

Abstract
Transcriptional regulation is a tightly regulated, vital process. The transcription factor cyclic AMP-response element-binding protein 1 (CREB1) controls ∼25% of the mammalian transcriptome by binding the CREB1 binding site consensus sequence (CRE) sequence (TGACGTCA). DNA lesions within CRE modulate CREB1 binding negatively and positively. Because appropriate DNA lesions also interact with base excision repair proteins, we investigated whether CREB1 and repair glycosylases compete with each other. We incubated 39-mer CRE-containing double-stranded oligonucleotides with recombinant CREB1 alone or with UNG2 or OGG1, followed by EMSA. The CpG islet within CRE was modified to contain a G/U or 8-oxoG (°G)/C mispair. OGG1 and CREB1 reversibly competed for CRE containing an °G/C pair. Also, OGG1 blocked CREB1 from dimerizing by 69%, even when total CREB1 binding was reduced only by 20–30%. In contrast, bound CREB1 completely prevented access to G/U-containing CRE by UNG2 and, therefore, to base excision repair, whereas UNG2 exposure prevented CREB1 binding. CREB1 dimerization was unaffected by UNG2 when CREB1 bound to CRE, but was greatly reduced by prior UNG2 exposure. To explore physiological relevance, we microinjected zebrafish embryos with the same oligonucleotides, as a sink for endogenous CREB1. As predicted, microinjection with unmodified or lesion-containing CRE, but not scrambled CRE or scrambled CRE with a G/U mispair, resulted in increased embryo death. However, only the G/U mispair in native CRE resulted in substantial developmental abnormalities, thus confirming the danger of unrepaired G/U mispairs in promoters. In summary, CREB1 and DNA glycosylases compete for damaged CRE in vitro and in vivo, thus blocking DNA repair and resulting in transcriptional misregulation leading to abnormal development.
Keywords: 8-Oxoguanine (8-oxoG), 8-Oxoguanine glycosylase (OGG1), cAMP-response element-binding protein (CREB), development, DNA repair, embryo, transcription, zebrafish, G/U mispair, uracil DNA glycosylase, DNA base excision repair, CREB1 transcription factor, glycosylase/transcription factor competition, DNA damage repair
Introduction
The study of DNA damage is usually focused on mutation and cellular misregulation that lead to aberrant development or cancer. However, oxidative damage is ongoing, and selective DNA oxidation is associated with transcription activation, such as estrogen-dependent and c-MYC-dependent transcriptional initiation (1, 2). Less well known is that DNA damage, specifically substrates and intermediates in the base excision repair (BER)2 pathway, can modulate transcription factor (TF) binding itself (3). The relative lack of nucleosomes in promoter regions ensures that these regions are especially prone to ongoing oxidative damage or accumulation of uracil/guanine or thymidine/guanine pairing resulting from demethylation immediately after fertilization.
Cyclic AMP-response element-binding protein (CREB1) is a critical TF that regulates ∼25% of the eukaryotic genome (4, 5). CREB1 binds to the palindromic cAMP-responsive element (CRE) site TGACGTCA or the half-CRE site CGTCA and acts as a required pleiotropic effector (6). CREB1 is also self-regulating with 13 CRE sites in its own promoter. It is responsible for initial development of the nervous system, memory formation, and neuronal protection (7, 8). Misregulation of CREB1 is implicated in a range of congenital and acquired central nervous system disorders, including Alzheimer disease and Parkinson disease (9). Methylation of the central CpG in the consensus sequence prevents binding of CREB1 and consequently prevents transcription of CREB1-dependent genes (10). Transcription activation of the central methylated CpG requires demethylation that involves AID/APOBEC or TET enzymes followed by DNA repair via the BER pathway (11, 12). Indeed, uracil DNA glycosylase 2 (UNG2) is so important in early zebrafish embryogenesis that knockdown is embryonic lethal, whereas overexpression is sufficient to reduce global DNA methylation (13).
Oxidative stress, an imbalance between the production and biological detoxification of reactive oxygen species (ROS), has also been linked with several CNS diseases, including Alzheimer disease and Parkinson disease (14). Although a variety of external sources can contribute to ROS levels, endogenous ROS are produced continuously as a byproduct of oxidative phosphorylation (15). Although ROS damage all cellular components, oxidatively damaged DNA must be repaired quickly or genomic instability results (14). Small, non-bulky DNA lesions such as those produced by ROS are recognized and repaired by the BER pathway (reviewed in Ref. 16). Several neurodegenerative diseases display increased levels of oxidative DNA damage, coupled with reduced DNA repair capability (17). Furthermore, many neurological disorders, including neurodegeneration, are associated with inherited defects of DNA repair pathways including BER (18). In a prior publication, we showed that specific DNA lesions within the CRE site that are repaired by BER can both positively and negatively modulate CREB1 recognition and binding to CRE, depending upon the specific site and type of damage (3). We therefore proposed that oxidative damage or repair intermediates in BER might modulate TF efficacy, a proposal consistent with that of others (14). In this study, we use both in vitro and in vivo methods to investigate whether and how BER enzymes and CREB1 compete for access to CRE site substrates containing DNA lesions typically repaired by BER. We show that CREB1 can block access of DNA glycosylases to DNA lesions and, in turn, DNA glycosylases can interfere with or prevent access of CREB1 to CRE. These results are relevant to studies of developmental biology, neurodegeneration, and aging.
Experimental Procedures
Oligonucleotides
39-mer double-stranded (ds) oligonucleotides (see Table 1) containing an unmodified or modified CRE site were sourced either from Midland Certified Reagent Co., Inc. (Midland, TX) or from Integrated DNA Technologies (Coralville, IA). Either upper or lower strands were labeled at the 5′ end with [γ-32P]ATP (PerkinElmer) by polynucleotide kinase (New England Biolabs, Ipswich, MA) as described previously (19, 20) and stored at −20 °C until use.
TABLE 1.
