

Abstract
The repertoire of peptides displayed in vivo by MHC II molecules derives from a wide spectrum of proteins produced by different cell types. Although intracellular endosomal processing in dendritic cells and B cells has been characterized for a few antigens, the overall range of processing pathways responsible for generating the MHC II peptidome are currently unclear. To determine the contribution of non-endosomal processing pathways, we eluted and sequenced over 3000 HLA-DR1-bound peptides presented in vivo by dendritic cells. The processing enzymes were identified by reference to a database of experimentally determined cleavage sites and experimentally validated for four epitopes derived from complement 3, collagen II, thymosin β4, and gelsolin. We determined that self-antigens processed by tissue-specific proteases, including complement, matrix metalloproteases, caspases, and granzymes, and carried by lymph, contribute significantly to the MHC II self-peptidome presented by conventional dendritic cells in vivo. Additionally, the presented peptides exhibited a wide spectrum of binding affinity and HLA-DM susceptibility. The results indicate that the HLA-DR1-restricted self-peptidome presented under physiological conditions derives from a variety of processing pathways. Non-endosomal processing enzymes add to the number of epitopes cleaved by cathepsins, altogether generating a wider peptide repertoire. Taken together with HLA-DM-dependent and-independent loading pathways, this ensures that a broad self-peptidome is presented by dendritic cells. This work brings attention to the role of “self-recognition” as a dynamic interaction between dendritic cells and the metabolic/catabolic activities ongoing in every parenchymal organ as part of tissue growth, remodeling, and physiological apoptosis.
Keywords: antigen presentation, antigen processing, dendritic cell, major histocompatibility complex (MHC), tolerance, lymph, proteomics, immunological tolerance
Introduction
During the last few years “omic” analyses have been performed on many biological fluids including plasma, serum, lymph, breast fluid aspirate, and urine, with the ultimate goal of identifying tissue-specific molecular signatures of physiological and pathological conditions. Indeed, under physiological conditions, comparative proteomic analyses have mapped common as well as divergent proteome components among these fluids and, under pathological conditions, identified diagnostic biomarkers associated with cancer, inflammatory response to pathogens, and autoimmunity (1–22). A common feature emerging from these studies is that all of the analyzed biological fluids contain a protein degradome composed of naturally processed protein fragments and peptides (1, 15, 17, 18, 23–28). Under physiological conditions the degradome/peptidome reflects the metabolic and catabolic activities ongoing in the different organs via which the biological fluid is drained. Under inflammatory or neoplastic conditions, additional peptides are observed, which reflect ongoing organ-specific pathological processes. Thus far over 6000 peptides and protein fragments have been mapped in the plasma and almost 1000 in the lymph (1, 6, 25–29).
Conventional dendritic cells (cDC)3 are a heterogeneous population of antigen-presenting cells involved in different aspects of tolerogenic and immunogenic T cell responses. They are subclassified as resident cDC, which reside in secondary lymphatic organs (spleen and lymph node), and migratory cDC, which traffic from peripheral tissues to the draining lymph nodes and the thymus (30–33). Both cDC subtypes constantly sample their environment; splenic and nodal cDC sample antigens in the lymph and bloodstream (34), whereas migratory cDC pick up antigens in the tissue of origin before migrating to the draining lymph node and the thymus (35). Although cDC are thought to play key roles in the regulation of immune responses, few studies have addressed the composition of the MHC II peptidome presented by cDC or the enzymes responsible for cleaving the MHC II-bound peptides. Many of the original studies identifying self-peptidomes were performed on splenic cells (36–38), in vitro expanded B cell lines (39), or cDC differentiated in vitro from monocyte populations (40). Many of these earlier studies found an MHC II peptidome mostly generated by cytosolic proteins derived from abundantly expressed proteins (36, 39, 41), although these studies may have been limited by instrumental constraints to detect only highly represented proteins. Indeed more recent studies performed on in vivo Flt3L-expanded mouse cDC and splenic B cells or in vitro expanded human B and T lymphocytes report the presence of an MHC II peptidome derived from proteins more homogenously distributed among different subcellular locations (42, 43). A comparative analysis of the sheep MHC II peptidome eluted from afferent lymph cDC or from peripheral blood APC also reports peptides derived from membrane, cytosolic, and extracellular proteins (44).
The cDC MHC II peptidome conventionally is understood to derive from antigens acquired through several sources including phagocytosis of exogenous antigens and autophagy of cytosolic antigens into endosomal compartments (41, 45–49). Recently, it has been shown that cDC can also capture peripheral antigens not only as proteins but also as preprocessed peptides found in biological fluids or delivered in the plasma, subcutaneously or in the peritoneal cavity, which directly drains in the peritoneal lymphatics (32, 33, 35, 50, 51). Antigens acquired through conventional phagocytosis or autophagy generate an MHC II peptidome processed mostly by endosomal cathepsins, whereas peptides found in biological fluids could derive from a greater variety of processing pathways (15, 27, 28, 52). Thus, in theory, the richness of naturally processed peptides found in biological fluids, including the lymph, could greatly expand the MHC II-presented self-peptidome.
This understanding prompted us to evaluate the contribution of the endogenous self-peptidome found in the lymph to the overall MHC class II peptidome transported by cDC. We found that lymph-carried self-antigens, processed by a variety of different proteases, contribute significantly to the MHC II self-peptidome presented by cDC in vivo and to the maintenance of central and peripheral tolerance.
Experimental Procedures
Chemicals
MG132 peptide aldehyde (carbobenzoxyl-l-leucyl-l-leucyl-l-leucine (C2211) was from Sigma-Aldrich and EZ-LinkTM sulfo-NHS-LC-biotin (catalog No. 21335) from Thermo Fisher Scientific. The streptavidin-horseradish peroxidase conjugate (catalog No. 21127B) was from Pierce and 4-methylumbelliferyl N-acetyl-β-d-glucosaminide dehydrate (substrate for β-N-acetylhexosaminidase) (M-2133) from Sigma-Aldrich. GM-6001 (catalog No. 364205), inhibitor of matrix metalloproteinases (MMPs), and cathepsin inhibitors (catalog No. 219393 for cathepsin S, 219385 for cathepsin B, 219427 for cathepsin L, and EI10 for cathepsin D) were purchased from EMD Millipore.
Proteins
Collagenase type I (catalog No. 195109) was from MP Biomedicals, and active human cathepsin B (SRP0289), cathepsin D (No. C8696), cathepsin L (SRP0291), and cathepsin S (SRP0292) were all purchased from Sigma Aldrich. Active human MMPs 1, 2, 3, and 9 (kit BML-AK013-0001), MMP-14 (BML-SE259), and MMP-7 (BML-SE181) were purchased from Enzo Life Sciences. Human complement C3 protein (P4945) was purchased from Novus Biologicals. Human collagen type II (CC052) and active caspase-3 (CC119) were purchased from EMD Millipore. Human recombinant gelsolin (HPG6-A) was purchased from Cytoskeleton, Inc.
Peptides
Complement C3 peptide SSKITHRIHWESASLLR was synthesized at Karebay Biochem, Inc. (Monmouth Junction, NJ) using standard solid-phase N-(9-fluorenyl)methoxycarbonyl (Fmoc) chemistry (HPLC-grade purity, >95%).
Mice and Treatment
Caspase-3 knock-out mice were purchased from The Jackson Laboratory. Complement 3 knock-out mice were a generous gift from M. Carroll (Harvard University). Transgenic HLA-DR1 mice (53) were bred at Albert Einstein College of Medicine (Einstein) to generate HLA-DR1+C3−/−I-A−/−I-E−/− mice. All mice were bred at Einstein, and tail specimens were sent to Transnetyx for genotyping using their established protocols. In some experiments, HLA-DR1 mice were injected subcutaneously with the B16-Flt3L-producing melanoma to expand the cDC population (54). To deplete regulatory T cells, mice were injected intraperitoneally with the rat-anti-mouse CD25 mAb (clone PC61, Imgenex, San Diego) at day 0. Inguinal node and spleen were collected at day 7 to verify CD25+ T cells deletion. In some experiments mice were injected intraperitoneally or intravenously with 50–100 nmol of soluble relevant peptides to induce thymic negative selection. Injections were performed at days 0, +2, and +4, and mice were sacrificed at day +6. In other experiments mice were immunized with complement C3 peptides (100 μg in 100 μl of PBS emulsified with 100 μl of complete Freund's adjuvant) at the base of the tail and back of the neck. Twenty-one days after immunization mice were sacrificed, and inguinal and axillary lymph nodes were collected. Animal euthanasia and tissue harvesting were conducted according to a protocol approved by the Institutional Animal Care and Use Committee at Albert Einstein College of Medicine.
Mouse cDC Preparation
HLA-DR1 (53) mice were injected subcutaneously with 40 × 106 B16-Flt3L-producing melanoma cells (54). At 12–14 days after injection, spleens were harvested and DCs were purified by density gradient centrifugation using 30% bovine albumin solution (Sigma-Aldrich). The preparation routinely contains 70–80% CD11c+ DCs. To further enrich the DC population, cells were purified using CD11c+ antibody-conjugated magnetic beads (Miltenyi Biotec). A total of 50 mice were used to generate a sufficient number of Flt3L-expanded cDC for HLA-DR1 immunoaffinity purification and peptide elution.
Primary Human Monocyte-derived DC
Peripheral blood mononuclear cells were purified from heparinized peripheral blood collected from healthy donors using Ficoll-Hypaque density gradient centrifugation. Monocyte subpopulations were further purified by separation on magnetic beads coated with CD14+ antibodies (CD14 MicroBeads, human, catalog No. 130-050-201, Miltenyi Biotec) using the manufacturer's instructions. After 7 days of culture with GM-CSF (10 ng/ml) and IL-4 (2 ng/ml) (R&D Systems), the monocyte-derived DC were collected for subcellular fractionation.
Plasma Membrane Isolation and Biotinylation
Primary monocyte-derived human DC (2 × 108) or mouse Flt3L-induced splenic DC were washed three times with ice-cold PBS (pH 8.0) to remove amine-containing media. Cells were then suspended at a density of 25 × 108 cells/ml in PBS (pH 8.0), and 8 mg of sulfo-NHS-LC-biotin reagent was added for each ml of cell suspension. The biotinylation reaction was performed at 4 °C for 1 h. Cells were then washed three times with ice-cold PBS containing 100 mm glycine to quench the unbound biotin and pelleted by centrifugation. DC were then suspended at 2 × 108 cells/ml in homogenizing buffer (0.25 m sucrose, 1 mm MgCl2, 1 mm EDTA, and a complete mixture of protease inhibitors in 10 mm Tris-HCl (pH 7.4)) and subjected to homogenization using a Teflon-glass homogenizer for 30 min. The homogenate of biotinylated DC was centrifuged at 280 × g in a Sorvall RT 6000B centrifuge for 10 min at 4 °C to pellet cellular debris and the nuclear fraction, which were discarded. The supernatant was further centrifuged at 1500 × g for 10 min at 4 °C to pellet all of the cellular membranes (plasma membrane and ER/Golgi). The supernatant was set aside for further purification of other intracellular organelles, and the pellet was processed to purify the plasma membrane fraction. The pellet was suspended in 2 ml of buffer A (0.25 m sucrose and 1 mm MgCl2 in 10 mm Tris-HCl (pH 7.4)) and mixed with an equal volume of buffer B (2.0 m sucrose and 1 mm MgCl2 in 10 mm Tris-HCl (pH 7.4)). The mixture was carefully layered on top of a 1-ml layer of sucrose and centrifuged at 11300 × g (30,0000 rpm in an SW41 rotor) for 1 h at 4 °C. The plasma membrane fraction was collected at the interface and washed with buffer A at 3000 × g (1700 rpm in a Sorval RT-6000B rotor) for 15 min at 4 °C. A second purification step was performed using streptavidin-conjugated beads. The purity of the plasma membrane was confirmed by Western blotting using the streptavidin-HRP conjugate to detect the biotinylated proteins and the absence of selective markers for other organelles using specific antibodies (p58 for endoplasmic reticulum, p130 for Golgi and LAMP1 for lysosomes/late endosomes).
Late Endosome Preparation
Monocyte-derived human DC and Flt3L-induced splenic murine DC (1–3 × 108) were pelleted, washed in PBS, and resuspended in PBS containing 0.25 m sucrose and 20 mm HEPES (pH 7.4). Late endosomes and lysosomes were isolated as reported previously (48). Briefly, the cells were homogenized in a Dounce homogenizer and spun at 150 × g for 10 min. The supernatant was loaded on a 27% Percoll gradient laid over a 2.5 m sucrose cushion and centrifuged for 1 h at 34,000 × g. The band above the sucrose cushion corresponds to the total lysosomal fraction. The band at the interface was enriched in late and early endosomes and was further separated on a 10% Percoll gradient by centrifugation at 34,000 × g for 1 h. The purity of the late endosomal fraction was confirmed by ultrastructural analysis and Western blotting for selected marker (48). In addition, the purity of the late endosomal fraction was confirmed by the levels of β-hexosaminidase using a sodium acetate buffer (pH 4.0) and 4-methylumbelliferyl N-acetyl-β-d-glucose-aminide as substrate (48).
Cytosol Preparation
Between 40 and 100 million cells (murine splenic Flt3L-induced DC from WT Casp3+/+ and Casp3−/− mice) were used in each experiment. Cells were collected and washed in PBS, and the pellet was resuspended in 2.5 ml of homogenization buffer (2.5 m sucrose, 20 mm HEPES, and dPBS (pH 7.2) containing complete protease inhibitor cocktail). Cells were homogenized with a fitted Dounce homogenizer for 15 min on ice and then centrifuged at 2000 × g for 10 min. The collected supernatant was centrifuged at 17,000 × g for 20 min to pellet the organelles. The collected supernatant was again centrifuged at 100,000 × g for 60 min to pellet the ER/Golgi and cytosolic vesicles. The supernatant corresponds to the soluble cytosol.
Induction of Apoptosis in Primary Murine DC
Tititrated amounts of the peptide aldehyde proteasome inhibitor MG132 (carbobenzoxyl-l-leucyl-l-leucyl-l-leucine, 6–30 μm) were used to induce apoptosis in murine primary dendritic cells as monitored by caspase-3 cleavage. Cytosol preparations were isolated from 10 mm MG132-treated DC.
