, Blain M, Glass DD, Lozano MM, O’Mara L, et al. Regulatory T-cell expansion during chronic viral infection is dependent on endogenous retroviral superantigens. PNAS. 2011;108:3677–82. doi: 10.1073/pnas.1100213108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Sugimoto K, Ikeda F, Stadanlick J, Nunes FA, Alter HJ, Chang KM. Suppression of HCV-specific T cells without differential hierarchy demonstrated ex vivo in persistent HCV infection. Hepatology. 2003;38:1437–48. doi: 10.1016/j.hep.2003.09.026. [DOI] [PubMed] [Google Scholar]
- 199.Zelinskyy G, Dietze KK, Husecken YP, Schimmer S, Nair S, et al. The regulatory T-cell response during acute retroviral infection is locally defined and controls the magnitude and duration of the virus-specific cytotoxic T-cell response. Blood. 2009;114:3199–207. doi: 10.1182/blood-2009-03-208736. [DOI] [PubMed] [Google Scholar]
- 200.Dietze KK, Zelinskyy G, Gibbert K, Schimmer S, Francois S, et al. Transient depletion of regulatory T cells in transgenic mice reactivates virus-specific CD8+ T cells and reduces chronic retroviral set points. PNAS. 2011;108:2420–25. doi: 10.1073/pnas.1015148108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Penaloza-MacMaster P, Kamphorst AO, Wieland A, Araki K, Iyer SS, et al. Interplay between regulatory T cells and PD-1 in modulating T cell exhaustion and viral control during chronic LCMV infection. J Exp Med. 2014;211:1905–18. doi: 10.1084/jem.20132577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Eggena MP, Barugahare B, Jones N, Okello M, Mutalya S, et al. Depletion of regulatory T cells in HIV infection is associated with immune activation. J Immunol. 2005;174:4407–14. doi: 10.4049/jimmunol.174.7.4407. [DOI] [PubMed] [Google Scholar]
- 203.Oswald-Richter K, Grill SM, Shariat N, Leelawong M, Sundrud MS, et al. HIV infection of naturally occurring and genetically reprogrammed human regulatory T-cells. PLOS Biol. 2004;2:e198. doi: 10.1371/journal.pbio.0020198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Kornfeld C, Ploquin MJ, Pandrea I, Faye A, Onanga R, et al. Antiinflammatory profiles during primary SIV infection in African green monkeys are associated with protection against AIDS. J Clin Investig. 2005;115:1082–91. doi: 10.1172/JCI23006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.MacDonald AJ, Duffy M, Brady MT, McKiernan S, Hall W, et al. CD4 T helper type 1 and regulatory T cells induced against the same epitopes on the core protein in hepatitis C virus-infected persons. J Infect Dis. 2002;185:720–27. doi: 10.1086/339340. [DOI] [PubMed] [Google Scholar]
- 206.Medzhitov R, Schneider DS, Soares MP. Disease tolerance as a defense strategy. Science. 2012;335:936–41. doi: 10.1126/science.1214935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Crotty S. T follicular helper cell differentiation, function, and roles in disease. Immunity. 2014;41:529–42. doi: 10.1016/j.immuni.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Ramiscal RR, Vinuesa CG. T-cell subsets in the germinal center. Immunol Rev. 2013;252:146–55. doi: 10.1111/imr.12031. [DOI] [PubMed] [Google Scholar]
- 209.Fahey LM, Wilson EB, Elsaesser H, Fistonich CD, McGavern DB, Brooks DG. Viral persistence redirects CD4 T cell differentiation toward T follicular helper cells. J Exp Med. 2011;208:987–99. doi: 10.1084/jem.20101773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Harker JA, Lewis GM, Mack L, Zuniga EI. Late interleukin-6 escalates T follicular helper cell responses and controls a chronic viral infection. Science. 2011;334:825–29. doi: 10.1126/science.1208421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Feng J, Hu X, Guo H, Sun X, Wang J, et al. Patients with chronic hepatitis C express a high percentage of CD4+CXCR5+ T follicular helper cells. J Gastroenterol. 2012;47:1048–56. doi: 10.1007/s00535-012-0568-1. [DOI] [PubMed] [Google Scholar]