Modifications made to CRE site used in this study
The Modification column shows the actual residue modification made to the CRE site. The residues and positions modified within the CRE site are denoted in the Location column. Modifications were made to, or opposite, the G2 residue within the CpG islet. The Strand column indicates the modified strand. T = top strand; B = bottom strand. The Sequence column shows the complete CRE site sequence including the modified residue(s). The full-length unmodified 39-mer CRE oligonucleotide is shown with the CRE site in bold in the bottom row. Numerals 1 and 2 indicate the guanine residues within the top strand of the CRE site. All modifications in this study were located at the G2 position or its complement. NA, not applicable.
| Modification | Location | Strand | Sequence |
|---|---|---|---|
| CRE | NA | T | T: 5′-TG ACG TCA- |
| B: 3′-AC TGC AGT- | |||
| 8-oxoguanine (oG) | G2 (CpG) | T | T: 5′-TG ACoG TCA- |
| B: 3′-AC TGC AGT- | |||
| G/U mispair (G/U) | G2 (CpG) | B | T: 5′-TG ACG TCA- |
| B: 3′-AC TGU AGT- | |||
| Scrambled CRE | Entire CRE-site | Both | T: 5′-GT CAT GAC- |
| B: 3′-CA GTA CTG- | |||
| Scrambled CRE (G/U) | Entire CRE-site U in bottom strand | Both | T: 5′-GT CAT GAC- |
| B: 3′-CA GTA UTG- | |||
| Full oligonucleotide sequence | 5′-TAC CAT GCC TTG1 ACG2 TCA GAG AGC ATT CGT AAT CAT GGT-3′ | ||
Source of Proteins
Purified recombinant human CREB1 protein was the gift of Dr. Jennifer Nyborg (Colorado State University, Fort Collins, CO), and purified recombinant UNG2 and 8-oxoguanine DNA glycosylase (OGG1) were the gifts of Dr. Nicole Noren Hooten (National Institute of Aging, Baltimore, MD). Purified recombinant mutant UNG2 (His-UNG2-D154N) was the gift of Dr. Bodil Kavli (Norwegian University of Science and Technology, Trondheim, Norway). AP endonuclease 1 (APEX1) was purified as described previously (19).
EMSA Assays
Competition EMSAs were modified from those described previously (3, 21). Briefly, 8.5 nm purified CREB1 protein was incubated with varying concentrations of purified UNG2 (0–64 nm) or OGG1 (0–64 nm) protein in a reaction buffer with 5 ng of poly(dA-dT), 0.25 μg/μl BSA, and 0.5 nm 32P end-labeled ds oligonucleotide probe, in a final volume of 20 μl. All incubations were carried out at 4 °C for 90 min, with variations in the order of addition of protein and oligonucleotide. In the first series, CREB1 and a DNA glycosylase were mixed together for 30 min followed by the addition of the appropriate oligonucleotide for 60 min. In the second series, CREB1 was allowed to bind with the appropriate oligonucleotide for 30 min after which the glycosylase was added for an additional 60 min. In the third series, the glycosylase was allowed to bind to the appropriate oligonucleotide for 30 min, after which time CREB1 was added for an additional 60 min. Fig. 1 is a representative EMSA showing resolution of CREB monomer and dimer as well as binding of OGG1 in the absence of CREB1. All experiments were performed independently a minimum of three times. To demonstrate specificity, °G-containing substrates were incubated with UNG2 and CREB1 and G/U-containing substrates were incubated with OGG1 and CREB1. No displacement of CREB1 was observed. Samples without tracking dye were resolved on pre-electrophoresed, native 5% (49:1 acrylamide:bisacrylamide) gels in buffer composed of 40 mm Tris-HCl (pH 8.5), 306 mm Tris-glycine, and 0.1% Nonidet P-40. Electrophoresis was performed at room temperature at 90 V for 2.5 h.
FIGURE 1.

Monomer and dimer CREB1 binding to the CRE site contained within a ds 39-mer oligonucleotide are cleanly resolved by EMSA and are differentially impacted by UNG and OGG1. In these representative gel images, UNG2 and CREB1, or OGG1 and CREB1, were mixed together followed by the addition of the labeled oligonucleotide (oligo) and incubated for 90 min at 4 °C prior to EMSA analysis. Note detectable OGG1 binding to the °G-containing oligonucleotide. Apparent reduction in unbound substrate at low concentrations of OGG1 is due to direct binding by the repair glycosylase. On the other hand, binding of UNG2 to U-containing CRE site is not sufficiently stable to detect in the absence of CREB1. Typical gels were obtained when UNG2 was added to substrate containing a G/U mispair (left) or when OGG1 was added to substrate containing a °G/C pair (right) in the CpG site of CRE.
Analysis
Distribution of isotope was visualized on a PhosphorImager screen with a GE StormTM PhosphorImagerTM (GE Healthcare, Little Chalfont, UK); data were quantified with GE Healthcare ImageQuant (19). CREB1 binding was determined by Reaction 1.
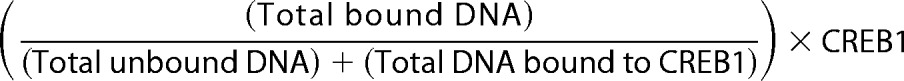 |
Cleavage Activity in the Presence and Absence of CREB1
Purified recombinant CREB1 (8.5 nm) was added to UNG2 (85 nm) or OGG1 (85 nm) in reaction buffer with 5 ng of poly(dA-dT) and 0.25 μg/μl BSA. The 32P end-labeled ds oligonucleotide probe (see Table 1) was added, and the reaction was incubated on ice for 1 h in a final volume of 20 μl. DNA was isolated by phenol:chloroform extraction as described previously (19) and resuspended in 5 μl of buffer containing 50 mm HEPES and 0.1 mm EDTA. Purified APEX1 protein (10 nm) was added to 2 μl of resuspended DNA samples in a final reaction volume of 5 μl and incubated for 30 min at room temperature. The cleavage assay was stopped by the addition of formamide loading buffer (80% formamide, 2% 0.5 m EDTA, and 10% xylene cyanol/bromphenol blue). Cleavage products were resolved by denaturing gel electrophoresis as described previously (19).