Complement 3, Collagen II, Gelsolin, and Thymosin Antigen Processing
We developed in vitro cell-free proteolytic assays to validate endosomal versus non-endosomal antigen processing and to characterize cleavage sites associated with different enzymatic activities reported previously by the MEROPS and CutB databases. We selected four proteins in which the endogenous peptides were found in the human lymph and/or eluted from HLA-DR1. The amounts of substrate proteins, cytosol, late endosomes, plasma membrane, and recombinant enzymes were titrated to determine the optimal experimental conditions for analyzing the processed peptidome. The experimental conditions are detailed below. A small aliquot of each reaction mixture (1 μg of protein/each time point) was stopped by the addition of Laemmli buffer (4% (m/v) SDS, 10% (v/v) glycerol, 0.01% (m/v) bromphenol blue, and 125 mm Tris-HCl (pH 6.8)) and run on SDS-PAGE. The rest of the reaction mixture was quenched with 0.5% acetonitrile and 5% trifluoroacetic acid and filtered through 10-kDa MWCO (molecular weight cut-off) devices to collect the peptidome for MS/MS analysis.
Collagen II Processing by Metalloproteases MMP1, -2, -3, -7, -9, and -14
Collagen II (10 μg) was incubated with each individual active MMP (300–500 nm) in 150 mm NaCl, 5 mm CaCl2, and 500 mm Tris-HCl (pH 7.4) buffer for 1, 2, 6, 8, and 24 h at 37 °C.
Collagen II and Complement C3 Processing by Cathepsins B, D, L, and S
Collagen II or complement C3 (10 μg) were incubated with each individual active cathepsin (B, D, L, and S) (500 nm) in low pH buffer (50 mm NaOAc, 2.5 mm EDTA, and 1 mm DTT (pH 4.0–5.5)) for 1, 2, 6, 8, and 24 h at 37 °C.
Processing of Collagen II, Complement C3, Thymosin β4, and Gelsolin by Late Endosomal Compartments
Ten μg of each protein was incubated with 1–3 μg of late endosomal compartments in 50 mm NaOAc, 2.5 mm EDTA, and 1 mm DTT (pH 5.5) for 1, 4, and 8 h at 37 °C.
Processing of Collagen II by Plasma Membrane Fractions
Ten μg of each protein was incubated with 1–3 μg of plasma membrane proteins in 150 mm NaCl, 5 mm CaCl2, and 500 mm Tris-HCl (pH 7.4) for 1, 4, and 8 h at 37 °C.
Processing of Gelsolin by Cytosolic Fractions
Ten μg of gelsolin was incubated with 3 μg of cytosol preparation from control or apoptotic DC prepared from wild type (caspase-3+/+) or KO (caspase-3−/−) mice for 1, 4, and 8 h at 37 °C.
Western Blotting Analysis
Purified organelles, cytosol, or total cells were lysed in lysis buffer (150 mm NaCl, 50 mm Tris, 1 mm EDTA, and 1% Nonidet P-40) for 1 h on ice. Samples were run on a 5–15% gradient SDS-PAGE prior to blotting onto nitrocellulose membranes and incubation with selected antibodies overnight at 4 °C followed by incubation with HRP-conjugated secondary antibody at room temperature for 2 h. Enhanced chemiluminescence assay (ECL, Thermo ScientificTM, Pierce) was used to develop the membrane.
Antibodies
The following antibodies were used: rabbit anti-LAMP-2A (Invitrogen), anti-MMP antibodies (kit SA-384 from BIOMOL International; the matrix metalloproteinase antibody set contained 10 μg of each of the following 11 rabbit polyclonal anti-MMP antibodies: SA102-9090 anti-MMP-1, SA103-9090 anti-MMP-2, SA104-9090 anti-MMP-3, SA105-9090 anti-MMP-7, SA370-9090 anti-MMP-8, SA106-9090 anti-MMP-9, SA434-9090 anti-MMP-10, SA371-9090 anti-MMP-11, SA107-9090 anti-MMP-12, SA372-9090 anti-MMP-13, and SA108-9090 anti-MMP-14; rabbit polyclonal anti-ERGIC-53/p58 endoplasmic reticulum marker (E1031, Sigma-Aldrich); rabbit polyclonal anti-LAMP1, lysosome/late endosome marker (Abcam ab62562); rabbit polyclonal anti-GM130, cis-Golgi marker (Abcam ab52649); rabbit polyclonal anti-cleaved caspase-3 (2305-PC-020, Trevigen); and rabbit polyclonal anti-caspase-3/CPP32 (AHZ0052, Invitrogen).
Thymic Deletion Assay
C57BL/6 congenic (CD90.2+) recipients were lethally irradiated on day 0 with two doses of 600 rads. Bone marrow was isolated from donor TEa mice (55) (CD90.1+), and CD34+ cells were separated using immunomagnetic separation with streptavidin-conjugated beads (Miltenyi Biotech). Five million of the purified CD34+ cells were injected intravenously into each irradiated recipient mouse. Four weeks later, CD90.1 and CD90.2 staining was performed on peripheral blood to determine successful chimerism. Mice were then injected intravenously or intraperitoneally with 50–100 nmol of the ASFEAQGALANIAVDKA peptide three consecutive times on days 0, +2, and +4 after the chimerism was determined. On day +6 the thymus was harvested and stained with the following fluorochrome-conjugated mAb to mouse CD4 (RM4–5) and CD8α (Ab clone number 53–6.7). C57BL/6 factor VIII KO mice were injected with 50 nmol of the SPSQARLHLQGRTNAWRPQVNDPKQWLQVD peptide on days 0, +2, and +4. On day +6 the thymus was harvested and stained as described above.
T Cell Proliferation
C57Bl6/J mice were depleted of regulatory T cells as described above. Inguinal node, skin, muscle, and lungs were collected on day 7. Single cell suspensions were generated following collagenase type IA (Sigma) digestion. Cells (2 × 105) were seeded in U-bottom 96-well plates with titrated amounts (0, 6, 12.5, 25, 50, and 100 μg/ml) of collagen XII α5, laminin α5, adlican, collagen VI α2, and peptides for 4 days. [3H]Thymidine (1 μCi/well) was added during the last 18 h of incubation to assay T cell proliferation. Plates were harvested, and the DNA [3H]thymidine incorporation was monitored using a Wallac liquid scintillation counter (PerkinElmer Life Sciences). The cell proliferation assays were performed on Microtest U-bottom 96-well polystyrene sterile plates with low evaporation lids (catalog No. 35-1177) from BD Biosciences. Proliferation experiments (reported in Fig. 10f) were performed using the cell proliferation ELISA BrdU colorimetric assay kit (catalog No. 11647229001, Roche Applied Science). Briefly, cells were incubated in the presence or absence of the complement C3 peptide (5, 25, 50, and 100 μm/200 μl/well) in quadruple replicates. Cells were incubated for 3 days at 37 °C in 5% CO2 and on the fourth day were labeled with BrdU and probed for BrdU incorporation following the manufacturer suggestions.
FIGURE 10.

Role of lymph peptides in central and peripheral tolerance. a, FACS analysis of thymocytes from chimeric mice generated by irradiating wild type C57Bl6 mice and reconstituting them with a mixture of bone marrow from TEα TCR transgenic mice (CD90.1) and C57Bl6 mice (CD90.2) in a 1:1 ratio. The percentage of CD4+ thymocytes is reported before and after injection of the I-Ab-restricted ASFEAQGALANIAVDK peptide. CD4+-CD90.1+ thymocytes derive from wild type C55Bl6 mice, whereas CD4+-CD90.2+ thymocytes derive from TEα TCR transgenic mice. b, bar graph showing the number of CD4+-CD90.1+ and CD4+-CD90.2+ thymocytes as determined in a. Mean ± S.D. of five separate experiments; ***, p < 0.01 for the depletion of TEα-TCR; *, p < 0.05 for the depletion of polyclonal TCR. c, FACS analysis of thymocytes from factor VIII knock-out mice. The number of CD4+ thymocytes is reported before and after injection of the I-Ab-restricted SPSQARLHLQGRTNAWRPQVNDPKQWLQVD peptide. d, bar graph of the percentage of CD4+ thymocytes, as determined in a. Mean ± S.D. of five separate experiments, *, p < 0.05 for the depletion of polyclonal TCR. e, nodal T cell proliferation of CD25+-competent or -depleted HLA-DR1+ mice. Inguinal lymph node were harvested and cultured for 4 days with titrated amounts of the reported peptides. f, T cell proliferation of CD25-competent or CD25-depleted HLA-DR1+ mice. Skin, muscle, and lung tissues were harvested and cultured for 4 days with titrated amounts of the same peptides.
Isolation of MHC II Complexes from Mouse DCs
MHC II complexes were isolated from mouse DCs using immunoaffinity chromatography of solubilized membranes. Briefly, cell pellets were resuspended in 10 mm Tris-HCl (pH 8) containing cOmplete protease inhibitor mixture (Roche Diagnostic Corp.), freeze-thawed, and homogenized, and the soluble fraction was recovered by centrifugation at 4000 × gmax/5 min. The extraction cycle was repeated five times, and supernatants were combined. Membranes were obtained from pooled supernatants by ultracentrifugation at 100,000 × gmax/1 h/4 °C. Crude membrane pellets were solubilized in buffer (50 mm Tris-HCl, 150 mm NaCl (pH 8), and protease inhibitors containing 5% octyl-β-glucopiranoside (OG, Sigma-Aldrich)) by mechanical homogenization and gentle stirring overnight at 4 °C. Nonsoluble material was separated by ultracentrifugation at 100,000 × gmax/1 h/4 °C and discarded.
Immunoaffinity Chromatography
Immunoaffinity beads were prepared as described (56)using HLA-DR1-specific monoclonal antibody (clone LB3.1) and immobilized rProtein A-agarose (IPA-400HC, Repligen Corp., Waltham, MA). Solubilized membranes were loaded onto tandem nonspecific and LB3.1-conjugated beads overnight at 4 °C (∼1 ml of beads/400 μg of MHC II). LB3.1 beads were washed with 5× bead volumes of Tris-HCl 50 mm, NaCl 150 mm + 5% octyl-β-glucopiranoside, 10× bead volumes of Tris-HCl 50 mm, NaCl 150 mm + 1% octyl-β-glucopiranoside, 15× bead volumes of PBS, and 20× bead volumes of HPLC-grade water.
Elution of Peptides
MHC-peptide complexes were denatured and eluted off of LB3.1 beads by incubating them five times with 200 μl of 0.2% TFA + 50 μl of 10% TFA for 10 min at room temperature followed by five washes with 200 μl of 0.2% TFA. To separate the MHC protein subunits from the peptides, samples were loaded onto Vydac C4 spin columns (The Nest Group, Inc., Southborough, MA). Briefly, the combined eluates and washes were adjusted to 25% acetonitrile and a fraction (∼40 μg of protein) was loaded onto the column followed by two washes with 100 μl of 25% acetonitrile/0.2% TFA. The flow-through containing the peptides was combined with the washes, dried, and resuspended in 5% acetonitrile/0.1 formic acid for LC-MS/MS analysis (56).
LC-MS/MS of MHC-bound Peptides
Eluted peptides were analyzed in triplicate by LC-MS/MS using a Waters NanoAcquity UPLC coupled to a Thermo Scientific Q Exactive hybrid quadrupole-Orbitrap mass spectrometer (Thermo Fisher Scientific). Peptides were loaded onto a 100-μm ID fused silica precolumn (with Kasil frit) packed with 2 cm of 100 Å, 3-μm C18AQ (Bruker-Michrom, Auburn, CA) particles at a flow rate of 4.0 μl/min for 4 min with 5% acetonitrile (0.1% (v/v) formic acid). Peptides were separated and eluted on a 75-μm ID fused silica analytical column with a gravity-pulled tip containing 25 cm of 100 Å, 3-μm C18AQ at a flow rate of 300 nl/min using a linear gradient from 0 to 35% B (A = 0.1% formic acid in water and B = 0.1% formic acid in acetonitrile) in 120 min followed by washing and re-equilibration to initial conditions. Q Exactive was operated in the positive electrospray ionization mode utilizing data-dependent acquisitions for the top 12 most abundant ions observed in each MS spectrum. One full MS scan (m/z 350–2000) was acquired in the Orbitrap followed by tandem mass spectra (MS/MS) obtained by higher energy collisional dissociation (HCD, 35% normalized collision energy) and acquired in the Orbitrap. Dynamic exclusion was utilized to minimize data redundancy and maximize peptide identifications.
Lymph Peptidome
At least 20 mg of total proteins from lymph (pooled from 18 patients) in 2 ml of sterile PBS buffer solution was treated with TFA (at 0.2% final concentration in the sample) to extract free peptides as well as peptides adsorbed on different proteins, chaperones, and albumin. Peptides were filtrated through Centriplus (Amicon) centrifugal filter devices (5000-Da cut-off) as described elsewhere (27). A mixture of protease inhibitors (Roche Applied Science) was added to the sample to avoid proteolysis before TFA treatment. Peptides were then lyophilized, resuspended in 0.2% TFA, and fractionated using a combination of reverse-phase FPLC and HPLC chromatographic methods. The lymph peptide filtrates were fractionated in the first dimension on a Superdex peptide 10/300 gel filtration column (Tricorn) in 30% acetonitrile in 0.1% TFA as solvent, and nine fractions were collected at 1 ml/min. Each FPLC fraction was further fractionated in the second dimension on a C18 column (100 Å) using a reverse-phase system on a Hewlett Packard HPLC unit (1100 series) and a gradient (0–100% buffer B) elution over 90 min (buffer A, 0.5% acetonitrile in 0.2% TFA; buffer B, 70% acetonitrile in 0.2% TFA). Each HPLC fraction (36 fractions in total) was further desalted on a pepClean C18 spin column (Pierce) and subjected to nanoLC-MS/MS.
LC MS/MS of Lymph Filtrates
One-dimensional nanoUPLC-MS/MS with combined HCD/ETD (higher energy collisional dissociation/electron transfer dissociation) analysis was performed on a Waters nanoAcquity UPLC coupled to a Thermo Scientific LTQ Orbitrap Velos hybrid mass spectrometer. About 18 μl of sample was loaded on a Symmetry® C18 trap column (180 μm × 20 mm, 5 μm, Waters) with a loading flow rate of 2 μl/min. After a 10-min wash with 0.2% formic acid in water at a flow rate of 2 μl/min, the trap column was switched on-line with a nanoAcquity UPLC column (75 μm × 250 mm, 1.7 μm, BEH300 C18, Waters), and a gradient was applied to separate the peptides. Mobile phase A consisted of 0.2% formic acid in water and mobile phase B of 0.2% formic acid in 100% acetonitrile. The mobile phase B gradient used was 2–5% in 2 min, 5–42% in 40 min, 42–65% in 10 min, 65–70% in 5 min, and 70–80% in 3 min, then held at 80% B for an additional 16 min. The MS parameter settings were nanospray voltage, 1.7 kV; capillary temperature, 270 °C; FTMS AGC target, 1 × 106 ions, with 1 microscan and maximum ion injection time of 500 ms; and FTMS/MS AGC target, 2 × 105 ions, with 1 microscan and maximum ion injection time of 500 ms. The ETD parameters were reagent vial ion injection time, 100 ms; and AGC target, 4 × 105 ions; the ETD activation time was set for 100 ms. The initial survey scan range was m/z 350–650 at a resolution of 60,000 (at m/z 400). For MS/MS a minimum signal of 2000 counts was required, and the isolation window was 2 m/z. The top 10 intensity ions obtained from the survey scan were selected, sequentially isolated, and fragmented to generate the MS/MS spectra.