- 212.Feng J, Lu L, Hua C, Qin L, Zhao P, et al. High frequency of CD4+CXCR5+ TFH cells in patients with immune-active chronic hepatitis B. PLOS ONE. 2011;6:e21698. doi: 10.1371/journal.pone.0021698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Lindqvist M, van Lunzen J, Soghoian DZ, Kuhl BD, Ranasinghe S, et al. Expansion of HIV-specific T follicular helper cells in chronic HIV infection. J Clin Investig. 2012;122:3271–80. doi: 10.1172/JCI64314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Petrovas C, Yamamoto T, Gerner MY, Boswell KL, Wloka K, et al. CD4 T follicular helper cell dynamics during SIV infection. J Clin Investig. 2012;122:3281–94. doi: 10.1172/JCI63039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Locci M, Havenar-Daughton C, Landais E, Wu J, Kroenke MA, et al. Human circulating PD-1+CXCR3−CXCR5+ memory Tfh cells are highly functional and correlate with broadly neutralizing HIV antibody responses. Immunity. 2013;39:758–69. doi: 10.1016/j.immuni.2013.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Pallikkuth S, Parmigiani A, Silva SY, George VK, Fischl M, et al. Impaired peripheral blood T-follicular helper cell function in HIV-infected nonresponders to the 2009 H1N1/09 vaccine. Blood. 2012;120:985–93. doi: 10.1182/blood-2011-12-396648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Cubas RA, Mudd JC, Savoye AL, Perreau M, van Grevenynghe J, et al. Inadequate T follicular cell help impairs B cell immunity during HIV infection. Nat Med. 2013;19:494–99. doi: 10.1038/nm.3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Boswell KL, Paris R, Boritz E, Ambrozak D, Yamamoto T, et al. Loss of circulating CD4 T cells with B cell helper function during chronic HIV infection. PLOS Pathog. 2014;10:e1003853. doi: 10.1371/journal.ppat.1003853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Planz O, Ehl S, Furrer E, Horvath E, Brundler MA, et al. A critical role for neutralizing-antibody-producing B cells, CD4+ T cells, and interferons in persistent and acute infections of mice with lymphocytic choriomeningitis virus: implications for adoptive immunotherapy of virus carriers. PNAS. 1997;94:6874–79. doi: 10.1073/pnas.94.13.6874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Thomsen AR, Johansen J, Marker O, Christensen JP. Exhaustion of CTL memory and recrudescence of viremia in lymphocytic choriomeningitis virus-infected MHC class II-deficient mice and B cell-deficient mice. J Immunol. 1996;157:3074–80. [PubMed] [Google Scholar]
- 221.Hangartner L, Zellweger RM, Giobbi M, Weber J, Eschli B, et al. Nonneutralizing antibodies binding to the surface glycoprotein of lymphocytic choriomeningitis virus reduce early virus spread. J Exp Med. 2006;203:2033–42. doi: 10.1084/jem.20051557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Bergthaler A, Flatz L, Verschoor A, Hegazy AN, Holdener M, et al. Impaired antibody response causes persistence of prototypic T cell-contained virus. PLOS Biol. 2009;7:e1000080. doi: 10.1371/journal.pbio.1000080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Bukh J, Thimme R, Meunier JC, Faulk K, Spangenberg HC, et al. Previously infected chimpanzees are not consistently protected against reinfection or persistent infection after reexposure to the identical hepatitis C virus strain. J Virol. 2008;82:8183–95. doi: 10.1128/JVI.00142-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Grady BP, Schinkel J, Thomas XV, Dalgard O. Hepatitis C virus reinfection following treatment among people who use drugs. Clin Infect Dis. 2013;57(Suppl. 2):S105–10. doi: 10.1093/cid/cit301. [DOI] [PubMed] [Google Scholar]
- 225.Euler Z, van Gils MJ, Bunnik EM, Phung P, Schweighardt B, et al. Cross-reactive neutralizing humoral immunity does not protect from HIV type 1 disease progression. J Infect Dis. 2010;201:1045–53. doi: 10.1086/651144. [DOI] [PubMed] [Google Scholar]
- 226.Wei X, Decker JM, Wang S, Hui H, Kappes JC, et al. Antibody neutralization and escape by HIV-1. Nature. 2003;422:307–12. doi: 10.1038/nature01470. [DOI] [PubMed] [Google Scholar]