Zebrafish Culture and Decoy CRE site Oligonucleotide Microinjections
Wild-type zebrafish (Danio rerio), purchased from Aquatica BioTech (Sun City Center, FL), were maintained and bred using standard protocols in accordance with approved Northeastern University Institutional Animal Care and Use Committee (IACUC) policies as described previously (22). Embryos were microinjected directly into the yolk prior to the 4-cell stage with 2 nl of 39-mer (0.18 nm) ds oligonucleotide substrate (see Table 1) in Danieau buffer. Phenol red (0.05%) was used as an injection indicator (23). Phenol red (0.05%) in Danieau buffer served as the control. Injected embryos were maintained at 29 °C until reaching desired developmental stages and fixed in 4% paraformaldehyde (24). Embryos were examined and photographed using a Leica MZ16FA stereomicroscope (Leica, Wetzlar, Germany).
Statistical Analyses
Data in Figs. 2 and 3 were analyzed by mixed model analyses of covariance using SPSS 19.0 (IBM, Armonk, NY). Prior to analysis, data were transformed using a natural logarithmic transformation to obviate heteroscedasticity. Homogeneity of variance was verified by Levene's test. Tukey's post hoc analysis was conducted on the factor in the absence of a significant interaction between factor and covariate. When a significant interaction was present between the factor and covariate, simple linear regression was performed.
FIGURE 2.

Both OGG1 and UNG2 impede CREB1 from binding to a CRE site substrate when the CRE site contains the specific DNA lesion recognized by each glycosylase. Interference depended on the lesion, concentration of glycosylase, and order of exposure. A–C, OGG1 in increasing concentrations competes with CREB1 for an °G-containing substrate. A, OGG1 added before CREB1; B, CREB1 added before OGG1; C, OGG1 and CREB1 added together. Total bound CREB1 measured in the control experiments was 0.15 ± 0.01 nm (n = 9). D–F, CREB1 and UNG2 each prevent binding of the other. D, UNG2 added before CREB1; E, CREB1 added before UNG2; F, UNG2 and CREB1 added together. Total CREB1 bound in these control experiments was 0.35 ± 0.01 nm (n = 9).
FIGURE 3.

CREB1 dimerization is heavily impacted by the presence of DNA repair glycosylases. As the concentration of glycosylase was increased, CREB1 dimerization decreased, except for when the G/U mispair-containing CRE site had prior exposure to CREB1. A–C, regardless of whether the substrate was exposed to OGG1 before or after CREB1, total CREB1 dimerization was reduced up to 69% ± 2% (n = 3) relative to control. A, OGG1 added before CREB1; B, CREB1 added before OGG1; C, OGG1 and UNG2 added together. CREB1 dimerization was significantly reduced when OGG1 and CREB1 had concurrent access to the substrate as compared with when OGG1 had prior access to the substrate (p = 0.012, 3). CREB1 dimer in the control experiments was 0.02 nm ± 0.003 nm (n = 9). D–F, dimerization of CREB1 on a G/U mispair-containing CRE site. D, UNG2 added before CREB1; E, CREB1 added before UNG2; F, UNG2 and CREB1 added together. Measured CREB1 dimer in control experiments was 0.14 ± 0.01 nm (n = 9).
Data in Figs. 4–7 were analyzed by one-way analysis of variance using SPSS 19.0. Before analysis was conducted, a Shapiro-Wilk's test for normality and a Levene's test for equal variance were conducted. When a significant difference was detected in any analysis, Tukey's post hoc analysis was performed to identify significantly different means. Significance for all statistical tests was set a priori at 0.05.
FIGURE 4.
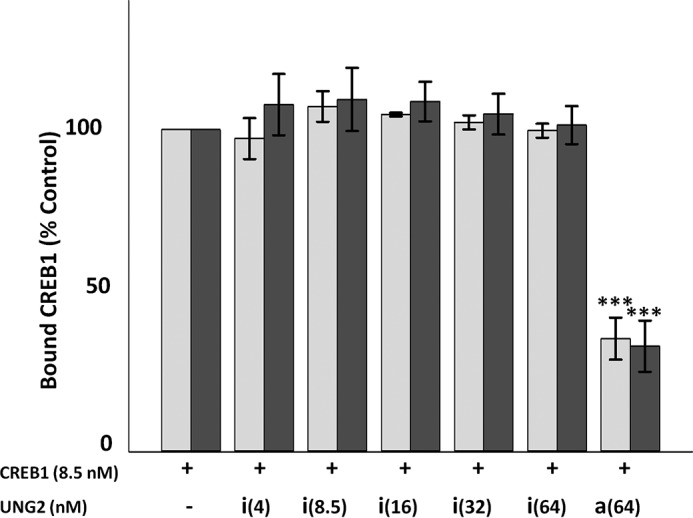
Inhibition of CREB1 binding by UNG2 depends upon UNG2 enzymatic activity. Catalytically inactive UNG2 (His-UNG2-D154N) had no effect on total CREB1 binding or dimerization on a CRE site substrate containing a G/U mispair. Wild type or mutant UNG2 was incubated with the substrate prior to the addition of CREB1. □, total bound CREB1 as compared with control; ■, total CREB1 as a dimer as compared with control. i = inactive mutant UNG2, a = active UNG2; numbers in parentheses represent UNG2 concentration. Error bars represent ± S.E., ***, p ≤ 0.001 relative to control.
FIGURE 5.
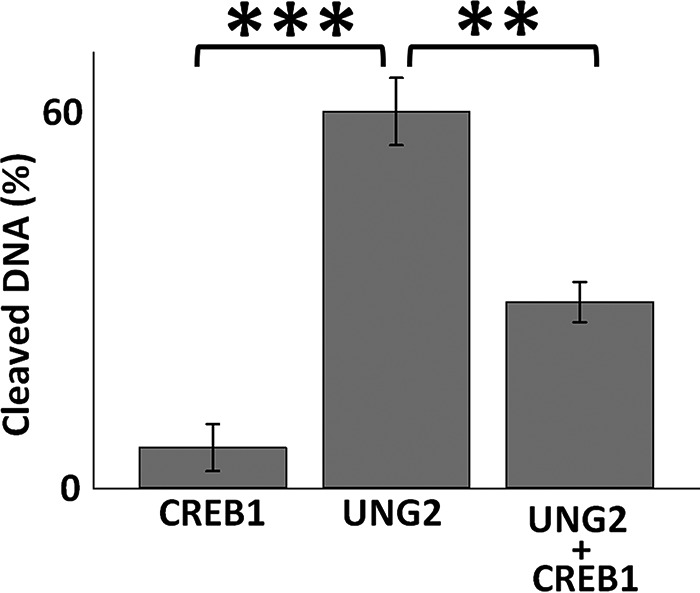
CREB1 interferes with UNG2 enzymatic activity when the CRE site contains a G/U mispair in the CpG islet. In the presence of 85 nm UNG2, 61% ± 5% (n = 3) of U residues within the CRE site were cleaved. However, the presence of 8.5 nm CREB1 together with UNG2 reduced cleavage by 50% ± 3% (n = 3). Error bars represent ± S.E., ***, p ≤ 0.001; **, p ≤ 0.005.