Data Processing and Database Search
Raw data from the three replicates was processed and searched using PEAKS DB, version 6.0 (Bioinformatic Solutions Inc., Waterloo, Ontario, Canada). A no-enzyme search was performed; parent mass tolerance was set to 10 ppm using monoisotopic mass, and fragment ion mass tolerance was set to 0.05 Da. Methionine oxidation (M+16) and pyro-Glu from glutamine (Glu-17) were set as variable modifications. A database containing all SwissProt entries for Mus musculus plus human DRA (P01903.1) and DRB1 (P04229.2) was used (20,847 entries). Data were validated using the FDR method available in PEAKS and filtered for <0.1% FDR for peptides.
A variety of post-translational modifications (PTM) were set as variable chemical modifications including acetylation, amidation, aminoadipic semialdehyde, arginine-glutamic semialdehyde, carboxylation, citrullination, cysteic acid, deamidation, dehydration, dimethylation, formylation, hexose, hexose NAc, kynurenin, oxidation, phosphopantetheine, phosphorylation, pyro-Glu, trimethylation, and 4-hydroxy nonenal. The percentage of unique peptides bearing one specific chemical modification was calculated by dividing the total number of unique peptides having a selected PTM to the total number of unique peptides retrieved from the searches conducted with both MASCOT and PEAKS DB engines.
Ingenuity Pathways Analysis software (IPA, Ingenuity Systems) was used to gain insight into the cellular location and functional characteristics of the identified proteins listed in supplemental Tables S1 and S3. An independent analysis of the biological pathways associated with each peptidome set was performed with DAVID (Database for Annotation, Visualization, and Integrated Discovery), version 6.7, which enabled the gene annotation enrichment analysis, functional annotation clustering, and BioCarta and KEGG pathway mapping of large proteomics data sets (57).
MEROPS and CutDB Analysis
The eluted DC and the lymph peptidomes were analyzed mainly by manually checking the proteolytic cleavage sites confined to each protein antigen substrate presented in supplemental Tables S1 and S3. The MEROPS database (58) was searched in combination with the PMAP CutDB proteolytic events database (59) to assign the proteases families responsible for the generation of the N- and C-terminal cleavage sites in the peptidome.
Bioinformatics Analysis of Human Lymph and HLA-DR1-eluted Peptidome
To find the corresponding human peptide orthologs, the mouse HLA-DR1-eluted peptides (Table S3b) were searched with the 'BLAST” tool against the UniProtKB/SwissProt database using the selection “human” as the targeted database. The “E-value” and “% identity” were recorded for each eluted mouse peptide.
The HLA-DR1 (DRB1*0101) binding affinity for all peptides identified in human lymph or eluted from dendritic cell HLA_DR1 MHC II proteins was predicted using the NetMHCIIPan 3.0 server (60). NetMHCIIpan 3.0 core epitopes were identified as the highest scoring 9-mer in each peptide. Using NetMHCIIPan 3.0, the predictor peptides were categorized as strong binders or weak binders according to the predicted IC50 value (a threshold of less than 50 nm was used to define strong binders and less than 500 nm to define weak binders).
IC50 Assay
A fluorescence polarization (FP) assay was used to measure the IC50 of each peptide as described (61, 62). Soluble recombinant MHC class II molecules DR1 (HLA-DRA*01:01;DRB1*01:01) and DM were expressed in Drosophila S2 cells and purified as described previously (63, 64). Peptides were obtained from 21st Century Biochemicals (Marlborough, MA). The N-terminally acetylated influenza HA-(306–318) analog peptide (Ac-PRFVKQNTLRLAT) was labeled with Alexa Fluor 488 tetrafluorophenyl ester (Invitrogen) through the primary amine of Lys-5 to be used as the probe peptide in the IC50 assay. Labeled peptides were purified by Jupiter C18 reverse-phase chromatography (Phenomenex, Torrance, CA). DR1 (100 nm) was incubated with probe peptide Alexa Fluor 488-HA-(306–318) (25 nm), together with a series dilution of test peptides, with a diluting factor of 5. Competition of each test peptide with the binding of the Alexa Fluor 488-HA-(306–318) probe to DR1 was measured by FP after incubation at 37 °C for 72 h. FP values were converted to % bound as (FP_sample-FP_free)/(FP_no_comp − FP_free), where FP_sample represents the FP values for sample well, FP_free represents the FP values for free Alexa Fluor 488-HA-(306–318), and FP_no_comp represents the FP values for wells without competitor peptides. We plotted % bound versus concentration of test peptide and fit the curve to the equation y = 1/(1 + [pep]/IC50), where [pep] is the concentration of the test peptide, y is the % of probe peptide bound at that concentration of test peptide, and IC50 is the 50% inhibition concentration of the test peptide. To measure DM susceptibility, 0.25 μm DM was included in the binding competition assay, and DM susceptibility was reflected by the ratio of IC50 measured in the presence and absence of DM after incubation at 37 °C for 24 h, as described elsewhere (61, 65).
Statistical Analysis
Statistical analysis was performed using Windows GraphPad Prism 6 (GraphPad Software, La Jolla, CA). A comparison between more than two groups was performed using two-tailed unpaired one-way analysis of variance. Results were considered statistically significant if p ≤ 0.05.
Results
Analysis of the Human Lymph Peptidome
We reported previously the first proteomic and degradomic/peptidomic analysis of the human lymph of healthy donors (4, 27). Increased capability in mass spectrometry, including utilization of high mass measurement accuracy, faster data acquisition speed, and overall improved sensitivity prompted us to perform additional analysis to identify peptides present in low abundance that previously were below the detection limit. In our original analyses, peptides generated from extracellular matrix proteins and housekeeping proteins were easily detected in lymph using low resolution (i.e. nominal mass) mass spectrometry, but peptides derived from low abundance proteins or cell-specific proteins were more difficult to map at a high confidence rate (4, 27). Using an LTQ Orbitrap Velos and two search engines (MASCOT and PEAKS DB), we now sequenced over 900 peptides, each assigned with <1% false discovery rate, which corresponds to a p value between 0.01 and 0.05 (Fig. 1 and supplemental Table S1, a–c, e, f, h, and j). Altogether, this analysis mapped naturally processed peptides deriving from over 480 proteins with different intra- and extracellular locations. These peptides derived from mitochondria, ER and Golgi enzymes, and other proteins, with cytosolic proteins, nuclear proteins, and peptides from extracellular proteins playing important roles as chaperones and in innate immune responses (Fig. 1 and supplemental Table S1, a–c, e–h, and j). Several additional naturally processed peptides derived from plasma membrane proteins and surface receptors were also identified (Fig. 1 and supplemental Table S1, a–c, e–h, and j).
FIGURE 1.

MS/MS characterization of the lymph peptidome. a, scattergram of the lymph peptidome. The x axis shows peptide amino acid length and the y axis the number of unique peptides for each of the amino acid lengths. Only peptides with a PEAKS score above identity (above a 1% FDR) are reported and analyzed further. b, examples of MS/MS fragmentation of peptides with a PEAKS score above identity. c, pie chart showing the ingenuity pathway analysis of the protein functions comprised in the lymph peptidome. d, list of the subcellular location of proteins comprised in the lymph peptidome, reported as a percentage of the total proteins.
The chemical diversity of the lymph peptidome was expanded by searching for a variety of PTMs. As shown in supplemental Table S1j the self-peptidome carried by the lymph and generated under physiological conditions contains many PTM, of which oxidation on many amino side chains (34%) is the leading chemical change followed by deamidation (22%), dehydration (12%), acetylation (7%), dimethylation (5%), citrullination (5%), amidation (4%), trimethylation (2%), and phosphorylation (1%). Although it is possible that some of these PTM are generated during sample preparations (Met and Trp oxidation, among others, and deamidation and dehydration), methylation, amidation, citrullination, and phosphorylation are likely to represent in vivo modifications (supplemental Table S1j). These PTM could further extend the diversity of self-peptides presented by MHC II molecules under physiological conditions.
Processing Enzymes
As a next step we sought to identify the proteolytic enzymes involved in the generation of the naturally processed peptides found in the lymph. Although algorithms are available to predict potential protease cleavage sites, we instead consulted two databases of experimentally determined cleavage sites and considered only sites where cleavage has been verified by N-terminal sequencing or MS/MS or SILAC-based experiments (58, 59). This analysis identified 45 peptides for which cleavage sites had been mapped previously (supplemental Table S1g). Several of these peptides are shown in Fig. 2a. Overall a wide variety of proteases were implicated in the generation of the lymph-bound peptidome (Fig. 2b), with MMPs and cathepsins highly represented followed by caspases and enzymes involved in the innate immune responses (angiotensin-converting enzyme, complement factor I, and granzymes) and enzymes of the coagulation cascade (thrombin, plasmin, and kallikreins).
FIGURE 2.

Processing pathways generating the lymph peptidome. a, examples of peptides for which the N- or C-terminal cleavage sites were identified using MEROPS and CutDB databases. b, pie chart showing the categories of enzymes involved in the processing of the lymph peptidome.
To determine the contribution of the lymph self-peptidome to the general MHC II peptidome displayed by antigen-presenting cells, we examined previously compiled lists of naturally processed peptides eluted from MHC II molecules (38, 40, 42–44, 66–71) to determine whether any of the peptides that we identified in the lymph had been eluted previously from MHC molecules. We found that 64 different peptides eluted previously from human MHC I (HLA-A) and human, murine, and chicken MHC II (HLA-DR, I-A, I-E, and B-L) shared either an MHC core epitope or an N- or C-terminal cleavage site. Eleven lymph peptides completely overlapped with MHC I- and MHC II-eluted peptides reported previously in the literature (supplemental Table S2).
Analysis of the in Vivo HLA-DR1-bound Peptidome of Dendritic Cells
Most of the studies published on MHC II peptidomes report a relatively small number of peptides (10–300) (38, 40, 42–44, 66–71) and most utilize cell populations expanded in vitro, where the antigen-processing machinery samples the in vitro culture medium rather than the native cellular environment. However, elutions from fresh cells recently also have been reported (43). To compare peptides present in human lymph and a human MHC II-peptidome, we wanted to identify the HLA-DR1-restricted self-peptidome presented by dendritic cells. To obtain sufficient material for analysis, we considered two options: either expand the human HLA-DR1+ monocytes in vitro or inject HLA-DR1-transgenic mice with an Flt3L-secreting tumor to greatly expand in vivo the number of cDC (31, 54, 72). The drawback of the in vitro expansion is that the HLA-DR1 peptidome is in part derived from phagocytosed proteins and would include peptides derived from bovine serum rather than endogenous self-proteins. On the other hand, the HLA-DR1 elution from in vivo expanded cDC would identify endogenous mouse peptides presented by the human MHC II protein. Because from previous experimental data we knew that there is a substantial overlap between murine and human MHC II-eluted peptide orthologues, we decided to go with the Flt3L-induced cDC approach, including a comparative sequence and HLA-DR1 binding affinity analysis between identified mouse and human peptides (Fig. 3 and supplemental Table S3b).
FIGURE 3.
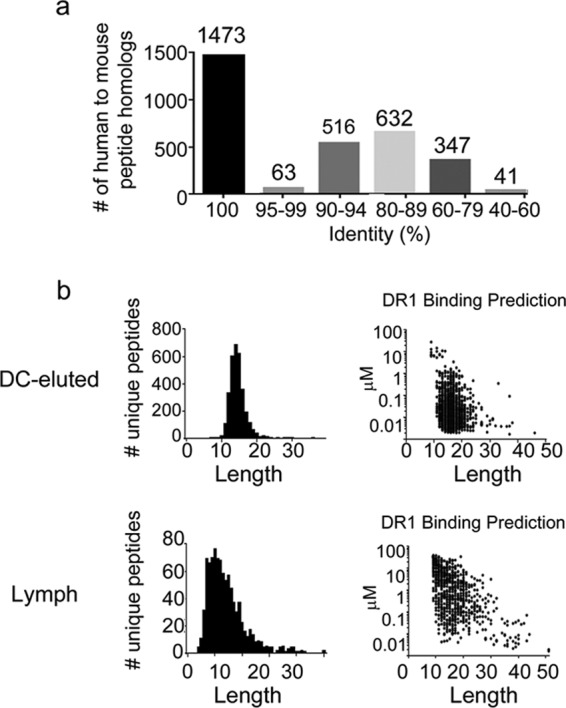
Comparative analysis between mouse HLA-DR1-eluted peptidome and human peptide orthologues. a, bar graph showing the number of human peptides identical to the mouse HLA-DR1-eluted peptidome. b, scattergram of the DC-eluted and lymph peptidome showing peptides number and length as well as prediction analysis of HLA-DR1 binding affinity the NetMHCIIPan 3.0 algorithms.
HLA-DR1 molecules were purified from isolated cDC by immunoaffinity chromatography, and MHC II-bound peptides were acid-eluted and analyzed by LC-MS/MS. Data from triplicate runs were processed and searched against a murine protein sequence database using a false discovery rate of 0.1%, resulting in a minimum peptide score, −10LgP of 34.00, corresponding to a p value of 0.0004. For a given score, this p value is the probability of having a false identification; the higher the score, the lower this probability (73). A total of 3216 unique peptides (supplemental Table S3, a and b) from 688 source proteins (supplemental Table S3c) were identified with an average score of 55 ± 12. The peptides have an average length of 16 residues (range: 7–44), which is consistent with the expected length distribution of class II peptides (Fig. 4a). Likewise, most of the peptides (85%) were identified as length variants (nested sets) consisting of a central core with N- and/or C-terminal extensions, which also is a characteristic feature of class II peptides. A mouse to human comparative amino acid sequence analysis for each of the HLA-DR1-eluted peptides (supplemental Table S3b, columns O, P, Q, and R), indicated that 1473 of the HLA-DR1-eluted peptidome overlap 100% between mouse and human homologs, and an additional 1211 peptides have 80–99% identity (Fig. 3a). More importantly, the sequence identity in the predicted HLA-DR1 core epitopes was 94% (Table S3b column L versus column S).