- 227.McMichael AJ, Borrow P, Tomaras GD, Goonetilleke N, Haynes BF. The immune response during acute HIV-1 infection: clues for vaccine development. Nat Rev Immunol. 2010;10:11–23. doi: 10.1038/nri2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.de Jong YP, Dorner M, Mommersteeg MC, Xiao JW, Balazs AB, et al. Broadly neutralizing antibodies abrogate established hepatitis C virus infection. Sci Transl Med. 2014;6:254ra129. doi: 10.1126/scitranslmed.3009512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Balazs AB, Chen J, Hong CM, Rao DS, Yang L, Baltimore D. Antibody-based protection against HIV infection by vectored immunoprophylaxis. Nature. 2012;481:81–84. doi: 10.1038/nature10660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Hong JJ, Amancha PK, Rogers K, Ansari AA, Villinger F. Spatial alterations between CD4+ T follicular helper, B, and CD8+ T cells during simian immunodeficiency virus infection: T/B cell homeostasis, activation, and potential mechanism for viral escape. J Immunol. 2012;188:3247–56. doi: 10.4049/jimmunol.1103138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Perreau M, Savoye AL, De Crignis E, Corpataux JM, Cubas R, et al. Follicular helper T cells serve as the major CD4 T cell compartment for HIV-1 infection, replication, and production. J Exp Med. 2013;210:143–56. doi: 10.1084/jem.20121932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Hufert FT, van Lunzen J, Janossy G, Bertram S, Schmitz J, et al. Germinal centre CD4+ T cells are an important site of HIV replication in vivo. AIDS. 1997;11:849–57. doi: 10.1097/00002030-199707000-00003. [DOI] [PubMed] [Google Scholar]
- 233.Xu Y, Weatherall C, Bailey M, Alcantara S, De Rose R, et al. Simian immunodeficiency virus infects follicular helper CD4 T cells in lymphoid tissues during pathogenic infection of pigtail macaques. J Virol. 2013;87:3760–73. doi: 10.1128/JVI.02497-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Fukazawa Y, Lum R, Okoye AA, Park H, Matsuda K, et al. B cell follicle sanctuary permits persistent productive simian immunodeficiency virus infection in elite controllers. Nat Med. 2015;21:132–39. doi: 10.1038/nm.3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Baumjohann D, Preite S, Reboldi A, Ronchi F, Ansel KM, et al. Persistent antigen and germinal center B cells sustain T follicular helper cell responses and phenotype. Immunity. 2013;38:596–605. doi: 10.1016/j.immuni.2012.11.020. [DOI] [PubMed] [Google Scholar]
- 236.Deenick EK, Chan A, Ma CS, Gatto D, Schwartzberg PL, et al. Follicular helper T cell differentiation requires continuous antigen presentation that is independent of unique B cell signaling. Immunity. 2010;33:241–53. doi: 10.1016/j.immuni.2010.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Kang SG, Liu WH, Lu P, Jin HY, Lim HW, et al. MicroRNAs of the miR-17~92 family are critical regulators of TFH differentiation. Nat Immunol. 2013;14:849–57. doi: 10.1038/ni.2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Crawford A, Angelosanto JM, Kao C, Doering TA, Odorizzi PM, et al. Molecular and transcriptional basis of CD4+ T cell dysfunction during chronic infection. Immunity. 2014;40:289–302. doi: 10.1016/j.immuni.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Quirion MR, Gregory GD, Umetsu SE, Winandy S, Brown MA. Cutting edge: Ikaros is a regulator of Th2 cell differentiation. J Immunol. 2009;182:741–45. doi: 10.4049/jimmunol.182.2.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Curran MA, Geiger TL, Montalvo W, Kim M, Reiner SL, et al. Systemic 4-1BB activation induces a novel T cell phenotype driven by high expression of Eomesodermin. J Exp Med. 2013;210:743–55. doi: 10.1084/jem.20121190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Speiser DE, Utzschneider DT, Oberle SG, Munz C, Romero P, Zehn D. T cell differentiation in chronic infection and cancer: functional adaptation or exhaustion? Nat Rev Immunol. 2014;14:768–74. doi: 10.1038/nri3740. [DOI] [PubMed] [Google Scholar]