FIGURE 6.
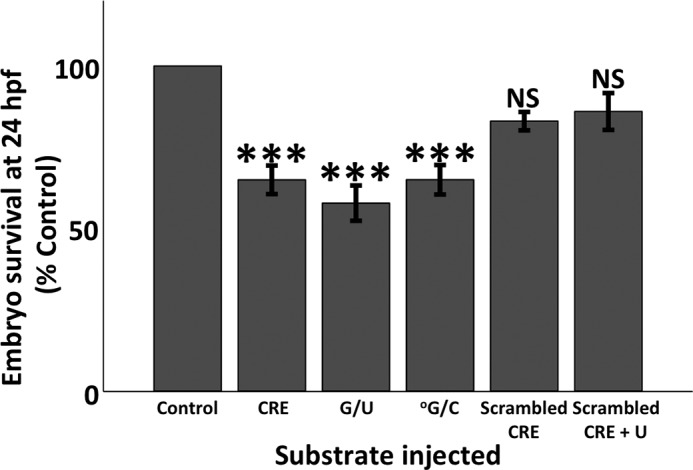
Microinjection of oligonucleotide substrates results in reduced survival of 24 hpf zebrafish embryos. Substrates consisted of 39-mer ds oligonucleotides encompassing an unmodified or modified CRE site (Table 1). Control consisted of 0.05% phenol red in 1× Danieau buffer. The presence of a U residue in CRE reduced survival by 42% ± 5% (n = 7), an °G reduced survival by 35% ± 5% (n = 5), and unmodified CRE reduced survival by 34% ± 4% (n = 13) of control values. There were no differences in survival among those three substrates. The scrambled CRE site substrate failed to reduce survival. Error bars represent ± S.E., ***, p ≤ 0.001 relative to control. NS, not significant.
FIGURE 7.

Only the G/U mispair within the CpG islet of the CRE site causes developmental defects in surviving 24 hpf zebrafish embryos. Embryos were microinjected with the indicated 39-mer ds oligonucleotide substrate (Table 1). Viability and developmental progress were determined at 24 hpf. No differences from control were noted except for those microinjected with the CRE site containing a G/U mispair (31% ± 11%, n = 8). Control consisted of phenol red in 1× Danieau buffer. Error bars represent ± S.E., **, p ≤ 0.01.
Results
Lesion-specific DNA Glycosylases Compete with CREB1
CRE has a CpG islet that, when oxidized or when containing a G/U mispair, either negatively or positively alters binding of CREB1, depending on the lesion (3). These lesions are repaired by BER. Because glycosylases are the primary entry point to the BER pathway, we asked here whether and how the two major glycosylases, OGG1 and UNG2, might affect CREB1 access to a CRE site that contains °G or U in the CpG islet portion of the CRE site, respectively. We also asked whether CREB1 might block access to these same glycosylases.
When the CRE site contained an °G lesion within the CpG position, total CREB1 binding to the CRE site was reduced by a maximum of 34% ± 9% (S.E., n = 3) in the presence of an 8-fold excess of OGG1 (Fig. 2, A–C). The decrease in CREB1 binding depended on the OGG1 concentration and the order in which OGG1 was exposed to the substrate. The ability of OGG1 to displace previously bound CREB1 was somewhat less efficient than its ability to prevent CREB1 from binding (p = 0.035, n = 3) (Fig. 2, A and B). Therefore, the presence of this glycosylase and the order of exposure to CRE had a small but substantial effect on total binding.
When the CRE site contained a U residue in the CpG islet, overall binding of CREB1 in the presence of UNG2 was reduced to a far greater extent by a maximum of 60% ± 2% (S.E., n = 3) (Fig. 2, D–F). However, the decrease depended strongly on the order in which the proteins were added. Bound CREB1 completely blocked access to UNG2 regardless of the presence of 8-fold excess of UNG2 (Fig. 2E). In contrast, total CREB1 binding was extremely sensitive to prior exposure of the CRE site to UNG2 (60% ± 2% inhibition decreases relative to control (S.E., n = 3)) (Fig. 2D) and somewhat sensitive to the simultaneous addition of UNG2 and CREB1 (23% ± 6% inhibition relative to control (S.E., n = 3)) (Fig. 2F). In short, unlike the case with °G/C, the order of addition made a substantial difference to total binding of CREB1 when the CRE site contained a G/U mispair in the CpG islet of CRE.
Dimerization of CREB1 on Its CRE Site Is Strongly Impacted by Lesion-specific DNA Glycosylases
CREB1 is transcriptionally active only as a dimer, which forms on the CREB·DNA monomer complex (25). The ability of CREB1 to dimerize on a CRE site substrate containing an °G lesion was heavily influenced by the presence of OGG1 regardless of the order of addition (Fig. 3, A–C). Thus, the presence of OGG1 prevented the addition of a second molecule of CREB1 to the °G-containing CRE site and displaced previously bound CREB1. We presume that the second CREB1 molecule was unable to bind efficiently because OGG1 binds its substrate in a flipped-out configuration that distorts the CRE site and is slow to dissociate (26). In fact, we could detect bound OGG1 on our gels, indicating that OGG1 remained associated with the substrate and physically blocked the second molecule of CREB1 from forming a dimer (Fig. 1). Consequently, the presence of the repair glycosylase was likely to prevent CREB1 from forming the dimer necessary to recruit TFs required to activate RNA polymerase II.