FIGURE 4.

MS/MS characterization of the MHC II peptidome eluted from cDC. a, scattergram of the MHC II-eluted peptidome. The x axis shows peptide amino acid length and the y axis the number of unique peptides for each amino acid length. Only peptides with a PEAKS score above identity are reported and analyzed further. b, list of the subcellular location of proteins comprising the MHC II-eluted peptidome, reported as a percentage of the total proteins. c, pie chart reporting the ingenuity pathway analysis of the protein functions comprised in the MHC II-eluted peptidome. d, pie chart showing the categories of enzymes involved in the processing of the MHC II-eluted peptidome.
To evaluate the effect of this sequence variation on HLA-DR1 binding, we used NetMHCIIpan to predict the binding affinity (65). (Fig. 3b and supplemental Table S3b, columns J– N versus columns S– W). Of the 895 independent core epitopes present in the eluted peptide pool, 784 were predicted by NetMHCIIpan to be strong binders for HLA-DR1 (IC50 < 50 nm). For the human orthologs of these 784 predicted strong binders, 748 also were predicted to be strong binders, with another 34 predicted to be weaker binders (IC50 < 500 nm). Only two human orthologs in this set (0.25%) were predicted not to bind to HLA-DR1. An additional 94 core epitopes from the eluted peptide pool were predicted to be weaker binders. For the human orthologs of these peptides, 11 were predicted to be strong binders and 76 weaker binders, with only 7 (0.9%) predicted not to bind.
To further characterize the HLA-DR1-eluted peptidome, we categorized source proteins according to cellular compartment and function (66). As expected, from a previously characterized MHC-II peptidome (43), the eluted peptides derived from a variety of intracellular compartments, including cytosol, mitochondria, endosomes/lysosomes, ER/Golgi, and nucleus, extracellular proteins comprised 11% of the total (Fig. 4b). As expected the peptides derived from proteins with diverse functions (Fig. 4c).
Protease cleavage sites, potentially responsible for generating these peptides, were identified as described above. Seventy-four peptide cleavage sites had been previously determined experimentally using either mass spectrometry or Edman degradation analysis of defined protease digestion reactions. Of these, roughly one-third of the peptides were processed by cathepsins at either the N or C terminus; another third was cleaved at the N terminus by a signal peptidase or another Ser-26 homolog, with the remainder cleaved by a metalloprotease, calpain, caspase, or other protease (Fig. 4d andsupplemental Table S3d). Thus, it appears that many proteases besides endosomal cathepsins can contribute to the formation of the MHC II-bound proteome.
Contribution of the Extracellular Fluid Degradome to the HLA-DR1 Peptidome
Next we asked whether the peptidome found in the lymph contributed to the overall HLA-DR1-bound peptidome (Fig. 3b). We selected the 39 proteins in which peptides were present in both the lymph and the eluted cDC peptidome (supplemental Table S4). Of the naturally processed peptides (391 MHC II-eluted and 208 in the lymph) derived from these 39 proteins, 17% of the dendritic cell-HLA-DR1-eluted peptides shared common sequences with peptides found in the lymph (supplemental Table S4). Among these peptides, 28 in the eluted peptidome shared a common N- or C-terminal cleavage site and three peptides completely overlapped (supplemental Table S4), further supporting the idea that some of the MHC II-bound peptides could be derived from the lymph (Fig. 5). In humans expressing a full complement of HLA-DP, HLA-DQ, and HLA-DR genes, an even greater overlap might be expected. One of the overlapping peptides derived from thymosin β4 (Fig. 5a), a soluble protein whose active peptide inhibits cell proliferation and is cleaved extracellularly by the angiotensin-converting enzyme. Another, derived from the serum complement protein C3 and known to be cleaved by the complement C1 cascade, was present as the identical peptide in lymph and eluted from HLA-DR (except for the S-I orthologous difference at the first position) (Fig. 5, b and c). Another well represented self-antigen was the HLA-DR1/I-E α-chain with 44 length variant peptides eluted from HLA-DR1 and six found in the human lymph (Fig. 5d). Peptides from the α-chain of MHC II molecules, reported previously to be highly present in the MHC II peptidome and even found in the urine (39, 42, 43, 74, 75), represent the target of the minor alloantigenic Y-Ae TCR.
FIGURE 5.
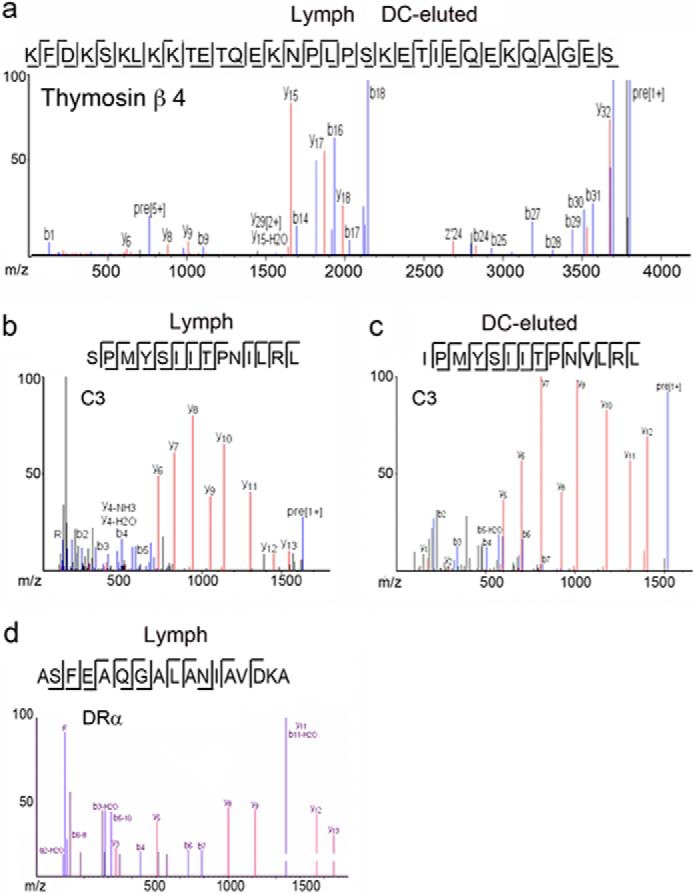
Comparison among the cDC, MHC II-eluted, and lymph peptidomes. a, MS/MS fragmentation of the thymosin β4 peptides found in the lymph and eluted from MHC II. b and c, MS/MS fragmentation of the C3 peptides found in the lymph and eluted from MHC II. d, MS/MS fragmentation of one of the I-Eα peptides found in the lymph and eluted from MHC II.
Validation of Complement 3, Collagen II, Gelsolin, and Thymosin β4 Peptide Processing
Three groups independently confirmed, in vitro (76–78) and in vivo (79, 80), that complement 1 is the protease that cleaves both the C and N termini of the R.SSKITHRIHWESASLL.R C3 peptide (Fig. 6, a and b). Additionally, it was also shown previously that in the absence of complement 1 (C1-KO mice) complement 3 is not cleaved (81). To determine whether endosomal proteases could produce the same C3 cleavage site, the following experiment was performed. Recombinant complement 3 was incubated for different time periods (1–3-6 h) with gradient-purified endosomal compartments isolated from dendritic cells (Fig. 6c) or with recombinant cathepsins B, D, L, and S, and the generated peptides were analyzed by mass spectrometry (Fig. 6d and supplemental Table S5). Altogether a total of 269 complement 3 peptides were sequenced; none of the analyzed sequences shared the same cleavage sites generated by complement 1 cleavage (supplemental Table S5). Additionally, because the R.SSKITHRIHWESASLL.R peptide was predicted to have a strong binding affinity (14.52 nm) for HLA-DR1 (supplemental Table S1h), a T cell proliferative assay was set up in HLA-DR1 transgenic mice crossed onto a I-A−/−, I-E−/−, and complement C3−/− KO background. Following C3 immunization, a proliferative response to the immunizing peptide (Fig. 6e) could be detected, confirming the presence of autoreactive T cells specific for different C3 determinants, as shown previously (82, 83).
FIGURE 6.

Analysis of complement 3 processing. a, sequence and MS/MS spectrum of the complement 3 peptide found in the human lymph. b, cleavage sites by complement 1 processing. c, β-hexosaminidase activity in fractions collected from a 27/10 Percoll gradient to identify endosomal compartments. d, Venn diagram of the total number of complement 3 peptides sequenced following processing by recombinant cathepsins or endosomal compartments. e, T cell proliferation in response to the complement 3 peptide following immunization of HLA-DR1 transgenic, I-A, I-E, and C3 knock-out mice.
An additional peptide that we had sequenced previously from the lymph peptidome (27) derives from the processing of collagen II (Fig. 7a). The N and C termini of this peptide were reported previously by two separate groups to be processed by MMPs (84, 85). Van den Steen et al. (84) report that MMP9, released from the supernatant of activated granulocytes, is the enzyme responsible for cleaving this collagen II peptide. Zhen et al. (85) also report that the same peptide was cleaved from fresh human cartilage upon processing by different MMPs. Additionally, the same peptide (Fig. 7a) was mapped as a B cell epitope capable of inducing an antibody response in a model of collagen-induced arthritis (86). To further determine that MMPs were responsible for cleaving the N terminus of the above referenced peptide, we prepared plasma membrane and endosomal compartments from freshly purified dendritic cells. Plasma membrane was purified with streptavidin beads following surface biotinylation (Fig. 7b). A series of Western blotting analyses was performed to verify: (i) surface biotinylation, (ii) lack thereof in the endosomal compartments, and (iii) the purity of the plasma membrane preparation as assessed by using a series of known ER and endosomal markers (Fig. 7, b and c). Additionally, the purified plasma membrane was also blotted for a series of MMPs known to be present in DC (Fig. 7d). Endosomal compartments were purified from the same dendritic cells using a 27/10 Percoll gradient (Fig. 6, c and d). Plasma membrane and endosomal preparations were incubated with collagen II for different time periods, and the digested collagen II peptides were analyzed by mass spectrometry (supplemental Table S6). Altogether 185 collagen II peptides were retrieved from either endosomal or plasma membrane processing. Among those, one peptide found previously in the lymph partially matched an endosomally cleaved peptide. On the other hand, four peptides previously mapped among the lymph peptidome matched to plasma membrane-cleaved peptides (supplemental Table S6), and among these was the MMP-processed peptide known to be a B cell epitope (84–86).
FIGURE 7.

Analysis of collagen II processing. a, sequence of the collagen II peptide found in the human lymph. b, Western blotting for membrane biotinylation in late endosomes (LE) and plasma membrane fractions (PM). c, Western blotting of ER, Golgi, and endosomal markers to check purity of plasma membrane preparation. Tot, total. d, Western blotting analysis for MMPs present in DC.
A third epitope that we analyzed is a gelsolin-derived peptide eluted from HLA-DR1 (Fig. 8a and supplemental Table S3) and reported previously to be cleaved by caspase-3 in apoptotic cells (87). To analyze gelsolin processing, dendritic cell cytosol, with our without active caspase-3, was fractionated (Fig. 8, b and c). As an additional control for gelsolin processing, late endosomes were also prepared from dendritic cells (Fig. 6, c and d). Gelsolin was incubated for 1, 4, and 8 h with the different cytosol preparations as well as late endosomal compartments, and processed peptides were mapped by mass spectrometry. A total of 69 peptides were retrieved following gelsolin processing by cytosolic caspase-3, and among the digested peptides the previously eluted HLA-DR1 peptide was also found (Fig. 8d and supplemental Table S7). On the other hand, the same peptide could not be retrieved following gelsolin digestion using cytosol preparation from caspase-3 knock-out mice (Fig. 8c and supplemental Table S7). 786 peptides were retrieved following gelsolin digestion by late endosomal compartment. None of the mapped peptides overlapped with the one cleaved by caspase-3 and eluted from HLA-DR1 (supplemental Table S8). Finally, a peptide from thymosin β4, found in the human lymph and eluted from the HLA-DR1 molecule (Fig. 5a), was validated to be processed in endosomal compartments (supplemental Table S8).
FIGURE 8.

Analysis of gelsolin processing. a, sequence of the gelsolin peptide eluted from HLA-DR1. b, phase contrast microscopy of control and apoptotic DC. c, Western blotting analysis of caspase-3 in the cytosolic fractions purified from control (Ctr) and apoptotic DC and caspase-3 knock-out dendritic cells. DMSO, dimethyl sulfoxide. d, MS/MS spectra of the gelsolin peptides found in the caspase-3-processed cytosol.
DM-sensitive and DM-resistant Peptides in the HLA-DR1 Peptidome
To further analyze the biological significance of the MHC II-eluted and the lymph-bound peptidome, we determined the HLA-DR1 affinity and susceptibility to HLA-DM editing of several high-abundance peptides from the MHC-eluted DC and lymph proteomes. DM susceptibility was assayed using a novel kinetic competition assay (61), where peptides susceptible to DM editing exhibit large ΔIC50 values when assayed early in the binding reaction. For example, IPMYSIIPNVLRL, derived from complement 3 and found in both lymph and MHC II-eluted peptides, was highly DM-resistant, with a ΔIC50 value near zero, whereas the SSKITHRIHWESASLLR peptide, derived from the same source but found only in lymph, was DM-sensitive with a ΔIC50 value of 0.06 μm (Fig. 9a and Table 1). Of the MHC II α-chain-derived peptides, both MHC II-eluted (EEFGRFASFEAQGALA) and lymph (ASFEAQGALANIAVDKA) peptides were DM-sensitive, but the lymph peptide was much more so, with ΔIC50 values of 0.07 and 3.4 μm, respectively (Fig. 9b and Table 1). These values can be compared with those of HA, a classic DM-resistant antigenic peptide from influenza HA (ΔIC50 = 0.03 μm) and CLIP, the canonical DM-sensitive peptide derived from the invariant chain MHC chaperone Ii (ΔIC50 = 0.12 μm) (Fig. 9c and Table 1). For 13 high-abundance peptides eluted from MHC II, DM susceptibility varied from lower than HA to higher than CLIP (Fig. 9d and Table 1). These data show that the MHC II peptidome is composed of both DM-sensitive and -resistant epitopes. For nine peptides found in lymph, there was no statistically significant difference in MHC binding affinity, whereas the lymph peptides were substantially more DM-sensitive.
FIGURE 9.

DM susceptibility of DR1 complexes with selected DC-eluted and lymph peptides. DM susceptibility was measured using a kinetic inhibition assay (113) and reported as ΔIC50, i.e. IC50 in the presence of DM minus IC50 in the absence of DM, measured after a 24-h incubation. DR1 binding inhibition was measured at 24 h in the presence (+) and absence (−) of DM for: a, C3 peptides, b, MHC IIa peptides, and c, classic DM-resistant (HA) and DM-sensitive (CLIP) peptides. Peptide sequences with core epitopes underlined are shown above the plots, with values for equilibrium binding affinity (IC50 at 72 h) and DM susceptibility (ΔIC50 at 24 h). d, summary of binding affinity (IC50) and DM susceptibility (ΔIC50) values for 13 peptides eluted from DCs, for nine peptides identified in lymph, and for replicate measurements of HA and CLIP. Statistical analysis was done using the Mann-Whitney test.
TABLE 1.
Binding affinities (IC50 in nm) for the recombinant DR1-B0101 MHC II of the selected peptides eluted from human MHC II (DR1-B0101) and from human lymph in the absence (−) or presence (+) of DM
The reported data sets were used to generate the graphs and statistical analyses in Fig. 6. Shown are the average ± S.D. and number of replicates (in parentheses). *, these peptides were reported as part of human the lymph peptidome sequenced at low resolution using the nanoLC-MS/MS on LTQ-ion trap (27).
| Peptide source | Peptide identified | Peptide synthesized | IC50 − DM | IC50 + DM | Protein ID |
|---|---|---|---|---|---|
| nm | nm | ||||
| DC | ALDFEQEMATAASSS | ALDFEQEMATAASSS | 116 ± 47 (3) | 112 ± 18 (3) | P60710 |
| DC | RDALNIETAVKTKG | RDALNIETAVKTKG | 283 ± 119 (3) | 274 ± 39 (3) | P07356 |
| DC | TGKLISLSAQNLVD | TGKLISLSAQNLVD | 18 ± 1 (3) | 14 ± 5 (3) | O70370 |
| DC | KGGSFQLLQGGQALE | GGSFQLLQGGQALE | 42 ± 8 (3) | 28 ± 9 (3) | P04186 |
| DC | SEKMFLSEESERSTDD | KMFLSEESERSTD | 896 ± 735 (3) | 721 ± 49 (3) | Q504P2 |
| DC | DENQFVAVTSTNAAK | NQFVAVTSTNAAK | 48 ± 9 (3) | 41 ± 6 (3) | O08553 |
| DC | GEFIKASSIEARQ | GEFIKASSIEARQ | 158 ± 52 (3) | 93 ± 18 (3) | P57759 |
| DC | NSNQFQTEVGKQLIS | NQFQTEVGKQLIS | 96 ± 21 (3) | 250 ± 90 (3) | P11835 |
| DC | SPNIVIALAGNKAD | NIVIALAGNKAD | 123 ± 34 (3) | 99 ± 34 (3) | P35278 |
| DC | VDKVIQAQTAYSANPA | KVIQAQTAYSANP | 53 ± 27 (3) | 87 ± 28 (3) | O08992 |
| DC | EAFQAMPPEELNK | EAFQAMPPEELNK | 292 ± 38 (3) | 452 ± 76 (3) | P82198 |
| DC | EEFGRFASFEAQGALA | EEFGRFASFEAQGALA | 50 ± 9 (2) | 120 ± 13 (2) | P01903.1 |
| DC | IPMYSIITPNVLRLESE | IPMYSIITPNVLRLESEET | 17 ± 1 (2) | 17 ± 3 (2) | P01027 |
| Lymph | ASFEAQGALANIAVDKA | ASFEAQGALANIAVDKA | 740 ± 109 (3) | 4142 ± 567 (3) | P01903 |
| Lymph | SSKITHRIHWESASLLR | SSKITHRIHWESASLLR | 53 ± 4 (3) | 110 ± 9 (3) | P01024 |
| Lymph | AGFKGEQGPKGEP | AGFKGEQGPKGEP | 4399 ± 243 (2) | 12641 ± 2897 (2) | P02458 |
| Lymph | MLHLLALFLH* | MLHLLALFLH | 7558 ± 3617 (2) | 19244 ± 1002 (2) | Q96CW9 |
| Lymph | PGALLGAPPPLVPAP* | PGALLGAPPPLVPAP | 2121 ± 152 (2) | 6809 ± 177 (2) | Q9HAH7 |
| Lymph | AWLDLEFISTVLGAP* | AWLDLEFISTVLGAP | 141 ± 7 (2) | 426 ± 48 (2) | P43026 |
| Lymph | RNMTLFSDLVAEKFI* | RNMTLFSDLVAEKFI | 2542 ± 203 (2) | 7413 ± 1225 (2) | P12110 |
| Lymph | EDTFAHLTPTPT* | EDTFAHLTPTPT | 221 ± 9 (2) | 783 ± 19 (2) | Q9NR99 |
| Lymph | KNFASVQGVSLESG* | KNFASVQGVSLESG | 242 ± 55 (2) | 341 ± 68 (2) | Q99715 |
| DM-resistant control | PKYVKQNTLKLAT | 63 ± 11 (4) | 88 ± 17 (4) | P03438 | |
| DM-susceptible control | VSKMRMATPLLMQ | 60 ± 12 (3) | 179 ± 28 (3) | P04233 |
Role of the HLA-DR1-bound Self-peptidome of Migratory cDC in Immunological Tolerance
Conventional DC comprise heterogeneous populations of DC that are divided into two major categories, resident and migratory cDC; the former reside in the spleen and lymph nodes, and the latter migrate from peripheral tissues to the draining lymph node and the thymus. cDC can load exogenously administered peptides for T cell presentation and induction of central and peripheral tolerance (32–35, 94–98, 122).
To evaluate a role for the HLA-bound self-peptidome presented by migratory cDC in central tolerance, we selected a self-peptide derived from the α-chain of MHC class II that contains binding registers for both I-Ab and HLA-DR1. The peptide sequence ASFEAQGALANIAVKDA is identical in I-Eα and HLA-DRα, is found in a common self-peptide eluted from I-Ab and HLA-DR1, and also is found in the human lymph. To study the role of this peptide in central tolerance, we used C57/Bl6 (H-2b) mice, which lack the Eα chain; thus T cells for this self-antigen escape thymic selection and are found in the periphery. We generated chimeric mice in which approximately half of the T cell population comprise endogenous T cells (CD90.2) and the other half (CD90.1) are T cells deriving from the bone marrow of a transgenic mouse bearing the TEα T cell receptor specific for the Eα peptide presented by I-Ab. We injected 50 nmol of the Eα peptide (intraperitoneally) and analyzed the thymus to evaluate deletion of T cells specific for the Eα peptide. A drastic reduction in the percentage of CD4+ T cells was observed in the transgenic TEα population (Fig. 10, a and b). As expected, the decrease in the endogenous polyclonal T cell population was smaller, yet still significant, due to the lower number of polyclonal T cells specific for the Eα antigen (Fig. 10, a and b). In a second set of experiments, we selected a peptide from factor VIII processed in the coagulation/fibrinolytic cascade, present in biological fluids, and shown previously to bind I-Ab. We injected this peptide in hemophilic mice deficient in factor VIII. As for the polyclonal T cell population, responsive to I-E peptide injection, the reduction of thymic CD4 T cells responsive to the factor VIII peptide was smaller but significantly present in all of the injected mice as compared with the controls (Fig. 10, c and d, and Supplement Fig. 1S).
The role of migratory DC is not limited to their ability to induce thymic negative deletion; they also are likely to be involved in generating CD25+ regulatory T cells. To test this hypothesis we selected four peptides known to bind HLA-DR1 and found previously in the human lymph. HLA-DR1 mice were depleted of CD25 regulatory T cells by CD25-specific antibody injection. This treatment is known to deplete regulatory T cells and induce an exaggerated autoimmune response, similar to what has been observed in FOXP3 knock-out mice. Indeed, nodal T cells from HLA-DR1 transgenic mice depleted of CD25+ regulatory T cells proliferated to each one of the selected peptides as compared with the nodal T cells from control mice (Fig. 10e). Proliferation was observed not only in nodal T cells but also among T cells that infiltrated parenchymal organs following depletion of the regulatory T cell population (Fig. 10f). These experiments indicate that naturally processed peptides found in biological fluids and eluted from MHC II molecules on cDC can play a role in generation and maintenance of immunological tolerance.
Discussion
During the last few years improved mass spectrometric analyses have determined that processed peptides are present in many biological fluids including serum, lymph, plasma, urine, and saliva (10, 11, 14, 21, 22, 88–91). The significant richness of these biological peptidomes reveals the ongoing metabolic processes of each parenchymal organ and has provided biomarkers of physiological and pathological processes (3, 5, 25, 27). Previously, our laboratory mapped the peptidome/degradome present in the human lymph under physiological conditions and showed that it consists of free peptides and peptides bound to different molecular chaperones (5, 27, 29). Here we report almost 1000 different peptide sequences found in human lymph, assigned with a false discovery rate of less than 1%.
For many of these peptides we were able to determine the processing enzymes involved in their generation by comparing our mapped peptidome with databases of experimentally characterized protein cleavage sites (58, 59). The processing pathways identified were involved in extracellular matrix degradation, surface receptor cleavage/editing, endosomal processing, and cellular apoptosis, revealing the overall diversity of the byproducts of parenchymal metabolic/catabolic processes collected in the lymph. Indeed, almost one-third of the mapped peptides contained one or more cleavage sites that were not derived from classical endosomal processing. Apoptotic cells are present in the lymph as byproducts of cellular turnover of different parenchymal organs, and caspase-processed peptides likely derive from these cells (52). Other peptides contained at least one matrix metalloprotease or calpain cleavage site, indicating their potential source from extracellular matrix remodeling processes. Peptides derived from blood proteolytic cascades, including complement processing, kinine systems, and coagulation/fibrinolytic systems, also were found.
Three peptides were validated by in vitro processing assays. The complement 3 peptide R.SSKITHRIHWESASLL.R was reported previously by several groups to be processed by complement 1 as part of the complement extracellular cascade (76–78, 80). It was also shown previously that complement 1 KO mice do not cleave this complement 3 epitope (81). Our experimental data confirm that the C3 peptide found in the human lymph and eluted from HLA-DR1 is cleaved by C1 but not by endosomal proteases. A collagen II peptide previously mapped in the human lymph (27) was originally reported by two separate groups to be processed by MMPs either released from activated granulocytes or present in the synovial fluid (84, 85). The same peptide was also mapped as a B cell epitope (86). Our experimental data confirmed that MMPs, but not endosomal proteases, were responsible for cleaving the formerly reported MMP-processed peptide. Finally, an HLA-DR1-eluted gelsolin-derived peptide was reported previously to be cleaved by caspase-3 in apoptotic cells (87). We confirmed experimentally that caspase-3, but not endosomal proteases, cleaved gelsolin to generate this peptide.
The snapshot presented here is limited by the paucity of experimentally determined protease cleavage sites and may be skewed by the fact that some processing pathways have been better characterized than others (e.g. caspases, cathepsins, and MMPs). However, it clearly indicates that several categories of protease/peptidase in additional to those present in endosomal/lysosomal compartments are involved in the formation of the lymph peptidome.
The present study adds substantially to our understanding of the spectrum of peptides that are naturally processed and presented in the MHC II pathway. Previous studies of mouse and human cultured cell lines have suggested the presence of more than 500–2000 peptides bound to MHC II, as indicated by the number of masses detected (35, 92–98), However, most masses could not be assigned to particular amino acid sequences. Here we have reported more than 3000 HLA-DR1-bound peptides. As we were not able to obtain sufficient amounts of human DC from lymph nodes or spleen to elute peptides from endogenous human HLA-DR1, we elected to use HLA-DR1 transgenic mice as the best alternative source of MHC II protein expressed in its natural milieu. This same approach was used previously by the Steinman group (43) to elute peptides from I-Ab+ dendritic cells. Another option would have been to differentiate in vitro cultures of HLA-DR1+ monocytes. However, in such case eluted peptides derived from phagocytosed proteins would have been from bovine serum and not representative of the in vivo cell milieu. In fact, bovine serum peptides have been identified in earlier studies of MHC II-bound peptides eluted from in vitro cell cultures, particularly where de novo sequencing rather than database matching was used for peptide identification (39, 74, 99).
A mouse to human comparative amino acid sequence analysis for each of the HLA-DR1-eluted peptides identified that almost 1500 HLA-DR1-eluted mouse peptides overlap 100% with their human orthologue. Additionally, the HLA-DR1 binding affinity for the human orthologues of each eluted mouse DC peptide indicates that even for many of the peptides that have only 80–90% mouse to human homology, the binding affinity is the same when the MHC II binding core is conserved. This analysis suggests that identification of self-antigens present in the HLA-DR1 peptidome from transgenic mice can inform our understanding of the native human HLA-DR1 peptidome.
As expected from previous work, the peptidome derives from both intracellular and extracellular sources. Additionally, as a new finding it appears that many proteases from various non-lysosomal intracellular and extracellular compartments contribute to the shaping of the MHC II peptidome.
An important novel finding of our research is the demonstration that the peptidome found in biological fluids can contribute to the surface MHC II-eluted peptidome. Indeed, we identified lymph-carried peptides sharing overlapping MHC-peptide binding registers and/or N- or C-terminal cleavage sites with peptides eluted previously from human, mouse, and ovine MHC molecules (38, 40, 42–44, 66–71). Additionally, the comparison between the HLA-DR1-eluted peptidome and the human lymph peptidome revealed 39 overlapping proteins, representing ∼10% of the total number of different source proteins for peptides eluted from HLA-DR1 or identified in lymph. Twenty-eight of the peptides derived from the processing of these proteins either contain an overlapping binding register or share the same N- and C-terminal cleavage site. Thus, extracellular protein processing can contribute to the overall MHC II-presented peptidome.
For lymph to serve as a potential source of antigens for MHC II processing and loading pathways, sufficient peptidic material must be present. For the most abundant peptides, we previously estimated a concentration in the 100 nanomolar range (27), comparable to the binding affinities determined herein for several high-abundance eluted peptides and predicted for many others. Recently, a number of reports have analyzed the half-life and overall stability of peptides in biological fluids and shown that most peptides are bound to carrier proteins, including albumin, lipoproteins, immunoglobulins, and transthyretin. The peptides exist in equilibrium between their free and bound form, and their half-life varies between 1 and 20 days, directly proportional to the half-life of the carrier protein (3, 25). The prolonged half-life and continuing production of these carrier-bound peptides support an available amount of biological fluids far superior to the steady-state concentration of free peptide.