The presence of UNG2 also negatively affected the ability of CREB1 to form a dimer on a G/U mispair-containing CRE site substrate. However, the competition kinetics were very different from those involving OGG1. The reduction depended on the order that the proteins were exposed to the substrate (Fig. 3, D–F). The effect was more selective and more severe than that on an °G-containing substrate. When UNG2 had first or equal exposure to the substrate relative to CREB1, the ability of CREB1 to form a dimer was markedly reduced up to 77% (Fig. 3, D and F). In contrast, if the CRE-containing substrate was allowed to bind CREB1 first, the ability of CREB1 to form a dimer was not altered relative to control regardless of an 8-fold excess of UNG2 (Fig. 3E). In other words, UNG2 interfered with the recruitment of a second molecule of CREB1 to the G/U-containing CRE site, but if CREB1 had already formed a dimer, then UNG2 could not displace the TF from the U-containing CRE site.
Catalytically inactive UNG2 at any concentration had no effect on the ability of CREB1 to bind to a G/U mispair-containing CRE site, whether measured as the total amount of CREB1 bound or CREB1 dimer formation (Fig. 4). Therefore, unlike exposure to OGG1 where we were unable to detect glycosylase activity, the reduction in CREB1 binding and dimerization to a CRE site containing a G/U mispair was due to excision of U through the catalytic action of UNG2 and not the physical presence of the enzyme blocking access. This result is consistent with the known rapid turnover of UNG2 (27).
CREB1 Impedes UNG2 from Excising a U Residue Present within a CRE site
We then asked whether CREB1 binding interfered with UNG2 enzymatic activity. Incubation of UNG2 alone with the oligomer containing the G/U mispair at the CRE site resulted in loss of 61% ± 5% (S.E., n = 3) of the U residues (Fig. 5). When CREB1 and UNG2 were co-incubated with this same substrate, the ability of UNG2 to remove U was reduced to 30% ± 3% (S.E., n = 3). Thus, when CREB1 binds to a CRE site containing a G/U mispair, it diminishes the ability of UNG2 to remove the U residue, which is the first step in BER. Furthermore, these data indicate that the presence of the U residue stabilized CREB1 binding because if CREB1 were constantly associating with and dissociating from the CRE site, then there would be renewed opportunity for UNG2 with its rapid turnover (27) to remove the U residue. Therefore, these data are consistent with prior results indicating that the presence of U in the CpG site of CRE stabilizes CREB1 binding to CRE (3).
°G and U in CREB1 CRE Affect Early Development
To establish the physiological relevance of the in vitro results, we explored whether the presence of DNA lesions repaired by BER in CRE affects normal embryonic development. For normal development to proceed, embryos need to activate select genes at the correct time and location. Because ∼25% of the vertebrate genome depends on functional CREB1, we hypothesized that exposure of developing zebrafish to a modified CRE site would result in abnormalities, depending on the affinity of CREB1 for the lesion. To that end, we microinjected the same oligonucleotides used in our in vitro studies (Table 1) into 2–4-cell-stage zebrafish embryos and harvested them at 24 hpf. Embryos at this stage contain ample OGG1 and UNG2 (19) and increasing amounts of CREB1 (23). Because CREB1 binds more strongly to a U-containing CRE site than to an unmodified CRE site (3), we predicted that the developing embryo would be sensitive to microinjection of an oligonucleotide containing a CRE site, but would be particularly sensitive to one containing a G/U mispair within the CpG of the CRE site. As expected, survival of zebrafish embryos to 24 hpf was reduced after microinjection of CRE site-containing oligonucleotides with or without BER-repairable modifications (Fig. 6). To explore whether the decreased survival of microinjected embryos was due to the presence of a modified or an unmodified CRE site, and not simply oligonucleotide toxicity, we also microinjected embryos with an oligonucleotide in which the CRE site sequence or the CRE site containing a uracil was scrambled. Embryos microinjected with the scrambled sequence or the scrambled U-containing sequence did not display any difference in survival from that of control (Fig. 6). Only microinjection of CRE site oligonucleotides containing the G/U mispair resulted in more developmental defects in 24 hpf embryos (Fig. 7). These included curvature of anterior-posterior axis, decreased axis length, and abnormal yolk extension formation (Fig. 8). When G/C in the scrambled site was replaced with a G/U mispair, there was no increase in developmental failure or defects in comparison with scrambled CRE. These data rule out the possibility that UNG2 was stably recruited to a random G/U mispair. In other words, CREB1 had a higher affinity for CRE containing a G/U mispair not only in the test tube but also in the developing embryo. This demonstrated that CRE containing a G/U mispair is capable of soaking up more endogenous CREB1 than the undamaged CRE so that proper development failed in a significant percentage of embryos.
FIGURE 8.
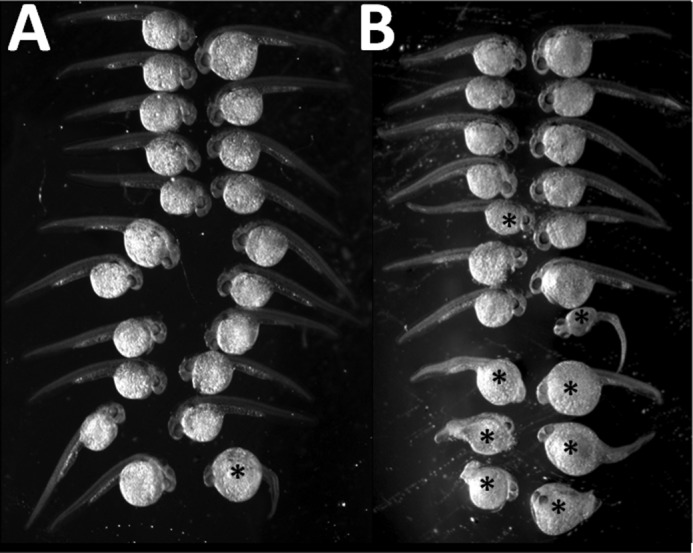
Developmental defects occur in zebrafish embryos microinjected with G/U mispair-containing CRE site oligonucleotide. A, control zebrafish embryos microinjected with 1× Danieau buffer and 0.05% phenol red dye displayed normal development; the lone exception (lower left in this image) displays curvature of the anterior-posterior axis. B, zebrafish embryos at 24 hpf displayed developmental defects of varying degrees after microinjection with a decoy CRE site oligonucleotide containing a G/U mispair in the CpG islet. Defects included kinked tail ends, curvature of anterior-posterior axis, and abnormal yolk extension. * denotes abnormal development.