MHC II loading of these peptides could occur by surface peptide exchange of invariant chain or previously loaded peptides, by binding onto peptide receptive surface proteins (as reported previously for both MHC class I and class II molecules in permissive haplotypes), or by loading in early recycling/sorting endosomes (100–109). In support of this idea, we found that some of the peptides presented by DC under physiological conditions were susceptible to DM-mediated exchange, suggesting that they were generated through a processing pathway that did not include substantial exposure to DM (107, 109, 110). Although most peptides tested were relatively resistant to DM action, with DM resistance factors similar to or even greater than a high-affinity immunodominant viral epitope, several peptides were highly sensitive to DM action, with susceptibility values near that of CLIP, the canonical DM-susceptible fragment of the MHC II associated invariant chain removed by DM. It is likely that DM-susceptible peptides presented by DC were loaded into cellular compartments with lower DM action, such as recycling vesicles, early endosomes, or the cell surface, which might easily be accessed by peptides taken up from extracellular fluid. Indeed, the eluted peptide with the highest ΔIC50 values among those tested (EAFQAMPPEELNK, ΔIC50 = 0.160 μm) derived from transforming growth factor-β-induced protein IG-H3 (P82198), a secreted TGFβ3-induced protein that binds collagen in the extracellular matrix. Peptides derived from extracellular sources might load onto MHC II at the cell surface or intracellular locations with low DM activity and could represent a source of antigens for recognition by type B T cells (110). HLA-DM sensitivity and the fact that many of the MHC II-eluted peptides correspond to sequences known to be cleaved by extracellular proteases, such as complement 1, meprin, granzyme, MMPs, and ADAMS, point to the fact that the extracellular milieu/lymph is the likely source of many of the peptides eluted from HLA-DR1.
MHC II loading of exogenously provided peptides has been shown previously to be an effective way of achieving central and peripheral tolerance (33, 35, 51, 96, 110–117). Indeed, in the last 20 years there has been a growing number of studies showing that intrathymic, intravenous, and intraperitoneal injection of exogenous peptides are efficient ways to achieve immunological tolerance through mechanisms that include thymic deletion (35, 93–98). In all of these studies, nanomoles of the relevant peptide were injected to achieve thymic cell deletion or induction of peripheral tolerance. Previously published and unpublished work from our laboratory using quantitative mass spectrometry indicates that circulating peptides are present in the high picomolar to high nanomolar range (27). In our experiments we injected 50 nmol of the relevant peptides, close to the expected physiological range.
Recently, the role of migratory DC in transporting exogenously administered peptides to the thymus to mediate negative selection and/or to lymph nodes to mediate peripheral T cell anergy and Treg differentiation has also been reported (33, 118). There is an extensive literature showing that migratory DC are the only exogenous APC that can enter the thymic organ (35, 119). Circulating free peptides cannot reach the thymus as such, as the thymus is the only immune organ that has efferent but not afferent lymphatic circulation (120, 121). We suggest that many or all of the self-peptides presented by DC and derived from exogenous sources after extracellular processing might participate in maintaining peripheral tolerance to these antigens. For proteins that are present predominately in the extracellular matrix or bodily fluids, this pathway might predominate over conventional intracellular processing pathways. Over 20 years ago, Stockinger and colleagues (51, 119) reported that a complement C5 peptide injected intraperitoneally could induce deletion of CD4+ thymocytes. Using C5 knock-out mice, the same group confirmed that the peptide was carried by antigen-presenting cells from the periphery to the thymus. We observed several complement peptides in both lymph-carried and MHC-eluted peptidomes. More recently many other groups (33–35) have demonstrated that migratory DC are responsible for loading exogenous peptides into the periphery and carrying them to the thymus to achieve central tolerance to proteins that are not normally expressed there. The Steinman group (122) demonstrated that the migratory DC are also important for peripheral tolerance. However, all of the above mentioned studies relied on exogenous administered peptides. Our work identifies, for the first time, the lymph peptidome as a likely source of endogenous antigens involved in central and peripheral tolerance. In general it seems reasonable that for maintenance of central and peripheral tolerance to the highest possible number of self-peptides, it would be important that different processing pathways be involved. Non-endosomal enzymes would add to the number of epitopes cleaved by cathepsins, generating a wider peptides repertoire. This, in conjunction with the HLA-DM-dependent and -independent loading pathways, ensures that a broader self-peptidome would be presented.
Extracellularly processed peptides also have also been implicated in the generation of autoimmunity (110, 116, 123). The difference between tolerance and autoimmunity outcome likely will be determined by differences in peptide MHC II binding affinity/stability, thymic selection, Treg induction, peptide generation by de novo processing pathways, and the nature of the presenting APC. MHC II-peptide binding affinity and stability vary substantially for different peptides, with high-affinity peptides having dissociation lifetimes of >100 h and low-affinity peptides rapidly lost in minutes. In the thymus it has long been shown that the high-affinity peptides are the ones that induce negative selection and generate regulatory T cells (124–126). The low-affinity peptides escape thymic selection. Thus it is likely that low-affinity peptides, even when loaded on migratory DC and transported to the thymus, will not be able to generate central tolerance. These may correspond to type B peptides that can drive autoimmunity when they are locally processed in high concentrations, thus overcoming the low MHC II binding affinity with a high copy number (116).
Another important aspect of the balance between tolerance and autoimmunity may be the activity of extracellular processing pathways. Herein we mapped the lymph peptidome and the extracellular pathways involved in its processing under non-inflammatory physiological conditions. However, under inflammatory conditions many proteolytic enzymes are up-regulated, and non-enzymatic oxidative cleavage has also been observed. Moreover, post-translational modifications associated with many pathological conditions occur during oxidative stress and would further expand the diversity of the peptidome available to the MHC II. Thus, the extracellular peptidome generated during inflammatory conditions can be quantitatively and qualitatively different from the one generated by metabolic activities during physiological conditions. Mapping the disease-associated post-translational modifications on the lymph peptidome and their additional processing pathways potentially could further expand our understanding of the immunological responses acquired under diverse pathological conditions.
Author Contributions
C. C. C., A. B., L. Y., V. Z., S. A. S., S. M., and A. F. performed the experiments; C. C. C., A. B., S. A. S., L. J. S., L. S., L. H., and A. F. analyzed the data; and C. C. C., A. B., L. J. S., and L. S. wrote the manuscript.
Supplementary Material
Acknowledgments
We acknowledge the support of the Laboratory for Macromolecular Analysis and Proteomic (under National Institutes of Health Grant NIH-1S10RR019352), Albert Einstein College of Medicine, and the Proteomics and Mass Spectrometry Facility, University of Massachusetts Medical School.
This work was supported by National Institutes of Health Grants NIH-AG045223 (to L. S.), NIH-AI38996 (to L. J. S.), and NIH-AI48833 (to L. J. S.). The authors declare that no competing financial interests exist. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Fig. S1 and Tables S1–S8.
- cDC
- conventional dendritic cell
- DC
- dendritic cell
- MMP
- matrix metalloproteinase
- ER
- endoplasmic reticulum
- UPLC
- ultra-performance liquid chromatography
- AGC
- automatic gain control
- PTM
- post-translational modification
- FP
- fluorescence polarization
- APC
- antigen presenting cells
- FDR
- false discovery rate
- TCR
- T cell receptor.
References
- 1. Ahn S. M., and Simpson R. J. (2007) Body fluid proteomics: prospects for biomarker discovery. Proteomics Clin. Appl. 1, 1004–1015 [DOI] [PubMed] [Google Scholar]
- 2. Anderson N. L., Polanski M., Pieper R., Gatlin T., Tirumalai R. S., Conrads T. P., Veenstra T. D., Adkins J. N., Pounds J. G., Fagan R., and Lobley A. (2004) The human plasma proteome: a nonredundant list developed by combination of four separate sources. Mol. Cell. Proteomics 3, 311–326 [DOI] [PubMed] [Google Scholar]
- 3. Antwi K., Hostetter G., Demeure M. J., Katchman B. A., Decker G. A., Ruiz Y., Sielaff T. D., Koep L. J., and Lake D. F. (2009) Analysis of the plasma peptidome from pancreas cancer patients connects a peptide in plasma to overexpression of the parent protein in tumors. J. Proteome Res. 8, 4722–4731 [DOI] [PubMed] [Google Scholar]
- 4. Clement C. C., Aphkhazava D., Nieves E., Callaway M., Olszewski W., Rotzschke O., and Santambrogio L. (2013) Protein expression profiles of human lymph and plasma mapped by 2D-DIGE and 1D SDS-PAGE coupled with nanoLC-ESI-MS/MS bottom-up proteomics. J. Proteomics 78, 172–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Clement C. C., Rotzschke O., and Santambrogio L. (2011) The lymph as a pool of self-antigens. Trends Immunol. 32, 6–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dzieciatkowska M., Wohlauer M. V., Moore E. E., Damle S., Peltz E., Campsen J., Kelher M., Silliman C., Banerjee A., and Hansen K. C. (2011) Proteomic analysis of human mesenteric lymph. Shock 35, 331–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fang J. F., Shih L. Y., Yuan K. C., Fang K. Y., Hwang T. L., and Hsieh S. Y. (2010) Proteomic analysis of post-hemorrhagic shock mesenteric lymph. Shock 34, 291–298 [DOI] [PubMed] [Google Scholar]
- 8. Goldfinch G. M., Smith W. D., Imrie L., McLean K., Inglis N. F., and Pemberton A. D. (2008) The proteome of gastric lymph in normal and nematode infected sheep. Proteomics 8, 1909–1918 [DOI] [PubMed] [Google Scholar]
- 9. Leak L. V., Liotta L. A., Krutzsch H., Jones M., Fusaro V. A., Ross S. J., Zhao Y., and Petricoin E. F. 3rd, and Fusaroa V. A.(2004) Proteomic analysis of lymph. Proteomics 4, 753–765 [DOI] [PubMed] [Google Scholar]
- 10. Li Y., Dang T. A., Shen J., Hicks J., Chintagumpala M., Lau C. C., and Man T. K. (2011) Plasma proteome predicts chemotherapy response in osteosarcoma patients. Oncol. Rep. 25, 303–314 [DOI] [PubMed] [Google Scholar]
- 11. Meng Z., and Veenstra T. D. (2007) Proteomic analysis of serum, plasma, and lymph for the identification of biomarkers. Proteomics Clin. Appl. 1, 747–757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mittal A., Phillips A. R., Middleditch M., Ruggiero K., Loveday B., Delahunt B., Cooper G. J., and Windsor J. A. (2009) The proteome of mesenteric lymph during acute pancreatitis and implications for treatment. JOP 10, 130–142 [PubMed] [Google Scholar]
- 13. Pieper R., Gatlin C. L., McGrath A. M., Makusky A. J., Mondal M., Seonarain M., Field E., Schatz C. R., Estock M. A., Ahmed N., Anderson N. G., and Steiner S. (2004) Characterization of the human urinary proteome: a method for high-resolution display of urinary proteins on two-dimensional electrophoresis gels with a yield of nearly 1400 distinct protein spots. Proteomics 4, 1159–1174 [DOI] [PubMed] [Google Scholar]
- 14. Ressom H. W., Varghese R. S., Abdel-Hamid M., Eissa S. A., Saha D., Goldman L., Petricoin E. F., Conrads T. P., Veenstra T. D., Loffredo C. A., and Goldman R. (2005) Analysis of mass spectral serum profiles for biomarker selection. Bioinformatics 21, 4039–4045 [DOI] [PubMed] [Google Scholar]
- 15. Shen Y., Liu T., Tolić N., Petritis B. O., Zhao R., Moore R. J., Purvine S. O., Camp D. G., and Smith R. D. (2010) Strategy for degradomic-peptidomic analysis of human blood plasma. J Proteome Res. 9, 2339–2346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shen Z., Want E. J., Chen W., Keating W., Nussbaumer W., Moore R., Gentle T. M., and Siuzdak G. (2006) Sepsis plasma protein profiling with immunodepletion, three-dimensional liquid chromatography tandem mass spectrometry, and spectrum counting. J. Proteome Res. 5, 3154–3160 [DOI] [PubMed] [Google Scholar]
- 17. Veenstra T. D., Conrads T. P., Hood B. L., Avellino A. M., Ellenbogen R. G., and Morrison R. S. (2005) Biomarkers: mining the biofluid proteome. Mol. Cell. Proteomics 4, 409–418 [DOI] [PubMed] [Google Scholar]
- 18. Xu X., and Veenstra T. D. (2008) Analysis of biofluids for biomarker research. Proteomics Clin. Appl. 2, 1403–1412 [DOI] [PubMed] [Google Scholar]
- 19. Zhao Y. S., Pang D., Wang F., Xue Y. W., Gao D. N., Li H., Li K., Wang B. Y., Wang D., and Li H. Y. (2009) Nipple aspirate fluid collection, related factors and relationship between carcinoembryonic antigen in nipple aspirate fluid and breast diseases in women in Harbin, PRC. Cancer Epidemiol. Biomarkers Prev. 18, 732–738 [DOI] [PubMed] [Google Scholar]
- 20. Zheng X., Wu S. L., Hincapie M., and Hancock W. S. (2009) Study of the human plasma proteome of rheumatoid arthritis. J. Chromatogr. A 1216, 3538–3545 [DOI] [PubMed] [Google Scholar]
- 21. Zhou M., Lucas D. A., Chan K. C., Issaq H. J., Petricoin E. F. 3rd, Liotta L. A., Veenstra T. D., and Conrads T. P. (2004) An investigation into the human serum “interactome.” Electrophoresis 25, 1289–1298 [DOI] [PubMed] [Google Scholar]
- 22. Zurawel A., Moore E. E., Peltz E. D., Jordan J. R., Damle S., Dzieciatkowska M., Banerjee A., and Hansen K. C. (2011) Proteomic profiling of the mesenteric lymph after hemorrhagic shock: differential gel electrophoresis and mass spectrometry analysis. Clin. Proteomics 8, 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhou M., Conrads T. P., and Veenstra T. D. (2005) Proteomics approaches to biomarker detection. Brief. Funct. Genomic. Proteomic. 4, 69–75 [DOI] [PubMed] [Google Scholar]
- 24. Ling X. B., Lau K., Deshpande C., Park J. L., Milojevic D., Macaubas C., Xiao C., Lopez-Avila V., Kanegaye J., Burns J. C., Cohen H., Schilling J., and Mellins E. D. (2010) Urine peptidomic and targeted plasma protein analyses in the diagnosis and monitoring Of systemic juvenile idiopathic arthritis. Clin. Proteomics 6, 175–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Geho D. H., Liotta L. A., Petricoin E. F., Zhao W., and Araujo R. P. (2006) The amplified peptidome: the new treasure chest of candidate biomarkers. Curr. Opin. Chem. Biol. 10, 50–55 [DOI] [PubMed] [Google Scholar]
- 26. Farrah T., Deutsch E. W., Omenn G. S., Campbell D. S., Sun Z., Bletz J. A., Mallick P., Katz J. E., Malmstrom J., Ossola R., Watts J. D., Lin B., Zhang H., Moritz R. L., and Aebersold R. (2011) A high-confidence human plasma proteome reference set with estimated concentrations in PeptideAtlas. Mol. Cell. Proteomics 10, M110 006353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Clement C. C., Cannizzo E. S., Nastke M. D., Sahu R., Olszewski W., Miller N. E., Stern L. J., and Santambrogio L. (2010) An expanded self-antigen peptidome is carried by the human lymph as compared to the plasma. PLoS One 5, e9863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shen Y., Tolić N., Liu T., Zhao R., Petritis B. O., Gritsenko M. A., Camp D. G., Moore R. J., Purvine S. O., Esteva F. J., and Smith R. D. (2010) Blood peptidome-degradome profile of breast cancer. PLoS One 5, e13133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Clement C. C., and Santambrogio L. (2013) The lymph self-antigen repertoire. Front. Immunol. 4, 424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liu K., and Nussenzweig M. C. (2010) Origin and development of dendritic cells. Immunol. Rev. 234, 45–54 [DOI] [PubMed] [Google Scholar]
- 31. Waskow C., Liu K., Darrasse-Jèze G., Guermonprez P., Ginhoux F., Merad M., Shengelia T., Yao K., and Nussenzweig M. (2008) The receptor tyrosine kinase Flt3 is required for dendritic cell development in peripheral lymphoid tissues. Nat. Immunol. 9, 676–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Randolph G. J., Ochando J., and Partida-Sánchez S. (2008) Migration of dendritic cell subsets and their precursors. Annu. Rev. Immunol. 26, 293–316 [DOI] [PubMed] [Google Scholar]
- 33. Goldschneider I., and Cone R. E. (2003) A central role for peripheral dendritic cells in the induction of acquired thymic tolerance. Trends Immunol. 24, 77–81 [DOI] [PubMed] [Google Scholar]
- 34. Sixt M., Kanazawa N., Selg M., Samson T., Roos G., Reinhardt D. P., Pabst R., Lutz M. B., and Sorokin L. (2005) The conduit system transports soluble antigens from the afferent lymph to resident dendritic cells in the T cell area of the lymph node. Immunity 22, 19–29 [DOI] [PubMed] [Google Scholar]
- 35. Bonasio R., Scimone M. L., Schaerli P., Grabie N., Lichtman A. H., and von Andrian U. H. (2006) Clonal deletion of thymocytes by circulating dendritic cells homing to the thymus. Nat. Immunol. 7, 1092–1100 [DOI] [PubMed] [Google Scholar]
- 36. Dongre A. R., Kovats S., deRoos P., McCormack A. L., Nakagawa T., Paharkova-Vatchkova V., Eng J., Caldwell H., Yates J. R. 3rd, and Rudensky A. Y. (2001) In vivo MHC class II presentation of cytosolic proteins revealed by rapid automated tandem mass spectrometry and functional analyses. Eur. J. Immunol. 31, 1485–1494 [DOI] [PubMed] [Google Scholar]
- 37. Delgado J. C., Escobar H., Crockett D. K., Reyes-Vargas E., and Jensen P. E. (2009) Identification of naturally processed ligands in the C57BL/6 mouse using large-scale mass spectrometric peptide sequencing and bioinformatics prediction. Immunogenetics 61, 241–246 [DOI] [PubMed] [Google Scholar]
- 38. Marrack P., Ignatowicz L., Kappler J. W., Boymel J., and Freed J. H. (1993) Comparison of peptides bound to spleen and thymus class II. J. Exp. Med. 178, 2173–2183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chicz R. M., Urban R. G., Lane W. S., Gorga J. C., Stern L. J., Vignali D. A., and Strominger J. L. (1992) Predominant naturally processed peptides bound to HLA-DR1 are derived from MHC-related molecules and are heterogeneous in size. Nature 358, 764–768 [DOI] [PubMed] [Google Scholar]
- 40. Röhn T. A., Boes M., Wolters D., Spindeldreher S., Müller B., Langen H., Ploegh H., Vogt A. B., and Kropshofer H. (2004) Upregulation of the CLIP self-peptide on mature dendritic cells antagonizes T helper type 1 polarization. Nat. Immunol. 5, 909–918 [DOI] [PubMed] [Google Scholar]
- 41. Muntasell A., Carrascal M., Serradell L., Veelen P., Verreck F., Koning F., Raposo G., Abián J., and Jaraquemada D. (2002) HLA-DR4 molecules in neuroendocrine epithelial cells associate to a heterogeneous repertoire of cytoplasmic and surface self-peptides. J. Immunol. 169, 5052–5060 [DOI] [PubMed] [Google Scholar]
- 42. Costantino C. M., Spooner E., Ploegh H. L., and Hafler D. A. (2012) Class II MHC self-antigen presentation in human B and T lymphocytes. PLoS One 7, e29805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bozzacco L., Yu H., Zebroski H. A., Dengjel J., Deng H., Mojsov S., and Steinman R. M. (2011) Mass spectrometry analysis and quantitation of peptides presented on the MHC II molecules of mouse spleen dendritic cells. J. Proteome Res. 10, 5016–5030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Muixí L., Contreras V., Collado J. A., Alexandre Y., Ballingall K., Bonneau M., Jaraquemada D., and Schwartz-Cornil I. (2012) Unraveling features of the natural MHC class II peptidome of skin-migrated dendritic cells. Int. Immunol. 24, 59–69 [DOI] [PubMed] [Google Scholar]
- 45. Savina A., and Amigorena S. (2007) Phagocytosis and antigen presentation in dendritic cells. Immunol. Rev. 219, 143–156 [DOI] [PubMed] [Google Scholar]
- 46. Anderson M. S., Venanzi E. S., Klein L., Chen Z., Berzins S. P., Turley S. J., von Boehmer H., Bronson R., Dierich A., Benoist C., and Mathis D. (2002) Projection of an immunological self-shadow within the thymus by the Aire protein. Science 298, 1395–1401 [DOI] [PubMed] [Google Scholar]
- 47. Strawbridge A. B., and Blum J. S. (2007) Autophagy in MHC class II antigen processing. Curr. Opin. Immunol. 19, 87–92 [DOI] [PubMed] [Google Scholar]
- 48. Sahu R., Kaushik S., Clement C. C., Cannizzo E. S., Scharf B., Follenzi A., Potolicchio I., Nieves E., Cuervo A. M., and Santambrogio L. (2011) Microautophagy of cytosolic proteins by late endosomes. Dev. Cell 20, 131–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dengjel J., Schoor O., Fischer R., Reich M., Kraus M., Müller M., Kreymborg K., Altenberend F., Brandenburg J., Kalbacher H., Brock R., Driessen C., Rammensee H. G., and Stevanovic S. (2005) Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. Proc. Natl. Acad. Sci. U.S.A. 102, 7922–7927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cavanagh L. L., and Von Andrian U. H. (2002) Travellers in many guises: the origins and destinations of dendritic cells. Immunol. Cell Biol. 80, 448–462 [DOI] [PubMed] [Google Scholar]
- 51. Zal T., Volkmann A., and Stockinger B. (1994) Mechanisms of tolerance induction in major histocompatibility complex class II-restricted T cells specific for a blood-borne self-antigen. J. Exp. Med. 180, 2089–2099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pang B., Neijssen J., Qiao X., Janssen L., Janssen H., Lippuner C., and Neefjes J. (2009) Direct antigen presentation and gap junction mediated cross-presentation during apoptosis. J. Immunol. 183, 1083–1090 [DOI] [PubMed] [Google Scholar]
- 53. Rosloniec E. F., Brand D. D., Myers L. K., Whittington K. B., Gumanovskaya M., Zaller D. M., Woods A., Altmann D. M., Stuart J. M., and Kang A. H. (1997) An HLA-DR1 transgene confers susceptibility to collagen-induced arthritis elicited with human type II collagen. J. Exp. Med. 185, 1113–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Dranoff G., Jaffee E., Lazenby A., Golumbek P., Levitsky H., Brose K., Jackson V., Hamada H., Pardoll D., and Mulligan R. C. (1993) Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc. Natl. Acad. Sci. U.S.A. 90, 3539–3543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Grubin C. E., Kovats S., deRoos P., and Rudensky A. Y. (1997) Deficient positive selection of CD4 T cells in mice displaying altered repertoires of MHC class II-bound self-peptides. Immunity 7, 197–208 [DOI] [PubMed] [Google Scholar]
- 56. Strug I., Calvo-Calle J. M., Green K. M., Cruz J., Ennis F. A., Evans J. E., and Stern L. J. (2008) Vaccinia peptides eluted from HLA-DR1 isolated from virus-infected cells are recognized by CD4+ T cells from a vaccinated donor. J Proteome Res. 7, 2703–2711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Huang da W., Sherman B. T., and Lempicki R. A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 [DOI] [PubMed] [Google Scholar]
- 58. Rawlings N. D., Morton F. R., and Barrett A. J. (2006) MEROPS: the peptidase database. Nucleic Acids Res. 34, D270–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Igarashi Y., Eroshkin A., Gramatikova S., Gramatikoff K., Zhang Y., Smith J. W., Osterman A. L., and Godzik A. (2007) CutDB: a proteolytic event database. Nucleic Acids Res. 35, D546–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Karosiene E., Rasmussen M., Blicher T., Lund O., Buus S., and Nielsen M. (2013) NetMHCIIpan-3.0, a common pan-specific MHC class II prediction method including all three human MHC class II isotypes, HLA-DR, HLA-DP, and HLA-DQ. Immunogenetics 65, 711–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yin L., Calvo-Calle J. M., Dominguez-Amorocho O., and Stern L. J. (2012) HLA-DM constrains epitope selection in the human CD4 T cell response to vaccinia virus by favoring the presentation of peptides with longer HLA-DM-mediated half-lives. J. Immunol. 189, 3983–3994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yin L., and Stern L. J. (2014) Measurement of peptide binding to MHC class II molecules by fluorescence polarization. Curr. Protoc. Immunol. 10.1002/0471142735.im0510s106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Busch R., Doebele R. C., von Scheven E., Fahrni J., and Mellins E. D. (1998) Aberrant intermolecular disulfide bonding in a mutant HLA-DM molecule: implications for assembly, maturation, and function. J. Immunol. 160, 734–743 [PubMed] [Google Scholar]
- 64. Stern L. J., and Wiley D. C. (1992) The human class II MHC protein HLA-DR1 assembles as empty αβ heterodimers in the absence of antigenic peptide. Cell 68, 465–477 [DOI] [PubMed] [Google Scholar]
- 65. Yin L., and Stern L. J. (2014) A novel method to measure HLA-DM-susceptibility of peptides bound to MHC class II molecules based on peptide binding competition assay and differential IC50 determination. J. Immunol. Methods 406, 21–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Fissolo N., Haag S., de Graaf K. L., Drews O., Stevanovic S., Rammensee H. G., and Weissert R. (2009) Naturally presented peptides on major histocompatibility complex I and II molecules eluted from central nervous system of multiple sclerosis patients. Mol. Cell. Proteomics 8, 2090–2101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bogunovic B., Srinivasan P., Ueda Y., Tomita Y., and Maric M. (2010) Comparative quantitative mass spectrometry analysis of MHC class II-associated peptides reveals a role of GILT in formation of self-peptide repertoire. PLoS One 5, e10599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Adamopoulou E., Tenzer S., Hillen N., Klug P., Rota I. A., Tietz S., Gebhardt M., Stevanovic S., Schild H., Tolosa E., Melms A., and Stoeckle C. (2013) Exploring the MHC-peptide matrix of central tolerance in the human thymus. Nat. Commun. 4, 2039. [DOI] [PubMed] [Google Scholar]
- 69. Espinosa G., Collado J. A., Scholz E., Mestre-Ferrer A., Kuse N., Takiguchi M., Carrascal M., Canals F., Pujol-Borrell R., Jaraquemada D., and Alvarez I. (2013) Peptides presented by HLA class I molecules in the human thymus. J. Proteomics 94, 23–36 [DOI] [PubMed] [Google Scholar]
- 70. Collado J. A., Alvarez I., Ciudad M. T., Espinosa G., Canals F., Pujol-Borrell R., Carrascal M., Abian J., and Jaraquemada D. (2013) Composition of the HLA-DR-associated human thymus peptidome. Eur. J. Immunol. 43, 2273–2282 [DOI] [PubMed] [Google Scholar]
- 71. Spencer C. T., Dragovic S. M., Conant S. B., Gray J. J., Zheng M., Samir P., Niu X., Moutaftsi M., Van Kaer L., Sette A., Link A. J., and Joyce S. (2013) Sculpting MHC class II-restricted self- and non-self peptidome by the class I Ag-processing machinery and its impact on Th-cell responses. Eur. J. Immunol. 43, 1162–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lindquist R. L., Shakhar G., Dudziak D., Wardemann H., Eisenreich T., Dustin M. L., and Nussenzweig M. C. (2004) Visualizing dendritic cell networks in vivo. Nat. Immunol. 5, 1243–1250 [DOI] [PubMed] [Google Scholar]
- 73. Zhang J., Xin L., Shan B., Chen W., Xie M., Yuen D., Zhang W., Zhang Z., Lajoie G. A., and Ma B. (2012) PEAKS DB: de novo sequencing assisted database search for sensitive and accurate peptide identification. Mol. Cell. Proteomics 11, M111 010587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Chicz R. M., Urban R. G., Gorga J. C., Vignali D. A., Lane W. S., and Strominger J. L. (1993) Specificity and promiscuity among naturally processed peptides bound to HLA-DR alleles. J. Exp. Med. 178, 27–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Sturm T., Leinders-Zufall T., Maček B., Walzer M., Jung S., Pömmerl B., Stevanović S., Zufall F., Overath P., and Rammensee H. G. (2013) Mouse urinary peptides provide a molecular basis for genotype discrimination by nasal sensory neurons. Nat. Commun. 4, 1616. [DOI] [PubMed] [Google Scholar]