Discussion
Here we demonstrate that a TF can alter access to repair of its consensus sequence containing damaged DNA; conversely, the presence of repair enzymes can block access to the TF. That is, the TF CREB1 competed with DNA repair glycosylases for its cognate CRE site when that sequence contained a DNA lesion in the CpG islet and vice versa. However, the type of repair glycosylase, in addition to the lesion, affected the binding ability of CREB1. When CREB1 was in competition with the repair glycosylase UNG2 for a CRE site that included a G/U mispair in the CpG islet, CREB1 entirely prevented UNG2 recognition and removal of the U lesion, whereas UNG2 removal of a U residue prevented CREB1 binding. In contrast, the repair glycosylase OGG1 competed with CREB1 for an °G-containing CRE site so that CREB1 binding and dimerization were reduced.
As UNG2 was largely incapable of displacing CREB1 after the TF had bound to a G/U mispair, these data are consistent with our previous results demonstrating that CREB1 binding and dimerization are enhanced when CRE contains a G/U mispair (3). However, when the G/U-containing CRE site was exposed to enzymatically active (but not inactive) UNG2 before exposure to CREB1, binding and dimerization were reduced in a concentration-dependent manner. This is unsurprising considering that UNG2 is extremely efficient in processing U lesions when they are mispaired with G, leading to a rapid and robust conversion of U to an apurinic/apyrimidinic site product to which CREB1 binds poorly (3, 27). Therefore, the TF interfered with entry into the repair pathway, and entry into the repair pathway was diminished by the binding and activation of the TF.
In contrast to results with UNG2, CREB1 binding and particularly dimerization were reduced as OGG1 concentration increased, regardless of the order in which CREB1 and OGG1 were added to the °G lesion-containing CRE site substrate. Moreover, OGG1 was capable of displacing CREB1 from CRE containing an °G lesion, despite the fact that °G within the CpG islet of the CRE site by itself did not affect total CREB1 binding or dimerization (3). Thus, OGG1 but not UNG2 was apparently capable of displacing CREB1 that had already bound to a CRE site. These results are consistent with the fact that OGG1 has a very low turnover number (28–30). This result suggests that OGG1-mediated reduction in CREB1 binding and dimerization was due to the physical presence of OGG1 blocking access to the CRE site itself rather than the existence of any apurinic/apyrimidinic site product that might have formed.
As noted above, CREB1 is a pleiotropic effector responsible for controlling ∼25% of the eukaryotic genome. During embryonic development, CREB1 regulates many genes and is essential for gastrulation (31). Decoy CRE site oligonucleotides are known to compete for CREB1 protein in vivo (32–34). Therefore, we microinjected CRE site oligonucleotides with or without a BER-repairable DNA lesion into zebrafish embryos to examine the in vivo effect of the lesion on CREB1 binding. Although all CRE-containing oligonucleotides acted as a sink and resulted in 30–40% failure to develop, only microinjection of those containing a G/U mispair resulted in developmental defects. These results are consistent with the critical role that CREB1 plays in early embryonic development and the ability of lesions repaired by BER to act in an epigenetic fashion. Therefore, the presence of a G/U mispair not only results in CREB1 binding its consensus sequence more tightly in vivo, but also, once bound, blocks access to the repair pathway, resulting in deleterious physiological consequences.
Most research conducted on the effects of DNA lesions, particularly oxidative damage or the presence of uracil, has focused on mutation within transcribed genomic regions (14, 35). However, several recent studies suggest that DNA lesions within the cognate sequences of activator protein 1 (AP-1), specificity protein 1 (Sp1), and the nuclear factor κ light chain enhancer of activated B cells (NF-κB) complex can also alter TF binding (36–38), indicating that DNA damage within promoter regions may have serious repercussions for cellular physiology. In addition, binding of the p50 subunit to the NF-κB sequence has been shown to shield °G lesions from recognition and repair by OGG1 and Fapy glycosylase (Fpg), suggesting that oxidative DNA damage may be able to persist long enough to affect gene expression (39). Furthermore, oxidative DNA damage to genomic promoter sites may be a common occurrence. For example, lysine-specific demethylase 1 (LSD1) generates hydrogen peroxide primarily at promoters as a byproduct of demethylation of histone H3 during the initiation of active transcription (2, 40). Peroxide in such close proximity to DNA is likely to result in oxidation.
The promoters for CREB1 itself and its modulating proteins including CREB-binding protein (CBP), cAMP-response element modulator (CREM), transducer of regulated CREB-binding proteins 1 and 3 (TORC1 and TORC3), and activating transcription factor (ATF) all contain CRE sites that could be subject to oxidative damage. AP-1 is a homodimer or heterodimer of FOS and JUN, both of which contain multiple CRE sites, as does specificity protein-1 (Sp-1). In addition, c-MYC, an important TF involved with metastasis and tumorigenesis, also contains a CRE site in its promoter, so that oxidation could alter MYC-dependent transcription and prevent DNA repair. Although NF-κB itself does not contain a CRE site, its promoter contains four FOS sites and one JUN site, which render it sensitive to BER-repaired damage. Thus, DNA damage to promoters, particularly CRE sites, can have profound, far-reaching consequences.
Our studies not only delineate alterations in both binding and displacement of CREB1 but also demonstrate the physiological relevance of unrepaired lesions normally repaired by BER. Small changes to gene expression of a major regulator like CREB1 can have major physiological ramifications. Gene expression is normally tightly regulated, but perturbations in expression are amplified either positively or negatively with profound consequences for cellular development and physiology. These results indicate that DNA repair enzymes can compete effectively with TFs and vice versa, further complicating the cellular response to damage repaired through BER.
Author Contributions
S. P. G. M. performed gel shift studies, analyzed data, participated in design and analysis of experiments, wrote the first draft of the manuscript, and participated in subsequent critical evaluation. J. K. performed and analyzed oligonucleotide digestions. K. J. T. performed gel shift studies and analyzed data. P. R. S. obtained funding and was responsible for conception and design of experiments, evaluation of data, and critical revision of the manuscript.
Acknowledgments
We thank Dr. Jennifer Nyborg (Colorado State University, Fort Collins, CO), for purified recombinant CREB1 protein, Dr. Nicole Noren Hooten (National Institute of Aging, Baltimore, MD) for purified recombinant uracil-DNA glycosylase (UNG2) and 8-oxoguanine DNA glycosylase (OGG1), and Dr. Bodil Kavli (Norwegian University of Science and Technology, Trondheim, Norway) for purified recombinant mutant UNG2 (His-UNG2-D154N). We also thank Dr. Tarik Gouhier (Marine Sciences Laboratory, Northeastern University, Boston, MA) for advice with statistical analyses and Benjamin Snow for technical review of this manuscript.
This work was supported by the G. Harold and Leila Y. Mathers Foundation, Aid for Cancer Research, and funds from Northeastern University. The authors declare that they have no conflicts of interest with the contents of this article.
- BER
- base excision repair
- CREB1
- cyclic AMP-response element-binding protein 1
- CRE
- CREB1 binding site consensus sequence
- ds
- double-stranded
- NF-κB
- nuclear factor κ light chain enhancer of activated B cells
- OGG1
- 8-oxoguanine DNA glycosylase
- ROS
- reactive oxygen species
- TF
- transcription factor
- UNG2
- uracil DNA glycosylase 2
- °G
- 8-oxoG
- hpf
- hours post-fertilization.
References
- 1. Perillo B., Ombra M. N., Bertoni A., Cuozzo C., Sacchetti S., Sasso A., Chiariotti L., Malorni A., Abbondanza C., and Avvedimento E. V. (2008) DNA oxidation as triggered by H3K9me2 demethylation drives estrogen-induced gene expression. Science 319, 202–206 [DOI] [PubMed] [Google Scholar]
- 2. Amente S., Bertoni A., Morano A., Lania L., Avvedimento E. V., and Majello B. (2010) LSD1-mediated demethylation of histone H3 lysine 4 triggers Myc-induced transcription. Oncogene 29, 3691–3702 [DOI] [PubMed] [Google Scholar]
- 3. Moore S. P. G., Toomire K. J., and Strauss P.R. (2013) DNA modifications repaired by base excision repair are epigenetic. DNA Repair (Amst.) 12, 1152–1158. [DOI] [PubMed] [Google Scholar]
- 4. Zhang X., Odom D. T., Koo S. H., Conkright M. D., Canettieri G., Best J., Chen H., Jenner R., Herbolsheimer E., Jacobsen E., Kadam S., Ecker J. R., Emerson B., Hogenesch J. B., Unterman T., Young R. A., and Montminy M. (2005) Genome-wide analysis of cAMP-response element binding protein occupancy, phosphorylation, and target gene activation in human tissues. Proc. Natl. Acad. Sci. U.S.A. 102, 4459–4464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Impey S., McCorkle S. R., Cha-Molstad H., Dwyer J. M., Yochum G. S., Boss J. M., McWeeney S., Dunn J. J., Mandel G., and Goodman R. H. (2004) Defining the CREB regulon: a genome-wide analysis of transcription factor regulatory regions. Cell 119, 1041–1054 [DOI] [PubMed] [Google Scholar]
- 6. Rudolph D., Tafuri A., Gass P., Hämmerling G. J., Arnold B., and Schütz G. (1998) Impaired fetal T cell development and perinatal lethality in mice lacking the cAMP response element binding protein. Proc. Natl. Acad. Sci. U.S.A. 95, 4481–4486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lonze B. E., and Ginty D. D. (2002) Function and regulation of CREB family transcription factors in the nervous system. Neuron 35, 605–623 [DOI] [PubMed] [Google Scholar]
- 8. Dworkin S., Heath J. K., deJong-Curtain T. A., Hogan B. M., Lieschke G. J., Malaterre J., Ramsay R. G., and Mantamadiotis T. (2007) CREB activity modulates neural cell proliferation, midbrain-hindbrain organization and patterning in zebrafish. Dev. Biol. 307, 127–141 [DOI] [PubMed] [Google Scholar]
- 9. Sakamoto K., Karelina K., and Obrietan K. (2011) CREB: a multifaceted regulator of neuronal plasticity and protection. J. Neurochem. 116, 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mayr B., and Montminy M. (2001) Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell Biol. 2, 599–609 [DOI] [PubMed] [Google Scholar]
- 11. Dominguez P. M., and Shaknovich R. (2014) Epigenetic function of activation-induced cytidine deaminase and its link to lymphomagenesis. Front. Immunol. 5, 642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ramiro A. R., and Barreto V. M. (2015) Activation-induced cytidine deaminase and active cytidine demethylation. Trends Biochem. Sci. 40, 172–181 [DOI] [PubMed] [Google Scholar]
- 13. Wu D., Chen L., Sun Q., Wu X., Jia S., and Meng A. (2014) Uracil-DNA glycosylase is involved in DNA demethylation and required for embryonic development in the zebrafish embryo. J. Biol. Chem. 289, 15463–15473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Evans M. D., Dizdaroglu M., and Cooke M. S. (2004) Oxidative DNA damage and disease: induction, repair and significance. Mutat. Res. 567, 1–61 [DOI] [PubMed] [Google Scholar]
- 15. Lindahl T. (1993) Instability and decay of the primary structure of DNA. Nature 362, 709–715 [DOI] [PubMed] [Google Scholar]
- 16. Iyama T., and Wilson D. M. 3rd (2013) DNA repair mechanisms in dividing and non-dividing cells. DNA Repair (Amst.) 12, 620–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Martin L. J. (2008) DNA damage and repair: Relevance to mechanisms of neurodegeneration. J. Neuropathol. Exp. Neurol 67, 377–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Caldecott K. W. (2008) Single-strand break repair and genetic disease. Nat. Rev. Genet. 9, 619–631 [DOI] [PubMed] [Google Scholar]