- 76. de Bruijn M. H., and Fey G. H. (1985) Human complement component C3: cDNA coding sequence and derived primary structure. Proc. Natl. Acad. Sci. U.S.A. 82, 708–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Tack B. F., Morris S. C., and Prahl J. W. (1979) Third component of human complement: structural analysis of the polypeptide chains of C3 and C3b. Biochemistry 18, 1497–1503 [DOI] [PubMed] [Google Scholar]
- 78. Davis A. E. 3rd, and Harrison R. A. (1982) Structural characterization of factor I mediated cleavage of the third component of complement. Biochemistry 21, 5745–5749 [DOI] [PubMed] [Google Scholar]
- 79. Davis A. E., 3rd. (1983) The efficiency of complement activation in MHC-linked diseases. Immunol. Today 4, 250–252 [DOI] [PubMed] [Google Scholar]
- 80. Davis A. E. 3rd, Harrison R. A., and Lachmann P. J. (1984) Physiologic inactivation of fluid phase C3b: isolation and structural analysis of C3c, C3d,g (α2D), and C3g. J. Immunol. 132, 1960–1966 [PubMed] [Google Scholar]
- 81. Tüzün E., Li J., Saini S. S., Yang H., and Christadoss P. (2008) Targeting classical complement pathway to treat complement-mediated autoimmune diseases. Adv. Exp. Med. Biol. 632, 265–272 [PubMed] [Google Scholar]
- 82. Seward R. J., Drouin E. E., Steere A. C., and Costello C. E. (2011) Peptides presented by HLA-DR molecules in synovia of patients with rheumatoid arthritis or antibiotic-refractory Lyme arthritis. Mol. Cell. Proteomics 10.1074/mcp.M110.002477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. De Groot A. S., Ross T. M., Levitz L., Messitt T. J., Tassone R., Boyle C. M., Vincelli A. J., Moise L., Martin W., and Knopf P. M. (2015) C3d adjuvant effects are mediated through the activation of C3d-specific autoreactive T cells. Immunol. Cell Biol. 93, 189–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Van den Steen P. E., Proost P., Brand D. D., Kang A. H., Van Damme J., and Opdenakker G. (2004) Generation of glycosylated remnant epitopes from human collagen type II by gelatinase B. Biochemistry 43, 10809–10816 [DOI] [PubMed] [Google Scholar]
- 85. Zhen E. Y., Brittain I. J., Laska D. A., Mitchell P. G., Sumer E. U., Karsdal M. A., and Duffin K. L. (2008) Characterization of metalloprotease cleavage products of human articular cartilage. Arthritis Rheum. 58, 2420–2431 [DOI] [PubMed] [Google Scholar]
- 86. Lindh I., Snir O., Lönnblom E., Uysal H., Andersson I., Nandakumar K. S., Vierboom M., 't Hart B., Malmström V., and Holmdahl R. (2014) Type II collagen antibody response is enriched in the synovial fluid of rheumatoid joints and directed to the same major epitopes as in collagen induced arthritis in primates and mice. Arthritis Res. Ther. 16, R143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Kothakota S., Azuma T., Reinhard C., Klippel A., Tang J., Chu K., McGarry T. J., Kirschner M. W., Koths K., Kwiatkowski D. J., and Williams L. T. (1997) Caspase-3-generated fragment of gelsolin: effector of morphological change in apoptosis. Science 278, 294–298 [DOI] [PubMed] [Google Scholar]
- 88. Peltz E. D., Moore E. E., Zurawel A. A., Jordan J. R., Damle S. S., Redzic J. S., Masuno T., Eun J., Hansen K. C., and Banerjee A. (2009) Proteome and system ontology of hemorrhagic shock: exploring early constitutive changes in postshock mesenteric lymph. Surgery 146, 347–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Omenn G. S., States D. J., Adamski M., Blackwell T. W., Menon R., Hermjakob H., Apweiler R., Haab B. B., Simpson R. J., Eddes J. S., Kapp E. A., Moritz R. L., Chan D. W., Rai A. J., Admon A., Aebersold R., Eng J., Hancock W. S., Hefta S. A., Meyer H., Paik Y. K., Yoo J. S., Ping P., Pounds J., Adkins J., Qian X., Wang R., Wasinger V., Wu C. Y., Zhao X., Zeng R., Archakov A., Tsugita A., Beer I., Pandey A., Pisano M., Andrews P., Tammen H., Speicher D. W., and Hanash S. M. (2005) Overview of the HUPO Plasma Proteome Project: results from the pilot phase with 35 collaborating laboratories and multiple analytical groups, generating a core dataset of 3020 proteins and a publicly available database. Proteomics 5, 3226–3245 [DOI] [PubMed] [Google Scholar]
- 90. Mörbt N., Mögel I., Kalkhof S., Feltens R., Röder-Stolinski C., Zheng J., Vogt C., Lehmann I., and von Bergen M. (2009) Proteome changes in human bronchoalveolar cells following styrene exposure indicate involvement of oxidative stress in the molecular-response mechanism. Proteomics 9, 4920–4933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Masuno T., Moore E. E., Cheng A. M., Sarin E. L., and Banerjee A. (2006) Bioactivity of postshock mesenteric lymph depends on the depth and duration of hemorrhagic shock. Shock 26, 285–289 [DOI] [PubMed] [Google Scholar]
- 92. Schilling O., and Overall C. M. (2008) Proteome-derived, database-searchable peptide libraries for identifying protease cleavage sites. Nat. Biotechnol. 26, 685–694 [DOI] [PubMed] [Google Scholar]
- 93. Clare-Salzler M. J., Brooks J., Chai A., Van Herle K., and Anderson C. (1992) Prevention of diabetes in nonobese diabetic mice by dendritic cell transfer. J. Clin. Invest. 90, 741–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Oluwole S. F., Jin M. X., Chowdhury N. C., Engelstad K., Ohajekwe O. A., and Hardy M. A. (1995) Evidence for the role of host antigen-presenting cells in the induction of specific unresponsiveness to allografts by intrathymic inoculation of allopeptides. Transplant. Proc. 27, 132–133 [PubMed] [Google Scholar]
- 95. Khoury S. J., Gallon L., Chen W., Betres K., Russell M. E., Hancock W. W., Carpenter C. B., Sayegh M. H., and Weiner H. L. (1995) Mechanisms of acquired thymic tolerance in experimental autoimmune encephalomyelitis: thymic dendritic-enriched cells induce specific peripheral T cell unresponsiveness in vivo. J. Exp. Med. 182, 357–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Liblau R. S., Tisch R., Shokat K., Yang X., Dumont N., Goodnow C. C., and McDevitt H. O. (1996) Intravenous injection of soluble antigen induces thymic and peripheral T-cells apoptosis. Proc. Natl. Acad. Sci. U.S.A. 93, 3031–3036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Murphy K. M., Heimberger A. B., and Loh D. Y. (1990) Induction by antigen of intrathymic apoptosis of CD4+CD8+TCRlo thymocytes in vivo. Science 250, 1720–1723 [DOI] [PubMed] [Google Scholar]
- 98. Mamalaki C., Norton T., Tanaka Y., Townsend A. R., Chandler P., Simpson E., and Kioussis D. (1992) Thymic depletion and peripheral activation of class I major histocompatibility complex-restricted T cells by soluble peptide in T-cell receptor transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 89, 11342–11346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Rötzschke O., Falk K., Deres K., Schild H., Norda M., Metzger J., Jung G., and Rammensee H. G. (1990) Isolation and analysis of naturally processed viral peptides as recognized by cytotoxic T cells. Nature 348, 252–254 [DOI] [PubMed] [Google Scholar]
- 100. Chou C. L., Mirshahidi S., Su K. W., Kim A., Narayan K., Khoruzhenko S., Xu M., and Sadegh-Nasseri S. (2008) Short peptide sequences mimic HLA-DM functions. Mol. Immunol. 45, 1935–1943 [DOI] [PubMed] [Google Scholar]
- 101. Nygard N. R., Giacoletto K. S., Bono C., Gorka J., Kompelli S., and Schwartz B. D. (1994) Peptide binding to surface class II molecules is the major pathway of formation of immunogenic class II-peptide complexes for viable antigen presenting cells. J. Immunol. 152, 1082–1093 [PubMed] [Google Scholar]
- 102. Ploegh H. L. (1992) MHC products: biosynthesis, intracellular traffic, and “empty” molecules. Cold Spring Harb. Symp. Quant. Biol. 57, 565–570 [DOI] [PubMed] [Google Scholar]
- 103. Schumacher T. N., Heemels M. T., Neefjes J. J., Kast W. M., Melief C. J., and Ploegh H. L. (1990) Direct binding of peptide to empty MHC class I molecules on intact cells and in vitro. Cell 62, 563–567 [DOI] [PubMed] [Google Scholar]
- 104. De Bruijn M. L., Schumacher T. N., Nieland J. D., Ploegh H. L., Kast W. M., and Melief C. J. (1991) Peptide loading of empty major histocompatibility complex molecules on RMA-S cells allows the induction of primary cytotoxic T lymphocyte responses. Eur. J. Immunol. 21, 2963–2970 [DOI] [PubMed] [Google Scholar]
- 105. Santambrogio L., Sato A. K., Carven G. J., Belyanskaya S. L., Strominger J. L., and Stern L. J. (1999) Extracellular antigen processing and presentation by immature dendritic cells. Proc. Natl. Acad. Sci. U.S.A. 96, 15056–15061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Santambrogio L., Sato A. K., Fischer F. R., Dorf M. E., and Stern L. J. (1999) Abundant empty class II MHC molecules on the surface of immature dendritic cells. Proc. Natl. Acad. Sci. U.S.A. 96, 15050–15055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Villadangos J. A., Driessen C., Shi G. P., Chapman H. A., and Ploegh H. L. (2000) Early endosomal maturation of MHC class II molecules independently of cysteine proteases and H-2DM. EMBO J. 19, 882–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Haque M. A., Hawes J. W., and Blum J. S. (2001) Cysteinylation of MHC class II ligands: peptide endocytosis and reduction within APC influences T cell recognition. J. Immunol. 166, 4543–4551 [DOI] [PubMed] [Google Scholar]
- 109. Pu Z., Lovitch S. B., Bikoff E. K., and Unanue E. R. (2004) T cells distinguish MHC-peptide complexes formed in separate vesicles and edited by H2-DM. Immunity 20, 467–476 [DOI] [PubMed] [Google Scholar]
- 110. Lovitch S. B., Esparza T. J., Schweitzer G., Herzog J., and Unanue E. R. (2007) Activation of type B T cells after protein immunization reveals novel pathways of in vivo presentation of peptides. J. Immunol. 178, 122–133 [DOI] [PubMed] [Google Scholar]
- 111. Campbell J. D., Buckland K. F., McMillan S. J., Kearley J., Oldfield W. L., Stern L. J., Grönlund H., van Hage M., Reynolds C. J., Boyton R. J., Cobbold S. P., Kay A. B., Altmann D. M., Lloyd C. M., and Larché M. (2009) Peptide immunotherapy in allergic asthma generates IL-10-dependent immunological tolerance associated with linked epitope suppression. J. Exp. Med. 206, 1535–1547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Chowdhury N. C., Jin M. X., Hardy M. A., and Oluwole S. F. (1995) Donor-specific unresponsiveness to murine cardiac allografts induced by intrathymic-soluble alloantigens is dependent on alternate pathway of antigen presentation. J. Surg. Res. 59, 91–96 [DOI] [PubMed] [Google Scholar]
- 113. Gallegos A. M., and Bevan M. J. (2004) Central tolerance to tissue-specific antigens mediated by direct and indirect antigen presentation. J. Exp. Med. 200, 1039–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Oluwole S. F., Jin M. X., Chowdhury N. C., Engelstad K., Ohajekwe O. A., and James T. (1995) Induction of peripheral tolerance by intrathymic inoculation of soluble alloantigens: evidence for the role of host antigen-presenting cells and suppressor cell mechanism. Cell. Immunol. 162, 33–41 [DOI] [PubMed] [Google Scholar]
- 115. Shimomura K., Hardy M. A., and Oluwole S. F. (1995) Tolerance induction to cardiac allografts by simultaneous or sequential intrathymic inoculation of disparate alloantigens. Transplantation 60, 806–811 [PubMed] [Google Scholar]
- 116. Strong B. S., and Unanue E. R. (2011) Presentation of type B peptide-MHC complexes from hen egg white lysozyme by TLR ligands and type I IFNs independent of H2-DM regulation. J. Immunol. 187, 2193–2201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Martin S., and Bevan M. J. (1997) Antigen-specific and nonspecific deletion of immature cortical thymocytes caused by antigen injection. Eur. J. Immunol. 27, 2726–2736 [DOI] [PubMed] [Google Scholar]
- 118. Donskoy E., and Goldschneider I. (2003) Two developmentally distinct populations of dendritic cells inhabit the adult mouse thymus: demonstration by differential importation of hematogenous precursors under steady state conditions. J. Immunol. 170, 3514–3521 [DOI] [PubMed] [Google Scholar]
- 119. Volkmann A., Zal T., and Stockinger B. (1997) Antigen-presenting cells in the thymus that can negatively select MHC class II-restricted T cells recognizing a circulating self-antigen. J. Immunol. 158, 693–706 [PubMed] [Google Scholar]
- 120. Odaka C., Morisada T., Oike Y., and Suda T. (2006) Distribution of lymphatic vessels in mouse thymus: immunofluorescence analysis. Cell Tissue Res. 325, 13–22 [DOI] [PubMed] [Google Scholar]
- 121. Takeya K. (1970) Thymus-lymphatic system and immunity. Iryo 24, 684–692 [PubMed] [Google Scholar]
- 122. Idoyaga J., Fiorese C., Zbytnuik L., Lubkin A., Miller J., Malissen B., Mucida D., Merad M., and Steinman R. M. (2013) Specialized role of migratory dendritic cells in peripheral tolerance induction. J. Clin. Invest. 123, 844–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Lovitch S. B., Walters J. J., Gross M. L., and Unanue E. R. (2003) APCs present Aβ(k)-derived peptides that are autoantigenic to type B T cells. J. Immunol. 170, 4155–4160 [DOI] [PubMed] [Google Scholar]
- 124. Kappler J. W., Roehm N., and Marrack P. (1987) T cell tolerance by clonal elimination in the thymus. Cell 49, 273–280 [DOI] [PubMed] [Google Scholar]
- 125. Marrack P., McDuffie M., Born W., Blackman M., Hannum C., and Kappler J. (1987) The T cell receptor: its repertoire and role in thymocyte development. Adv. Exp. Med. Biol. 213, 1–12 [DOI] [PubMed] [Google Scholar]
- 126. Yin L., Scott-Browne J., Kappler J. W., Gapin L., and Marrack P. (2012) T cells and their eons-old obsession with MHC. Immunol. Rev. 250, 49–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.