- 19. Fortier S., Yang X., Wang Y., Bennett R. A. O., and Strauss P. R. (2009) Base excision repair in early zebrafish development: evidence for DNA polymerase switching and standby AP endonuclease activity. Biochemistry 48, 5396–5404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Strauss P. R., Beard W. A., Patterson T. A., and Wilson S. H. (1997) Substrate binding by human apurinic/apyrimidinic endonuclease indicates a Briggs-Haldane mechanism. J. Biol. Chem. 272, 1302–1307 [DOI] [PubMed] [Google Scholar]
- 21. Lopez D. I., Mick J. E., and Nyborg J. K. (2007) Purification of CREB to apparent homogeneity: removal of truncation products and contaminating nucleic acid. Protein Expr. Purif. 55, 406–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang Y., Shupenko C. C., Melo L. F., and Strauss P. R. (2006) DNA repair protein involved in heart and blood development. Mol. Cell. Biol. 26, 9083–9093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pei D.-S., Yang X. J., Liu W., Guikema J. E., Schrader C. E., and Strauss P. R. (2011) A novel regulatory circuit in base excision repair involving AP endonuclease 1, Creb1 and DNA polymerase β. Nucleic Acids Res. 39, 3156–3165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nusslein-Volhard C., and Dahm R. eds (2002) Zebrafish: A Practical Approach, 1st Ed., Oxford University Press Inc., New York [Google Scholar]
- 25. Shaywitz A. J., and Greenberg M. E. (1999) CREB: A stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu. Rev. Biochem. 68, 821–861 [DOI] [PubMed] [Google Scholar]
- 26. Chen L., Haushalter K. A., Lieber C. M., and Verdine G. L. (2002) Direct visualization of a DNA glycosylase searching for damage. Chem. Biol. 9, 345–350 [DOI] [PubMed] [Google Scholar]
- 27. Kavli B., Sundheim O., Akbari M., Otterlei M., Nilsen H., Skorpen F., Aas P. A., Hagen L., Krokan H. E., and Slupphaug G. (2002) hUNG2 is the major repair enzyme for removal of uracil from U:A matches, U:G mismatches, and U in single-stranded DNA, with hSMUG1 as a broad specificity backup. J. Biol. Chem. 277, 39926–39936 [DOI] [PubMed] [Google Scholar]
- 28. Hill J. W., Hazra T. K., Izumi T., and Mitra S. (2001) Stimulation of human 8-oxoguanine-DNA glycosylase by AP-endonuclease: potential coordination of the initial steps in base excision repair. Nucleic Acids Res. 29, 430–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sidorenko V. S., Grollman A. P., Jaruga P., Dizdaroglu M., and Zharkov D. O. (2009) Substrate specificity and excision kinetics of natural polymorphic variants and phosphomimetic mutants of human 8-oxoguanine-DNA glycosylase. FEBS J. 276, 5149–5162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zharkov D. O., Rosenquist T. A., Gerchman S. E., and Grollman A. P. (2000) Substrate specificity and reaction mechanism of murine 8-oxoguanine-DNA glycosylase. J. Biol. Chem. 275, 28607–28617 [DOI] [PubMed] [Google Scholar]
- 31. Sundaram N., Tao Q., Wylie C., and Heasman J. (2003) The role of maternal CREB in early embryogenesis of Xenopus laevis. Dev. Biol. 261, 337–352 [DOI] [PubMed] [Google Scholar]
- 32. Park Y. G., Nesterova M., Agrawal S., and Cho-Chung Y. S. (1999) Dual blockade of cyclic AMP response element- (CRE) and AP-1-directed transcription by CRE-transcription factor decoy oligonucleotide: gene-specific inhibition of tumor growth. J. Biol. Chem. 274, 1573–1580 [DOI] [PubMed] [Google Scholar]
- 33. Lee Y. N., Park Y. G., Choi Y. H., Cho Y. S., and Cho-Chung Y. S. (2000) CRE-transcription factor decoy oligonucleotide inhibition of MCF-7 breast cancer cells: cross-talk with p53 signaling pathway. Biochemistry 39, 4863–4868 [DOI] [PubMed] [Google Scholar]
- 34. Hara T., Hamada J., Yano S., Morioka M., Kai Y., and Ushio Y. (2003) CREB is required for acquisition of ischemic tolerance in gerbil hippocampal CA1 region. J. Neurochem. 86, 805–814 [DOI] [PubMed] [Google Scholar]
- 35. Cooke M. S., Evans M. D., Dizdaroglu M., and Lunec J. (2003) Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J. 17, 1195–1214 [DOI] [PubMed] [Google Scholar]
- 36. Ghosh R., and Mitchell D. L. (1999) Effect of oxidative DNA damage in promoter elements on transcription factor binding. Nucleic Acids Res. 27, 3213–3218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ramon O., Sauvaigo S., Gasparutto D., Faure P., Favier A., and Cadet J. (1999) Effects of 8-oxo-7,8-dihydro-2′-deoxyguanosine on the binding of the transcription factor Sp1 to its cognate target DNA sequence (GC box). Free Radic. Res. 31, 217–229 [DOI] [PubMed] [Google Scholar]
- 38. Abraham J., and Brooks P. J. (2011) Divergent effects of oxidatively induced modification to the C8 of 2′-deoxyadenosine on transcription factor binding: 8,5′ (S)-cyclo-2′-deoxyadenosine inhibits the binding of multiple sequence specific transcription factors, while 8-oxo-2′-deoxyadenosine increases binding of CREB and NF-κB to DNA. Environ. Mol. Mutagen. 52, 287–295 [DOI] [PubMed] [Google Scholar]
- 39. Hailer-Morrison M. K., Kotler J. M., Martin B. D., and Sugden K. D. (2003) Oxidized guanine lesions as modulators of gene transcription: altered p50 binding affinity and repair shielding by 7,8-dihydro-8-oxo-2′-deoxyguanosine lesions in the NF-κB promoter element. Biochemistry 42, 9761–9770 [DOI] [PubMed] [Google Scholar]
- 40. Li J., Braganza A., and Sobol R. W. (2013) Base excision repair facilitates a functional relationship between guanine oxidation and histone demethylation. Antioxid. Redox. Signal. 18, 2429–2443 [DOI] [PMC free article] [PubMed] [Google Scholar]